
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient biosynthesis of 2,4-dihydroxybutyric acid by a methanol assimilation pathway in engineered Escherichia coli†
Xianjuan
Dong‡
a,
Chao
Sun‡
 abc,
Jing
Guo
abc,
Xiangyu
Ma
ad,
Mo
Xian
*abc and
Rubing
Zhang
*abc
abc,
Jing
Guo
abc,
Xiangyu
Ma
ad,
Mo
Xian
*abc and
Rubing
Zhang
*abc
aCAS Key Laboratory of Bio-Based Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, China. E-mail: zhangrb@qibebt.ac.cn; xianmo@qibebt.ac.cn
bShandong Energy Institute, Qingdao 266101, China
cQingdao New Energy Shandong Laboratory, Qingdao 266101, China
dUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 18th August 2023
Abstract
2,4-Dihydroxybutyric acid (2,4-DHB) is an important fine chemical, which can be used as a precursor in the production of important chemicals. Methanol, as an attractive C1 compound, is an ideal nonfood sugar feedstock for biomanufacturing. In this study, we reported for the first time the biosynthesis of 2,4-DHB in engineered Escherichia coli from a mixture of methanol and glucose. In this process, methanol dehydrogenase was used to convert methanol into formaldehyde, and the key intermediate 2-keto-4-hydroxybutyric acid was synthesized through the condensation of formaldehyde and pyruvate derived from glycolysis by pyruvate-dependent aldolase, which was subsequently transformed into the target product 2,4-DHB by an enzymatic hydrogenation reaction. The new pathway demonstrated the feasibility of producing 2,4-DHB from methanol and glucose in E. coli. For improving the production of 2,4-DHB step by step, dehydrogenases and aldo–keto reductases with higher activity towards 2,4-DHB were screened, and the pyruvate degradation pathway genes were knocked out to enhance the flux from pyruvate towards 2,4-DHB biosynthesis, resulting in the strain DHB9 with about 10-fold improvement compared to the initial strain DHB4. Additionally, the methanol dehydrogenase MDHCn with higher enzymatic activity was obtained by screening. The mutant MDHCn (G48F) with improved activity in vitro was obtained by rational analysis of MDHCn, and the results showed that the enzymatic activity of MDHCn (G48F) increased 3-fold in vitro compared with that of the wild type. Finally, on the basis of the above work, the best engineered strain DHB13 was constructed by knocking out the gene (frmRAB) responsible for the conversion of formaldehyde to formic acid, and the target product 2,4-DHB was accumulated up to 14.6 g L−1 in a 5 L bioreactor. To date, the highest concentration of 2,4-DHB has been achieved in this work, which is promising to make the bioprocess feasible from an economic perspective. Moreover, one glucose molecule and two methanol molecules can be bio-transformed into two 2,4-DHB molecules with 100% conversion of carbon atoms in theory, showing the high carbon atom economy of the novel synthetic metabolic pathway.
Introduction
In recent years, with the depletion of fossil fuels and the increase in greenhouse effect induced by the burning of fossil fuels, C1 compounds (such as methane, methanol and methylamine) have attracted increasing attention due to their low cost, accessibility, sustainability and environmental friendliness.1,2 Methyltrophs have the ability to utilize C1 compounds as carbon and energy sources, with great potential to convert C1 compounds into a variety of applicable chemicals and materials.3,4 Methanol, as a single carbon compound, is a promising substrate for use as a fuel, energy storage material and feedstock,5 and it has great potential to be a carbon source for microbial fermentation processes.3,6,7 From the perspective of biomanufacturing, methanol is expected to be a key feedstock for the next generation of biomanufacturing as a non-food feedstock that is widely available, inexpensive, a pure substrate, and highly controllable, and using low-cost carbon-based materials facilitates the commercialization process of industrial biotechnology.8–10 From the perspective of biotechnology and bulk chemicals, methanol is a highly exploitable C1 compound that can be used directly in fermentation as a pure substrate.11 The use of synthetic biology technology to construct recombinant microorganisms that can efficiently utilize methanol to realize the biotransformation from methanol to high-value chemicals has become a hot research topic at home and abroad.12Methanol is first converted into formaldehyde in vivo by enzymatic catalysis and then further oxidized to CO2 through anabolic pathways or converted into energy and substances required for the vital activities of living organisms through certain assimilation pathways.4,13 It is difficult to construct a new methanol metabolic pathway in methylotrophic bacteria for the production of commercial chemicals or biofuels using techniques such as genetic engineering because most natural methylotrophic bacteria lack well-established genetic tools. At present, bioavailability platforms such as E. coli have been widely used for industrial production of bulk chemicals,11 so we constructed a methanol utilization pathway in E. coli with a view to realizing the bioavailability of methanol. However, due to the specificity and complexity of the methanol metabolic process, it is difficult for the current artificially designed methylotrophic bacteria to realize methanol as the sole carbon source, and methanol-dependent growth with glucose as the auxiliary substrate is the most important case. Glucose is more abundant and cheaper than ribose and xylose.14 Therefore, it is an ideal co-substrate for expanding the industrial application of methanol-dependent strains.
2,4-Dihydroxybutyric acid (2,4-DHB) is a compound with high potential for economic benefits as an important precursor for the chemical synthesis of the methionine analogue 2-hydroxy-4-(methylthio) butyric acid (HMTB).15,16 HMTB is widely used as animal rations for nutritional supplementation to improve the nutritional value of animal feed.13,14 However, there is no fermentation process for synthesizing HMTB and metabolic costs will dramatically increase due to the introduction of sulfur.17 A two-stage process for the production of HMTB is envisaged: first using fermentation to biosynthesize the precursor 2,4-DHB of HMTB, and then converting 2,4-DHB and methanethiol into HMTB by an established chemical method with 100% carbon yield.16 However, petrochemical synthesis of 2,4-DHB is not economically viable, and no natural metabolic pathways exist for the biochemical production of 2,4-DHB. Li et al. constructed a three-step synthetic pathway using malic acid as a raw material to obtain 2,4-DHB by activating the terminal carboxyl group of malic acid via CoA, followed by reduction to an aldehyde group and further reduction to a hydroxyl group.18 However, this pathway was reported to yield only trace amounts of 2,4-DHB (<10 mg L−1). Then, a three-step metabolic pathway for 2,4-DHB synthesis starting from the natural metabolite malate was similarly constructed.15 They used malate kinase, malate semialdehyde dehydrogenase and malate semialdehyde reductase, along with a rational design for malate semialdehyde reductase, and the pathway was expressed in an engineered E. coli strain, producing 1.8 g L−1 of 2,4-DHB. To further increase the production of 2,4-DHB, Walther et al. developed a two-step synthetic metabolic pathway to synthesize 2,4-DHB by converting the natural amino acid homoserine into 2,4-DHB.16 In their study, 2-oxo-4-hydroxybutyric acid (OHB) was obtained through homoserine deamination catalyzed by homoserine transaminase and then reduced to 2,4-DHB by OHB reductase. Finally, 5.3 g L−1 of 2,4-DHB can be produced by batch culture of the engineered E. coli. Research indicated that the accumulation of intracellular homocysteine strongly inhibits its own biosynthesis,19 and researchers believed that the elevated intracellular homoserine concentration was the main reason for the rather limited production of 2,4-DHB by the engineered strain. Although engineered strains can efficiently synthesize homoserine, the low activity of homoserine aminotransferase which catalyzes the deamination of homoserine to produce OHB results in an increase in intracellular homoserine concentration. Therefore, the development of more efficient aminotransferases represents an important strategy to increase DHB production in the homoserine-dependent pathway.
