
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advancements and challenges in the production of low-carbon fuels via catalytic fast pyrolysis of biomass through refinery integration and co-product generation†
Matthew M.
Yung
a,
Calvin
Mukarakate
 a,
Kristiina
Iisa
a,
A. Nolan
Wilson
a,
Mark R.
Nimlos
a,
Susan E.
Habas
a,
Abhijit
Dutta
a,
Kinga A.
Unocic
b,
Joshua A.
Schaidle
a and
Michael B.
Griffin
*a
a,
Kristiina
Iisa
a,
A. Nolan
Wilson
a,
Mark R.
Nimlos
a,
Susan E.
Habas
a,
Abhijit
Dutta
a,
Kinga A.
Unocic
b,
Joshua A.
Schaidle
a and
Michael B.
Griffin
*a
aCenter for Catalytic Carbon Transformation and Scale-up, National Renewable Energy Laboratory, Golden, CO 80401, USA. E-mail: Michael.Griffin@nrel.gov
bCenter for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
First published on 7th August 2023
Abstract
The production of advanced biofuels represents a near-term opportunity to decarbonize the heavy vehicle transportation sector. However, important barriers must be overcome and successful deployment of these technologies will require (i) catalyst and process development to reduce cost and improve carbon utilization and (ii) industry-relevant validation of operability to de-risk scale-up. Herein, we seek to address these challenges for an integrated two-step process involving catalytic fast pyrolysis (CFP) followed by co-hydrotreating of bio-oil with refinery streams. Technoeconomic and lifecycle analysis based on the data presented herein reveal the potential to generate low-carbon transportation fuels and chemical co-products with a modelled selling price of $2.83 gasoline gallon equivalent (2016$) and a 78% reduction in greenhouse gas emissions compared to fossil-based pathways. The feedstock for this research was a blend of 50 wt% loblolly pine and 50 wt% waste forest residues, and the CFP step was performed using an ex situ fixed bed of Pt/TiO2 with co-fed H2 at atmospheric pressure. Compared to previous state-of-technology benchmarks, advancements in catalyst design and synthesis methodology enabled a four-fold reduction in Pt loading and a 400% increase in time on stream without negatively impacting upgrading performance. Additionally, a first-of-its-kind integrated assessment of waste gas adsorption showed near quantitative recovery of acetone and 2-butanone, which collectively represent approximately 5% of the biomass carbon. The valorization of these co-products opens opportunities to support decarbonization of the chemical sector while simultaneously improving the overall process carbon efficiency to >40%. After condensation, the CFP-oil was co-hydrotreated with straight run diesel (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 vol%) to achieve 95% biogenic carbon incorporation. The oxygen content of the hydrotreated oil was below detection limits, and the diesel fraction exhibited a cetane number and cloud point suitable for a finished fuel. This manuscript concludes by highlighting remaining research needs associated with improving thermal management during catalyst regeneration, mitigating catalyst deactivation due to inorganic deposition, and demonstrating the durability of biomass feeding systems when operated in hydrogen-rich environments.
:90 vol%) to achieve 95% biogenic carbon incorporation. The oxygen content of the hydrotreated oil was below detection limits, and the diesel fraction exhibited a cetane number and cloud point suitable for a finished fuel. This manuscript concludes by highlighting remaining research needs associated with improving thermal management during catalyst regeneration, mitigating catalyst deactivation due to inorganic deposition, and demonstrating the durability of biomass feeding systems when operated in hydrogen-rich environments.
1.0 Introduction
The development of low carbon energy technologies is critical to meet the increasing demand of fuels and chemicals as a growing number of corporate and global entities commit to reducing greenhouse gas emissions. These trends in decarbonization are exemplified by the >140 nations pledging to reach net-zero carbon emissions during the 2021 United Nations Climate Change Conference (COP26) as well as commitment by the International Air Transport Association (IATA) to fly net zero carbon by 2050. Increasing the capacity of renewable electricity generation can reduce utility and light-duty vehicle emissions as electric vehicle adoption continues to grow. However, the heavy-vehicle transportation sector (e.g., aviation, trucking, marine) is more difficult to electrify, and achieving near-term emission reductions will require the production of low carbon liquid fuels with high energy densities.In 2022, the United States consumed ∼20 million barrels of petroleum per day, with two-thirds of that total used in the transportation sector.1 Due to these high volumes, identifying an appropriately sized feedstock resource is critical to technology deployment. The U.S. Department of Energy has estimated a sustainable, annual U.S. resource potential of one billion tons of biomass which could produce 50–60 billion gallons of low-carbon biofuels and offset 30% of U.S. petroleum consumption.2 As shown in Fig. 1, the ability of the U.S. biomass resource potential to satisfy the market demand for aviation and distillate fuels makes the development of biomass conversion technologies well-suited for these sectors.
 | ||
| Fig. 1 United States petroleum consumption in 2022 and estimated U.S. biomass potential for low-carbon biofuel production to offset petroleum usage.1,2 | ||
Within the slate of biomass conversion technologies, refinery hydroprocessing of biogenic oils represents a promising pathway to produce renewable distillate-range fuels for the aviation and heavy-vehicle sectors. This approach includes stand-alone processing of 100% biogenic oils as well as co-processing biogenic- and fossil-derived oils in a single unit. Both scenarios have potential to achieve near-term emission reductions by leveraging existing infrastructure, workforces, and capital. The value proposition of this refinery integration strategy is clearly demonstrated though recent growth in the commercial-scale production of renewable diesel (RD) and sustainable aviation fuel (SAF) via hydroprocessing of esters and fatty acids (HEFA), exemplified by ongoing projects from multinational petrochemical companies such as Phillips 66 and Royal Dutch Shell. Namely, Phillips 66 is reconfiguring its San Francisco Refinery in Rodeo, California to produce renewable fuels from cooking oil, fats, greases, and soybean oil.3 The Phillips 66 facility is proposed to leverage existing refinery assets and a trained workforce to produce ∼800 million gallons per year of renewable diesel, gasoline, and sustainable aviation fuel (SAF), with production slated to begin in 2024. Similarly, Royal Dutch Shell is investing to construct a ∼250 million gallon per year biorefinery at the Shell Energy and Chemicals Park Rotterdam, the Netherlands, formerly known as the Pernis refinery.4 The Rotterdam facility is expected to be operational in 2024 and will utilize waste feedstocks (waste animal fats, used cooking oils, and greases) as well as certified sustainable vegetable oils, such as rapeseed, to supplement the waste feedstocks.4 Despite growing demand, further expansion of these hydroprocessing technologies will require feedstock diversification, including the development of new conversion pathways to produce biogenic intermediates from a wide range of renewable carbon sources.4 Towards this goal, lignocellulosic biomass is an abundant feedstock that does not compete with food production.
Fast pyrolysis is a commercially proven pathway for the direct liquefaction of lignocellulosic biomass. The bio-oil generated from fast pyrolysis, however, has several undesirable qualities stemming from its high oxygen content, including low heating value, high acidity, and chemical instability.5–11 These issues can be mitigated by catalytically deoxygenating the pyrolysis vapors prior to condensation. The upgraded bio-oil exhibits improved properties that can be tailored for refinery hydroprocessing en route to a finished transportation fuel. Catalytic upgrading of biomass pyrolysis vapors can occur within the pyrolysis reactor or externally in a separate down-stream upgrading unit, which are referred to as in situ and ex situ catalytic fast pyrolysis (CFP), respectively. In situ CFP is advantaged in terms of the comparatively low capital requirements since pyrolysis and catalytic upgrading occur in a single unit.12 Alternatively, ex situ CFP offers greater flexibility since each unit operation can be optimized independently. Likewise, ex situ CFP reduces catalyst exposure to inorganic element-containing materials that are present in the pyrolysis reactor, which has the potential to prolong catalyst lifetime and increase process durability. In all cases, CFP is conducted near atmospheric pressure, differentiating it from hydropyrolysis in which hydrogen pressures of up to 4 MPa are utilized during upgrading.13–15
Several classes of catalysts have been investigated for ex situ CFP. Zeolites, which are typically utilized in a fluidized bed or riser configuration in inert environments, have been widely demonstrated to be effective for producing aromatic hydrocarbons from biomass pyrolysis vapors.16–31 Alternatively, bifunctional metal-acid catalysts operated with co-fed hydrogen have been reported to improve carbon yields by reducing coke formation and promoting oxygen rejection as water instead of carbon containing light gases.5,31–35 In previous research, our team demonstrated a biomass-to-fuels process based on ex situ CFP using a fixed bed Pt/TiO2 catalyst with co-fed hydrogen.31 This work demonstrated that Pt-based catalysts can achieve 37–42% carbon yields with a bio-oil oxygen content of 16–18 wt% on a dry basis. Comparatively, previous studies using zeolite catalysts under inert conditions have reported carbon yields of 21–33% for oils with 18–24 wt% oxygen.20,26,27 The upgraded Pt/TiO2 CFP-oil was successfully hydroprocessed in a single stage to produce gasoline and diesel-range blendstocks with <0.5 wt% oxygen content on a dry basis. This integrated data set informed a modelled MFSP of $3.86–3.91 GGE and provided a compelling proof-of-concept for the approach.
