
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn innovative catalytic pathway for the synthesis of acyl furans: the cross-ketonization of methyl 2-furoate with carboxylic acids†
Jacopo
De Maron
 a,
Davide
Cesari
a,
Sabra
Banu Rameesdeen
a,
Tommaso
Tabanelli
*ab,
Andrea
Fasolini
a,
Francesco
Basile
ab and
Fabrizio
Cavani
ab
a,
Davide
Cesari
a,
Sabra
Banu Rameesdeen
a,
Tommaso
Tabanelli
*ab,
Andrea
Fasolini
a,
Francesco
Basile
ab and
Fabrizio
Cavani
ab
aDipartimento di Chimica Industriale “Toso Montanari”, Alma Mater Studiorum - Università di Bologna, Viale del Risorgimento 4, 40136 Bologna, Italy. E-mail: tommaso.tabanelli@unibo.it
bCenter for Chemical Catalysis-C3, Università di Bologna, Viale del Risorgimento 4, 40136 Bologna, Italy
First published on 17th August 2023
Abstract
The usefulness of ketonization reactions for the valorization of a wide plethora of biomass-derived carboxylic acids is widely recognized; however, the full potential of this reaction has yet to be suitably exploited for the production of high added-value aromatic and/or aliphatic asymmetric ketones. For this reason, herein we report for the first time in the literature the continuous-flow, gas-phase synthesis of 2-acetyl furan (AF) by means of the catalytic cross-ketonization of bio-based 2-methyl furoate (2-MF) and acetic acid (AA) over a simple and cheap ZrO2 catalyst. Interestingly, AF is considered a valuable food additive and a pharmaceutical intermediate for the synthesis of antibiotics. The optimization of the molar ratio between reactants at 350 °C allowed us to achieve 87% AF selectivity at 90% 2-MF conversion, with a space–time yield of 0.152 h−1, a value that is similar to the ones obtained by following the traditional Friedel–Crafts acylation and Wacker oxidation routes. On the other hand, the E-factor of the herein-proposed process is several times lower compared to the aforementioned traditional routes performed in the liquid phase and under batch conditions. Finally, the versatility of the cross-ketonization synthetic approach was successfully demonstrated and applied for the selective synthesis of other valuable acyl furans (i.e., propionyl furan and butyryl furan).
Introduction
In recent years, ketonization has emerged as a very promising way to valorise multiple biomass-derived products such as carboxylic acids, esters, aldehydes and alcohols.1–3During the last two decades, the ketonization of light carboxylic acids such as acetic4,5 and propionic acids,6,7 which are typically found in relatively high amounts in the liquid product of the flash-pyrolysis of lignocellulosic biomass,8 has been extensively investigated to produce biofuels, as an intermediate upgrading step prior to a final refining with molecular H2 (hydrodeoxygenation, HDO).9 In fact, since the coupling of two acids into a ketone coproduces CO2 and H2O, ketonization can be exploited to remove a significant fraction of the oxygen contained in pyrolysis oils before HDO, thus diminishing substantially the H2 demand of the final refining process.1 On top of that, ketonization enhances the stability and the calorific power of bio-oils by reducing their acidity and creating new C–C bonds. Similarly, a multistep catalytic process involving ketonization followed by hydrogenation of the resulting ketones has been recently proposed for the upgrading of wet waste-derived volatile fatty acids (VFAs) to a renewable jet fuel.10 VFAs (C2–C8 carboxylic acids) are obtained by the fermentation (arrested methanogenesis)11 of food waste, animal manure, and wastewater sludge. Considering the environmental impact of organic waste landfill (due to the release of CH4 in the atmosphere), this multistep process is considered promising for decarbonizing air transport. The usefulness of ketonization has also been recognized as a way to produce valuable bio-based waxes by coupling the C12–C18 fatty acids found in waste animal fat or vegetable oils.12 It has been pointed out that the availability of renewable waxes is highly desirable because the production volume of their petroleum-based paraffin counterparts is decreasing due to the increased interest in hydro-cracking and hydro-isomerization towards lower molecular weight products (e.g., fuels and lubricants).
Despite the recent advancements in ketonization-based processes for the production of renewable fuels and oleochemicals, the full potential of this reaction has yet to be suitably exploited. This is especially true considering the wide plethora of high added-value aromatic, aliphatic and/or allylic asymmetric ketones used as intermediates for the preparation of fine chemicals,13–16 the production of which mainly relies on the Friedel–Crafts acylation and oxidation reactions, while the application of a selective, continuous-flow, cross-ketonization in the gas-phase is relatively underdeveloped. Actually, the coupling of binary mixtures of model acids has been extensively investigated to uncover the reaction mechanism,17–20 but the literature specifically focusing on the selective synthesis of valuable asymmetric ketones in the gas-phase is limited to a few model reactions, such as the preparation of heptanoyl benzene,21 2-acetyl pyridine,22 acetyl cyclopropane,23 2-undecanone,24,25 pinacolone, propiophenone and cyclohexyl phenyl ketone.26 On the other hand, a liquid-phase process for the production of 2-dodecanoyl furan (a valuable precursor of bio-based surfactants) by means of the cross-ketonization of furoic acid with lauric acid was recently reported.27,28 The main hurdle limiting the widespread application of this synthetic approach is that the cross-ketonization between two non-identical acids usually competes with the unwanted coupling of each reactant with itself (the so-called “homo-ketonization”), resulting in unsatisfactory selectivities, as shown in Fig. 1. Therefore, selectively obtaining the desired cross-ketonization product is crucial to find a suitable strategy to reduce the extent of the parasitic homo-ketonization. Depending on the catalyst used, gas-phase ketonization can occur following two distinct mechanisms. The so-called “surface mechanism” is typical of materials possessing high lattice energies1 such as zeolites, ZrO2, TiO2 and Al2O3 and postulates that the coupling of chemisorbed reactants is confined to the surface of the catalyst, following the Langmuir Hinshelwood model.10,29,30 Despite the large body of research available on this matter,5,17,20,31–33 the exact mechanism is still the object of debate and the participation of several intermediates has been proposed, such as carboxylic anhydrides,34 carboxylate anions,35 ketenes36 or via β-ketoacids,37 the latter being the most probable one.1,2 In the mechanism via a β-ketoacid, a basic site on the catalyst surface activates an adsorbed carboxylate anion (the so-called enolic component19) by the abstraction of an acidic α-hydrogen, forming a nucleophilic 1-hydroxy enolate anion; then, this nucleophilic intermediate reacts with an adjacent electrophilic carboxylate anion (the so-called carbonyl component19) forming a β-ketoacid, which finally decomposes into CO2 and a ketone. Compelling evidence from several authors indicates that the reactivity of carboxylic acids decreases with a decreasing number of α-H17 and with increasing steric hindrance of the α-H.19,30 In the extreme case of acids completely lacking α-H, homo-ketonization cannot occur.20 As a consequence, the product distribution of the cross-ketonization of an equimolar mixture of two acids depends on their difference in reactivity, so that the coupling of two acids with similar reactivity results in a binomial distribution (e.g., B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :A:B′ = 1/2/1) between the symmetric (B and B′) and asymmetric ketones (A). Instead, significant deviations in such products distribution can be obtained by either change the molar ratio of the reagents in the feed or in the presence of a recalcitrant, less reactive acid.18 Remarkably, cross-ketonization with surface ketonization catalysts requires the presence of an α-H in just one of the two reacting acids, which acts as the enolic component: therefore, even if unable to undergo homo-ketonization, acids completely lacking α-H can still participate in cross-ketonization as the carbonyl component,19,20 thus reducing the possible products from three to two.
:A:B′ = 1/2/1) between the symmetric (B and B′) and asymmetric ketones (A). Instead, significant deviations in such products distribution can be obtained by either change the molar ratio of the reagents in the feed or in the presence of a recalcitrant, less reactive acid.18 Remarkably, cross-ketonization with surface ketonization catalysts requires the presence of an α-H in just one of the two reacting acids, which acts as the enolic component: therefore, even if unable to undergo homo-ketonization, acids completely lacking α-H can still participate in cross-ketonization as the carbonyl component,19,20 thus reducing the possible products from three to two.
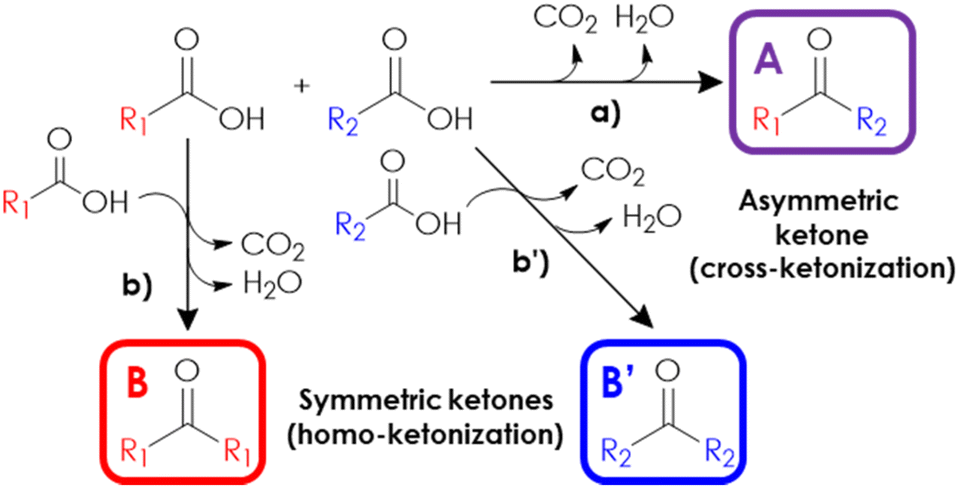 | ||
| Fig. 1 Reactions involved in the coupling of a mixture of two non-identical carboxylic acids: (a) cross-ketonization towards an asymmetric ketone; (b) and (b′) homo-ketonization towards two different symmetrical ketones. | ||
The so-called “bulk mechanism” is typical of metal oxides with low lattice energy such as alkaline-earth oxides31 and rare-earth oxides4 and involves a bulk acid–base reaction between the oxide and carboxylic acid vapours that produces carboxylate salts and water.20 High temperatures trigger the thermal decomposition of carboxylate salts and the resulting fragments undergo a radical recombination38 that produces a ketone, leaving behind a metal carbonate/oxycarbonate. Finally, a catalytic cycle is established when the reaction temperature is high enough to decompose the carbonate/oxycarbonate, producing CO2 and regenerating the pristine metal oxide. On one hand, bulk homo-ketonization can be used to target symmetric ketones that cannot be obtained by surface homo-ketonization (e.g., by coupling acids lacking α-H such as benzoic acid to produce benzophenone38). On the other hand, bulk ketonization is less suitable than surface ketonization for the selective production of asymmetric ketones because the extent of homo-ketonization cannot be limited by the absence of α-H.