In this study, with the aim of increasing the carbon atom economy of 2,4-DHB biosynthesis, a novel pathway was developed to produce 2,4-DHB using glucose and methanol as the feedstocks. The pathway consists of three main enzymatic reactions: (I) oxidation of methanol to formaldehyde using methanol dehydrogenase (MDH); (II) condensation of formaldehyde and pyruvate to 2-keto-4-hydroxybutyric acid using aldolase; and (III) reduction of 2-keto-4-hydroxybutyric acid to 2,4-DHB using dehydrogenase. First, the 2,4-DHB biosynthesis module from formaldehyde was created and proofed using 13C-labeled formaldehyde with glucose as the co-substrate. Then, the full pathway of 2,4-DHB biosynthesis from methanol and glucose was established in an engineered E. coli. The low oxidation efficiency of methanol in heterologous microorganisms is one of the major barriers to methanol utilization, and the activity of NAD+-dependent MDH is crucial for the conversion of methanol into formaldehyde. The MDHCn with higher activity was obtained by screening, and rational modification of the more active MDH showed higher activity in vivo and in vitro to a certain extent. Finally, the optimized strain was used for high-density fermentation, and the concentration of 2,4-DHB reached 14.6 g L−1. In this system, pyruvic acid (a three-carbon metabolite from glucose) and formaldehyde (a one-carbon metabolite from methanol) were converted into the final product without carbon loss, showing the possibility of being a promising platform for the bioconversion of methanol to valuable chemicals.
Materials and methods
Strains and plasmids
All strains and plasmids used in this work are shown in Table S1.†E. coli DH5α was used as a host for recombinant DNA manipulation and plasmid maintenance. E. coli BL21(DE3) was used for protein overexpression and purification, and E. coli W3110 was used as the starting strain to be metabolically engineered to produce 2,4-DHB. The aldolase gene khb (Sequence ID: CP102711.1) involved in the aldol condensation of pyruvate and formaldehyde was amplified directly from the genome DNA of E. coli K-12. The MDH genes mdh2Bm (Sequence ID: CP007739.1) and mdhCn (Sequence ID: CP002878.1) involved in methanol metabolism were synthesized after codon optimization for E. coli. The encoding genes par, pgcr, cpar4, ldh, 2ldh and mdh involved in 2,4-DHB biosynthesis were synthesized after codon optimization for E. coli with the corresponding sequences reported in Gene Sequence ID: AB020760.3, XM_001482396.1, JX512915.1, CP039707.1, CP013921.1, and CP110410.1, respectively. The genes were codon optimized and synthesized by Suzhou GENEWIZ Biotechnology Co., Ltd (Suzhou, China). The plasmids required for gene overexpression were constructed using pETDuet-lac as the backbone, and a promoter Plac,1-6 was cloned into plasmid pETDuet-1 instead of promoter PT7 to generate the plasmid pETDuet-lac. The DNA sequence and the intensity of transcription of Plac,1-6 are derived from a previous research study.20 The strains and plasmids used in this study are summarized in Table S1.†The plasmid vector pETDuet-lac was also used to construct the novel pathway for 2,4-DHB production. The polymerase chain reaction (PCR) primers used for gene cloning and plasmid construction are summarized in Table S2.† PCR was performed using PrimeSTAR Max DNA polymerase (Takara, Dalian, China) according to the manufacturer's instructions for the amplification of DNA fragments. All plasmids were constructed with the ClonExpress Ultra One Step Cloning Kit (Vazyme Biotech Co., Ltd, China) protocol.
The chromosomal poxB, pflB, adhE and pta genes of the E. coli W3110 strain encoding for pyruvate formate-lyase, pyruvate oxidase, ethanol dehydrogenase and phosphate transacetylase, respectively, were knocked out using the P1 transduction method with the selected Keio collection strain.21 All transduction donor strains were taken from the E. coli Keio knockout collection (Open Biosystems, Huntsville, AL). The frmRAB genes encoding for formaldehyde dehydrogenase for the conversion of formaldehyde to formic acid were also knocked down sequentially as described above. The recombinant plasmids were transformed into the corresponding hosts for 2,4-DHB biosynthesis.
Media and culture conditions
To determine the ability of different engineered strains to produce 2,4-DHB, shake flask culture experiments were used for evaluation. The seed culture preparation was performed in LB medium (tryptone 10 g L−1, yeast extract 5 g L−1, and NaCl 10 g L−1), which was incubated at 37 °C with shaking at 200 rpm. For the production of 2,4-DHB, strains are grown in M9 salt medium (Na2HPO4·12H2O 15.2 g L−1, KH2PO4 3 g L−1, NH4Cl 1 g L−1, and NaCl 0.5 g L−1) containing 20 g L−1 of glucose and 1 mM MgSO4. Ampicillin (100 mg L−1) and kanamycin (50 mg L−1) were added to culture media as required. The production culture was initially induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and 50 mM formaldehyde (or methanol) when the OD600 reached ∼0.8 and incubated for 48 h with 200 rpm shaking in a rotary shaker at 30 °C.To obtain a higher concentration of 2,4-DHB, fed-batch fermentations were conducted in a Biostat B plus MO5L bioreactor (Sartorius, Germany) containing 2 L of fermentation broth. The engineering strain was cultured for 12 h at 37 °C in a 250 mL shake flask containing 50 mL of M9 liquid medium. Then, the seed culture was transferred into 2 L fermentation medium (9.8 g L−1 K2HPO4·3H2O, 2.1 g L−1 citric acid·H2O, 0.3 g L−1 ferric ammonium citrate, 3 g L−1 (NH4)2SO4) supplemented with 20 g L−1 glucose and 1 mL of trace elements (2.5 g L−1 CuSO4·5H2O, 2.9 g L−1 ZnSO4·7H2O, 3.7 g L−1 (NH4)6Mo7O24·4H2O, 15.8 g L−1 MnCl2·4H2O, and 24.7 g L−1 H3BO3). The temperature was set at 37 °C, and the pH value was adjusted to 7.0 by an automatic addition of 26% ammonia solution. The dissolved oxygen (DO) level was maintained at 20% by automatically adjusting the stirring speed. When the OD600 reached 30 (initial glucose was exhausted), 0.5 mM IPTG was added and the temperature was set at 30 °C for 85 h. The feeding speed was variable for the induction time with 60% glucose (2.0–5.0 g L−1 h−1) and 20% methanol (0.6–1.2 g L−1 h−1) supplements. The sample was collected for analysis after incubation for a period of time depending on the needs of the experiment.