In this manuscript, we report recent advancements in the state of technology (SOT) of the Pt/TiO2 CFP pathway (Fig. 2) to increase efficiency and reduce costs. These advancements include (1) optimization of synthesis methods and support morphology to achieve a 4× reduction in Pt loading and a 4× increase in time on stream, (2) continuous co-hydrotreating of CFP oil with straight run diesel to achieve >95% biogenic carbon incorporation, oxygen content below detection limits, and a diesel fraction with an indicated cetane number and cloud point meeting ASTM criteria for a finished fuel, and (3) an first-of-its kind integrated assessment of waste gas adsorption showing near quantitative recovery of acetone and 2-butanone, which open opportunities to support decarbonization of the chemical sector while simultaneously improving the overall process carbon yield to >40%. These advancements represent important steps to improve process economics, and the data directly informed a process model that revealed a 36% reduction in the MFSP compared to previous literature reports ($4.34 per GGE to $2.83 per GGE).31,36,37 Further, these integrated experimental campaigns provide insight into remaining technical challenges, and we provide a perspective on ongoing research needs to de-risk process scale-up and deployment.
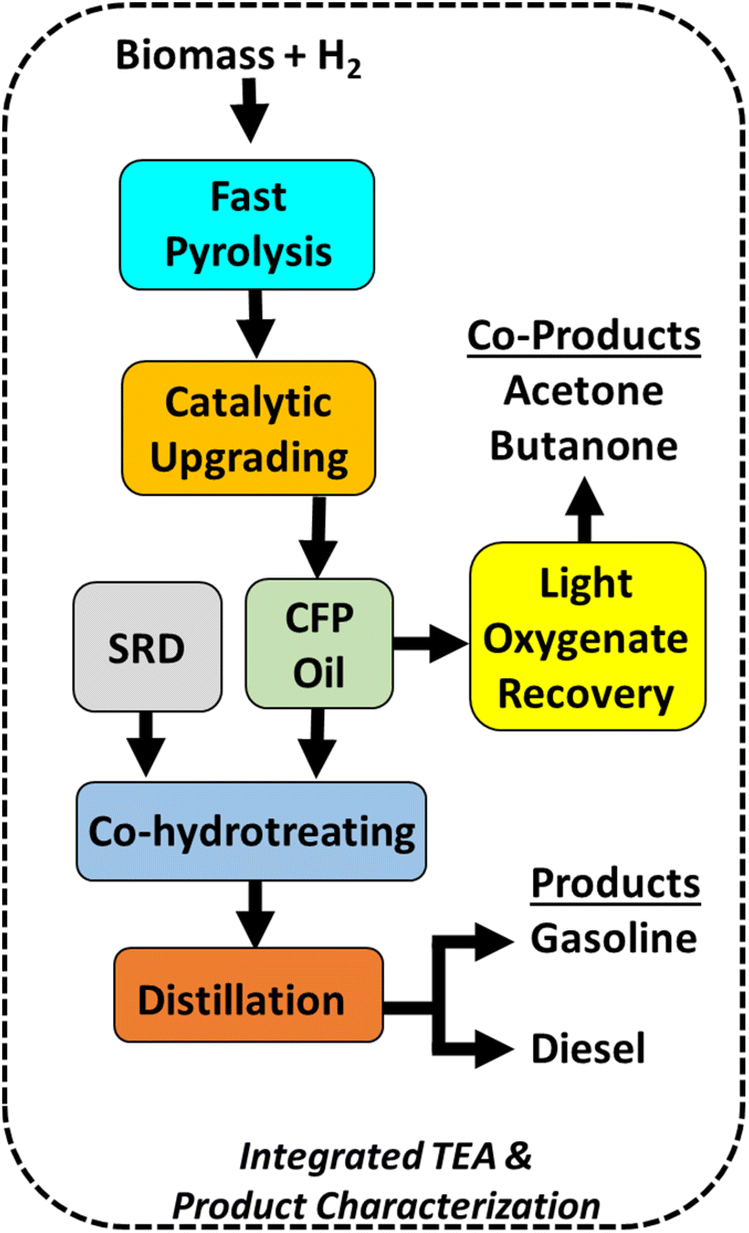 | ||
| Fig. 2 Schematic of integrated bench-scale experimental data with TEA for renewable fuel production via co-processing of CFP oil with straight-run diesel (SRD). | ||
2.0 Results & discussion
2.1 Biomass characterization
For the conversion of a raw material into a finished commodity product, the cost and availability of the feedstock play an important role. For the CFP process, a high quality, finely ground, low ash feedstock is desirable, and our previous work on the CFP pathway focused on the conversion of clean pine.31 A recent woody feedstocks state of technology report by Idaho National Laboratory provided a detailed analysis of the logistics involved in supplying the woody feedstock for CFP and identified a 50:50 wt% blend of clean pine and forest residues as a preferred feedstock.38 Forest residues are a harvest waste that consist of branches, tops, and logging residues. All costs associated with growth, handling, and processing, are included in the cumulative delivered feedstock costs. In changing from clean pine to a 50:50 wt% blend of clean pine and forest residues, the delivered feedstock cost was reduced by 15% from $79 per dry ton to $67 per dry ton.36,37 Among the technical challenges associated with incorporating forest residues into the feedstock stream is the higher ash content as compared to clean pine, which typically has about 0.3 wt% ash (Table 1). This ash is comprised of a variety of inorganic elements including Al, Si, Ca, Fe, K, Na, Mg, and S (Table SI.1†). Several of these elements have the potential to act as catalyst poisons, and while the ability to utilize this lower cost waste feedstock would represent a significant advancement, it also carries considerable risks.
:50 wt% blend of clean pine and forest residues used as a feedstock for the biomass CFP experiments, with modeled cost of $67 per dry ton (ref. 39)
| wt% | wt%, dry basis | Feed composition |
|---|---|---|
| 49.6% | 50.7% | C |
| 6.6% | 6.5% | H |
| 0.1% | 0.1% | N |
| 0.03% | 0.03% | S |
| 0.8% | 0.8% | Ash |
| 2.1% | — | H2O |
| 43.0% | 42.0% | O (*by difference) |
2.2 CFP catalyst properties
A 0.5 wt% Pt/TiO2 catalyst was synthesized using the strong electrostatic adsorption (SEA) method, as described in section 4.2 Catalyst preparation, herein referred to as 0.5% Pt/TiO2. A summary of the catalyst properties as well as the modelled cost is given in Table 2. The titrated acid and metal site densities confirm the bifunctional nature of this material, which has been identified as an important design criterion for CFP catalysts that need to activate H2 for hydrogenation on metallic sites and perform dehydration for deoxygenation on acidic sites. Cost estimates were performed using the publicly available CatCost™ tool,40 as has been reported elsewhere.38 The catalyst cost is largely driven by the precious metal content and decreases proportionally with the Pt-loading. As such, the catalyst reported here was approximately four times less expensive than the 2% Pt/TiO2 catalyst mainly utilized in a previous report.31| Catalyst properties | |
|---|---|
| TiO2 support geometry | 0.5 mm spheres |
| Modelled cost | $203 per kg (ref. 36) |
| TiO2 acidity, NH3-TPD, μmol g−1 | 156 |
| TiO2 surface area, m2 g−1 | 54 |
| TiO2 pore volume, cm3 g−1 | 0.37 |
| TiO2 median pore diameter, Å | 328 |
| 0.5% Pt/TiO2 CO binding site density, μmol g−1 | 19 |
A scanning electron microscopy (SEM) image of the 0.5% Pt/TiO2 catalyst is shown in Fig. 3. Characterization by X-ray diffraction (XRD) revealed the presence of mixed anatase and rutile TiO2 in the fresh 0.5% Pt/TiO2 catalyst, as shown in Fig. 4. No reflections associated with platinum were observed on the fresh or spent samples, Fig. 4, which is attributed to both the low metal loading (0.5% Pt) and relatively small Pt crystallites (∼2 nm as determined by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) coupled with energy dispersive X-ray spectroscopy (EDS), Fig. 5). The effect of regeneration temperature on the properties of the support was evaluated by XRD and N2 physisorption measurements (Fig. 4). For regeneration temperatures up to 500 °C, the 0.5% Pt/TiO2 catalyst had negligible phase transformation and minor changes in surface area and porosity. When the 0.5% Pt/TiO2 catalyst was regenerated at 600 °C and 700 °C, however, a significant phase change in TiO2 from anatase to rutile was observed, which was calculated using Rietveld analysis. The change from anatase to rutile TiO2 was accompanied by decreases in surface area and pore volume that became more pronounced with higher regeneration temperatures and complete transformation to the rutile TiO2 phase following regeneration at 700 °C was observed. These results indicate the importance of limiting the maximum temperature that the catalyst experiences, and resulted in the development of a controlled oxidation process to carefully remove coke and limit the maximum catalyst bed temperature to 500 °C during bench-scale CFP experiments.