Starting from these considerations, herein is proposed a synthetic strategy aimed at maximizing the cross-selectivity of ketonization by targeting asymmetric ketones obtainable from one acid lacking α-H in combination with a surface ketonization catalyst (thus reducing the number of possible products from 3 to 2) and by feeding a moderate excess of one of the two reactants. As a proof of concept, 2-acetyl furan (AF) was selected as the target product because it is considered both a valuable food additive39 and chemical intermediate in the synthesis of the antibiotic cefuroxime.40 Moreover, this compound can be obtained by the coupling of furoic acid (FA) or its esters with acetic acid (AA) or ethyl acetate (EA), which are all obtainable from renewable platform molecules such as furfural and bio-ethanol (Fig. 2).
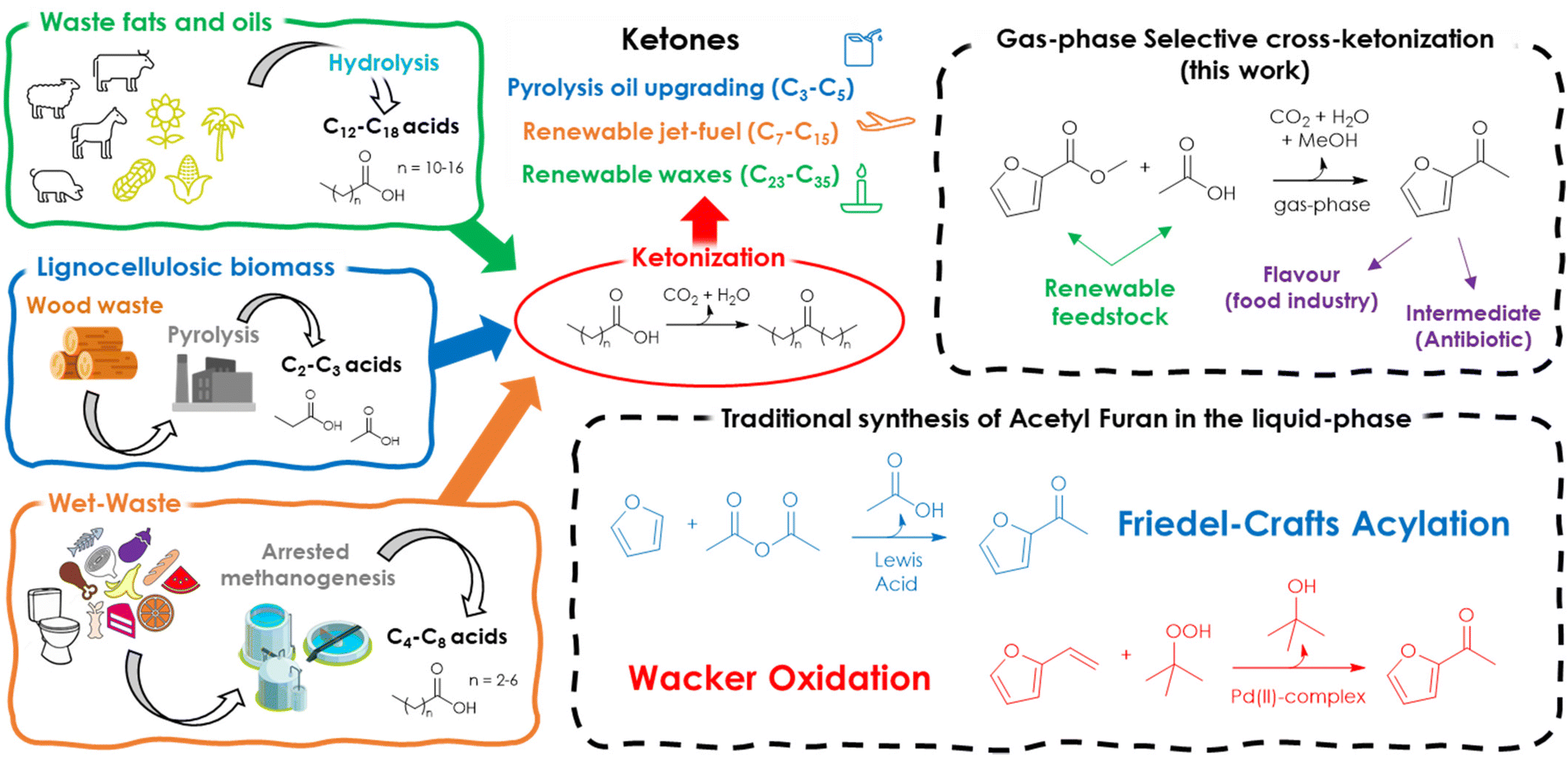 | ||
| Fig. 2 Overview of the valorization of different renewable feedstocks via ketonization towards biofuels, jet fuels, bio-based waxes and bio-based asymmetric ketone intermediates. Production routes to acetyl furan (AF). | ||
To the best of our knowledge, the synthesis of AF in the gas-phase by means of cross-ketonization is reported herein for the first time. Finally, with respect to the traditional processes in the liquid phase (namely, Friedel–Crafts acylation of furan (F) with acetic anhydride (AAN) catalysed by Lewis acids or zeolites and the Wacker oxidation of vinyl furan (VF) with tert-butyl hydroperoxide (TBHP) catalysed by Pd(II) complexes), it represents an innovative alternative that could benefit from continuous operation, lower formation of wastes and easier work-up operation.
Experimental
Synthesis and characterization of catalysts
Ceria (CeO2) is a commercial reference material (Rhodia Actalys HAS 5) and was calcined in air at a heating rate of 5 °C min−1 up to 400 °C for 3 hours before use. Zirconia (ZrO2) was synthesized by means of precipitation, adapting a method reported elsewhere;6 ceria–zirconia (Ce/Zr/O) was synthesized adapting the method used for the synthesis of ZrO2. A more detailed description of catalysts’ preparation can be found in the ESI.†Powder X-ray diffraction (XRD) patterns of ZrO2, CeO2 and Ce/Zr/O were collected on a Philips X'Pert diffractometer with the Bragg–Brentano geometry and the Cu Kα radiation as the X-ray source (λ = 1.54178 Å, Ni-filtered). Raman spectra were collected between 100–2000 cm−1 using a Renishaw InVia Raman spectrometer configured with a Leica DM LM microscope and equipped with an Ar+ laser (514.5 nm) and a diode laser (785.0 nm). Energy dispersive spectrometry (EDS) was carried out with a scanning electron microscope (SEM) Zeiss EP EVO 50 equipped with an INCA X-Act penta FET precision detector (Oxford Instruments Analytical). Spectra were recorded with an accelerating voltage of 20 kV for 60 seconds.
The specific surface area (SSA) of all catalysts was measured using a single-point BET Fisons Sorpty 1750 instrument.
The reducibility of catalysts was assessed by means of temperature-programmed reduction (TPR) with hydrogen, using a Micromeritics AutoChem II 2920 instrument equipped with a TCD detector. Thermogravimetric analyses (TGA) were carried out using a NETZSCH TG 209 F1 instrument. A more detailed description of temperature ramps used to carry out TPR and TGA characterization can be found in Chapter S2 in the ESI.†
Catalytic tests
All catalytic tests were carried out in a gas-phase plant operating at atmospheric pressure (Fig. S1†) and the catalyst (1 mL) was charged into a fixed bed down-flow quartz reactor in the form of pellets with granulometry between 30 and 60 mesh.The two reactants (2-MF, 98% Sigma-Aldrich and AA, 99% Sigma-Aldrich or EA, 99% Sigma-Aldrich) were mixed in the desired molar ratio and fed together from the same syringe using a KD Scientific Legacy 100 volumetric pump; the LHSV and the flow of the N2 carrier were adjusted in order to obtain the desired molar fractions in the gas-phase always maintaining a contact time τ (at T = 350 °C) = 1 s, equivalent to a GHSV (at T = 25 °C) = 1722 h−1. Before each catalytic test, the catalyst was heated up to reaction temperature at 10 °C min−1 under a flow of N2 and the final temperature was maintained for 30 min before starting to feed the liquid reactant. The effluent from the reactor was bubbled through two cold traps in series kept at 0 °C with an ice bath and filled with acetonitrile (AcCN, Sigma-Aldrich, 99.8%) to absorb the condensable products. At regular intervals of time the content of the two cold traps was collected, added with 1 g of a dodecane (Sigma-Aldrich, 99%) internal standard solution (4 × 10−5 mol g−1). All catalytic tests were carried out for at least 6 hours and the average conversion and yields were calculated only once a stationary catalytic performance was achieved. A control experiment was repeated 3 times under the same conditions to check for reproducibility and conversion and yields differed by less than 5% from one reaction to another.
