
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoupled immobilized bi-enzymatic flow reactor employing cofactor regeneration of NAD+ using a thermophilic aldehyde dehydrogenase and lactate dehydrogenase†
Kim
Shortall
a,
Simin
Arshi
a,
Simon
Bendl
a,
Xinxin
Xiao
 b,
Serguei
Belochapkine
a,
Denise
Demurtas
a,
Tewfik
Soulimane
a and
Edmond
Magner
*a
b,
Serguei
Belochapkine
a,
Denise
Demurtas
a,
Tewfik
Soulimane
a and
Edmond
Magner
*a
aDepartment of Chemical Sciences, Bernal Institute, University of Limerick, V94 T9PX, Ireland. E-mail: edmond.magner@ul.ie
bDepartment of Chemistry and Bioscience, Aalborg University, Fredrik Bajers Vej 7H, 9220 Aalborg, Denmark
First published on 26th May 2023
Abstract
The use of enzymes in biochemical processes is of interest due to their ability to work under mild conditions while attaining high reaction rates. A limitation in the use of enzymes such as oxidoreductases on a large scale lies with their requirement for costly cofactors, e.g. NAD+, in stoichiometric quantities. Cofactor regeneration mechanisms using bienzymatic recycling systems is an attractive way to increase productivity and efficiency. The thermophilic enzyme aldehyde dehydrogenase (ALDHTt) was immobilized directly from E. coli cell lysate, containing the expressed enzyme, onto Ni2+ activated Sepharose®. The system displayed a rate of conversion of approx. 63% NAD+ with reuse achievable for up to 5 cycles and residual activity of the enzyme upon storage of 93% after 7 days. L-Lactate dehydrogenase was immobilized in a second reactor module downstream of ALDHTtvia two different methods, electrochemical entrapment in poly(3,4-ethylenedioxypyrrole) (PEDOP) and covalent attachment on glyoxyl agarose. Both reactors allowed for up to 100% conversion of NADH, however LDH@agarose proved superior in terms of reuse and storage. LDH@agarose displayed no reduction in activity after 6 cycles of use and retained 98% activity following 56 days storage. A coupled reactor containing immobilized ALDHTt–LDH was operated with the substrates hexanal, benzaldehyde, terephthalaldehyde and p-tolualdehyde. A particular advantage of the system is its ability to preferentially oxidise a single aldehyde group in substrates containing two aldehyde functional groups. The reactor demonstrated efficient cofactor regeneration under continual operation for up 24 h, with enhanced product yields.
1. Introduction
Enzyme based biocatalysts play an important role in an array of applications1–5 that include the synthesis of fine chemicals and pharmaceutical materials and the manufacture of food, textiles, and cosmetics. The use of enzymes in biochemical processes is of interest due to their ability to operate under mild conditions while attaining high reaction rates. In solution, enzymes suffer from poor stability, while separation from reaction media and reuse can be difficult. Immobilization of the enzyme can help overcome these drawbacks, while also increasing ease of use.6 While enzymes such as hydrolases, lipases and laccases are widely used in industrial processes,7,8 the use of dehydrogenase enzymes is limited by their requirement for expensive cofactors e.g., NAD+, that are required in stoichiometric quantities. Cofactor regeneration systems are essential when using these enzymes, and are employed to ensure sufficient supply of cofactor, minimising the amount of cofactor required.9,10 Regeneration can be performed chemically,11 electrochemically,12 photochemically13 and enzymatically.14,15 Chemical methods are relatively simple to use, however, they can suffer from a lack of specificity in the production of the active form of the cofactor and are incompatible with many enzymatic systems. Electrochemical and photochemical methods can be used to regenerate the cofactor but can suffer from similar drawbacks as chemical methods.14 Electrochemical approaches possess the advantage that a second enzyme or reagent is not required and additional by-products are not generated.16 While progress has been made, the use of electrochemical methods in biocatalysis on a large scale has yet to be realized. Enzymatic cofactor regeneration utilises two enzymes in tandem to continuously recycle the cofactor and increase the amount of desired product. A range of different enzymes have been co-immobilized to enable cofactor regeneration in single reactor modules.17 This is an attractive approach for the use of dehydrogenase enzymes in biocatalytic reactors, enabling increased stability and productivity.The aldehyde dehydrogenase from Thermus thermophilus (ALDHTt) is a thermophilic, non-substrate specific enzyme that converts aldehydes to the corresponding carboxylic acids using NAD+ as a cofactor.18,19 ALDHTt can be utilised at temperatures up to 50 °C and can catalyse the oxidation of aliphatic (acetaldehyde, propanal and hexanal) and aromatic (benzaldehyde, terephthalaldehyde and p-tolualdehyde)19 substrates. Terephthalic acid (TPA) is an important precursor in the manufacture of polyethylene terephthalate (PET) plastics. Synthesis routes for TPA typically require high temperatures and pressures, acidic conditions and chemical catalysts. TPA is synthesised via the oxidation of p-xylene, utilising acetic acid and a cobalt–manganese-bromide catalyst in a titanium lined reactor operating at 200 °C and 290 psi.20p-Toluic acid is an intermediate in the synthesis of TPA and is an important raw material in the manufacture of dyes, anticorrosive materials, agricultural chemicals and pharmacueticals.20
L-Lactate dehydrogenase (LDH)21,22 is an NADH-dependent enzyme that converts pyruvate to L-lactate. LDH has previously been employed as a cofactor regenerating enzyme and possesses the advantage that both the enzyme and its substrate are readily available and at low cost. In a recent example, the incorporation of an LDH cofactor regeneration system significantly increased the conversion efficiency of a glucose dehydrogenase based reactor from 10 to virtually 100%.21
An array of methods and supports are available for the design of enzymatic reactors. However, when utilising NAD+-dependent enzymes the effect of diffusion limitations of the relatively bulky cofactor must be considered. Biocatalytic flow systems can alleviate these effects and also possess advantages23 such as improved temperature control and mixing, reduced mechanical stress, increased productivity and higher surface to volume ratios. Two immobilization strategies are typically used in flow biocatalysis; the enzymes are attached to the wall of the reactor or immobilized onto a solid carrier material that is then integrated into the reactor.24
Polypyrrole (PPy) and poly(3,4-ethylenedioxypyrrole) (PEDOP) are conductive polymers (CP) that can be prepared by applying an appropriate electrochemical potential.25 Enzymes can be entrapped in the polymer films via a single step electrodeposition process.26,27 However, significant over-oxidation of PPy can occur due to the formation of hydroxyl radicals, resulting in loosely bound polymer with subsequent leaching of the enzyme.28,29 In contrast, PEDOP possesses high stability to over-oxidation during electropolymerization.30 While the use of CP has primarily focused on electrochemical applications such as bioelectrosynthesis,31 biosensors,32 and biofuel cells,33 they can also be used to immobilize enzymes on conductive supports. For example, lipase was immobilized electrochemically in silica films on nanoporous gold (NPG) supports and incorporated into a flow reactor for the hydrolysis of 4-nitrophenyl butyrate.34 Recently, we demonstrated that glucose oxidase (GOx) can be immobilized in PPy films on graphite rods for the generation of hydrogen peroxide at a rate of 42 μM h−135 in a flow reactor.
Packed bed reactors possess advantages such as the ability to immobilize higher amounts of enzyme when compared to wall coated reactors and with the use of porous supports, the possibility of improved mixing via turbulent flow. Agarose beads are highly porous, mechanically resistant, chemically and physically inert, and highly hydrophilic, making them good supports for immobilization of enzymes.36 Their extensive use in chromatography19,37,38 also enables easy transfer for use as a packed bed biocatalytic reactor.17,39 The outer surface of agarose can be functionalized to assist with enzyme immobilization e.g. with metal ions for affinity binding with a His-tagged protein or preparation of glyoxyl agarose via addition of an aldehyde coated surface.6,40,41 These supports enable affinity binding (reversible) and covalent attachment immobilization (irreversible) respectively and have previously been used to prepare highly stable biocatalysts.17,42
The use of ALDHs in biocatalysis has not been investigated in detail. ALDHTt has the potential to provide mild, biocatalytic routes for the synthesis of compounds such as TPA and are attractive candidates for applications in biocatalysis.43 We describe the immobilization of ALDHTt together with LDH for use in a biocatalytic flow reactor employing cofactor regeneration for the production of carboxylic acid derivatives (Scheme 1). ALDHTt was immobilized directly from cell lysate on Ni2+ activated Sepharose® using an engineered His-tag, eliminating the need for purification. This system displayed cofactor conversion rates of approx. 63% with reuse achievable for up to 5 cycles and residual activity upon storage of 93% after 7 days. LDH was immobilized in a second reactor module via two different methods; covalent attachment on glyoxyl agarose and entrapment in PEDOP electropolymerized on a graphite rod (GRE). Both reactors yielded close to 100% conversion of cofactor, however the LDH@agarose reactor retained higher levels of activity after reuse and storage. LDH@agarose displayed no reduction in activity upon recycling for up to 6 cycles and following 56 days storage, 98% activity was retained. To the best of our knowledge this is the highest storage stability reported for both enzymes in a flow reactor to date. The coupled reactor demonstrated efficient cofactor regeneration under constant operation for 8–24 h, showing no accumulation of NADH. Significantly increased conversion of substrates to products (up to 7-fold) were obtained.