 | ||
| Fig. 3 SEM image of fresh 0.5% Pt/TiO2 catalyst on 0.5 mm TiO2 spheres. | ||
 | ||
| Fig. 4 Characterization of fresh and post-reaction 0.5% Pt/TiO2 catalysts following simulated CFP reaction/regeneration cycling with oxidative regeneration at various temperatures. (a) XRD patterns with the anatase (25.2°) and rutile (27.3°) TiO2 phases denoted with dashed lines and estimated wt% of rutile TiO2 denoted on each diffraction pattern, and (b) N2 physisorption surface area and pore volume measurements. | ||
 | ||
| Fig. 5 HAADF-STEM images with corresponding EDS elemental maps (Ti, O, and Pt) showing TiO2 and Pt particle morphology for fresh 0.5% Pt/TiO2 (Pt, davg = 1.8 nm). | ||
2.3 CFP reaction testing
To assess the impact of the feedstock on the CFP oil properties, screening studies were performed in a bench-scale fixed bed reactor to a cumulative biomass-to-catalyst ratio (B:C) of 3 using a 0.5% Pt/TiO2 catalyst (Fig. 6). A full description of the reaction testing methods is given in section 4.4. In these experiments a comprehensive suite of analytical methods was applied to achieve average mass and carbon balance closures of 102% and 99%, respectively. This level of detail is important to accurately map material flux into various process streams (Table 3) and minimize the uncertainty of process models that are informed by these data.36,37 Feedstocks consisted of (1) clean pine, (2) forest residues, and (3) 50:50 wt% blended material. Amongst the three tested feedstocks, the carbon yield and oxygen content were the most desirable (highest C yield and lowest O content) from the clean pine, while the pure forest residues produced the oil with the least desirable qualities. The oil produced from the 50:50 wt% blend had properties that fell between those of the two isolated feedstocks in terms of carbon yield and oxygen content. Because forest residues are a less expensive feedstock than clean pine, the reduction in the modeled cost in supplying the blended feedstock, which does not require extensive separation of components such as branches, needles, and bark, overcomes the slightly lower C yield and higher oxygen content as compared to CFP of pure clean pine. For this reason, the 50:50 wt% blend of clean pine and forest residues was selected for additional experiments.39
 | ||
| Fig. 6 Oil yields and oxygen content from ex situ CFP over 0.5% Pt/TiO2 using clean pine (squares), forest residues (circles), or a blend of 50 wt% clean pine and 50 wt% forest residues (triangles). Experiments were run to a cumulative B:C of 3. | ||
:C of 12 in each cycle
| Mass yield, g g−1 dry biomass | Carbon yield, g C per g C biomass | |
|---|---|---|
| Organic phase | 24.8 ± 1.3% | 35.7 ± 1.3% |
| Aqueous phase | 22.2 ± 2.3% | 1.7 ± 0.7% |
| Condensable light gases | 10.8 ± 0.4% | 12.9 ± 0.2% |
| Non-condensable gases | 29.7 ± 2.4% | 28.1 ± 3.2% |
| Char | 11.1 ± 0.2% | 17.2 ± 0.9% |
| Coke | 1.7 ± 0.5% | 3.3 ± 0.0% |
| Total | 102.4 ± 0.5% | 98.9 ± 1.5% |
One advancement in the CFP technology was the use of a lower-cost catalyst, driven by the reduction in Pt-loading. Triplicate CFP experiments were conducted with the 0.5% Pt/TiO2 catalyst in which the CFP reaction cycle reached a cumulative biomass-to-catalyst (B:C) of 12:1. During CFP experiments, the fixed-bed flow reactor was operated at 400 °C and 0.08 MPa H2 with a continuous biomass feed at a weight hourly space velocity (WHSV) of 1.5 g biomass g per catalyst per h. The CFP-oil oxygen content and carbon yield for these experiments is provided in Fig. 7 and compared against results from a 2% Pt/TiO2 catalyst,31 a commercial ZSM-5 catalyst, and non-catalytic fast pyrolysis. There was no statistically significant difference in the quality of the CFP oils produced from the two Pt catalysts (2% Pt/TiO2 and 0.5% Pt/TiO2). Compared to a conventional zeolite (ZSM-5) catalyst, the Pt/TiO2 catalysts produced oils with higher carbon yields and lower oxygen contents. The non-catalytic fast pyrolysis oil has a significantly higher organic oil yield and also a higher oxygen content.
 | ||
| Fig. 7 Comparison of CFP organic oil carbon yield and oxygen content of the oil for experiments conducted on 0.5% Pt/TiO2 (B:C = 12), 2% Pt/TiO2 pellets (B:C = 3),31 ZSM-5 (B:C = 3), and with no catalyst. Error bars represent standard deviation from triplicate experiments on 0.5% Pt/TiO2. | ||
The data show that the 0.5% Pt/TiO2 catalyst produces CFP oil with similar quality and carbon efficiency as the 2% Pt/TiO2 catalyst, despite a four-fold reduction in Pt loading and 400% increase in the amount of biomass processed prior to regeneration. In addition to the difference in Pt content, the two catalysts also differed in the TiO2 support: the support for the 0.5% Pt/TiO2 consisted of spheres with a particle diameter of 0.5 mm whereas the 2% Pt/TiO2 support had a nominal diameter 1.7 mm. A comparison of the performance of 0.5% Pt on supports of different sizes at the same B:C showed a dependence on particle support size (Fig. SI.1†). The performance of the 0.5% Pt/TiO2 is also consistent with literature values for CFP performed with co-fed hydrogen, in which various catalysts have been reported to achieve 30–43% carbon yield with <20 wt% oil oxygen content.41 Comparatively, CFP performed with zeolite catalysts operated without co-fed hydrogen have been reported to achieve 20–30% carbon efficiency and 22–37 wt% oxygen in the oil.
The elemental composition of the CFP oil is shown in Table 4 and is compared to the oil produced over 2% Pt/TiO2, further illustrating the consistency in bio-oil quality. Given the reduction in Pt loading, the increase in cycle length, and the inclusion of waste feedstock materials, the ability to make an oil with equivalent properties represents a significant advancement in the state of technology that enhances the commercial viability of this approach.
31
| Process details | ||
| Catalyst | 0.5% Pt/TiO2 | 2% Pt/TiO2 |
| TiO2 support | 0.5 mm spheres | 1.6 mm pellets |
| Feedstock | 50:50 Pine + Forest residues |
Pine |
| B:C (g g−1) |
12 | 3 |
| Biomass fed before regeneration (g) | 1200 | 300 |
| Oil elemental composition | ||
| C, wt% dry basis | 77% | 76% |
| H, wt% dry basis | 7.0% | 7.7% |
| N, wt% dry basis | 0.2% | 0.2% |
| O, wt% dry basis | 15% | 16% |
| H2O, wt% | 2.2% | 4.2% |
| H:C, mol/mol dry basis |
1.21 | 1.23 |
| Mass yield of oil | 25% | 27% |
| Carbon yield of oil | 36% | 38% |
To better understand the factors that impact catalyst performance, mass spectrometry was utilized for online monitoring of the CFP process stream during the course of an experiment. Due to the chemical complexity of pyrolysis vapors, individual mass fragments are typically derived from multiple species. However, the relative intensity of select mass signals can be compared to provide a qualitative assessment of catalyst performance. As has been reported previously,31 the mass fragment m/z = 91 is attributed primarily to alkylated aromatics and is generally representative of deoxygenated hydrocarbons, whereas the mass fragment m/z = 60 is attributed primarily to carboxylic acids and is generally representative of primary, unreacted biomass pyrolysis vapors. In Fig. 8 the intensity of these peaks is compared according to eqn (1) in order to provide an assessment of relative deoxygenation activity over the course of an experiment.
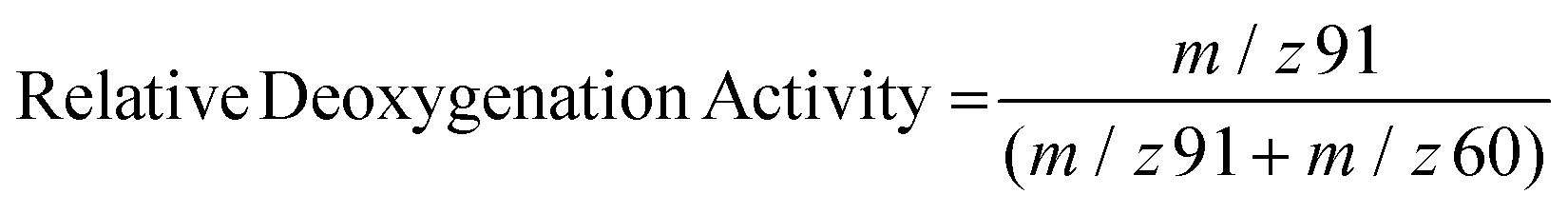 | (1) |
 | ||
| Fig. 8 Comparison of relative deoxygenation activity of 0.5% Pt/TiO2 (orange) and 2% Pt/TiO2 (blue) for biomass CFP at 400 °C, WHSV = 1.5 h−1, and ambient pressure. Relative deoxygenated activity is determined from online mass spectrometry measurements as described in eqn (1). | ||
According to these data, the 2.0% Pt/TiO2 deactivates rapidly, retaining only ca. 50% of its original activity after 100 min time on stream. Conversely, the 0.5% Pt/TiO2 catalyst retains 55–60% of its original activity after 550 min time on stream. The extended cycle length of 0.5% Pt/TiO2 may be associated with the smaller support particle size (0.5 mm vs. 1.7 mm nominal diameter), which decreases internal diffusional distance and mitigates the negative effects of pore blockage that could occur on larger-sized catalyst supports. Importantly, the extended cycle time using the smaller, spherical support advances the technology towards an operational regime in which a swing bed reactor configuration can be utilized. In this cyclic process, one reactor is removed from service for regeneration and a freshly regenerated reactor is simultaneously returned to service. The commercial feasibility of swing bed reactor systems has been demonstrated for propylene production from fossil-based steam cracker or fluid catalytic cracking (FCC) effluents (e.g., TechnipFMC OMEGA technology). However, to the best of our knowledge, this approach has not been applied for upgrading of biomass pyrolysis vapors and allows the use of a fixed-bed reactor(s) which expands the options for catalyst materials, as it alleviates mechanical property constraints associated with circulating and fluidized bed reactors. Process models for fixed bed CFP operated in a swinging bed configuration have shown that extending the reaction cycle length such that the catalyst uptime (the ratio of time the catalyst is under reaction conditions to the time being regenerated) increases from 0.4 to 2.0 translates to a 10% reduction in the MFSP due to a decrease in the number of reactors required.34,35,42 The ability to effectively restore catalyst activity through oxidative regeneration is critical to the success of this approach and has been previously demonstrated by our project team.31,34
2.4 Continuous co-hydrotreating of CFP oil with straight-run diesel
Straight run diesel is produced directly from distillation and must be hydrotreated prior to use for on-road applications. As such, co-hydrotreating biogenic oils with SRD represents a promising strategy for refinery integration. In this work a continuous hydrotreating experiment was performed using a 9:1 volumetric ratio of SRD and CFP oil, respectively, and the results were compared to hydroprocessing of 100% SRD under the same conditions. The SRD and CFP oil were fed by separate syringe pumps and combined with the hydrogen flow before entering the reactor. The conditions for these experiments are described in Table 5 and were informed by industrial practices for SRD hydrotreating operations. Additional experimental details are provided in section 4.5. Over the course of the experimental campaigns, there were no operational challenges associated with the introduction of CFP oil that would preclude the co-processing of SRD with CFP oil at a 9:1 ratio, and the resulting product was compositionally similar on an elemental basis to the product from pure SRD. Namely, the SRD and co-hydrotreated products had similar C and H content, with O, N, and moisture below detection limits (Table 5). However, the sulfur contents of both oils were above the limit of 15 ppm of ultra-low sulfur fuels, which suggests that process modifications such as additional polishing, more severe hydrotreating conditions, or a special ultra-low sulfur catalyst could be employed to minimize sulfur migration from the catalyst into the finished product.