Product analysis
The outlet from the cold traps was connected to an Agilent 5890 Series II GC instrument equipped with FID and TCD detectors. The quantification of the condensed products was carried out offline with an Agilent J and W DB-1701 capillary column (25 m × 530 μm × 1.05 μm) connected to the FID detector. The quantification of the gaseous products was carried out online using an Agilent CP-Molsieve 5A capillary column (25 m × 530 μm × 50 μm,) and an Agilent CP-SilicaPLOT capillary column (30 m × 530 μm × 6 μm). Unknown products were identified by means of an Agilent Technologies 6890 GC instrument equipped with an Agilent Technologies 5973 quadrupole mass analyser (GC-MS) and an Agilent HP-5 capillary column (30 m × 250 μm × 1.05 μm). The following equations were used to calculate the conversions of the two reactants (XR, eqn (1)), the yields (YRi, eqn (2)), the selectivities (SRi, eqn (3)), and the sum of yields (YSR, eqn (4)).| XR = (molRIN − molROUT)/(molRIN) × 100 | (1) |
| YRi = (moliOUT)/(molRIN) × 100 | (2) |
| SRi = (Yi)/(XR) × 100 | (3) |
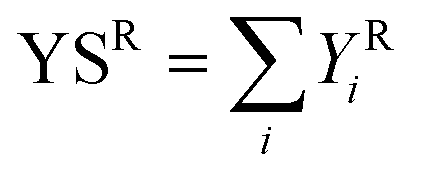 | (4) |
Results and discussion
Fresh catalysts characterization
The powder XRD diffractograms of the fresh catalysts are reported in ESI, Fig. S2;† the XRD diffractogram of ZrO2 is characterized by the typical reflections of the most stable monoclinic phase, plus a very weak reflection (2θ = 30.3 degrees) attributable to the presence of a small impurity of the tetragonal phase; this material is the one possessing the lowest specific surface area (SSA) of 55 m2 g−1. Instead, both the commercial CeO2 and the mixed metal oxide Ce/Zr/O possess a fluorite-type cubic lattice and a higher SSA of 193 m2 g−1 and 124 m2 g−1, respectively. However, the diffractogram of the synthesized Ce/Zr/O mixed oxide is characterized by broader reflections, and each one is systematically shifted towards higher values of 2θ degrees with respect to those of pure CeO2, indicating that the substitution of the bulkier Ce4+ cation (0.97 Å) by the smaller Zr4+ cation (0.84 Å) results in a contraction of the interplanar distances in the fluorite-type lattice, in agreement with previous literature.41,42 The actual Ce/Zr atomic ratio in Ce/Zr/O measured by means of SEM-EDS is equal to 1.05, corresponding to the composition Ce0.51Zr0.49O2.2-Acetyl furan production via continuous-flow cross-ketonization
Preliminary solubility/miscibility tests of furoic acid (FA), methyl 2-furoate (2-MF) or ethyl 2-furoate (2-EF) with acetic acid (AA) or ethyl acetate (EA) led us to select 2-MF as the most favourable reactant. In fact, both FA and 2-EF are solids at room temperature and must be dissolved in EA or AA to be vaporized and fed to the reactor. Unfortunately, FA solubility in both AA and EA is low, while 2-EF is sufficiently soluble (e.g., 0.71 g mL−1 equal to a 1:1 molar ratio) only in EA. On the other hand, 2-MF, being the only furoic derivative liquid at room temperature, is miscible with both AA and EA.
Prior to investigating the cross-ketonization, the stability of 2-MF under typical reaction conditions was investigated by feeding only 2-MF (1 mol%) diluted in N2 in the absence of any catalyst at 350 °C by keeping the same total volumetric flow compared to the following catalytic tests. The outcome of this run is shown in Fig. S3a† and the results are expressed in terms of conversion and yields as a function of the time on stream (TOS). The conversion of 2-MF, initially as high as 80%, decreased down to less than 5% after 6 hours on stream; the main products of the reactions were furan (F), methanol (MeOH) and carbon dioxide (CO2), together with other unknown compounds obtained in low yield (others). Since F, MeOH and CO2 were obtained in a near to 1:1:1 molar ratio under steady-state conditions, they are likely to be formed by the same reaction (e.g., hydrolysis of 2-MF in the presence of traces of water followed by decarboxylation). When 2-MF was fed over 1 mL of ZrO2 in a decomposition test (Fig. S3b†) carried out under the same conditions as the blank run, a very similar product distribution was obtained, but the conversion (≈10%) was slightly higher. These results suggest that 2-MF is quite stable under the reaction conditions investigated even in the presence of ZrO2. The synthesis of acetyl furan (AF) by means of the gas-phase cross ketonization between 2-MF and AA was initially investigated by feeding an equimolar mixture of the two reactants over the ZrO2 catalyst at 350 °C with a contact time τ of 1 second. The results of this catalytic test in terms of conversion and yields with respect to 2-MF are reported in Fig. 3a. Both 2-MF conversion and product selectivities changed significantly with TOS. At the beginning of the reaction 2-MF conversion was complete, then decreased down to 54% over the course of 11 hours on stream. The desired AF was obtained as the main product with 53% yield after 2–5 hours on stream, but then its yield decreased down to 37% during the following 6 hours. The main by-product was F with a constant yield of 10%, which is higher than the one obtained during the previous blank run; this fact was ascribed to the occurrence of the homo-ketonization of AA to acetone (AC), CO2 and water, which fostered the hydrolysis and decarboxylation of 2-MF to F, MeOH and CO2. The results of the same catalytic test expressed with respect to the other reactant (AA) are shown in Fig. 3b. Interestingly, AA conversion was complete over the course of the entire reaction, while the yield of AC increased steadily exceeding the AF yield after 5 hours on stream and reaching 63% after 11 hours. Therefore, the incomplete conversion of 2-MF has to be ascribed to the occurrence of AA homo-ketonization to AC, which limited the amount of AA available in the reaction environment reducing the extent of the desired cross-ketonization between 2-MF and AA to AF. Starting from these results, in the attempt of achieving a complete conversion of the furoic reactant, it was decided to increase the molar excess of AA with respect to 2-MF while keeping constant the total % of organics in the feed. It is worth noting that according to the synthetic approach described in the introduction, the reactant lacking α-H (e.g., 2-MF) should be the one fed in excess due to its lack of reactivity toward homo-ketonization; however, in the case of AF synthesis this is not economically favourable, because of the higher cost of 2-MF compared to AA. The results of the gas-phase cross-ketonization carried out by feeding a mixture of 2-MF/AA/N2 = 0.4/1.6/98 over ZrO2 at 350 °C with a contact time τ of 1 second are shown in Fig. 4a (with respect to 2-MF) and in Fig. 4b (with respect to AA).