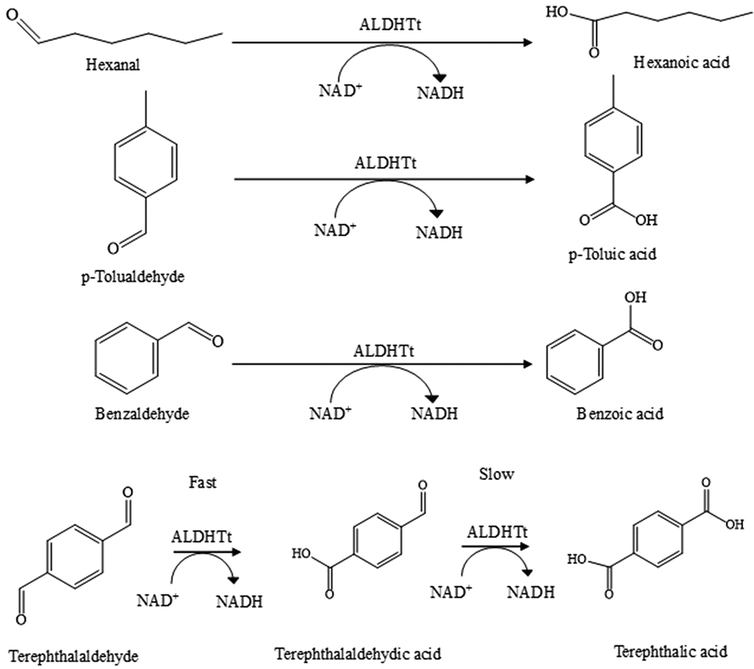 | ||
| Scheme 1 Oxidation reactions catalyzed by ALDHTt. | ||
2. Materials and methods
2.1 Materials
Sodium hydroxide (NaOH >95%), hexanal (98%), terephthalaldehyde (99%), benzaldehyde (≥99%), hexanoic acid (99%), terephthalic acid (98%), benzoic acid (≥99.5%), p-toluic acid (analytical standard), L-lactate dehydrogenase from rabbit muscle (800–1200 U mg−1), β-nicotinamide adenine dinucleotide, reduced disodium salt hydrate (NADH), β-nicotinamide adenine dinucleotide sodium salt (NAD+), sodium pyruvate (≥99%), ethylenediaminetetraacetic acid (EDTA) (99%), nickel(II) sulfate hexahydrate (≥98%), imidazole (≥99%), sodium acetate (≥99%) Trizma™ base, sodium phosphate monobasic, sodium phosphate dibasic, potassium chloride, potassium phosphate monobasic, potassium phosphate dibasic, hydrochloric acid (37%), phosphoric acid (85 wt%), methanol for HPLC (≥99.9), tridecane, dichloromethane for HPLC (≥99.9), sodium chloride, glacial acetic acid, Sepharose™ CL-6B, sodium borohydride (≥99%), glycidol (96%), sodium periodate (≥99%), sodium carbonate, sodium bicarbonate, ethanol, acetone and 3,4-ethylenedioxypyrrole (EDOP) 2% (w/v) in THF were purchased from Sigma-Aldrich, Ireland. HiTrap® Chelating Sepharose® High Performance columns (1 ml) were purchased from Cytiva Lifesciences. p-Tolualdehyde (98%) was purchased from Fisher Scientific, Ireland. FPLC columns, G-TRAP™ were purchased from VWR. Nylon membrane filters PES (pore size: 0.45 μm) was purchased from Agilent Technologies. All reagents were used as received without further purification. Milli-Q water (18.2 MΩ cm) was used for the preparation of all aqueous solutions.2.2 Instrumentation
A Nanodrop ND-1000 spectrophotometer was used for the determination of the concentration of ALDHTt using an extinction coefficient (1%) of 16.71 M−1 cm−1 at 280 nm. Cell culture was performed in a New Brunswick Scientific Inova 40 shaking incubator with cell collection by centrifugation in a Thermo Fisher Scientific Sorvall RC6 Plus Centrifuge. Sonication of cells was performed using a Bandelin Sonoplus sonicator. Biorad Mini Protein Tetra System was utilized for SDS-PAGE analysis. pH measurements were performed using a Thermo Scientific Orion 2-star benchtop pH meter. A Cary 60 UV-vis spectrophotometer equipped with a temperature controller was utilized for all spectrophotometric measurements. Pharmacia LKB-Pump P1 or Instech P720 pumps were used in the flow reactors. Electrochemical experiments were performed with a CHI630A potentiostat (CH Instruments, Austin, Texas). A conventional three-electrode cell using a graphite rod (GRE, 4.77 mm OD) (Graphite Store), stainless-steel mesh and Ag/AgCl (KCl, 4 M) as the working, counter and reference electrodes, respectively. Before electrode modification, GREs were polished with sandpaper (P2000), cleaned by sonication in solutions of water (5 min) and of water–ethanol–acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) (10 min) and then dried in the oven. HPLC was carried out on an Agilent 1260 Infinity Series (Agilent Technologies, Palo Alto, USA) using an ACE 5 C18 column (250 × 4.6 mm). Gas chromatography was performed on a Perkin-Elmer GC with flame ionization detection (GC-FID) with DB-Wax bonded-phase fused silica capillary column (30 m × 0.25 mm I.D.) (Agilent Technologies).
:1:1) (10 min) and then dried in the oven. HPLC was carried out on an Agilent 1260 Infinity Series (Agilent Technologies, Palo Alto, USA) using an ACE 5 C18 column (250 × 4.6 mm). Gas chromatography was performed on a Perkin-Elmer GC with flame ionization detection (GC-FID) with DB-Wax bonded-phase fused silica capillary column (30 m × 0.25 mm I.D.) (Agilent Technologies).
2.3 ALDHTt expression and purification
ALDHTt from T. thermophilus was expressed and purified as described previously.182.4 Immobilization of ALDHTt on Ni-Sepharose® column
Prior to immobilization of ALDHTt the column was pre-equilibrated consisting of a series of wash steps, with 1 column volume (CV) equating to 1 ml. The colμμns used contained chelating Sepharose® and required loading of the desired metal ion, in this case Ni2+. The pre-equilibration steps were as follows, 2 CV of H2O, 0.5 CV of 0.2 M EDTA, 0.5 M NaCl pH 7, 2 CV of 0.5 M NaCl, 2 CV of H2O, 0.5 CV of 0.2 M Ni(II)SO4, 5 CV of H2O, 5 CV of 20 mM sodium acetate, 0.5 M NaCl pH 4 and finally 2 CV of binding buffer, 20 mM Tris–HCl pH 7.5, 5 mM β-mercaptoethanol, 10 mM imidazole and 200 mM NaCl. Crude cell extracts were filtered (0.45 μm) and loaded onto the 1 ml column equilibrated with binding buffer. Non-specifically bound proteins were removed with a further 5 ml wash with binding buffer. Previous purification experiments demonstrated that ALDHTt can elute at 200 mM imidazole and all unspecific proteins are removed at 10 mM imidazole.192.5 Enzymatic activity of ALDHTt-Ni-Sepharose® in a flow system
The assay solution consisted of 10 mM potassium phosphate buffer pH 8, 250 μM NAD+ and 5.7 mM hexanal in a reaction volume of 10 ml and was continuously flowed through a ALDHTt-Ni-Sepharose® reactor in a closed loop configuration. The reaction was monitored for a period of 40 min at room temperature at a flow rate of 2 ml min−1 unless otherwise stated. At pre-determined time intervals, a sample was taken and the absorbance read at 340 nm. The concentration of NADH produced over time was determined by comparison to a calibration curve at 340 nm (Fig. S1†).2.6 Optimisation of ALDHTt-Ni-Sepharose® reactor
The concentration of NAD+ used was optimized over the range 21–833 μM using a cell lysate loading of 7 ml. The cell lysate loading was then optimized over the range 0.5–12 ml using an NAD+ concentration of 250 μM. The flow rate for the biocatalytic reactor was then optimised over the range 0.5–2.0 ml min−1 using a cell lysate loading of 2 ml and a concentration of 250 μM [NAD+].2.7 Quantification of immobilized ALDHTt
ALDHTt was immobilized using 2 ml cell lysate loading. The column was subsequently washed with 10 ml of 20 mM Tris–HCl pH 7.5, 5 mM β-mercaptoethanol, 200 mM imidazole and 200 mM NaCl to elute the bound protein. The concentration of eluted protein was determined using the Nanodrop Spectrophotometer. Flow through and elution samples were also analysed by SDS-PAGE for determination of specific binding of ALDHTt.2.8 Stability of immobilized ALDH-Ni-Sepharose® reactor
ALDHTt Ni-Sepharose® was assayed as above after periods of storage (1–56 days) to determine the storage stability of the enzymatic reactor. ALDHTt-Ni-Sepharose® was stored at 4 °C in binding buffer and assayed for 30 min following storage and the residual activity reported. The recycling stability of the reactor was performed for 5 reaction cycles using hexanal as substrate followed by a 10 ml wash with binding buffer in between cycles for regeneration and removal of excess substrate and cofactor.2.9 Surface modification and immobilization of LDH on GRE
PEDOP-LDH-GRE was prepared in a solution of EDOP (10 mM) and LDH (0.75 mg ml−1) in phosphate buffer (0.1 M, pH 7). The solution was stirred at 60 rpm for 15 minutes at 20 °C. PEDOP-LDH-GRE was prepared by applying a constant potential of 0.85 V (vs. Ag/AgCl) for 300 s. The electrodes were stored in humid air at 4 °C overnight before use. Modified GREs were immersed in phosphate buffer 10 mM pH 8 for 1 h before incorporation into the reactor. The loading of enzyme loading was measured by using the Bradford assay before and after the immobilization process.2.10 PEDOP-LDH-GRE flow system set-up and operation
A flow reactor was designed to incorporate the GRE as described previously.35 Briefly, the reactor was constructed using acrylate-based material and consists of two caps, a cylinder (inner radius: 5 mm), and a graphite rod with an accessible surface area of 5.08 cm2. An Instech P720 peristaltic pump was used to pump the solution through the channel at a flow rate of 0.37 or 0.56 ml min−1. Fig. S2† shows a schematic diagram of the assembled reactor. One or more reactors were used in series as required.2.11 Preparation of glyoxyl agarose