Determination of 13C-labeled intracellular metabolites
For the proof of the new biosynthetic pathway of 2,4-DHB using E. coli W3110, a single colony was inoculated into 3 mL of LB medium and cultivated at 200 rpm for 10 h at 37 °C. Then, the fermentation broth was transferred (1% inoculum) into a 250 mL flask containing 50 mL of M9 medium supplemented with 100 μg mL−1 ampicillin and incubated at 37 °C. When the OD600 reached about 0.8, 0.5 mM IPTG and 5 mM formaldehyde or 13C-HCHO were added to induce gene expression. After induction at 30 °C for 12 h, 50 mM formaldehyde, 13C-HCHO was added as a substrate. Batch cultivation was performed at 30 °C and 200 rpm for 48 h. The entire shake flask fermentation was carried out in two independent experiments.Site-directed saturation mutagenesis of mdhCn
The gene mdhCn was cloned into pET28a(+) via BamH I and Sac I double digestion to create the plasmid pET28a-mdhCn expressing an N-terminal fusion protein with a His6-tag. MDHCn mutants were derived from pET28a-mdhCnvia site-directed saturation mutagenesis, and the site-directed saturation mutagenesis was carried out on pET-28a-mdhCn plasmids using the primers listed in Table S2.† The mutated plasmids were treated using Mut Express®IIFast Mutagenesis Kit V2 (Vazyme Biotech Co., Ltd, China) according to the manufacturer's instructions. All mutated plasmids were verified to carry the desired mutations by DNA sequencing.Enzyme purification and enzymatic assays
The plasmids pET28a-mdh2Bm and pET28a-MDHCn were transformed into E. coli BL21(DE3) for expression, resulting in the strains DHB14 and DHB15. DHB14 and DHB15 were separately inoculated into 100 mL of LB medium supplemented with 100 μg mL−1 kanamycin and cultivated at 37 °C with shaking at 200 rpm. After the OD600 reached 0.6, 0.5 mM IPTG was added and the temperature was set at 16 °C for 18 h. Cells were harvested by centrifugation at 4 °C, washed twice with 50 mM Tris–HCl (pH 7.0) and resuspended in the buffer containing 50 mM Tris-HCl (pH 7.0). The recombinant protein was affinity purified through a Ni-NTA column. The purity of the purified enzyme was checked by SDS-PAGE and the protein concentration was determined using the Takara Bradford Protein Assay Kit (Takara, Dalian, China). Other mutant proteins (MDHCn(G48F), MDHCn(M52F), and MDHCn(C369N)) were similarly prepared.MDH activity was defined as 1 μmol of NAD+ reduced to NADH per minute at 37 °C.6 The assay mixture contained 0.5 mM NAD+, 1 M methanol, 5 mM MgSO4 and buffer A (2.69 g L−1 KH2PO4, 18.24 g L−1 K2HPO4·3H2O, pH 7.4), and an appropriate amount of purified enzyme. The enzymatic assays were carried out at 37 °C. The reactions were followed by the characteristic absorption of NADH at 340 nm in a microplate reader (Tecan Spark, Switzerland).
Analytical methods
Quantification of glucose, pyruvate and a high concentration of 2,4-DHB was performed using an LC-20AT high-performance liquid chromatography (HPLC) system (Shimadzu, Japan) equipped with an Aminex HPX-87H analytical column (Bio-Rad, USA), a differential refractive index detector, and a UV detector at 210 nm. The mobile phase was 5 mM H2SO4 with a flow rate of 0.5 mL min−1. The column temperature was 50 °C with an RID detector temperature of 35 °C.For the quantification of the low concentration of 2,4-DHB, gas chromatography-mass spectrometry (GC-MS) was conducted using a slightly modified method.22 A brief description is as follows: 500 μL of filtered and dried fermentation liquid sample (pH > 10) was redissolved in N,N-dimethylformamide, followed by the addition of 80 μL of the derivatization reagent bis(trimethylsilyl)trifluoroacetamide (BSTFA) with 1% trimethylchlorosilane, and incubated at 70 °C for 30 min. The supernatant was tested by GC-MS. 1 μL of sample was injected with a split ratio of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 into an SH-Rtx-5MS Cap. column (30 m, 0.25 mm ID, 0.25 μm) using helium as the carrier gas. The inlet temperature and flow rate were set at 280 °C and 3.0 mL min−1, respectively. The oven temperature gradient program was set as follows: initially held at 60 °C for 0 min, raised by 10 °C min−1 to 270 °C, and held for 9 min. The total run time was 30 min. The MS conditions for identification of 2,4-DHB were as follows: for Q1 scan mode, 30–500 m/z mass range. For Q1 SIM, the selected ions were set at 103, 129, 219 and 232 m/z. Quantification was based on the peak area ratios of the different compounds to standard chemicals.
:1 into an SH-Rtx-5MS Cap. column (30 m, 0.25 mm ID, 0.25 μm) using helium as the carrier gas. The inlet temperature and flow rate were set at 280 °C and 3.0 mL min−1, respectively. The oven temperature gradient program was set as follows: initially held at 60 °C for 0 min, raised by 10 °C min−1 to 270 °C, and held for 9 min. The total run time was 30 min. The MS conditions for identification of 2,4-DHB were as follows: for Q1 scan mode, 30–500 m/z mass range. For Q1 SIM, the selected ions were set at 103, 129, 219 and 232 m/z. Quantification was based on the peak area ratios of the different compounds to standard chemicals.
Results and discussion
Development and evaluation of the 2,4-DHB biosynthesis pathway
As shown in Fig. 1, the biosynthetic pathway of 2,4-DHB from glucose and formaldehyde consisted of three enzymatic reactions. Gibbs free energy was used to evaluate the feasibility of the artificial synthetic pathway. The calculated total standard Gibbs free energy change of the three reactions for the biosynthesis of 2,4-DHB from glucose and formaldehyde was −12.8 kcal mol−1 (Fig. S1†). The negative standard Gibbs free energy for the designed pathway attests to its thermodynamic feasibility. | ||
| Fig. 1 Biosynthetic pathway of 2,4-DHB from glucose and formaldehyde in E. coli. Abbreviations: MDH, methanol dehydrogenase; KHB, aldolase; PAR, aldo–keto reductase; DH, dehydrogenase. | ||
In order to determine the successful construction of the 2,4-DHB pathway, the engineered E. coli strain DHB1 expressing the two enzymes KHB and PAR was cultured with the co-substrates formaldehyde and glucose. GC-MS and LC-MS were employed to determine the 2,4-DHB production in the fermentation culture, respectively (Fig. S2A and Fig. S2B†), and the analytical results proved that the product was successfully synthesized. Furthermore, 2,4-DHB was obtained by performing liquid chromatography and analyzed using nuclear magnetic resonance (NMR) (Fig. S3†).