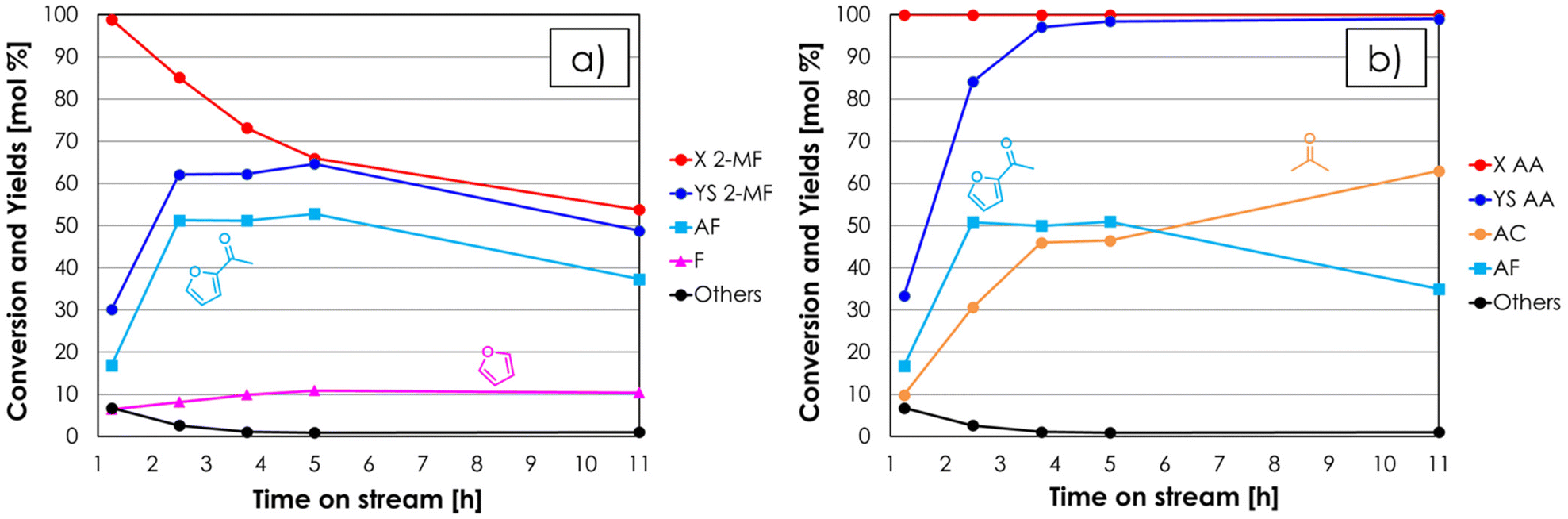 | ||
| Fig. 3 Results of the cross-ketonization between 2-MF and AA as a function of TOS with respect to 2-MF (a) and AA (b). Reaction conditions: temperature = 350 °C, 2-MF/AA/N2 = 1/1/98 mol%, contact time = τ = 1 s, GHSV = 1722 h−1, 1 mL of ZrO2. Symbols: (a) 2-methyl furoate conversion (X 2-MF), sum of yields (YS 2-MF), acetyl furan yield (AF), furan yield (F), sum of yields of unknown by-products (Others); (b) acetic acid conversion (X AA), sum of yields (YS AA), acetone yield (AC), acetyl furan yield (AF), sum of yields of unknown by-products (Others). | ||
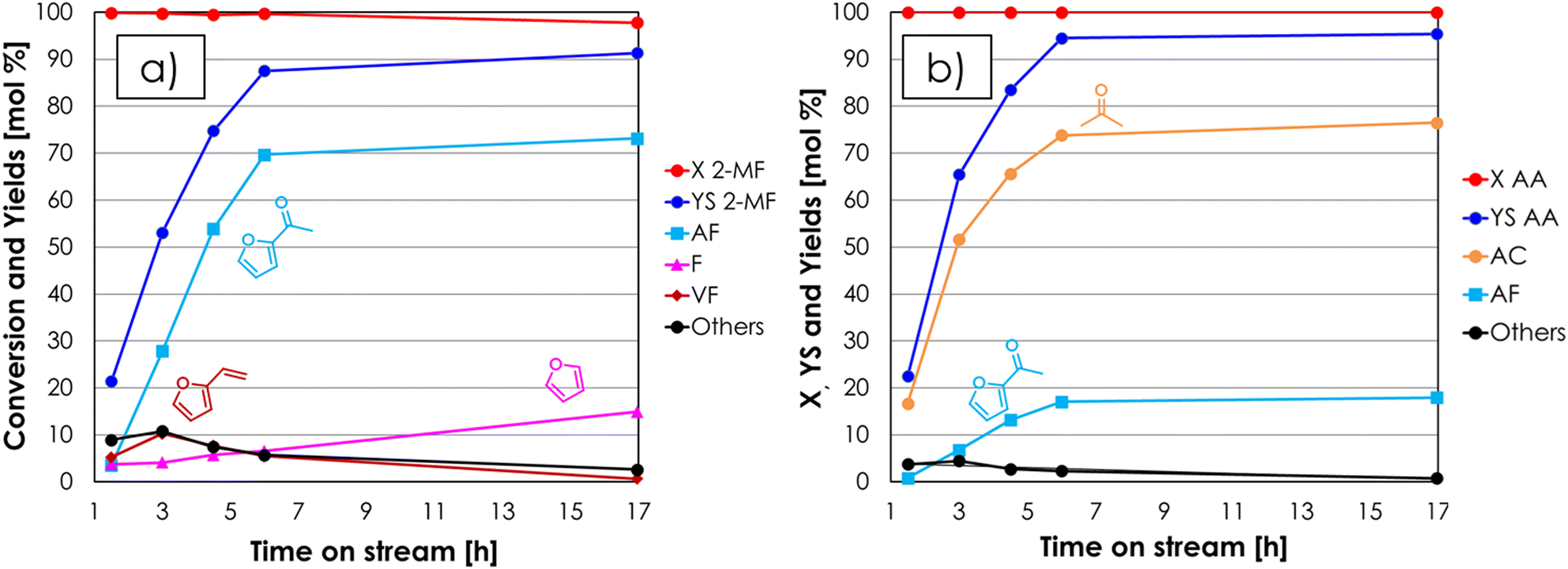 | ||
| Fig. 4 Results of the cross-ketonization between 2-MF and AA as a function of the time on stream with respect to 2-MF (a) and AA (b). Reaction conditions: temperature = 350 °C, 2-MF/AA/N2 = 0.4/1.6/98 mol%, contact time = τ = 1 s, GHSV = 1722 h−1, 1 mL ZrO2. Symbols: (a) 2-methyl furoate conversion (X 2-MF), sum of yields (YS 2-MF), acetyl furan yield (AF), furan yield (F), vinyl furan yield (VF), sum of yields of unknown by-products (Others); (b) acetic acid conversion (X AA), sum of yields (YS AA), acetone yield (AC), acetyl furan yield (AF), sum of yields of unknown by-products (Others). | ||
Increasing the excess of AA was very effective, because under these conditions the conversion 2-MF was complete and the yield in AF was 70% after 6 hours on stream, a value which slightly increased for the next 10 hours. Indeed, at the beginning of the reaction also vinyl furan (VF) was found in the reaction mixture: its yield reached a maximum (10%) after a TOS of 3 hours and then decreased down to zero at the end of the reaction. The formation of this compound can be explained by the consecutive H-transfer reduction of the carbonyl group of AF by the MeOH co-produced during the cross-ketonization acting as an H-donor.43–46 The resulting secondary alcohol immediately dehydrates under the given reaction conditions leading to the formation of VF, as shown in Scheme 1. The main drawback of feeding a molar excess of AA is related to its high reactivity, which results in complete conversion of this reactant and the formation of significant amounts of AC. Still, the separation of AC from AF by distillation is easier than the work-up operation required for the traditional Friedel–Crafts acylation and Wacker oxidation methods, and AC is a product of interest both as a solvent and as a chemical intermediate (e.g., in the production of methyl methacrylate, bisphenol A, methyl isobutyl ketone and diacetone alcohol47). Recently, the use of AC has also been proposed for the gas-phase synthesis of 2-undecanone by means of the so-called reketonization reaction.24,48
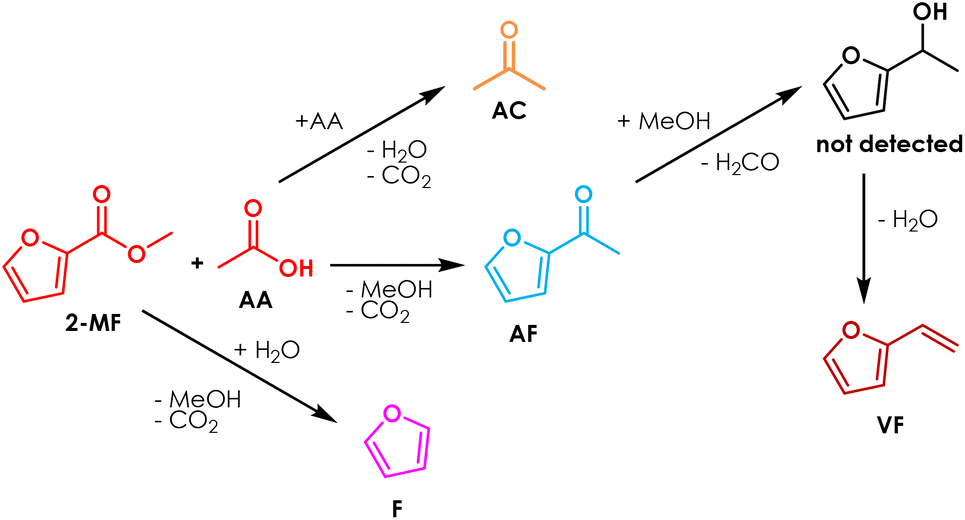 | ||
| Scheme 1 Proposed reaction scheme for the cross-ketonization of 2-MF with AA. | ||
Once favourable reaction conditions for the synthesis of AF were identified over ZrO2, it was decided to extend the study also to other materials often reported as very active and selective ketonization catalyst in the literature (i.e., CeO217 and Ce/Zr/O49). The catalytic activity of these materials was compared under the same conditions of the test shown in Fig. 4 and the results for CeO2 and Ce/Zr/O with respect to 2-MF as a function of TOS are reported in Fig. S4a and S4b†, respectively. Both catalysts at the beginning of the reaction were very active and 2-MF conversion was complete, but the carbon loss was significant (i.e., the sum of the yields of all detected products did not exceed ≈ 40–50%). Then, after several hours on stream the conversion dropped down, roughly matching the sum of yields. This trend, unseen over ZrO2, was attributed to the well-known redox activity CeO2 and Ce/Zr/O,50–52 which fostered the decomposition of 2-MF into gaseous products such as propylene and butylene, which were identified by means of GC-MS but could not be quantified with the online GC-TCD system used. The H2-TPR characterization of the 3 catalysts shown in Fig. S5† substantially confirms that in the temperature range relevant for ketonization ZrO2 is a non-reducible oxide,53 while the reduction of both CeO2 and Ce/Zr/O calcined at moderate temperature (e.g., 400 °C) starts at temperatures below 350 °C, in agreement with previous literature.54,55 ZrO2, CeO2 and Ce/Zr/O after reaction were also characterized by means of XRD and Raman spectroscopy. The XRD characterization showed that the crystal structure of all three materials is unchanged after reaction (Fig. S6†) with no segregation of bulk metal carboxylates, in agreement with the surface ketonization mechanism; on the other hand, two bands attributable to the presence of amorphous coke (e.g., Raman shift = 1605 and 1385 cm−1) were found in the Raman spectra of CeO2 and Ce/Zr/O but not in the one of ZrO2 (Fig. S7a†), indicating that carbon deposits were formed over the surface of the Ce-containing catalyst during the reaction. Therefore, a thermogravimetric analysis under an air flow (TGA, Fig. S7b†) was carried out to quantify the carbonaceous deposit, showing a weight loss of 2.9, 8.5 and 13.4% for ZrO2, Ce/Zr/O and CeO2, respectively. Noteworthily, these carbon deposits can justify only 15.4% and the 22% of the carbon loss observed respectively for CeO2 and Ce/Zr/O during the reaction, further suggesting that the formation of light alkenes was the major contributor to the carbon loss. Finally, the SSAs of ZrO2, CeO2 and Ce/Zr/O after the reaction were 39 m2 g−1 (−29%), 87 m2 g−1 (−55%) and 3 (−98%) m2 g−1 respectively, suggesting that the fouling catalyst surface and blockage of pores was more serious for Ce-containing catalysts. The bar chart shown in Fig. 5 summarize the main results of the catalyst screening, comparing the mean values of conversion and yields obtained over ZrO2, Ce/Zr/O and CeO2, calculated once a steady performance of the catalyst was achieved (in the case of CeO2 and Ce/Zr/O, conversion and yields were calculated once the conversion of 2-MF dropped down matching the sum of yields). The results obtained strongly suggest that ZrO2 is the most active and selective catalyst for the gas-phase synthesis of AF by means of cross-ketonization because it does not foster the unwanted decomposition of 2-MF. On the other hand, the AC produced by the homo-ketonization of AA was stable over all 3 materials, which were more or less equally active and selective (Fig. 5b), in agreement with the literature.17,49
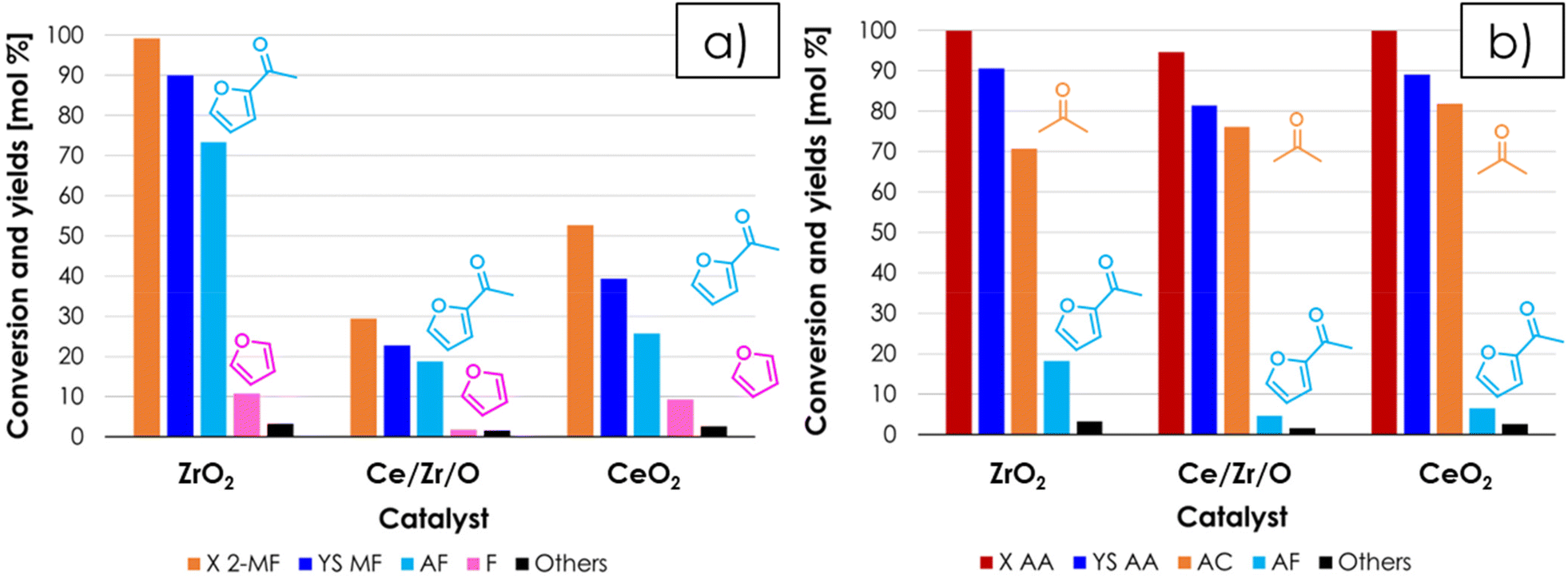 | ||