:1 volumetric blend of SRD and CFP oil, and the calculated CFP oil. Results are averages and standard deviation for 12 h steady-state periods
| Hydrotreating process conditions and oil properties | ||
|---|---|---|
| Temperature, °C | 340 | |
| Pressure | 8.3 MPa (1200 psi) | |
| Catalyst | NiMo/Al2O3 | |
| WHSV, h−1 | 1 | |
| Liquid feed for SRD, mL h−1 | 10 | |
| Liquid feed for SRD + CFP, mL h−1 | 9.0 + 1.0 | |
| H2 feed, mL min−1 | 100 | |
| H2:liquid, mL mL−1 |
600 | |
| Liquid yields, g g −1 feed | SRD | SRD + CFP |
| Oil, % | 102% ± 3% | 101 ± 3% |
| Aqueous, % | n.a. | 2.8% ± 0.6% |
| Product composition | SRD | SRD + CFP |
| Density, g mL−1 | 0.79 | 0.78 |
| C, wt% dry basis | 85.2 | 85.6 |
| H, wt% dry basis | 14.8 | 14.4 |
| N, wt% dry basis | 0.01 | 0.01 |
| O, wt% dry basis | <0.5 | <0.5 |
| S, ppm | 77 | 78 |
| Water, wt% | <0.1 | <0.1 |
| H:C, mol/mol |
2.08 | 2.02 |
The products were analyzed using gas chromatography-vacuum ultraviolet spectroscopy (GC-VUV) and simulated distillation, and these results are shown in Fig. 9. Simulated distillation shows that the hydrotreated product from the 9:1 SRD:CFP blend contained more low-boiling compounds as compared to the SRD product, but the distillation curves converge at the 50% point at a temperature of 290 °C. The GC-VUV analysis shows that the hydrotreated product from the SRD:CFP blend was enriched in aromatic compounds at the expense of paraffins and isoparaffins. The GC-VUV analysis corroborated a low oxygen content in the hydrotreated product (0.7% phenolics suggesting ≤0.1 wt% oxygen).
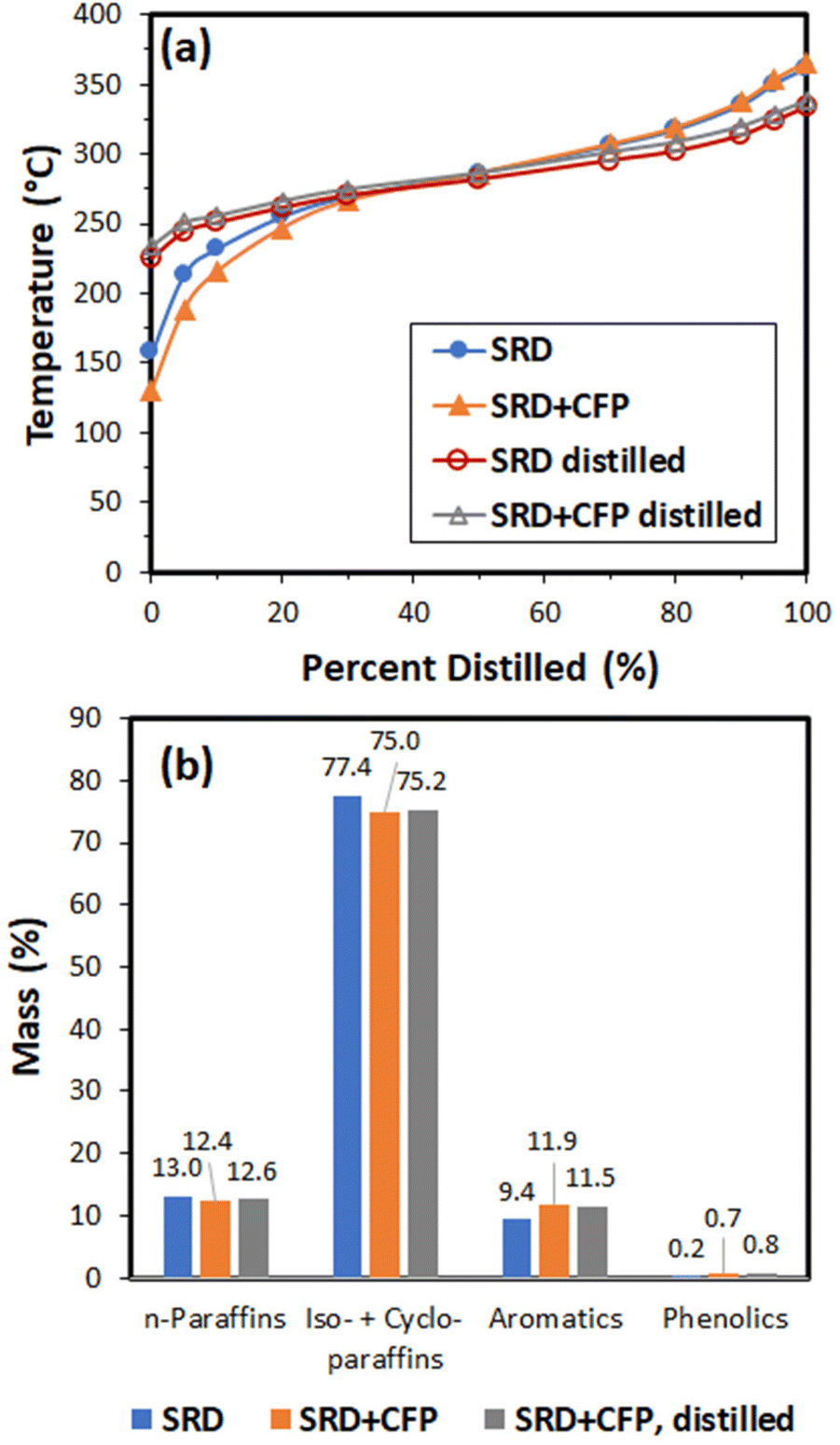 | ||
| Fig. 9 Analysis of hydrotreated products using (a) simulated distillation and (b) GC-VUV. SRD = product from hydrotreating of straight-run diesel only, SRD + CFP = product from co-hydrotreating of 90% straight-run diesel + 10% CFP oil, and SRD + CFP dist. = diesel-range cut from SRD + CFP. | ||
The hydrotreated product was distilled into gasoline, diesel, and residue cuts (Table 6). The diesel-range product had an indicated cetane number (50) and cloud point (−19 °C) that are suitable for a finished diesel fuel.43,44 Upon distillation, the diesel cut from the co-hydrotreated product stayed enhanced in aromatic hydrocarbons and phenolics (Fig. 9). The diesel cut simulated distillation curves for the SRD and SRD:CFP blend were indistinguishable. In order to determine the distribution of biogenic carbon within the distillation fractions, the co-hydrotreated product and fuel cuts were analyzed using 14C analysis (Table 6). The technique differentiates carbon atoms from the biomass (arising from plants <100 years old) and those from petroleum (arising from plants ∼millions of years old). 14C analysis indicated 9.5% biogenic carbon in the hydrotreated SRD/CFP product, which suggests that 95% of the carbon in the CFP oil was incorporated in the hydrotreated product. The biogenic carbon was split roughly equally between the gasoline and diesel fractions, indicating the ability to incorporate biogenic carbon into multiple types of transportation fuel using the CFP pathway and co-hydrotreating with the appropriate blendstock.
| Gasoline | Diesel | Residue | |
|---|---|---|---|
| Atmospheric equivalent temperature, °C | <182 | 182–330 | >330 |
| Mass fraction by distillation | 10% | 82% | 9% |
| Biogenic carbon distribution | 49% | 44% | 7% |
| Indicated cetane number (ICN) | n.a. | 50 | n.a. |
| Cloud point | n.a. | −19 °C | n.a. |
Overall, these results suggest that the blending of 10 vol% CFP oil with SRD results in only minor changes to the hydrotreated product properties as compared to hydrotreating of SRD only. The hydrotreating gave a high incorporation of biogenic carbon from the CFP oil, and the diesel range product had an indicated cetane number and cloud point suitable for a finished diesel fuel. These experiments were of a short duration, and longer experiments are needed to evaluate the impact of CFP oil addition on catalyst deactivation. However, these data provide a basis for developing routes to use renewable feedstocks to aid in decarbonization of the transportation sector.
2.5 Light oxygenate co-products
The efficient utilization of biomass carbon is critical to reduce cost and lifecycle GHG emissions for this process. Moreover, the generation of high-value chemical co-coproducts represents a leading strategy to offset fuel production costs. During CFP experiments analysis of the effluent gas revealed that an average of 13% of the biomass carbon was contained in volatile “light oxygenates” that are difficult to condense in the bio-oil (Table 3). The composition of the condensable, light oxygenates is shown in Table 7 and consists primarily of three compounds: acetaldehyde, acetone, and 2-butanone. Detailed speciation of the components of the condensable stream is provided in ESI Table SI.3,† with the minor components in the condensable stream consisting of linear ketones, cycloketones, and furans. Although industrial-scale quenching systems may be more effective for condensing these molecules, their low boiling point and inherent volatility preclude utilization as distillate-range fuels. As such, an adsorption process was developed and evaluated in order to capture acetaldehyde, acetone, and 2-butanone for chemical applications. Previous research has identified the presence of light oxygenates in condensed bio-oil, but to our knowledge this represents the first demonstration of light oxygenated recovery directly from the gas phase.45:C of 3 and product recovery via desorption using a silica gel adsorbent
| Product yield | Product recovery | % Recovered | |
|---|---|---|---|
| (Adsorber offline) | (Adsorber online) | ||
| g g−1 dry biomass | g C per g C biomass | ||
| Acetaldehyde | 3.2% | 3.5% | 44.7% |
| Acetone | 3.5% | 4.3% | 96.8% ± 4.5% |
| 2-Butanone | 1.1% | 1.4% | 103.6% ± 4.5% |
| Other | 2.5% | 3.5% | n.d. |
Through a series of model compound screening experiments, silica gel was identified as a promising adsorption media due to its high capacity, low cost, and ability to be fully regenerated. To validate this approach, a silica gel adsorption column was installed downstream of the condensation unit to capture and concentrate the condensable light oxygenates from experiments with biomass feedstocks. The experimental apparatus allows the adsorption unit to be bypassed to obtain accurate quantification of the light oxygenates without interference. In two experiments, the adsorber was used during CFP runs to adsorb acetone and 2-butanone and then recover them as co-products during desorption. The adsorber was online until acetone was detected in the effluent, and this took approximately two hours after which the adsorber was bypassed for the remaining six hours of the CFP run. The molecules captured on the adsorber were then recovered by desorption with 2.5 slm of N2, which is equal to 15% of the total flow used during adsorption. The desorption lasted ∼3 hours (one hour at 150 °C and two hours at 200 °C) and 97% of acetone and 100% of 2-butanone that were captured during adsorption were recovered during desorption (Table 7). A typical concentration profile of acetone during adsorption/desorption experiments is shown in Fig. 10. It was observed that the co-product recoveries were maximized by starting desorption at a lower temperature and gradually increasing it to a maximum of 200 °C. A conceptual process for purification to chemical grade is discussed in a previous report.36 The integrated adsorption and recovery of acetone and butanone during biomass bench-scale CFP experiments provides promising proof-of-concept for this strategy, which would have considerable positive impacts on the economics and sustainability of the process.
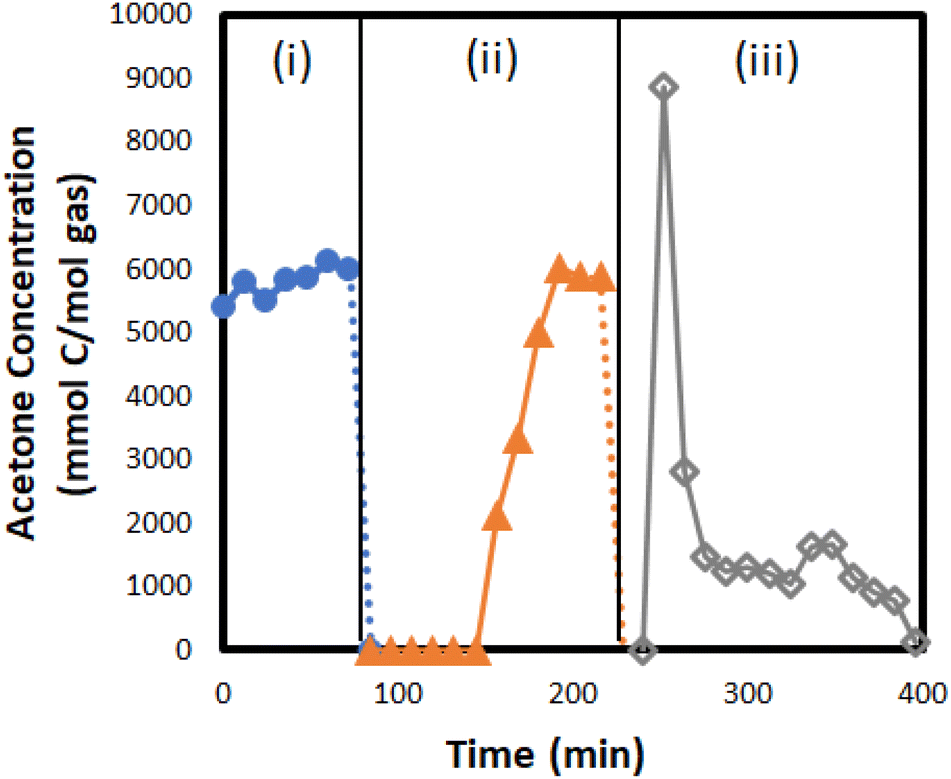 | ||
| Fig. 10 Acetone concentrations during CFP adsorption/desorption experiments involving a silica gel adsorption unit. Labeled regions show: (i) adsorber is offline/bypassed; (ii) adsorption over silica gel; (iii) thermal desorption. Experiment consisted of CFP of a 50 wt% forest residues and 50 wt% clean pine blend over a packed-bed reactor containing 0.5% Pt/TiO2. Reaction conditions: pyrolysis at 500 °C, catalytic upgrading at 400 °C, 0.08 MPa H2 in N2, WHSV 1.5 (g g−1) h−1, and B:C of 3 for adsorption. | ||
2.6 Technoeconomic analysis and life cycle assessment
A detailed State-of-Technology (SOT) report was prepared by Dutta et al. and incorporates the bench-scale experimental findings from this contribution into a detailed process model for a conceptual 2000 dry-ton-per-day woody biomass conceptual process.36,37 The process model and SOT report build upon improvements from earlier SOT reports30,36 to assess the economic and environmental significance of key metrics and advancements such as carbon yield, catalyst lifetime, co-product valorization, co-hydroprocessing, and type of feedstock. The modelled 2020 SOT MFSP was $2.83 per GGE with a 78% GHG (greenhouse gas) reduction as compared to the baseline petroleum scenario. Key advances included (1) improvements in understanding the potential to leverage existing refinery infrastructure, (2) experimentally obtained data on the previously proposed concept of including chemical coproducts along with primary fuels production, and (3) high overall carbon utilization with the combined slate of fuels and coproducts.36 Future research needs include further study of hydrogen utilization at scale, full demonstration/integration of light oxygenate capture/purification and extended-duration operations for both catalytic fast pyrolysis and refinery coprocessing. It should be also noted that the modelled costs are based on data from bench-scale experiments and challenges and risks remain to be addressed during potential future scale-up. Additional information on the technoeconomic and lifecycle analysis is available in previous reports.36,372.7 Research needs
The data presented in this manuscript highlight opportunities to produce distillate-range hydrocarbon fuels and chemical co-products from woody biomass via catalytic fast pyrolysis and co-hydroprocessing. This work has also revealed specific technical challenges that must be overcome to de-risk scale-up and deployment. Among these challenges for CFP is poor thermal management during fixed bed catalyst regeneration which can lead to localized exotherms resulting in process disruption by irreversible catalyst deactivation. Namely, coke deposited during CFP experiments (Fig. SI.2†) must be removed to regenerate the catalysts. The oxidative regeneration of these materials is highly exothermic, and finite element computational modelling based on this system has shown the potential for severe exotherms and sharp temperature gradients46 due to the limited ability to dissipate heat in fixed-bed configurations which could result in Pt sintering (Fig. SI.3†), TiO2 phase change, and physical degradation of catalyst particles (Fig. SI.4†). The degradation of the catalyst particles (Fig. SI.4†) was most pronounced at the inlet of the catalyst bed, where the carbon content from coke was the highest and would result in the largest localized exotherms during regeneration – highlighting the importance of thermal management during coke removal as a research need.It should be noted that the regeneration in this study occurred in a top-down fashion, by introducing oxygen to the reactor inlet where CFP vapors are introduced. Reversing the flow direction to perform a bottom-up regeneration may provide an opportunity to partially alleviate temperature excursions caused by the high concentration of coke near the reactor top/inlet that led to high exotherms during regeneration and could also physically blow any loosely held debris or deposits away/off of the top of the reactor bed, which could be routed out of the reactor system to slow the effects of long term fouling and deposition or inorganic contaminants. Considering that these issues are likely to be amplified at larger scales, identifying optimal reactor configurations to improve thermal management is an important step to de-risk process scale up and deployment. Additional thermal management strategies include integration with a heat exchanger to provide active cooling/heat-removal, use of a catalyst bed diluent, and slow introduction of oxygen during regeneration to limit the rate of coke burnoff.
Another technical challenge for CFP is associated with inorganic element-containing materials that are intrinsic to pyrolysis of biomass and waste carbon feedstocks. Even at relatively low concentrations, the presence of these impurities can become an issue at commercial scale. For example, the 0.8 wt% ash generated from the feedstock utilized in these experiments translates to a daily production rate of 16 tons of ash for a commercial CFP plant that processes 2000 dry tons per day of biomass. The alkali and alkaline earth metals contained in the ash (Table SI.1†) have been widely ascribed as an important cause of catalyst deactivation during biomass conversion.47 In experiments consisting of 13 reaction/regeneration cycles, consisting of 50 h time-on-stream, and 7.4 kg of biomass, K deposition was detected at the highest concentrations reaching a value of ∼0.25 wt% along the leading edge of the bed (Fig. SI.5†). This mechanistic impact of K deposition was probed by our project through a series of complementary experiments in which the poisoning effect of K on Pt/TiO2 catalysts was systematically investigated.47 This work, which has been published elsewhere, revealed preferential poisoning of Lewis acid Ti sites on the TiO2 support and at the metal–support interface. Regeneration by washing the catalyst with water was found to successfully remove 85% of the deposited K species and restore catalytic performance for dehydration of isopropyl alcohol.47
To mitigate the negative effects of inorganic deposition on process performance, there is an option to remove inorganics from the feedstock via acid washing or to perform periodic washing of the catalyst. A cost comparison of these approaches has not been conducted and the long-term impact of inorganic deposition over the catalyst lifetime remains an open question and additional research is needed to establish catalyst tolerance ranges and inform feedstock pre-processing requirements. In addition to experiments at longer time on stream, accelerated catalyst aging experiments for the upgrading of CFP vapors performed on the bench-scale with pre-poisoned catalyst at varying levels of potassium could be employed to determine the transferability of lab-scale research47 and effectiveness of regeneration processes.
In addition to the technical challenges described above, there is also a need to develop solutions for biomass feeding systems that improve safety and durability when operated in hydrogen rich environments, especially at elevated pressure.48 The benchmark technology for continuous feeding into pressurized systems are gravity fed sealed lock hoppers.48–50 However, these systems are not ideal for hydrogen operations due to potential for seal failures resulting from fabrication tolerances, materials of construction, and wear; these issues are relevant with all gate and valve types of feeders. While piston-type and plug-screw type feeders have been adopted by some manufacturers and system integrators, biomass plugs are inherently porous and the ability of these systems to prevent hydrogen backflow remains an open question.51,52 In this context, there is a need to establish performance requirements, engineering data, and experimental results that enable commercial design, manufacture, and long-term use of biomass feeding systems appropriate for commercial-scale deployment of hydropyrolysis and CFP performed with co-fed hydrogen.
3.0 Conclusions
This research highlights opportunities to produce renewable hydrocarbon fuels from woody biomass via CFP and co-hydroprocessing. This approach opens a pathway to support decarbonization of the heavy-vehicle transportation sector (e.g., aviation, trucking, marine) through near-term emission reductions that can be achieved by leveraging existing infrastructure, workforces, and capital. A series of experiments were performed to establish end-to-end performance metrics spanning the entirety of the biomass-to-fuel process. Co-hydroprocessing of CFP-oil with SRD demonstrated the potential to produce a high-quality distillate fuel with an oxygen content below detection limits and a cetane value of 50. Isotopic analysis indicated that 95% of the biogenic carbon fed into the co-hydroprocessing step was accounted for in the liquid product, with approximately equal distribution between gasoline- and diesel-range fuels. Notably, a first-of-its-kind integrated assessment of light oxygenate adsorption from real biomass pyrolysis vapors showed near quantitative recovery of acetone and 2-butanone. These early-stage data provide an opportunity to improve the overall process efficiency through the valorization of co-products that are too volatile to be effectively captured in the CFP-oil. Collectively, these data informed technoeconomic and lifecycle models, which revealed a MFSP of $2.83 GGE ($2016) and a 78% reduction in GHG emissions compared to fossil-based pathways. The integrated approach utilized in these experiments confirms the technical feasibility of the chemistry and potential benefits of the process. It also revealed important risks and data gaps that need to be addressed prior to scale up. Among these challenges are poor thermal management during regeneration of fixed bed catalysts, the potential for irreversible catalyst deactivation due to inorganic contaminants, and questionable durability of biomass feeding systems when operated in hydrogen-rich reaction environments. Moving forward, there is much to be gained by applied R&D efforts to de-risk process scale-up by addressing these important issues.4.0 Experimental
4.1 Feedstock
A commercially harvested biomass feedstock was supplied by Idaho National Laboratory and consisted of 50 wt% loblolly pine and 50 wt% forest residues (composition and ash speciation provided in Table 1 and ESI Table SI.1,† respectively).4.2 Catalyst preparation
The 0.5% Pt/TiO2 catalyst was prepared using strong electrostatic adsorption31 at a pH of 11.5 controlled by NaOH using a Pt(NH3)4(NO3)2 precursor to produce a nominal target Pt loading of 0.5% Pt on the 0.5 mm microspheres (Johnson Matthey). The catalyst support was immersed for >24 h in an aqueous solution with a surface loading of 250 m2 L−1 (2.85 × 10−4 M Pt). Following impregnation, the catalyst was dried for >24 h at room temperature and then dried at 60 °C for >12 h. The dried catalyst was reduced in flowing 5% H2/N2 flow at 450 °C and then cooled to room temperature prior to being passivated in 1% O2/N2 before being loaded into the reactor.The 2% Pt/TiO2 catalyst was prepared using the incipient wetness impregnation technique as previously described31 by adding Pt(NH3)4(NO3)2 (Strem Chemicals) to deionized water and heating 40 °C and sonicating the aqueous solution to fully dissolve the precursor. The solution was then added dropwise to the TiO2 support (Evonik, 1.6 mm pellets, Aerolyst 7711) with an incipient wetness point of 0.52 mL g−1. The impregnated support was allowed to dry at room temperature for >48 h. The catalyst was then heated at 5 K min−1 to 450 °C under 5% H2/N2 flow and reduced for 2 h and then cooled to room temperature and passivated in 1% O2/N2 flow prior to reaction experiments. The hydrotreating catalyst was a NiMo/alumina catalyst prepared by Johnson Matthey.
4.3 Catalyst characterization
The structural characterization was conducted by X-ray diffraction (XRD) on a Rigaku Ultima IV diffractometer using Cu Kα radiation generated at 40 kV and 44 mA. The data was collected in the diffraction angle range 2θ = 20–80° at a scan rate of 4° min−1. In a typical experiment, 10–20 mg of catalyst sample was supported and well-packed on a glass sample holder with a 0.5 mm recessed sample area to obtain a uniform solid surface. Phase identification and composition were analyzed and calculated by using reference card files from the International Center for Diffraction Data (Pt, 00-004-0802; TiO2-anatase, 00-001-0562, TiO2-rutile, 00-001-1292).Density of metal active sites and metal dispersion of Pt/TiO2 catalysts were measured from CO pulse chemisorption performed on Altamira AMI-390 microflow reactor system equipped with a thermal conductivity detector (TCD). About 250 mg of catalyst sample was loaded in a quartz U-tube reactor and held between quartz wool plugs to keep the catalyst bed stabilized. Before each chemisorption experiment, the sample was pre-treated by heating under 10% H2/Ar flow to 450 °C by ramping at 5 K min−1 and held for 2 h. After the reduction step, the catalyst sample was flushed with He (50 mL min−1) for 30 min to remove adsorbed hydrogen. The sample was then cooled to 30 °C and 500 μL pulses of a 10% CO/He mixture were sent through the catalyst bed until the catalyst surface reached saturation. The saturation of the sample was achieved when the peak areas of two consecutive pulses were within ±4%. The site density and average particle size were calculated, assuming that one CO molecule will adsorb on an active site with 1:1 stoichiometric ratio and the metal particle is hemi-sphere. It should be noted that the measurements conducted on bare TiO2 supports did not show any CO uptake.
The acid sites density was identified by NH3 temperature-programmed desorption (NH3-TPD) on Altamira Instruments AMI-390 system with gas flow rates of 25 mL min−1. About 300–350 mg of catalyst sample was loaded into a quartz U-tube reactor and held between two layers of quartz wool to keep the catalyst bed stabilized. Before the TPD experiment, the Pt/TiO2 catalyst sample was pre-treated at 500 °C with ramping rate of 5 °C min−1 for 4 h under flowing 10% O2/Ar. After pretreatment, the sample was cooled to 120 °C under He flow, followed by flowing 10% NH3/He through the catalyst bed at the same temperature for 30 min to saturate the catalyst surface. Excess and physisorbed NH3 were removed by flowing He through the sample at 120 °C for 1 h. The TPD experiment was started by ramping the reactor temperature from 120 to 500 °C with ramping rate of 30 K min−1 and then holding for 30 min under He flow. The signal of desorbed NH3 with increasing temperature was measured with a thermal conductivity detector (TCD) and calibration was performed after each experiment by introducing 10 pulses of 10% NH3/He from a 5.0 mL sample loop into a stream of flowing He.
Pt particle sizes on the fresh (reduced) and unregenerated spent 0.5% Pt/TiO2 catalysts were determined by analyzing high-angle annular dark-field STEM (HAADF-STEM) images with a custom-developed Python code.47 The boundaries of particles were defined using a geodesic active contour algorithm and overlapping particles were subsequently segmented using a watershed method. Particle sizes were reported as effective diameters by assuming the particle is circular in projection and utilizing the areas measured. The measurements included at least 250 particles per condition.
To assess the effect of thermal stability during oxidative regeneration, simulated CFP experiments were conducted. In the micro-scale reaction tests, the 0.5% Pt/TiO2 catalyst was evaluated for hydrodeoxygenation (HDO) of acetic acid, used as a surrogate for biomass pyrolysis vapors, and underwent a series of durability tests lasting ∼30 hours that consisted of total of five (5) one-hour reaction cycles at a WHSV ∼20 h−1, separated by a 2-hour oxidation/reduction regeneration protocol, which resulted in a cumulative reactant-to-catalyst mass ratio of 100. The regeneration protocols were varied from experiment-to-experiment to determine the impact on the process and catalyst. The regeneration consisted of heating from the HDO reaction temperature (400 °C) in inert prior to 1 h of oxidation in 1% O2 at temperatures of 400, 500, 600, or 700 °C, followed by a common reduction at 400 °C in 85% H2 to activate the catalyst prior to the acetic acid HDO activity measurements. The acetic acid conversion in each of these five cycles, at the various regeneration temperatures are shown in ESI Fig. SI.6† and the spent catalysts, following oxidative regeneration were characterization using XRD and N2 physisorption to determine surface area and pore volume.
4.4 Catalytic fast pyrolysis and light oxygenate recovery
A schematic of the 2-inch fluidized bed (2FBR) system coupled to an adsorber is provided in Fig. 11 and a detailed discussion of a similar reactor system has been presented in previous studies.31 The <0.5 mm blend of 50 wt% clean pine and 50 wt% forest residues was introduced and pyrolyzed in a 5.2 cm inner diameter bubbling fluidized bed reactor, char and fine particulates were separated using a combination of a cyclone and a hot-gas filter, after which the pyrolysis vapors and gases were passed through the fixed-bed reactor containing the 0.5% Pt/TiO2 catalyst. The upgraded vapors from the fixed-bed reactor were introduced into a fractional condensation unit comprising (1) an air-cooled or ethylene glycol/water mixture-cooled vessel, followed by (2) an electrostatic precipitator (ESP), (3) two catch pots in dry-ice traps, (4) an iced coalescing filter, and (5) a third catch pot in a dry-ice trap. Oil and aqueous phase yields were determined gravimetrically from the mass increase in the condensation train. | ||
| Fig. 11 A schematic of the major components of the 2FBR system used for CFP and light oxygenate capture experiments comprising (1) feeding, (2) fluidized-bed pyrolysis, (3) char and fine particulate separations, (4) pyrolysis products catalysis, (5) fractional condensation, (6) co-product recovery, and (7) on-line product analyses. | ||
The condensable stream after the fractional condensation unit was passed through an adsorber containing 200 g of grade 40 silica gel to capture acetone and 2-butanone. Light gas compositions in the exit gases were measured by NDIR analyzers (CO, CO2, and CH4) and by a micro-GC (N2, H2, CO, CO2, and C1–C4 hydrocarbons) and composition of light condensable compounds in the exit gas was measured by an online GC-MS/Polyarc-FID, and the gas flow by a dry gas meter. The catalyst mass in the upgrading reactor was 100 g. Biomass was fed at a rate of 150 g h−1 and gases with a combined composition of 85% H2/15% N2 were fed at a rate 16 slm during CFP upgrading. The total amount of biomass fed prior to regeneration for experiments with a cumulative B:C ratio of 3 was 300 g. The total amount of biomass fed before regeneration for experiments with a cumulative B:C ratio of 12 was 1200 g. The pyrolysis temperature was 500 °C and the upgrading temperature set point was 400 °C. Prior to each upgrading cycle, the catalyst was pre-reduced for one hour in a gas flow of 85% H2/15% N2. After CFP cycles with the adsorber online, 2.5 slm of N2 was used to desorb coproducts from the adsorber set at 150–200 °C and an online GC-MS/Polyarc-FID was used for analysis. After desorption, the catalyst was regenerated via oxidation in a mixture of air and nitrogen to remove coke with a reactor setpoint of 400 °C. The air flow was adjusted during oxidation to keep the catalyst temperature below 500 °C and the regeneration process took four hours. The amount of coke was determined from the gas flows and the CO2 and CO concentrations. It should be noted that all temperatures for upgrading and coke oxidation refer to the set points. Due to exothermic reactions, the measured catalyst temperatures were higher, up to ∼50 °C higher during upgrading and up to ∼100 °C higher during coke oxidation.
4.5 Co-hydrotreating and distillation of CFP oils
The CFP oil from CFP Run #3 (Table SI.2†) was hydrotreated together with straight-run diesel (SRD) in a continuous trickle-bed hydrotreater located at National Renewable Energy Laboratory (NREL). The reactor was fed either SRD or SRD and CFP oil fed in the ratio 90 vol%:10 vol%, respectively. The liquids were fed separately by ISCO syringe pumps, combined with the hydrogen flow and fed through an injector located at the top of the reactor. The hydrotreating temperature was 340 °C, pressure 83 bar, the catalyst was crushed NiMo/alumina prepared by Johnson Matthey, and the nominal weight hourly space velocity (WHSV) was 1.0 (g g−1) h−1 based on catalyst located in the isothermal zone; however, there was additional catalyst in the temperature transition zone. The total liquid feed rate was 10 mL h−1 in each experiment, and the H2:liquid ratio was 600 standard mL mL−1. The H2 gas contained 150 ppm H2S to maintain the catalyst in a sulfided state after presulfidation. The catalyst was presulfided in a flow of hydrogen and sulfiding agent (35% ditert-butyldisulfide in decane) for 1 h at 150 °C, during ramping of temperature to 385 °C at 1.5 °C min−1 and holding at final temperature for 4 h. After the sulfidation, SRD was hydrotreated for 73 h after which the feed was switched to the mixture of CFP oil and SRD and continued for 54 h. Product liquids were collected approximately every 12 h, and the hydrotreated products from each period were analyzed for CHNO by elemental analysis, S by ICP, and water by Karl Fischer titration. The results reported are averages for steady-state periods at each condition (37–73 h for SRD and 97–124 h for the SRD/CFP blend).
The co-hydrotreated products from steady state periods were combined and distilled in a microdistillation unit to produce gasoline- and diesel-range cuts, and residue. The overall co-hydrotreated products, and the distillation fractions were analyzed for biogenic carbon content by 14C analysis at UC Irvine. The diesel cuts were analyzed for composition by gas chromatography-vacuum ultraviolet spectroscopy (GC-VUV), and for boiling point range by simulated distillation, indicated cetane number and cloud point by ASTM standards D2887, D8183, and D5773, respectively.
Disclaimer
The views expressed in the article do not necessarily represent the views of the U.S. Department of Energy or the United States Government.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding for this research, provided by the U.S. Department of Energy (DOE), Office of Energy Efficiency and Renewable Energy (EERE), Bioenergy Technologies Office (BETO). This work was performed in collaboration with the Chemical Catalysis for Bioenergy Consortium (ChemCatBio), a member of the Energy Materials Network, at Oak Ridge National Laboratory (ORNL) under Contract No. DE-AC05-00OR22725, and at the National Renewable Energy Laboratory (NREL) under Contract No. DE-AC36-08-GO28308. Microscopy was performed at ORNL through a user project supported by ORNL's Center for Nanophase Materials Sciences (CNMS), which is sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy. Part of the microscopy research was also supported by the Office of Nuclear Energy, Fuel Cycle R&D Program and the Nuclear Science User Facilities. Authors thank S. K. Reeves for assistance with TEM sample preparation.References
- U.S. Energy, Information Administration (EIA), Monthly Energy Review, April 2022, https://www.eia.gov.
- M. H. Langholtz, B. J. Stokes and L. M. Eaton, 2016 Billion-Ton Report: Advancing Domestic Resources for a Thriving Bioeconomy, Volume I: Economic Availability of Feedstocks, U.S. Department of Energy Oak Ridge National Laboratory, 2016 Search PubMed.
- Phillips 66 press release, 2020-08-12, https://s22.q4cdn.com/128149789/files/doc_news/2020/Rodeo-Renewed-Press-Release.pdf.
- Shell press release, 2021-09-60, https://www.shell.com/media/news-and-media-releases/2021/shell-to-build-one-of-europes-biggest-biofuels-facilities.html.
- D. A. Ruddy, J. A. Schaidle, J. R. Ferrell III, J. Wang, L. Moens and J. E. Hensley, Green Chem., 2014, 16, 454–490 RSC.
- A. V. Bridgwater, Fast Pyrolysis of Biomass: A Handbook, CPL Press, Newbury, UK, 1999 Search PubMed.
- A. V. Bridgwater, Fast Pyrolysis of Biomass: A Handbook Volume 2, CPL Press, Newbury, UK, 2002 Search PubMed.
- A. V. Bridgwater, Fast Pyrolysis of Biomass: A Handbook Volume 3, CPL Press, Newbury, UK, 2005 Search PubMed.
- A. d. R. Pinho, M. B. B. de Almeida, F. L. Mendes, L. C. Casavechia, M. S. Talmadge, C. M. Kinchin and H. L. Chum, Fuel, 2017, 188, 462–473 CrossRef CAS.
- A. d. R. Pinho, M. B. B. de Almeida, F. L. Mendes, V. L. Ximenes and L. C. Casavechia, Fuel Process. Technol., 2015, 131, 159–166 CrossRef CAS.
- S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 18, 590–598 CrossRef CAS.
- D. M. Santosa, C. Zhu, F. A. Agblevor, B. Maddi, B. Q. Roberts, I. V. Kutnyakov, S.-J. Lee and H. Wang, ACS Sustainable Chem. Eng., 2020, 8(13), 5156–5164, DOI:10.1021/acssuschemeng.9b07439.
- D. C. Dayton, J. Carpenter, J. Farmer, B. Turk and R. Gupta, Energy Fuels, 2013, 27(7), 3778–3785, DOI:10.1021/ef400355t.
- D. C. Dayton, J. Hlebak, J. R. Carpenter, K. Wang, O. D. Mante and J. E. Peters, Energy Fuels, 2016, 30(6), 4879–4887, DOI:10.1021/acs.energyfuels.6b00373.
- M. Z. Stummann, M. Høj, J. Gabrielsen, L. R. Clausen, P. A. Jensen and A. D. Jensen, Renewable Sustainable Energy Rev., 2021, 143, 110960, DOI:10.1016/j.rser.2021.110960.
- T. R. Carlson, Y.-T. Cheng, J. Jae and G. W. Huber, Energy Environ. Sci., 2011, 4, 145–161 RSC.
- C. Mukarakate, M. J. Watson, J. ten Dam, X. Baucherel, S. Budhi, M. M. Yung, H. Ben, K. Iisa, R. M. Baldwin and M. R. Nimlos, Green Chem., 2014, 16, 4891–4905 RSC.
- J. Jae, R. Coolman, T. Mountziaris and G. W. Huber, Chem. Eng. Sci., 2014, 108, 33–46 CrossRef CAS.
- N. L. Hammer, R. A. Garrido, J. Starcevich, C. G. Coe and J. A. Satrio, Ind. Eng. Chem. Res., 2015, 54, 10629–10637 CrossRef CAS.
- K. Iisa, R. J. French, K. A. Orton, M. M. Yung, D. K. Johnson, J. ten Dam, M. J. Watson and M. R. Nimlos, Energy Fuels, 2016, 30, 2144–2157 CrossRef CAS.
- C. Engtrakul, C. Mukarakate, A. K. Starace, K. A. Magrini, A. K. Rogers and M. M. Yung, Catal. Today, 2016, 269, 175–181 CrossRef CAS.
- M. M. Yung, A. R. Stanton, K. Iisa, R. J. French, K. A. Orton and K. A. Magrini, Energy Fuels, 2016, 30, 9471–9479 CrossRef CAS.
- M. M. Yung, A. K. Starace, M. B. Griffin, J. D. Wells, R. E. Patalano, K. R. Smith and J. A. Schaidle, Catal. Today, 2019, 323, 76–85 CrossRef CAS.
- B. Luna-Murillo, M. Pala, A. L. Paioni, M. Baldus, F. Ronsse, W. Prins, P. C. A. Bruijnincx and B. M. Weckhuysen, ACS Sustainable Chem. Eng., 2021, 9(1), 291–304, DOI:10.1021/acssuschemeng.0c07153.
- V. Paasikallio, C. Lindfors, E. Kuoppala, Y. Solantausta, A. Oasmaa, J. Lehto and J. Lehtonen, Green Chem., 2014, 16, 3549 RSC.
- V. Paasikallio, K. Kalogiannis, A. Lappas, J. Lehto and J. Lehtonen, Energy Technol., 2017, 5, 94–103 CrossRef CAS.
- K. Iisa, R. J. French, K. A. Orton, S. Budhi, C. Mukarakate, A. R. Stanton, M. M. Yung and M. R. Nimlos, Top. Catal., 2015, 59, 94–108 CrossRef.
- M. S. Talmadge, A. Dutta, J. R. Ferrell and E. Tan, NREL Thermochemical Platform Analysis, BETO, Denver, CO, 2017 Search PubMed.
- U.S. Department, of Energy Bioenergy Technologies Office Multi-Year Program Plan March 2016, https://energy.gov/sites/prod/files/2016/03/f30/mypp_beto_march2016_2.pdf.
- A. Dutta, A. Sahir, E. Tan, D. Humbird, L. Snowden-Swan, P. Meyer, J. Ross, D. Sexton, R. Yap and J. Lukas, Process Design and Economics for the Conversion of Lignocellulosic Biomass to Hydrocarbon Fuels—Thermochemical Research Pathways with In Situ and Ex Situ Upgrading of Fast Pyrolysis Vapors, National Renewable Energy Laboratory, Golden, CO, NREL/TP-5100-62455, PNNL-23823, 2015, https://www.nrel.gov/docs/fy15osti/62455.pdf Search PubMed.
- M. B. Griffin, K. Iisa, H. Wang, A. Dutta, K. A. Orton, R. J. French, D. M. Santosa, N. Wilson, E. Christensen, C. Nash, K. M. Van Allsburg, F. G. Baddour, D. A. Ruddy, E. C. D. Tan, H. Cai, C. Mukarakate and J. A. Schaidle, Energy Environ. Sci., 2018, 11, 2904–2918, 10.1039/C8EE01872C.
- S. Wan, T. Pham, S. Zhang, L. Lobban, D. Resasco and R. Mallinson, AIChE J., 2013, 59, 2275–2285, DOI:10.1002/aic.14038.
- P. Cross, K. Wang, J. Weiner, E. Reid, J. Peters, O. Mante and D. C. Dayton, Energy Fuels, 2020, 34(4), 4678–4684, DOI:10.1021/acs.energyfuels.0c00320.
- R. J. French, K. Iisa, K. A. Orton, M. B. Griffin, E. Christensen, S. Black, K. Brown, S. E. Palmer, J. A. Schaidle, C. Mukarakate and T. D. Foust, ACS Sustainable Chem. Eng., 2021, 9(3), 1235–1245, DOI:10.1021/acssuschemeng.0c07025.
- K. Wang, D. C. Dayton, J. E. Peters and O. Dante, Green Chem., 2017, 19, 3243–3251, 10.1039/C7GC01088E.
- A. Dutta, C. Mukarakate, K. Iisa, H. Wang, M. Talmadge, D. Santosa, K. Harris, F. Baddour, D. Hartley, H. Cai, L. Ou, J. Schaidle and M. Griffin, Ex Situ Catalytic Fast Pyrolysis of Lignocellulosic Biomass to Hydrocarbon Fuels: 2020 State of Technology, National Renewable Energy Laboratory, Golden, CO, NREL/TP-5100-80291, 2021. https://www.nrel.gov/docs/fy21osti/80291.pdf Search PubMed.
- A. Dutta, H. Cai, M. S. Talmadge, C. Mukarakate, K. Iisa, H. Wang, D. M. Santosa, L. Ou, D. S. Hartley, A. N. Wilson, J. A. Schaidle and M. B. Griffin, Chem. Eng. J., 2023, 451, 138485, DOI:10.1016/j.cej.2022.138485.
- D. Hartley, D. Thompson, H. Hu and H. Cai, Woody Feedstocks 2018 State of Technology Report, Idaho National Laboratory, Idaho Falls, ID, INL/EXT-18-51655, 2018 Search PubMed.
- D. S. Hartley, D. N. Thompson and H. Cai, Woody Feedstocks 2020 State of Technology Report, Idaho National Laboratory, Idaho Falls, ID, INL/EXT-20-59976, 2021. DOI:10.2172/1782211.
- K. M. Van Allsburg, E. C. D. Tan, J. D. Super, J. A. Schaidle and F. G. Baddour, Nat. Catal., 2022, 5, 342–353, DOI:10.1038/s41929-022-00759-6.
- K. Wang, D. C. Dayton, J. E. Peters and O. D. Mante, Green Chem., 2017, 19, 3243–3251, 10.1039/C7GC01088E.
- F. G. Baddour, L. Snowden-Swan, J. D. Super and K. M. Van Allsburg, Org. Process Res. Dev., 2018, 22(12), 1599–1605, DOI:10.1021/acs.oprd.8b00245.
- ASTM D8183-18, Standard Test Method for Determination of Indicated Cetane Number (ICN) of Diesel Fuel Oils using a Constant Volume Combustion Chamber—Reference Fuels Calibration Method, ASTM International, West Conshohocken, PA, 2018. DOI:10.1520/D8183-18.
- ASTM D5773-21, Standard Test Method for Cloud Point of Petroleum Products and Liquid Fuels (Constant Cooling Rate Method), ASTM International, West Conshohocken, PA, 2021. DOI:10.1520/D5773-21.
- S. Ghysels, N. Acosta, A. Estrada, M. Pala, J. D. Vrieze, F. Ronsse and D. Rabaey, Sustain. Engery Fuels, 2020, 4, 3712–3725 RSC.
- M. B. Pecha, K. Iisa, M. Griffin, C. Mukarakate, R. French, B. Adkins, V. S. Bharadwaj, M. Crowley, T. D. Foust, J. A. Schaidle and P. N. Ciesielski, React. Chem. Eng., 2021, 6, 125–137, 10.1039/D0RE00339E.
- F. Lin, Y. Lu, K. A. Unocic, S. E. Habas, M. B. Griffin, J. A. Schaidle, H. M. Meyer III, Y. Wang and H. Wang, ACS Catal., 2022, 12(1), 465–480, DOI:10.1021/acscatal.1c02368.
- J. Dai, H. Cui and J. R. Grace, Prog. Energy Combust. Sci., 2012, 38(5), 716–736, DOI:10.1016/j.pecs.2012.04.002.
- K. R. Cummer and R. C. Brown, Biomass Bioenergy, 2002, 23(2), 113–128 CrossRef CAS.
- K. W. Kwant, Status of Gasification in countries participating in the IEA Bio-energy gasification activity, IEA Bioenergy Gasification Country reports, Netherlands, 2001 Search PubMed.
- S. K. Gangwal, Demonstration of a Piston Plug feed System for Feeding Coal/Biomass Mixtures across a Pressure Gradient for Application to a Commercial CBTL System, DOE Award Number: DE-NT0006523, Final Scientific Technical Report, 2011, https://www.osti.gov/servlets/purl/1035865 Search PubMed.
- A. V. Bridgwater, A. J. Toft and J. G. Brammer, Renewable Sustainable Energy Rev., 2002, 181–248 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01574b |
| This journal is © The Royal Society of Chemistry 2023 |