Interestingly, we found that 2,4-DHB and α-hydroxy-γ-butyrolactone can be interconverted under different pH conditions. As shown in Fig. S4,† α-hydroxy-γ-butyrolactone is the main component under low pH conditions, and 2,4-DHB is the major component under high pH conditions, especially, pH > 10. Thus, 10 M KOH was used to keep the pH > 10 in order to obtain a large amount of 2,4-DHB during fermentation.
This is the first time our study has successfully created a biosynthetic pathway through the overexpression of aldolase and carbonyl reductases to produce 2,4-DHB in E. coli. This pathway is not only much shorter and simpler than the recently proposed homoserine dependent pathway, but also avoids the complex regulations involving essential amino acid precursors.16 This work constructed a novel unnatural biosynthetic pathway by designing the production of 2,4-DHB from glucose and formaldehyde, which has created the possibility of producing 2,4-DHB from cheaper substrates.
Validation and optimization of the formaldehyde-based 2,4-DHB production module
In order to demonstrate that the exogenously added formaldehyde was condensed into 2,4-DHB, 13C-HCHO was used as the substrate for performing the biosynthesis. As shown in Fig. 2, the ratios of the peaks at around m/z 103 and m/z 104 are almost the same with the unlabeled standard sample and the 13C-HCHO samples. This finding indicated that formaldehyde is an essential precursor in the synthesis of 2,4-DHB, and the coupling of formaldehyde with endogenous pyruvate derived from glycolysis provides an excellent opportunity for the synthesis of the valuable chemical 2,4-DHB. | ||
| Fig. 2 The biosynthesis of 2,4-DHB using glucose and formaldehyde as co-substrates. (A) Metabolic routes for 2,4-DHB production from glucose and formaldehyde in E. coli. (B) Verification of 2,4-DHB formation from 13C-HCHO and glucose using GC-MS. (C) Verification of 2,4-DHB formation from formaldehyde and glucose using GC-MS. | ||
To strengthen the conversion from 2-keto-4-hydroxybutyrate to 2,4-DHB, the key enzyme was screened from three kinds of dehydrogenases (Ldh, 2Ldh and Mdh) and three kinds of aldo–keto reductases (PAR, PgCR and CPAR4). These candidates were co-expressed with Khb in E. coli W3110, named DHB1-6. These results are shown in Fig. 3; the production of 2,4-DHB in the strains DHB3 and DHB4 was approximately 2.4-fold and 3-fold higher than that in the strain DHB1, respectively. Thus, strain DHB4 with the highest production of 2,4-DHB was selected for the subsequent study.
 | ||
| Fig. 3 Effect of different aldo–keto reductases and dehydrogenases on the production of 2,4-DHB. | ||
The above results indicate that both dehydrogenase and carbonyl reductase can convert 2-keto-4-hydroxybutyric acid into 2,4-DHB by catalytic hydrogenation to reduce carbonyl groups to hydroxyl groups. However, the catalytic mechanisms were different between them. Carbonyl reductase, as part of the short-chain dehydrogenase/reductase family, belongs to the oxidoreductase protein.23 These aldo–keto reductases primarily reduce aldehydes and ketones to primary and secondary alcohols.24 The PAR enzyme was considered as a new member of long-chain alcohol dehydrogenases, and the optimal substrates for PAR are medium-chain (C5–C10) common aldehydes and 2-ketoalkanes, as well as aryl aldehydes and aryl ketones.25 As for PgCR, it is more likely to be interested in substrates with substituents adjacent to the carbonyl group or those with substituents on the para position of the phenyl ring,26 and CPAR4 was active towards the selected substrates, especially 2-hydroxyacetophenone and ethyl 4-chloro-3-oxobutyrate.27 However, the target product, 2,4-DHB, is a polyhydroxy short-chain acid, so the selected aldo–keto reductase shows a low catalytic activity for it. Among them, lactate dehydrogenase (Ldh), which has a great influence on 2,4-DHB production, can catalyze the interconversion between 2,4-DHB and its corresponding 2-keto acid using the NAD+/NADH coenzyme system.28 Ldh shows strong selectivity towards monocarboxylic acid and the compensatory stabilizing loop interactions in Ldh contribute to the conversion of 2-keto acid into 2,4-DHB.29,30
Effects of weakening pyruvate catabolism