| Fig. 5 Results of the cross-ketonization between 2-MF and AA as a function of the catalytic material with respect to 2-MF (a) and AA (b). Reaction conditions: temperature = 350 °C, 2-MF/AA/N2 = 0.4/1.6/98 mol%, contact time = τ = 1 s GHSV = 1722 h−1, 1 mL of catalysts. Symbols: (a) 2-methyl furoate conversion (X 2-MF), sum of yields (YS 2-MF), acetyl furan yield (AF), furan yield (F), sum of yields of unknown by-products (Others); (b) acetic acid conversion (X AA), sum of yields (YS AA), acetone yield (AC), acetyl furan yield (AF), sum of yields of unknown by-products (Others). | ||
Comparison of acetic acid and ethyl acetate as ketonization agents
The reaction between 2-MF and ethyl acetate (EA) for the synthesis of AF was investigated to assess whether aliphatic esters may be suitable reactants as well as the corresponding carboxylic acids, in view of an extension of the synthetic method proposed in this work to other substrates. In fact, some reactants may not be soluble/miscible with carboxylic acids (protic, polar solvents), thus requiring the use of the respective aprotic, less polar esters. The results of a catalytic test carried out by feeding a mixture of 2-MF/EA/N2 = 0.4/1.6/98 over ZrO2 at 350 °C with a contact time τ of 1 second is shown in Fig. S8a† (with respect to 2-MF) and in Fig. S8b† (with respect to EA). Under these conditions, significant amounts of highly reactive ethanol (EtOH) were co-produced by the homo-ketonization of EA, as depicted in Scheme 2. As a result, the product selectivities changed significantly over the course of 18 hours on stream, due to the occurrence of 3 main reactions: (1) the desired cross-ketonization between 2-MF and EA to give AF; (2) the consecutive H-transfer reduction and dehydration of AF to VF with the more reactive EtOH as the H-donor;56,57 (3) the transesterification of the reactant 2-MF with EtOH to give 2-ethyl furoate (2-EF). In particular, during the first hours ZrO2 fostered the consecutive H-transfer reduction of AF so that the main product was VF (Y = 29%); then, after 9 hours of TOS, AF became the main product of the reaction (Y = 51%), possibly due to the deactivation of the active sites of ZrO2 responsible for the H-transfer reductions. A similar behaviour was previously observed in the case of the H-transfer reduction of methyl levulinate with EtOH over ZrO2 and was ascribed to the fouling of Lewis acidic sites by carbon deposits.56,57 Between the 5th and the 9th hour on stream the active sites of ZrO2 responsible for ketonization (i.e., Lewis acid–base pairs1 as in the case of H-transfer reactions58) started deactivating as well, so that the yield of AF progressively decreased down to 27% after 18 hours on stream while at the same time the one in 2-EF increased up to 31%. The results shown in Fig. S8b† indicate that EA is less reactive than AA under the same conditions, because its conversion is significantly lower (≈50% after 18 hours on stream against 100% after 17 hours on stream for AA, Fig. 4b). Apart from reacting with the reactant 2-MF and with the product AF, the EtOH co-produced by the homo-ketonization of EA also underwent intramolecular dehydration to ethylene over the acidic sites of ZrO2 and dehydrogenation to acetaldehyde (AAL) over its basic sites, in agreement with previous reports.59–61 The AAL resulting from EtOH dehydrogenation reacted further with AC and EA leading to the formation of small amounts of 2-pentanone (2-P) and ethyl butyrate (EB), according to the multistep hydroxy-alkylation/dehydration/H-transfer reduction pathway shown in Scheme 2. This reaction pathway is analogous to the one proposed previously by our group to explain the formation of methyl isobutyrate (MIB) by the coupling of formaldehyde (FAL, produced by in situ dehydrogenation of MeOH) with methyl propionate (MP) and the formation of mono- and dimethyl pentanones from the coupling of 3-pentanone (3-P) and FAL.62 Remarkably, in spite of the selectivity issues related to the use of EA as the aliphatic reactant, the results shown in Fig. S9a and S9b† demonstrates that the yield towards AF can be greatly improved (up to 71%) by simply feeding 2-EF instead of 2-MF as the furoic reactant, so that the two esters possess the same aliphatic chain. Still, this approach does not limit the extent of the unwanted H-transfer reduction of AF to VF during the first hours on stream, nor reduces the progressive deactivation of the active sites responsible for ketonization that lead to a decrease of both the furoic reactant conversion and AF yield for longer TOS. | ||
| Scheme 2 Proposed reaction scheme for the cross-ketonization of 2-MF with EA. | ||
Optimization of reaction conditions and synthesis of other alkanoyl furans
The results obtained so far indicated that ZrO2 and AA are respectively the most effective catalyst and reagent for the synthesis of AF; therefore, it was decided to investigate more in-depth the effect of the molar ratio between AA and 2-MF on the product selectivity, by keeping the molar percentage of 2-MF constant and equal to 1% in the feed and varying AA mol % from 1 to 4 to 9% (Fig. 6a and b). Comparing the results shown in Fig. 5 (2-MF/AA/N2 = 0.4/1.6/98 mol%) and in Fig. 6 (2-MF/AA/N2 = 1/4/95 mol%) one can see that no adverse effects on 2-MF conversion or AF selectivity occurred by increasing the concentration of the reactants while maintaining the molar AA/2-MF ratio equal to 4, this way boosting the space–time yield of AF (STY, gAF h−1 gCAT−1) by roughly 2.5 times (e.g., from 0.0168 to 0.0427 h−1). On the other hand, by increasing the molar ratio between AA and 2-MF up to 9 by feeding a gas mixture consisting of 1/9/90 = 2-MF/AA/N2 mol% a remarkably stable AF yield of 85% was maintained for over 12 hours on stream (Fig. S10†). However, the improvement in AF selectivity obtained by increasing the AA/AF molar ratio in the feed from 4 to 9 (e.g., the AF yield increased from 80% to 85%) was altogether moderate and does not justify the use of such a large excess of AA; therefore, the molar ratio 2-MF/AA = 1/4 was selected as the best compromise between achieving a high AF selectivity and avoiding excessive waste of the AA reactant. Finally, the STY of AF was further increased by roughly a factor of 4 (e.g., from 0.0413 to 0.152 h−1) by feeding a 2-MF/AA/N2 = 4/16/80 mol% mixture over ZrO2 at 350 °C and τ = 1 second without adverse effects on the AF yield. The results of the catalytic tests carried out with a molar ratio of AA/AF = 4/1 with increasing concentrations are summarized in Fig. S11.†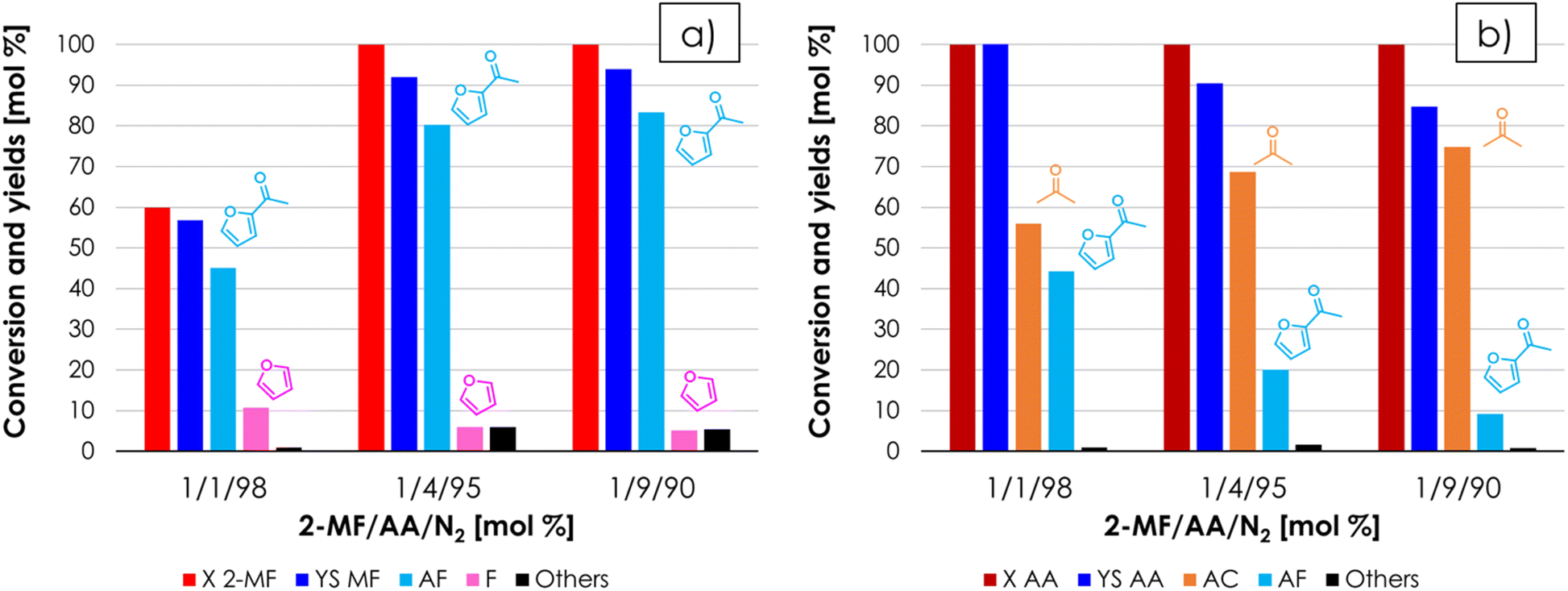 | ||