Glyoxyl agarose was prepared as previously reported with some adaptation.39 Sepharose® CL-6B (10 g) was filtered via vacuum filtration to remove any ethanol present and washed times with 10 ml deionised water. A 2.8 ml aliquot of deionised water was added to the collected agarose and resuspensed by magnetic stirring at room temperature. A 4.8 ml aliquot of 1.7 M NaOH, 14.3 mg ml−1 sodium borohydride was added to the agarose suspension on ice, and the formation of the ether linkage was by dropwise addition of 3.4 ml of glycidol. The reaction was allowed to go to completion (18 h on ice). The carrier was filtered using vacuum filtration and washed (3 × 10 ml) with deionised water. Oxidation was initiated by the addition of 68 ml of 100 mM NaIO4 and the reaction was carried out for 2 h at room temperature. The carrier was then filtered, washed with deionised water and stored at 4 °C until further use.2.12 Immobilization of LDH on glyoxyl agarose
A 1 mg sample of LDH was incubated with 1 g of glyoxyl agarose in 10 ml of buffer (50 mM sodium carbonate, pH 10) for 3 h at 4 °C under magnetic stirring of approx. 60 rpm. Chemical reduction of the imine groups was carried out by the addition of 10 mg of NaBH4 at 4 °C under stirring for 30 min. The immobilized enzyme was filtered under vacuum, washed thoroughly with deionised water (3 × 10 ml), followed by washing (20 ml) with the assay buffer (either 100 mM sodium phosphate pH 7.5 or 10 mM potassium phosphate, pH 8). The immobilized enzyme (LDH@agarose) was stored at 4 °C until further use.2.13 Activity of LDH@agarose
The enzymatic activity of LDH@agarose was determined spectrophotometrically at 340 nm by measuring the decrease in absorbance due to the removal of NADH. The batch reaction mixture consisted of 100 mM sodium phosphate buffer, pH 7.5, 120 μM NADH, 2.3 mM sodium pyruvate and 20 mg of LDH@agarose in a final reaction volume of 3 ml. Enzymatic assays were continuously monitored for 2 min at 37 °C, with magnetic stirring at 100 rpm. A sample (1 g) of LDH@agarose was resuspended in 800 μL of assay buffer and 1 ml of the suspension was packed into an empty FPLC column. The flow reactor was operated at 2 ml min−1 for 20 min in a closed loop configuration at room temperature, using 10 ml of assay solution containing 0.12 mM NADH and 2.3 mM sodium pyruvate.2.14 Stability of immobilized LDH@agarose reactor
The activity of an LDH@agarose column was assayed after periods of storage (up to 56 days) to determine the storage stability of the enzymatic reactor. The LDH@agarose column was stored at 4 °C, room temperature and 37 °C in 10 mM potassium phosphate pH 8 and assayed for 20 min following storage and the residual activity reported. The stability of the reactor was examined for 6 reaction cycles. The activity was assayed for 20 min using sodium pyruvate and NADH followed by a 10 ml wash with 10 mM potassium phosphate pH 8 in between cycles.2.15 ALDHTt–LDH coupled bi-enzymatic immobilized reactor
The ALDHTt-Ni-Sepharose® and LDH@agarose components were connected in series and assayed using a flow rate of 2 ml min−1 at room temperature (Fig. S3†). The activity of ALDHTt was evaluated using a solution of 5.7 mM hexanal and 250 μM NAD+ in 10 mM potassium phosphate pH 8. Cofactor regeneration was realised by addition of 5.7 mM hexanal, 250 μM NAD+, 2.3 mM sodium pyruvate in 10 mM potassium phosphate pH 8. The activity of the reactor was examined using the substrates, hexanal (5.7 mM), terephthalaldehyde (0.85 mM), benzaldehyde (1.69 mM) and p-tolualdehyde (1.69 mM). Formation of the carboxylic acid products was performed for the ALDHTt reactor (40 min) and ALDHTt–LDH reactor (8 h) using GC-FID (hexanoic acid) and HPLC (TPA, benzoic acid and p-toluic acid).2.16 Quantification of carboxylic acids
Hexanoic acid was quantified using GC-FID. A Perkin-Elmer GC system with flame ionization detection (GC-FID) was used with a DB-Wax bonded-phase fused silica capillary column (30 m × 0.25 mm I.D.) (Agilent Technologies). The oven temperature profile was 60 °C (4 min), increasing (10 °C min−1) to 180 °C where it was maintained for 30 min. The injector and detector temperatures were 220 and 250 °C, respectively, while the N2 gas flow rate was 1.8 ml min−1. Hexanoic acid was extracted by liquid/liquid extraction method with dichloromethane. Tridecane was used as the internal standard.The concentrations of terephthalic, benzoic and p-toluic acids were quantified by HPLC using an Agilent 1260 Infinity Series (Agilent Technologies, Palo Alto, USA) using an ACE 5 C18 column (250 × 4.6 mm). The mobile phase, comprised of A: 0.1% phosphoric acid, and B: HPLC grade methanol (Sigma Aldrich, Ireland) (A:B, 70:30, v/v) was delivered to the column at a flow rate of 0.8 ml min−1, which yielded a column back pressure of ∼150 bar. The concentration of TPA (60 μL injections) was analysed in 50% MeOH at 30 °C using a run time of 20 min. The concentration of benzoic acid (20 μL injections) was determined at 40 °C with a run time of 40 min. The concentration of p-toluic acid (30 μL injections) was analysed in 50% MeOH at 40 °C with a run time of 75 min. Samples were filtered through 0.45 μm nylon filters prior to injection while UV detection was conducted at a wavelength of 240 nm.
3. Results and discussion
3.1 Optimisation of the ALDHTt-Ni-Sepharose® flow reactor
Immobilization techniques such as covalent binding or cross-linking can lead to decreased enzyme activity while encapsulation and entrapment methods may give rise to diffusion limitations, especially with bulky cofactors such as NAD+, reducing enzymatic activity. Affinity binding immobilization can overcome these drawbacks, as the active site of the enzyme can remain accessible, while also allowing for reversible immobilization in a specific orientation. For example, the hexa-histidine tag in ALDHTt is located at the N-terminus,18 ensuring that orientation of the enzyme on a surface can be tailored so that the active site is not involved in the immobilization process. The use of affinity immobilization using a histidine tag, can be considered as moderate in terms of its effect on the enzyme structure, with the potential advantages of higher immobilization yields and increased enzyme activity in comparison with other immobilization methods.4 Immobilization of ALDHTt from crude E. coli cell lysate expressing the enzyme was achieved via specific binding of a His-tag to Ni2+-activated Sepharose® beads in a 1 ml column. This approach resulted in catalytically active enzyme, with 50–70% conversion (of cofactor) achieved (compared to 82% in 2 h in solution) in a stable format in a flow reactor at relatively high flow rates up to 2 ml min−1. Moreover, this method allows for concomitant, selective isolation and immobilization of the expressed enzyme in one-step,17,44,45 reducing the time required from 48 to 3 h, yielding a pure, active immobilized enzyme ready for use in a packed bed reactor.The amount of NADH produced increased as the concentration of NAD+ was increased (Fig. 1A). As the cofactor is often the most expensive reagent in dehydrogenase based reactors,12 the use of lower concentrations of cofactor together with efficient regeneration systems is desirable. It is not feasible to add the cofactor in stoichiometric amounts, typically the ratio of cofactor to substrate ranges from 0.03–3%.17,46,47 A concentration of 250 μM was selected, a level that enabled the highest rate of conversion while minimizing the concentration of cofactor (Fig. 1A), with a second enzyme (LDH) used to regenerate the cofactor resulting in a cofactor–substrate ratio of 4.4% for hexanal.
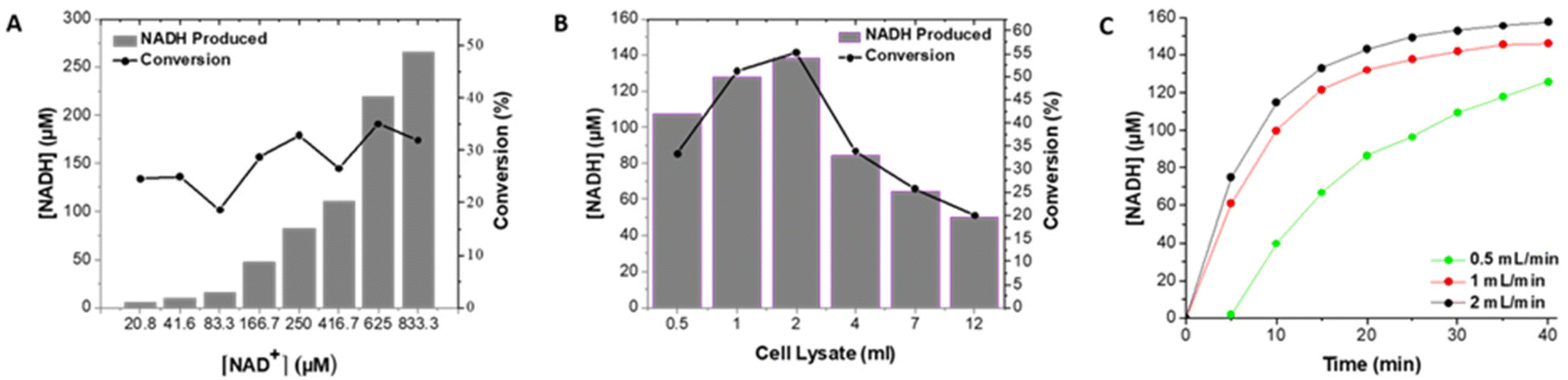 | ||
| Fig. 1 Plot of [NADH] produced and % conversion in 5.7 mM hexanal as a function of (A) [NAD+] at a flow rate of 2 ml min−1, (B) cell lysate loading; plot of [NADH] produced in 250 μM NAD+ and a flow rate of 2 ml min−1 (C) as a function of flow rate in 250 μM NAD+. Each determination was performed once for optimization purposes. | ||
The volume of cell lysate, and consequently the quantity of ALDHTt, was varied to optimise the degree of conversion of NAD+. As the loading was increased from 0.5 to 2 ml of lysate, the degree of conversion increased, with an optimum response obtained at a cell lysate loading of 2 ml. The amount of NADH decreased with increased loadings (4, 7 and 12 ml) (Fig. 1B), a decrease that can be attributed to excess loading of enzyme on the column. For loadings of 0.5 and 1 ml, the rate of production of NADH was slightly lower (Fig. S4†) as less enzyme was present. Loadings greater than 2 ml resulted in low conversion rates (<35%) in agreement with previous studies demonstrating that blocking of some of the enzyme can occur at high enzyme loadings.17,48 Using the optimum cell lysate loading of 2 ml resulted in a NADH concentration of 138 μM (conversion of 55.3%).