Pyruvate is an important central metabolic intermediate in the glycolysis or the Embden–Meyerhof–Parnas pathway in E. coli,31 and the key metabolic node from which target products can be derived as well as other byproducts. Pyruvate can be naturally converted into acetyl-CoA, lactate, acetate or ethanol by endogenous pathways. Among them, pyruvate oxidase (encoded by poxB) is a peripheral membrane flavoprotein that catalyzes the decarboxylation of pyruvate to acetate and CO2 combined with the reduction of the flavin adenine dinucleotide FAD and furthermore contributes to pyruvate catabolism and aerobic growth of E. coli.32,33 The pyruvate formate-lyase (encoded by pflB) catalyzes the reaction from pyruvate to acetyl-CoA and pyruvate accumulation can be achieved when it is blocked.34 Ethanol dehydrogenase (encoded by adhE) plays a role in producing ethanol from pyruvate via acetyl-CoA, and is responsible for the accumulation of acetyl-CoA.35,36 Since in this study pyruvate is a key precursor of 2,4-DHB biosynthesis, an E. coli chassis for the accumulation of pyruvate is essential for improving the production. Hence, the pyruvate catabolism related genes poxB, pflB and adhE were sequentially knocked out in E. coli W3110 to inhibit the metabolic branch flux of pyruvate, resulting in E. coli D2, D4 and D6 (Fig. 4A). The recombinant plasmid pETDuet-lac-khb-ldh was translated into these strains, resulting in the strains DHB7, DHB8 and DHB9, respectively. As shown in Fig. 4B, the concentration of 2,4-DHB in strain DHB9 with simultaneous knockdown of poxB, pflB and adhE was approximately 10-fold higher than that in strain DHB4. In addition, the concentration of 2,4-DHB in shake flask fermentation varied with culture time with glucose and formaldehyde as co-substrates, and the highest concentration of 2,4-DHB was reached at 20 h (Fig. S5A†). Based on the above result, the E. coli chassis with high pyruvate accumulation exhibited high potential to be an ideal chassis for the biosynthesis of 2,4-DHB.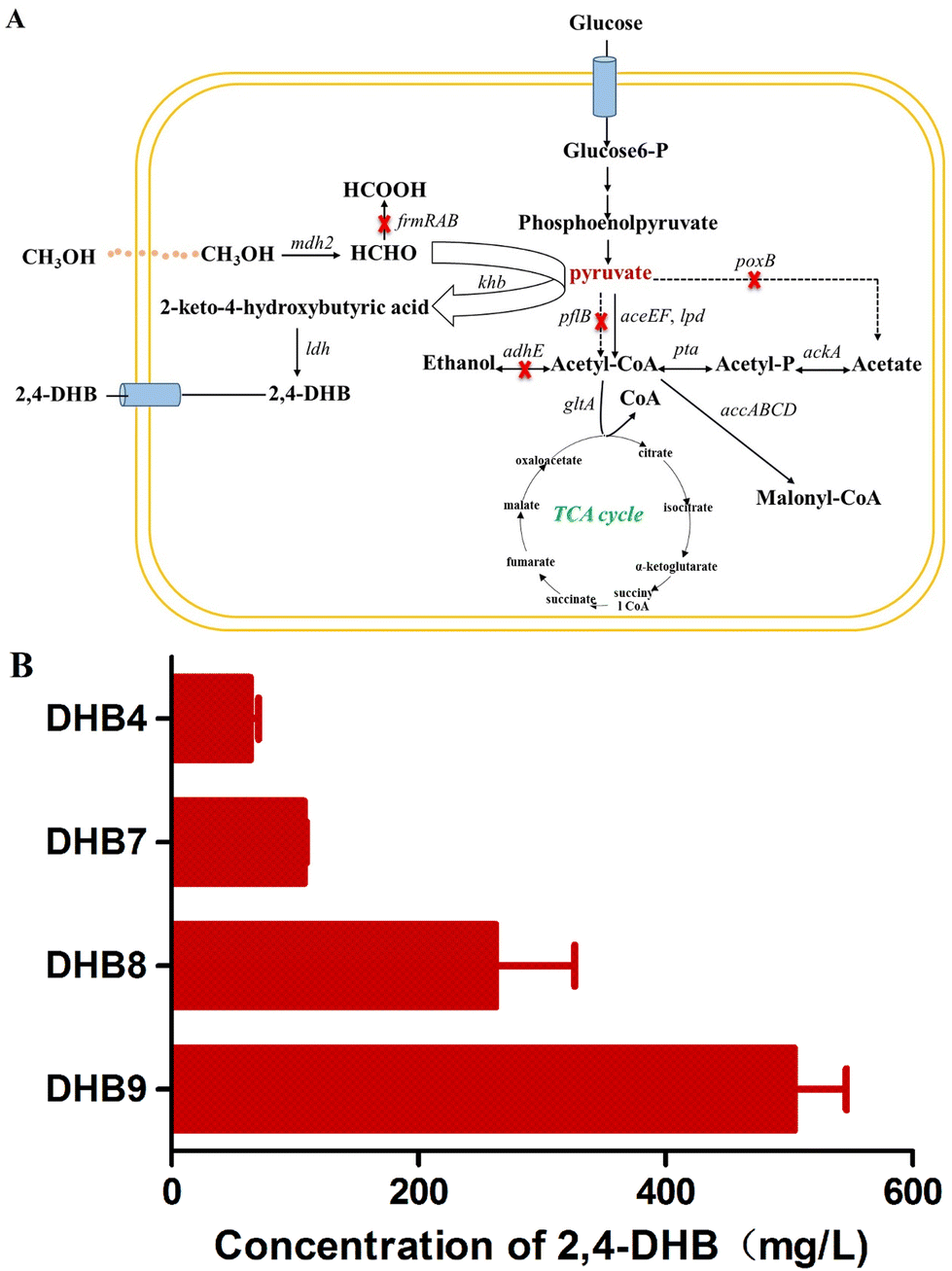 | ||
| Fig. 4 Building a chassis cell to produce 2,4-DHB using multiple gene regulation strategies. (A) Pyruvate degradation pathway and the gene knockout. (B) Effect of different E. coli chassis on the production of 2,4-DHB. | ||
Evaluation of the methanol supply module for 2,4-DHB biosynthesis
The research results presented above provide the optimal pathway for 2,4-DHB biosynthesis from formaldehyde and glucose. To further develop the methanol supply module, methanolic dehydrogenase derived from Bacillus methanolicus (MDH2Bm) was selected, the strain DHB10 was constructed, and the production of 2,4-DHB reached its maximum at 8 h in shake flask fermentation with glucose and methanol as co-substrates (Fig. S5B†). The Gibbs free energy for the synthesis of formaldehyde from methanol is 10.45 kcal mol−1, combined with the Gibbs free energy for the synthesis of 2,4-DHB from glucose and formaldehyde (−12.8 kcal mol−1), so the Gibbs free energy is estimated to be negative (−2.35 kcal mol−1) (Fig. S1†). A novel synthetic metabolic pathway for the biosynthesis of 2,4-DHB using glucose and methanol as raw materials was established. As shown in Fig. 5, it was found that the concentration of 2,4-DHB produced with methanol and glucose as co-substrates was significantly lower than that of 2,4-DHB from the mixture of formaldehyde and glucose. The concentration of 2,4-DHB with methanol alone was only 110 mg L−1 in the shake flask, while the concentration of 2,4-DHB obtained with formaldehyde alone was as high as 500 mg L−1. Furthermore, since formaldehyde was added based on the addition of the substrate methanol, the concentration of 2,4-DHB also reached 500 mg L−1 with the mixture of formaldehyde and methanol, the same as that with formaldehyde alone. The result indicates that the activity of MDH may be the bottleneck in this metabolic pathway. Krog et al. reported that MDHs generally have higher activity and affinity towards formaldehyde compared to methanol,37 which is consistent with our results. Moreover, their catalytic activity towards methanol is considerably lower than that towards other alcohols generally. | ||
| Fig. 5 Effect of different substrates on the production of 2,4-DHB. Methanol: shake flask fermentation with glucose and methanol as substrates; formaldehyde: shake flask fermentation with glucose and formaldehyde as substrates; and methanol + formaldehyde: shake flask fermentation with glucose, methanol and formaldehyde as substrates. | ||
The NAD+-dependent methanol dehydrogenase MDH2Bm, which is derived from Bacillus methanolicus, has the highest electron retention in the catalytic oxidation of methanol and facilitates the production of reduced metabolites.38 However, the reaction is thermodynamically unfavorable to the oxidation of methanol, and the affinity and catalytic activity of these enzymes towards methanol are low.39 Meng et al. also reported that MDH is the bottleneck in the bioconversion using methanol and ethanol as co-substrates.40 Therefore, mining and designing MDHs would be important for enhancing the rate-limiting step for the transformation of methanol to formaldehyde and significant for methanol-based biomanufacturing of fuels and chemicals.
Selection and modification of methanol dehydrogenase
Since the MDH responsible for the first step in methanol utilization has low activity in heterologous microorganisms, enzyme screening and protein engineering of MDH are expected to improve the efficiency of this rate-limiting step. The literature also reports that the NAD+-dependent MDHs have broad substrate specificities, with high activity for 1-propanol or n-butanol and low activity for methanol.37 The MDH, catalyzing the formation of formaldehyde from methanol, has poor thermodynamic properties (Fig. S1†). Therefore, in order to screen for a MDH with high activity towards methanol as an alternative to MDH2Bm, we cloned a MDH derived from Cupriavidus necator N-1, which was named MDHCn. As shown in Fig. 6A, the enzyme activity of MDHCn is six-fold higher than that of MDH2Bmin vitro, and the specific activities of MDHCn and MDH2Bm are shown in Table S3.† | ||
| Fig. 6 Activity and the effect of MDH and its mutants on 2,4-DHB production. (A) Comparison of the activity of two methanol dehydrogenases in vitro. (B) Comparison of the enzymatic activity of MDHCn and its mutants in vitro. (C) Effect of DHB11 and DHB12 fermentation on the production of 2,4-DHB. (D) MDHCn AlphaFold-predicted protein structures. (E) Relative position of G48 and NAD+ with MDHCn before mutation, and green represents glycine. (F) Relative position of F48 to NAD+ with MDHCn after mutation, and green represents phenylalanine. | ||
To obtain methanol dehydrogenase with higher activity, enzymatic modification was conducted based on the obtained MDHCn. Although the crystal structure of MDHCn has not been solved, the structural analysis and sequence alignment confirmed that MDHCn belongs to group Adh.41 Then, we employed computational docking simulations to improve its catalytic activity by using AutoDock Vina and AlphaFold-predicted protein structures to identify protein–ligand interactions between the substrate and the protein of MDHCn.42,43 AutoDock Vina relies on empirical free energy scoring functions to evaluate docking poses.44 AlphaFold-predicted protein structures are shown in Fig. 6D. The homology model of the enzyme MDHCn with the substrate methanol was established. We subjected the MDHCn mutants to mutation operation manually using the wild-type MDHCn as the template. Interestingly, we found that the 3D structure of the AlphaFold simulated MDHCn is similar to the crystal structure of iron-dependent alcohol dehydrogenase 2 (ZmADH2) from Zymomonas mobilis ZM4, so we refer to the crystal structure of ZmADH2 complexed with the NAD+ cofactor (named 3OX4).45 The 3OX4 dimer structure is formed by a limited interaction between the two subunits, and the bound NAD+ at the cleavage formed along the structural domain interface. By analyzing the morphology and relative positions of the interaction between MDHCn and NAD+ in the crystal structure of 3OX4, we identified G48, M52, and C369 as modification sites. As a result, compared with the wild type, the three mutants obtained from the screening showed high activity toward methanol in vitro, and the activities of G48F, M52F and C369N mutants are about 3-fold, 1.5-fold and 2-fold higher toward methanol than the wild type in vitro, respectively (Fig. 6B). The specific activities of the wild-type enzyme and mutants of the MDHCn are shown in Table S4.†
The wild-type MDHCn and the G48F mutant were constructed into plasmids pETDuet-lac-mdhCn-khb-ldh and pETDuet-lac-mdhCn(G48F)-khb-ldh, respectively, and then transformed into strain D6 to obtain strains DHB11 and DHB12. DHB11 and DHB12 were subjected to shake flask cultivation to analyze whether the mutant G48F also exhibited high activity in vivo. As shown in Fig. 6C, the concentration of 2,4-DHB in the corresponding strain DHB12 was increased by 17% compared to that in DHB11, and this suggests that the site mutation G48F of MDHCn can increase methanol oxidation activity significantly. Based on this study, the G48F mutant is the best NAD+-dependent methanol dehydrogenase with respect to methanol specificity. By comparing MDHCn with the crystal structure of 3OX4, we found that the active center of MDHCn is not conducive to the entry of NAD+ and the docking posture was poor after docking of wild-type MDHCn with NAD+ (Fig. 6E); therefore, we speculate that this may be the reason for the lower activity of MDHCn. MDH, one of the common enzymes, requires NAD+ as a cofactor for accepting or donating electrons in many cellular oxidation and reduction reactions. MDHCn, as one of the MDHs, also requires NAD+ as a cofactor; thus, adjusting the posture of NAD+ inside the active center for better binding to the substrate is essential for the improvement of MDHCn activity. By mutating the glycine residue to the phenylalanine residue at position 48, the purine ring on NAD+ forms a π–π conjugation or hydrophobic interactions that promote NAD+ steering and better contact between the cofactor groups involved in the reaction and methanol (Fig. 6F). Hydrophobic F48 residues may spatially regulate the exact location of NAD+ in the substrate binding pocket. The cofactor NAD+ binds to the substrate and then enters the active center, and the F48 residue determines the exact direction of NAD+, ensuring that NAD+ can fully enter the active site and bind well to the substrate. It is worth noting that, when the complex structure of the substrate and NAD+ cofactor is elucidated, their interactions can be clearly defined, which is more conducive to the modification of MDHs and the search for more active sites. Then the detailed molecular mechanisms will be better explained.