| Fig. 6 Results of the cross-ketonization between 2-MF and AA as a function of the molar ratio between 2-MF and AA, with respect to 2-MF (a) and AA (b). Reaction conditions: temperature = 350 °C, 2-MF/AA/N2 = variable, contact time = τ = 1 s, GHSV = 1722 h−1, 1 mL ZrO2. Symbols: (a) 2-methyl furoate conversion (X 2-MF), sum of yields (YS 2-MF), acetyl furan yield (AF), furan yield (F), sum of yields of unknown by-products (Others); (b) acetic acid conversion (X AA), sum of yields (YS AA), acetone yield (AC), acetyl furan yield (AF), sum of yields of unknown by-products (Others). | ||
The versatility of the cross-ketonization approach for the gas-phase synthesis of alkanoyl furans was investigated by expanding the substrate scope for the preparation of propionyl furan (PF) and butyryl furan (BF), by reacting 2-MF with propionic acid (PA) and butyric acid (BA), respectively. This study was carried out by feeding a 2-MF/acid/N2 gas mixture with a 1/4/95 mol% composition over ZrO2 at 350 °C and at a contact time of 1 second. The results shown in Fig. 7a indicate that the method has broad applicability and can be used for the synthesis of alkanoyl furans with C1–C3 aliphatic chains with minimal loss of performance in terms of both 2-MF conversion (100%, 96%, and 91% with AA, PA, and BA respectively) and selectivity of the target product (80%, 76% and 76% for AF, PF and BF respectively). Interestingly, the results shown in Fig. 7b suggests that as the length of the alkyl chain of the carboxylic acid used increases the formation of a methyl ester by means of its trans-esterification with the MeOH co-produced by the target reaction (and by the decomposition of 2-MF into F) becomes more and more favoured competing with the homo-ketonization reaction toward the symmetric, aliphatic ketones, which still remain the main by-products of this synthetic strategy. Noteworthily, 3-pentanone finds application as a solvent for paints, as well as in fragrances formulation and as an intermediate for the production of vitamin E.63 On the other hand, the possible applications of 4-heptanone are much less investigated in the literature. Nonetheless, it can be considered an intermediate for the production of 2,6-dimethyl-4-heptanone, which is again a known solvent for paints, dyes, and adhesives, and as an extraction agent and a solvent for recrystallization of organic compounds.63 Finally, the best results obtained in this work in terms of conversion, yield and STY are shown in Table 1, where they are compared to those calculated from the data published by other authors, via traditional synthetic strategies such as the Friedel–Crafts acylation of F with acetic anhydride (AAN) or AA in the presence of Lewis acid catalysts, the Wacker oxidation of VF with tert-butyl hydroperoxide (TBHP) catalysed by Pd(II) complexes and the oxidation of ethyl furan (EF) with TBHP catalysed by heteropolyacids. Despite the different target products and reactor configurations, the performances of the recently reported liquid-phase process for the production of 2-dodecanoyl furan by means of the cross-ketonization of furoic acid with lauric acid were added to the comparison as well, because it provides an example of cross ketonization with furoic acid and fatty acids.27,28 To the best of our knowledge, only one continuous gas-phase process has been proposed so far in the literature for the production of AF:64 it involves the use of zeolites as catalysts for the acylation of F with AAN, and it is outperformed by our method both in terms of selectivity and STY. On the other hand, the STY calculated for most liquid-phase batch processes reported in the literature are higher than ours. It is worth noting, however, that only the reaction time (which is the only information usually published) was considered in the calculation of the STY in Table 1, therefore these values are overestimated to some extent. In fact, the total time required to prepare a batch of AF is actually longer due to the extra time needed to charge the liquid reactants into the batch reactor, to heat them up to reaction temperature, to unload the reactor, to separate the catalyst and so on. Moreover, several batch processes require either the use of CRM (critical raw materials)-based catalysts (e.g., Pd, Sc, W), or harmful reactants (TBHP, AAN, CH3SO3H, trifluoroacetic anhydride = TFAAN) or large amounts of solvents. Therefore, the sustainability of our method was compared to those of the traditional methods reported in Table 1, by calculating the E-factor (kgWASTE kgAF−1) on the basis of the information available in each reference about the waste produced due to unreacted reagents, by-products formation, auxiliaries (solvents, catalysts, and co-catalysts) and, whenever possible, work-up operation related to catalyst separation. These results (together with detailed information on how E-factors were calculated) are reported in Table S1 of the ESI.† The comparison shows that the amount of waste produced by our method (E-factor equal to 1.74) is lower compared to every liquid-phase batch process, even considering the co-produced AC and F as waste, owing to the advantages of gas-phase operation, which avoids both the use of solvents and catalyst separation.
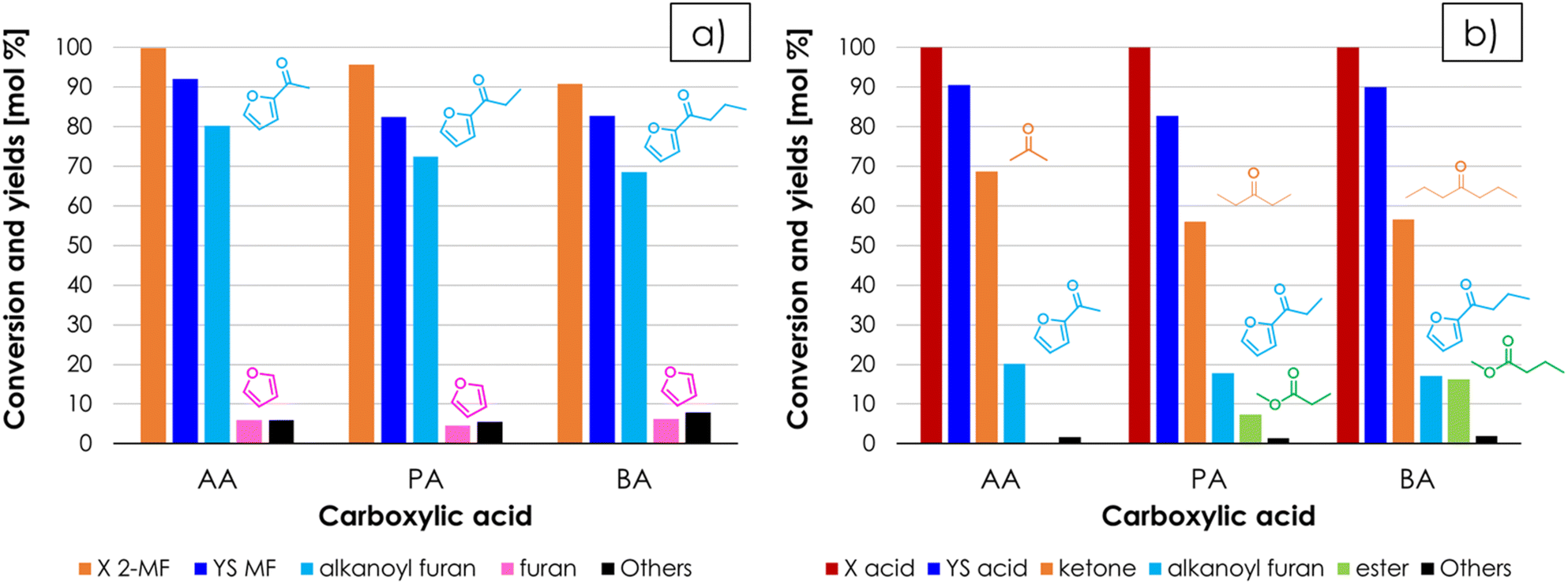 | ||
| Fig. 7 Results of the cross-ketonization between 2-MF and different carboxylic acids with respect to 2-MF (a) and the carboxylic acid (b). Reaction conditions: temperature = 350 °C, 2-MF/AA/N2 = 1/4/95 mol%, contact time = τ = 1 s, GHSV = 1722 h−1, 1 mL ZrO2. Symbols: (a) 2-methyl furoate conversion (X 2-MF), sum of yields (YS 2-MF), alkanoyl furan yield, furan yield, sum of yields of unknown by-products (Others); (b) carboxylic acid conversion (X acid), sum of yields (YS acid), ketone yield, alkanoyl furan yield, ester yield, sum of yields of unknown by-products (Others). | ||
| Synthetic pathway | Process type | Catalyst | Solvent | Time [h] | Temp. [°C] | τ [s] | Reactants molar ratio | Y AF (XR) [mol %] | STYAF [h−1] | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| C = continuous, B = batch, GP = gas-phase, LP = liquid-phase, Ht = heterogeneous catalyst, Hm = homogeneous catalyst, τ = contact time, YAF = AF yield, XR = furanic reactant conversion, STYAF = AF space–time yield = g h−1 of AF divided by g of catalyst, TBHP = tert-butyl hydroperoxide, LA = lauric acid, FA = furoic acid, EF = ethyl furan, AAN = acetic anhydride, TFAAN = trifluoroacetic anhydride, values reported for ref. 27 and 28 refer to the synthesis of dodecanoyl-furoate (DF) from furoic and lauric acid. | ||||||||||
| Cross-ketonization | C, GP, Ht | ZrO2 | — | — | 350 | 1 | 4 (AA/2-MF) | 77 (90) | 0.152 | This work |
| B, LP, Ht | FeO | C12H26 | 1.5 | 315 | — | 0.2 (LA/FA) | 8.2 (85) | 0.51 | 27 | |
| MgO | C12H26 | 1.5 | 350 | — | 0.6 (LA/FA) | 50 (100) | 2.49 | 28 | ||
| Wacker oxidation | B, LP, Hm | Pd(II)(aPmic)(CH3CN)2(BF4)2 | CH3CN | 12 | 45 | — | 3 (TBHP/VF) | 90 | 0.664 | 65 |
| Oxidation | B, LP, Hm | Cu(OOCCH3)2 + H3PMo12O40 | CH3CN/H2O | 10 | 90 | — | 2.5 (TBHP/EF) | 66 | 0.3 | 66 |
| B, LP, Ht | FePW12O40/g-C3N4 | CH3CN | 30 | 25 | — | 2.5 (TBHP/EF) | 65 | 0.239 | 67 | |
| Acylation | B, LP, Hm | Sn(CF3SO3)2 | CH3NO2 | 4 | 25 | — | 2 (AAN/F) | 86 | 1.13 | 68 |
| Zn(CF3SO3)2 | CH3NO2 | 4 | 25 | — | 1.5 (AAN/F) | 75 | 0.438 | 69 | ||
| B, LP, Ht | Sc(CF3SO3)3/terpyridine dendrimer ligand | CH3CN | 0.25 | 130–160 | — | 2 (AAN/F) | 99 | 4.624 | 70 | |
| HBEA (Si/Al = 12) | Neat | 2 | 60 | — | 5 (AAN/F) | 91 | 2.63 | 71 | ||
| graphite/CH3SO3H | Neat | 0.33 | 0–10 | — | 1 (AA/F) | 98 | 0.668 | 72 | ||
| AlPW12O40/TFAAN | Neat | 1.25 | 0 | — | 1 (AA/F) | 94 | 0.354 | 73 | ||
| Fe3+ exchanged K10 montmorillonite | Neat | 8 | 40 | — | 0.25 (AAN/F) | 22.5 | 0.248 | 74 | ||
| Al2O3/TFAAN | Neat | 3 | 0–5 | — | 1 (AA/F) | 75 | 0.0211 | 75 | ||
| Sulphated TiO2 | Neat | 40 | 50 | — | 1.5 (AAN/F) | 95 | 0.0131 | 76 | ||
| C, LP, Ht | HBEA (Si/Al = 27.6) | Neat | — | 60 | 8280 | 5 (AAN/F) | 80 | 0.17 | 39 | |
| C, GP, Ht | HZSM-5 | — | — | 150 | 7200 | 1 (AAN/F) | 42 (93) | 0.135 | 64 | |
Conclusions
In this work, the synthesis of the valuable food additive and pharmaceutical intermediate 2-acetyl furan (AF) by means of the continuous-flow, gas-phase catalytic cross-ketonization of reactants obtainable from renewable platform molecules (e.g., 2-methyl furoate (2-MF) from furfural and acetic acid (AA) from bio-ethanol) is reported for the first time in the literature. This reaction is effectively promoted over a simple and cheap ZrO2 catalyst, which exhibited higher catalytic performance and stability with respect to other well-known ketonization catalysts (e.g., CeO2 and Ce/Zr/O).Catalytic tests showed that AA, being able to participate in both the desired cross-ketonization with 2-MF and the unwanted homo-ketonization toward acetone (AC), behaves as a limiting reactant, so that a complete 2-MF conversion cannot be achieved unless AA is fed with a slightly higher molar excess with respect to 2-MF. In these conditions (e.g., AA/2-MF molar ratio = 4), 2-MF conversion became quantitative and the AF selectivity was 80%, the main by-product being furan (F), while all the AA exceeding the stoichiometry of cross-ketonization was converted into AC, which however is a product of interest and is easily separable. The use of other reactants such as 2-ethyl furoate (2-EF) and ethyl acetate (EA) was investigated as well, but it was found that it is less favourable. In fact, the ketonization of both these reactants co-produces ethanol (EtOH) in the reaction environment, fostering unwanted parasitic reactions such as the H-transfer reduction of AF to the corresponding alcohol (which immediately dehydrates leading to the formation of vinyl furan, VF) and the transesterification of 2-MF that results in the formation of 2-EF. The optimization of the reaction parameters (e.g., the molar ratio between reactants and the concentration of the feed) allowed us to achieve an STY of 0.152 h−1 at 90% of 2-MF conversion and 86% AF selectivity, which, to the best of our knowledge, are the best values reported so far for the synthesis of AF in the gas-phase. The substrate scope of the synthetic approach also was extended to the preparation of 2-propionyl and 2-butyryl furan (which were both obtained with 2-MF conversion >90% and product selectivity >75%) by reacting 2-MF with propionic acid and butyric acid, respectively. Finally, the environmental impact of the synthetic strategy towards AF proposed in this work was assessed by calculating its E-factor and it was found that the amount of waste produced is several times lower than that of traditional methods carried out batchwise in the liquid phase (e.g., Friedel–Crafts acylation and Wacker oxidation), thanks to the gas-phase operation that avoids both the use of solvents and catalyst separation.
Author contributions
JDM: data curation, conceptualisation, and writing the original draft; DC: investigation; SBR: investigation and validation; TT: conceptualisation, methodology, and writing the original draft; AF: methodology and data curation; FB: writing review & editing: FC: funding acquisition, conceptualisation, and writing review & editing.Conflicts of interest
There are no conflicts of interest to declare.References
- T. N. Pham, T. Sooknoi, S. P. Crossley and D. Resasco, ACS Catal., 2013, 3, 2456–2473 CrossRef CAS.
- B. Boekaerts and B. F. Sels, Appl. Catal., B, 2021, 283, 119607 CrossRef CAS.
- R. Kumar, N. Enjamuri, S. Shah, A. S. Al-Fatesh, J. J. Bravo-Suarez and B. Chowdhury, Catal. Today, 2018, 302, 16–49 CrossRef CAS.
- Y. Yamada, M. Segawa, F. Sato, T. Kojima and S. Sato, J. Mol. Catal. A: Chem., 2011, 346, 79–86 CrossRef CAS.
- T. N. Pham, D. Shi, T. Sooknoi and D. E. Resasco, J. Catal., 2012, 295, 169–178 CrossRef CAS.
- J. De Maron, L. Bellotti, A. Baldelli, A. Fasolini, N. Schiaroli, C. Lucarelli, F. Cavani and T. Tabanelli, Sustainable Chem., 2022, 3, 58–75 CrossRef CAS.
- M. Glinski, G. Zalewski, E. Burno and A. Jerzak, Appl. Catal., A, 2014, 470, 278–284 CrossRef CAS.
- C. A. Mullen and A. A. Boateng, Energy Fuels, 2008, 22, 2104–2109 CrossRef CAS.
- L. Qu, X. Jiang, Z. Zhang, X. Zhang, G. Song, H. Wuang, Y. Yuan and Y. Chang, Green Chem., 2021, 23, 9348 RSC.
- J. H. Miller, G. R. Hafenstine, H. H. Nguyen and D. R. Vardon, Ind. Eng. Chem. Res., 2022, 61, 2997–3010 CrossRef CAS.
- A. T. Giduthuri and B. K. Ahring, Fermentation, 2023, 9, 1–20 Search PubMed.
- B. Boekaerts, M. Vandeputte, K. Navare, J. Van Aelst, K. Van Acker, J. Cocquyt, C. Van Caneyt, P. Van Puyvelde and B. F. Sels, Green Chem., 2021, 23, 7137–7161 RSC.
- D. J. Foley and H. Waldmann, Chem. Soc. Rev., 2022, 51, 4094–4120 RSC.
- M. Stompor, D. Broda and A. Bajek-bil, Molecules, 2019, 24, 4468 CrossRef CAS PubMed.
- J. Panten and H. Sturburg, Flavors and Fragrances, 3. Aromatic and Heterocyclic Compounds, in Ullmann's Encyclopedia of Industrial Chemistry, 2015 Search PubMed.
- M. Roper, E. Gehrer, T. Narbeshuber and W. Siegel, Acylation and Alkylation, in Ullmann's Encyclopedia of Industrial Chemistry, 2012 Search PubMed.
- O. Nagashima, S. Sato, R. Takahashi and T. Sodesawa, J. Mol. Catal. A: Chem., 2005, 227, 231–239 CrossRef CAS.
- A. V. Ignatchenko, J. S. DeRaddo, V. J. Marino and A. Mercado, Appl. Catal., A, 2015, 498, 10–24 CrossRef CAS.
- A. V. Ignatchenko and E. I. Kozliak, ACS Catal., 2012, 2, 1555–1562 CrossRef CAS.
- R. Pestman, R. M. Koster, A. van Duijne, J. A. Z. Pieterse and V. Ponec, J. Catal., 1997, 168, 265–272 CrossRef CAS.
- M. Glinki and J. Szudybill, React. Kinet., Mech. Catal., 2002, 77, 335–340 CrossRef.
- V. I. Yakerson, L. I. Lafer and A. M. Rubinshtein, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1966, 15, 280–285 CrossRef.
- S. D. Randery, J. S. Warren and K. M. Dooley, Appl. Catal., A, 2002, 226, 265–280 CrossRef CAS.
- O. Marie, A. V. Ignatchenko and M. Renz, Catal. Today, 2021, 367, 258–267 CrossRef CAS.
- A. M. Jackson and S. C. Cermak, Appl. Catal., A, 2012, 431–432, 157–163 CrossRef.
- C. Schommer, K. Ebel, T. Dockner, M. Irgang, W. Hoelderich and H. Rust, US4950763, 1990.
- H. Nguyen, Y. Wang, D. Moglia, J. Fu, W. Zheng, M. Orazov and D. G. Vlachos, Catal. Sci. Technol., 2021, 11, 2769 Search PubMed.
- T. Goculdas, S. Deshpande, W. Zheng, R. J. Ghorte, S. Sadula and D. G. Vlachos, Green Chem., 2023, 25, 614–626 RSC.
- S. Rajadurai, Catal. Rev., 2006, 36, 385–403 CrossRef.
- T. N. Pham, D. Shi and D. E. Resasco, J. Catal., 2014, 314, 149–158 CrossRef CAS.
- V. I. Yakerson, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1963, 12, 914–921 CrossRef.
- A. Pulido, O. T. Borja, M. Renz, M. Boronat and A. Corma, ChemSusChem, 2013, 6, 141–151 CrossRef CAS PubMed.
- A. V. Ignatchenko, J. Phys. Chem. C, 2011, 115, 16012–16018 CrossRef CAS.
- E. Bamberger, Chem. Ber., 1910, 43, 3517 CrossRef.
- M. Renz, Eur. J. Org. Chem., 2005, 979–988 CrossRef CAS.
- F. Gonzales, G. Munuera and J. A. Prieto, J. Chem. Soc., Faraday Trans., 1978, 74, 1547 Search PubMed.
- O. Neunhoeffer and P. Paschke, Ber. Dtsch. Chem. Ges., 1939, 72, 919–929 CrossRef.
- Y. Sakata, A. van Tol-Koutstaal and V. Ponec, J. Catal., 1997, 169, 13–21 CrossRef CAS.
- Y. Xiong, W. Chen, J. Ma, Z. Chen and A. Zeng, RSC Adv., 2015, 5, 103695 RSC.
- J. Dhuguru, E. Zviagin and R. Skouta, Pharmaceuticals, 2022, 15, 66 CrossRef CAS PubMed.
- V. Sánchez Escribano, E. Fernández López, M. Panizza, C. Resini, J. M. Gallardo Amores and G. Busca, Solid State Sci., 2003, 5, 1369–1376 CrossRef.
- F. Basile, R. Mafessanti, A. Fasolini, G. Fornasari, E. Lombardi and A. Vaccari, J. Eur. Ceram. Soc., 2019, 39, 41–52 CrossRef CAS.
- L. Grazia, T. Della Rosa, D. Bonincontro, T. Tabanelli, N. Schiaroli, F. Cavani, C. Lucarelli and S. Albonetti, Catal. Today, 2023, 114036 CrossRef CAS.
- T. Tabanelli, Curr. Opin. Green Sustainable Chem., 2021, 29, 100449 CrossRef CAS.
- L. Grazia, D. Bonincontro, A. Lolli, T. Tabanelli, C. Lucarelli, S. Albonetti and F. Cavani, Green Chem., 2017, 19, 4412–4422 RSC.
- M. S. Gyngazova, L. Grazia, A. Lolli, G. Innocenti, T. Tabanelli, M. Mella, S. Albonetti and F. Cavani, J. Catal., 2019, 372, 61–73 CrossRef CAS.
- M. Weber, W. Pompetzki, R. Bonmann and M. Weber, Acetone, in Ullmann's Encyclopedia of Industrial Chemistry, 2013 Search PubMed.
- A. V. Ignatchenko, T. J. DiProspero, H. Patel and J. R. LaPenna, ACS Omega, 2019, 4, 11032–11043 CrossRef CAS PubMed.
- H. Dou and Y. Roman-Leshkov, US8748670B1, 2014 Search PubMed.
- A. Fasolini, R. Mafessanti, S. Abate, P. Gramazio, J. De Maron, G. Centi and F. Basile, Catal. Today, 2023, 418, 114047 CrossRef CAS.
- J. De Maron, R. Mafessanti, P. Gramazio, E. Orfei, A. Fasolini and F. Basile, Nanomaterials, 2023, 13, 53 CrossRef CAS PubMed.
- A. Fasolini, S. Ruggieri, C. Femoni and F. Basile, Catalysts, 2019, 9, 800 CrossRef CAS.
- A. Ruiz Puigdollers, S. Toson and G. Pacchioni, J. Phys. Chem. C, 2016, 120, 15329–15337 CrossRef CAS.
- S. Watanabe, X. Ma and C. Song, J. Phys. Chem. C, 2009, 113, 14249–14257 CrossRef CAS.
- S. M. Schimming, O. D. LaMont, M. Konig, A. K. Rogers, A. D. D'Amico, M. M. Yung and C. Sievers, ChemSusChem, 2015, 8, 2073–2083 CrossRef CAS PubMed.
- T. Tabanelli, E. Paone, P. Blair Vasquez, R. Pietropaolo, F. Cavani and F. Mauriello, ACS Sustainable Chem. Eng., 2019, 7, 9937–9947 CrossRef CAS.
- P. Blair Vasquez, T. Tabanelli, E. Monti, S. Albonetti, D. Bonincontro, N. Dimitratos and F. Cavani, ACS Sustainable Chem. Eng., 2019, 7, 8317–8330 CrossRef.
- M. J. Gilkey and B. Xu, ACS Catal., 2016, 6, 1420–1436 CrossRef CAS.
- G. Balestra, J. De Maron, T. Tabanelli, F. Cavani and J. M. López Nieto, Catal. Today, 2023, 423, 114013 CrossRef CAS.
- A. Chieregato, J. Velasquez Ochoa, C. Bandinelli, G. Fornasari, F. Cavani and M. Mella, ChemSusChem, 2015, 8, 377–388 CrossRef CAS PubMed.
- L. Izzo, T. Tabanelli, F. Cavani, P. Blair Vasquez, C. Lucarelli and M. Mella, Catal. Sci. Technol., 2020, 10, 3433–3449 RSC.
- J. De Maron, M. Eberle, F. Cavani, F. Basile, N. Dimitratos, P. J. Maireles-Torres, E. Rodriguez-Castellon and T. Tabanelli, ACS Sustainable Chem. Eng., 2021, 9, 1790–1803 CrossRef CAS.
- H. Siegel and M. Eggersdorfer, Ketones in Ullmann's Encyclopedia of Industrial Chemistry, 2012 Search PubMed.
- P. Ram Reddy, M. Subrahmanyam and S. J. Kulkarni, Catal. Lett., 1998, 54, 95–100 CrossRef.
- S. Sayantani, Y. Suman, R. Noor U Din, D. Indranil, K. Sooraj and J. K. Bera, ACS Catal., 2020, 19, 11385–11393 Search PubMed.
- P. Li, Y. Wang, X. Wang, Y. Wang, Y. Liu, K. Huang, J. Hu, L. Duan, C. Hu and J. Liu, J. Org. Chem., 2020, 85, 3101–3109 CrossRef CAS PubMed.
- Y. Wang, P. Li, J. Wang, Z. Liu, Y. Wang, Y. Lu, Y. Liu, L. Duan, W. Li, S. Sarina, H. Zhu and J. Liu, Catal. Sci. Technol., 2021, 11, 4429–4438 RSC.
- I. Komoto, J. Matsuo and S. Kobayashi, Top. Catal., 2002, 19, 43–47 CrossRef CAS.
- F. He, H. Wu, J. Chen and W. Su, Synth. Commun., 2008, 38, 255–264 CrossRef CAS.
- A. Perrier, M. Keller, A. M. Caminade, J. P. Majoral and A. Ouali, Green Chem., 2013, 15, 2075–2080 RSC.
- V. F. D. Alvaro, A. F. Brigas, E. G. Derouane, J. P. Lourenço and B. S. Santos, J. Mol. Catal. A: Chem., 2009, 305, 100–103 CrossRef CAS.
- M. Hosseini Sarvari and H. Sharghi, Synthesis, 2004, 2165–2168 Search PubMed.
- H. Firouzabadi, N. Iranpoor and F. Nowrouzi, Tetrahedron Lett., 2003, 44, 343–5345 Search PubMed.
- B. M. Choudary, M. Sateesh, L. Kantam, K. V. S. Ranganath and K. V. Raghavan, Catal. Lett., 2001, 76, 231–233 CrossRef CAS.
- B. C. Ranu, K. Ghosh and U. Jana, J. Org. Chem., 1996, 61, 9546–9547 CrossRef CAS.
- M. Hosseini-Sarvari and E. Safary, J. Sulfur Chem., 2011, 32, 463–473 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed description of the reactor scheme, catalyst synthesis and characterisation (i.e. H2-TPR, XRD, Raman and TGA analyses), additional catalytic tests and blank runs, as well as a comparison of the E-factors for the synthesis of AF by means of ketonization and other methods from the literature. See DOI: https://doi.org/10.1039/d3gc01992f |
| This journal is © The Royal Society of Chemistry 2023 |