A bind and elute method was used to examine the amount immobilized. The enzyme was immobilized and then washed with buffer (10 ml) containing 200 mM imidazole. SDS-PAGE analysis demonstrated that specific immobilization of ALDHTt (lane 2) from the crude cell lysate had occurred at a loading of 2.5 ± 0.23 mg ml−1 without binding of contaminating E. coli proteins to the support (lanes 3 and 4, (Fig. S5†)). The column was washed with an additional aliquot (1 ml) of buffer containing 200 mM imidazole to ensure that complete elution of ALDHTr had occurred. Negligible quantities of protein were detected in this wash (30 μg, 1.2% of total immobilized enzyme). After washing of the column with an excess volume (10 ml) of buffer containing imidazole (200 mM) and removing Ni2+ with EDTA (200 mM), the column could be regenerated, recharged with metal for reuse for at least 10 immobilization cycles demonstrating that the immobilization process is reversible, allowing for multiple reuses of the support.
Increased residence time can lead to higher concentrations of product.34,49 On comparing flow rates, the highest % conversion was achieved at a flow rate of 2 ml min−1 (Fig. 1C and Table S1†) with a residence time of 0.5 min. Previous studies have demonstrated that the use of multiple reactors can increase the overall yield obtained when compared to a single reactor. ω-Transaminase was immobilized on silica monoliths and used in an enantioselective transamination reaction in continuous flow mode.50 However, in order to achieve 100% conversion of the amine, 4-bromo-α-methylbenzylamine, 8 silica monoliths in series were required, with a residence time of 200 min through the reactor. Similarly, through use of 4 NPG-lipase modified electrodes, mounted into a bespoke flow reactor, 100% conversion of p-nitrophenyl butyrate to p-nitrophenol and butyric acid was achieved over 8 flow cycles.34 Three reactor columns were combined in series to increase the overall quantity of enzyme while retaining the optimum enzyme loading of 2.5 mg ml−1 of resin. However, only a small increase in the amount of NADH was achieved, indicating that the amount of enzyme required had attained saturation levels (Fig. S6†). Cofactor regeneration was then used to further increase the amount of product.
3.2 Storage stability and reuse of the ALDHTt-Ni-Sepharose® reactor
On storage in buffer at 4 °C (Fig. 2), the immobilized biocatalyst retained good residual activity (93%, using hexanal as substrate), with 35% activity retained after 56 days. In contrast the soluble enzyme retained less than 10% activity after 48 hours. Previous reports of immobilization on Ni-activated agarose utilising affinity binding showed similar enhancements in storage stability.17,45,51 A cofactor regenerating enzymatic reactor utilising co-immobilized His-tagged ketoreductase and glucose dehydrogenase on Ni-Sepharose® remained stable retaining 100% activity following 50 days storage at 4 °C, and 50% activity after 100 days.17 Similarly metal affinity co-immobilized amine dehydrogenase and formate dehydrogenase demonstrated no loss in activity following 30 days storage.45 Transaminase immobilized on Ni activated polyvinyl magnetic microbeads retained 50% activity after 38 days storage at 4 °C, whereas the soluble enzyme lost all activity after 18 days.52 To determine the capability of reusing the reactor, the catalytic activity was monitored with intermittent washes to remove traces of substrate, cofactor and product. The catalytic activity remained stable with no decrease observed (Fig. 2C) after 5 cycles. The ability to reuse the reactor over multiple cycles with no loss in activity can be attributed to a combination of increased enzyme stability upon immobilization and to strong enzyme–support interactions that minimize leaching. The amount of ALDHTt leached in the first reaction cycle (38 μg), corresponded to 1.5% of the total amount of immobilized enzyme. As no leaching was detected in subsequent cycles, the protein leached in the first cycle likely arose from loosely bound protein that was not completely removed during the washing procedure. Previous studies on affinity immobilization in flow biocatalysis also reported miniscule rates of enzyme leaching during reactor operation.45,53 Given the stability of this reactor system, it was then combined with an enzymatic cofactor regeneration system.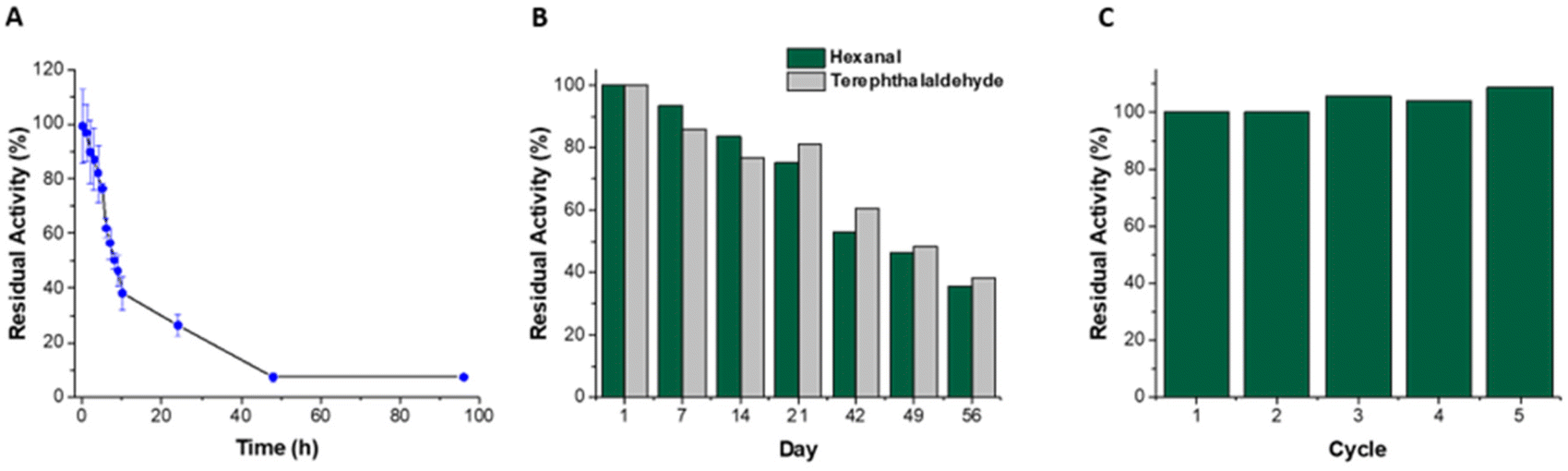 | ||
| Fig. 2 Storage stability and reuse of ALDHTt; residual activity of (A) ALDHTt in solution on storage in buffer at 4 °C, error bars are ±standard deviation obtained from triplicate measurements (some error bars are too small to be visible). (B) ALDHTt-Ni-Sepharose® reactor on storage in buffer at 4 °C, (single determination for each substrate), (C) ALDHTt-Ni-Sepharose® reactor using hexanal as substrate over 5 reaction cycles (for one reactor system). | ||
3.3 LDH as a cofactor regeneration enzyme
LDH has been previously employed for the regeneration of NAD+.54,55 It is favourable for use here as the rate of catalytic conversion of pyruvate to L-lactate by LDH is higher than the ALDHTt catalysed oxidation of aldehydes. In comparison to another commonly employed NAD+ regenerating enzyme, glutamate dehydrogenase (GluDH)14,56,57 (€5.90 per U, Sigma), LDH is less expensive (≤€0.01 per U, Sigma) and possesses higher specific activity (GluDH: approx. 20 U mg−1, LDH: approx. 800 U mg−1 (Fig. S7†)). The Michaelis–Menten constants for LDH for both pyruvate and NADH were 0.34 mM and 24 μM respectively (Fig. S7 and Table S2†). The specific activity and kcat for LDH is significantly higher than ALDHTt,19 ensuring that regeneration of NADH is not the limiting step in the overall rate of reaction. Additionally, the values of KM and kcat for NADH as a substrate of LDH are 24 μM and 3333 s−1 (Table S2†) indicating that low concentrations of NADH are sufficient for adequate rates of cofactor regeneration.When using two enzymes within a single reactor system, the selection of the temperature and buffer must be optimised to allow both enzymes to operate efficiently. Upon increasing the temperature, the activity of soluble LDH increased slightly over the temperature range of 37–50 °C (Fig. S8†). However, when LDH activity was monitored in 10 mM potassium phosphate pH 8 (ALDHTt buffer), the activity was significantly decreased when the temperature was increased above 37 °C (Fig. S8†). Notably, at 50 °C in 10 mM potassium phosphate pH 8, LDH lost all activity after 1 min of assaying indicative of enzyme stability issues. The specific activity of LDH in both buffers was comparable at 37 °C. In solution, the optimal conditions of 37 °C in 10 mM potassium phosphate pH 8 were selected to maintain the highest activity for both enzymes while retaining enzyme stability.
3.4 PEDOP-LDH-GRE immobilized flow reactor
As LDH did not possess an associated His-tag, it could not be co-immobilized on the same support as ALDHTt, a strategy that is commonly employed for cofactor regeneration systems. Different methods were explored, with varying degrees of strength of attachment to obtain active and stable enzyme. LDH was immobilized via two methods, entrapment in a polymer via electrodeposition on GREs and by covalent attachment on glyoxyl agarose. The entrapment method, using a rod coated reactor set-up, uses milder conditions in comparison to the covalent attachment method. We compared both reactors in terms of activity, stability and practical use.Previously we have shown that electrochemical methods can be used for the immobilization of enzymes in biocatalysis.34,35 LDH was immobilized on GREs in a single step using PEDOP to produce PEDOP-LDH-GRE. The activity of PEDOP-LDH-GRE was tested in a batch system prior to flow reactor operation (Fig. S9†). The enzyme loading on PEDOP-LDH-GRE was 0.36 ± 0.19 mg. PEDOP-LDH-GRE was incorporated into the reactor and the reactor system was optimized at a flow rate of 0.36 ml min−1. The conversion of NADH increased when the number of reactors was increased from 1 to 3. Using three PEDOP-LDH-GRE reactors in series was optimal, displaying conversion of 97% (Fig. 3A).
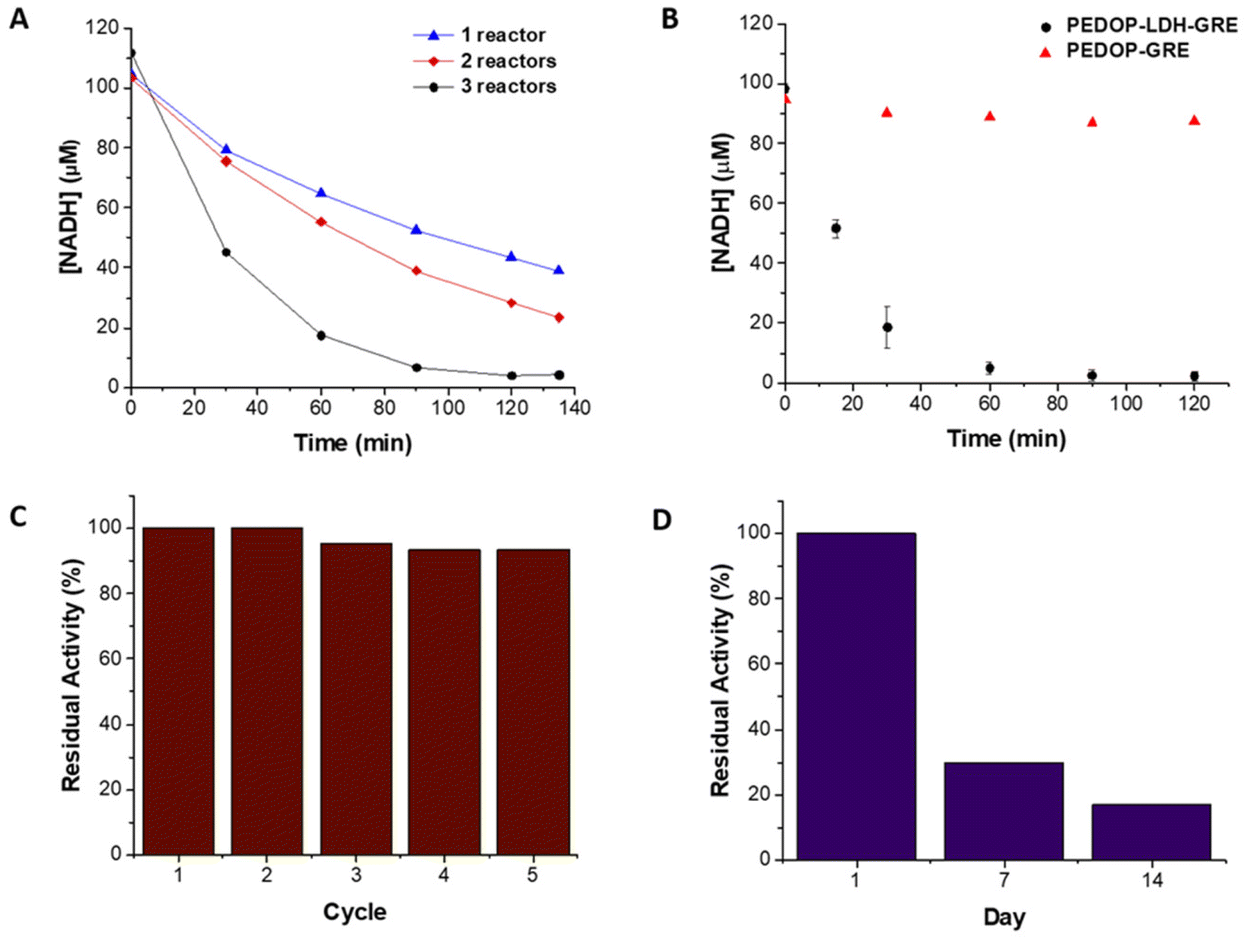 | ||
| Fig. 3 Plot of [NADH] as a function of time (A) using multiple LDH-GRE reactor stages at a flow rate of 0.36 ml min−1 (each optimization condition was performed once) and (B) in PEDOP-LDH-GRE and PEDOP-GRE flow reactors at a flow rate of 0.56 ml min−1 (error bars are ±standard deviation obtained from triplicate immobilizations and subsequent measurement, some error bars are too small to be visible); residual catalytic activity of PEDOP-LDH-GRE flow reactor (C) as a function of reaction cycle (flow rate of 0.56 ml min−1) and (D) on storage for 14 days in humid air at 4 °C. Data from recycling and storage are each from single reactors. | ||
On using control systems comprised of only GRE or of PEDOP-GRE, a decrease in the concentration of NADH was observed, indicating that adsorption (ca. 20%) of NADH had occurred on the support materials. Increasing the flow rate from 0.36 to 0.56 ml min−1 decreased the amount of adsorbed NADH to 7% on the support. When the flow rate was increased to 0.56 ml min−1, matching the flow rate used with ALDHTt-Ni-Sepharose®, 92% conversion was achieved (Fig. 3B). On reuse (5×) of the PEDOP-LDH-GRE reactor, a decrease in residual activity (to 93%) was observed at a flow rate of 0.56 ml min−1 (Fig. 3C). The residual activity was similar (91%) at a lower flow rate of 0.36 ml min−1 (Fig. S10†). In our previous study, glucose oxidase (GOx) was immobilized in Ppy electrodeposited on graphite rods.
The immobilized GOx lost 27% of its initial activity after 6 hours of continuous operation in a flow system,35 indicating that LDH immobilized in PEDOP was more stable. On examining the activity of the PEDOP-LDH-GRE in batch mode, the specific activity of immobilized LDH in PEDOP was 0.026 ± 0.004 U mg−1, compared with a value of 3.6 ± 0.3 U mg−1 for LDH@agarose, indicating that diffusional limitation of the cofactor in the polymer layer significantly reduced the activity of PEDOP-LDH-GRE. The storage stability of PEDOP-LDH-GRE was low, retaining 30 and 7% residual activity after 7 and 14 days respectively, when stored at 4 °C (Fig. 3D). The decrease in activity upon reuse and storage could be attributed to the immobilization method and enzyme stability and thus a covalent attachment method was investigated, as a more stable biocatalyst is required.
3.5 LDH@agarose immobilized flow reactor
As the activity of LDH in solution was significantly higher than that of ALDHTt, any decreases in the activity of LDH upon immobilization should not impact on the overall catalytic activity of a combined LDH–ALDHTt reactor. Using PyMOL,58 LDH possesses a number of surface accessible lysines (Fig. S11†) available for immobilization on agarose. Immobilization of enzymes on glyoxyl agarose usually results in highly active enzymes that retain 50–100% activity.41 However, a recent study reported a residual activity of 3% on immobilization of amine transaminase.39 The activity was low as immobilization was performed at room temperature using the strong reducing agent, NaBH4. The residual activity increased to 30% when immobilization was performed using the milder reagent, NaCNBH3 at 4 °C.Two concentrations of LDH (1 and 2 mg) were examined for immobilization on glyoxyl agarose. The amount of immobilized LDH was 1 and 1.55 mg, respectively (100 and 77.5% immobilization yield) (Table S3†). SEM images demonstrated that smooth micron sized beads were observed with unmodified agarose while with LDH@agarose the beads possessed uniformly rougher surfaces (Fig. S12†), indicating that complete coverage of enzyme on the particles had been achieved. On examining the activity of the LDH@agarose reactor in batch mode, the highest specific activity was achieved when 1 mg of enzyme was immobilized (3.6 ± 0.27 U mg−1) with a lower specific activity when 2 mg was used (2.62 ± 0.15 U mg−1) (Fig. S13 and Table S3†). On this basis a loading of 1 mg LDH was chosen for further study, a loading sufficient to ensure that the activity was significantly higher than that of ALDHTt (∼0.58 U mg−1 at 37 °C).19
The flow reactor was assayed in a closed loop configuration at a flow rate of 2 ml min−1 in 100 mM sodium phosphate pH 7.5 (LDH buffer) and 10 mM potassium phosphate pH 8 (ALDHTt buffer). The reaction was complete in 20 min with >99% conversion of NADH achieved in both buffers (Fig. 4). This time period was ca. 50% of that required for the ALDHTt-Ni-Sepharose® reactor and significantly shorter than the time required using PEDOP-LDH-GRE.
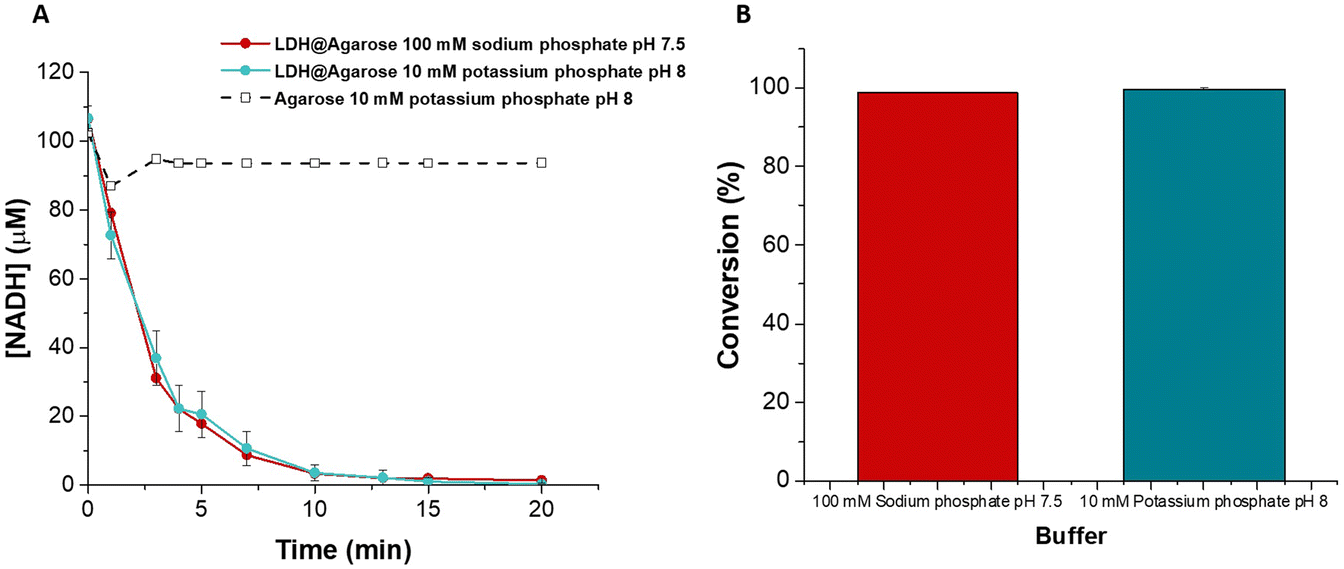 | ||
| Fig. 4 (A) Plot of the concentration of NADH as a function of time using an LDH@agarose flow reactor in different buffer solutions (error bars are ±standard deviation obtained from triplicate measurement) and (B) % conversion of NADH by the LDH@agarose reactor in different buffer solutions after 20 min. Error bars (±std. dev.) are included but are too small to be visible. | ||
While both LDH reactors showed close to complete conversion of NADH, the covalent immobilization method was superior in terms of stability and reuse. Covalent binding immobilization on agarose generally results in improvements in the stability of enzymes, allowing for very good levels of reuse of the reactors.42,59,60 A β-xylosidase biocatalyst on glyoxyl agarose support run in batch mode was used for 10 reaction cycles without any decrease in conversion and enzyme activity.60 In a previous report, when LDH was immobilized on glyoxyl agarose, no decrease in the conversion of pyruvate to L-lactate was observed after 15 reaction cycles.42 The storage stability of the biocatalyst was not reported, nor was the biocatalyst used in a continuous flow system. Similarly, a xylanase-glyoxyl agarose batch reactor could be reused for 10 cycles without any decrease in residual activity.59 While this system was used in a continuous flow packed bed reactor, its stability was not reported. Here, LDH@agarose was reused for up to 6 cycles, retaining full activity upon reuse (Fig. 5).
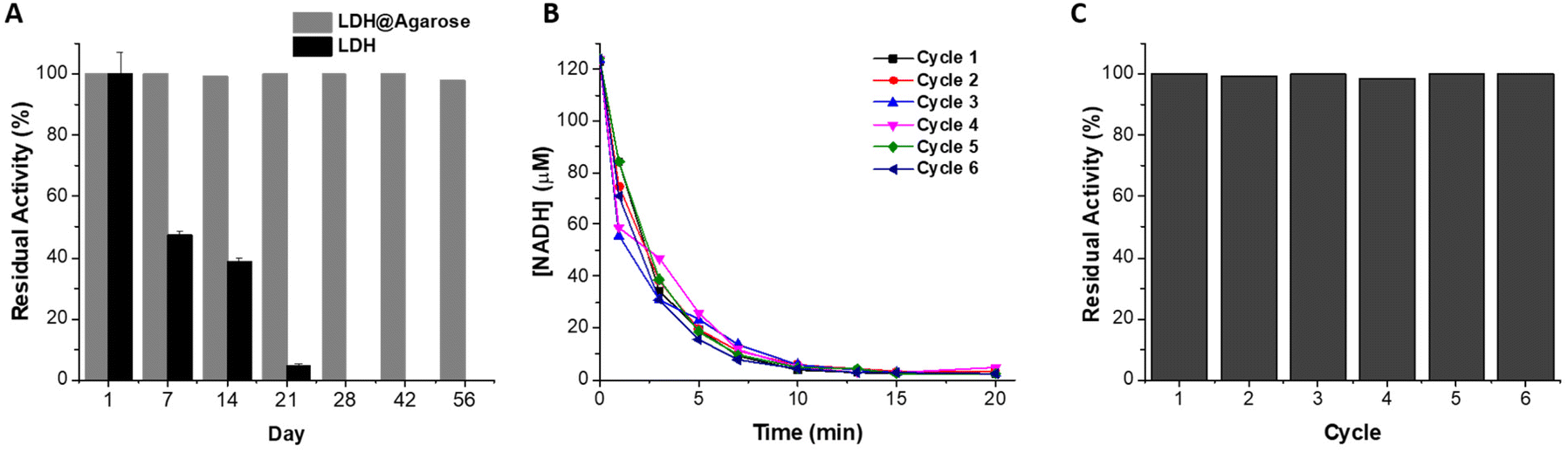 | ||
| Fig. 5 (A) Plot of residual activity of LDH (error bars are ±standard deviation obtained from triplicate measurements) and LDH@agarose as a function of storage time in 10 mM potassium phosphate pH 8 at 4 °C, (B) plot of the concentration of NADH versus time for 6 consecutive reaction cycles and (C) residual activity of LDH@agarose reactor for 6 reaction cycles. Data from recycling and storage are each from single reactors. | ||
The immobilization process also resulted in a significantly increased storage stability when compared to PEDOP-LDH-GRE. Over a 56-day period at 4 °C, 98% residual activity was maintained, whereas only 47% and 5% residual activity was observed after 7 and 21 days, respectively, for LDH in solution (Fig. 5). Excellent storage stability was observed at room temperature and 37 °C, maintaining residual activities of 99.4 and 98.7%, respectively after 28 days storage (Fig. S14†). To the best of our knowledge this is the most stable system yet reported in terms of the reuse and storage stability of LDH in a flow reactor. As mentioned previously, continuous processing in flow reactors can lead to enzyme leaching, however covalent binding to the support via multipoint attachment of the tetrameric LDH enabled increased stability and recyclability.
3.6 ALDHTt–LDH co-immobilized enzymatic reactor
The oxidation of the substrates, hexanal, benzaldehyde, p-tolualdehyde and terephthalaldehyde in the ALDHTt reactor was examined. The increase in the concentration of NADH was monitored for all substrates and resulted in conversions of 58–72% (Fig. 6), demonstrating the broad substrate range of the enzyme. The production of carboxylic acids was determined by GC and HPLC and presented in Table 1 (discussed below).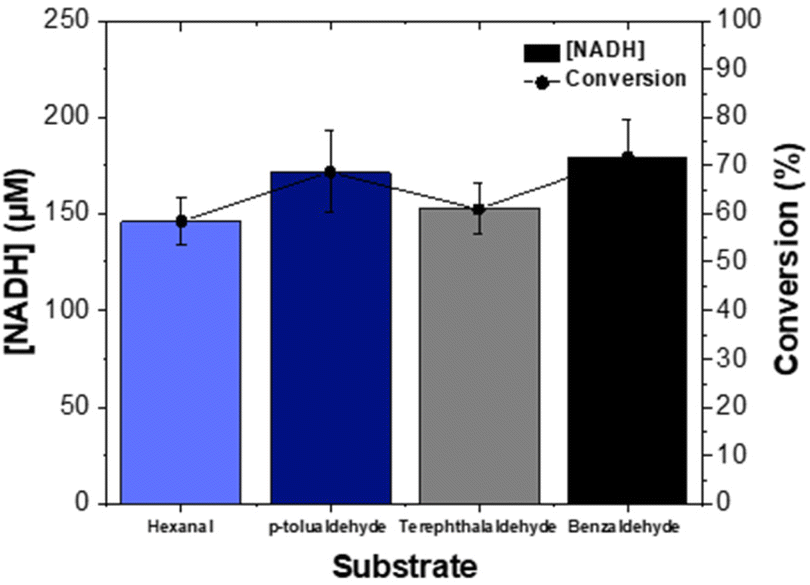 | ||
| Fig. 6 Conversion of NAD+ to NADH as a function of substrate; hexanal (5.7 mM), p-tolualdehyde (1.69 mM), terephthalaldehyde (0.85 mM) and benzaldehyde (1.69 mM) utilising 250 μM NAD+ and a flow rate of 2 ml min−1. Error bars are ±standard deviation obtained from triplicate measurements. | ||
| Substrate (mM) | Product | Method | ALDHTt reactor (40 min) | ALDHTt–LDH reactor (8 h) | ||
|---|---|---|---|---|---|---|
| [Product] (μM) | Conv. (%) | [Product] (μM) | Conv. (%) | |||
| a Not detected. | ||||||
| Hexanal (5.7) | Hexanoic acid | GC | 130 | 2.27 | 907 | 15.9 |
| Benzaldehyde (1.69) | Benzoic acid | HPLC | 168 | 9.96 | 965 | 57.1 |
| p-Tolualdehyde (1.69) | p-Toluic acid | HPLC | 223 | 13.2 | 922 | 54.6 |
| Terephthalaldehyde (0.85) | TPAA | HPLC | 142 | 16.7 | NDa | NDa |
The ALDHTt-Ni-Sepharose® and LDH@agarose reactors were combined to develop a bi-enzymatic reactor for the oxidation of aldehydes to the corresponding carboxylic acid products with regeneration of the cofactor. In a control experiment, hexanal and NAD+ were added to the reactor to monitor the increase in the amount of NADH produced with time in the absence of pyruvate. The concentration of NADH reached a plateau (59% conversion) after 40 min (Fig. 7), consistent with the data obtained with the single ALDHTt-Ni-Sepharose® reactor described above. As expected, this demonstrates that immobilized LDH did not interfere with the ALDHTt reaction. On addition of hexanal, NAD+ and pyruvate, no accumulation of NADH was observed for a period of up to 3 h, indicative of highly efficient cofactor regeneration. Additionally, when the ALDHTt–LDH reactor was operated with benzaldehyde as an example substrate, no accumulation was evident for a period of 8 h (Fig. S15†).
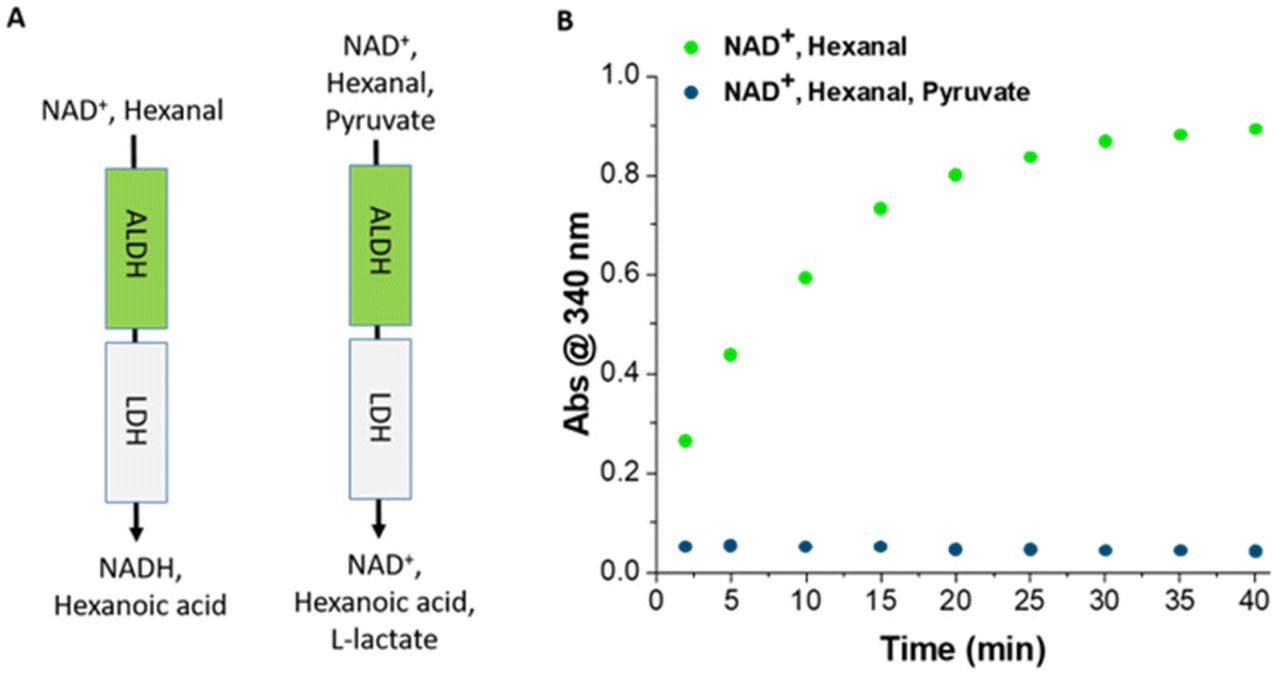 | ||
| Fig. 7 (A) Schematic diagram of ALDHTt–LDH@agarose coupled reactor and (B) plot of A340nm as a function of time in the presence and absence of cofactor recycling. One coupled reactor was prepared and operated for each condition. | ||
The stability of a co-immobilized system is determined by the least stable component. Re-use of the ALDHTt–LDH reactor did not show significant changes in activity, but after a period of 7 days, a decrease in activity arising from the instability of ALDHTt-Ni-Sepharose® would be expected. Alanine dehydrogenase (AlaDH) from Bacillus subtilis immobilized on glyoxyl agarose, and formate dehydrogenase (FDH) from Candida boidinii immobilized by covalent attachment to polyethylenimine coating the agarose support were combined in a reactor for the production of L-alanine with regeneration of NADH.61 Immobilized AlaDH retained 100% of its catalytic activity after eight cycles, whereas immobilized FDH retained only 20% activity, limiting the stability of the reactor.61 When the activities of ALDHTt-Ni-Sepharose® and LDH@agarose were examined individually, no decrease in activity was observed after constant operation for 2 h, demonstrating that continuous operation using both enzymes was feasible. Moreover, after 24 h of continuous operation of the ALDHTt–LDH reactor no accumulation of NADH was observed. LDH@agarose was also run in an open loop configuration for 24 h displaying complete conversion of cofactor.
The oxidation of aldehydes was examined using the combined reactor and a low concentration (250 μM) of NAD+. Significant increases in the concentrations of carboxylic acid products were obtained following 8 h continuous operation (Table 1), with no further increases when the reactor was run for 24 h. A 4 to 7-fold increase in production was achieved with cofactor regeneration, in its absence, the maximum conversion of product was 17%. In the synthesis of benzoic acid with NAD+ regeneration, 57% conversion was obtained. In comparison (Geotrichum candidum, GcALDH), immobilized on organic–inorganic nanocrystals resulted in 61% conversion62 (10 mM substrate and cofactor) and a 70% conversion when immobilized on montmorillonite (2.5 mM substrate and cofactor),63 demonstrating the efficiency of the combined by ALDHTt and LDH reactor described here.
Oxidation of terephthalaldehyde by ALDHTt resulted in the production of terephthalaldehydic acid (TPAA) as well as TPA. A previous study demonstrated that the ALDH catalysed rate of oxidation of terephthalaldehyde was much higher than the rate of oxidation of the aldehydic acid intermediate,64 indicating that the selective oxidation of terephthalaldehyde is possible.64 Selective oxidation of dialdehydes to aldehydic acids is of interest due to the high reactivity of the aldehyde group, rendering synthesis of the intermediate difficult. Following operation of the ALDHTt-Ni-Sepharose® reactor for 40 min, only TPAA was produced, demonstrating that the system was capable of preferentially oxidizing a single aldehyde functional group in a substrate containing two aldehyde groups. After 8 h of operation, both intermediate and product were present due to further oxidation of TPAA. Different systems have shown varying results in the oxidation of terephthalaldehyde, to selectively produce TPAA or a mixture TPAA and TPA,64–66 that may be attributed to the differing affinities of the enzyme for the dialdehyde and aldehydic acid. ALDH from Geotrichum candidum showed selective oxidation to TPAA, while TPA was not detected in the reaction,64 whereas Serratia liquefaciens whole cells produced 177 and 87 mM TPAA and TPA, respectively, from a 330 mM solution of terephthalaldehyde.65
4. Conclusion
A coupled bi-enzymatic flow reactor was realised utilising immobilized ALDHTt and LDH for the production of carboxylic acids while employing cofactor regeneration. The use of ALDHTt eliminates the need for high temperatures and pressures normally required in the synthesis of these products. ALDHTt was efficiently immobilized via affinity interactions, using the associated N-terminal His-tag, on Ni-activated Sepharose®, displaying good conversion and storage stability with recycling abilities realised for at least 5 cycles. In order to increase productivity, LDH was used in a cofactor regeneration system. Two immobilization techniques were analysed; enzyme entrapment in electropolymerized EDOP on a graphite rod electrode and covalent attachment on glyoxyl agarose. On examining the activity of immobilized enzymes, the specific activity of LDH@agarose (3.6 ± 0.27 U mg−1) was considerably higher than that of PEDOP-LDH-GRE (0.026 ± 0.004 U mg−1), demonstrating that diffusional limitations associated with the PEDOP film restricted the enzymatic activity observed. While both systems demonstrated high levels of NADH conversion and could be reused, the LDH@agarose reactor was superior due to a higher conversion rate and storage stability (98% following 56 days of storage at 4 °C). Combining the two enzyme reactors enabled efficient NAD+ regeneration within a flow system, with no accumulation of NADH evident for at least 24 h of continual use. This allowed for increased productivity (up to 7-fold) of the reactor, for the production of hexanoic, benzoic and p-toluic acid. The selective oxidation of terephthalaldehyde at short reaction times was possible, demonstrating the utility of the system.Author contributions
The manuscript was written with contributions from all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge funding from the European Union's Horizon 2020 research and innovation program, OYSTER (grant agreement no. 760827), the Marie Skłodowska-Curie MSCA-ITN “ImplantSens” (813006), the Science Foundation Ireland Research Centre for Pharmaceuticals (grant no. 12/RC/2275), The Higher Education Authority, Ireland and the Department of Chemical Sciences, University of Limerick. We thank Bridget Hogan, Department of Chemical Sciences, for the use of HPLC and GC columns.References
- G. Hughes and J. C. Lewis, Chem. Rev., 2018, 118, 1–3 CrossRef CAS PubMed.
- E. D. Yushkova, E. A. Nazarova, A. V. Matyuhina, A. O. Noskova, D. O. Shavronskaya, V. V. Vinogradov, N. N. Skvortsova and E. F. Krivoshapkina, J. Agric. Food Chem., 2019, 67, 11553–11567 CrossRef CAS PubMed.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- R. A. Sheldon, A. Basso and D. Brady, Chem. Soc. Rev., 2021, 50, 5850–5862 RSC.
- M. Bilal, Y. Zhao, S. Noreen, S. Z. H. Shah, R. N. Bharagava and H. M. Iqbal, Biocatal. Biotransform., 2019, 37, 159–182 CrossRef CAS.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- A. Basso and S. Serban, Mol. Catal., 2019, 479, 110607 CrossRef CAS.
- D. Singh and N. Gupta, Biologia, 2020, 75, 1183–1193 CrossRef CAS.
- W. Böhmer, T. Knaus and F. G. Mutti, ChemCatChem, 2018, 10, 731–735 CrossRef PubMed.
- W. Liu and P. Wang, Biotechnol. Adv., 2007, 25, 369–384 CrossRef CAS PubMed.
- J. B. Jones, D. W. Sneddon, W. Higgins and A. J. Lewis, J. Chem. Soc., Chem. Commun., 1972, 856–857 RSC.
- Y. S. Lee, R. Gerulskis and S. D. Minteer, Curr. Opin. Biotechnol., 2022, 73, 14–21 CrossRef CAS PubMed.
- A. Singh, R. K. Yadav, U. Yadav and T. W. Kim, Photochem. Photobiol., 2022, 98, 412–420 CrossRef CAS PubMed.
- H. K. Chenault, E. S. Simon and G. M. Whitesides, Biotechnol. Genet. Eng. Rev., 1988, 6, 221–270 CrossRef CAS PubMed.
- S. Mordhorst and J. N. Andexer, Nat. Prod. Rep., 2020, 37, 1316–1333 RSC.
- M. Yuan, M. J. Kummer, R. D. Milton, T. Quah and S. D. Minteer, ACS Catal., 2019, 9, 5486–5495 CrossRef CAS.
- M. Plž, T. Petrovičová and M. Rebroš, Molecules, 2020, 25, 4278 CrossRef PubMed.
- K. Hayes, M. Noor, A. Djeghader, P. Armshaw, T. Pembroke, S. Tofail and T. Soulimane, Sci. Rep., 2018, 8, 1–14 CAS.
- K. Shortall, E. Durack, E. Magner and T. Soulimane, Cells, 2021, 10, 3535 CrossRef CAS PubMed.
- R. A. Tomás, J. C. Bordado and J. F. Gomes, Chem. Rev., 2013, 113, 7421–7469 CrossRef PubMed.
- S. M. Jo, F. R. Wurm and K. Landfester, Angew. Chem., Int. Ed., 2021, 60, 7728–7734 CrossRef CAS PubMed.
- W. Liu, S. Zhang and P. Wang, J. Biotechnol., 2009, 139, 102–107 CrossRef CAS PubMed.
- C. Wiles and P. Watts, Green Chem., 2012, 14, 38–54 RSC.
- A. I. Benítez-Mateos, M. L. Contente, D. R. Padrosa and F. Paradisi, React. Chem. Eng., 2021, 6, 599–611 RSC.
- D. Czerwińska-Główka, M. Skonieczna, A. Barylski, S. Golba, W. Przystaś, E. Zabłocka-Godlewska, S. Student, B. Cwalina and K. Krukiewicz, Bioelectrochemistry, 2022, 144, 108030 CrossRef PubMed.
- Ö. Türkarslan, S. K. Kayahan and L. Toppare, Sens. Actuators, B, 2009, 136, 484–488 CrossRef.
- P. Bartlett and R. Whitaker, J. Electroanal. Chem. Interfacial Electrochem., 1987, 224, 37–48 CrossRef CAS.
- J.-C. Vidal, E. Garcia and J.-R. Castillo, Biosens. Bioelectron., 1998, 13, 371–382 CrossRef CAS.
- C. Debiemme-Chouvy and T. T. M. Tran, Electrochem. Commun., 2008, 10, 947–950 CrossRef CAS.
- C. L. Gaupp, K. Zong, P. Schottland, B. C. Thompson, C. A. Thomas and J. R. Reynolds, Macromolecules, 2000, 33, 1132–1133 CrossRef CAS.
- M. A. Komkova, A. K. Orlov, A. A. Galushin, E. A. Andreev and A. A. Karyakin, Anal. Chem., 2021, 93, 12116–12121 CrossRef CAS PubMed.
- A. Ghoorchian, Z. Amouzegar, M. Moradi, S. Khalili, A. Afkhami, T. Madrakian and M. Ahmadi, Conductive Polymers in Analytical Chemistry, ACS Publications, 2022, pp. 165–184 Search PubMed.
- A. Kausaite-Minkstimiene, A. Kaminskas, A. Popov, A. Ramanavicius and A. Ramanaviciene, Sci. Rep., 2021, 11, 1–11 CrossRef PubMed.
- X. Xiao, T. Siepenkoetter, R. Whelan, U. Salaj-Kosla and E. Magner, J. Electroanal. Chem., 2018, 812, 180–185 CrossRef CAS.
- S. Arshi, X. Xiao, S. Belochapkine and E. Magner, ChemElectroChem, 2022, 9(17), e202200319 CrossRef CAS PubMed.
- P. Zucca, R. Fernandez-Lafuente and E. Sanjust, Molecules, 2016, 21, 1577 CrossRef PubMed.
- V. Grazu, L. Betancor, T. Montes, F. Lopez-Gallego, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2006, 38, 960–966 CrossRef CAS.
- J. C. Janson and J. Å. Jönsson, Protein Purification: Principles, High Resolution Methods, and Applications, 2011, pp. 23–50 Search PubMed.
- R. Semproli, G. Vaccaro, E. E. Ferrandi, M. Vanoni, T. Bavaro, G. Marrubini, F. Annunziata, P. Conti, G. Speranza and D. Monti, ChemCatChem, 2020, 12, 1359–1367 CrossRef CAS.
- Y. Asanomi, H. Yamaguchi, M. Miyazaki and H. Maeda, Molecules, 2011, 16, 6041–6059 CrossRef CAS PubMed.
- C. Mateo, J. M. Palomo, M. Fuentes, L. Betancor, V. Grazu, F. López-Gallego, B. C. Pessela, A. Hidalgo, G. Fernández-Lorente and R. Fernández-Lafuente, Enzyme Microb. Technol., 2006, 39, 274–280 CrossRef CAS.
- E. Jackson, F. López-Gallego, J. Guisan and L. Betancor, Process Biochem., 2016, 51, 1248–1255 CrossRef CAS.
- T. Knaus, V. Tseliou, L. D. Humphreys, N. S. Scrutton and F. G. Mutti, Green Chem., 2018, 20, 3931–3943 RSC.
- T. Ivannikova, F. Bintein, A. Malleron, S. Juliant, M. Cerutti, A. Harduin-Lepers, P. Delannoy, C. Augé and A. Lubineau, Carbohydr. Res., 2003, 338, 1153–1161 CrossRef CAS PubMed.
- R. D. Franklin, J. A. Whitley, A. A. Caparco, B. R. Bommarius, J. A. Champion and A. S. Bommarius, Chem. Eng. J., 2021, 407, 127065 CrossRef CAS.
- T. Petrovičová, K. Markošová, Z. Hegyi, I. Smonou, M. Rosenberg and M. Rebroš, Catalysts, 2018, 8, 168 CrossRef.
- D. Kalaitzakis, J. D. Rozzell, S. Kambourakis and I. Smonou, Org. Lett., 2005, 7, 4799–4801 CrossRef CAS PubMed.
- V. Gascón, C. Carucci, M. B. Jiménez, R. M. Blanco, M. Sánchez-Sánchez and E. Magner, ChemCatChem, 2017, 9, 1182–1186 CrossRef.
- K. Szymańska, M. Pietrowska, J. Kocurek, K. Maresz, A. Koreniuk, J. Mrowiec-Białoń, P. Widłak, E. Magner and A. Jarzębski, Chem. Eng. J., 2016, 287, 148–154 CrossRef.
- L. Van den Biggelaar, P. Soumillion and D. P. Debecker, Catalysts, 2017, 7, 54 CrossRef.
- A. A. Halim, N. Szita and F. Baganz, J. Biotechnol., 2013, 168, 567–575 CrossRef PubMed.
- S. M. Dold, L. Cai and J. Rudat, Eng. Life Sci., 2016, 16, 568–576 CrossRef.
- W. Böhmer, T. Knaus, A. Volkov, T. K. Slot, N. R. Shiju, K. E. Cassimjee and F. G. Mutti, J. Biotechnol., 2019, 291, 52–60 CrossRef PubMed.
- A. S. Demir, F. N. Talpur, S. B. Sopaci, G.-W. Kohring and A. Celik, J. Biotechnol., 2011, 152, 176–183 CrossRef CAS PubMed.
- H. K. Chenault and G. M. Whitesides, Bioorg. Chem., 1989, 17, 400–409 CrossRef CAS.
- S. Ren, Z. Wang, M. Bilal, Y. Feng, Y. Jiang, S. Jia and J. Cui, Int. J. Biol. Macromol., 2020, 155, 110–118 CrossRef CAS PubMed.
- A. Weckbecker, H. Gröger and W. Hummel, Biosystems Engineering I, 2010, pp. 195–242 Search PubMed.
- C. DeLano , Scientific LLC, Palo Alto, USA, unpublished work.
- C. C. Aragon, A. F. Santos, A. I. Ruiz-Matute, N. Corzo, J. M. Guisan, R. Monti and C. Mateo, J. Mol. Catal. B: Enzym., 2013, 98, 8–14 CrossRef CAS.
- F. A. Corradini, T. S. Milessi, V. M. Gonçalves, R. Ruller, C. R. Sargo, L. A. Lopes, T. C. Zangirolami, P. W. Tardioli, R. C. Giordano and R. L. Giordano, Enzyme Microb. Technol., 2021, 145, 109725 CrossRef CAS PubMed.
- S. Velasco-Lozano, E. S. da Silva, J. Llop and F. López-Gallego, ChemBioChem, 2018, 19, 395–403 CrossRef CAS PubMed.
- K. Ogura, M. A. Hawari and T. Matsuda, Enzyme Microb. Technol., 2021, 150, 109866 CrossRef PubMed.
- O. Argaman, Z. B.-B. Zelas, A. Fishman, G. Rytwo and A. Radian, Appl. Clay Sci., 2021, 212, 106216 CrossRef CAS.
- T. Hoshino, E. Yamabe, M. A. Hawari, M. Tamura, S. Kanamaru, K. Yoshida, A. A. Koesoema and T. Matsuda, Tetrahedron, 2020, 76, 131387 CrossRef CAS.
- K. Mitsukura, Y. Sato, T. Yoshida and T. Nagasawa, Biotechnol. Lett., 2004, 26, 1643–1648 CrossRef CAS PubMed.
- H. Y. Jia, M. H. Zong, G. W. Zheng and N. Li, ChemSusChem, 2019, 12, 4764–4768 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01536j |
| This journal is © The Royal Society of Chemistry 2023 |