Deleting the competing formaldehyde-consuming pathway and fed-batch fermentation for 2,4-DHB biosynthesis
Formaldehyde might be converted into formic acid and CO2 by the native glutathione-dependent formaldehyde detoxification system (frmRAB) in E. coli.46 To test the effects of FrmRAB expression levels on the by-product formic acid, the frmR, frmA and frmB genes were deleted to construct strain D8 by using the P1 transduction method (Fig. 4A and Table S1†). Then, the plasmid pETDuet-lac-mdhcn(G48F)-khb-ldh was transformed into D6 and D8, to form different recombinant strains DHB12 and DHB13, and both strains were cultivated in fermentation media under the same shake-flask conditions. As shown in Fig. 7A, the concentration of formic acid in strain DHB12 was as high as 100 mg l−1, while the level of formic acid in strain DHB13, which knocked out frmRAB, was only 59 mg L−1. The production of formic acid was decreased in DHB13 compared with that in the strain DHB12, indicating that the gene deletion inhibited the conversion of formaldehyde into formic acid to a certain degree. Chen et al. reported that the deletion of the FrmRAB manipulator is critical for methanol-dependent growth, possibly by preserving formaldehyde to promote cell growth.47 By knocking down the frmRAB gene, oxidation of formaldehyde to CO2 was prevented and formaldehyde fixation was promoted in this study, which in turn provided an additional source of substrate for 2,4-DHB biosynthesis. Guo et al. increased the flux of formaldehyde into the D-allulose synthesis pathway by knocking down frmRAB, thereby increasing the D-allulose titer and yield.48 Therefore, the results indicate that knockdown of frmRAB can not only improve formaldehyde fixation, but also increase the titer and yield of the target product. | ||
| Fig. 7 Time profiles of 2,4-DHB produced in the engineered strain DHB13. (A) Comparison of the formic acid content of DHB12 and DHB13 after fermentation. (B) Fermentation process curve of strain DHB13 in a 5 L bioreactor. | ||
In order to further evaluate the fermentation potential of the best strain DHB13 under aerobic fed-batch conditions, fed-batch culture was put into effect in a 5 L bioreactor with a working volume of 2 L. After IPTG induction, the residual glucose in the bioreactor was maintained at about 0.5 g L−1, in order to avoid the accumulation of acetate during the whole fermentation process and to ensure the supply of sufficient substrate. Glucose consumption, cell density, and 2,4-DHB concentration were monitored during the fermentation process. The time profiles of cell growth and 2,4-DHB accumulation are shown during the fed-batch process (Fig. 7B). The cell growth rate was relatively stable without significant fluctuations during the high-density culture. During the fermentation process of strain DHB13, the concentration of 2,4-DHB was low in the first few hours after induction but accelerated from about 8 h after induction. Then, the product maintained a high rate of accumulation until about 37 h, reaching a maximum concentration of 14.6 g L−1 within 79 h after induction. During the fed-batch fermentation, the maximum biomass reached an OD600 of 134 at 29 h and then the cell density stopped increasing. The highest recorded level of 2,4-DHB production was achieved in E. coli, much higher than 5.3 g L−1 reported in the previous study.16 In addition, this is the first report of a technique for the efficient production of 2,4-DHB from methanol and glucose.
Conclusions
In this work, a new pathway for the biosynthesis of 2,4-DHB was successfully developed with glucose and methanol as co-substrates in engineered E. coli, and a highly efficient biosynthesis network was also constructed to improve 2,4-DHB production in E. coli. In brief, we first identified the biosynthetic pathway for the synthesis of 2,4-DHB from formaldehyde and glucose by screening Ldh with higher activity towards 2,4-DHB; however, the production of 2,4-DHB was low. In order to increase the production of 2,4-DHB, we created a chassis cell for 2,4-DHB synthesis through multiple gene regulation strategies, which in turn increased the production of 2,4-DHB. During the biosynthesis of 2,4-DHB using methanol and glucose as substrates, we found that methanol dehydrogenase was the rate-limiting enzyme. In order to increase the activity of methanol dehydrogenase, we screened and modified methanol dehydrogenase to obtain a methanol dehydrogenase with relatively high activity. For the first time, the final strain of DHB13 achieved the highest 2,4-DHB production ever reported in engineered E. coli. This novel pathway is the shortest pathway for the biosynthesis of 2,4-DHB using glucose and methanol as co-substrates at present, which in theory opens up the possibility to synthesize an appealing C4 compound from a C1 and a C3 compound without carbon loss, providing an attractive and environment-friendly synthesis route. Additionally, this systematic metabolic engineering work on host strains should be of great interest for the direct use of C1 compounds for the production of value-added products in synthetic biology.Author contributions
Rubing Zhang and Mo Xian conceived and initiated the project. Xianjuan Dong, Chao Sun, Xiangyu Ma and Rubing Zhang wrote the paper and performed the experiments. Jing Guo reviewed this paper and provided valuable suggestions. All the authors read, edited, and approved the final manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (No. 2022YFC2104700), the National Natural Science Foundation of China (No. 22007091), and the Key R&D Program of Shandong Province, China (2023JMRH0201).References
- M. Guo, W. Song and J. Buhain, Renewable Sustainable Energy Rev., 2015, 42, 712–725 CrossRef CAS.
- C. Cao and Q. Wu, Chem Catal., 2023, 3, 100494 CrossRef CAS.
- A. M. Ochsner, F. Sonntag, M. Buchhaupt, J. Schrader and J. A. Vorholt, Appl. Microbiol. Biotechnol., 2015, 99, 517–534 CrossRef CAS PubMed.
- W. Zhang, T. Zhang, S. Wu, M. Wu, F. Xin, W. Dong, J. Ma, M. Zhang and M. Jiang, RSC Adv., 2017, 7, 4083–4091 RSC.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed.
- J. E. N. Müller, T. M. B. Heggeset, V. F. Wendisch, J. A. Vorholt and T. Brautaset, Appl. Microbiol. Biotechnol., 2015, 99, 535–551 CrossRef PubMed.
- C. A. Henard, I. R. Akberdin, M. G. Kalyuzhnaya and M. T. Guarnieri, Green Chem., 2019, 21, 6731–6737 RSC.
- W. Zhang, M. Song, Q. Yang, Z. Dai, S. Zhang, F. Xin, W. Dong, J. Ma and M. Jiang, Biotechnol. Biofuels, 2018, 11, 260 CrossRef CAS PubMed.
- G. A. Olah, Chem. Eng. News Archive, 2003, 81, 5 CrossRef.
- O. K. Lee, D. H. Hur, D. T. N. Nguyen and E. Y. Lee, Biofuels, Bioprod. Biorefin., 2016, 10, 848–863 CrossRef CAS.
- W. L. Zhu, J. Y. Cui, L. Y. Cui, W. F. Liang, S. Yang, C. Zhang and X. H. Xing, Appl. Microbiol. Biotechnol., 2016, 100, 2171–2182 CrossRef CAS PubMed.
- G. A. Olah, Angew. Chem., Int. Ed., 2013, 52, 104–107 CrossRef CAS PubMed.
- S. Zhang, F. Guo, Q. Yang, Y. Jiang, S. Yang, J. Ma, F. Xin, T. Hasunuma, A. Kondo, W. Zhang and M. Jiang, Green Chem., 2023, 25, 183–195 RSC.
- R. K. Bennett, M. Dillon, J. R. Gerald Har, A. Agee, B. von Hagel, J. Rohlhill, M. R. Antoniewicz and E. T. Papoutsakis, Metab. Eng., 2020, 60, 45–55 CrossRef CAS PubMed.
- T. Walther, C. M. Topham, R. Irague, C. Auriol, A. Baylac, H. Cordier, C. Dressaire, L. Lozano-Huguet, N. Tarrat, N. Martineau, M. Stodel, Y. Malbert, M. Maestracci, R. Huet, I. André, M. Remaud-Siméon and J. M. François, Nat. Commun., 2017, 8, 15828 CrossRef CAS PubMed.
