
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO2-derived non-isocyanate polyurethanes (NIPUs) and their potential applications
Rita
Turnaturi
 a,
Chiara
Zagni
*a,
Vincenzo
Patamia
a,
Vincenzina
Barbera
b,
Giuseppe
Floresta
a and
Antonio
Rescifina
*a
a,
Chiara
Zagni
*a,
Vincenzo
Patamia
a,
Vincenzina
Barbera
b,
Giuseppe
Floresta
a and
Antonio
Rescifina
*a
aDepartment of Drug and Health Sciences, University of Catania, V.le A. Doria 6, 95125, Catania, Italy. E-mail: chiara.zagni@unict.it; arescifina@unict.it
bDepartment of Chemistry, Materials and Chemical Engineering “G. Natta”, Politecnico di Milano, Via Mancinelli 7, 20131 Milano, Italy
First published on 1st November 2023
Abstract
Using CO2 as feedstock to fabricate valuable products has become essential to green and sustainable chemistry and represents a rewarding challenge. Among the noticeable routes used to convert CO2 into synthetic polymers, this review highlights the reactions concerning the cycloaddition of epoxides with CO2 in cyclic carbonate as precursors for various forms of chemical synthesis, such as polycarbonates and polyurethanes (PUs). It is a fundamental challenge in polymer production to exploit biomass and CO2 as feedstock. PUs are one of the most versatile classes of polymeric materials that exhibit excellent properties. PUs are usually synthesized by a route involving the reaction of diols with diisocyanates derived from toxic phosgene gas. Non-isocyanate-derived polyurethane (NIPU) has been produced from cyclic carbonates and diamines without isocyanate. NIPU usually displays increased chemical resistance, lower permeability, improved water absorption, and thermal stability. In this review, we report the synthesis of several NIPUs for different applications in a more environmentally friendly manner, employing CO2 as a reagent and/or fully biobased reactants.
 Rita Turnaturi | Rita Turnaturi is a Researcher at the CNR in Catania. In 2013, she completed her doctoral studies in Pharmaceutical Sciences (medicinal chemistry, University of Catania). She discussed the thesis NEW PLAYERS IN AN OLD GAME: Design and Synthesis of Conformationally Constrained Compounds as New Tramadol-like Candidates. During the postdoc, she focused on designing, synthesizing, and characterizing compounds with a benzomorphan core. She carried out research at the Department of Chemical Sciences (Catania University), dealing with the synthesis of new systems for tumor cell targeting and the synthesis and characterization of nanosystems. She was a post-doctoral fellow for the Functionalization of cyclodextrins using substrates capable of increasing the CO2 absorption capacity. |
 Chiara Zagni | Chiara Zagni is a Researcher in Organic Chemistry at the University of Catania (Italy). She holds a master's degree in Chemistry and Pharmaceutical Technologies and a Ph.D. in Pharmaceutical Science. Her past research activity primarily focused on synthesizing novel compounds with potential applications as antiviral and anticancer drugs. She was a research fellow at the University of Michigan, where she studied the molecular mechanisms underlying the development and progression of cancer. She also joined the IPCB Institute of CNR, where she specialized in developing new polymeric materials for drug release, water purification, and CO2 absorption applications. Her primary research focus is creating innovative heterogeneous catalysts for organic reactions. |
 Vincenzo Patamia | Vincenzo Patamia completed his master's in Materials Chemistry in 2018 at the University of Catania. In 2022, he obtained his international Ph.D. in Chemical Sciences at the University of Catania, during which he worked on synthesizing supramolecular systems and porous materials for the adsorption and conversion of carbon dioxide and the catalysis of organic reactions in water. In mid-2022, the Italian Chemical Society selected him as a finalist for the Best Doctoral Thesis Award in Organic Chemistry. He is the author of 29 peer-reviewed publications. He is currently a Research Associate at the University of Catania, where he works on synthesizing chelating materials for catalytic and pharmaceutical applications. |
 Vincenzina Barbera | Vincenzina Barbera is a Professor in the Chemistry and Environmental Chemistry courses at Politecnico di Milano. She got her Ph.D. in Pharmaceutical Chemistry at the University of Catania with a thesis on Domino reactions. From 2014, she was a Postdoc Research Fellow at the Department of Chemistry, Material, and Chemical Engineering, “G. Natta”, Politecnico di Milano. She is the author of 44 peer-reviewed publications, inventor of 20 patent applications, and 15 families of patents. Her research topics range from synthetic organic chemistry to single-atom catalysis, materials for tires, and sustainable, flexible electronics in projects granted by Europe and companies. |
 Giuseppe Floresta | Giuseppe Floresta completed his PharmD in 2014 at the University of Catania. He then completed a Ph.D. in Chemistry at the University of Catania; during this period, he spent 11 months at the King's College London, where he started his interest in chelating agents, peptides, and radiochemistry. After one year of postdoc research at the University of Catania, he moved to King's College London, where he has been a postdoc research associate since September 2019. In May 2020, he was granted a Marie Curie post-doctoral Fellowship at King's College London. He is the author of more than 80 peer-reviewed publications. Today, he is a Lecturer and Researcher at the University of Catania, where he is involved in many scientific projects to synthesize novel pharmaceutical and imaging tools. |
 Antonio Rescifina | Antonio Rescifina received his M.Sc. degrees in Chemistry and Pharmaceutical Technologies (1989) and in Pharmacy (1991) at the University of Messina (Italy) and his Ph.D. in Chemical Sciences (1995) at the University of Catania (Italy). Subsequently, he was awarded a nine-month research contract by the University of Messina as part of the project BROMETHEIA. In 1996, he joined the University of Catania, where he is currently a Full Professor of Organic Chemistry. His current scientific interests focus on developing supramolecular nanoreactors for green and sustainable organocatalysis, studying the reactivity and the reaction mechanisms by computational chemistry, and CO2 recycling and utilization. |
1. Introduction
Polyurethanes (PUs) are versatile materials widely used in every field of daily life, from the building sector to medicine, thanks to their outstanding mechanical, ranging from flexible to hard materials, and physical properties.1 They are suitable for producing foams, adhesives, highly durable protective coatings, elastics, paints, and biomedical products.2At the industrial level, PUs are synthesized through the polyaddition reaction of various polyols, isocyanates, and chain extenders, which may also contain urethane bond linkages in the central skeleton structure, as well as other aliphatic and aromatic hydrocarbons, ethers, esters, imides, urea, and isocyanate groups.3 In 1849 was discovered that the urethane functional group (carbamate) could be obtained by reacting an isocyanate with a hydroxylated molecule.4 Although PUs are useful and versatile polymers, the starting materials usually used for their preparation are connected to global warming and health issues. Isocyanate derivatives, one of the two building blocks, are phosgene derivatives. Phosgene and isocyanate per se are highly toxic and harmful compounds. On the other hand, the common precursors for synthesizing polyols, ethylene, and propylene oxides are usually derived from petroleum resources.5 Finding greener approaches to PUs is now crucial given the REACH regulation, which establishes limitations on using chemicals containing free isocyanates known to cause asthma and dermatitis.6–8 Non-isocyanate polyurethane (NIPU) has attracted great interest from the industrial sector as a far more eco-friendly and nontoxic alternative to standard PU derived from petroleum resources and isocyanates.9 In this view, researchers have spent much effort finding a new synthetic process to produce these widely used materials in an eco-friendly and sustainable manner.
In 1957, Dyer and Scott discovered a method to produce PUs without moisture-sensitive isocyanates.10 Avoiding the isocyanate approach by reacting cyclic carbonates with amines to create the urethane group is feasible. Traditionally, diols and phosgene have been employed to make cyclic carbonate.11 Being phosgene, the chemical filrouge of the carbonate and carbamate preparations, its replacement is a crucial step. The possibility of replacing it with CO2 may be the right way because of its abundance, easy availability, and, most importantly, its low toxicity in nature. One of the most efficient examples of artificial CO2 fixation is the 100% atom-efficient production of cyclic carbonate via CO2 cycloaddition to epoxides. Converting CO2 into chemical products for which there is significant commercial demand represents one of the solutions to reduce CO2 atmospheric emissions responsible for increasing global warming.12,13 The International Panel on Climate Change (IPCC) predicts that, by the year 2100, the atmosphere may contain up to 570 ppm CO2, triggering a rise in the mean global temperature of around 1.9 °C. Consequently, there will be an increase in the mean sea level of up to 1 m by 2100, leading to increased desert formation and species extinction.14
Bio-based polyols, including propoxylated sugars, castor oil,15 and polyol intermediates provided by biorefineries, have been used to create PUs with high bio-based component contents. The possibility of increasing this sustainable character is currently being explored with the development of ways from biological CO2 fixation in biomass conversion to direct chemical fixation of CO2. Unlike isocyanates, cyclic carbonates are much less sensitive to moisture, enabling their facile long-term storage and manipulation.
The first part of this review will focus on synthesizing NIPU from cyclic carbonates formed by the reaction of epoxide with CO2 in the presence of various catalysts. The second section will focus on the synthesis of NIPU using entirely bio-based reagents, such as polyol precursors. In addition, the qualities, benefits, and applications of PUs in numerous disciplines were explored.
2. Synthesis of organic cyclic carbonate as the precursor of polyhydroxyurethanes
In the 1970s, CO2-based PUs were obtained by reacting CO2 with diamines under the catalysis of phosphate.16,17 The synthesis of PUs has now been described using several innovative CO2 utilization approaches, including the CO2-based cyclic carbonate route.18 However, CO2 is a thermodynamically stable molecule (ΔG = 394 kJ mol−1).19 Consequently, the pathways for the early preparation of CO2-based cyclic carbonate precursors need a catalyst and a specific temperature or pressure.It is essential to consider the source of the CO2 to be used for the production first of the carbonates and then of the NIPU. Aresta reported that the industrial applications of CO2 (quantity and source of CO2 used per year and duration of products) are in ever-increasing quantities.11 This infers that in a business as usual scenario, the use of CO2 in the chemical market will expand. In particular, for the production of carbonates (monomers and polycarbonates), a market of 2 Mt was estimated, and the quantity used until 2010 was only 0.2 Mt. The sources of CO2 were various; for example, CO2 recovered from power plants or industrial or natural reservoirs. Enhanced oil recovery (EOR) is a practice that can store CO2 for a long time while producing an economic benefit (improved oil extraction). EOR could easily double the total amount of CO2 used in just a few years. It is essential to match the question with the source, considering that the CO2 could be less pure than that extracted from a natural reservoir (i.e., a well) or produced from Ca and Mg carbonates. The cost of purifying CO2 can be high if traces of toxic substances need to be removed. The amount of CO2 used in industrial applications by compound class or use option varies from kt to Mt. There is no direct relationship between emissions reduction and the amount of CO2 used. In all cases, CO2 will be released with a different delay depending on the nature and use of each product. The topic of direct air capture (DAC) has recently received the scientific community's attention. Nearly a decade ago, the cost estimate for capturing CO2 from the air using traditional sorbents such as monoethanolamine (MEA) was approximately $600/tCO2, which remains highly uncertain.20 This highlights the need for research into high-performance sorbents that can dramatically reduce DAC costs. Researching to establish a practical lower limit for DAC is vitally important. The most commonly investigated approaches are the direct capture of CO2 from the air and subsequent conversion to produce fuels.21 However, other avenues of use are also possible, including chemical conversion to produce synthetic intermediates for pharmaceuticals.22
Many researchers have attempted a new approach with lower temperatures, lower pressures, and no catalysis in recent years. Only by going in this direction would it be possible to produce PUs with lower costs, less energy, and higher yields.
A solvent-free and catalyst-free CO2 indirect utilization route was settled for the synthesis of PUs starting from the alkyl carbonate salt.23 Alkyl carbonate salt of CO2 storage materials (CO2SMs) with the structure of –NH3+–O–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–O− could be simply prepared by fixation of CO2 in diamine and glycol systems (mole ratio = 1
O)–O− could be simply prepared by fixation of CO2 in diamine and glycol systems (mole ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at room temperature under atmospheric pressure.24 This easily prepared carbonate can be converted into carbamate groups (–N(H)–C(O)–O–) by inner-molecular dehydration (Fig. 1).23,25 Several PUs have been successfully synthesized using these starting materials at 90 °C and atmospheric pressure, employing DIC as a dehydrating agent. PUs produced by this method may have a certain potential in waterproof coatings as they are water-insoluble.23 The average molecular weight of the PUs produced by this method was between 10400 g mol−1 and 12700 g mol−1, and all of them were thermally stable below 142 °C while completely decomposed above 409 °C. The pre-capture and immobilization approach of CO2 avoids the need for its activation, significantly reducing energy consumption and enabling the synthesis of CO2-based PU at low temperatures and atmospheric pressure. This represents a crucial point for energy savings and emissions reductions.
:1) at room temperature under atmospheric pressure.24 This easily prepared carbonate can be converted into carbamate groups (–N(H)–C(O)–O–) by inner-molecular dehydration (Fig. 1).23,25 Several PUs have been successfully synthesized using these starting materials at 90 °C and atmospheric pressure, employing DIC as a dehydrating agent. PUs produced by this method may have a certain potential in waterproof coatings as they are water-insoluble.23 The average molecular weight of the PUs produced by this method was between 10400 g mol−1 and 12700 g mol−1, and all of them were thermally stable below 142 °C while completely decomposed above 409 °C. The pre-capture and immobilization approach of CO2 avoids the need for its activation, significantly reducing energy consumption and enabling the synthesis of CO2-based PU at low temperatures and atmospheric pressure. This represents a crucial point for energy savings and emissions reductions.
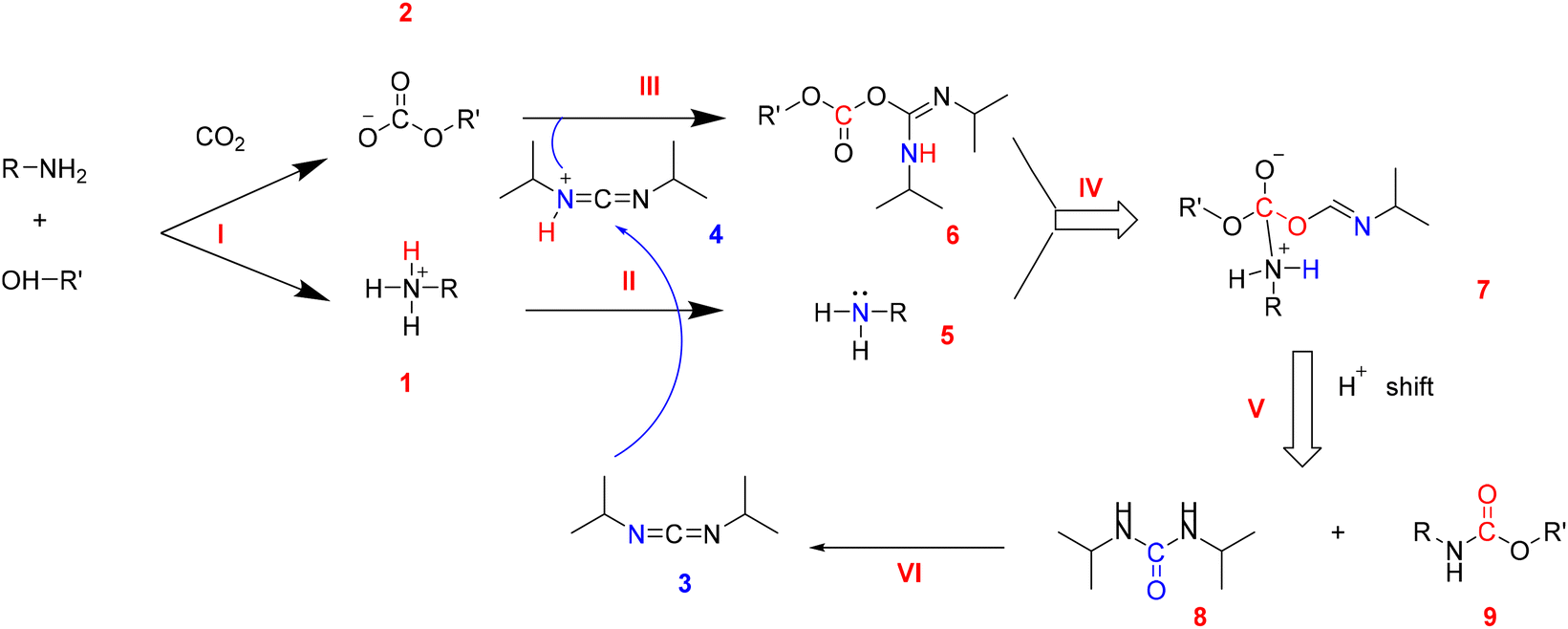 | ||
| Fig. 1 Reaction mechanism of PU by intermolecular dehydration via CO2-storage materials and DIC. | ||
Wulf et al.26 reported the preparation of a series of cyclic carbonates as monomers for NIPUs. They compared the efficiency of a Ca-based system and a bifunctional ammonium salt as catalysts for converting polyfunctional epoxides. Both catalytic systems have been shown to provide effective conversions; however, the Ca-based system27 is more active even at moderate temperatures, and the organocatalytic system is more moisture-resistant.28 The Ca-based system screening was performed to evaluate the catalytic efficiency of CaI2 in combination with six different crown ethers to convert the silyl-bridged bisepoxide to the corresponding biscarbonate. The combination of CaI2 with 18-crown-6 (L1, Fig. 2A), named Ca/L1, led to the desired product with a yield of 93% in ambient conditions (6 h, 23 °C, p(CO2) = 1 atm) that increases up to 98%, prolonging the reaction time at 24 h. Also, the catalytic performance of the organocatalyst tri-n-butyl-(2-hydroxyethyl)ammonium iodide (Fig. 2B) gave the silyl-bridged biscarbonate with a yield of 96%.
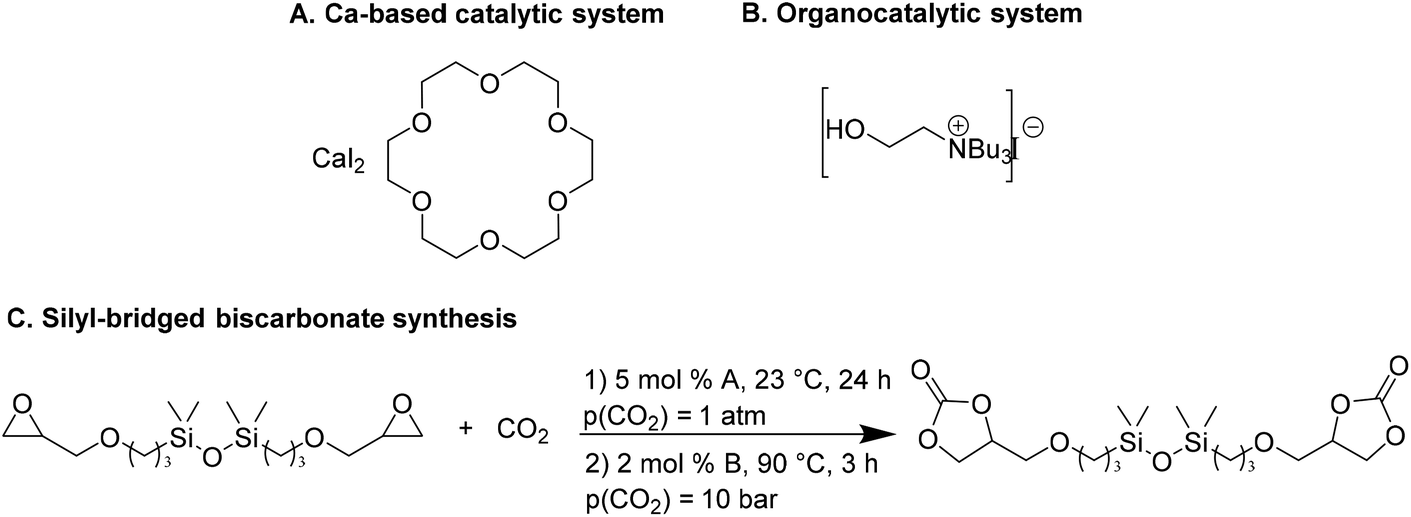 | ||
| Fig. 2 Structures of Ca-based (A) and organocatalytic (B) systems. (C) Silyl-bridged biscarbonate formation of Ca/L1 (A) and tri-n-butyl-(2-hydroxyethyl)ammonium iodide (B) catalyzed. | ||
The influence of both catalytic systems in converting several functionalized bis-epoxides (Fig. 3) to the corresponding carbonates was explored. Both catalytic systems gave high yields (90–98%) of the desired carbonates 11b, 11c, and 11f, with minor modifications in their reaction conditions. Unlike the Ca/L1 system, yields up to 90% were achieved for products 11d and 11e using the organocatalyst (95–98% and 68–79%, respectively).
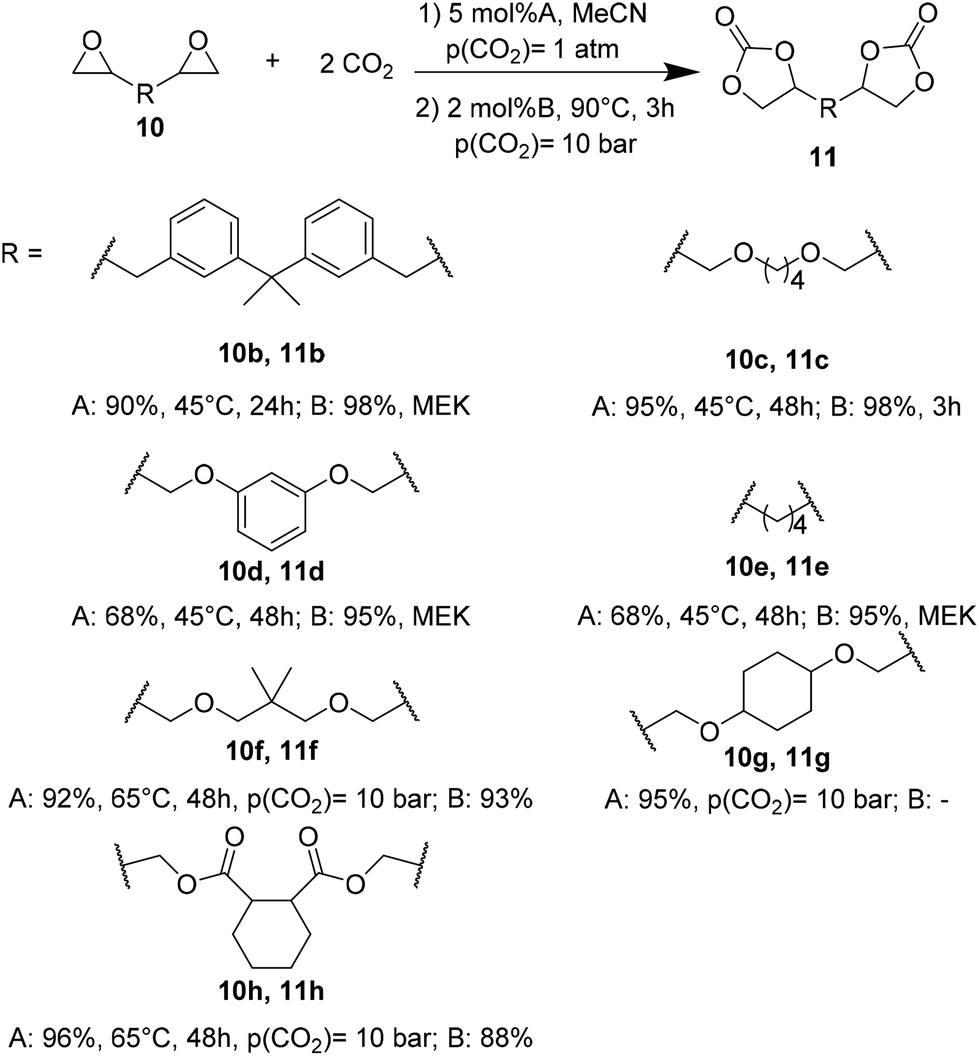 | ||
| Fig. 3 Structures of bis-epoxides and their corresponding bis-carbonates. | ||
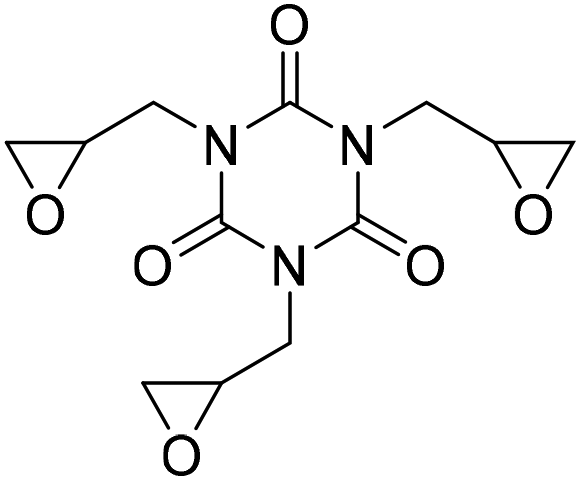
Ca/L1 and tri-n-butyl-(2-hydroxyethyl)ammonium iodide catalytic efficiencies were compared to prepare cyclic carbonates with polymeric backbones and polyfunctional carbonates (Fig. 4). The polyether-bridged biscarbonates 13i and 13j as well as the isocyanurate-bridged triscarbonate 13l (94–95%), bearing three carbonate moieties, were obtained with high yields (93–99% and 94–95%, respectively) by both catalytic systems. While the polypropylene-glycol diglycidyl ether and tetraglycidyl-substituted bisamine conversion in the corresponding carbonates 13k and 13n was addressed by adding only the organocatalyst, probably due to residual water in the respective substrates that deactivates CaI2.
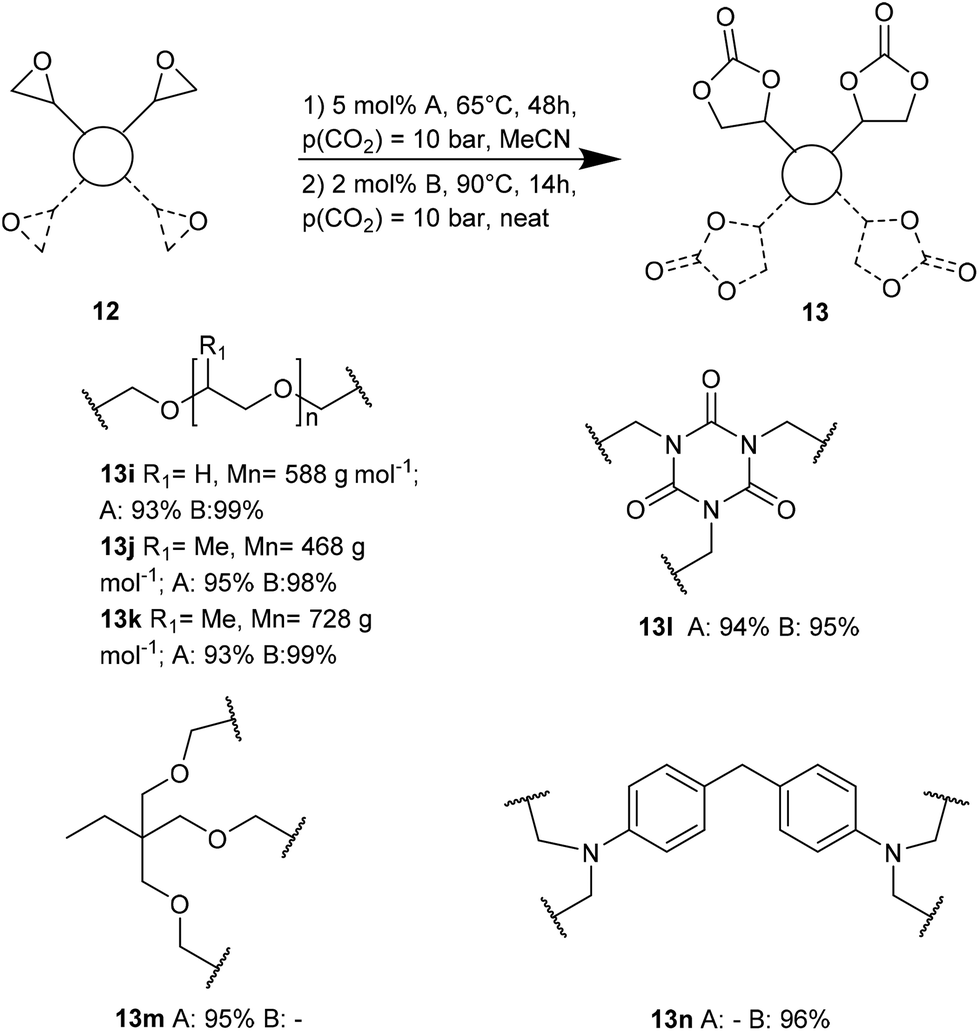 | ||
| Fig. 4 Cyclic carbonates with polymeric backbone and polyfunctional carbonates. | ||
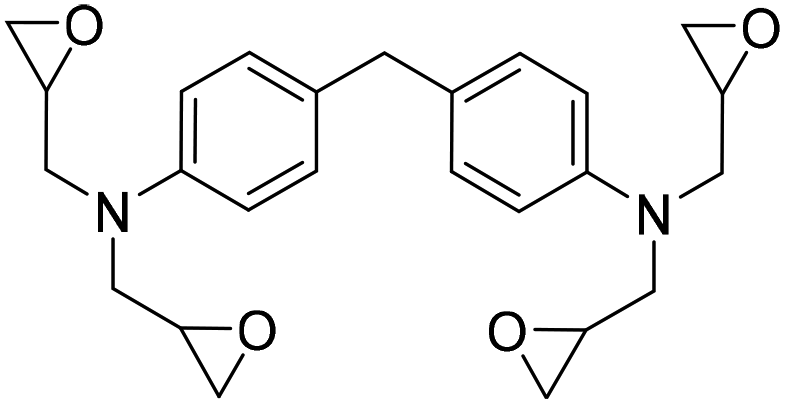
The authors also evaluated the influence of the catalyst systems on the two- and one-pot synthesis of NIPUs. In this regard, bisphenol A diglycidyl carbonate 11b, according to the literature,29,30 was chosen as a precursor for the NIPU synthesis and allowed to react with 1,4-diaminobutane (1,4-DAB, 14) in DMSO in the presence and absence of catalysts (Fig. 5). Compared to the uncatalyzed reaction, Ca/L1 and tri-n-butyl-(2-hydroxyethyl)ammonium iodide generated NIPU with a lower molecular mass, a low polydispersity index, and a minor glass transition temperature. Because of the difficulty of removing DMSO from the polymers, the same reactions were conducted using MeCN as the solvent. Also, in this case, the obtained polymers presented a molecular mass lower than that obtained with the uncatalyzed reaction.
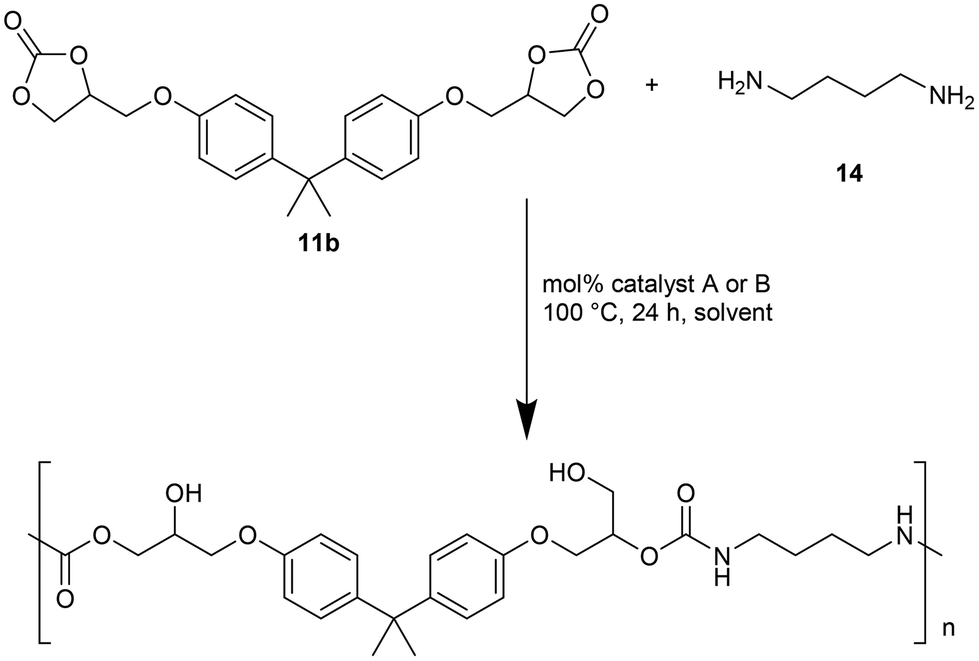 | ||
| Fig. 5 Two-pot process for the synthesis of NIPU. | ||
The Ca-based catalytic system and organocatalyst allowed a successful NIPU one-pot synthesis. The results were obtained by combining carbonate synthesis with polymerization. Specifically, the aryl-bridged and alkyl-bridged carbonates (2b and 2c, Fig. 6), obtained by the respective epoxides (10b and 10c, Fig. 6), reacted with 1,4-DAB (14, Fig. 6) and 1,8-diaminooctane (5, Fig. 6), in MeCN (100 °C, 24 h), to give NIPUs with molecular masses up to 14 kg mol−1.
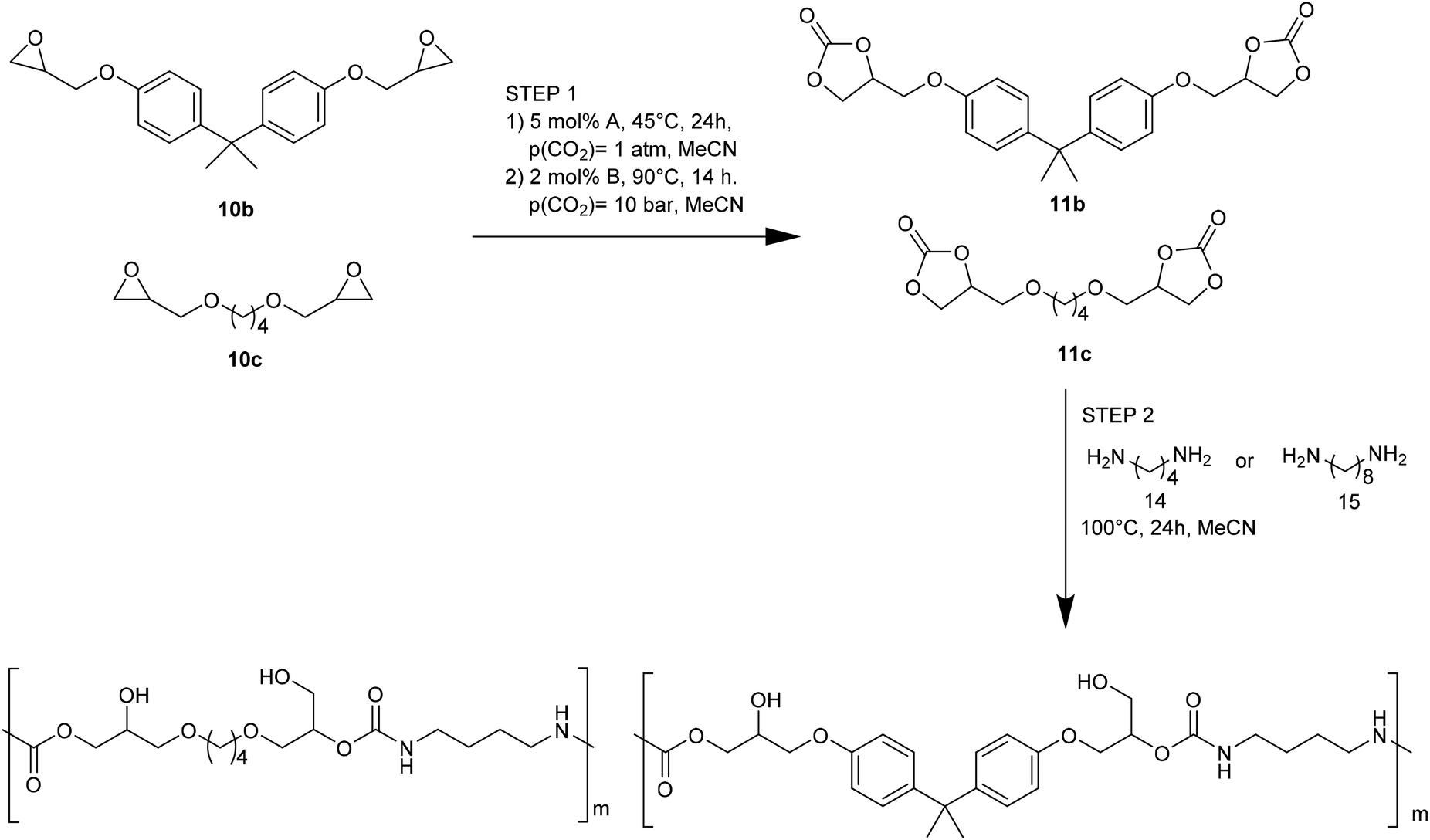 | ||
| Fig. 6 One-pot process for the synthesis of NIPU. | ||
NIPUs can also be obtained by polycarbonate polyols such as poly(propylene carbonate) (17, PPC), synthesized from the copolymerization of CO2 and propylene oxide (16, Fig. 7). PPC is a polymer constituted by carbonate linkages.31 Some catalysts have demonstrated the capability to promote ether linkage formation in propylene oxide polymerization.32 Catalysts such as double metal cyanides (DMC), dinuclear Zn-complexes or Cr-bishydroxy-chinoline complexes, and a poly-alcohol, such as polyethylene glycol as a starter, led to polyethercarbonate polyols by copolymerization of propylene oxide and CO233 (Fig. 7).
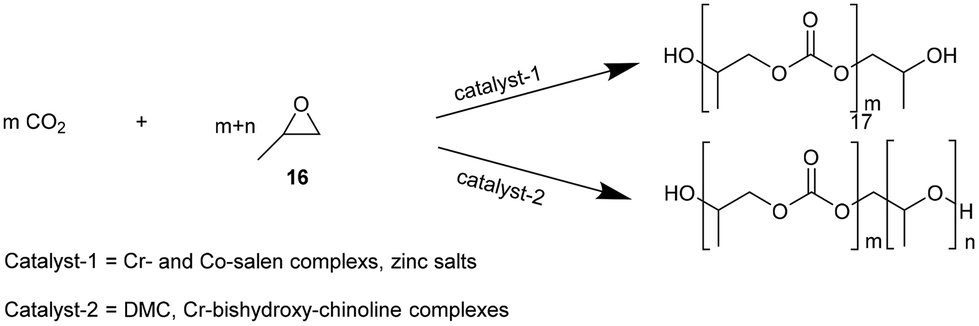 | ||
| Fig. 7 Catalyzed copolymerization of propylene oxide and CO2 to PPC: alternating polycarbonates (top) and polyethercarbonates (bottom). | ||
In this regard, NIPUs with different carbonate linkage content were synthesized34 by reaction of PPCn (99% carbonate linkages, PPCn-RHMPA), PPCz (with 65% carbonate linkages, PPCz-RHMPA), and a mixture of PPCn and PPCz with a molar ratio of 1:1 (PPCnz-RHMPA) with PHA in the presence of MDI as catalyst (Fig. 8).
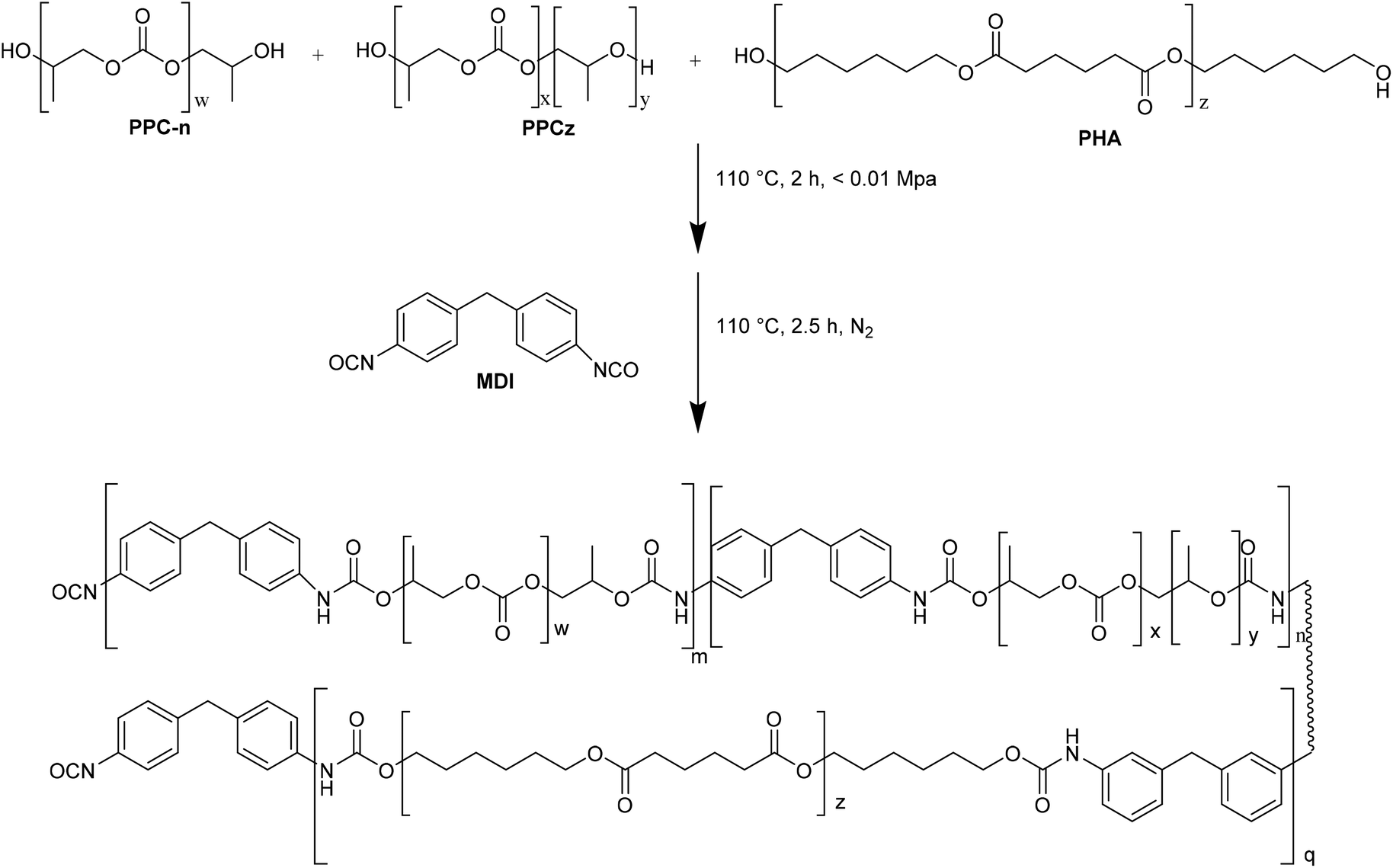 | ||
| Fig. 8 Synthesis of PPCn-RHMPA (m = 1, n = 0, q = 0), PPCz-RHMPA (m = 0, n = 1, q = 1) and PPCnz-RHMPA (m = 0.5, n = 0.5, q = 1). | ||
1H-NMR and FT-IR spectroscopies were performed to confirm the carbonate linkage content. A nearly 100% polycarbonate backbone was confirmed since the spectra showed negligible signals corresponding to the strong ether linkage in PPCz-RHMPA and PPCnz-RHMPA. Different carbonate linkage content in the resulting NIPUs reflects thermal property differences. For instance, as established by DSC, the cold crystallization temperature decreases as the carbonate linkage content increases, thanks to the abundant hydrogen bonding between carbonate and urethane groups that limits PHA fragment mobility. A similar trend was also discovered for TG and DTG analysis where T5 (the temperature of the sample with the weight loss of 5%) has values of 287.0 °C, 285.4 °C, and 282.6 °C for PPCz-RHMPA, PPCnz-RHMPA, and PPCn-RHMPA, respectively. The carbonate linkage content also affects the mechanical properties of these polymers. Longer linkage increases (i) tensile strength (σb of 15.69, 18.42 and 21.91 for PPCz-RHMPA, PPCnz-RHMPA, and PPCn-RHMPA, respectively); (ii) tensile modulus (Et of 28.00, 41.35 and 245.65 for PPCz-RHMPA, PPCnz-RHMPA, and PPCn-RHMPA, respectively); (iii) tensile strength at 100% elongation (σ100 of 2.85, 4.65 and 13.72 for PPCz-RHMPA, PPCnz-RHMPA, and PPCn-RHMPA). An inverted trend was seen for the breaking elongation that decreases as the carbonate linkage content increases (εb of 1007.51, 876.04, and 484.52 for PPCz-RHMPA, PPCnz-RHMPA, and PPCn-RHMPA, respectively). Through a single lap-shear test, the adhesion properties of NIPUs were evaluated and compared to those of LOCTITE 3542, a well-known petroleum-based polyol. All NIPUs showed higher adhesion properties than LOCTITE 3542. Strong adhesion properties were detected for PPCn-RHMPA, followed by PPCnz-RHMPA, PPCz-RHMPA, and LOCTITE®3542, as a consequence of the difference in carbonate linkage content.
NIPUs, applicable as waterborne coatings and adhesives, were also synthesized from organic cyclic decarbonates.35 Through a circulating gas-flow system, CO2 addition to bisepoxides reported in Fig. 9 using different solvents (acetone, THF, methyl isobutyl ketone iBMK, toluene) and tributyl ammonium bromide (TBAB) as the catalyst, in mild conditions (1 atm, T < 100 °C), gave the respective organic cyclic dicarbonates.36
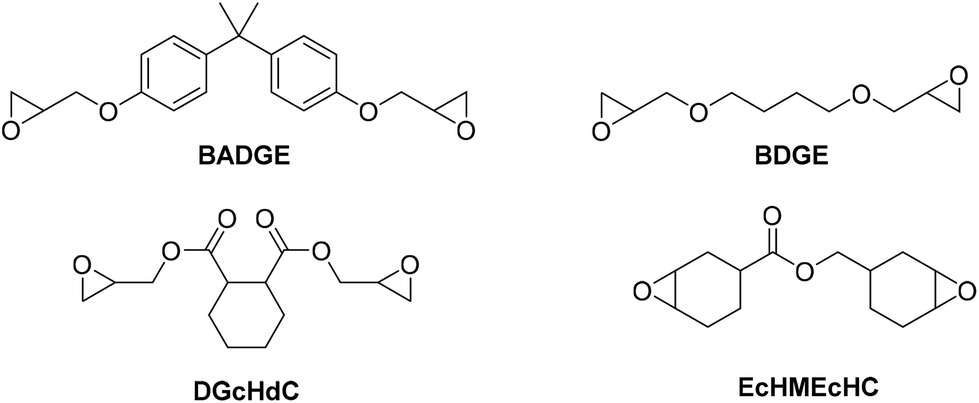 | ||
| Fig. 9 Structures of cyclic dicarbonates BADGE (bisphenol A diglycidyl ether), BDGE (1,4-butanediol diglycidyl ether), DGcHdC (diglycidyl 1,2-cyclohexanedicarboxylate), and EcHMEcHC (3,4-epoxycyclohexylmethyl 3,4-epoxycyclohexane carboxylate). | ||
The reactions have been carried out with a circulating gas-flow system that allows homogeneous continuous distribution of the CO2 within the solvent and aerosol formation for all the tested solvents (even at temperatures near 20 °C). In this way, the concentration of all components in the reaction mixture increases with a consequent increment of the reaction conversion or decrement of the reaction time. Moreover, homogenous CO2 distribution within the solvent improves the conversion time catalyzed by TBAB. Among tested solvents, iBMK resulted in the highest conversion within the shortest time, whereas acetone resulted in the slowest reaction rate. Cyclic dicarbonates prepared by the developed gas-flow system were used as intermediates for NIPUs synthesis by aminolysis with n-butyl amine and 1,6-diamino hexane (8 and 9, Fig. 10) by adding the amine to the same reaction mixture.
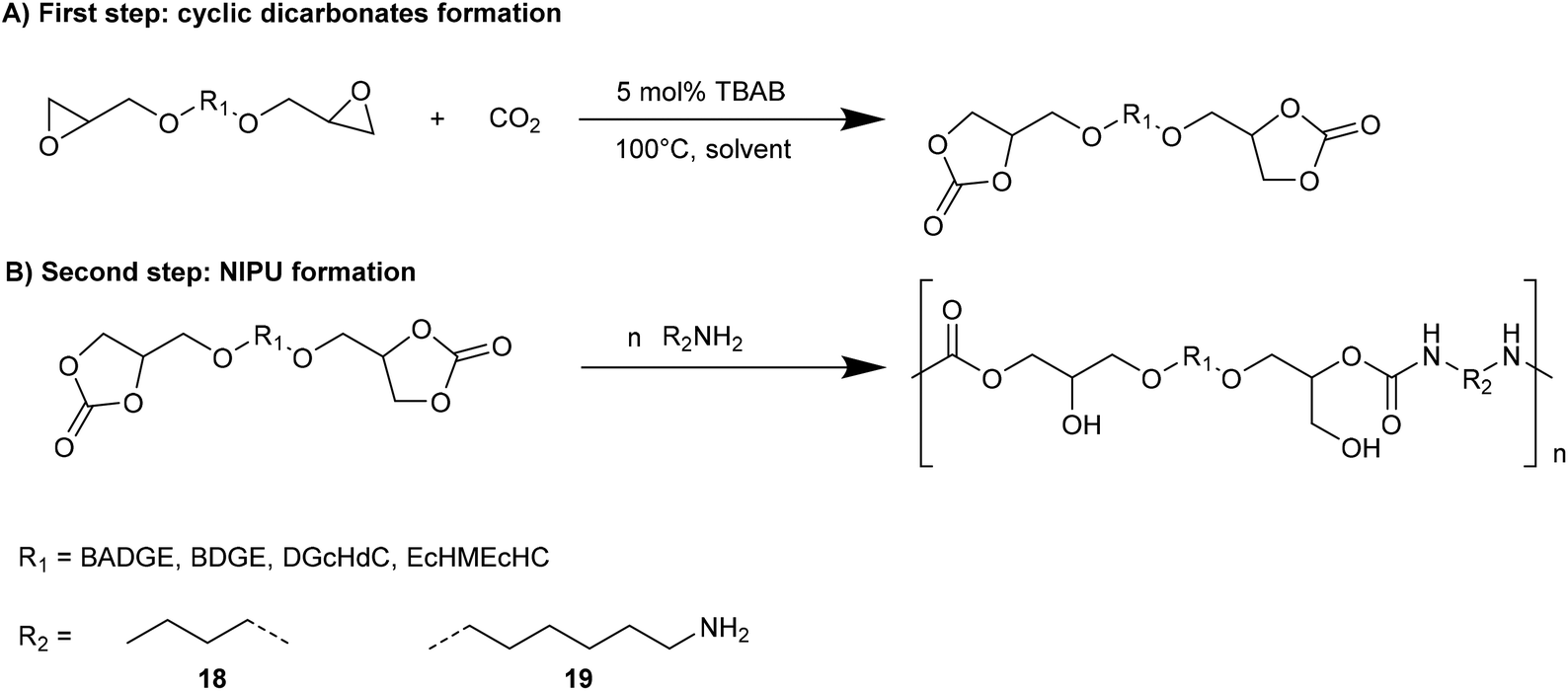 | ||
| Fig. 10 One-pot synthetic pathway for NIPUs. | ||
The CO2-based synthesis of 5-membered dicyclic carbonates and amino-terminal polyurea oligomers as monomers for poly-hidroxyurethane-urea (PHUU) was reported.37 The optimized reaction conditions used (150 °C, TBAB, Fig. 11A) led to about 98% of epoxy group conversion as monitored by 1H-NMR spectroscopy. Indeed, the disappearance of two signals of methylene protons linked to the oxygen atom of epoxy groups and the appearance of a signal corresponding to the methine protons of the cyclic carbonates unequivocally indicated the formation of dicyclic carbonates. This evidence fitted with that obtained by FT-IR where the C–O–C stretching vibration of the epoxy group disappeared and the CO stretching vibration of cyclic carbonate appeared. Then, according to the synthetic pathway reported in Fig. 11B, amino-terminal polyurea oligomers D230* and TTD* were prepared and characterized by 1H-NMR and FT-IR spectroscopies.
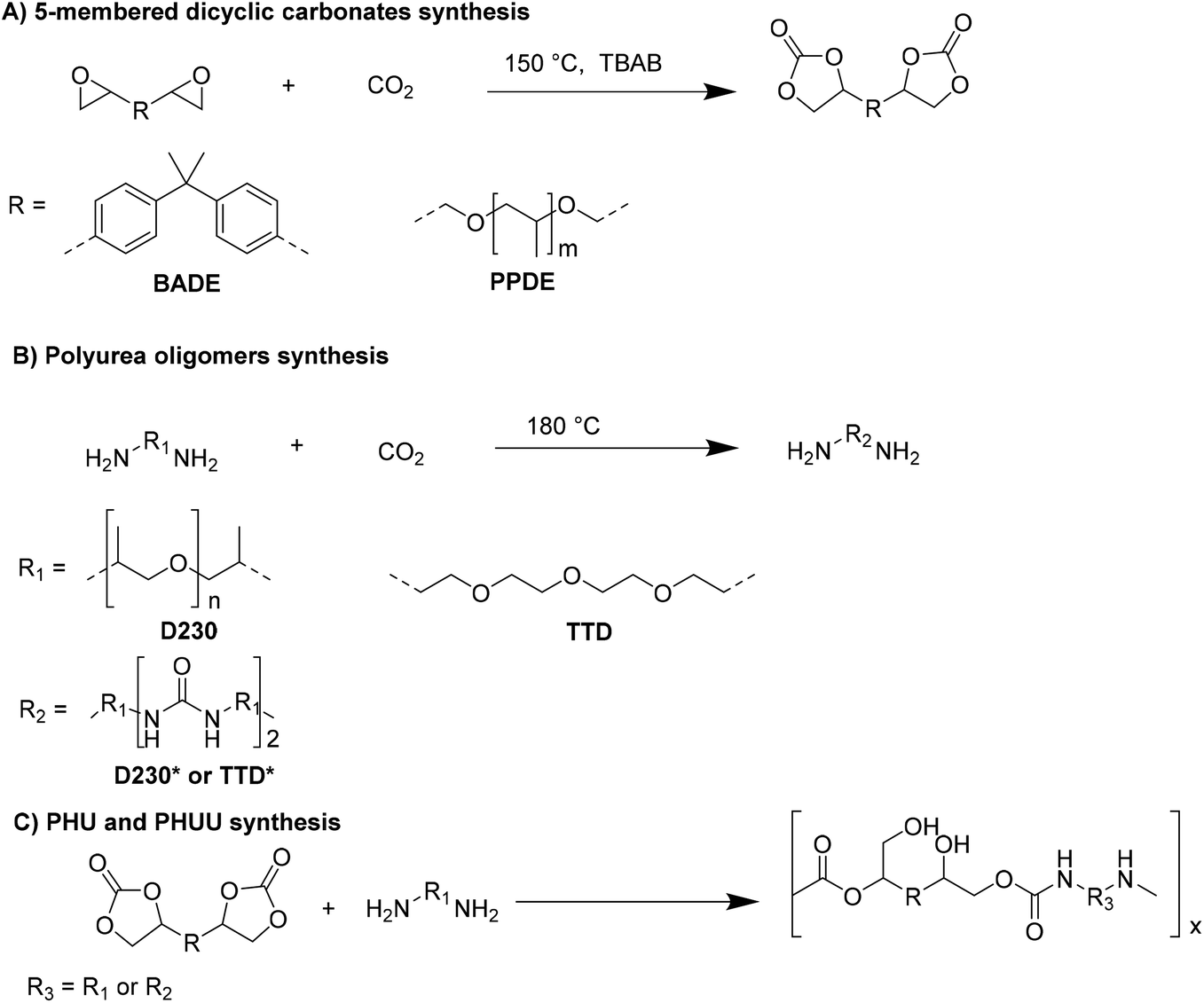 | ||
| Fig. 11 Polyhydroxiuethane (PHU) and PHUU synthetic route. | ||
Finally, the polyaddition of amino-terminal polyurea oligomers to 5-membered dicyclic carbonates was conducted on several polyhydroxiuethane-urea derivatives, PHUU-1, PHUU-2, and PHUU-3 (Fig. 11C).
The urethane/urea ratio, calculated by 1H-NMR, indicated a higher urea content for PHUU-2 and PHUU-3 (0.88 and 0.86) than PHUU-1 (0.58). This last was also characterized by a lower molecular weight (3896 g mol−1vs. 19340 and 16357 for PHUU-2 and PHUU-3, respectively) due to the more considerable steric hindrance of the amino group in D230* hampering dicyclic carbonate aminolysis. Introducing urea functionality in PHUU-2 and PHUU-3 determined stronger hydrogen bond formation and interchain interactions, reflecting improved thermal properties and surface hydrophobicity. Indeed, the strong interchain interactions restrict the water penetration as evaluated by the water contact angle measurement. As evaluated by TGA, the thermal degradation temperature of PHUU-2 and PHUU-3 was higher than the corresponding PHU-2 and PHU-3, synthesized from 5-membered dicyclic carbonates and diamines. It is possible to attribute this peculiarity to the influence of a different diamine sequence that led to the incorporation of urea moieties. PHUUs exhibited different mechanical properties as detected by tensile tests. For example, in comparison to PHU-2, a decreased Young's module (20.7 vs. 5.0 MPa) and tensile strength (6.9 vs. 0.3 MPa) and an increased elongation at break were reported for PHUU-2 (258 vs. 914%, respectively). After the urea moiety's introduction, PHU-3, a viscous oil, became a solid (PHUU-3). This physical property change confers to PHUU-3 improved cohesive energy and mechanical properties (Young's module 8.6 MPa, tensile stress 1.3 MPa, and elongation at break of 31.3%).
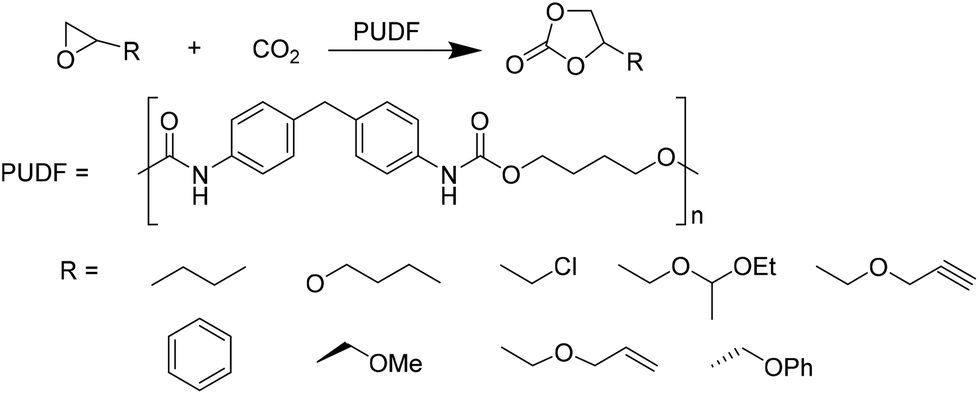
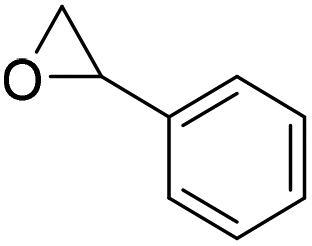
Motokucho et al.38 examined the activity of PUDF (Fig. 12) as a catalyst in the cycloaddition reaction between epoxide and CO2 to give the corresponding 5-membered cyclic carbonate. Firstly, the reaction was carried out by heating the phenyl glycidyl ether (PGE) at 150 °C for 42 h under 9.0 MPa pressure of CO2. The same reaction performed in the absence of PUDF didn't occur, whereas the presence of PUDF led to the forming of the carbonate with a yield of 49% and 99% selectivity, demonstrating its effectiveness as a catalyst. Moreover, the author demonstrated that PUDF could be easily separated from the reaction mixture and reused for further catalysis. The reaction conditions were systemically optimized by varying reaction time, CO2 pressure, temperature, and PUDF/epoxide ratio. Increasing the reaction time increases the conversion of PGE into its carbonate. The reactions become quantitative after 48 h. Increasing CO2 pressure increases the yield of the reaction, however, reaching a plateau at 4.9 MPa. Analogously, the temperature range of 120–180 °C affects the yield of the reaction, but the highest temperature causes a little loss of selectivity. PGE/carbonate conversions of 73% and 98% were achieved for the PUDF/PGE ratio of 0.1 and 0.4, respectively. The optimized reaction conditions were then used to evaluate the reusability and recyclability of PUDF as a catalyst for recycling the insoluble solid for several cycles of polyaddition PGE/CO2. After ten cycles, the reaction yields were almost constant (45–50%), indicating the reusability of the PUDF catalyst.
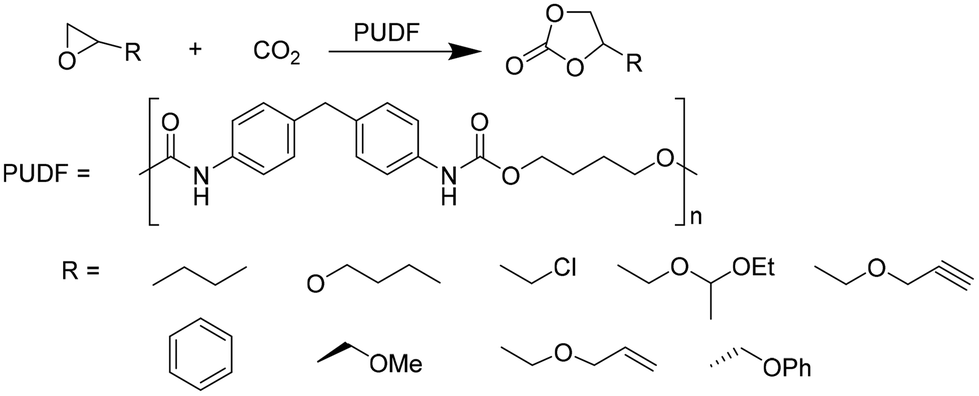 | ||
| Fig. 12 PUDF-catalyzed cycloaddition of CO2 to various epoxides. | ||
Finally, PUDF was tested as a catalyst for the cycloaddition of CO2 to various epoxides (Fig. 12), demonstrating its versatility since the PUDF-catalyzed reaction can be applied to a broad spectrum of epoxides.
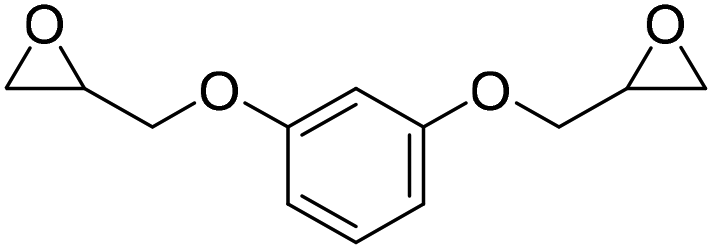
Based upon the PUs’ non-processability concerns due to their permanently fixed topology, several efforts were focused on preparing the PUs’ covalent adaptable networks (CANs).39 These last are cross-linked networks capable, under external stimuli such as temperature or light, of activating exchange reactions between dynamic bonds, thus allowing the rearrangement of their topology network.40,41 In this context, Pronoitis et al.42 synthesized PHUs CAN by exploiting the urethane bond dynamicity or by creating dual dynamic networks by combining the urethane bond with other dynamic motifs such as imines and disulfides. In the reaction catalyzed by DBU·I2 complex, poly(ethylene glycol) diglycidyl ether (PEGDE), 1,4-butanediol diglycidyl ether (BDGE) and resorcinol diglycidyl ether (RDGE) reacted with CO2 to give the corresponding cyclic carbonates (PEGDC, BDC; RDC) with 80–97% yields (Fig. 13). Then, the reaction between cyclic carbonates and cysteamine at a 1:1 ratio was conducted on corresponding PHU CANs designated by using the dicarbonate mol% with respect to the TMPTC cross-linker: TMPTC100 (only TMPTC and cysteamine), PEGDC30 (30 mol% PEGDC and 70 mol% TMPTC), PEGDC50 (50 mol% PEGCD and 50 mol% TMPTC), PEGDC70 (70 mol% PEGCD and 30 mol% TMPTC), BDDC30 (30 mol% BDDC and 70 mol% TMPTC) and RDC30 (30 mol% RDC and 50 mol% TMPTC). Their polymerization was monitored through the characteristic FTIR bands of the urethane group.
 | ||
| Fig. 13 Chemical structure of a network's segment. | ||
The properties of the resulting PHU CANs can be controlled by varying the networks’ chemical structure and cross-link density. For instance, the aliphatic and long ether spacers of BDDC and PEGDC, respectively, conferred flexibility to the network, contrary to the aromatic RDC, which instead provided stiffness and rigidity. The decarbonate/TMPTC ratio also modulated the properties of the networks. Consequently, PHU CANs networks showed variegated thermal, mechanical, and viscoelastic properties. As detected by DSC, Tg of PHU CANs, as-synthesized, ranged from −7 to 36 °C, with a slight decrease after their hot-pressed treatment attributed to a moisture-induced plasticizing effect exerted by the presence of the OH groups. Indeed, contact angle values of TMPTC100, BDDC30, and RDC30 – featured by the higher density of the pendant OH groups – were lower than those of PEGDC30 and PEGDC70. Moreover, when PHU-CANs were exposed to a 50% relative humidity for 20 days, the measured water uptake was fast and higher for the PEG-based polymer because of both the pendant −OH groups and the hydrophilicity of PEG spacers. Instead, RDC exhibited the slowest water uptake with most hydrophobic aromatic groups. Also, viscoelastic properties are a function of varying the chemical structure and cross-link density of PHU CANs networks. Finally, the reprocessing of PHU CANs networks has been evaluated. The adapt hot-pressing conditions were screened to select a temperature (100 °C for 20 min) that enabled a fast S–S exchange without inducing the urethane bond dissociation and the reversible cyclic carbonate-aminolysis side reactions. At this temperature, no cyclic carbonate was formed as detected by FT-IR, whereas at 140–160 °C, a band at 1790 cm−1 attributed to cyclic carbonate's stretching appeared and progressively increased. Vacuum-dried PHU CANs tensile testing evaluation established the stiffness and strength of TMPTC100, BDDC30, and RDC30 with Young's moduli (E) above 1 GPa and small deformation at break (εb = 1–2%) and the softness and flexibility of PEGDC-based CANs.
Through the terpolymeration of propylene oxide, CO2, and [3-(2,3-epoxypropoxy)-propyl]-trimethoxysilane (KH-560), siloxane-functionalized poly(ether carbonate)s (Si-PECs, Fig. 14) were synthesized and used as the soft segments of waterborne polyurethane (WPU, Fig. 14).43
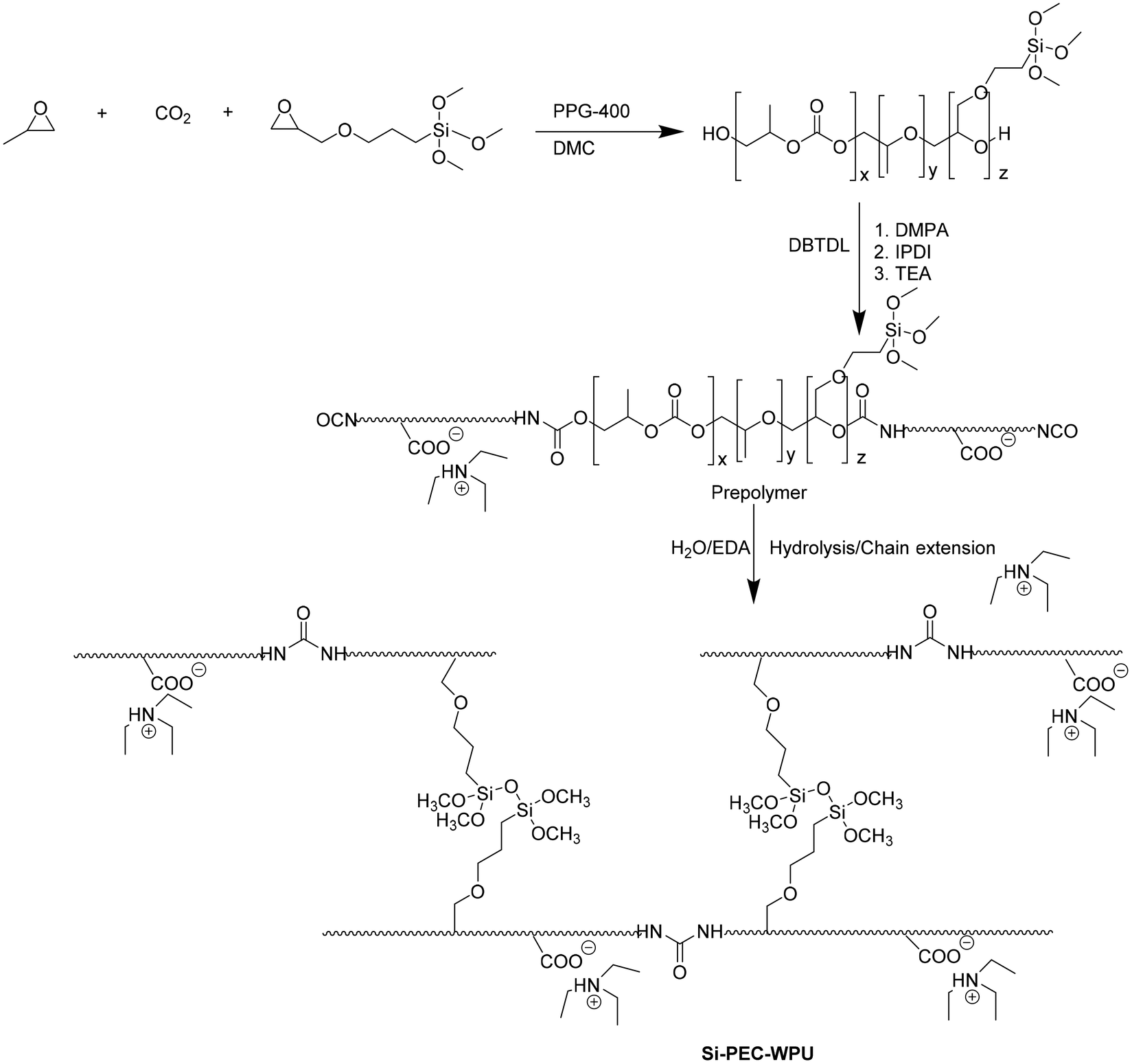 | ||
| Fig. 14 Synthetic pathway for Si-PEC-WPU preparation. | ||
It has been demonstrated that (1) siloxane moieties increase water resistance and heat resistance of the urethane bond, (2) CO2 introduction confers rigidity, and (3) KH-560 promotes temperature performance. Thus, PPG-WPU, PEC-WPU, 5% Si-PEC-WPU, and 10% Si-PEC-WPU were obtained from PPG-2000, PEC-2000, 5% Si-PEC-2000, and 10% Si-PEC-2000, respectively. As emerged by DSC investigations, flexible side chains and the hydrolytic cross-linking of the siloxane groups affect the Tg. Indeed, the rigid carbonate units in PPG-WPU confer a higher Tg degree than Si-PEC–WPU with soft side chains. A similar trend was rescued for the two-step degradation process (the first step was at about 295, 313, and 322 °C; the second step at about 352, 360, and 367 °C for PPG-WPU, PEC-WPU, and Si-PEC-WPU, respectively). The carbonate content and the siloxane presence affect the mechanical performance. The carbonate content in PEC-2000 increases the tensile strength but reduces the elongation at break, whereas the carbonate and siloxane unit introduction in Si-PEC-WPUs leads to good tensile strength and elongation at break. As expected, Si-PEC-WPU showed a lower swelling ratio (36.6%) than PPG-WPU and PEC-WPU (55.2 and 49.3%, respectively).
In summary, the carbonate unit improved the mechanical resistance of WPU, whereas the siloxane units offered higher cross-linking density and thermal stability.
Another green, sustainable route for the synthesis of NIPU consists of the reaction between [4,4′-bi(1,3-dioxolane)]-2,2′-dione (BDC, 21, Fig. 15) and aliphatic diamines.44 To this purpose, CO2 cycloaddition to 1,3-butadiendiepoxide (20, Fig. 15) using a bivalent catalytic system consisting of metal–organic frameworks (MOFs) as catalysts and cetyltrimethylammonium bromide (CTAB, d, Fig. 15) as co-catalyst, was performed.
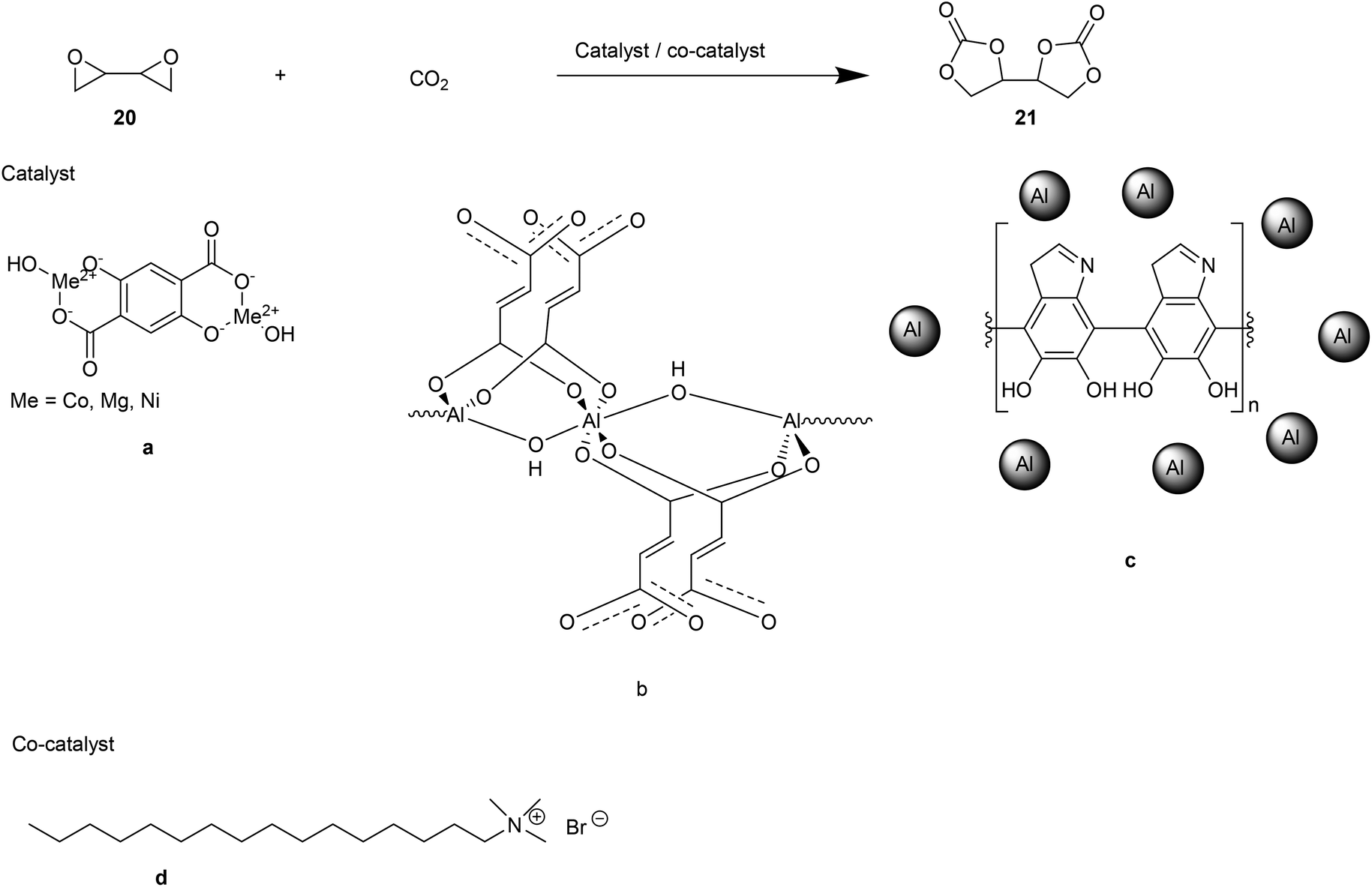 | ||
| Fig. 15 Synthesis of BDC. | ||
MOFs are homogeneous catalysts created from metals and organic linker compounds with high chemical, thermal, and mechanical stability. In this context, Benedito et al. selected and tested a series of MOFs based on their selectivity, capability to capture CO2 (Co- or Mg- or Ni- or GO-Ni-MOF-74, a, Fig. 15), and long catalytic life (Al-OH-fumarate (b) and Al-PDA (c), Fig. 15). The authors focused on finding optimal reaction conditions for the highest BDC yields.
To achieve this aim, the carbonation of 1,3-butadiendiepoxide was monitored through GC-MS and 1H-NMR. The epoxide conversion into BCD was truly efficient when the reaction was conducted in the presence of a co-catalyst (catalyst/co-catalyst ratio 1:1). Their synergic action reflects on the higher percentage of epoxide/BDC conversion (44–95% yield) than the ones detected in the absence of the co-catalyst (0–6% yield). In addition to being solvent-free, the synthetic strategy adopted to obtain BDC does not require the removal of by-products.
In Table 1, the epoxide used, as well as the synthesized cyclic carbonates, and some remarks on the reaction are summarized.
| Starting epoxide | Cyclic carbonate prepared | Fixation of CO2 conditions | Ref. | ||||
|---|---|---|---|---|---|---|---|
| Catalyst | T | t | Pressure | Solvent | |||
|
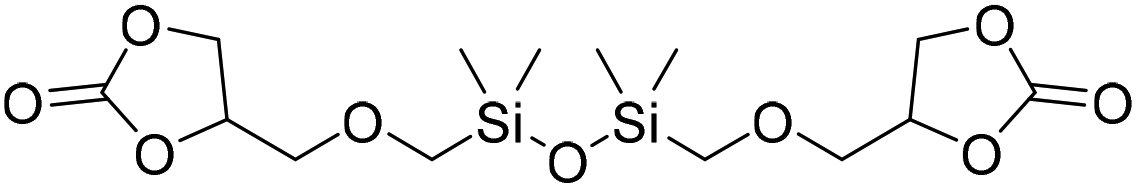
|
(A) CaI2 with 18-crown-6 | (A) 23 °C | (A) 24 h | (A) 1 atm | — | Wulf et al.26 |
| (B) Tri-n-butyl-(2-hydroxyethyl)ammonium iodide | (B) 90 °C | (B) 3 h | (B) 10 bar | ||||
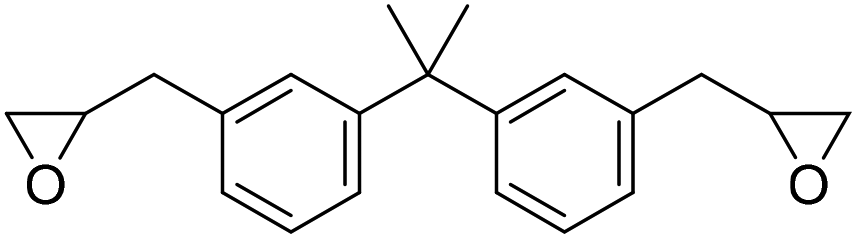
|
|
(A) CaI2 with 18-crown-6 | (A) 45 °C | (A) 24 h | (A) 1 atm | (A) MeCN | Wulf et al.26 |
| (B) Tri-n-butyl-(2-hydroxyethyl)ammonium iodide | (B) 90 °C | (B) 3 h | (B) 10 bar | (B) MEK | |||
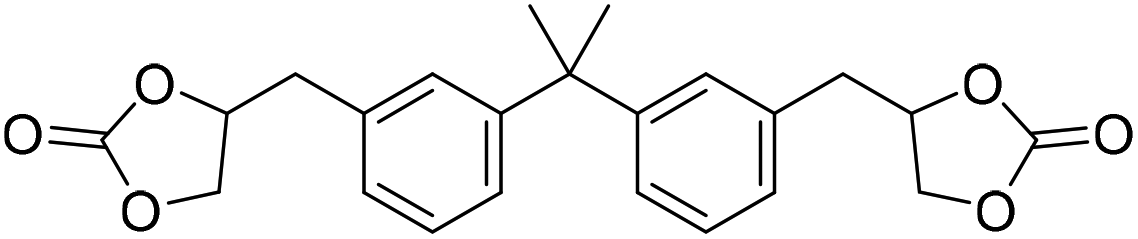
|
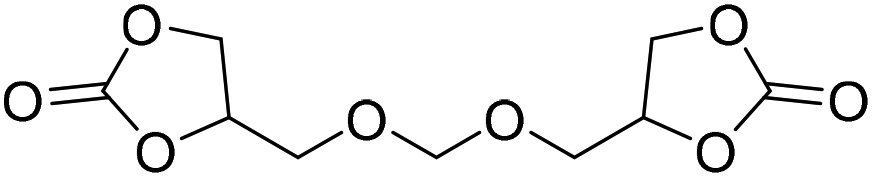
|
(A) CaI2 with 18-crown-6 | (A) 45 °C | (A) 48 h | (A) 1 atm | (A) MeCN | Wulf et al.26 |
| (B) Tri-n-butyl-(2-hydroxyethyl)ammonium iodide | (B) 90 °C | (B) 3 h | (B) 10 bar | (B) — | |||
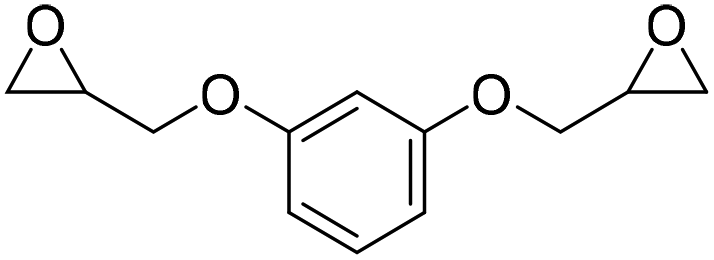
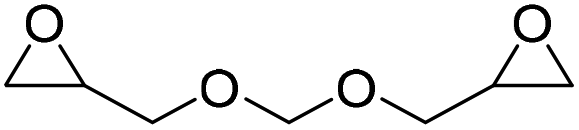
|
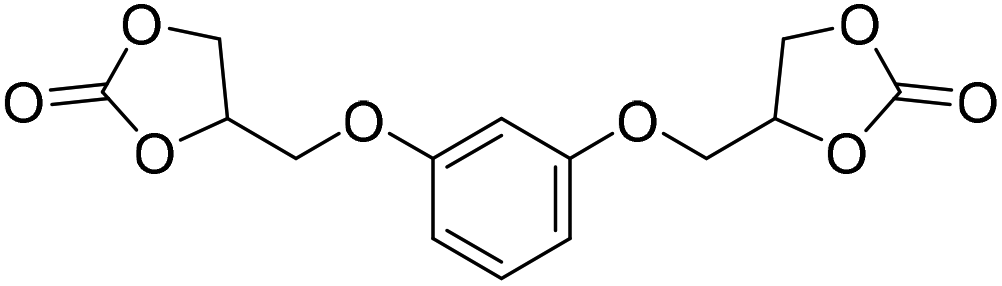
|
(A) CaI2 with 18-crown-6 | (A) 45 °C | (A) 48 h | (A) 1 atm | (A) MeCN | Wulf et al.26 |
| (B) Tri-n-butyl-(2-hydroxyethyl)ammonium iodide | (B) 90 °C | (B) 3 h | (B) 10 bar | (B) MEK | |||
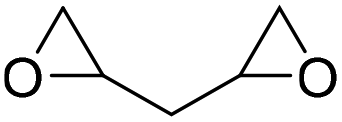
|
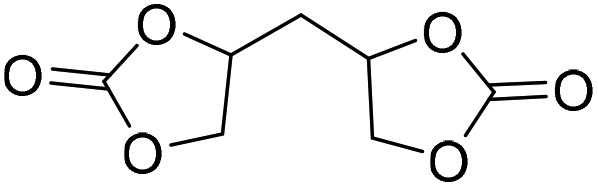
|
(A) CaI2 with 18-crown-6 | (A) 45 °C | (A) 48 h | (A) 1 atm | (A) MeCN | Wulf et al.26 |
| (B) Tri-n-butyl-(2-hydroxyethyl)ammonium iodide | (B) 90 °C | (B) 3 h | (B) 10 bar | (B) MEK | |||
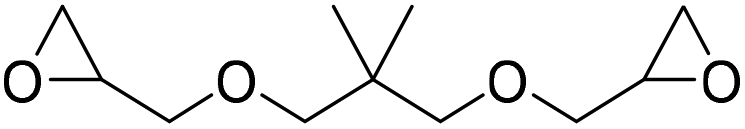
|
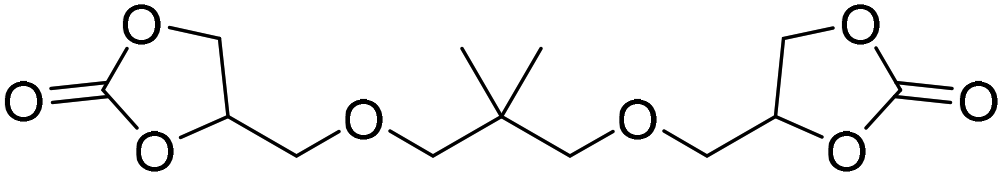
|
(A) CaI2 with 18-crown-6 | (A) 65 °C | (A) 48 h | (A) 10 bar | (A) MeCN | Wulf et al.26 |
| (B) Tri-n-butyl-(2-hydroxyethyl)ammonium iodide | (B) 90 °C | (B) 3 h | (B) 10 bar | (B) — | |||
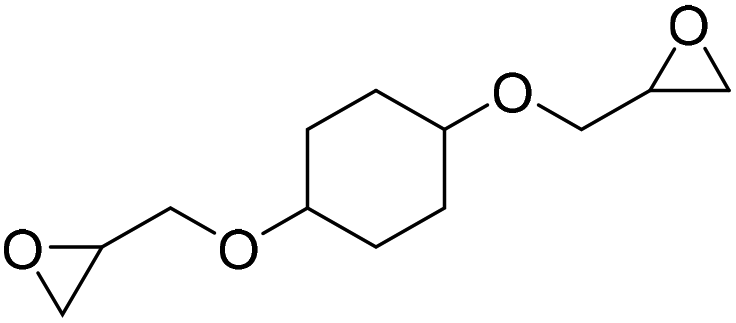
|
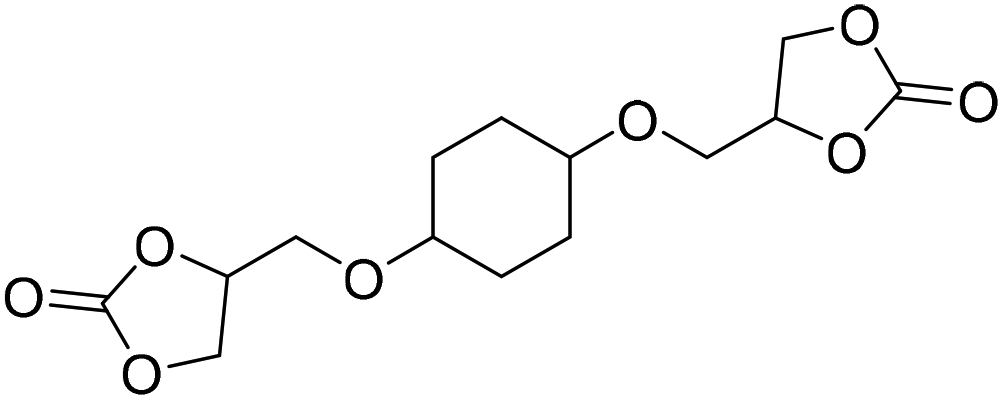
|
(A) CaI2 with 18-crown-6 | (A) 65 °C | (A) 48 h | (A) 10 bar | (A) MeCN | Wulf et al.26 |
| (B) Tri-n-butyl-(2-hydroxyethyl)ammonium iodide | (B) — | (B) — | (B) — | (B) — | |||
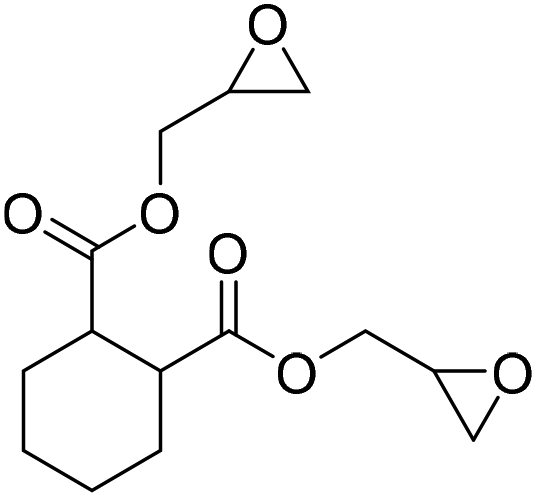
|
|
(A) CaI2 with 18-crown-6 | (A) 65 °C | (A) 48 h | (A) 10 bar | (A) MeCN | Wulf et al.26 |
| (B) Tri-n-butyl-(2-hydroxyethyl)ammonium iodide | (B) 90 °C | (B) 3 h | (B) 10 bar | (B) — | |||
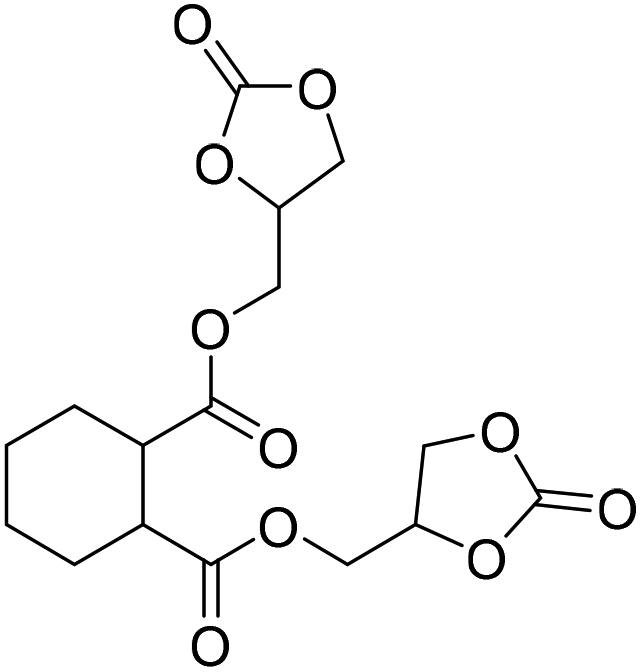
|
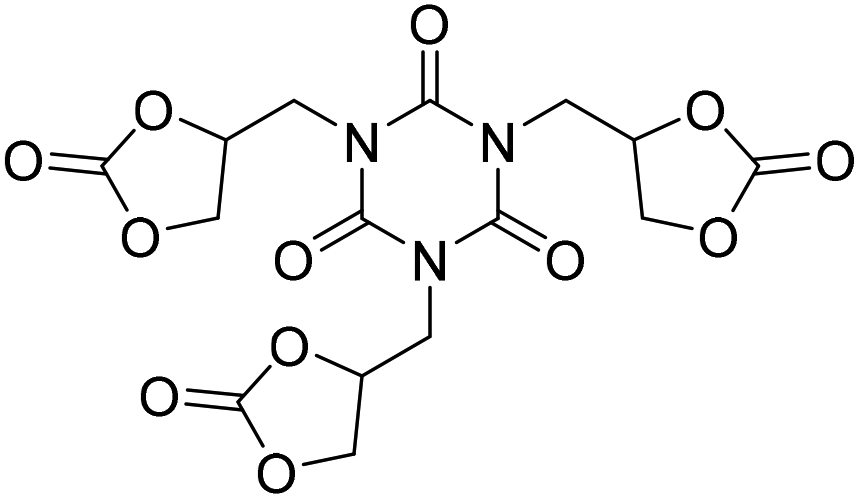
|
(A) CaI2 with 18-crown-6 | (A) 65 °C | (A) 48 h | (A) 10 bar | (A) MeCN | Wulf et al.26 |
| (B) Tri-n-butyl-(2-hydroxyethyl)ammonium iodide | (B) 90 °C | (B) 14 h | (B) 10 bar | (B) — | |||
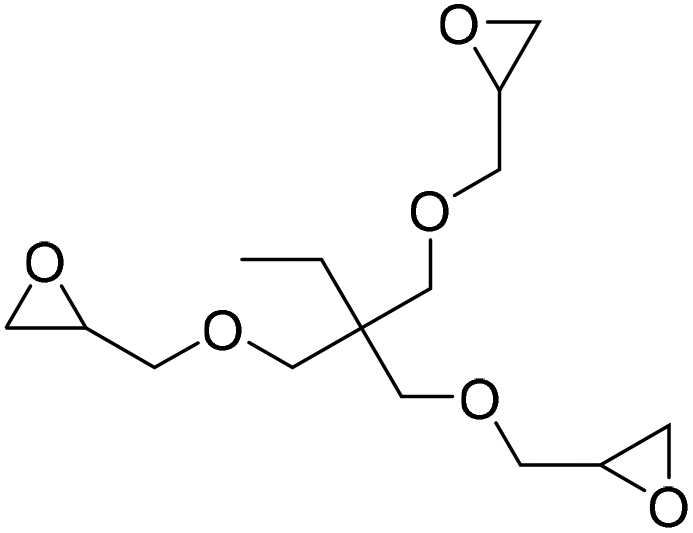
|
|
(A) CaI2 with 18-crown-6 | (A) 65 °C | (A) 48 h | (A) 10 bar | (A) MeCN | Wulf et al.26 |
| (B) Tri-n-butyl-(2-hydroxyethyl)ammonium iodide | (B) — | (B) — | (B) — | (B) — | |||
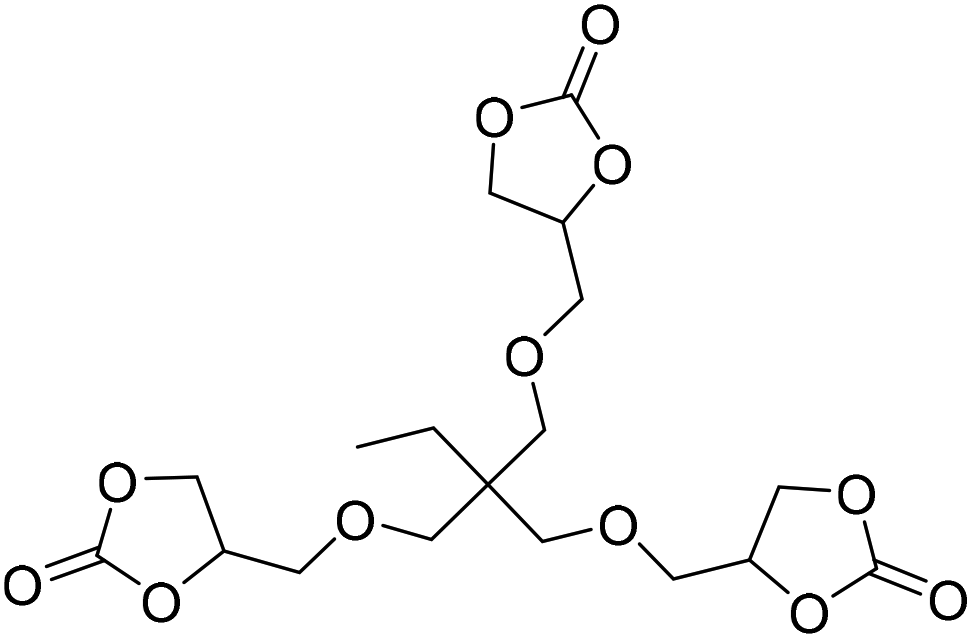
|
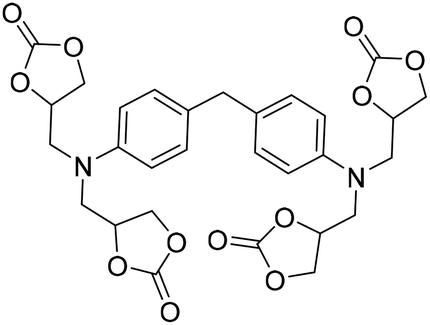
|
(A) CaI2 with 18-crown-6 | (A) — | (A) — | (A) — | (A) — | Wulf et al.26 |
| (B) Tri-n-butyl-(2-hydroxyethyl)ammonium iodide | (B) 90 °C | (B) 14 h | (B) 10 bar | (B) — | |||
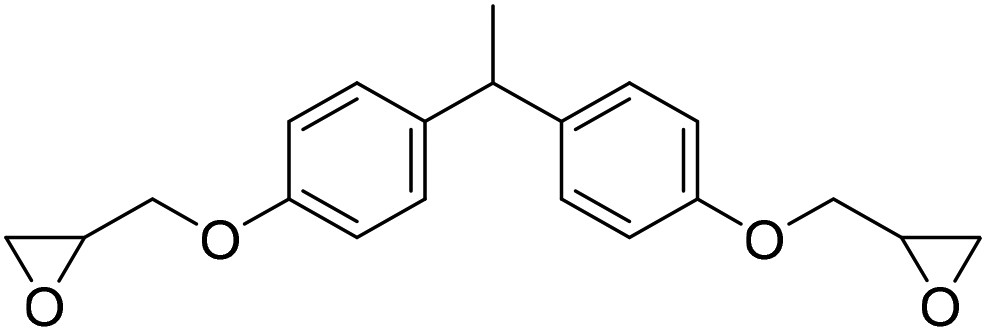
|
|
TBAB | <100 °C | 4 h | 1 atm | iBMK | Schmitt et al.36 |
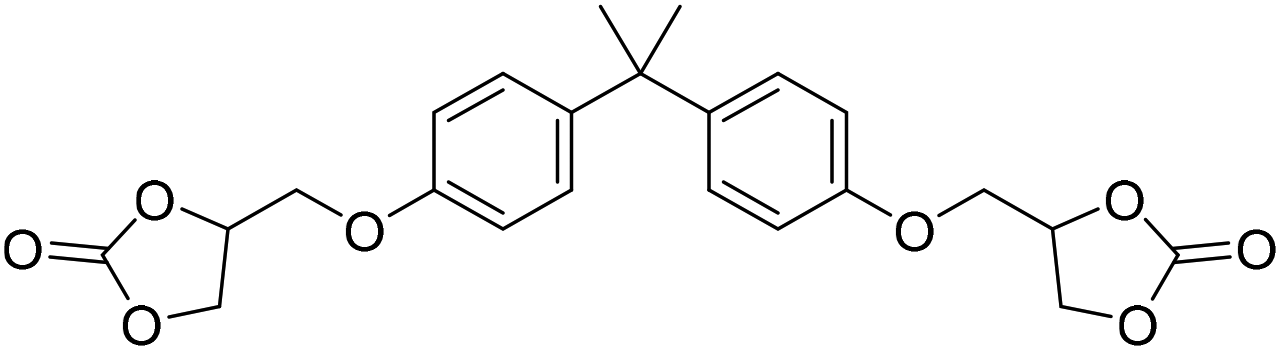
|
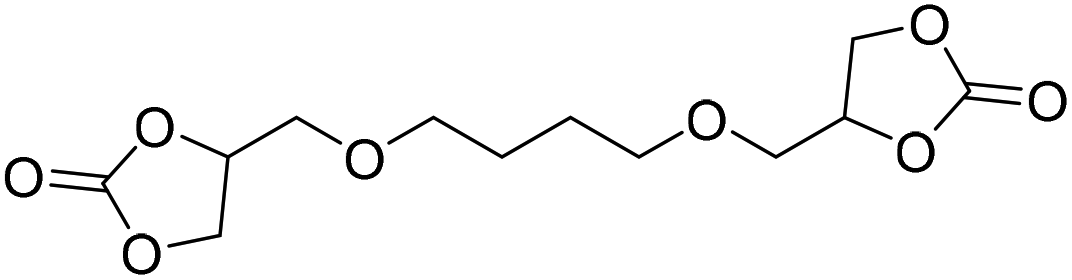
|
TBAB | <100 °C | 5 h | 1 atm | iBMK | Schmitt et al.36 |
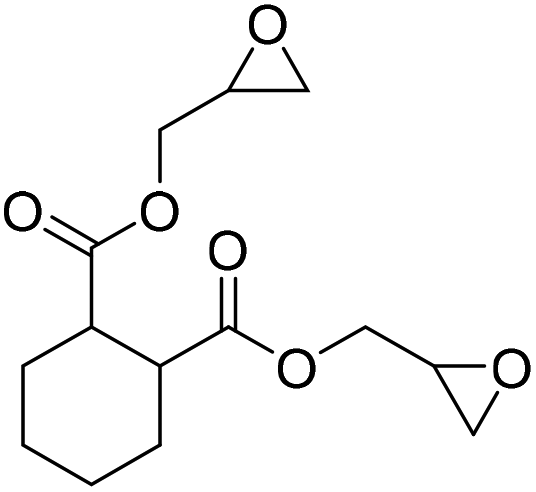
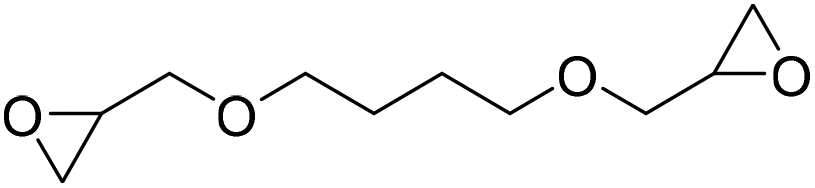
|
|
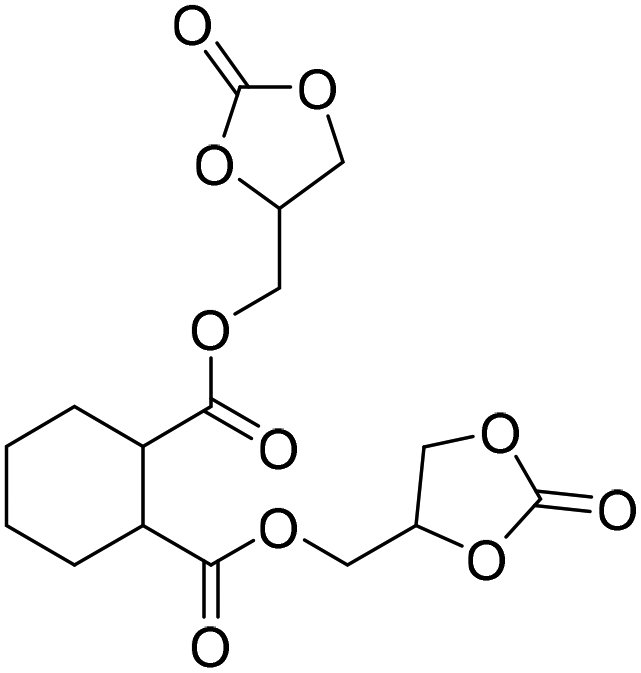
TBAB | <100 °C | 3 h | 1 atm | iBMK | Schmitt et al.36 |
|
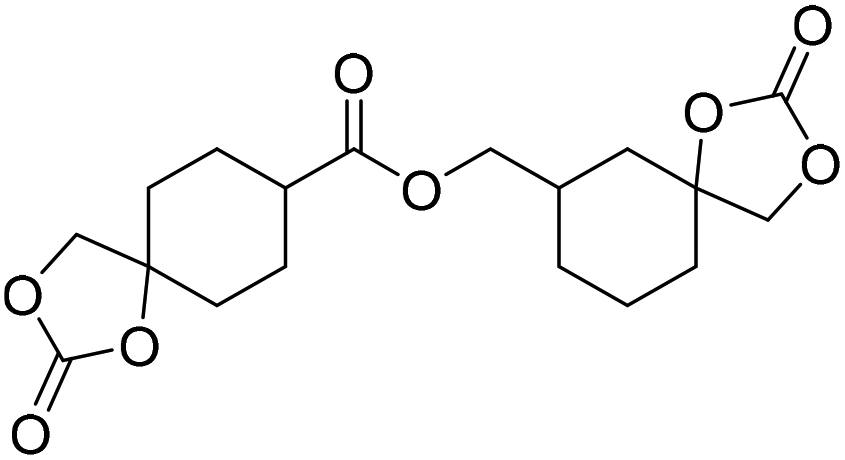
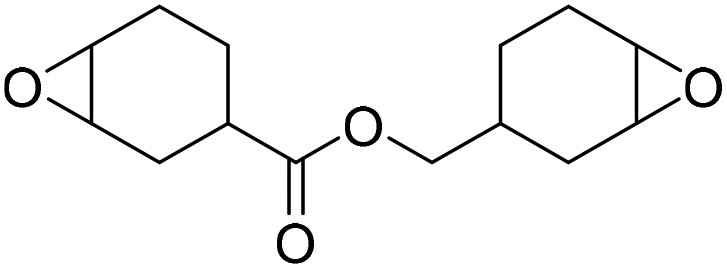
|
TBAB | <100 °C | 6.9 h | 1 atm | iBMK | Schmitt et al.36 |
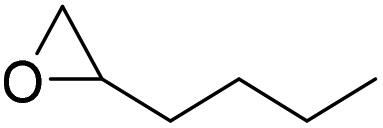
|
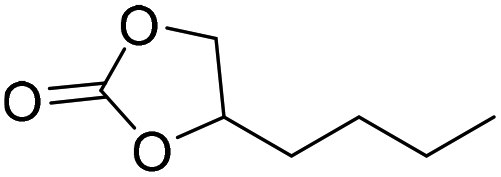
|
PUDF | 150 °C | 42–48 h | 9 MPa | — | Motokucho et al.38 |
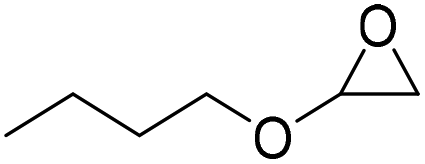
|
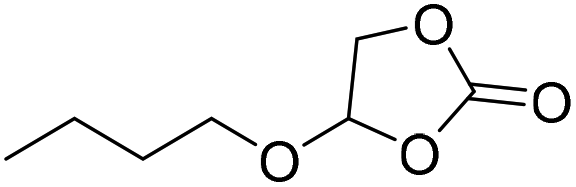
|
PUDF | 150 °C | 42–48 h | 9 MPa | — | Motokucho et al.38 |
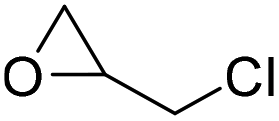
|
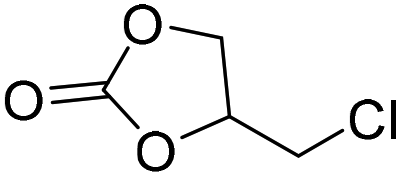
|
PUDF | 150 °C | 42–48 h | 9 MPa | — | Motokucho et al.38 |
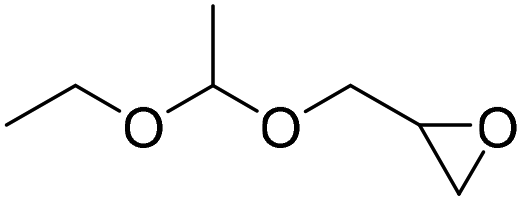
|
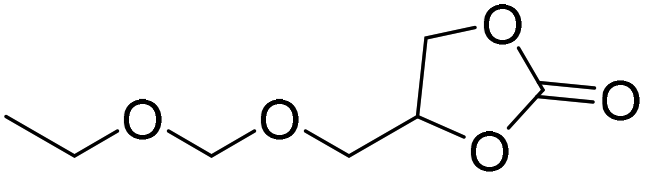
|
PUDF | 150 °C | 42–48 h | 9 MPa | — | Motokucho et al.38 |
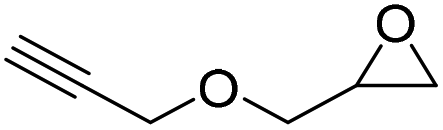
|
|
PUDF | 150 °C | 42–48 h | 9 MPa | — | Motokucho et al.38 |
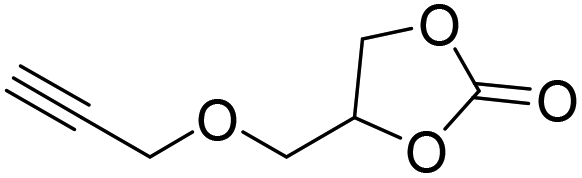
|
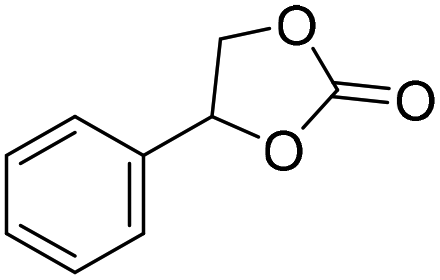
|
PUDF | 150 °C | 42–48 h | 9 MPa | — | Motokucho et al.38 |
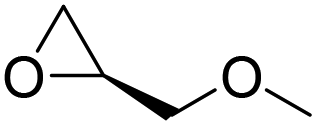
|
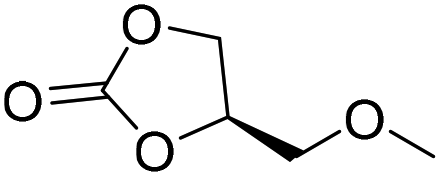
|
PUDF | 150 °C | 42–48 h | 9 MPa | — | Motokucho et al.38 |
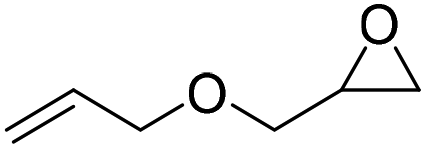
|
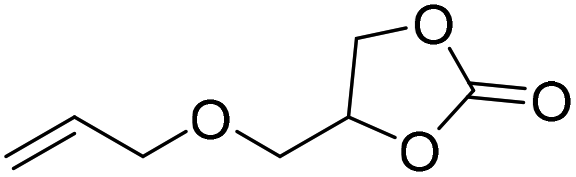
|
PUDF | 150 °C | 42–48 h | 9 MPa | — | Motokucho et al.38 |
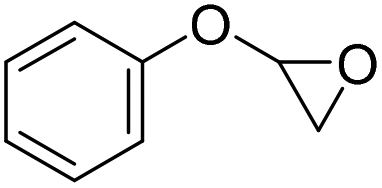
|
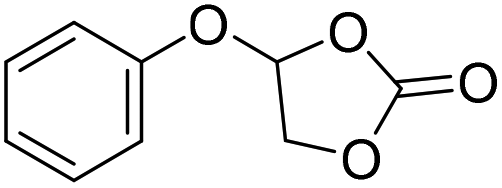
|
PUDF | 150 °C | 42–48 h | 9 MPa | — | Motokucho et al.38 |
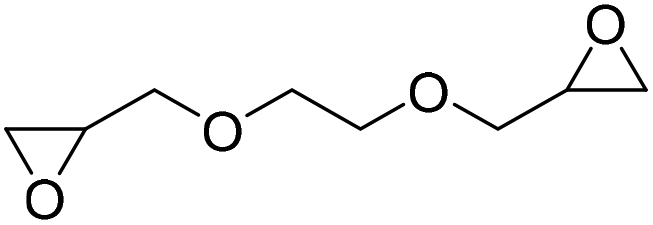
|
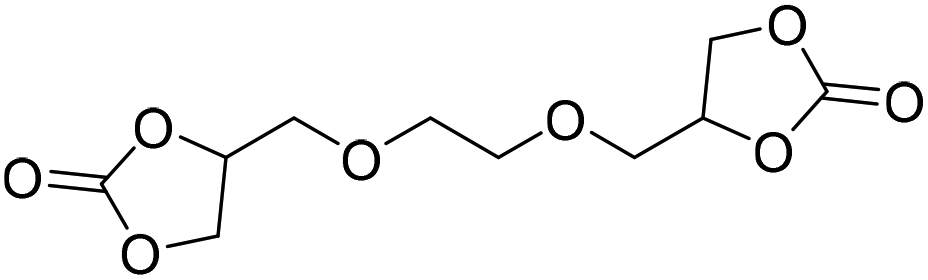
|
DBU·I2 complex | 100 °C | 92 h | 1 atm | — | Pronoitis et al.42 |
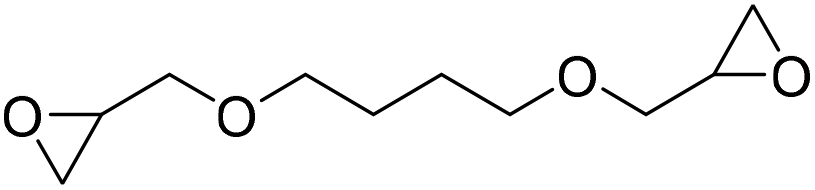
|
|
DBU·I2 complex | 100 °C | 92 h | 1 atm | — | Pronoitis et al.42 |
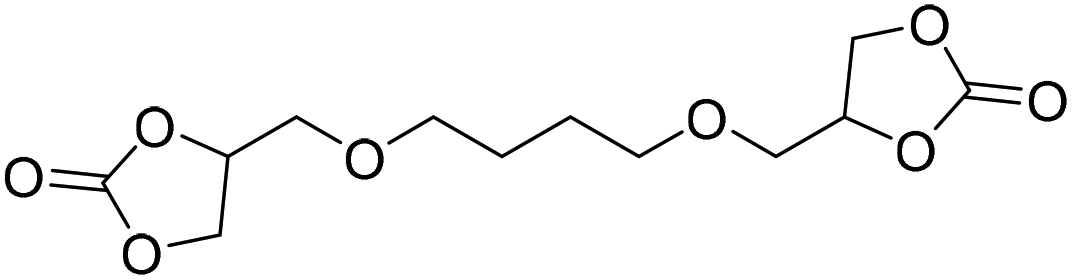
|
|
DBU·I2 complex | 100 °C | 92 h | 1 atm | — | Pronoitis et al.42 |
|
|
MOF/CTAB | 120 °C | 6 h | 30 bar | — | Benedito et al.44 |
|
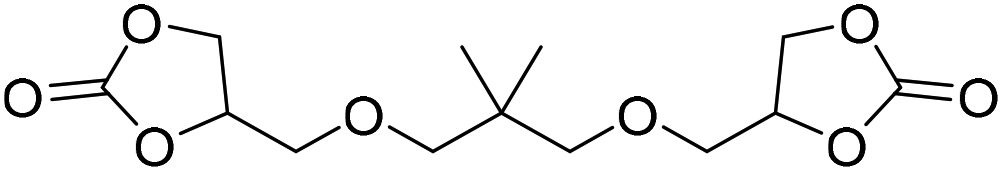
|
LiBr | 80 °C | 48 h | 1 bar | DMF | Hwang et al.45 |
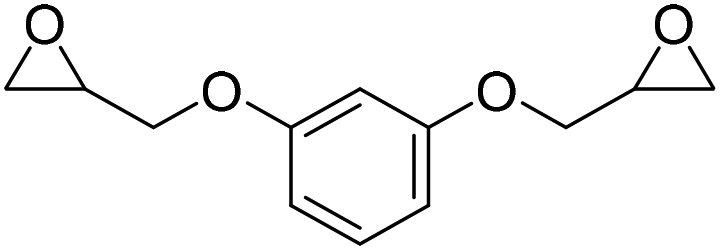
|
|
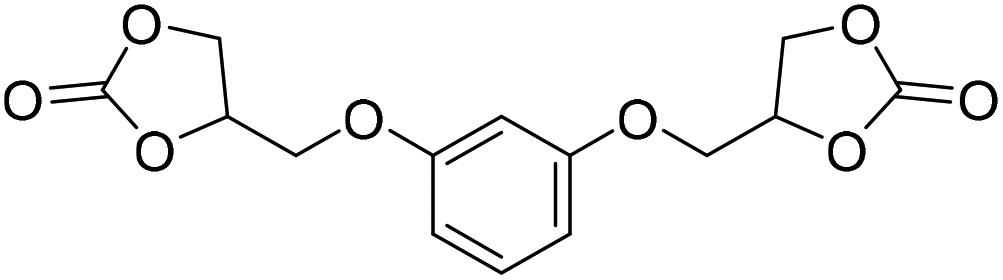
LiBr | 80 °C | 48 h | 1 bar | DMF | Hwang et al.45 |
|
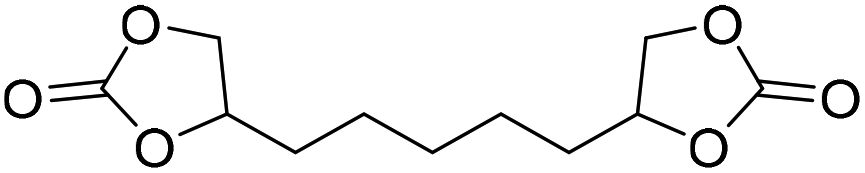
|
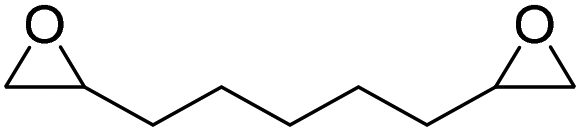
LiBr | 80 °C | 48 h | 1 bar | DMF | Hwang et al.45 |
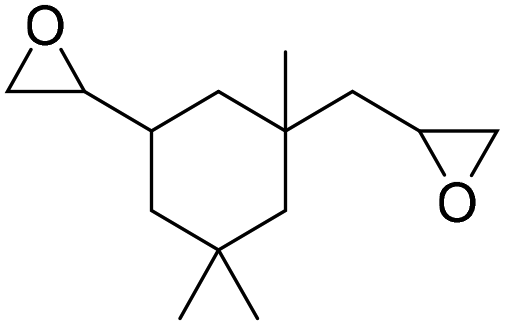
|
|
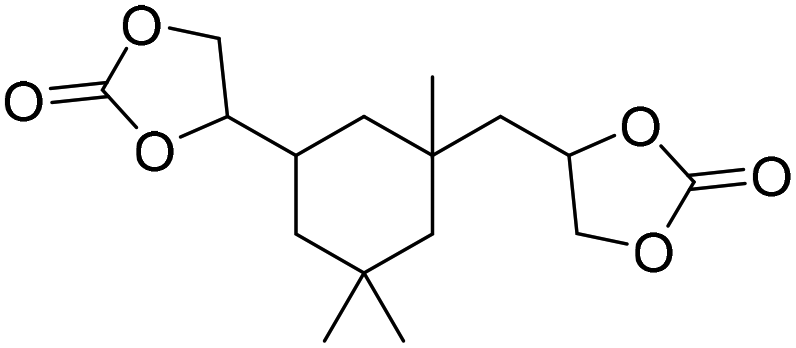
LiBr | 80 °C | 48 h | 1 bar | DMF | Hwang et al.45 |
|
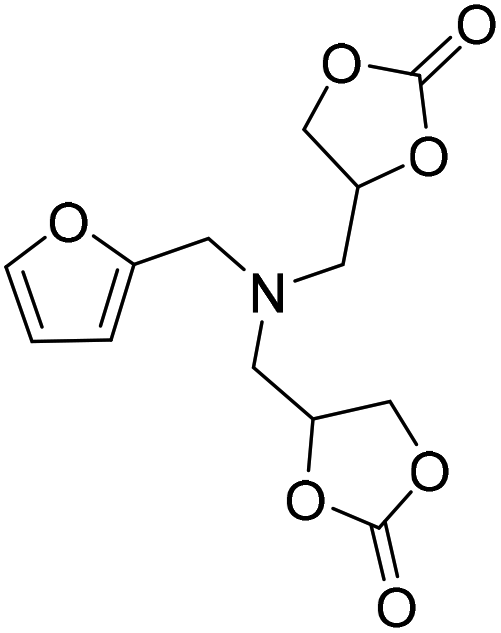
|
LiBr | 100 °C | 4 h | 5 MPa | DMF | Wulf et al.26 |
A synthetic route to produce PUs without isocyanate was reported by Chun et al., which described the usage of bis(cyclic carbonate)s and diamines to synthesize PHU compounds.45 Neopentyl glycol diglycidyl ether (NPGDGE) and resorcinol diglycidyl ether (RDGE) were initially converted into bis(cyclic carbonate)s through the simple reaction of the epoxides with CO2 (Fig. 16). The subsequent reactions used either isophorone diamine (IPDA) or 1,4-DAB, and each bis(cyclic carbonate). The production of all four different PHUs was quantitative. Then, using the DSC apparatus, the Tg of the non-isocyanate PHUs (NI-PHUs) were determined in order to evaluate their thermal properties. When RDGE was used as the diglycidyl ether rather than NPGDGE, the Tg of the NI-PHU was higher. Additionally, when IPDA was employed as the diamine as opposed to 1,4-DAB, the Tg of the NI-PHU was higher. As a result, the NI-PHUs created by IPDA displayed greater thermal stability, making them potential candidates for coating applications.
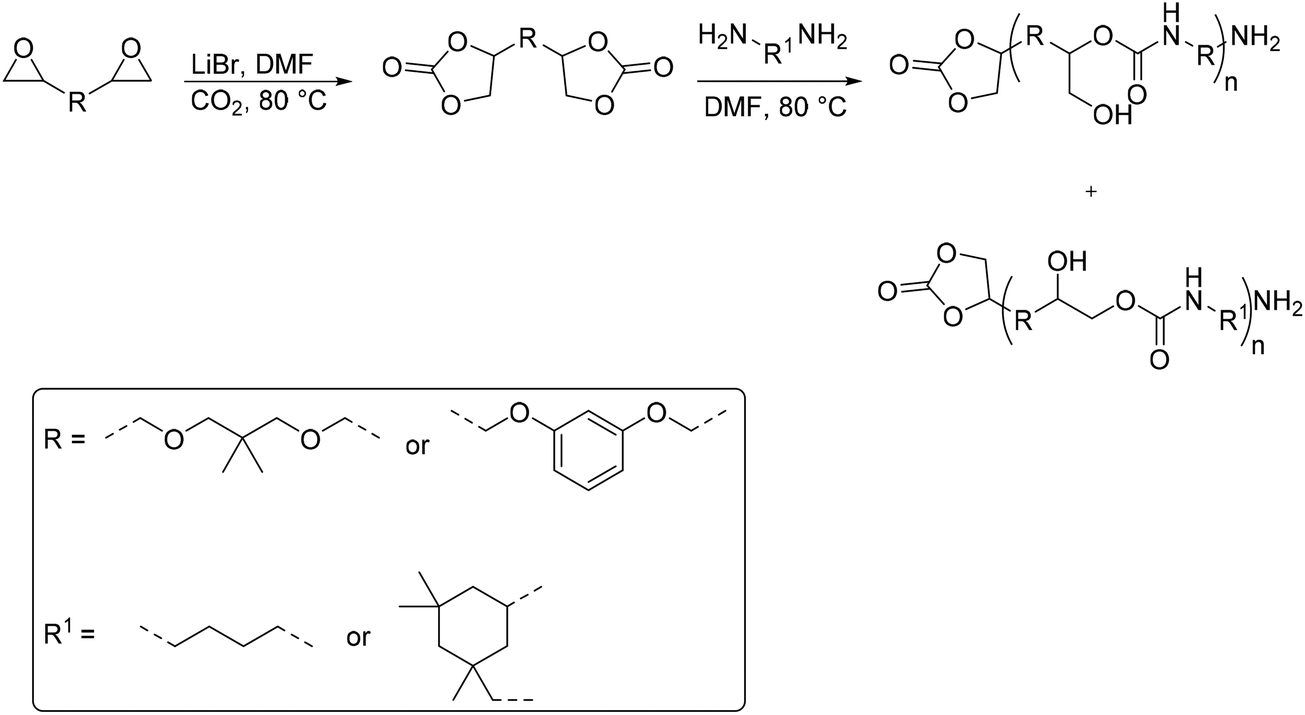 | ||
| Fig. 16 Preparation of the NI-PHUs. | ||
Among the most suitable polymers are epoxy resins, which have excellent mechanical strength, chemical resistance, heat resistance, and bonding ability qualities.46,47 Of course, epoxy resins have limitations, and to overcome these, several strategies have been employed, such as using reactive and non-reactive additives.48 Recently, a flexible aliphatic epoxy with urethane linkages was created using CO2 in a non-isocyanate, environmentally friendly method (Fig. 16). The synthesized aliphatic epoxy was cross-linked with rigid aromatic epoxy (DGEBA) to adjust its mechanical properties.49 Simply changing the ratio of two epoxies allowed for a wide range of mechanical property tuning, and the resulting epoxy composites displayed three distinct natures: a toughened polymer, an elastomer, and a pressure-sensitive adhesive (PSA). The viscoelastic and adhesive capabilities of the PSA epoxy were evaluated, and single lap shear, cyclic tensile, and Izod impact tests were performed for toughened or elastomeric epoxy composites. It has been proven that epoxy composites that have been toughened, made of elastomers, and resemble PSA tapes can be used in place of traditional structural adhesives, elastomers, and PSA tapes, respectively. Two kinds of aromatic and aliphatic epoxides were employed to provide adjustable mechanical qualities. An environmentally friendly technique was used to create aliphatic epoxy with urethane linkage (C-UME), as reported in Fig. 17. Polymer-supported imidazolium salts containing zinc were utilized for the manufacture of propylene carbonate from CO2.50,51
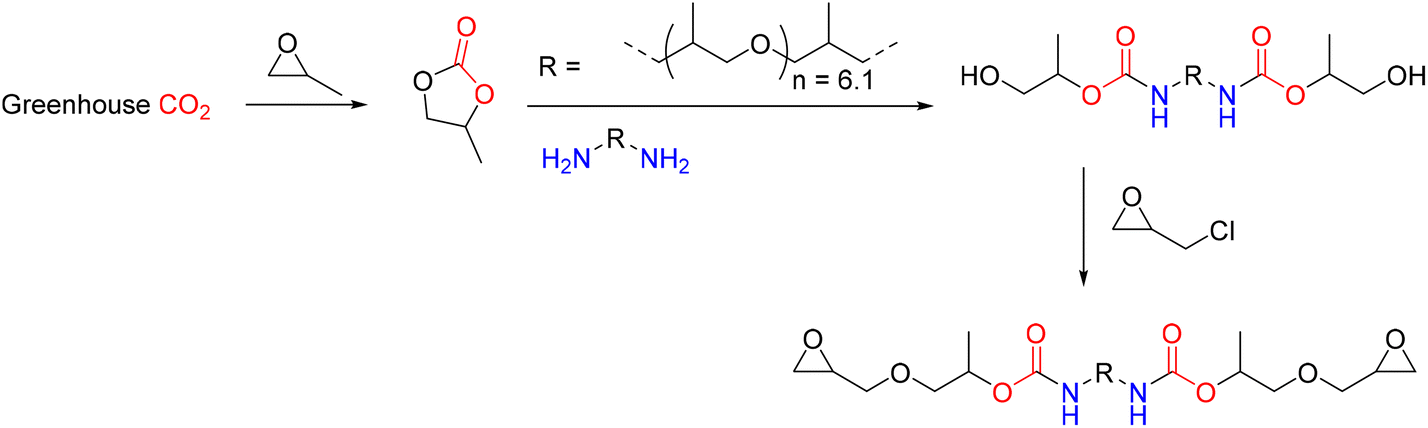 | ||
| Fig. 17 Synthesis of urethane-modified epoxy (C-UME) through an eco-friendly route. | ||
The propylene carbonate was allowed to react with polyoxypropylenediamine, and a brown-colored hydroxyurethane-terminated aliphatic diol incorporating urethane linkage was quantitatively obtained. This molecule was treated with epichlorohydrin, sodium hydroxide, and a catalytic amount of TBAB to obtain the C-UME. Bisphenol A diglycidyl ether (DGEBA), dicyandiamide, and 3,3′-(4-methyl-1,3-phenylene) bis(1,1-dimethylurea) were used as the aromatic epoxy, curing agent, and accelerator, respectively. To balance flexibility and rigidity, the content of C-UME and DGEBA was adjusted while keeping the mole ratio of the difunctional epoxy resin, curing agent, and accelerator constant at 100:28.6:1.43. The most flexible epoxy among the composites that had been made had PSA characteristics, and its adhesion performance (peel strength = 6.8 N cm−1) matched that of traditional PSAs. The prepared PSA's viscoelastic window implied it might be used as a detachable or all-purpose PSA.
A new family of PUs known as poly(oxazolidone) is readily accessible via an isocyanate-free method by step-growth copolymerizing primary diamines with CO2-based monomers, e.g., bis(-alkylidene cyclic carbonates), at room temperature (Fig. 18). Detrembleur et al. explored the scope of this procedure by looking at how the diamine used, and the reaction conditions affect the polymer's structure and macromolecular characteristics.52 The effects of different amine structures on the reaction's speed, yield, and selectivity were first examined using model reactions on small molecules (Fig. 18).
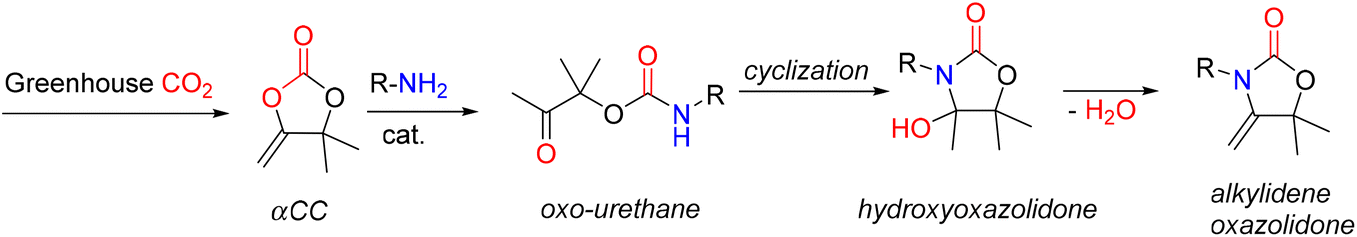 | ||
| Fig. 18 Model reaction of αCC. | ||
Both the ring opening of the cyclic carbonate and the intramolecular cyclization of the oxo-urethane intermediate into the hydroxyoxazolidone were shown to be affected by the amine structure. The hydroxyoxazolidone was primarily provided by aliphatic and benzylic amines, which explains why the intramolecular cyclization of the oxo-urethane happened quickly. However, cycloaliphatic amines produced both the oxo-urethane and the hydroxyoxazolidone products; it was thought that the steric hindrance of these amines caused the cyclization step to proceed more slowly. However, the addition of DBU as a catalyst sped up this process. The subsequent polyaddition of bis-a-alkylidene cyclic carbonate (aCC) with the diamines produced many poly(hydroxyoxazolidone)s (Fig. 19). According to the model reaction, the more sterically hindered cycloaliphatic diamines produced polymers with hydroxyoxazolidone and oxo-urethane links. Higher molar mass polymers were more likely to form when DBU was added. Aliphatic and benzylic diamines are provided exclusively as poly(hydroxyoxazolidone)s. Simply refluxing poly(hydroxyoxazolidone)s in glacial acetic acid made obtaining poly(alkylidene oxazolidone)s possible. Importantly, depending on the steric hindrance of the poly(hydroxyoxazolidone), two forms of oxazolidone connections with exocyclic olefin moieties were found. α-Alkylidene oxazolidone linkages were generated preferentially for the sterically hindered ones, but the less bulky ones showed a mixture of α- and β-alkylidene oxazolidone nearly in equimolar proportions. The unsaturated poly(oxazolidone)s showed a high thermal degradation temperature (Td > 370 °C) with a high Tg (90–130 °C), indicating that this novel family of polymers is highly appealing for applications requiring high temperatures, even though the Tg of the poly(hydroxyoxazolidone)s could not be measured due to low polymer degradation temperature.
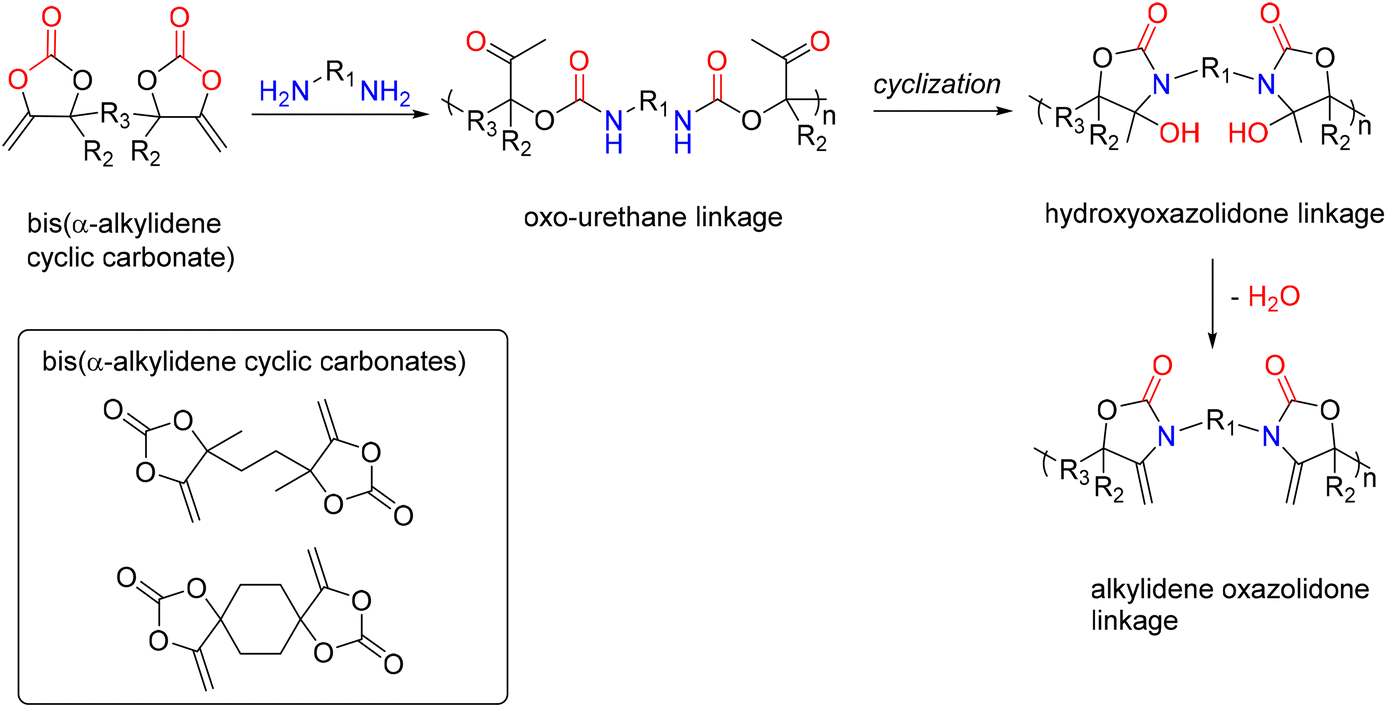 | ||
| Fig. 19 Polyaddition of bis(α-alkylidene cyclic carbonate)s to diamine. | ||
3. Fully biobased polyester
As mentioned, another strategy often used to produce PUs is by reacting polyols and polyisocyanates. Although many biobased polyols are offered commercially, very few bio-based isocyanates are accessible.53 Thus, PUs are only partially bio-based as they are synthesized from bio-based polyols and petroleum-based isocyanates. However, the transition to plastics and polymers made without fossil fuels brings new structures to market for various purposes.54 Bio-based polyols are typically synthesized from vegetable oils, carbohydrates, lignocellulose, proteins, or fatty acids.55,56Castor-based PUs were obtained through a non-isocyanate route using oligomeric ricinoleic acid (ORA) with variable average polymerization degrees depending on the catalytic conditions.57 Following esterification, epoxidation, cycloaddition with CO2, and subsequent curing with diamine, ORA intermediates allow the generation of various PHUs with various mechanical and thermal characteristics. Ricinoleic acid-based NIPUs were also obtained using aliphatic diamine, incorporating octa(aminopropyl) polyhedral oligomeric silsesquioxane (OAP-POSS).58 The integration of POSS units into the polymer consent to control the characteristics of the resulting material, improving its thermal stability.
By using two biscarbonates made from oleic acid methyl ester, internal carbonated fatty acid diester (ICFAD) and terminal carbonated fatty acid diester (TCFAD), and two diamines in bulk polyaddition, novel linear PUs with molecular weights up to 13500 g mol−1 and Tg of about −15 °C were obtained.59
From jojoba and castor oils, cyclic carbonate monomers with diverse functionalities were produced using thiol–ene coupling with thioglycolic acid and esterification with glycerin carbonate.60 By reacting the bifunctional cyclic oil-based carbonate with different amines, linear and cross-linked PHUs were obtained with Tg ranging from −45 to 20 °C.
An alternative approach involves polyols combined with sugar-derived 2,5-furandicarboxylic acid to produce fully biobased poly-hydroxylurethanes.61 By inserting urethane links into the polyester's backbone, it is possible to obtain thermoplastic polyhydroxy(ester-urethane)s (NIPHEUs), entirely made of bio-based non-isocyanate materials. This technique effectively improves these materials’ poor physical and mechanical qualities.62 The synthetic route started with glycerol carbonate (GC), 1,8-diaminooctane, and 1,12-diaminododecane, two aliphatic diamines that can be produced from renewable feedstocks through biotechnological processes. It led to the formation of two hydroxyurethane (HU)-tetraols, entirely bio-based (Fig. 20). Consequently, the dimethyl ester of 2,5-furandicarboxylic acid was reacted with HU-tetraols to produce NIPHEUs with vitrimer-like behavior. The as-obtained polyester-HU exhibited outstanding thermal behavior, thermally induced bond exchange driven by the transcarbamoylation reaction, and a hydrogen bonding network. This new class of NIPHEUs exhibits peculiarities that may open the door to developing a new assortment of valuable green materials.
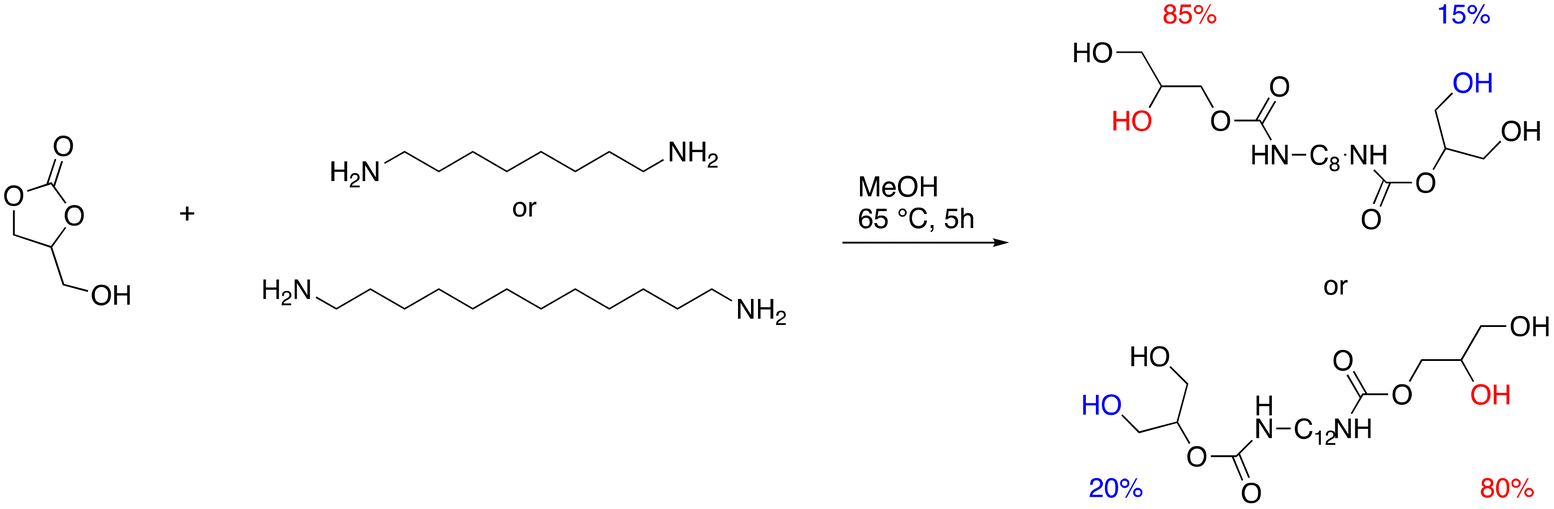 | ||
| Fig. 20 Synthetic route of HU-tetraol-C8 and HU-tetraol-C12. | ||
In addition to glycerol carbonate, pentaerythritol (PC) and trimethylolpropane (TMC) carbonates have been prepared to study carbonate functionality's role in NIPU formation.63 Since GC has a very low carbonate functionality and hinders amine-mediated cross-linking, it may be added to PEC as a reactive diluent, reducing resin viscosity and significantly enhancing the material's thermal and mechanical characteristics. Therefore, a blend of blends of cyclic carbonates with different carbonate functionalities improves Tg, temperature, stiffness, and strength.
NIPU derivatives have also been prepared using an environmentally friendly and safe method that employs CO2 and soybean oil.64 Epoxidized soybean oil (ESBO) (Fig. 21) was reacted with CO2 in the presence of a catalytic system consisting of TBAB and CaCl2 to synthesize cyclic carbonate (CSBO). Subsequently, CSBO was reacted with tetraethylenepentamine to obtain the corresponding NIPU. The free secondary amine product was treated with epichlorohydrin to produce cross-linked azetidinium networks (Fig. 21). The material was designed as wound dressing thanks to the 100% antibacterial performance offered by the cationic azetidinium groups against various bacteria. Neither the dressings nor their extracted leachates showed cytotoxicity against fibroblasts. The dressings can preserve damaged skin tissue from external forces, as they show the desired tensile properties.
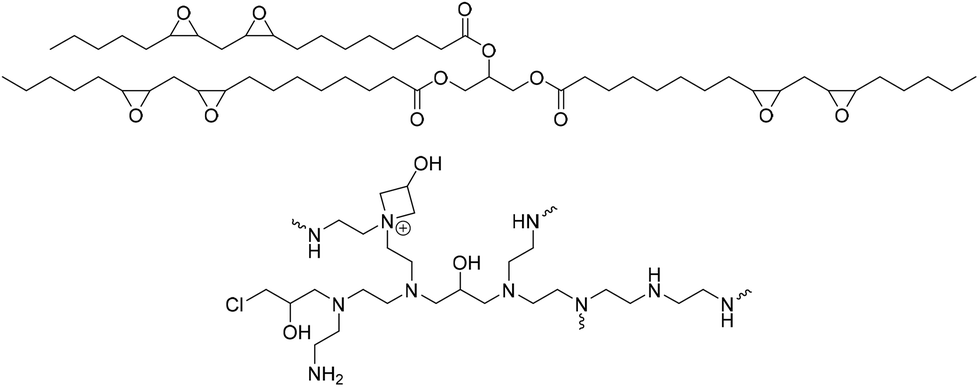 | ||
| Fig. 21 Structures of ESBO and NIPU with azetidimium groups. | ||
Epoxy soybean oil (ESO) has also been employed to produce partially bio-based NIPUs with a double dynamic network to reprocess and self-healing materials under mild conditions.65 Cyclic carbonates were synthesized from ESO by reactions with CO2, thus obtaining carbonate soybean oil (CSBO). The latter was then reacted with a diamine (4,4′-diaminodiphenyl methane (DDM) or 4,4′-diaminodiphenyl disulfide (DDS)) for the preparation of partially bio-based NIPU (CSBO-DDM and CSBO-DDS) without using catalysts or solvents; the reaction scheme is reported in Fig. 22. Furthermore, ESO was directly reacted with DDS containing disulfide bonds to prepare a material (ESBO-DDS) to explore the dynamic properties of the network formed by the disulfide bond. Relaxation experiments with external stimuli were performed to correlate the different bond exchange capacities to the different dynamic structures of the covalent network underneath. To quantify the self-repair capacity of NIPUs, scratches on the material surface were observed after several different heating or UV irradiation times. The repair of severely damaged materials was done thanks to the synergistic effect of the exchange of disulfide and carbamate bonds with a controlled heat press to obtain rework.
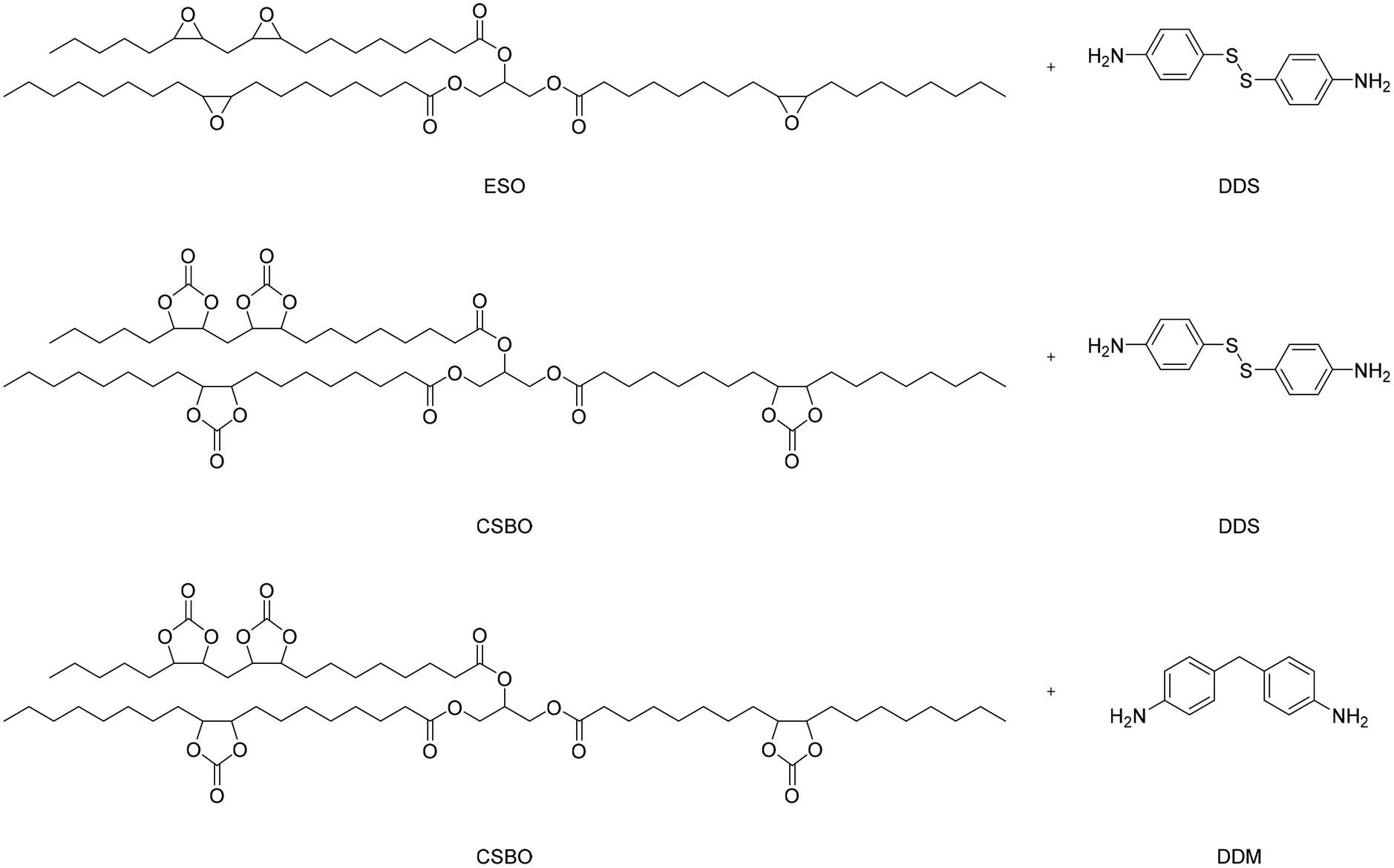 | ||
| Fig. 22 Reaction scheme for obtaining poly-oil (ESO-DDS) and partially bio-based NIPUs (CSBO-DDM and CSBO-DDS). | ||
The composition of NIPUs materials has a significant bearing on their characteristics.66 The poor mechanical characteristics of the NIPUs composed of CSBO and short diamines were attributed to cross-linking because of the high cyclic carbonate functionality of CSBO. By varying the degree of carbonation, it is possible to create NIPUs with tunable cross-linking densities, realizing material with improved mechanical properties. Moreover, by changing the curing time and material ratio for CSBO–DDM, NIPUs with improved mechanical and thermal properties can be obtained. It was found that a curing time of 3 days and a 1:1 ratio yielded the best results, giving rise to a material with suitable tensile strength and elongation at break. However, cross-linking density affects the Tg, and excessive rigid ring introduction causes issues.67
On the other hand, it was simple to create entirely bio- and CO2-sourced NIPUs with increased elongation break using a melt copolymerization process between CSBO and amino-telechelic oligoamide. The amount of cyclic carbonate in CSBO was correlated with its mechanical characteristics, particularly its tensile strength.
Rigid PU foam (PUF) was produced from Soybean meal biomass.68 Once the percentage of soluble carbohydrates in the biomass was determined, they were separated from the other components, such as protein, ash, and moisture. Then, the entire soybean meal (soymeal) was converted into bio-based polyol through two reaction steps. Initially, soymeal was reacted with ethanolamine in different ratios, resulting in various amino derivatives. The amino derivatives were then transformed into polyols by reacting with propylene carbonate without a catalyst. The process can be completed in 2–4 hours, saving money and avoiding problems with contamination or catalyst recovery problems. The reactions were conducted sequentially in the same reactor without further purification. Different mass ratios of soybean meal and ethanolamine (1:2, 1:3, 1:5) produced polyols with different hydroxyl values. In synthesizing bio-based rigid PUFs comprising 20% and 50% biobased polyols, soy flour polyol has been successfully employed. The compressive strength of a biobased PUF was greater than 200 kPa for a foam with a density of 40 kg m−3. Soymeal polyols may be produced at costs between $0.6 and $0.7 per pound, making their usage in commerce feasible. The performance characteristics of rigid PUF made from biomaterials were comparable to those of PUF made from commercial polyol. This offers a significant cost advantage and makes these soybean meal-based polyols the lowest-cost biobased polyols on the market.
Another biological resource used as the starting material for NIPUs was linseed oil (LO) (Fig. 23).69 Pouladi et al. conducted a study to exploit LO as a biological resource for synthesizing a resin with anti-corrosive properties. LO unsaturations were first converted into epoxy groups, followed by polycondensation and carbonation reactions to form cyclocarbonate groups in the presence of CO2 and TBAB as catalyst. The polymerization performance of the resins synthesized with the hardener diethylenetriamine (DETA) in different ratios was evaluated using various techniques such as DSC, TGA, and rheological tests. As the carbonation content increased, more hydroxyl groups were formed in the final polyhydroxylated PU structure. This was due to the reaction of the cyclocarbonate with the amine, which increases the polymer's adhesion strength and mechanical properties.
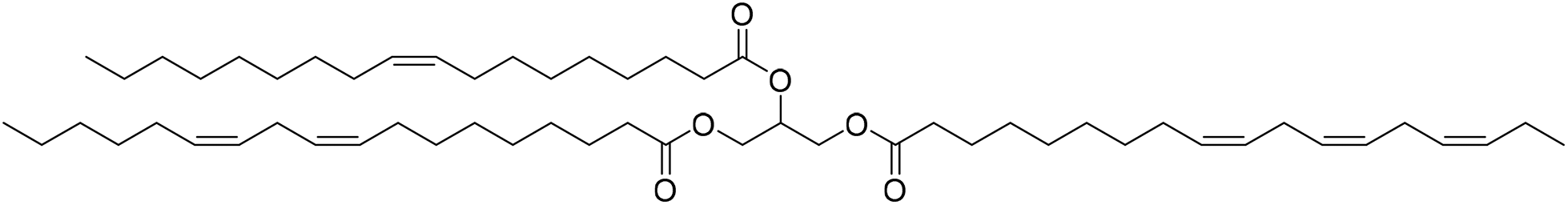 | ||
| Fig. 23 Chemical structure of linseedoil (LO). | ||
However, the conventional method that involves the production of NIPU from bicyclic carbonate and polyamine does not naturally produce foaming agents.70 Researchers recently used a new blowing agent to make high-density NIPU foams with large interconnected cells.71 Common blowing agents can worsen fire behavior, so there is interest in using nonflammable alternatives. Producing low-density NIPU foams with small, closed pores for thermal insulation remains challenging. In this view, low-density microcellular foams derived from bio and CO2 sources have been produced through supercritical carbon dioxide foaming technology.72 CO2-sourced monomers were created from poly(ethylene glycol) diglycidyl ether (PEG) and epoxidized soybean oil (ESBO) using a bicomponent organocatalyst. NIPUs were then synthesized through melt step-growth polymerization. The resulting CO2-blown microcellular NIPU foams exhibited low density, small pores, and low thermal conductivity. Also, obtaining NIPU with tailored cross-linking density is a challenge. However, Catalá et al. generated bio-based NIPU from triglyceride carbonates, produced in scCO2, and different diamines by using water or bioalcohols as capping substances to tailor the cross-linking density for specific applications.73 To improve the mechanical properties and thermal stability of bio-based NIPUs, isoeugenol-based 6-membered cyclic carbonate (iEbcc) has been chosen as a monomer thanks to the presence of rigid benzene ring structure and no flexible linkage groups between benzene ring.74 In this way, it is possible to obtain biobased and degradable NIPU with thermal stability up to 340 °C tensile stress of 64 MPa that can be reprocessed via transcarbamoylation reactions under elevated temperatures.
Tailor-made bis(cyclic carbonate)s cross-linked with the commercially available bio-based amine hardener have been used to produce, through a green path, NIPUs.75 A two-step process involving the epoxidation of bio-based polyether polyols (PO3G) with different molecular weights and the subsequent chemical fixation of CO2 allowed the production of a series of five-member bis(cyclic carbonates), subsequently used as monomers for the synthesis of NIPU. Initially, the polyether polyols were reacted with epichlorohydrin in the presence of BF3, after which the product obtained was reacted with NaOH to obtain the respective epoxides. The cycloaddition reaction with CO2 was then carried out in the presence of TBAB as a catalyst at 110 °C. Finally, the bis(cyclic carbonates) were converted to NIPUs by reaction with a bio-based amine, Priamine 1071, a mixture of dimer fatty diamine and trimer fatty triamine (Fig. 24). The synthetic methods employed focus on the design of processes and products that minimize negative environmental impact. In fact, the syntheses were attempted under solvent-free conditions with a view to green and sustainable polymer chemistry.
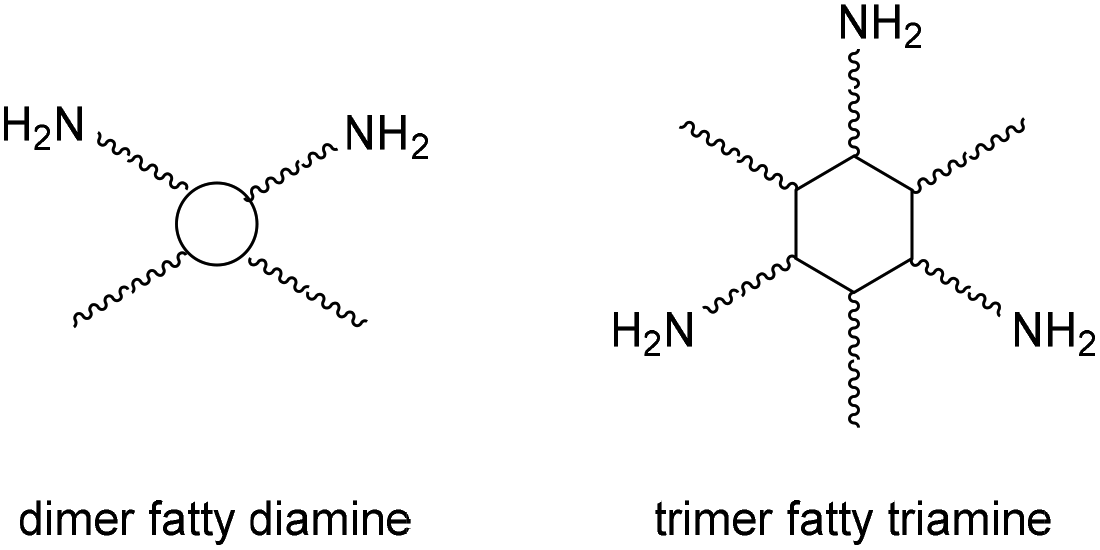 | ||
| Fig. 24 Structure of Priamine 1071. | ||
Western red cedar bark was also exploited as a raw material for the synthesis of NIPUs. The main constituents of the bark are lignin, cellulose, and tannins which are used as sources of hydroxyl functions.76 Experimentally, the bark was first subjected to the oxypropylation reaction in the presence of propylene oxide KOH, resulting in an OH-activated bark oil (HABO). The oil was then epoxidized, first by reaction with BF3 and epichlorohydrin and later with NaOH, to give Epox-HABO. Subsequently, the cycloaddition reaction with CO2 was carried out to obtain Carbonated-HABO. Finally, the latter was reacted with various amines, such as ethylenediamine (EDA), hexamethylene diamine (HMDA), isophorone diamine (IPDA), DETA, and tris(2-aminoethyl)amine (TAEA), to produce bark-based NIPUs. Because so many OH groups were produced when urethane was synthesized, NIPUs typically exhibit substantial moisture absorption.77 Thermal experiments showed that NIPUs were thermally stable up to 250 °C before the polyether segments and urethane connections began deteriorating.76
Epoxidation of PBDs, polymers with a high content of double bonds, generates PE-PBDs by reaction with hydrogen peroxide.78 Using bis(triphenylphosphine)iminium (PPN) or tetra-n-butylammonium (TBA) salts with chloride, bromide, and iodide as counter ions and CO2 as reagent the cycloaddition reaction is performed to produce the corresponding carbonates PC-PBDs. They are used to achieve cross-linking reactions of NIPUs using bio-derived diamines.
Finally, PBD-based NIPUs [P(NIPU)-PBDs] were synthesized by the reaction of PC-PBD samples with 1,5-pentanediamine (PDA) or 1,8-octanediamine (ODA). PC-PBD was mixed with diamine (in a molar ratio of 1:1 or 1:2 relative to the epoxide content) and polymerized by heating in a nitrogen atmosphere for 16 hours (Fig. 25). In the PUs obtained, the carbonate units contained in the incompletely polymerized networks show stabilizing effects, presumably through interactions between urethane and carbonate.
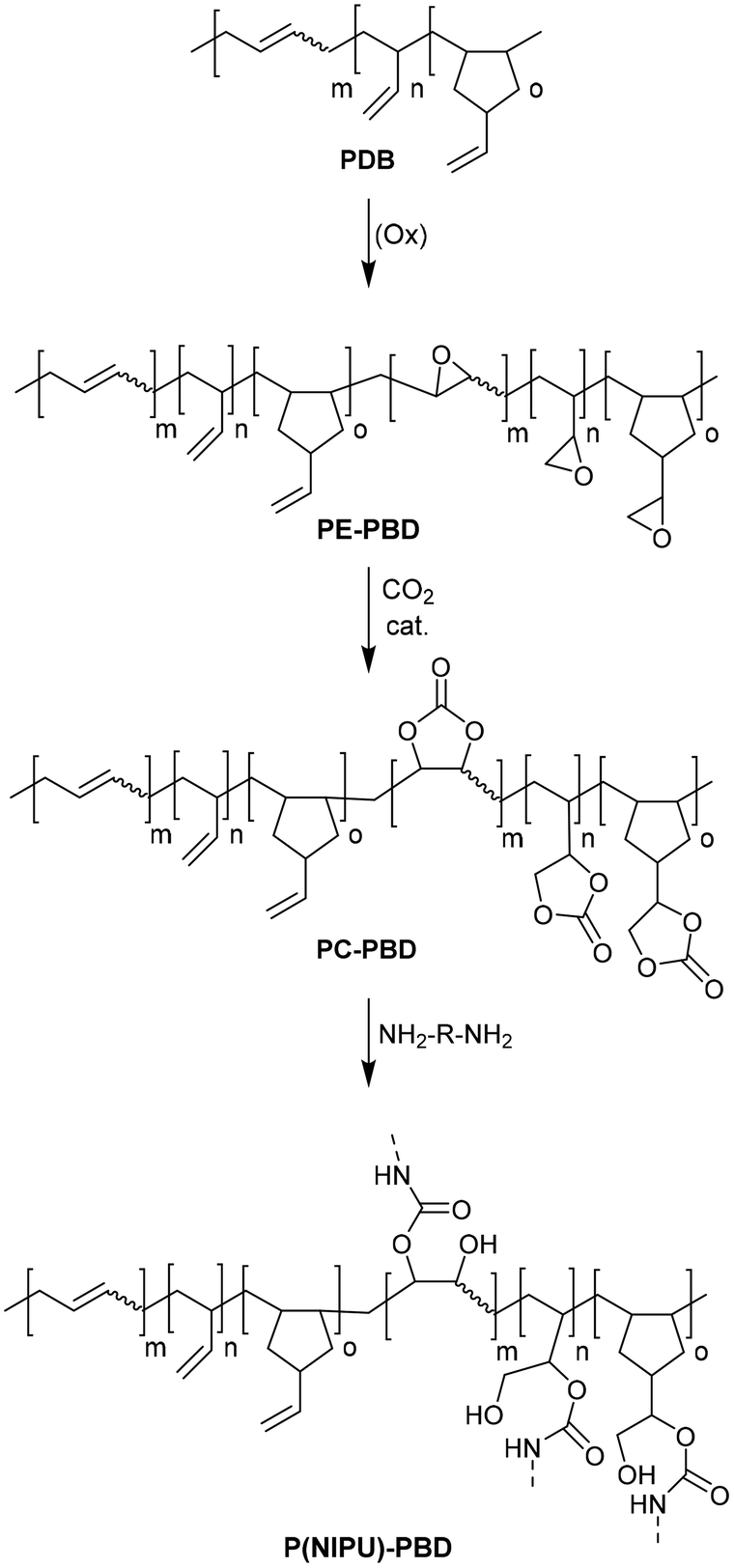 | ||
| Fig. 25 Reaction sequence for the preparation of P(NIPU)-PBD polymers. | ||
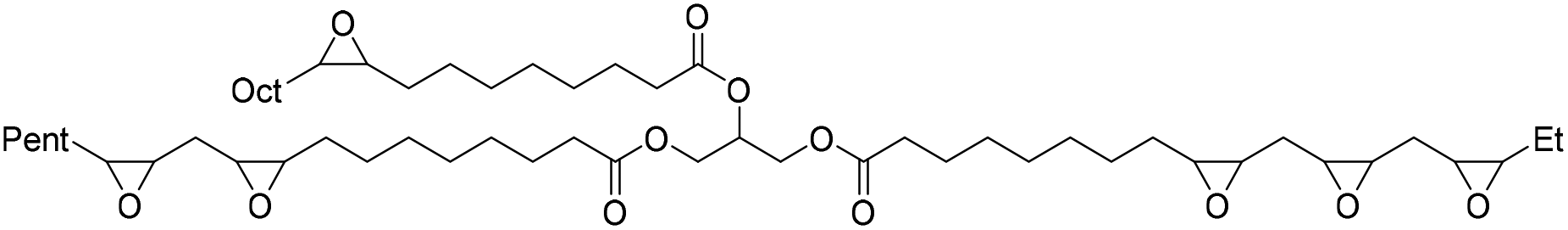
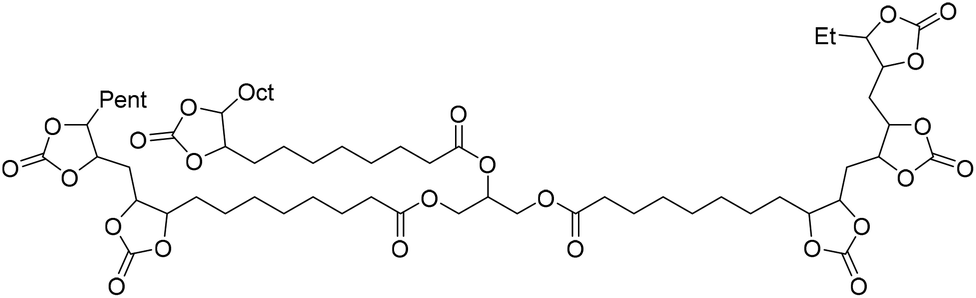
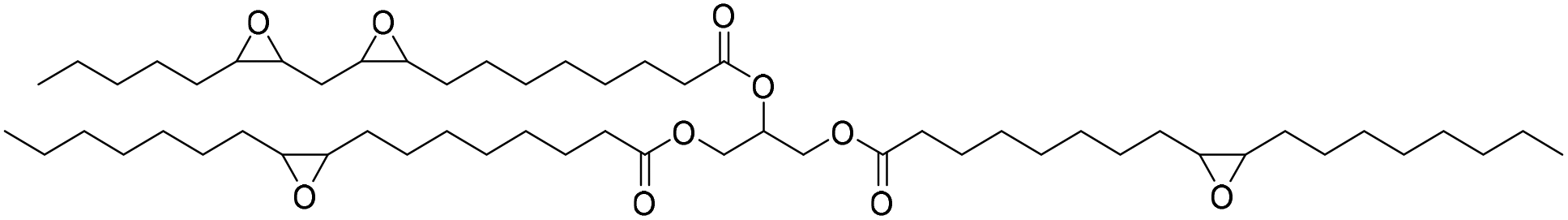
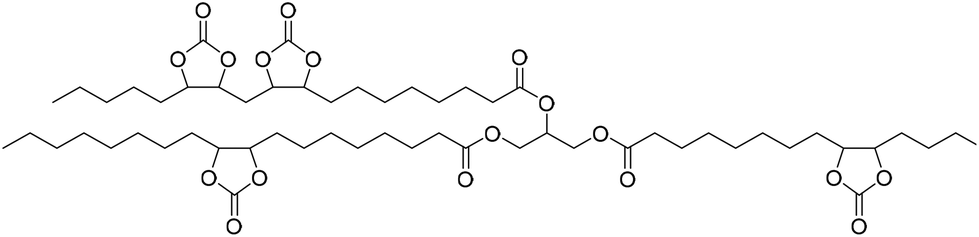
Fish oil (FO), derived from aquaculture waste, is a potential alternative to vegetable-based oils. The FO is extracted from by-products produced in fish processing plants, including heads, bones, skin, and entrails. The United Nations has emphasized aquaculture as a sustainable method of producing protein. However, this is only true if the entire fish is used, not just the expensive fillets sold as food. Epoxidized FO can react with CO2 to produce cyclic carbonates.79 Three different methods are used for the epoxidation of FO: (i) oxidation with 3-chloroperoxybenzoic acid, (ii) oxidation with hydrogen peroxide and acetic acid, catalyzed by sulphuric acid, and (iii) oxidation with hydrogen peroxide and acetic acid and (iv) oxidation with hydrogen peroxide catalyzed by formic acid. The synthesized FO epoxides are reacted with CO2 in the presence of TBAB as the catalyst and ascorbic acid to produce cyclic FO carbonates with high conversions (Fig. 26). Subsequently, the obtained carbonates are reacted with a biomass-derived amine (NC-540 or 4,7,10-trioxa-1,13-tridecanediamine) to give bio-derivative amine PU materials.
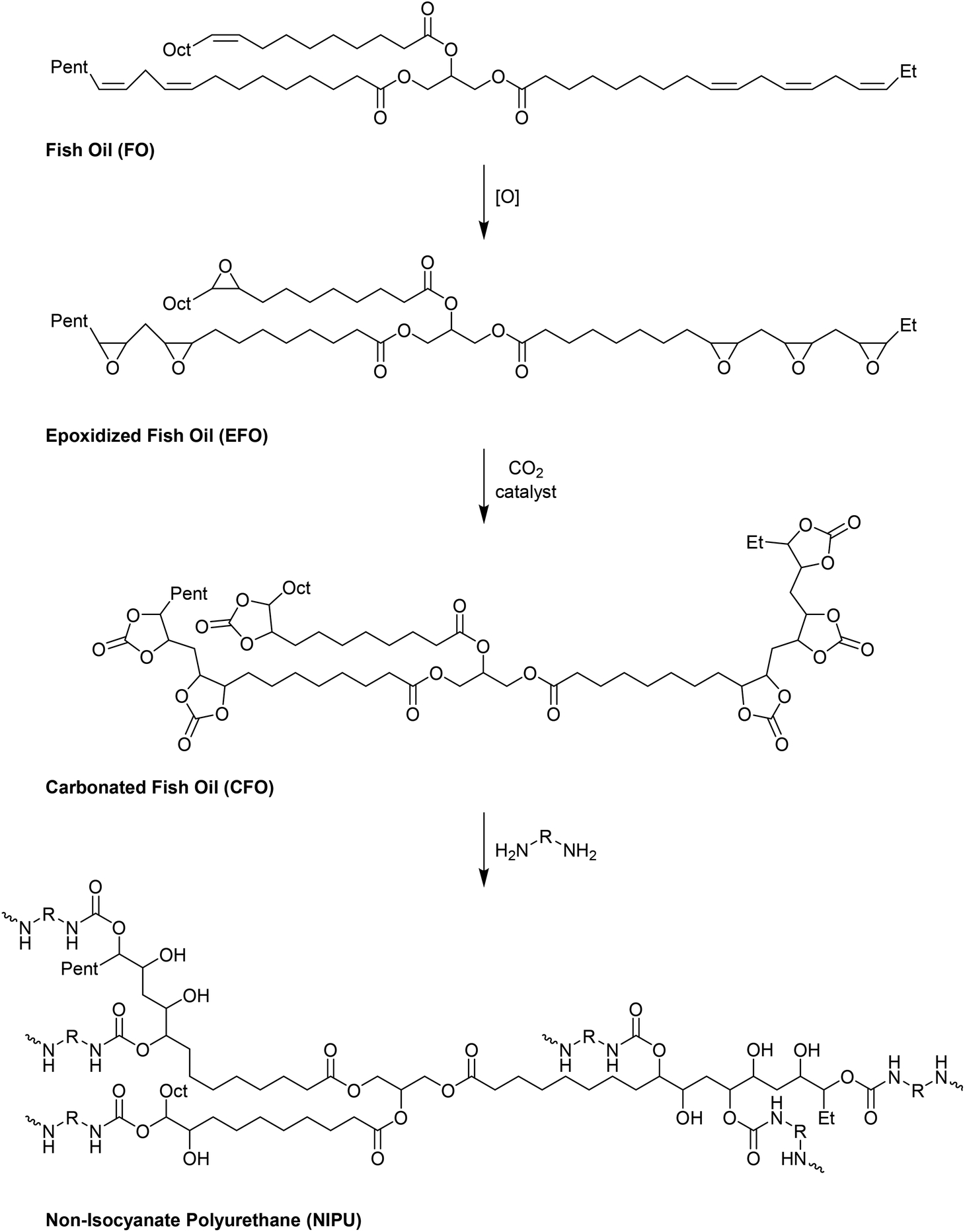 | ||
| Fig. 26 Synthesis of carbonated fish oil (CFO) from waste-derived fish oil (FO) and preparation of FO-based NIPU from CFO and diamines. | ||
The starting epoxide used and the corresponding synthesized cyclic carbonate, with some remarks on the reaction, are summarized in Table 2.
| Starting epoxide | Cyclic carbonate prepared | Fixation of CO2 conditions | Ref. |
|---|---|---|---|
|
|
Catalyst TBAB/ascorbic acid | Laprise et al.79 |
| T 110 °C | |||
| t 48 h | |||
| Pressure 10 bar | |||
| Solvent neat | |||
|
|
Catalyst TBAB/CaCl2 | Yang et al.67 |
| T 140 °C | |||
| t 32 h | |||
| Pressure 40 atm | |||
| Solvent neat | |||
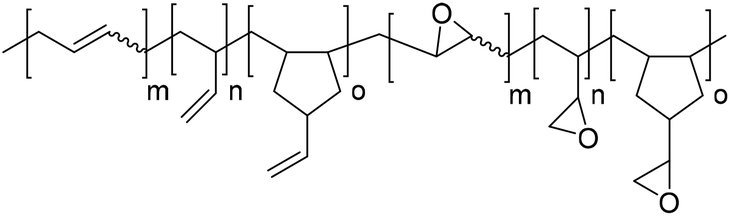
|
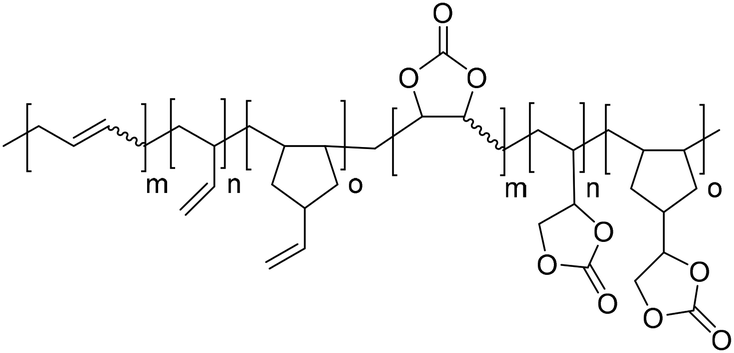
|
Catalyst bis(triphenylphosphine)iminium chloride (PPN-Cl) | Dechent et al.78 |
| T r.t. | |||
| t 24–72 h | |||
| Pressure 20 bar | |||
| Solvent MEK |
4. NIPU from oxidative carbonylation
Another possible route for the non-phosgene production of isocyanate precursors was reported as the oxidative carbonylation of toluene-2,4-diamine (TDA) with methyl formate (MF), which can be produced from CO2.80 The findings of this study demonstrate that a green non-phosgene route to PUs is indeed possible by the oxidative carbonylation of industrially important diamines using CO2-based MF as a reagent. In this context, a thorough study has been done on the Pd-catalyzed oxidative carbonylation of TDA with MF, identifying the most crucial side products aside from toluene-2,4-dicarbamate (TDC) as the target product. Three distinct routes that result in side products have been identified (Fig. 27).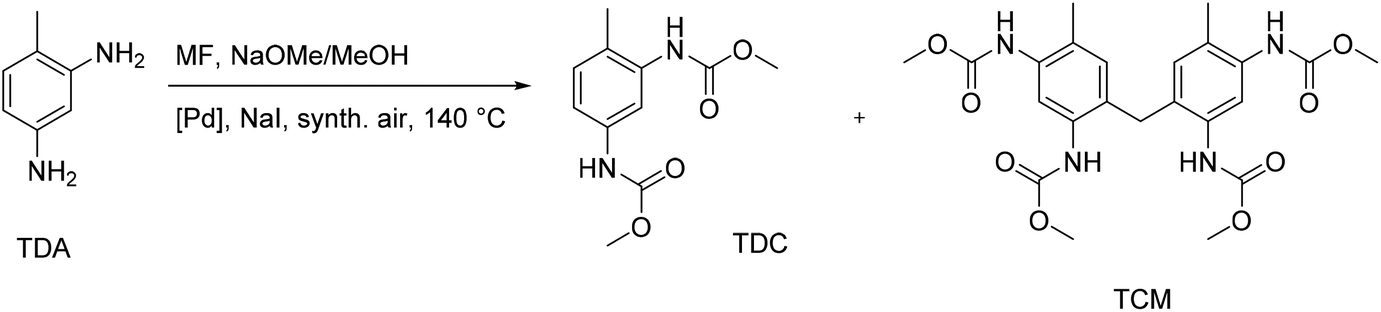 | ||
| Fig. 27 Oxidative carbonylation of TDA with MF and major subproduct (TCM). | ||
First, formamides are produced via a nucleophilic substitution at the MF carbonyl group without using a Pd catalyst or oxygen. Second, the formaldehyde produced in situ underwent electrophilic aromatic substitution/condensation to give the methylene-bridged tetracarbamate (TCM).81 The TCM dimer is particularly interesting for industrial use due to its potential and makes an appealing target product. Thirdly, unintended N-methylations were brought on by a reaction with the dimethyl carbonate also created during the oxidative carbonylation with MF. PdCl2/CuCl2 and several heterogeneous Pd-catalysts were investigated for the catalyst's impact on product distribution. The discovery of partial suppression of the undesirable side reactions by the oxidic support materials ZrO2, CeO2, and SiO2 led to better yields of TDC and TCM. The yields of TDC and TCM together were up to about 50%, and higher TDC yields were correlated with lower TCM yields.
The method of oxidative carbonylation with MF was extended to other diamines, 4,4′-methylenedianiline (MDA) and 2,4-diaminomesitylene (DAS), and the identified products of the reaction are reported in Fig. 28 and 29.
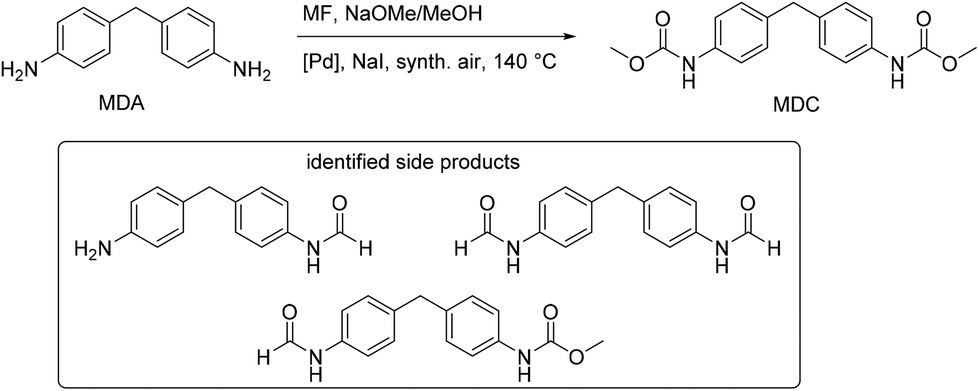 | ||
| Fig. 28 Oxidative carbonylation of MDA with MF. | ||
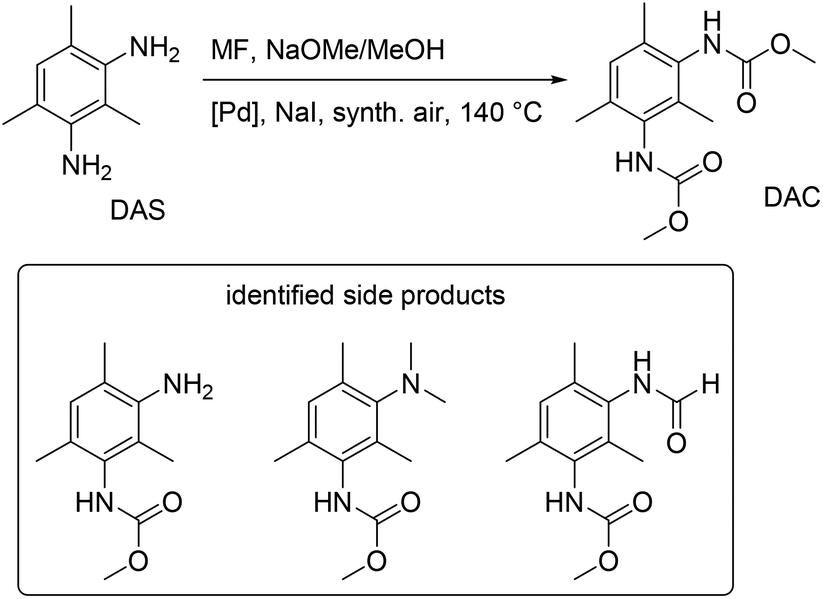 | ||
| Fig. 29 Oxidative carbonylation of DAS with MF. | ||
5. Self-healing NIPUs
Artificial materials are vulnerable to deterioration in the form of cracking or microcracking.82 Microcracks can expand and merge, which shortens the material service life and increases the risk of catastrophic breakdown of the materials. Early microcrack diagnosis and repair prevent these issues and extend the service life.In these situations, self-healing materials can be used since they can identify cracks early and automatically fix them.83 Additionally, these materials may increase material dependability, lengthen service life, lower replacement costs, and increase product safety. Numerous research investigations on the creation of self-healing systems are carried out each year due to these appealing characteristics, and polyurethane chemistry is involved in several ways84 since the first studies in self-healing materials.85 A comprehensive review of the field is out of the scope of this review, but an examination of the most advanced polyurethane-based self-healing material will be provided.
One of the approaches to give a material self-healing properties is to exploit a reversible covalent bond formation. Diels–Alder reactions have been used with recyclable PU nanocomposites,86,87 and photochemical [2 + 2] cycloaddition have been used with coumarin-based PU.88,89 Disulfide (S–S) exchange reactions and S-aromatic thiourethane exchange reaction have been reported with PU nanocomposite90 and polythiourethane networks,91 respectively. Transesterification is another example of a reversible reaction employed for self-healing thermoplastic PU.92 Hydrogen bonds can be used in addition to tuning the mechanical properties of materials. In a few examples, they have been used to the self-healing extent in PU elastomer93 and to control host–guest interactions between cyclodextrin and adamantane in thermoplastic PU prepared from hexamethylene diisocyanate and tetraethylene glycol polymer hydrogel of poly(N-isopropylacrylamide).93–95 Hydrogen bonds control has also been coupled with the already discussed Diels–Alder, where multi-arm UV-cured PU, N,N′(4,4′-methylenediphenyl)bismaleimide, and furfuryl alcohol have been reacted.96
Polymeric materials can also recover their properties through physical interactions. Polymer blends and remote self-healing have been used as healing mechanisms for thermosetting vitrimer with thermoplastic PU,97 PU nanocomposite, and carbon nanotubes,98 respectively. Microvascular networks and nanoparticle-based self-healing have also been reported for PU-based materials.99,100
In 2016, Caillol et al. created a thermo-responsive CO2-based PU by converting furfuryl glycidyl ether into a cyclic carbonate derivative and combining it with dimaleimide-terminated polypropylene glycol and the necessary diamine. Due to the breakdown of Diels–Alder adducts, the resulting PU may shift from a solid to a liquid state, which is desired for many thermo-reversible material applications.101 According to the literature, one of the most promising reactions for self-healing polymer materials is the Diels–Alder process.102 Based on this consideration, Zhan et al. created a self-healing and recyclable CO2-based polymer with exceptional strength qualities by establishing stronger cross-linking bonds via the Diels–Alder reaction from CO2 and furfuryl amine. The more excellent dynamic covalent connections, the combination of ordered and disordered hydrogen bonding interactions, and the suitable amounts of cross-linked Diels–Alder groups would provide higher mechanical strength and stability to the newly synthesized material.103 The synthetic green route to recyclable polymer PU-urea (PUUa) synthesis started from the polyaddition reaction between the furfuryl amine cyclocarbonate derivative and oligourea. Then PUUa was cross-linked by the Diels–Alder reaction between furfuryl and 4,4-bismaleimidodiphenylmethane (BIM). Meanwhile, the PUUa Diels–Alder adduct showed excellent self-healing capability and recyclability. The present green route is effective at building strong and tough self-healing and recyclable polymer PUUa started from bis(cyclic carbonate) furfuryl amine (BCCFA) and oligourea (OUa) through a polyaddition reaction, as illustrated in Fig. 30. It was worth noting that BCCFA or OUa were synthesized using CO2 as a reagent. Finally, the reaction between furan and maleimide groups of PUUa generated the Diels–Alder adduct PUUa-DA. The PUUa-DA adduct is a challenging and self-healing thermosetting material compared with PUUa. The retro-Diels–Alder reaction, which takes place at 120 °C, converts PUUa-DA into PUUa, a thermoplastic polymer that melts at that temperature. Then, the broken hydrogen bond in PUUa was recombined, and the cross-linking groups through the Diels–Alder reaction were restructured at 60 °C. Since the retro-Diels–Alder reaction is inefficient at a healing temperature lower than 108 °C, PUUa-DA can be considered a thermosetting material without melting and cannot be healed efficiently at a temperature below 108 °C.
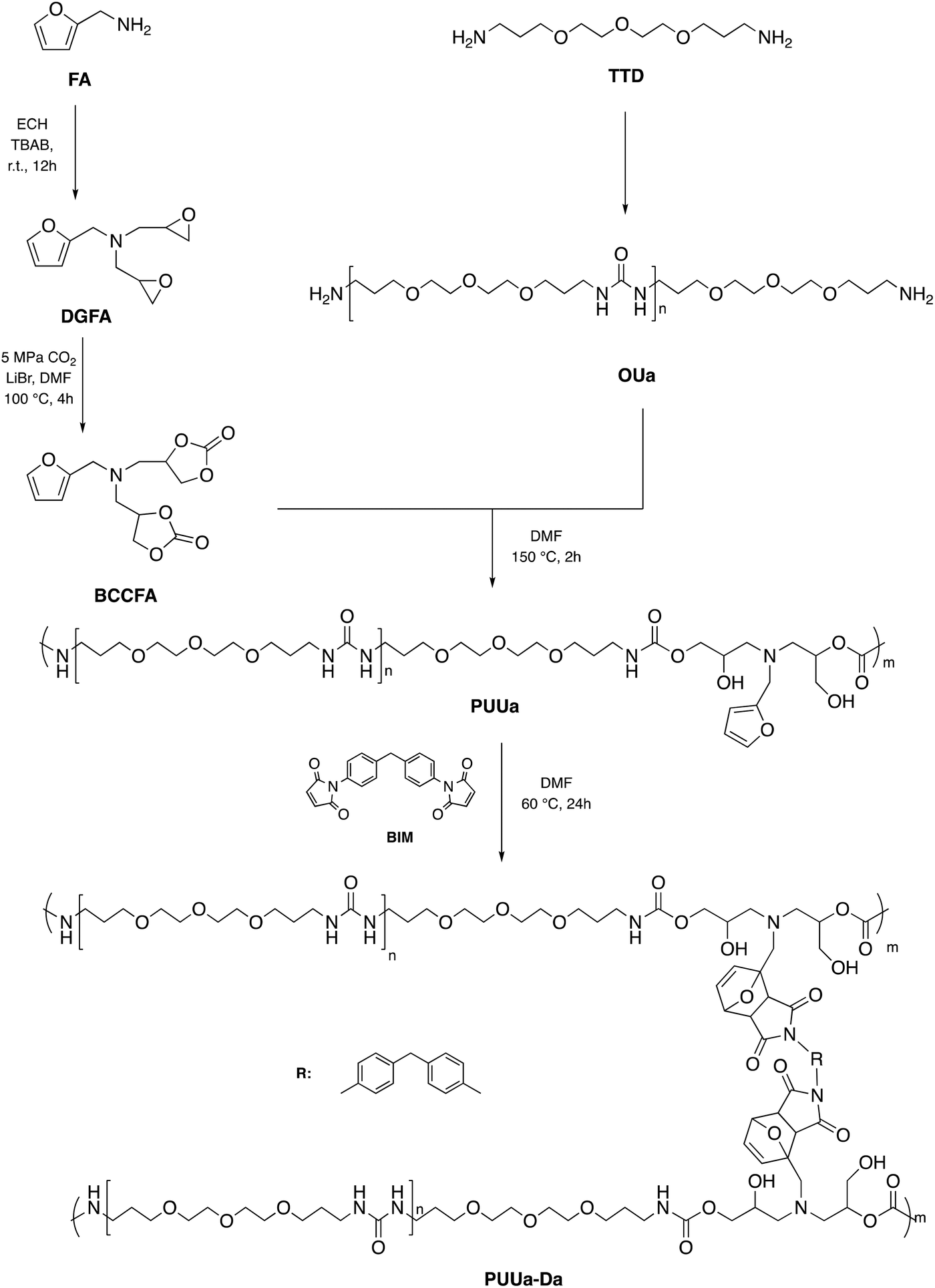 | ||
| Fig. 30 Synthesis of non-isocyanate polyurethane-urea (PUUa) from CO2, and sequential reaction of PUUa with 4,4-bismaleimidodi-phenylmethane (BIM) to the polyurethane-urea diels-alder adduct (PUUa-DA). | ||
NIPU coatings, which are recyclable and healable via three different healing mechanisms, have recently been developed from bio-CO2-derived materials.103 Four different novel NIPUs, namely, poly(BBC-FBA), poly(FBC-FBA), poly(FBC-DAP), and poly(SuBC-FBA), were synthesized by the reaction of phenoxycarbonyloxymethyl ethylene carbonate (BBC) or hydroxymethylfuran bis(cyclic carbonate) (FBC) or succinate bis(cyclic carbonate) (SuBC) and commercially available 2,5-bis(aminomethyl)furan (FBA) or 1,5-diaminopentane (DAP) in DMF at 70 °C (Fig. 31).104,105
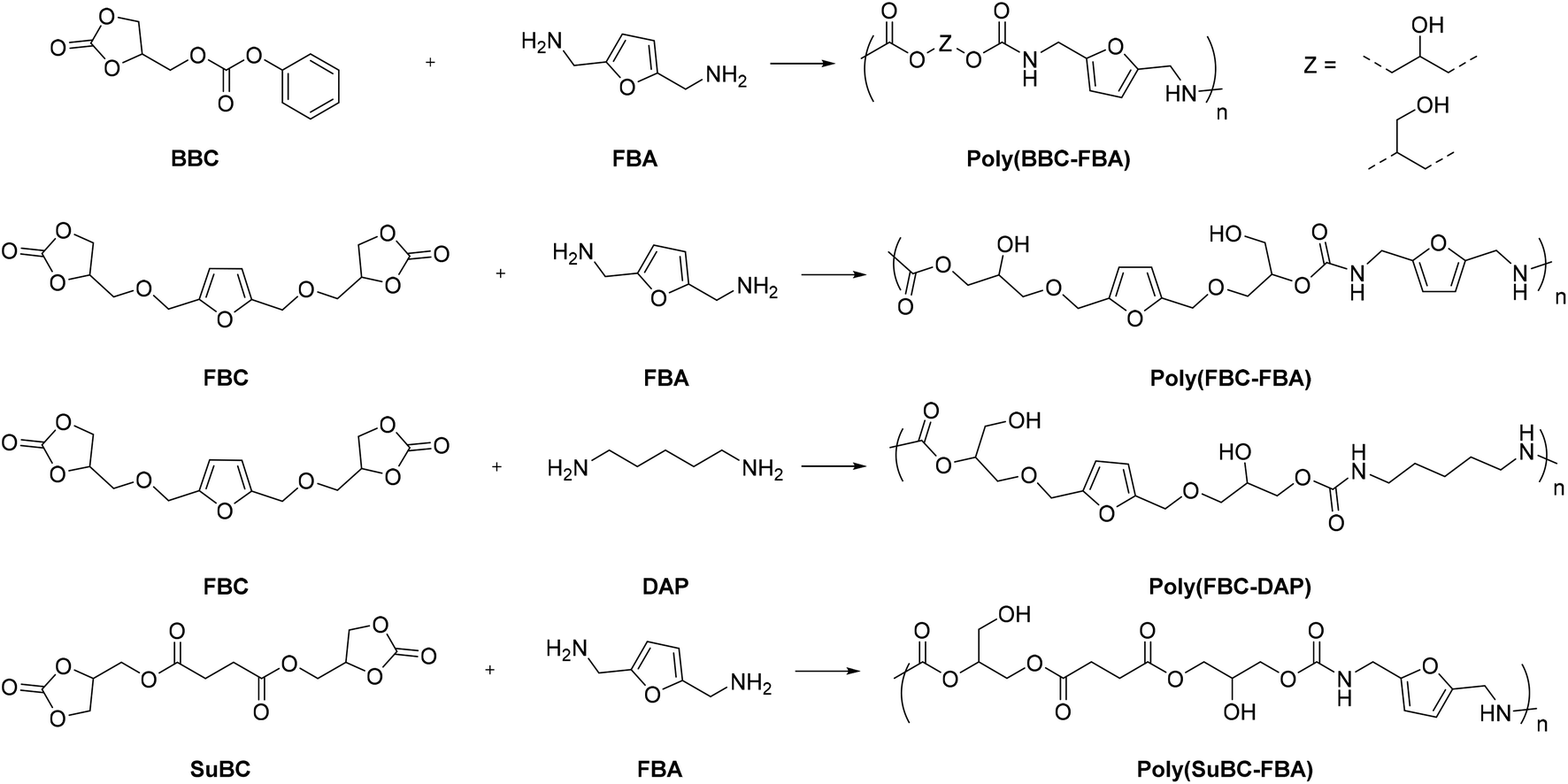 | ||
| Fig. 31 Synthesis of main-chain furan-containing NIPUs by utilizing furan-based bis(cyclic carbonate) and diamine. Reaction conditions: DMF, 70 °C, up to 48 h. | ||
Interestingly, these polymers could react with bismaleimides (BM) via a furan-maleimide Diels–Alder reaction to produce a cross-linked network containing a DA adduct since furan was present in their major chains (Fig. 32).
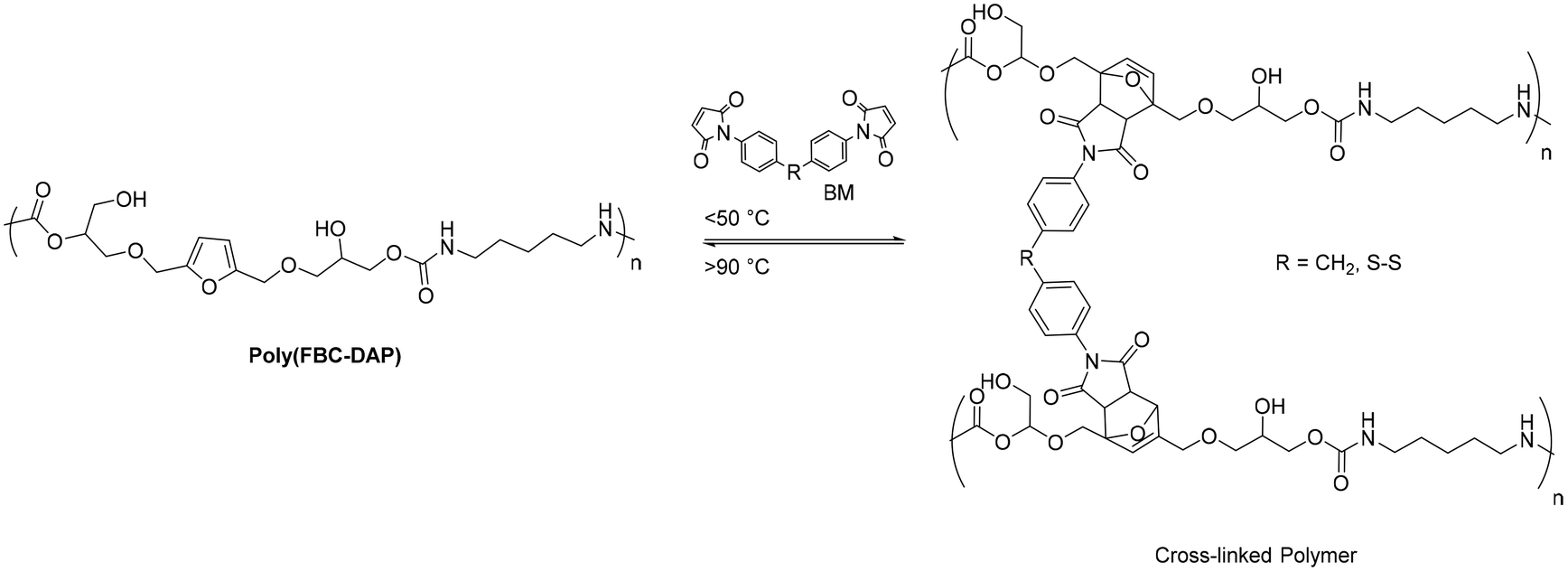 | ||
| Fig. 32 Diels–Alder and retro-Diels–Alder thermoreversible reactions of poly(FBC-DAP) cross-linked with bismaleimide. | ||
The primary chain of these NIPUs, when cross-linked with bismaleimides, produces organogels with thermo-reversible sol–gel transition and solvent-borne coatings with enhanced characteristics. By carefully choosing the bismaleimide cross-linker structure, it was possible to create recyclable coatings that naturally heal using heat, moisture, and, more intriguingly, dry circumstances at room temperature with the structure reported in Fig. 33.
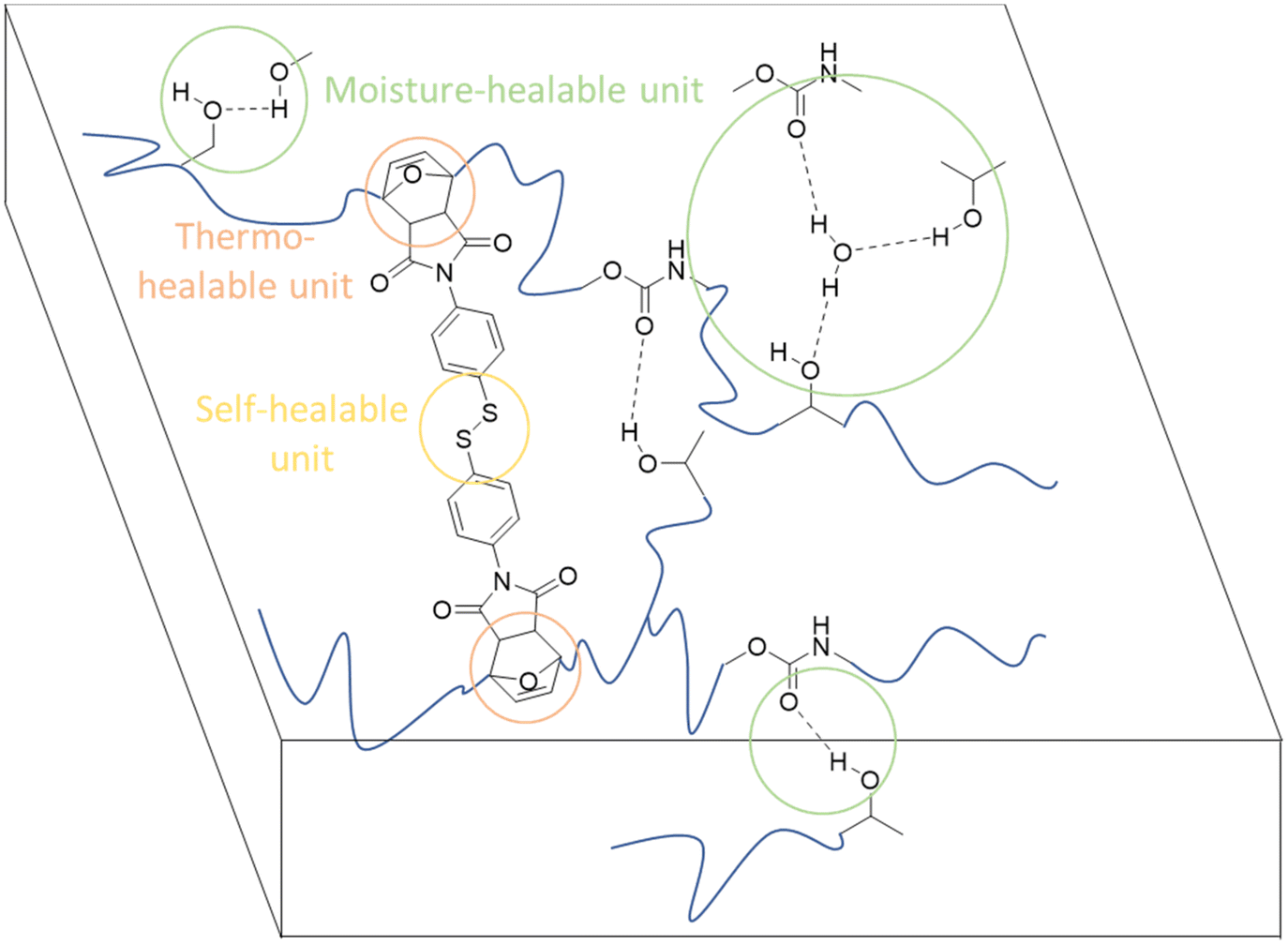 | ||
| Fig. 33 Cross-linked NIPU coatings have qualities that allow them to be thermo-, moisture-, and self-healing. | ||
The hydro-plasticization and moisture-healing properties of coatings based on NIPUs were characterized for the first time. These NIPUs properties, which set them apart from more traditional PUs, are principally due to the presence of hydroxyl groups in their structure. A careful examination also reveals how Tg and chain flexibility impact the healing efficiency.
These results highlight the necessity of combining various dynamic, reversible cross-linking processes to create practical coating materials.
6. Conclusion
The present review highlights recent years’ considerable efforts to (i) produce PU without using toxic reagents such as isocyanates and (ii) improve their circular character by following different strategies. This fundamental shift in PU chemistry positions NIPUs as a sustainable choice, a facet that sets them apart from conventional PUs. NIPUs offer a paradigm shift with improved safety profiles, lower toxicity, and a reduced environmental footprint. Their exceptional mechanical properties, thermal stability, and versatility open doors to diverse adhesives, coatings, foams, and more applications.A circular design seems mandatory; it ranges from the synthesis of monomers to the PU's use-life and end-of-life, closing the loop on a greener and more sustainable PU industry. In particular, the creation of sustainable value chains in the PU field requires a multidisciplinary approach with respect to the selection of starting materials (of biological origin), the processes (synthesis), and the dismantling of the polymer at the end of its life (recycling). Introducing biobased, compostable, and renewable building blocks and optimizing PU production processes are necessary to reduce the environmental footprint. One option discussed in this review involves chain extension as a way to meet the molecular weight requirements of PUs. Alternatives to isocyanates are known to provide low-molecular-weight polymers. Among the various strategies for PU cross-linking, using Diels-Alder and retro Diels–Alder reactions represents a great opportunity and a real option.
Another deeply discussed point is increasing the green character through environmentally friendly processes. A significant advancement in this effort has been the utilization of CO2 as the C1 building block for the synthesis of organic molecules. CO2 has been used as the C1 unit for synthesizing organic molecules. CO2 represents an ideal reagent due to its beneficial properties, such as its high abundance, non-toxicity, and renewability, all of which align with the principles of green chemistry and sustainability.
The coupling reaction of carbonates and aliphatic amines has emerged as the most promising approach for producing NIPUs with enhanced green credentials. Several catalysts and processes have been described, but only the combination of using CO2 to obtain cyclic carbonates, their ring opening, and cross-linking via [4 + 2] cycloaddition could provide PUs with mechanical, thermal, morphological, and chemical properties comparable to the classic PUs obtained via the isocyanate route. By making this circular connection from CO2 utilization in monomer synthesis to the end-of-life of NIPU products, we create a holistic and sustainable framework that improves the environmental footprint of polyurethanes and aligns with the broader goals of reducing carbon emissions and promoting a circular economy.
Abbreviations
| 1H-NMR | Proton nuclear magnetic resonance |
| aCCs | Bis-a-alkylidene cyclic carbonates |
| BADGE | Bisphenol A diglycidyl ether |
| BBC | Phenoxycarbonyloxymethyl ethylene carbonate |
| BCCFA | Bis(cyclic carbonate) furfuryl amine |
| BDC | [4,4′-Bi(1,3-dioxolane)]-2,2′-dione |
| BDGE | 1,4-Butanediol diglycidyl ether |
| BIM | 4,4-Bismaleimidodiphenyl-methane |
| C-UME | Urethane-modified epoxy |
| CANs | Covalent adaptable networks |
| CFO | Carbonated fish oil |
| CSBO | Carbonated soybean oil |
| CTAB | Cetyltrimethyl-ammonium bromide |
| D230 | Poly(propylene glycol) bis(2-aminopropyl ether) |
| DAP or PDA | 1,5-Diaminopentane |
| DAS | 2,4-Diaminomesitylene |
| DBU·I2 | 1,8-Diazabicyclo[5.4.0]undec-7-ene iodine |
| DDM | 4,4′-Diaminodiphenyl methane |
| DDS | 4,4′-Diaminodiphenyl disulphide |
| DETA | Diethylenetriamine |
| DGcHdC | Diglycidyl 1,2-cyclohexanedicarboxylate |
| DGEBA | Bisphenol A diglycidyl ether |
| DIC | N,N-Diisopropylcarbodiimide |
| DMC | Double metal cyanides |
| DMSO | Dimethylsulfoxide |
| DSC | Differential scanning calorimetry |
| DTG | Differential thermogravimetry |
| EcHMEcHC | 3,4-Epoxycyclohexylmethyl 3,4-epoxycyclohex-anecarboxylate |
| EDA | Ethylenediamine |
| ESBO | Epoxidized soybean oil |
| ESO | Epoxy soybean oil |
| FBA | 2,5-Bis(aminomethyl)furan |
| FBC | Hydroxymethylfuran bis(cyclic carbonate) |
| FO | Fish oil |
| FT-IR | Fourier-transform infrared spectroscopy |
| GC | Glycerol carbonate |
| HABO | OH-activated bark oil |
| HMDA | Hexamethylene diamine |
| HU | Hydroxyurethane |
| iBMK | Methyl isobutyl ketone |
| ICFAD | Internal carbonated fatty acid diester |
| IPCC | International panel on climate change |
| IPDA | Isophorone diamine |
| KH-560 | [3-(2,3-Epoxypropoxy)-propyl]-trimethoxysilane |
| LO | Linseed oil |
| MDA | 4,4′-Methylenedianiline |
| MDI | 2′,4′/4,4′-Diphenyl-methane diisocyanate |
| MeCN | Acetonitrile |
| MEK | Methyl ethyl ketone |
| MF | Methyl formate |
| MOFs | Metal–organic frameworks |
| NI-PHUs | Non-isocyanate polyhidroxyurethanes |
| NIPHEUs | Polyhydroxy(ester-urethane)s |
| NIPUs | Non-isocyanate polyurethanes |
| NPGDGE | Neopentyl glycol diglycidyl ether |
| ORA | Oligomeric ricinoleic acid |
| ODA | 1,8-Octanediamine |
| OUa | Oligourea |
| P(NIPU)-PBDs | Polybutadienes-based non-isocyanate polyurethanes |
| PC | Pentaerythritol |
| PBDs | Polybutadienes |
| PDA | 1,5-Pentanediamine |
| PEGDE | Poly(ethylene glycol) diglycidyl ether |
| PGE | Phenyl glycidyl ether |
| PHA | Polyhydroxyalkanoate |
| PHUU | Polyhydroxiuethane-urea |
| PPC | Poly(propylene carbonate) |
| PPDE | Poly(propylene glycol) diglycidyl ether |
| PPN | Bis(triphenylphosphine)iminium |
| PSA | Pressure-sensitive adhesive |
| PU | Polyurethane |
| PUF | Polyurethane foam |
| PUUa | Polyurethane-urea |
| PUUa-DA | Polyurethane-urea Diels–Alder adduct |
| RDGE | Resorcinol diglycidyl ether |
| REACH | Registration, evaluation, authorisation, and restriction of chemicals |
| RHMPA | Reactive hot-melt polyurethane adhesives |
| Si-PECs | Siloxane-functionalized poly(ether carbonate)s |
| SuBC | Succinate bis(cyclic carbonate) |
| TAEA | Tris(2-aminoethyl)amine |
| TBA | Tetra-n-butylammonium |
| TBAB | Tributyl ammonium bromide |
| TCFAD | Terminal carbonated fatty acid diester |
| TCM | Methylene-bridged tetracarbamate |
| TDA | Toluene-2,4-diamine |
| TDC | Toluene-2,4-dicarbamateTG thermogravimetric |
| TMC | Trimethylolpropane carbonates |
| TMPTC | Trimethylolpropane tris-carbonate |
| TTD | 4,7,10-Trioxa-1,13-tridecanediamine |
| WPU | Waterborne polyurethane |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results has received funding from the EU-funded PON REACT project Azione IV.6 – “Contratti di ricerca su tematiche green” del nuovo Asse IV del PON Ricerca e Innovazione 2014–2020 “Istruzione e ricerca per il recupero – REACT – EU”; Progetto “CO2 as C1 renewable source for innovative sustainable synthetic approaches: from small molecules to materials”, (Antonio Rescifina and Chiara Zagni), CUP: E61821004320005.References
- H. Khatoon, S. Iqbal, M. Irfan, A. Darda and N. K. Rawat, Prog. Org. Coat., 2021, 154, 106124 CrossRef CAS.
- A. Das and P. Mahanwar, Adv. Ind. Eng. Polym. Res., 2020, 3, 93–101 Search PubMed.
- M. Ates, S. Karadag, A. A. Eker and B. Eker, Polym. Int., 2022, 71, 1157–1163 CrossRef CAS.
- C. Prisacariu, in Polyurethane Elastomers, Springer, 2011, pp. 1–22 Search PubMed.
- H. Khatoon and S. Ahmad, J. Ind. Eng. Chem., 2017, 53, 1–22 CrossRef CAS.
- M. H. Karol and J. A. Kramarik, Toxicol. Lett., 1996, 89, 139–146 CrossRef CAS PubMed.
- D. Bello, C. A. Herrick, T. J. Smith, S. R. Woskie, R. P. Streicher, M. R. Cullen, Y. Liu and C. A. Redlich, Environ. Health Perspect., 2007, 115, 328–335 CrossRef CAS PubMed.
- M. Barbhuiya, S. Bhunia, M. Kakkar, B. Shrivastava, P. K. Tiwari and S. Gupta, J. Cytol., 2014, 31, 20 CrossRef PubMed.
- A. Gomez-Lopez, F. Elizalde, I. Calvo and H. Sardon, Chem. Commun., 2021, 57, 12254–12265 RSC.
- E. Dyer and H. Scott, J. Am. Chem. Soc., 1957, 79, 672–675 CrossRef CAS.
- M. Aresta and E. Quaranta, CHEMTECH, 1997, 27, 32–40 CAS.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- G. Yuan, C. Qi, W. Wu and H. Jiang, Curr. Opin. Green Sustain. Chem., 2017, 3, 22–27 CrossRef.
- H. Yang, Z. Xu, M. Fan, R. Gupta, R. B. Slimane, A. E. Bland and I. Wright, J. Environ. Sci., 2008, 20, 14–27 CrossRef CAS PubMed.
- D. S. Ogunniyi, Bioresour. Technol., 2006, 97, 1086–1091 CrossRef CAS PubMed.
- N. Yamazaki, T. Iguchi and F. Higashi, Tetrahedron, 1975, 31, 3031–3034 CrossRef CAS.
- N. Yamazaki, F. Higashi and T. Iguchi, J. Polym. Sci., Polym. Lett. Ed., 1974, 12, 517–521 CrossRef CAS.
- Z. Chen, N. Hadjichristidis, X. Feng and Y. Gnanou, Macromolecules, 2017, 50, 2320–2328 CrossRef CAS.
- S. Sujith, J. K. Min, J. E. Seong, S. J. Na and B. Y. Lee, Angew. Chem., Int. Ed., 2008, 47, 7306–7309 CrossRef CAS PubMed.
- Direct Air Capture of CO2 with Chemicals: A Technology Assessment for the APS Panel on Public Affairs, ed. R. Socolow, M. Desmond, R. Aines, J. Blackstock, O. Bolland, T. Kaarsberg, N. Lewis, M. Mazzotti, A. Pfeffer, K. Sawyer, J. Siirola, B. Smit and J. Wilcox, American Physical Society, 2011 Search PubMed.
- V. Nikulshina, D. Hirsch, M. Mazzotti and A. Steinfeld, Energy, 2006, 31, 1715–1725 CrossRef CAS.
- Y. Takeda, S. Okumura, S. Tone, I. Sasaki and S. Minakata, Org. Lett., 2012, 14, 4874–4877 CrossRef CAS PubMed.
- L. Zhao, F. Sha, Y. Li and J. Zhang, J. Environ. Chem. Eng., 2021, 9, 105309 CrossRef CAS.
- T. Zhao, B. Guo, L. Han, N. Zhu, F. Gao, Q. Li, L. Li and J. Zhang, ChemPhysChem, 2015, 16, 2106–2109 CrossRef CAS PubMed.
- J. Wang, H. Zhang, Y. Miao, L. Qiao, X. Wang and F. Wang, Polymer, 2016, 100, 219–226 CrossRef CAS.
- C. Wulf, M. Reckers, A. Perechodjuk and T. Werner, ACS Sustainable Chem. Eng., 2020, 8, 1651–1658 CrossRef CAS.
- L. Longwitz, J. Steinbauer, A. Spannenberg and T. Werner, ACS Catal., 2018, 8, 665–672 CrossRef CAS.
- G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC.
- G. Rokicki, P. G. Parzuchowski and M. Mazurek, Polym. Adv. Technol., 2015, 26, 707–761 CrossRef CAS.
- M. Janvier, P.-H. Ducrot and F. Allais, ACS Sustainable Chem. Eng., 2017, 5, 8648–8656 CrossRef CAS.
- X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- J. Langanke, A. Wolf, J. Hofmann, K. Böhm, M. A. Subhani, T. E. Müller, W. Leitner and C. Gürtler, Green Chem., 2014, 16, 1865–1870 RSC.
- G. Trott, P. K. Saini and C. K. Williams, Philos. Trans. R. Soc., A, 2016, 374, 20150085 CrossRef PubMed.
- Z. Liu, J. Huang, S. Chen, Y. Huang, F. Ding, W. Guo, D. Lei, L. Yang, F.-L. Qing and Z. You, Int. J. Adhes. Adhes., 2020, 96, 102456 CrossRef CAS.
- C. Carré, Y. Ecochard, S. Caillol and L. Avérous, ChemSusChem, 2019, 12, 3410–3430 CrossRef PubMed.
- M. Schmitt and V. Strehmel, Org. Process Res. Dev., 2020, 24, 2521–2528 CrossRef CAS.
- S. Jiang and L. Liu, Polymer, 2022, 244, 124652 CrossRef CAS.
- S. Motokucho, Y. Takenouchi, R. Satoh, H. Morikawa and H. Nakatani, ACS Sustainable Chem. Eng., 2020, 8, 4337–4340 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, G. X. De Hoe, R. L. Snyder, W. R. Dichtel and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2018, 6, 11145–11159 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed.
- P. I. Kordomenos and J. E. Kresta, Macromolecules, 1981, 14, 1434–1437 CrossRef CAS.
- C. Pronoitis, M. Hakkarainen and K. Odelius, ACS Sustainable Chem. Eng., 2022, 10, 2522–2531 CrossRef CAS.
- Z. Ma, X. Zhang, X. Zhang, N. Ahmed, H. Fan, J. Wan, C. Bittencourt and B. Li, Ind. Eng. Chem. Res., 2020, 59, 3044–3051 CrossRef CAS.
- A. Benedito, E. Acarreta and E. Giménez, Catalysts, 2021, 11, 628 CrossRef CAS.
- J. Hwang, B. Y. Jeong, J. Cheon, H. Jun, P. Huh, W.-K. Lee and J. H. Chun, Mol. Cryst. Liq. Cryst., 2020, 706, 136–140 CrossRef CAS.
- S. Huo, S. Yang, J. Wang, J. Cheng, Q. Zhang, Y. Hu, G. Ding, Q. Zhang and P. Song, J. Hazard. Mater., 2020, 386, 121984 CrossRef CAS PubMed.
- S. Huo, P. Song, B. Yu, S. Ran, V. S. Chevali, L. Liu, Z. Fang and H. Wang, Prog. Polym. Sci., 2021, 114, 101366 CrossRef CAS.
- A. Rudawska and M. Frigione, Polymers, 2022, 14, 2277 CrossRef CAS PubMed.
- J.-H. Back, C. Hwang, D. Baek, D. Kim, Y. Yu, W. Lee and H.-J. Kim, Composites, Part B, 2021, 222, 109058 CrossRef CAS.
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- T. Habets, F. Siragusa, B. Grignard and C. Detrembleur, Macromolecules, 2020, 53, 6396–6408 CrossRef CAS.
- C. Zhang, R. Ding and M. R. Kessler, Macromol. Rapid Commun., 2014, 35, 1068–1074 CrossRef CAS PubMed.
- L. Hojabri, X. Kong and S. S. Narine, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3302–3310 CrossRef CAS.
- D. Kyriacos, Biobased Polyols for Industrial Polymers, John Wiley & Sons, 2020 Search PubMed.
- R. Morales-Cerrada, R. Tavernier and S. Caillol, Polymers, 2021, 13, 1255 CrossRef CAS PubMed.
- F.-Y. Ren, F. You, S. Gao, W.-H. Xie, L.-N. He and H.-R. Li, Eur. Polym. J., 2021, 153, 110501 CrossRef CAS.
- H. Li, F.-Y. Ren, H.-R. Li and L.-N. He, Polym. Eng. Sci., 2023, 63, 1507–1515 CrossRef CAS.
- A. Boyer, E. Cloutet, T. Tassaing, B. Gadenne, C. Alfos and H. Cramail, Green Chem., 2010, 12, 2205–2213 RSC.
- C. Mokhtari, F. Malek, A. Manseri, S. Caillol and C. Negrell, Eur. Polym. J., 2019, 113, 18–28 CrossRef CAS.
- B. Quienne, N. Kasmi, R. Dieden, S. Caillol and Y. Habibi, Biomacromolecules, 2020, 21, 1943–1951 CrossRef CAS PubMed.
- S. Ye, X. Xiang, S. Wang, D. Han, M. Xiao and Y. Meng, ACS Sustainable Chem. Eng., 2020, 8, 1923–1932 CrossRef CAS.
- M. Fleischer, H. Blattmann and R. Mülhaupt, Green Chem., 2013, 15, 934–942 RSC.
- H. Gholami and H. Yeganeh, Eur. Polym. J., 2021, 142, 110142 CrossRef CAS.
- X. Yang, S. Wang, X. Liu, Z. Huang, X. Huang, X. Xu, H. Liu, D. Wang and S. Shang, Green Chem., 2021, 23, 6349–6355 RSC.
- L. Poussard, J. Mariage, B. Grignard, C. Detrembleur, C. Jérôme, C. Calberg, B. Heinrichs, J. De Winter, P. Gerbaux, J.-M. Raquez, L. Bonnaud and Ph. Dubois, Macromolecules, 2016, 49, 2162–2171 CrossRef CAS.
- X. Yang, C. Ren, X. Liu, P. Sun, X. Xu, H. Liu, M. Shen, S. Shang and Z. Song, Mater. Chem. Front., 2021, 5, 6160–6170 RSC.
- S. D. Bote and R. Narayan, Ind. Eng. Chem. Res., 2021, 60, 5733–5743 CrossRef CAS.
- J. Pouladi, S. M. Mirabedini, H. Eivaz Mohammadloo and N. G. Rad, Eur. Polym. J., 2021, 153, 110502 CrossRef CAS.
- S. El Khezraji, H. Ben Youcef, L. Belachemi, M. A. Lopez Manchado, R. Verdejo and M. Lahcini, Polymers, 2023, 15, 254 CrossRef CAS PubMed.
- A. Cornille, S. Dworakowska, D. Bogdal, B. Boutevin and S. Caillol, Eur. Polym. J., 2015, 66, 129–138 CrossRef CAS.
- B. Grignard, J.-M. Thomassin, S. Gennen, L. Poussard, L. Bonnaud, J.-M. Raquez, P. Dubois, M.-P. Tran, C. B. Park, C. Jerome and C. Detrembleur, Green Chem., 2016, 18, 2206–2215 RSC.
- J. Catalá, I. Guerra, J. M. García-Vargas, M. J. Ramos, M. T. García and J. F. Rodríguez, Polymers, 2023, 15, 1589 CrossRef PubMed.
- J. Liu, P. Miao, X. Leng, J. Che, Z. Wei and Y. Li, Macromol. Rapid Commun., 2023, 44, 2300263 CrossRef CAS PubMed.
- K. Błażek, H. Beneš, Z. Walterová, S. Abbrent, A. Eceiza, T. Calvo-Correas and J. Datta, Polym. Chem., 2021, 12, 1643–1652 RSC.
- H. Chen, P. Chauhan and N. Yan, Green Chem., 2020, 22, 6874–6888 RSC.
- B. Nohra, L. Candy, J.-F. Blanco, C. Guerin, Y. Raoul and Z. Mouloungui, Macromolecules, 2013, 46, 3771–3792 CrossRef CAS.
- S.-E. Dechent, A. W. Kleij and G. A. Luinstra, Green Chem., 2020, 22, 969–978 RSC.
- C. M. Laprise, K. A. Hawboldt, F. M. Kerton and C. M. Kozak, Macromol. Rapid Commun., 2021, 42, 2000339 CrossRef CAS PubMed.
- C. Hussong, J. Langanke and W. Leitner, Green Chem., 2020, 22, 8260–8270 RSC.
- W. Leitner, G. Franciò, M. Scott, C. Westhues, J. Langanke, M. Lansing, C. Hussong and E. Erdkamp, Chem. Ing. Tech., 2018, 90, 1504–1512 CrossRef CAS.
- S. Islam and G. Bhat, Mater. Adv., 2021, 2, 1896–1926 RSC.
- Y. Yang, X. Ding and M. W. Urban, Prog. Polym. Sci., 2015, 49–50, 34–59 CrossRef CAS.
- S. Islam and G. Bhat, Mater. Adv., 2021, 2, 1896–1926 RSC.
- B. Ghosh and M. W. Urban, Science, 2009, 323, 1458–1460 CrossRef CAS PubMed.
- P. Wu, L. Liu and Z. Wu, Macromol. Mater. Eng., 2020, 305, 2000359 CrossRef CAS.
- C. Lin, H. Ge, T. Wang, M. Huang, P. Ying, P. Zhang, J. Wu, S. Ren and V. Levchenko, Polymer, 2020, 206, 122894 CrossRef CAS.
- J. Ling, M. Z. Rong and M. Q. Zhang, Polymer, 2012, 53, 2691–2698 CrossRef CAS.
- R. Gunckel, B. Koo, Y. Xu, B. Pauley, A. Hall, A. Chattopadhyay and L. L. Dai, ACS Appl. Polym. Mater., 2020, 2, 3916–3928 CrossRef CAS.
- H. Jia and S.-Y. Gu, J. Polym. Res., 2020, 27, 298 CrossRef CAS.
- A. Erice, A. Ruiz de Luzuriaga, I. Azcune, M. Fernandez, I. Calafel, H.-J. Grande and A. Rekondo, Polymer, 2020, 196, 122461 CrossRef CAS.
- J.-Y. Liang, S.-R. Shin, S.-H. Lee and D.-S. Lee, Polymers, 2020, 12, 1011 CrossRef CAS PubMed.
- Z. Liang, D. Huang, L. Zhao, Y. Nie, Z. Zhou, T. Hao and S. Li, J. Inorg. Organomet. Polym., 2021, 31, 683–694 CrossRef CAS.
- J. Park, S. Murayama, M. Osaki, H. Yamaguchi, A. Harada, G. Matsuba and Y. Takashima, Adv. Mater., 2020, 32, 2002008 CrossRef CAS PubMed.
- C. Jin, G. Sinawang, M. Osaki, Y. Zheng, H. Yamaguchi, A. Harada and Y. Takashima, Polymers, 2020, 12, 1393 CrossRef CAS PubMed.
- J. Liu, Z. Zhou, X. Su, J. Cao, M. Chen and R. Liu, Prog. Org. Coat., 2020, 146, 105699 CrossRef CAS.
- Z. Chen, Y.-C. Sun, J. Wang, H. J. Qi, T. Wang and H. E. Naguib, ACS Appl. Mater. Interfaces, 2020, 12, 8740–8750 CrossRef CAS PubMed.
- H. Jia and S.-Y. Gu, Eur. Polym. J., 2020, 126, 109542 CrossRef CAS.
- S. Xu, J. Li, H. Qiu, Y. Xue and J. Yang, Compos. Commun., 2020, 19, 220–225 CrossRef.
- S. Yang, X. Du, Z. Du, M. Zhou, X. Cheng, H. Wang and B. Yan, Polymer, 2020, 190, 122219 CrossRef.
- E. Dolci, V. Froidevaux, G. Michaud, F. Simon, R. Auvergne, S. Fouquay and S. Caillol, J. Appl. Polym. Sci., 2017, 134, 44408 CrossRef.
- S. Islam and G. Bhat, Mater. Adv., 2021, 2, 1896–1926 RSC.
- P. Wu, H. Cheng, X. Wang, R. Shi, C. Zhang, M. Arai and F. Zhao, Green Chem., 2021, 23, 552–560 RSC.
- R. Jaratrotkamjorn, A. Nourry, P. Pasetto, E. Choppé, W. Panwiriyarat, V. Tanrattanakul and J.-F. Pilard, J. Appl. Polym. Sci., 2017, 134, 45427 CrossRef.
- J. F. G. A. Jansen, A. A. Dias, M. Dorschu and B. Coussens, Macromolecules, 2003, 36, 3861–3873 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |