
Green synthesis of sulfur-containing polymers by carbon disulfide-based spontaneous multicomponent polymerization†
Xu
Chen
abd,
Anjun
Qin
 *ab and
Ben Zhong
Tang
*bce
*ab and
Ben Zhong
Tang
*bce
aState Key Laboratory of Luminescent Materials and Devices, Guangdong Provincial Key Laboratory of Luminescence from Molecular Aggregates, South China University of Technology, Guangzhou 510640, China. E-mail: msqinaj@scut.edu.cn
bCenter for Aggregation-Induced Emission, AIE Institute, South China University of Technology, Guangzhou 510640, China
cSchool of Science and Engineering, Shenzhen Institute of Aggregate Science and Technology, The Chinese University of Hong Kong, Shenzhen (CUHK-Shenzhen), Guangdong 518172, China. E-mail: tangbenz@cuhk.edu.cn
dDepartment of Infectious Diseases, the Fifth Affiliated Hospital, Sun Yat-sen University, 52 East Meihua Road, Zhuhai 519000, Guangdong Province, China
eHong Kong Branch of the Chinese National Engineering Research Centre for Tissue Restoration and Reconstruction, The Hong Kong University of Science & Technology, Kowloon 999077, Hong Kong, China
First published on 29th November 2023
Abstract
Sulfur-containing polymers have gained much attention in polymer science due to their unique properties. However, their preparation has posed considerable challenges, particularly in diversifying their structures and achieving highly efficient polymerizations. This is especially true for polymers derived from CS2, a readily available one-carbon (C1) feedstock, as their synthesis often requires the use of harsh conditions or results in unexpected by-products. In this work, we established a regio-selective and atom-economical spontaneous multicomponent polymerization based on carbonyl or ester group-activated internal alkynes, commercially available amines, and CS2. Similar to the angled half lap joint of two plates in a scarf joint of ancient Chinese “mortise and tenon” architecture, internal ethynyl and amino groups cannot be readily linked at room temperature, whereas added CS2 acts as a “wedge” to make the monomers spontaneously expand into tight linkages of polymer chains with satisfactory molecular weights (up to 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600) in high yields (up to 97%). The polymers exhibited exceptional optical characteristics (refractive indices of up to 1.7471 at 632.8 nm) and good film-forming capabilities. This polymerization opens up a new avenue for the green and efficient synthesis of functional sulfur-containing polymers with low energy consumption, minimized waste generation and efficient use of CS2 resources.
600) in high yields (up to 97%). The polymers exhibited exceptional optical characteristics (refractive indices of up to 1.7471 at 632.8 nm) and good film-forming capabilities. This polymerization opens up a new avenue for the green and efficient synthesis of functional sulfur-containing polymers with low energy consumption, minimized waste generation and efficient use of CS2 resources.
1. Introduction
Sulfur-containing polymers have gained much attention due to their superior electrical, optical, biological or mechanical properties, as well as distinct features like stabilization of free radicals or adhesion to heavy metals.1–5 Condensation or ring-opening polymerizations are frequently used to prepare sulfur-containing polymers, such as poly(thioether)s, poly(thiourethane)s, poly(thiocarbonate)s, poly(trithiocarbonate)s, poly(thioester)s, and poly(sulfur-random-styrene)s.6 Other reactions, such as copolymerizations, are generally difficult to perform due to their poor selectivity.7 Thus, it is still urgently necessary to expand the copolymerizations for preparing sulfur-containing polymers.Abundant sulfur resources, including mercaptans, elemental sulfur, carbon disulfide (CS2), carbonyl sulfide, and other sulfides, can be utilized as monomers to prepare sulfur-containing polymers.8–11 Among them, CS2 stands out as a readily available sulfur-rich one-carbon (C1) feedstock, which exists in more transportable liquid form and shows strong nucleophilicity.12,13 However, CS2-based polymerizations are rather limited. As shown in Scheme 1, the salenCrX-based system has been developed as the most popular catalyst for the copolymerization of episulfide/epoxide with CS2 toward alternating polymers since it was first reported by Nozaki et al. in 2007.14 The catalyst was further expanded by Darensbourg et al. to a heterogeneous zinc–cobalt double cyanide complex or a homogeneous (salen)CrCl complex.15
 | ||
| Scheme 1 Previous CS2-involving polymerizations. | ||
Recently, a catalyst-free strategy was proposed for polythiourea preparation from diamines and CS2 at 45 °C. However, the toxic hydrogen sulfide by-product was also generated.16 Liu et al. also reported that CS2, dihalides, and electron-withdrawing group-activated methylene derivatives could be polymerized using K2CO3 as a catalyst at room temperature. However, the reaction took up to 7 days to complete at high monomer concentration.17 Therefore, efficient and green synthesis of polymers based on CS2 at room temperature, without catalysts and byproducts, remains a challenge in response to minimizing the adverse impacts of chemical production and energy consumption on environmental sustainability and human health.
The principles of “green synthesis” involve promoting sustainability, reducing energy consumption, minimizing toxic reagents and products, minimizing harm to the ecosystem, mitigating the risk of global warming, utilizing naturally available resources, generating byproducts in a rational manner, etc.18,19 Our groups have established a series of alkyne-based spontaneous click polymerizations, such as spontaneous amino–yne click polymerizations, which just comply with the above principles, and from which regio- and stereoregular polymers were efficiently prepared.20,21 The spontaneousness of the reaction relies on the activation of ethynyl groups by covalently connecting with electron-withdrawing ester, carbonyl, or sulfonyl groups.22,23 However, the reactivity of activated internal alkynes is greatly lower compared to that of the aforementioned terminal ones, although they are more stable and could be synthesized more facilely. For example, the polymerization between activated internal alkynes and amines could not occur spontaneously and has to be conducted at elevated temperature or in the presence of catalysts.24
As an ancient architectural culture of excavating the inherent characteristics of materials, Chinese “mortise and tenon” can be traced back to approximately 7000 years ago and has gained significant international attention to date. As shown in Scheme 2, inspired by the design of two angled half lap joint plates in a scarf joint of “mortise and tenon”, which relies on the use of a wedge for a secure connection, we speculated that the introduction of another monomeric component into the polymerization system might realize more efficient polymerization of activated internal alkynes.25–31 In other words, the multicomponent polymerizations (MCPs) might be ideal candidates although the one- and two-component polymerizations are well-established.32–42 Indeed, it is reported that CS2 is capable of efficiently reacting with secondary amines to form more nucleophilic dithiocarbamic acid derivatives,43,44 which could react with activated internal alkynes in a spontaneous manner.45 Thus, CS2 acts as a “wedge” in this reaction. As a result, S-acylvinyl-N,N-dialkyl dithiocarbamates (ADDCs) could be produced more facilely than previously reported reactions.46–49
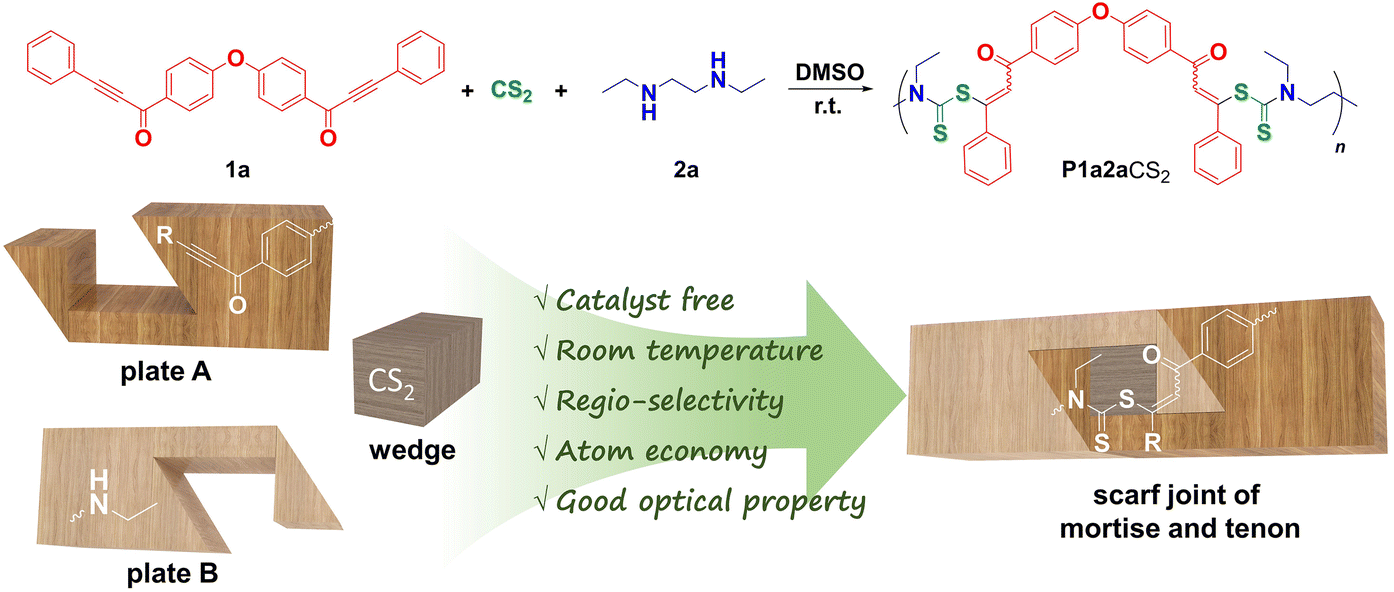 | ||
| Scheme 2 Spontaneous polymerization of activated internal diyne 1a, secondary diamine 2a and CS2. The two-angled half lap joint plates of internal ethynyl and amino groups cannot spontaneously react, but with the assistance of a CS2 “wedge”; these two functional groups can be tightly linked to form stable polymer chains in a spontaneous manner. | ||
Based on the above reports and the “mortise and tenon” strategy, in this work, we successfully developed a spontaneous MCP of activated internal alkynes, secondary amines, and CS2. The atom-economical polymerization proceeded smoothly at room temperature, and regioregular poly(S-acylvinyl-N,N-dialkyldithiocarbamate)s (PADDCs) with weight-average molecular weights of up to 31600 were produced in high yields of up to 97%. PADDCs show excellent processability and film-forming ability as well as high transparency and refractivity. Thus, we established a green polymerization reaction for facilely preparing functional sulfur-containing polymers with low energy consumption, minimized waste generation, and efficient use of CS2 resources.
2. Results and discussion
2.1 Polymerization
The polymerization of activated internal diyne 1a, secondary diamine 2a and CS2, as shown in Scheme 2, was firstly conducted to understand the effects of solvent, monomer concentration, CS2 equivalent and reaction time on its efficiency. The polymerization was initially conducted in chloroform (Table 1, entry 1), but the formed intermediate dithiocarbamic acid derivative from 2a and CS2 was poorly soluble, making it difficult to generate the final product. Hence, we screened solvents with higher polarity than chloroform, including tetrahydrofuran (THF), dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF) and N,N-dimethylacetamide (DMAc) for the polymerization (entries 2–5). Eventually, the polymer with the highest weight-average molecular weight (Mw) was obtained in DMSO, but the yield is lower than that in DMAc, which might be due to product loss during the purification process. The use of a mixed solvent of DMSO and DMAc (entry 6) did not significantly improve the yield. Since the loss of product during extraction could be reduced with enhanced Mw, the green solvent DMSO was selected as the optimal reaction solvent for further studies.a
| Entry | Solvent | Yield (%) |
M
wb |
Đ |
|---|---|---|---|---|
| a Carried out under nitrogen at 25 °C for 12 h; [1a] = [2a] = 0.05 M, [CS2] = 0.15 M. b Determined by gel-permeation chromatography (GPC) in DMF containing 0.05 M LiBr using linear polymethylmethacrylate (PMMA) for calibration. | ||||
| 1 | CHCl3 | — | — | — |
| 2 | THF | 29 | 1700 | 1.14 |
| 3 | DMSO | 54 | 7500 | 1.42 |
| 4 | DMF | 49 | 5900 | 1.29 |
| 5 | DMAc | 64 | 5500 | 1.27 |
| 6 | DMAc + DMSO | 57 | 7100 | 1.35 |
Secondly, we studied the impact of monomer concentration on the polymerization results. We further increased the monomer concentration to improve the Mw and yield of product because they are low when the polymerization was performed at a low concentration of 0.05 M (entry 1, Table 2). As shown in Table 2 (entries 2–5), polymers with significantly higher Mw values were obtained when the monomer concentration was increased to 0.3 M. A further increase in the concentration to 0.4 M resulted in a slight decrease of Mw of the product, which might be due to the uneven stirring of the high viscosity system.50 Therefore, the optimal monomer concentration was selected to be 0.3 M.
a
| Entry | [1a] and [2a] (mol L−1) | Yield (%) |
M
wb |
Đ |
|---|---|---|---|---|
| a Carried out under nitrogen in anhydrous DMSO at 25 °C for 12 h, [CS2] = 3[1a] = 3[2a]. b Determined by GPC in DMF containing 0.05 M LiBr using linear PMMA for calibration. | ||||
| 1 | 0.05 | 54 | 7500 | 1.42 |
| 2 | 0.1 | 53 | 8400 | 1.44 |
| 3 | 0.2 | 56 | 11300 |
1.62 |
| 4 | 0.3 | 73 | 27900 |
1.93 |
| 5 | 0.4 | 74 | 24600 |
1.83 |
Thirdly, we varied the CS2 content during the polymerization. The data listed in Table S1† revealed that the concentration of CS2 does not significantly affect the efficiency of the polymerization. Using a 3-fold higher concentration of CS2 compared to the other monomers is sufficient to ensure efficient propagation of the polymerization. Finally, the time course of the polymerization was followed (Table S2†). The Mw values of the products increase gradually at the early reaction stage, and the highest Mw value was recorded at 8 h. Continuing the reaction for a longer duration did not yield a higher Mw value. Thus, the optimized polymerization reaction conditions were obtained as being 0.3 M 1a with a feed ratio of [1a]:[2a]:[CS2] = 1:1:3 at room temperature for 8 h. All the Mw values of the polymers were measured by GPC, and the traces are provided in the ESI (Fig. S1†).
After establishing the optimal polymerization conditions, we further investigated the scope of activated alkynes by using them to react with diethyldithiocarbamic acid (Scheme 3). The experiments were conducted by first mixing diethylamine with CS2 to form the diethyldithiocarbamic acid (4) intermediate, followed by the addition of alkynes to the reaction system. According to a previous report,43 phenylacetylene 5 can react with 4 to yield the sulfur-containing adduct 6 at 80 °C. However, a neat reaction was required as the use of solvent does not yield product. Notably, at room temperature, the carbonyl- or ester-activated internal alkynes 7 or 9 in solutions can spontaneously react with 4 to yield ADDC derivatives 8 and 10, respectively, while the most reactive carbonyl-activated terminal alkyne 11 will extract diethylamine from 4 to generate β-enaminone 12via the spontaneous amino-yne click reaction.23 It is worth noting that the binding ability of primary amines toward CS2 is weaker than that of secondary aliphatic amines.51 For example, in the presence of n-Bu3P, primary amines can react with activated internal alkynes and CS2 to produce a five-membered cyclic product (15).52 However, the low yield and the presence of side products make it unsuitable for polymerization development.
 | ||
| Scheme 3 Reaction of different alkynes with diethyldithiocarbamic acid (4). | ||
The above results suggested that the carbonyl- or ester-activated internal alkynes precisely meet the demands for the spontaneous reaction with diethyldithiocarbamic acid. Therefore, we investigated the universality of the polymerization of carbonyl- or ester-activated internal diynes and secondary aliphatic diamine monomers (Scheme 4 and Table 3). The results demonstrated that all the used diynes could readily polymerize with secondary diamines and CS2 in a spontaneous manner, furnishing soluble polymers with high Mw (up to 31600) and high yields (up to 97%). Notably, the polymerization carried out in air had lesser effect on the results compared with that under nitrogen, suggesting that moisture or oxygen did not significantly affect the efficiency of the spontaneous MCP. The robust adaptability of this polymerization to its environment means that maintaining the polymerization progress requires only low energy consumption.
 | ||
| Scheme 4 Spontaneous multicomponent polymerization of activated internal diynes 1, aliphatic secondary diamines 2 and CS2. | ||
a
| Entry | Monomers | Yield (%) |
M
wb |
Đ |
|---|---|---|---|---|
| a Carried out under nitrogen in anhydrous DMSO at 25 °C for 8 h, [CS2] = 3[1] = 3[2] = 0.9 M. b Determined by GPC in DMF containing 0.05 M LiBr using linear PMMA for calibration. c Conducted under open air. | ||||
| 1c | 1a/2a/CS2 | 75 | 21700 |
1.94 |
| 2 | 1a/2a/CS2 | 76 | 31600 |
2.13 |
| 3 | 1a/2b/CS2 | 92 | 21200 |
1.81 |
| 4 | 1a/2c/CS2 | 55 | 7100 | 1.39 |
| 5 | 1b/2a/CS2 | 55 | 11200 |
1.59 |
| 6 | 1b/2b/CS2 | 80 | 14900 |
1.67 |
| 7 | 1c/2a/CS2 | 65 | 10600 |
1.52 |
| 8 | 1c/2b/CS2 | 97 | 31400 |
1.93 |
2.2 Structural characterization
The resultant polymers generated by the spontaneous MCPs were characterized using Fourier transform infrared (FT-IR) and nuclear magnetic resonance (NMR) spectroscopy techniques. Herein, the characterization of P1a2aCS2 was given as a representative example.The FT-IR spectra of diyne 1a, diamine 2a, model compound 8, and polymer P1a2aCS2 are displayed in Fig. 1. The spectra of 8 and P1a2aCS2 are quite similar. The peak corresponding to the stretching vibration of the ethynyl group in 1a appeared at 2195 cm−1. This peak was almost absent in the spectrum of P1a2aCS2, indicating the consumption of diyne 1a by the MCP. The remaining weak signal might be ascribed to the terminated ethynyl group of the polymer chains. Furthermore, the peaks corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond vibration in 8 and P1a2aCS2 appeared at 1245 cm−1 and 1230 cm−1, respectively.53 Notably, neither N–H nor S–H peak was detected in compound 8 and P1a2aCS2, and the positions of the other characteristic peaks were consistent with the structures of ADDC derivatives. These results indicated that the MCP proceeded as expected. The FT-IR spectra of other polymers were similar to that of P1a2aCS2 (Fig. S2†).
S bond vibration in 8 and P1a2aCS2 appeared at 1245 cm−1 and 1230 cm−1, respectively.53 Notably, neither N–H nor S–H peak was detected in compound 8 and P1a2aCS2, and the positions of the other characteristic peaks were consistent with the structures of ADDC derivatives. These results indicated that the MCP proceeded as expected. The FT-IR spectra of other polymers were similar to that of P1a2aCS2 (Fig. S2†).
 | ||
| Fig. 1 FT-IR spectra of (A) monomer 1a, (B) monomer 2a, (C) model compound 8, and (D) polymer P1a2aCS2. | ||
After obtaining a clear picture of the IR stretching peaks, we proceeded to monitor the polymerization process using in situ IR spectral analysis (Fig. S3†). When CS2 was added to the mixture of 1a and 2a at 0 h, the intermediate dithiocarbamic acid was rapidly formed, resulting in an increase in the characteristic C–N peak at a wavenumber of 1519 cm−1. Subsequently, the peak gradually decreased as the intermediate was consumed. The peak of CO gradually shifted from 1635 cm−1 to 1661 cm−1, indicating the step-growth reaction of the intermediate with the activated diyne. Meanwhile, the generation of the CC bond (1588 cm−1) was also observed. These results demonstrated that polymerization initially involved the rapid formation of the dithiocarbamic acid intermediate from CS2 and amines, followed by a gradual reaction with diyne monomers. The peaks showed a steady trend within 8 h, which is well consistent with the optimized reaction time.
1H and 13C NMR spectroscopy techniques were used to further confirm the structures of the polymers. The 1H NMR spectra of 1a, 2a, 8, and P1a2aCS2 are shown in Fig. 2 as examples. The proton resonance of the vinyl group in 8 mainly appeared at 7.62 and 7.28 ppm due to the existence of E/Z-isomers.54,55 Since these two isomers are difficult to separate by crystallization or column chromatography, they were then tested by 1H–1H nuclear Overhauser effect spectroscopy (NOESY). As shown in Fig. 3, a peak corresponding to the correlation between H1 and H2 was identified, while a prominent resonance related to the correlation between H1′ and H2′ was not observed. This result indicated that H2 is associated with the vinyl group in a Z-configuration and H2′ represents the proton resonance of the vinyl group in an E-configuration. Therefore, the E/Z ratio of model compound 8 was calculated from the integral areas as about 1:5, and the Z-configuration was the dominant structure.
 | ||
| Fig. 2 1H NMR spectra of (A) diyne 1a, (B) diamine 2a, (C) model compound 8, and (D) polymer P1a2aCS2 in DMSO-d6. The solvent peaks are marked with asterisks. | ||
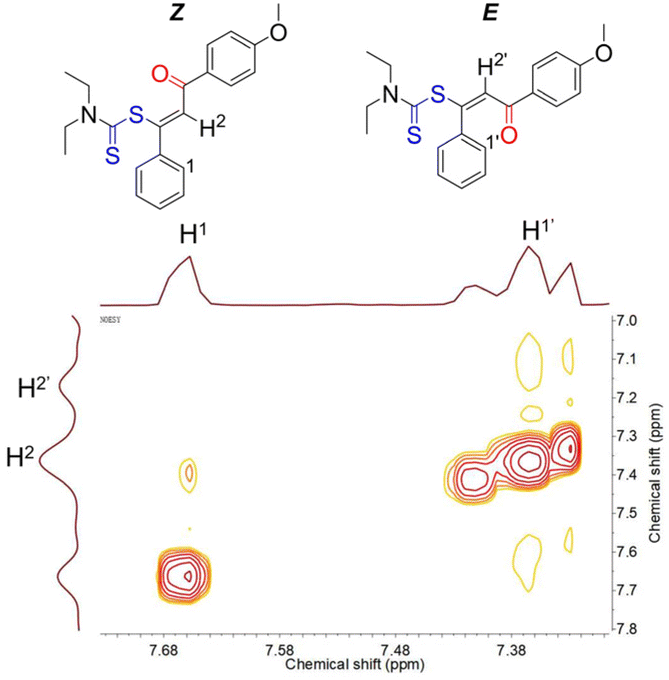 | ||
| Fig. 3 1H–1H NOESY spectra of model compound 8 in CDCl3. | ||
To exclude the possible hydroamination between the secondary amine and the alkyne, a control experiment was carried out by mixing compound 7 with diethylamine in THF, DCM, chloroform, or methanol. No product was found when the reaction was carried out at room temperature. Only when the mixture was heated to above 60 °C was β-enaminone derivative S9 detected and confirmed by NMR spectra (Fig. S4 and S5†). The proton resonance values of the vinyl group in S9 at about 5.5–6.0 ppm were not found in the 1H NMR spectrum of model compound 8, which rules out the possibility of a side reaction occurring between the secondary amine and 7.
The 1H NMR spectrum of P1a2aCS2 is similar to that of model compound 8, but with broad resonance peaks. The characteristic peak of the hydroamination product in the range of 5.5–6.0 ppm was also not observed. By comparing the polymer spectrum with that of 8, we could find that both E- and Z-units existed in P1a2aCS2 with characteristic hydrogen peaks at 7.37 and 7.17 ppm. The integral of Hg and Hg′ indicated that the E/Z ratio is about 3:4.
The structure of the polymers could be further analyzed by 13C NMR spectra (Fig. 4). Two sets of the carbon resonant peaks were observed in the spectrum of 8, attributed to the presence of E- and Z-isomers. The peaks corresponding to the ethynyl carbons of 1a appeared at 93.32 and 86.86 ppm. These peaks were absent in the spectra of 8 and P1a2aCS2, proving the consumption of ethynyl groups by the reaction. Meanwhile, the carbon resonant peak of the CS unit appeared at 190 ppm. These characterization results demonstrated the successful synthesis of ADDC derivatives. The NMR spectra of the other monomers, polymers, and model compounds are presented in the ESI (Fig. S6–S25†) and similar conclusions could be drawn.
 | ||
| Fig. 4 13C NMR spectra of (A) monomer 1a, (B) monomer 2a, (C) model compound 8, and (D) polymer P1a2aCS2 in DMSO-d6. The solvent peaks are marked with asterisks. | ||
2.3 Thermal stability of polymers
To study the thermal properties of PADDCs, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were conducted (Fig. 5). As shown in Fig. 5A, the decomposition temperature (Td) values with 5% weight loss of polymers are all lower than 250 °C. This might be attributed to the presence of dithiocarbamate groups, which are prone to being decomposed at elevated temperatures. This property holds the potential to endow these polymers with high-performance and recycling abilities.56 The polymer products require only a low decomposition temperature for waste treatment, alleviating the burden of disposal. Interestingly, the Td values of the polymers generated from 1b are higher than those from 1a and 1c, suggesting that the electron-withdrawing groups in the polymer chains could cause a remarkable effect on their thermal stability. In addition, the TGA curves of P1a2aCS2 and P1c2aCS2 showed two decomposition stages. The first decomposition temperature was approximately 175 °C, and the weight loss during this stage corresponds to the loss of CS2 content in the polymers. The second stage was not prominent and mainly corresponded to the degradation of other more stable units in these polymers. The high char yields of P1a2aCS2, P1c2aCS2 and P1c2bCS2 could be ascribed to their higher content of aromatic units than that in other polymers. | ||
| Fig. 5 (A) TGA thermograms and (B) DSC thermograms of PADDCs under nitrogen. | ||
The glass transition temperature (Tg) values of the polymers are in the range of 80–120 °C (Fig. 5B). These temperatures are significantly lower than those of the initial decomposition, ensuring that the polymers remain intact during thermo-processing. Interestingly, the subtle change of the co-monomers during the MCP led to a distinct difference in the Tg values of polymers. For example, the polymers produced from 2a generally have higher Tg values than the others except when the co-monomer is 1a. Although 2b contains aromatic phenyl rings, it also has the flexible hexyl group, which resulted in lower Tg values of the products. The higher Tg value of P1a2aCS2 compared to P1a2bCS2 might be due to better chain packing in the former.
2.4 Light transmittance of polymers
The PADDCs contain large numbers of sulfur atoms, carbonyl groups, and aromatic groups, making them potentially beneficial as optical materials.57,58 First, the light transmittance of polymer films, fabricated by spin-coating of their solutions with a concentration of 50 mg mL−1, was measured using an ultraviolet-visible (UV-vis) spectrophotometer in the wavelength range of 300–900 nm. As shown in Fig. 6, the light transmittance of most polymer films (except P1c2aCS2) was higher than 85% beyond 400 nm, and the transmittance recorded for P1b2aCS2 and P1b2bCS2 films was as high as 90% at 400 nm. The transmittance decreases in the wavelength range of 300–500 nm might be caused by the absorption of polar chromophoric units in the polymers (Fig. S26†). In conventional optical polymers, elemental sulfur of poor solubility is often used to improve the refractive index, which leads to the formation of long polymeric sulfur chains and decreases their visible light transmittance. This work used liquid carbon disulfide to increase sulfur content in the polymers while ensuring a uniform distribution of sulfur atoms, thereby allowing for the simultaneous enhancement of the refractive index and transmittance.59 | ||
| Fig. 6 Light transmission spectra of the PADDC films using quartz as the basement. | ||
2.5 Light refraction properties of polymers
It is reported that the polymers containing polarizable aromatic rings and sulfur atoms might possess high refractive indices (n).60,61 The n values of PADDCs were measured in the wavelength range of 400–1700 nm (Fig. 7 and Table S3†). The results showed that the n values of the polymers at 632.8 nm are in the range of 1.65–1.75, which are significantly higher than those of commodity polymers, such as PMMA (n = 1.49) and polycarbonate (n = 1.59). Notably, the polymers generated from 1c, characterized by a high sulfur content (FS), showed outstanding n values. The highest n value of 1.7471 was recorded in P1c2aCS2 with an FS value of 27.7 wt%. Meanwhile, the optical dispersion property shows a positive correlation with the n value. P1a2bCS2 with the lowest n value among the resultant polymers possesses a low optical dispersion (D′) of 0.004, which helps reduce the propagation loss of light. Thus, the polymerization in this work effectively fixed readily available CS2 into PADDCs, resulting in excellent optical transmittance, high refractivity, and low dispersion, demonstrating good potential in the field of optical signal transmission. | ||
| Fig. 7 Wavelength dependence of refractive indices of the PADDC films using silica as their base. | ||
2.6 Ultrathin membranes of polymers
It is reported that the surface tension, which is caused by the difference in surface free energies between water and polymer solutions, could drive the polymers to spread on the surface of water as thin films.62 Compared with spin coating and deposition techniques, this on-water spreading is an easier method to fabricate large and thickness-controlled membranes, which could be used by transfer printing them to fabricate optoelectronic devices.63,64 Thanks to their excellent solubility in commonly used organic solvents, PADDCs could be easily spread to form high-quality ultrathin membranes or fibers (Fig. S27†).Experiments were conducted using P1a2aCS2 as a representative example. The contact angle between the polymer membrane and water was found to be 83° (Fig. 8A). The specific surface free energy corresponding to P1a2aCS2 was calculated to be approximately 33 J m−2. As the free energy of water (72 J m−2) is higher, it can be inferred that the surface tension is adequately high to facilitate the spontaneous spread of the polymer solution on the surface of water (Fig. 8B). As shown in Fig. 8C, the large area ultrathin polymer membrane floating on the water's surface displayed interference patterns under white-light irradiation. The confocal laser scanning microscopy (CLSM) and atomic force microscopy (AFM) characterizations of the P1a2aCS2 membrane (Fig. 8D–F) suggested that its surface was smooth with a thickness of approximately 62.2 nm, and the average surface roughness (RA) was as low as 1.72 nm. The excellent film-forming properties of the polymers make them suitable for application in diverse fabrication processes.
 | ||
| Fig. 8 (A) Water contact angle of the P1a2aCS2 membrane on silica. (B) Preparation diagram of the polymer membrane and (C) the large area membrane of P1a2aCS2 spread on water. (D) Optical image of the ultrathin P1a2aCS2 membrane on quartz recorded by CLSM under a bright field. (E) AFM image at the edge of the P1a2aCS2 membrane on silica and its (F) three-dimensional image. | ||
3. Conclusions
In this work, we successfully developed a spontaneous multicomponent polymerization of activated internal diynes, secondary diamines and low-cost CS2. Notably, CS2 acts like a “wedge” in the “mortise and tenon” structure to connect the diynes and diamines in a spontaneous reaction manner, which has generally required elevated temperature to occur. The regioregular PADDCs with high Mw values (up to 31600) were prepared at room temperature in high yields (up to 97%). The PADDCs possess good light transmittance values and high refractive indices. In addition, the polymers are processible and could be fabricated into high-quality ultrathin membranes by a method of spreading on the surface of water. The spontaneous MCP described in this study demonstrates green and sustainable characteristics, such as efficient CS2 fixation, a straightforward reaction process, the absence of toxic by-products, and energy savings, holding great potential to advance the utilization of sulfur-containing C1 monomers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21788102 and 21525417), the Natural Science Foundation of Guangdong Province (2019B030301003), and the Innovation and Technology Commission of Hong Kong (ITC-CNERC14SC01).References
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef PubMed.
- D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55, 15486 CrossRef CAS PubMed.
- A. Kausar, S. Zulfiqar and M. I. Sarwar, Polym. Rev., 2014, 54, 185 CrossRef CAS.
- R. S. Glass, Top. Curr. Chem., 2018, 376, 22 CrossRef PubMed.
- J. Zhang, Q. Zang, F. Yang, H. Zhang, J. Z. Sun and B. Z. Tang, J. Am. Chem. Soc., 2021, 143, 3944–3950 CrossRef CAS PubMed.
- A. S. Narmon, E. Leys, I. Khalil, G. Ivanushkin and M. Dusselier, Green Chem., 2022, 24, 9709 RSC.
- T.-J. Yue, W.-M. Ren, L. Chen, G.-G. Gu, Y. Liu and X.-B. Lu, Angew. Chem., Int. Ed., 2018, 57, 12670 CrossRef CAS PubMed.
- K. F. Long, N. J. Bongiardina, P. Mayordomo, M. J. Olin, A. D. Ortega and C. N. Bowman, Macromolecules, 2020, 53, 5805 CrossRef CAS.
- W. Cao, F. Dai, R. Hu and B. Z. Tang, J. Am. Chem. Soc., 2020, 142, 978 CrossRef CAS PubMed.
- M. Luo, Y. Li, Y. Y. Zhang and X. H. Zhang, Polymer, 2016, 82, 406 CrossRef CAS.
- Y. Fan, H. Ben, L. Li, S. Meng, S. Zhang, X. Zheng, J. Zhang, L. Yin and S. Chen, Appl. Catal., B, 2020, 274, 119073 CrossRef CAS.
- J. L. Yang, Y. Wang, X. H. Cao, C. J. Zhang, Z. Chen and X. H. Zhang, Macromol. Rapid Commun., 2021, 42, e2000472 CrossRef PubMed.
- B. Ochiai and T. Endo, Prog. Polym. Sci., 2005, 30, 183 CrossRef CAS.
- K. Nakano, G. Tatsumi and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 15116 CrossRef CAS PubMed.
- M. Luo, X.-H. Zhang and D. J. Darensbourg, Acc. Chem. Res., 2016, 49, 2209 CrossRef CAS PubMed.
- S. Wu, M. Luo, D. J. Darensbourg and X. Zuo, Macromolecules, 2019, 52, 8596 CrossRef CAS.
- C. Zhao, X. Meng, R. Lu, H. Xie and J. Liu, Polym. Chem., 2019, 10, 5333 RSC.
- R. Noyori, Chem. Commun., 2005, 14, 1807 RSC.
- M. Sharma and M. Sharma, in Advances in Green Synthesis, Advances in Science, Technology & Innovation, ed. Inamuddin, R. Boddula, M. I. Ahamed and A. Khan, Springer, Cham, 2021 Search PubMed.
- X. Fu, A. Qin and B. Z. Tang, Aggregate, 2023, 4, e350 CrossRef CAS.
- X. Chen, T. Bai, R. Hu, B. Song, L. Lu, J. Ling, A. Qin and B. Z. Tang, Macromolecules, 2020, 53, 2516 CrossRef CAS.
- H. Kuroda, I. Tonita and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 1597 CrossRef CAS.
- S. Liu, J. Liu, Q. Wang, J. Wang, F. Huang, W. Wang, C. Sun and D. Chen, Org. Chem. Front., 2020, 7, 1137 RSC.
- B. He, S. Zhen, Y. Wu, R. Hu, Z. Zhao, A. Qin and B. Z. Tang, Polym. Chem., 2016, 7, 7375 RSC.
- R. Hu and B. Z. Tang, in Multi-Component and Sequential Reactions in Polymer Synthesis, ed. P. Theato, Springer International Publishing, Cham, 2015, vol. 17 Search PubMed.
- J. Wang, A. Qin and B. Z. Tang, Macromol. Rapid Commun., 2021, 42, 2000547 CrossRef CAS PubMed.
- X. Wang, B. Li, J. Peng, B. Wang, A. Qin and B. Z. Tang, Macromolecules, 2021, 54, 6753 CrossRef CAS.
- Z. Zhang, Y. You and C. Hong, Macromol. Rapid Commun., 2018, 39, 1800362 CrossRef PubMed.
- X. Wu, J. He, R. Hu and B. Z. Tang, J. Am. Chem. Soc., 2021, 143, 15723 CrossRef CAS PubMed.
- C. Wang, B. Yu, W. Li, W. Zou, H. Cong and Y. Shen, Mater. Today Chem., 2022, 25, 100948 CrossRef CAS.
- N. Zheng, H. Gao, Z. Jiang and W. Song, Sci. China: Chem., 2023, 66, 870 CAS.
- H. K. J. Hall, Angew. Chem., Int. Ed., 1983, 22, 440 CrossRef.
- Q. Li, S. Ma, N. Lu, J. Qiu, J. Ye, Y. Liu, S. Wang, Y. Han, B. Wang, X. Xu, H. Feng and J. Zhu, Green Chem., 2020, 22, 7769 RSC.
- P. A. J. M. de Jongh, D. M. Haddleton and K. Kempe, Prog. Polym. Sci., 2018, 87, 228 CrossRef CAS.
- X. Chen, R. Hu, C. Qi, X. Fu, J. Wang, B. He, D. Huang, A. Qin and B. Z. Tang, Macromolecules, 2019, 52, 4526 CrossRef CAS.
- B. He, J. Zhang, J. Wang, Y. Wu, A. Qin and B. Z. Tang, Macromolecules, 2020, 53, 5248 CrossRef CAS.
- L. Dong, W. Fu, P. Liu, J. Shi, B. Tong, Z. Cai, J. Zhi and Y. Dong, Macromolecules, 2020, 53, 1054 CrossRef CAS.
- W. Fu, G. Zhu, J. Shi, B. Tong, Z. Cai and Y. Dong, Chin. J. Polym. Sci., 2019, 37, 981 CrossRef CAS.
- R. Hu, X. Chen, T. Zhou, H. Si, B. He, R. T. K. Kwok, A. Qin and B. Z. Tang, Sci. China: Chem., 2018, 62, 1198 CrossRef.
- O. S. Fenton, J. L. Andresen, M. Paolini and R. Langer, Angew. Chem., Int. Ed., 2018, 57, 16026 CrossRef CAS PubMed.
- Y. Liu, A. Qin and B. Z. Tang, Prog. Polym. Sci., 2018, 78, 92 CrossRef CAS.
- B. Song, D. Lu, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2022, 144, 1672 CrossRef CAS PubMed.
- F. Aryanasab and M. R. Saidi, Monatsh. Chem., 2014, 145, 521 CrossRef CAS.
- A. McClain and Y. L. Hsieh, J. Appl. Polym. Sci., 2003, 92, 218 CrossRef.
- V. N. Elokhina, A. S. Nakhmanovich, A. E. Aleksandrova, B. I. Bishnevskii and I. D. Kalikhman, Pharm. Chem. J., 1986, 20, 1061 CAS.
- J. Yan and Z. Chen, Synth. Commun., 1999, 29, 2867 CrossRef CAS.
- Y. Liu and W. Bao, Tetrahedron Lett., 2007, 48, 4785 CrossRef CAS.
- M. R. Wood, D. J. Duncalf, S. P. Rannard and S. Perrier, Org. Lett., 2006, 8, 553 CrossRef CAS PubMed.
- N. Azizi, F. Aryanasab and M. R. Saidi, Org. Lett., 2006, 8, 5275 CrossRef CAS PubMed.
- W.-F. Su, Step Polymerization, in Principles of Polymer Design and Synthesis, Lecture Notes in Chemistry, Springer, Berlin, Heidelberg, 2013, vol. 82 Search PubMed.
- A. Z. Halimehjani and Y. L. Nosood, Org. Lett., 2017, 19, 6748 CrossRef PubMed.
- S. Gabillet, D. Lecerclé, O. Loreau, M. Carboni, S. Dézard, J. M. Gomis and F. Taran, Org. Lett., 2007, 9, 3925 CrossRef CAS.
- Z. Chen, Y. Jin and P. J. Stang, J. Org. Chem., 1987, 52, 4117 CrossRef CAS.
- H. Deng, Z. He, J. W. Y. Lam and B. Z. Tang, Polym. Chem., 2015, 6, 8297 RSC.
- M. Pramanik, K. Choudhuri, S. Chakraborty, A. Ghosh and P. Mal, Chem. Commun., 2020, 56, 2991 RSC.
- Y. Wei and S. A. Hadigheh, Composites, Part B, 2023, 260, 110786 CrossRef CAS.
- K. S. Kang, C. Olikagu, T. Lee, J. Bao, J. Molineux, L. N. Holmen, K. P. Martin, K. J. Kim, K. H. Kim, J. Bang, V. K. Kumirov, R. S. Glass, R. A. Norwood, J. T. Njardarson and J. Pyun, J. Am. Chem. Soc., 2022, 144(50), 23044 CrossRef CAS PubMed.
- M. Lee, Y. Oh, J. Yu, S. G. Jang, H. Yeo, J. J. Park and N. H. You, Nat. Commun., 2023, 14, 2866 CrossRef CAS PubMed.
- D. H. Kim, W. Jang, K. Choi, J. S. Choi, J. Pyun, J. Lim, K. Char and S. G. Im, Sci. Adv., 2020, 6(28), eabb5320 CrossRef CAS PubMed.
- A. Nishant, K.-J. Kim, S. A. Showghi, R. Himmelhuber, T. S. Kleine, T. Lee, J. Pyun and R. A. Norwood, Adv. Opt. Mater., 2022, 10, 2200176 CrossRef CAS.
- D. Xin, A. Qin and B. Z. Tang, Polym. Chem., 2019, 10, 4271–4278 RSC.
- H. Kuhn, Thin Solid Films, 1989, 178, 1 CrossRef CAS.
- S. Tang, J. Gong, Y. Shi, S. Wen and Q. Zhao, Nat. Commun., 2022, 13, 3227 CrossRef PubMed.
- S. Xiong, J. Li, J. Peng, X. Dong, F. Qin, W. Wang, L. Sun, Y. Xu, Q. Lin and Y. Zhou, Adv. Opt. Mater., 2022, 10, 2101837 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc02415f |
| This journal is © The Royal Society of Chemistry 2024 |