- T. Walther, F. Calvayrac, Y. Malbert, C. Alkim, C. Dressaire, H. Cordier and J. M. François, Metab. Eng., 2018, 45, 237–245 CrossRef CAS PubMed.
- J. O. Krömer, C. Wittmann, H. Schröder and E. Heinzle, Metab. Eng., 2006, 8, 353–369 CrossRef PubMed.
- X. Li, Z. Cai, Y. Li and Y. Zhang, Sci. Rep., 2014, 4, 5541 CrossRef CAS PubMed.
- H. Li, B. Wang, L. Zhu, S. Cheng, Y. Li, L. Zhang, Z. Y. Ding, Z. H. Gu and G. Y. Shi, Process Biochem., 2016, 51, 1973–1983 CrossRef CAS.
- M. Liu, M. Tolstorukov, V. Zhurkin, S. Garges and S. Adhya, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6911–6916 CrossRef CAS PubMed.
- S. D. Moore, in Strain Engineering: Methods and Protocols, ed. J. A. Williams, Humana Press, Totowa, NJ, 2011, pp. 155–169 Search PubMed.
- X. Li, Z. Cai, Y. Li and Y. Zhang, Sci. Rep., 2015, 4, 5541 CrossRef PubMed.
- G. L. Forrest and B. Gonzalez, Chem.-Biol. Interact., 2000, 129, 21–40 CrossRef CAS PubMed.
- Y. Jin and T. M. Penning, Annu. Rev. Pharmacol. Toxicol., 2007, 47, 263–292 CrossRef CAS PubMed.
- J. C. Wang, M. Sakakibara, J. Q. Liu, T. Dairi and N. Itoh, Appl. Microbiol. Biotechnol., 1999, 52, 386–392 CrossRef CAS PubMed.
- G. C. Xu, H. L. Yu, X. Y. Zhang and J. H. Xu, ACS Catal., 2012, 2, 2566–2571 CrossRef CAS.
- R. Guo, Y. Nie, X. Q. Mu, Y. Xu and R. Xiao, J. Mol. Catal. B: Enzym., 2014, 105, 66–73 CrossRef CAS.
- P. Minárik, N. Tomášková, M. Kollárová and M. Antalík, Gen. Physiol. Biophys., 2002, 21, 257–265 Search PubMed.
- D. Madern, J. Mol. Evol., 2002, 54, 825–840 CrossRef CAS PubMed.
- C. J. R. Frazão, C. M. Topham, Y. Malbert, J. M. François and T. Walther, Biochem. J., 2018, 475, 3887–3901 CrossRef PubMed.
- T. Matsumoto, T. Tanaka and A. Kondo, Bioresour. Technol., 2017, 245, 1362–1368 CrossRef CAS PubMed.
- A. M. Abdel-Hamid, M. M. Attwood and J. R. Guest, Microbiology, 2001, 147, 1483–1498 CrossRef CAS PubMed.
- Y. Fang, S. Zhang, J. Wang, L. Yin, H. Zhang, Z. Wang, J. Song, X. Hu and X. Wang, Metabolites, 2021, 11, 30 CrossRef CAS PubMed.
- L. Zhou, Z. R. Zuo, X. Z. Chen, D. D. Niu, K. M. Tian, B. A. Prior, W. Shen, G. Y. Shi, S. Singh and Z. X. Wang, Curr. Microbiol., 2011, 62, 981–989 CrossRef CAS PubMed.
- J. Zhou, X. Lu, B. Tian, C. Wang, H. Shi, C. Luo, X. Zhu, X. Yuan and X. Li, 3 Biotech, 2019, 9, 343 CrossRef PubMed.
- T. Matsumoto, K. Higuma, R. Yamada and H. Ogino, Biochem. Eng. J., 2023, 191, 108772 CrossRef CAS.
- A. Krog, T. M. B. Heggeset, J. E. N. Müller, C. E. Kupper, O. Schneider, J. A. Vorholt, T. E. Ellingsen and T. Brautaset, PLoS One, 2013, 8, e59188 CrossRef CAS PubMed.
- N. Arfman, H. J. Hektor, L. V. Bystrykh, N. I. Govorukhina, L. Dijkhuizen and J. Frank, Eur. J. Biochem., 1997, 244, 426–433 CrossRef CAS PubMed.
- N. Arfman, E. M. Watling, W. Clement, R. J. van Oosterwijk, G. E. de Vries, W. Harder, M. M. Attwood and L. Dijkhuizen, Arch. Microbiol., 1989, 152, 280–288 CrossRef CAS PubMed.
- H. Meng, C. Wang, Q. Yuan, J. Ren and A. P. Zeng, ACS Synth. Biol., 2021, 10, 799–809 CrossRef CAS PubMed.
- T. Y. Wu, C. T. Chen, J. T. J. Liu, I. W. Bogorad, R. Damoiseaux and J. C. Liao, Appl. Microbiol. Biotechnol., 2016, 100, 4969–4983 CrossRef CAS PubMed.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed.
- M. Varadi, S. Anyango, M. Deshpande, S. Nair, C. Natassia, G. Yordanova, D. Yuan, O. Stroe, G. Wood, A. Laydon, A. Žídek, T. Green, K. Tunyasuvunakool, S. Petersen, J. Jumper, E. Clancy, R. Green, A. Vora, M. Lutfi, M. Figurnov, A. Cowie, N. Hobbs, P. Kohli, G. Kleywegt, E. Birney, D. Hassabis and S. Velankar, Nucleic Acids Res., 2022, 50, D439–D444 CrossRef CAS PubMed.
- F. Wong, A. Krishnan, E. J. Zheng, H. Stärk, A. L. Manson, A. M. Earl, T. Jaakkola and J. J. Collins, Mol. Syst. Biol., 2022, 18, e11081 CrossRef CAS PubMed.
- J. H. Moon, H. J. Lee, S. Y. Park, J. M. Song, M. Y. Park, H. M. Park, J. Sun, J. H. Park, B. Y. Kim and J. S. Kim, J. Mol. Biol., 2011, 407, 413–424 CrossRef CAS PubMed.
- J. E. N. Müller, F. Meyer, B. Litsanov, P. Kiefer, E. Potthoff, S. Heux, W. J. Quax, V. F. Wendisch, T. Brautaset, J. C. Portais and J. A. Vorholt, Metab. Eng., 2015, 28, 190–201 CrossRef PubMed.
- C. T. Chen, F. Y. H. Chen, I. W. Bogorad, T. Y. Wu, R. Zhang, A. S. Lee and J. C. Liao, Metab. Eng., 2018, 49, 257–266 CrossRef CAS PubMed.
- Q. Guo, M. M. Liu, S. H. Zheng, L. J. Zheng, Q. Ma, Y. K. Cheng, S. Y. Zhao, L. H. Fan and H. D. Zheng, J. Agric. Food Chem., 2022, 70, 14255–14263 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc02083e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |