
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aerobic oxidation of alcohols using a slurry loop membrane reactor†
Baldassarre
Venezia
 and
Asterios
Gavriilidis
*
and
Asterios
Gavriilidis
*
Department of Chemical Engineering, University College London, Torrington Place, London WC1E 7JE, UK. E-mail: a.gavriilidis@ucl.ac.uk
First published on 28th June 2023
Abstract
The use of molecular oxygen is unquestionably the green path to the selective oxidation of alcohols to aldehyde and ketones. However, this reaction class poses safety problems associated with mixing oxygen with organic substrates. Continuous membrane reactors offer an attractive solution, owing to their ability to keep the oxygen phase separated from the liquid substrate, while controlling the dosing of oxygen during reaction. In this work, we demonstrate a slurry loop membrane reactor for continuous oxidations as well as hydrogenations. The catalyst slurry was circulated around a loop, to which a saturator containing a flat Teflon AF-2400 membrane was connected, along with a crossflow filter to keep the catalyst particles within the loop. Under a recycle flowrate 100 times higher than the inlet, the residence time distribution was found to be comparable to that of an ideal CSTR. A remarkably high kLa of 1.2 s−1 was achieved under a moderate specific power input of 2.4 kW m−3 during styrene hydrogenation. Continuous aerobic oxidations of various primary and secondary alcohols were carried out for 6–7 h at 90–120 °C and 2–6 bar, using a 1 wt% Au-Pd/TiO2 powder catalyst, leading to conversions between 17% and 75%. The reactor could also be operated in batch mode, achieving higher conversions, while scaled-up operations produced aldehyde yields of 0.4–19 g with only 88 mg of catalyst. Overall, the slurry loop membrane reactor provides significant advantages in terms of catalyst usage and process safety for aerobic oxidations.
Introduction
Clean and atom-efficient organic syntheses are becoming pressing requirements in industrial chemical transformations amid current stringent ecological standards.1–5 The selective oxidation of alcohols to aldehydes and ketones represents an important synthetic route that introduces carbonyl functionalities for the manufacture of fragrances and valuable intermediates for fine chemicals.6–9 Toxic inorganic oxidants have been traditionally employed to achieve this transformation, including chromium and manganese dioxide, however, they are generally expensive and produce large amounts of waste.10,11In recent years, there has been an attempt to replace these stochiometric oxidants with molecular oxygen and selective catalysts, as oxygen is inexpensive, readily available and its reactions exhibit a high atom economy.12–21 However, while being used in academic research, there is hesitation to use oxygen in industrial liquid phase oxidations, owing to the safety concern of mixing it with flammable organics.22,23 To drive a paradigm shift towards greener industrial oxidation of alcohols, this process safety hurdle needs to be overcome.
The recent emergence and increase in popularity of flow chemistry has provided researchers with different tools to perform hazardous reactions in a controlled fashion,24–28 including aerobic oxidations.29–32 Among flow reactors, continuous membrane reactors have the advantage of separating the gaseous oxidant from the organic phase, allowing for a controlled dosing of molecular oxygen, and this approach has been successfully adopted and reported in recent works.33–36 Teflon amorphous fluoroplastic (AF) is a material with remarkable properties, including high permeability to light gases, chemical inertness and a high liquid breakthrough pressure.37,38 First introduced by the group of Ley,39 Teflon AF has been employed as a gas saturator in several continuous reactors, for gas-mediated reactions.40–46
As stated in the twelve guiding principles of green chemistry, catalysis is an important tool to achieve sustainable syntheses,4,47 and unlike their homogeneous counterparts, heterogeneous catalysts have the extra benefit of being easily recovered and reused after reaction, meeting the recyclability objective of green chemistry.13,47 A common way of using solid catalysts is in high-pressure packed-bed reactors, which have been extensively used in aerobic oxidation of alcohols.48–50 We have previously demonstrated the use of a membrane packed-bed reactor for the continuous and scalable aerobic oxidation of benzyl alcohol, using a flat Teflon AF-2400 membrane and 1 wt% Au-Pd/TiO2 as a catalyst.46 However, the need for large catalyst particles in order to reduce pressure drop inevitably leads to a reduction in the catalyst efficiency. Moreover, packed-bed reactors can suffer from non-uniform temperature distributions which can cause degradation of temperature-sensitive functional groups in pharmaceutical intermediates. Mo et al. demonstrated a thin membrane reactor for aerobic oxidations and hydrogenations in flow.51 The Teflon AF-2400 membrane was sandwiched between two porous carbon cloth layers, where the gas and the liquid were flowing separately. For heterogeneous reactions the authors embedded a catalyst in the carbon cloth and scale-up was achieved by stacking membranes one over another. The catalyst amount was limited by the surface area available on the carbon cloth and high pressures had to be employed to achieve high yields.
The use of fine catalyst particles suspended in the reaction medium can represent an alternative and attractive solution. In fact, slurry reactors offer better temperature control, moderate pressure drops, a boost in catalyst activity owing to the reduction of diffusional resistances, and enhanced mixing properties between the catalyst particles and the liquid.52–54 In our previous work, we demonstrated a slurry loop reactor with a tubular membrane saturator for the aerobic oxidation of benzyl alcohol in flow.55 The catalyst slurry was continuously pumped in a loop, to which a tubular Teflon AF-2400 membrane was connected and provided oxygen for the reaction. It operated safely and effectively, and achieved similar turnover frequencies to those achieved with a conventional autoclave reactor. Furthermore, it showed superior performance compared to a flat membrane packed-bed reactor, using only small amounts of catalyst and moderate oxygen pressures.46 However, safe and broadly applicable continuous flow reactor designs for gas-liquid-solid reactions are still required for small-scale applications.
In this work, we present a novel slurry loop membrane reactor, with a Teflon AF-2400 membrane film as a saturator and a plug-and-play configuration, which can be used for heterogeneous aerobic oxidations of various primary and secondary alcohols and hydrogenation reactions using different powder catalysts.
Materials and methods
Slurry loop membrane reactor design and setup
The slurry loop membrane reactor (SLMR) was comprised of three main units: a gas saturator, a recirculation pump and a crossflow filter. These were connected in a loop, and catalyst powder together with liquid substrate and reaction products circulated continuously. The heart of the loop was the saturator (see Fig. 1). Here, the slurry flowed in a serpentine channel above which a flat Teflon AF-2400 membrane (0.023′′ thickness, Biogeneral) was placed, separating the slurry flow from the gaseous reactant pressurised on the other side.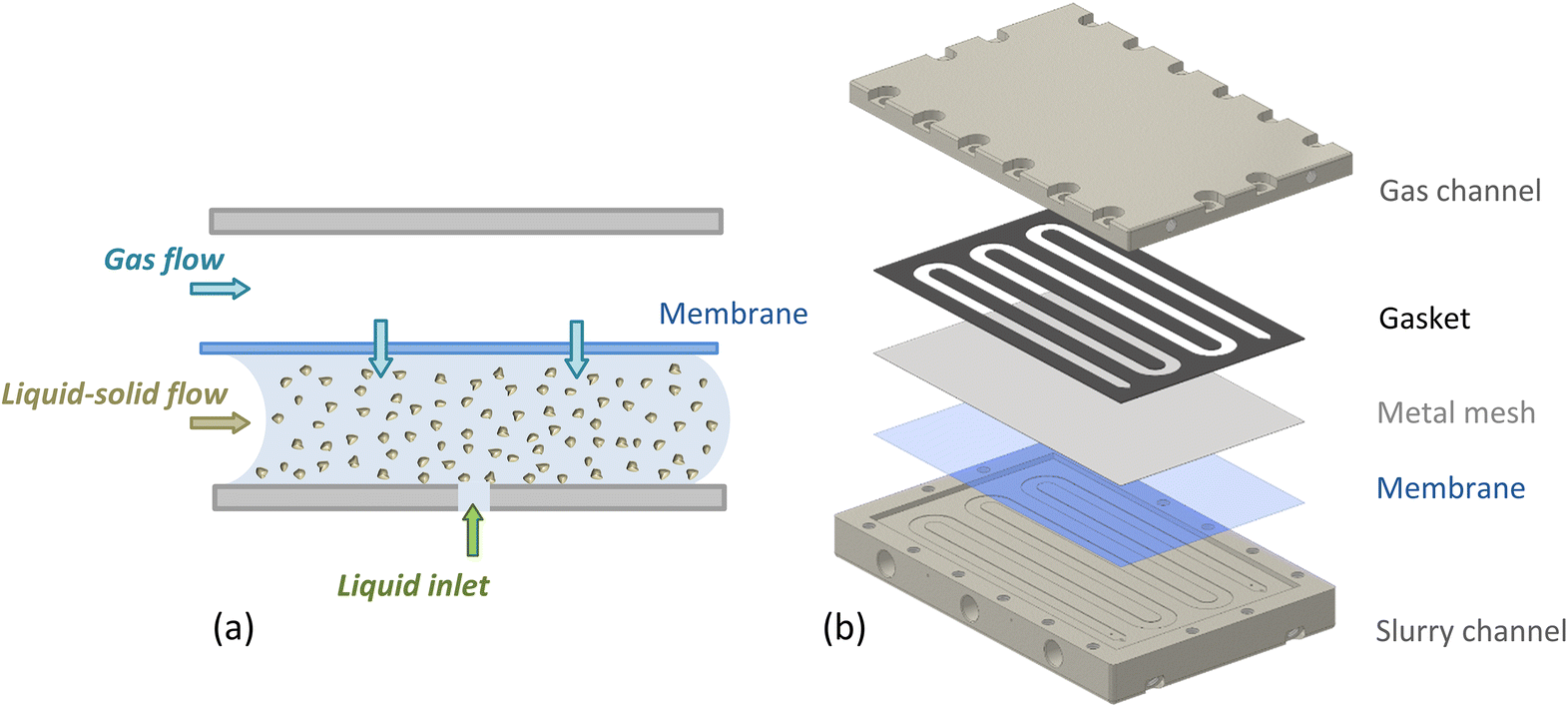 | ||
| Fig. 1 (a) Schematic showing the Teflon AF-2400 membrane inside the saturator, separating the catalyst slurry from the gas phase and allowing controlled dosing of the gas during reaction. (b) Exploded assembly of the saturator unit (see ESI for details†). | ||
The slurry channel had a depth and width of 0.3 and 6 mm respectively, and a length of 808 mm, with 4850 mm2 surface area exposed to the membrane. Fig. 1b shows the saturator assembly, comprising of a membrane, a 0.05 mm thick mesh (Industrial Netting) made of 304 stainless steel for mechanical support and a 0.5 mm-thick Kalrez® gasket (DuPont), sealing the liquid and the gas channel (see ESI for details†). The membrane's selective permeability to gases allowed gas molecules to diffuse through the membrane and into the slurry mixture where they reacted on the flowing catalyst particles. The saturator also included an inlet port for the continuous feeding of liquid substrate during continuous operation.
The schematic of the setup is shown in Fig. 2. Connected to the saturator, the recirculation pump was a micro annular gear pump (mzr-4605, HNP Mikrosysteme) that pumped the slurry around the loop at a flowrate ranging between 5 and 20 mL min−1. A crossflow filter was connected between the pump and the saturator and enabled the continuous withdrawal of pure liquid during continuous reactions, while keeping the catalyst flowing inside the loop. Polyfluoroalkoxy alkane (PFA) tubing (1/16′′ OD × 1 mm ID, Idex) provided connection to these three units. The details on the design of the saturator and the filter are reported in the ESI.† The total volume of the slurry flowing in the loop reactor was 1.4 cm3, which could be increased to 8.8 cm3 for scaled-up reactions that were performed to achieve higher productivity. This was done by adding extra tubing (PFA, 1/8′′ OD × 0.062′′ ID, volume 7.4 cm3) to the loop (“Loop extension” in Fig. 2).
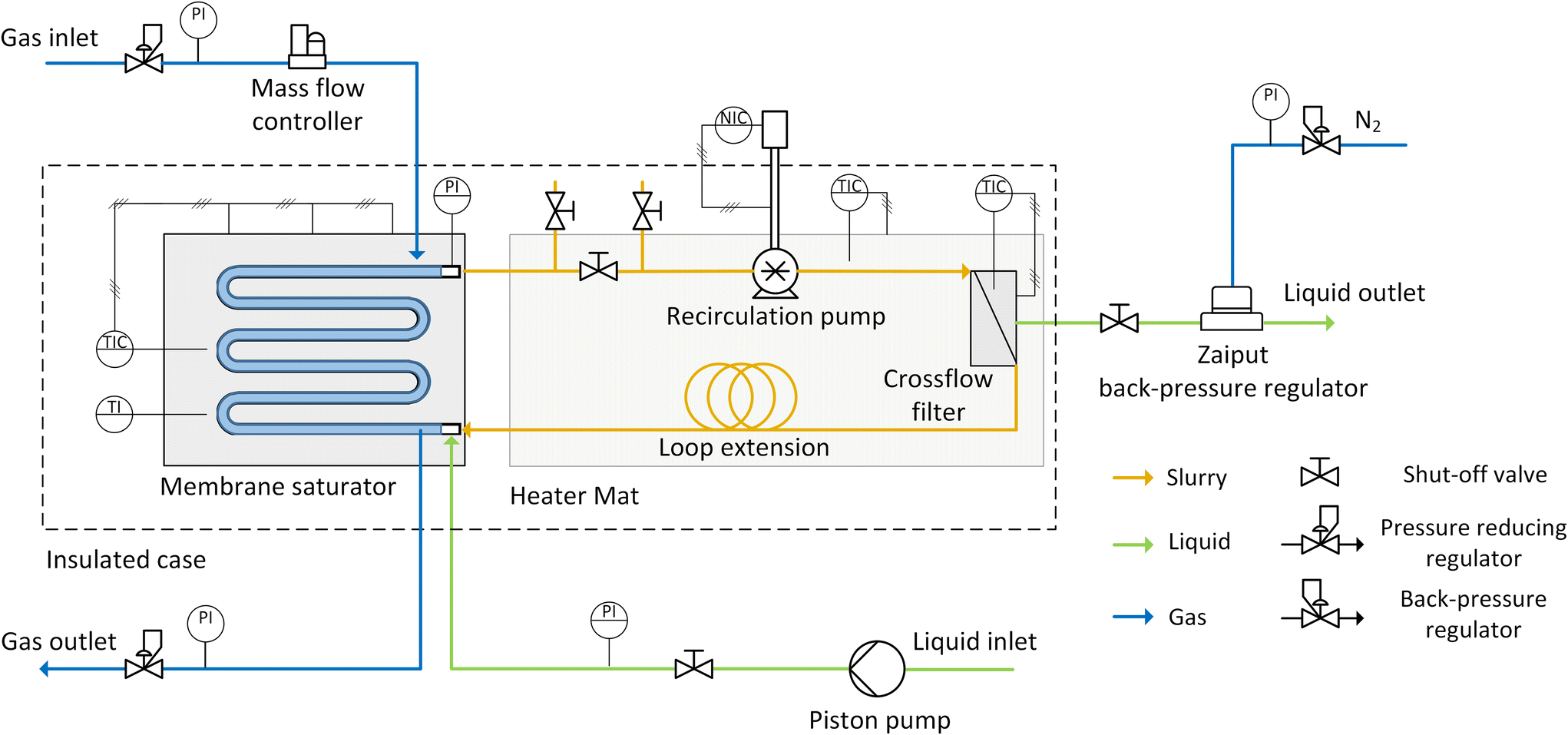 | ||
| Fig. 2 Schematic diagram of the slurry loop membrane reactor setup. PI: pressure indicator, TI: temperature indicator, TIC: temperature indicator and controller, NIC: pump speed indicator and controller. | ||
Continuous and batch operation modes
The setup could be operated either in batch or continuous mode. In both cases the catalyst slurry mixture was prepared, prior to addition to the reactor, inside a glass bottle (100 mL, Schott) with ca. 50 mL of substrate solution stirred together with the desired catalyst amount. Between the slurry outlet of the saturator and the slurry inlet of the recirculation pump, a series of three shut-off PEEK valves (0.04′′ ID hole, Kinesis) was installed to facilitate the introduction of the catalyst slurry in the loop prior to any reaction. This was fed into the loop using a micro annular gear pump, through the system of valves, until the whole loop was full of the slurry mixture.For continuous and scaled-up reactions, the substrate inlet flowrate was controlled by a dual piston pump (Azura P2.1S, Knauer) which pumped the substrate solution to the inlet port in the slurry channel of the saturator. The outlet liquid flow was delivered from the crossflow filter, which was connected to a back-pressure regulator (BPR-01, Zaiput) that ensured a constant pressure upstream. This could be regulated using the pressure reducer on the nitrogen line that was connected to the liquid back-pressure regulator.
For batch reactions, once the slurry mixture was fed into the loop, the piston pump was used to deliver the liquid substrate (e.g., 100 μL min−1 for 2 min) and pressurise the slurry loop up to the desired pressure. After reaction, the system of valves between the pump and the saturator was used to withdraw the product mixture from the loop.
The slurry pressure across the saturator channel was measured using two pressure sensors (PX309, Omega), one connected to the liquid inlet and a second one to the other end of the serpentine channel. This was done to ensure that the gas pressure was always 0.5 bar lower than that of the slurry at any point in the saturator, in order to prevent oxygen breakthrough via the membrane into the flowing slurry.
The Teflon AF-2400 membrane film is capable of withstanding a broad pressure difference between the liquid and the gas. Its resistance to high transmembrane pressures has been demonstrated in a packed-bed membrane reactor developed in our previous work, using a similar membrane thickness (0.07 mm) to that employed in this work.46 The membrane was able to withstand a pressure difference of 20 bar between the liquid that was flowing in a 75 mm × 3 mm large channel and the gas phase, without leaking or showing a noticeable change of shape when used with the same metal mesh as used in this work.
Concerning the reacting gas, this was depressurised from the gas line using a gas pressure regulator (KPR, Swagelok) and its flowrate controlled by a mass flow controller (4850, Brooks). During experiments when the gas was stagnant, the mass flow controller was disconnected and the pressure reducer was directly connected to the saturator. The gas pressure in the saturator was controlled at the gas outlet by a gas back-pressure regulator (BP301, Pressure Tech) for both flowing and stagnant gas operation modes. Stainless steel tubes of 1/16′′ outer and 0.04′′ internal diameter were used to deliver the gas at the inlet and vent it at the outlet. In order for the reactor to operate up to 120 °C, the saturator and the crossflow filter were heated by electric heaters and the reactor covered in calcium-magnesium silicate thermal insulating sheets (6 mm thick, RS Components) and placed inside a Delrin® acetal box to reduce heat losses (see ESI for details†).
Macromixing study
Macromixing was characterised by a pulse-input injection of a tracer under different recycle-to-inlet flowrate ratios (recycle ratios). A volume of 100 μL of tracer (Basic Blue 3, Sigma Aldrich) was injected at the inlet, while Ultraviolet-Visible (UV-Vis) spectroscopy (DH-2000BAL+UV-VIS-ES, Ocean Optics) was used in the loop to monitor the tracer absorbance. Experimental details and derivation of the residence time distribution are presented in the ESI.† The recycle flowrate was set to 10 mL min−1, while the inlet flowrate was varied in order to have recycle ratios, R, of 10, 20, 50 and 100.Mass transfer resistance study
The hydrogenation of styrene to ethylbenzene (Scheme 1) was performed to demonstrate the applicability of the SLMR in hydrogenation reactions and to study the mass transport rate of hydrogen from the gas to the bulk liquid in the flowing slurry. Reactions were conducted using 2 M styrene (99%, containing 4-tert-butylcatechol as stabiliser) dissolved in methanol (99.8%), and 0.5 M decane (99%) was used as internal standard. All reagents were purchased from Sigma Aldrich. The catalyst powder employed in the hydrogenation of styrene was a 5 wt% Pd/C (type 487, Alfa Aesar). Laser diffraction analysis (LS 13 320, Beckman Coulter) showed a bimodal distribution of the catalyst particles with two peaks, at 25.0 and 52.6 μm, and an overall mean particle size of 23.7 μm with a standard deviation of 17.0 μm (Fig. S8, ESI†).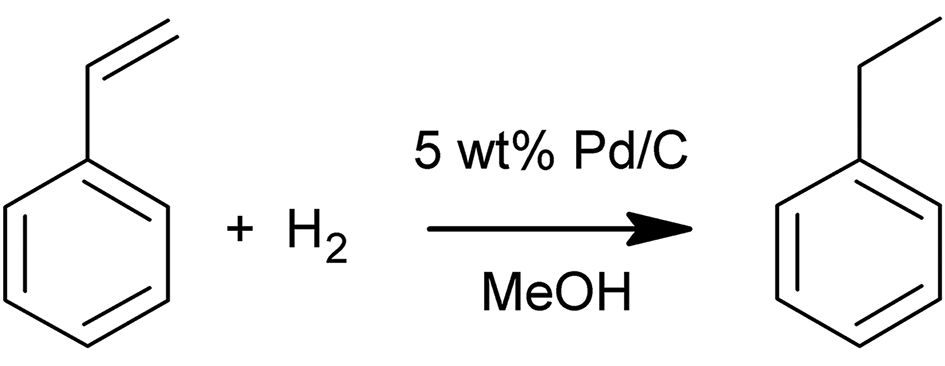 | ||
| Scheme 1 Styrene hydrogenation to ethylbenzene in methanol using 5 wt% Pd/C catalyst. | ||
Hydrogenations were performed at 21 °C and under constant inlet flowrate of 0.050 mL min−1. The gas was delivered from a hydrogen generator (PH200, Peak Scientific) and was kept stagnant at 4 bar above the Teflon AF-2400 membrane.
An initial set of experiments was performed to investigate the effect of the recycle flowrate on styrene conversion under constant inlet flowrate and catalyst concentration of 0.60 g L−1. The recycle flowrate was set to 5, 10 and 20 mL min−1, corresponding to a R of 100, 200 and 400, respectively. In a second set of experiments, the effect of catalyst concentration ranging from 0.05 to 0.60 g L−1 was explored under a recycle flowrate of 10 mL min−1. This was done to determine the volumetric gas-liquid mass transfer coefficient, kLa. Eqn (1) shows the ratio between the hydrogen concentration at the membrane-liquid interface, CH2,i, and the hydrogen reaction rate, rH2, being equal to the sum of the gas-liquid, ΩGL, liquid-solid, ΩLS, and the combined catalyst internal diffusion and surface reaction resistance, ΩR. By plotting CH2,i/rH2 against the inverse of the catalyst concentration, ρcat, the volumetric gas-liquid mass transfer coefficient, kLa, can be determined by taking the inverse of the intercept on the graph. Details on the full derivation and the definition of the terms of eqn (1) are reported in the ESI.†
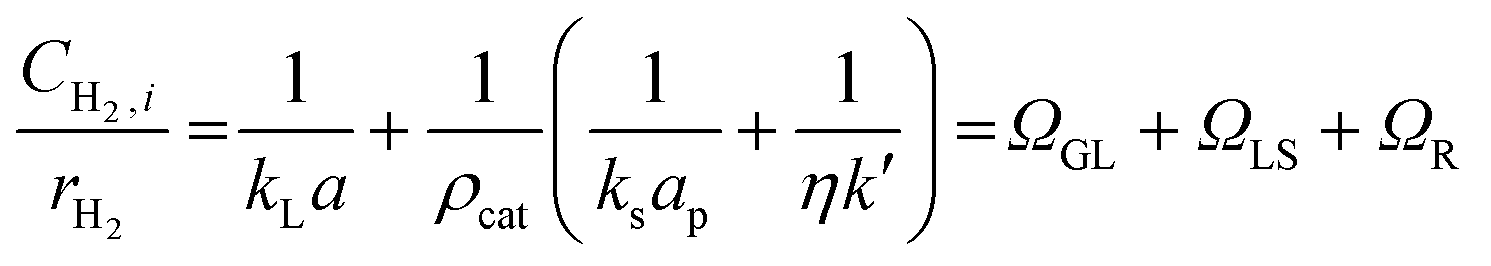 | (1) |
Aerobic oxidation of alcohols
Various primary and secondary alcohols were oxidised in flow and batch using molecular oxygen (see Scheme 2). Benzyl alcohol (99.8%), cinnamyl alcohol (98%), geraniol (98%), 1-phenylethanol (98%), 1-phenyl-1-propanol (97%), piperonyl alcohol (98%) and 4-methylbenzyl alcohol (98%) were oxidised using tert-butylbenzene (99%) as a solvent, starting from a concentration of 0.1 M. Mesitylene (98%) was used as internal standard at a concentration of 0.05 M. All compounds were purchased from Sigma Aldrich. The solvent was chosen based on its high boiling point and lower toxicity compared to other solvents (e.g., toluene56). Furthermore, to demonstrate a solvent-free aerobic oxidation, benzyl alcohol was also oxidised without solvent. Batch and continuous reactions were conducted for 6–8 h using molecular oxygen (N5.5, BOC) pressurised and kept stagnant above the membrane, except during continuous scaled-up experiments where oxygen was fed via a mass flow controller at a constant flowrate of 30 NmL min−1. With the aim of achieving high conversions, a catalyst concentration of 10 g L−1 was employed for all aerobic oxidations, higher than those used in the hydrogenation of styrene. The reaction temperature ranged between 90 and 120 °C and the inlet flowrate was varied between 0.020 to 0.360 mL min−1 in flow experiments. Higher flowrates would require a larger filter area to keep a constant pressure drop across the filter, since it was observed that at high outlet flowrates particle fines progressively deposit on the filter causing a gradual increase of the loop liquid pressure. | ||
| Scheme 2 Aerobic oxidation of primary and secondary alcohols to aldehydes and ketones using 1 wt% Au-Pd/TiO2 catalyst. | ||
Choice of the catalyst
Aerobic oxidations of alcohols were performed using a 1 wt% Au-Pd/TiO2 catalyst (Scheme 2). Other works report the use of this catalyst,46,55,57 including Hutchings’ group, who found that the addition of Au to Pd nanocrystals enhanced the selectivity to aldehydes in the aerobic oxidation of alcohols, owing to the core-shell structure that Au creates with Pd.57 The catalyst was supplied by Johnson Matthey and was prepared by co-impregnating HAuCl4·3H2O (Johnson Matthey) and PdCl2 (Johnson Matthey) onto TiO2 (Evonik, P25) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19 Au-to-Pd weight ratio. The resulting slurry mixture was spray-dried at a nozzle temperature of 220 °C and calcined in static air at 400 °C for 3 h. Inductively coupled plasma atomic emission spectroscopy established a content of gold and palladium of 0.05 wt% and 0.85 wt% respectively. Transmission electron microscopy showed that the metal particle size was in the range of 1–2 nm, while laser diffraction (Fig. S12, ESI†) showed that the catalyst particle size distribution had two peaks at 8.1 μm and 27.4 μm, with an overall mean particle size of 9.3 μm and a standard deviation of 8.4 μm.
:19 Au-to-Pd weight ratio. The resulting slurry mixture was spray-dried at a nozzle temperature of 220 °C and calcined in static air at 400 °C for 3 h. Inductively coupled plasma atomic emission spectroscopy established a content of gold and palladium of 0.05 wt% and 0.85 wt% respectively. Transmission electron microscopy showed that the metal particle size was in the range of 1–2 nm, while laser diffraction (Fig. S12, ESI†) showed that the catalyst particle size distribution had two peaks at 8.1 μm and 27.4 μm, with an overall mean particle size of 9.3 μm and a standard deviation of 8.4 μm.
Analysis of the products
Reaction products were analysed using a gas chromatograph (7820A, Agilent Technologies) equipped with an automatic liquid sampler, a HP-INNOWAX (19091-133) capillary column and a flame ionisation detector. Before the analysis of batch samples, the slurry mixture was filtered by means of glass microfibre filters (Grade GF/B, Whatman). For samples from continuous oxidations, filtration was continuously performed using the crossflow filter.Results and discussion
Macromixing
The slurry loop membrane reactor was first characterised by studying its macromixing. The resulting normalised residence time distributions (RTD), E(θ), of the tracer are plotted in Fig. 3 against the dimensionless time, θ. The RTDs are plotted along with that of an ideal CSTR as a reference (orange line) and the deviation between the two is calculated using the normalised residual sum of squares (RSS). Under a recycle ratio, R, of 100, the RTD matches well with that of an ideal CSTR (RSS = 0.001). However, when decreasing the recycle ratio, the RTD started exhibiting more intense oscillations soon after the injection, and a growing deviation from the exponential trend of an ideal CSTR. This is clear for the case of a recycle ratio of 10, in which periodic spikes of tracer concentrations were visible at earlier times for θ < 0.5, indicating inhomogeneous macromixing. The ESI† reports details of the determination of the E(θ) curves from the experimental data and the RSS.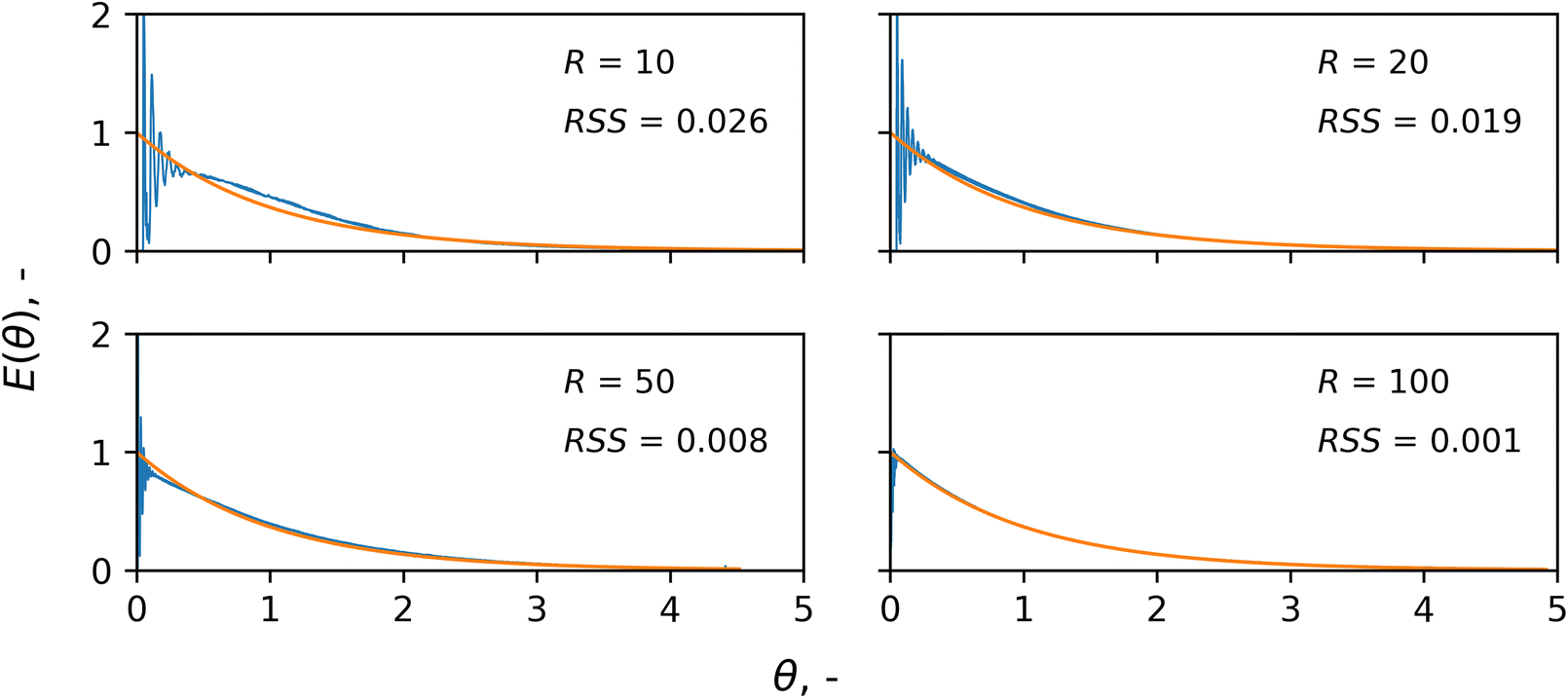 | ||
| Fig. 3 Normalised RTDs, E(θ), of a tracer in the slurry loop membrane reactor as a function of the dimensionless time, θ, under a constant recycle flowrate of 10 mL min−1 and different recycle ratios, R: 10, 20, 50, 100. The orange line represents the RTD of an ideal CSTR and its deviation from the experimental data (blue line) is calculated using the normalised residual sum of squares, RSS. | ||
Mass transfer resistances
A series of experiments was conducted to study the role of the recycle ratio and catalyst loading in the hydrogenation of styrene over a 5 wt% Pd/C catalyst. The first results showed an increase in styrene conversion when the recycle flowrate was changed from 5 mL min−1 (28%) to 20 mL min−1 (45%), despite operating in a CSTR macromixing regime (R > 100) (see Fig. S9, ESI†). This can possibly be attributed to an increased mass transfer of hydrogen in the slurry flow and to an enhanced shear rate between the catalyst particles and the membrane surface. However, from a 10 mL min−1 flowrate, conversion started to plateau. Under this recycle flowrate conversion was 40%, only ca. 10% lower than that obtained with double the recycle flowrate.The catalyst concentration was varied to estimate the average gas-liquid mass transfer coefficient under a recycle flowrate of 10 mL min−1 (Fig. S10, ESI†). The ratio between CH2,i and rH2 was calculated and plotted against the reciprocal of the catalyst concentration, and a linear regression was achieved with R2 of 0.991 (Fig. S11, ESI†). From the intercept of the fitting line, the gas-liquid resistance to hydrogen mass transfer, ΩGL, can be derived, as shown in eqn (1), and its value was equal to 0.87 s. To understand the limiting factor in the hydrogenation of styrene, ΩGL needs to be compared to the sum of the liquid-solid resistance, ΩLS, and the combined internal diffusion and surface reaction resistance, ΩR. These can be estimated from the slope of the fitting line and from the catalyst concentration. Table 1 shows the results. At low catalyst concentrations, the reaction is limited by the amount of catalyst, reflected by the higher value of the sum of ΩLS and ΩR compared to ΩGL. However, by increasing the catalyst concentration above 0.50 g L−1, the gas-liquid mass transfer resistance (ΩGL = 0.87 s) overcomes the sum of the liquid-solid and the combined internal diffusion and surface reaction resistance, ΩLS + ΩR, thus becoming the limiting factor.
| ρ cat, g L−1 | Ω LS + ΩR, s (ΩGL = 0.87 s) |
|---|---|
| 0.05 | 7.64 |
| 0.07 | 5.46 |
| 0.20 | 1.91 |
| 0.50 | 0.76 |
| 0.60 | 0.64 |
Stamatiou and Muller determined an overall volumetric mass transfer resistance for hydrogen during styrene hydrogenation in methanol inside a 0.6 L batch stirred autoclave reactor, of ca. 42 s, at 32 °C, 4 bar H2, 1200 rpm, using 0.05 g L−1 of a 5 wt% Pd/C catalyst powder.54 The catalyst particle size (20 μm) was in the same range as that employed in this work (23.7 μm). At 2 bar (ceteris paribus) the gas-liquid mass transfer resistance was found to be approximately 11 s. The authors state that a change in pressure would not impact gas-liquid mass transfer, hence it can be deduced that at 4 bar hydrogen pressure the autoclave achieved a ΩGL of 11 s and a ΩLS + ΩR equal to 31 s.
Despite the higher temperature and the intense agitation speed in the autoclave, the SLMR achieved approximately 13 times lower ΩGL and 4 times lower ΩLS + ΩR than that in the autoclave. The lower gas-liquid resistance can be ascribed to the large (48.5 cm2) available membrane surface area, while the shear rate experienced by the catalyst particles in the SLMR's loop may be responsible for the lower solid-liquid mass transfer resistance achieved in our work.
By taking the inverse of the gas-liquid mass transfer resistance, it is possible to estimate the volumetric mass transfer coefficient, kLa, to be 1.2 s−1. It is worth comparing this value with that of other gas-liquid contactors reported in the literature. Mo et al. reported a membrane reactor consisting of a Teflon AF-2400 membrane film sandwiched between two carbon cloth sheets, which separated the flowing liquid from the gas phase.51 Simulations of the hydrogen transport through the membrane and into the liquid phase were performed to understand the role of the carbon cloth thickness in the gas-liquid mass transfer. It was found that a kLa of 0.3 s−1 was achieved with a 0.3 mm thick carbon cloth and to obtain a kLa of 1.2 s−1 a thickness smaller than 0.2 mm would be necessary. In comparison, the SLMR could deliver a higher volumetric gas-liquid mass transfer, with only a 0.3 mm deep saturator channel and the slurry being pumped at 10 mL min−1. To assess the efficiency of mixing, the specific power input, ε, that the pump had to provide during the operation was calculated. This is equal to the power dissipated by circulating the catalyst slurry and can be determined using eqn (2) (see ESI for the derivation†), where Δppump is the pressure drop across the recirculation pump (0.2 bar) at a specific recycle flowrate, vrec (10 mL min−1), and VR is the reactor volume (1.4 cm3). This leads to a specific power input of 2.4 kW m−3.
 | (2) |
Table 2 shows the volumetric gas-liquid mass transfer coefficients and the specific power inputs for various gas-liquid contactors. The kLa in the SLMR lies within the upper range of kLa achieved in a stirred slurry reactor,58 or a static mixer,59 but at a much lower volumetric power input. At a comparable specific power input, the SLMR outperformed the reported loop reactor,60 bubble column61 and Taylor-Couette reactor62 in terms of gas-liquid mass transfer. Only the reported microreactor could deliver a much higher kLa. This was a micropacked-bed reactor in which gas-liquid mass transfer was investigated in the hydrogenation of cyclohexene over 50 μm Pt/Al2O3 catalyst particles under trickle-bed conditions.63 The reactor had a relatively high dissipation power, due to pressure drop across the packed bed that the pump had to overcome.
| Gas-liquid contactor | k L a, s−1 | ε, kW m−3 |
|---|---|---|
| Baffled stirred slurry reactor with sparger58 | 0.01–0.8 | 0.1–10 |
| Bubble column reactor61 | 0.02–0.15 | 0.5–3 |
| Fixed-bed silicon-glass microreactor63 | 5–15 | 2–5 |
| Loop reactor with downflow ejector60 | 0.01–0.1 | 0.1–10 |
| Static mixer in a horizontal pipe59 | 0.1–5 | 10–100 |
| Taylor-Couette reactor (horizontal)62 | 0.002–0.005 | 1–7 |
| This work (SLMR) | 1.2 | 2.4 |
Continuous aerobic oxidations
To demonstrate the versatility in the operation and the applicability of the slurry loop membrane reactor to the aerobic oxidation of alcohols in flow, various primary and secondary alcohols were oxidised. Steady-state conversion and product selectivity were achieved between 1 and 4 h, given the broad range of inlet flowrates that was explored (see ESI† for time to reach steady-state for each reaction). Oxygen pressure was varied between 2 and 6 bar, while temperature ranged between 90 and 120 °C. For each temperature set point, all the units of the loop reactor operated nearly isothermally. As an example, during the solvent-free oxidation of benzyl alcohol performed at a temperature set point of 110 °C, the maximum temperature difference in the loop was 4 °C (Table S2, ESI†).
Table 3 shows the alcohol conversion, X, the corresponding aldehyde or ketone selectivity, S, and the average turnover frequency, TOF, for each alcohol oxidation reaction. From the inverse of the turnover frequency, it is possible to estimate an average characteristic reaction time, which ranged from 0.14 s (25000 h−1) for the benzyl alcohol to 133 s (27 h−1) for the piperonyl alcohol oxidation. Assuming a similar order of magnitude for the characteristic gas-liquid mass transfer time found in the hydrogenation of styrene, 0.87 s, it is possible to infer that, except for the solvent-free oxidation of benzyl alcohol, the reactions studied here were not limited by gas-liquid mass transfer, but presumably by the combined internal diffusion and surface reaction resistance.
| Entry | Product | v, μL min−1 | CCT , gcat min gROH−1 | X, % (S, %) | TOF , h−1 | |
|---|---|---|---|---|---|---|
| a CCT = catalyst mass divided by alcohol inlet mass flowrate. b TOF = moles of product per hour per moles of Au-Pd. c 110 °C, 6 bar. d 100 °C, 2 bar. e 6 bar. f Estimated using GC-MS (see ESI†). | ||||||
| 1 |
|
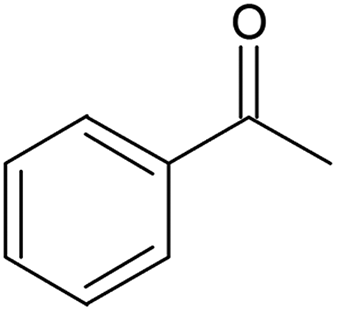
Solvent-freec | 100 | 0.13 | 50 (63) | 25000 |
| Diluted | 100 | 13 | 74 (84) | 385 | ||
| 2d |
|
100 | 10 | 75 (60) | 390 | |
| 3e |
|
20 | 45 | 28 (28) | 30 | |
| 4e |
|
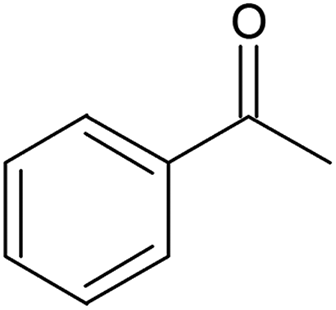
80 | 14 | 31 (100) | 130 | |
| 5 |
|
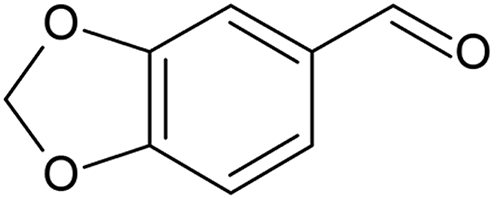
30 | 31 | 17 (100f) | 27 | |
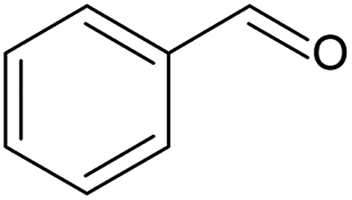
In the case of the benzyl alcohol solvent-free oxidation (Table 3, entry 1), a conversion of 50% was achieved with a selectivity of 63% to benzaldehyde at a catalyst contact time of 0.13 gcat min gROH−1, corresponding to an alcohol inlet flowrate of 0.100 mL min−1 contacting 14 mg of catalyst. In our previous study where benzyl alcohol was oxidised with oxygen in a slurry loop tube-in-tube membrane reactor using the same catalyst, 50% conversion and 66% benzaldehyde selectivity were achieved at 110 °C, 5 bar oxygen pressure and under a higher catalyst contact time (0.28 gcat min gROH−1), with a turnover frequency of 12200 h−1.55 Conversion was similar to that achieved in this work, possibly due to a combination of lower oxygen pressure and higher catalyst contact time. Moreover, an isothermal temperature profile could not be attained in that work, owing to the recirculation pump being outside the oil bath where the reactor was placed. As a result of the different catalyst contact time and a lower average loop temperature, turnover frequency was also lower than 25000 h−1 achieved by the SLMR. More interestingly, no deactivation of the catalyst was observed during the solvent-free oxidation of benzyl alcohol (Fig. S14, ESI†). Using an inlet concentration of 0.1 M in tert-butylbenzene and at 120 °C, benzyl alcohol conversion and benzaldehyde selectivity stabilised to 74% and 84%, respectively. The use of diluted benzyl alcohol hindered the disproportionation reaction to form toluene, which is enhanced at high concentrations of benzyl alcohol.64 Selectivity to toluene was in fact below 3%, while the selectivity to benzoic acid, resulting from the oxidation of benzaldehyde, was around 5% (Fig. S15, ESI†).
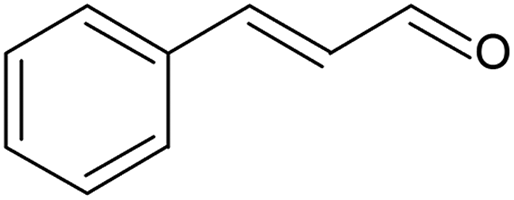
Cinnamyl alcohol was oxidised at 100 °C and 2 bar oxygen pressure for 7 h (Table 3, entry 2). No deactivation was observed in the last 5 h of the reaction and conversion was stable at 75%, while selectivity to cinnamaldehyde was ca. 60% (Fig. S18, ESI†). Under a similar catalyst contact time (7.5 gcat min gROH−1), 0.5 M cinnamyl alcohol in toluene was pre-mixed with flowing oxygen at 4 bar and fed into a packed-bed capillary microreactor containing 10 mg of the same Au-Pd/TiO2 catalyst.65 At 100 °C, conversion dropped from 40% (24 mmol h−1 gcat−1) to 20% (12 mmol h−1 gcat−1) during 7 h of continuous reaction, while selectivity to cinnamaldehyde was equal to 65%. On the other hand, despite operating at a lower oxygen pressure (2 bar), the SLMR could achieve a stable conversion, and a productivity of cinnamaldehyde of 32 mmol h−1 gcat−1. This difference can be ascribed to the use of smaller catalyst particles that decrease intraparticle diffusional resistances and increase the contact surface area between the catalyst and the reacting liquid. Concerning selectivity, the lower cinnamaldehyde selectivity of 60% is due to the lower pressure than that used in the packed-bed reactor.
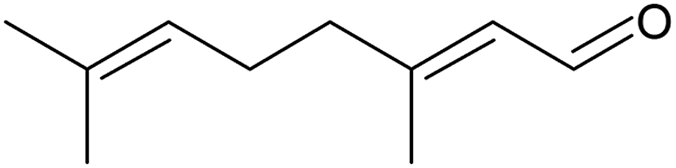
The oxidation of the terpene geraniol (Table 3, entry 3) was more challenging. Selectivity to geranial was found to decrease and conversion to increase from the start to the end of the reaction until they both stabilised to 28% in the last three hours of the reaction (Fig. S20, ESI†). Enache et al. found a similar selectivity-conversion trend using Au/SiO2 catalyst in the oxidation of geraniol with and without solvent.66 Regarding the catalyst activity, under steady-state the average turnover frequency was approximately 30 h−1. This was lower than, but of a similar order of magnitude to that reported by Li et al. (71 h−1) who performed solvent-free batch oxidation of geraniol using Pd catalyst on NaX zeolite.67
1-Phenylethanol oxidation (Table 3, entry 4) proceeded with 100% selectivity to acetophenone for 7 h with a conversion of 31% (Fig. S21, ESI†). In their batch oxidation of primary alcohols Enache et al. reported an average turnover frequency of 269000 h−1 for 1-phenylethanol using a Au-Pd /TiO2 catalyst.57 This is orders of magnitude higher than that achieved in this work, however the temperature employed was 160 °C. Pascanu et al. demonstrated the use of 2–3 nm palladium nanoparticles supported on metal organic frameworks, enclosed in a coating of silica nanoparticles for the continuous aerobic oxidation of 1-phenylethanol.68 The reaction was conducted for 7 days inside a packed-bed reactor under a flowrate of 50 μL min−1 and 80% of 0.1 M of 1-phenylethanol in toluene was continuously converted at 110 °C and 1 bar air pressure using 0.0385 mmol of Pd metal. The TOF achieved by Pascanu et al. was 6 h−1 which is two orders of magnitude smaller than that reported in our work (130 h−1), possibly ascribed to a lower temperature and oxygen pressure than those employed in our work.
Piperonyl alcohol (Table 3, entry 5) was continuously oxidised for 6.5 h with a conversion of 17% and 100% selectivity to piperonal, determined using GC-MS (Fig. S23, ESI†). Zotova et al. reported the aerobic oxidation of alcohols in toluene over 5% Ru/Al2O3 under 5 bar and 90 °C.48 Reactions were carried out in an XCube™ flow reactor where 0.29 g of catalyst was packed in a cartridge. The alcohol solution was pumped into the cartridge and recirculated from the outlet to the inlet in a differential batch reactor mode, allowing to achieve higher conversions. Both piperonyl alcohol and 1-phenylethanol were oxidised to 98% and >99% in 1 h, respectively, however the average TOF was ca. 10 h−1 in both cases, lower than that achieved in our work. This could be ascribed to a larger amount of catalyst employed in their work.
Batch aerobic oxidations
To achieve higher conversions, the slurry loop membrane reactor was operated under batch mode using longer catalyst contact times (see Table 4).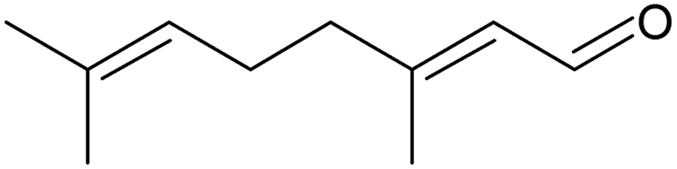
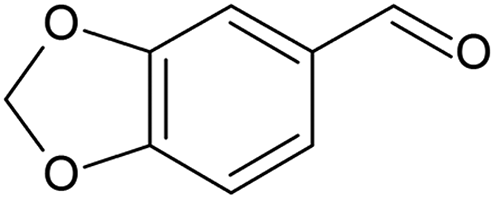
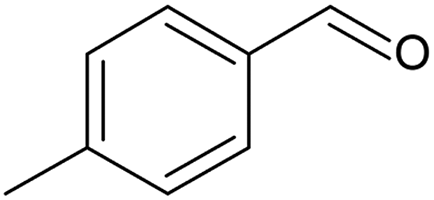

Geraniol (Table 4, entry 1) was oxidised for 7.8 h to 91% conversion, however with a low selectivity (5%) to geranial. Different by-products can result from the oxidation of geraniol, most of which are ascribed to isomerisation reactions.66,69 Selectivity to geranial could be improved at high conversion by using different catalysts like chromium supported on mesoporous molecular sieves, as presented by Dapurkar et al.,70 or by using small Pd nanoparticles as highlighted by Li et al.67 1-Phenylethanol was oxidised to completion after 6 h of batch reaction (Table 4, entry 2), with a selectivity of 72% to acetophenone, lower than that achieved in the continuous mode (Table 3, entry 4). Similarly, piperonyl alcohol was oxidised to almost completion (Table 4, entry 3), at a 6.6 times higher catalyst contact time with respect to the continuous reaction. Selectivity to piperonal was around 65% and components like piperonylic acid and 3,4-methylenedioxy toluene were detected among the products (Fig. S24, ESI†). Two further substrates were oxidised in batch: 4-methylbenzyl alcohol (Table 4, entry 4) and 1-phenyl-1-propanol (Table 4, entry 5). The former oxidation resulted in 72% conversion, 63% of which to 4-methylbenzaldehyde, while the secondary alcohol reached a conversion of 31% with a selectivity to propiophenone of 22%.
Scaled-up continuous aerobic oxidations
Productivity increase was demonstrated by using a larger reactor volume and higher inlet flowrates in the benzyl and cinnamyl alcohol aerobic oxidations (see Table 5).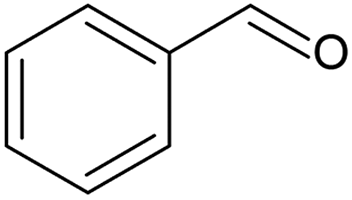
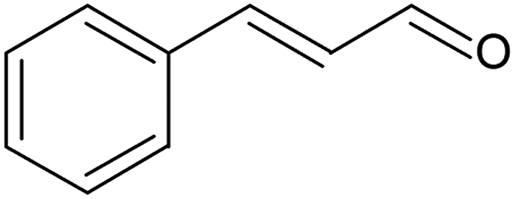
The 7 h long solvent-free oxidation of benzyl alcohol (Table 5, entry 1) proceeded at a conversion of 35% and a selectivity to benzaldehyde of 59% (Fig. S16, ESI†). The reaction was performed at a CCT of 0.38 gcat min gROH−1, 2.9 times higher than that in the smaller reactor (Table 3, entry 1). However, despite the longer catalyst contact time, a lower conversion was realised (vide supra). This could have been caused by a lower oxygen pressure employed in the reaction and by an inadequate insulation of the loop extension, which in turn could have lowered the average reaction temperature. Nevertheless, a 19 g yield of benzaldehyde was achieved after 7 h. The benzaldehyde productivity resulting from this reactor can be compared to that from a scaled-up packed-bed membrane reactor developed in our laboratory, where the same catalyst was used for the solvent-free aerobic oxidation of benzyl alcohol.46 At 120 °C and 8.2 bar oxygen pressure, Wu et al. reported a 58% conversion with a 69% benzaldehyde selectivity using a flowrate of 50 μL min−1 and 1 g of catalyst. Comparing these results with the conversion, selectivity and flowrate employed in this work, the SLMR achieved 2.3 times higher benzaldehyde yield than that of the scaled-up packed-bed membrane reactor. Furthermore, it produced 26 times higher yield of benzaldehyde per mg of Au-Pd/TiO2 catalyst compared to that achieved by the packed-bed membrane reactor. This was due to the smaller amount of catalyst employed in this work, which shows again the superior performance of the SLMR in terms of catalyst usage, compared to a membrane packed-bed reactor.
As there are aerobic oxidations that can require longer catalyst contact times, scaled-up continuous oxidation was also performed in tert-butylbenzene using an inlet concentration of 0.1 M. Benzyl alcohol reacted with 84% conversion and 86% selectivity (Fig. S17, ESI†), providing 1.2 g yield to benzaldehyde after 6 h reaction. Similarly, cinnamyl alcohol was oxidised (Table 5, entry 2) with a 65% conversion, 51% of which to 0.4 g cinnamaldehyde after 6.5 h continuous reaction (Fig. S19, ESI†).
Flammability hazard considerations
Despite the presence of a membrane that prevented the direct contact between the gaseous oxidant and the organic substrates, flammable mixtures can form in the gas phase due to the pervaporation of organic molecules from the slurry, diffusing through the membrane and into the oxygen phase. During scaled-up oxidation experiments, oxygen was fed at 30 NmL min−1 and the organic vapours pervaporating through the membrane were condensed from the gas outlet using a cold trap, to estimate the pervaporation rate. Using tert-butylbenzene as solvent in both the benzyl and cinnamyl alcohol oxidations, no condensed organics were detected at the gas outlet. However, in the scaled-up solvent-free oxidation of benzyl alcohol a total of 341 mg of condensed organics was collected at the gas outlet after 6 h reaction. This corresponded to 57 mg h−1 of organics pervaporating on average, throughout the reaction time. Table 6 shows the average mass flowrate of each component and their concentration in oxygen. Details of the estimation of the lower flammability limit in oxygen for each component, LFLoxy,i, and for the mixture are reported in the ESI.†| Component | ṁ i , mg h−1 | y i , mol% | LFL oxy,i, mol% |
|---|---|---|---|
| Benzyl alcohol | 24 | 0.08 | 1.25 |
| Benzaldehyde | 17 | 0.06 | 1.35 |
| Toluene | 16 | 0.06 | 1.01 |
| Total | 57 | 0.20 | 1.18 |
The total average concentration of organics in the flowing oxygen equated to approximately 0.20 mol%, 6 times lower than the organics mixture LFLoxy of 1.18 mol% (see ESI†). Therefore, the continuous aerobic oxidation of benzyl alcohol operated outside the flammability window, owing to the sweeping oxygen flowrate which guaranteed a low concentration of organics in the vapour phase and that no pockets of condensate were forming in the gas channel, reducing the risk of creating flammable mixtures.
Summary and conclusions
This study demonstrated a novel slurry loop membrane reactor for aerobic oxidations and hydrogenation reactions. A Teflon AF-2400 membrane film was used within a saturator connected to a loop where the catalyst slurry was circulated and a crossflow filter allowed continuous flow, while keeping the catalyst contained inside the loop. Homogeneity and perfect mixing behaviour were achieved at recycle ratios higher than 100, and an isothermal temperature profile was accomplished during reaction. Gas-liquid mass transfer was characterised in the reactor by hydrogenating styrene to ethylbenzene in methanol with a 5 wt% Pd/C catalyst. Gas-liquid mass transfer resistance was found to be the least rate-limiting resistance for catalyst concentrations lower than 0.50 g L−1, and the kLa was estimated to be 1.2 s−1. This was achieved under a specific power consumption of 2.4 kW m−3; moderate compared to other gas-liquid contactors. Continuous alcohol oxidations showed enhanced turnover frequencies compared to packed-bed reactors where oxygen was pre-mixed with the organic substrate, due to the smaller amount of catalyst employed and the reduction of intraparticle diffusion resistances resulting from the use of smaller size catalyst particles in a slurry form. Solvent-free continuous oxidation was demonstrated with benzyl alcohol, achieving a turnover frequency of 25000 h−1. Using batch operation and longer catalyst contact times, higher conversions could be attained, yet this resulted in lower selectivities. Increased productivity was demonstrated by enlarging the loop volume by a factor of 6.3 and using higher inlet flowrates. A maximum 19 g of aldehyde was obtained in the solvent-free oxidation of benzyl alcohol after 7 h of continuous reaction. Safety hazards associated with the pervaporation of the organics were overcome by using 30 NmL min−1 of flowing oxygen, which reduced the concentration of volatile organics below their lower flammability limit in oxygen. The excellent performance of the flowing slurry catalyst, combined with the membrane's ability to safely handle oxygen, and a plug-and-play configuration, make this reactor a promising platform for shifting the paradigm towards green large-scale aerobic oxidations.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank EPSRC for funding (grant EP/L003279/1) and for BV's DTP studentship. We also thank Simon Dawes and Adam Maney for machining the saturator and the crossflow filter, Qingning Yang for performing GC-MS analyses and Dr Peter Ellis (Johnson Matthey) for providing the Au-Pd/TiO2 catalyst. Dr Carsten Damerau (HNP Mykrosysteme) is gratefully acknowledged for his support with the micro annular gear pump.References
- M. Poliakoff and P. Licence, Nature, 2007, 450, 810–812 CrossRef CAS
.
-
P. T. Anastas and J. C. Warner, Green chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed
- J. H. Clark, Green Chem., 1999, 1, 1–8 RSC
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC
-
P. J. Dunn, A. S. Wells and M. T. Williams, Green Chemistry in the Pharmaceutical Industry, Wiley-VCH, Weinheim, 2010 Search PubMed
-
M. Hudlicky, Oxidations in Organic Chemistry, American Chemical Society, Washington DC, 1990 Search PubMed
-
J.-E. Bäckvall, Modern Oxidation Methods, John Wiley & Sons, Weinheim, 2011 Search PubMed
- S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC
-
G. Cainelli and G. Cardillo, in Chromium Oxidations in Organic Chemistry, Springer, Berlin, 1st edn, 1984, vol. 19, pp. 118–216 Search PubMed
- R. A. Lee and D. S. Donald, Tetrahedron Lett., 1997, 38, 3857–3860 CrossRef CAS
- R. A. Sheldon, I. W. C. E. Arends, G.-J. ten Brink and A. Dijksman, Acc. Chem. Res., 2002, 35, 774–781 CrossRef CAS
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS
- T. Matsumoto, M. Ueno, N. Wang and S. Kobayashi, Chem. – Asian J., 2008, 3, 196–214 CrossRef CAS PubMed
- F. Cavani and J. H. Teles, ChemSusChem, 2009, 2, 508–534 CrossRef CAS PubMed
- C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547–564 RSC
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17–45 RSC
-
S. S. Stahl and P. L. Alsters, Liquid Phase Aerobic Oxidation Catalysis, Wiley–VCH, New York, 2016 Search PubMed
- Y.-C. Son, V. D. Makwana, A. R. Howell and S. L. Suib, Angew. Chem., Int. Ed., 2001, 40, 4280–4283 CrossRef CAS
- Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480–3524 RSC
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed
- C. A. Hone, D. M. Roberge and C. O. Kappe, ChemSusChem, 2017, 10, 32–41 CrossRef CAS
- P. M. Osterberg, J. K. Niemeier, C. J. Welch, J. M. Hawkins, J. R. Martinelli, T. E. Johnson, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2015, 19, 1537–1543 CrossRef CAS PubMed
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed
- M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC
- H. P. L. Gemoets, Y. Su, M. Shang, V. Hessel, R. Luque and T. Noël, Chem. Soc. Rev., 2016, 45, 83–117 RSC
- C. Wiles and P. Watts, Green Chem., 2012, 14, 38–54 RSC
- D. Dallinger and C. O. Kappe, Curr. Opin. Green Sustainable Chem., 2017, 7, 6–12 CrossRef
-
B. Pieber and C. O. Kappe, in Organometallic Flow Chemistry, ed. T. Noël, Springer, Cham, 2016, vol. 15, pp. 97–136 Search PubMed
- A. Gavriilidis, A. Constantinou, K. Hellgardt, K. K. Hii, G. J. Hutchings, G. L. Brett, S. Kuhn and S. P. Marsden, React. Chem. Eng., 2016, 1, 595–612 RSC
- X. Ye, M. D. Johnson, T. Diao, M. H. Yates and S. S. Stahl, Green Chem., 2010, 12, 1180–1186 RSC
- J. F. Greene, J. M. Hoover, D. S. Mannel, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2013, 17, 1247–1251 CrossRef CAS
- C. A. Hone and C. O. Kappe, Top. Curr. Chem., 2019, 377, 2 CrossRef PubMed
- C. J. Mallia and I. R. Baxendale, Org. Process Res. Dev., 2016, 20, 327–360 CrossRef CAS
- A. Constantinou, G. Wu, B. Venezia, P. Ellis, S. Kuhn and A. Gavriilidis, Top. Catal., 2019, 62, 1126–1131 CrossRef CAS
- B. Venezia, D. C. Morris and A. Gavriilidis, AIChE J., 2022, 69, e17880 Search PubMed
-
T. C. Merkel, I. Pinnau, R. Prabhakar and B. D. Freeman, in Materials Science of Membranes for Gas and Vapor Separation, ed. Y. Yampolskii, I. Pinnau and B. D. Freeman, John Wiley & Sons, Chichester, 2006, pp. 251–270 Search PubMed
- I. Pinnau and L. G. Toy, J. Membr. Sci., 1996, 109, 125–133 CrossRef CAS
- A. Polyzos, M. O'Brien, T. P. Petersen, I. R. Baxendale and S. V. Ley, Angew. Chem., Int. Ed., 2011, 50, 1190–1193 CrossRef CAS PubMed
- B. Venezia, L. Panariello, D. Biri, J. Shin, S. Damilos, A. N. P. Radhakrishnan, C. Blackman and A. Gavriilidis, Catal. Today, 2021, 362, 104–112 CrossRef CAS
- M. O'Brien, I. R. Baxendale and S. V. Ley, Org. Lett., 2010, 12, 1596–1598 CrossRef PubMed
- M. O'Brien, N. Taylor, A. Polyzos, I. R. Baxendale and S. V. Ley, Chem. Sci., 2011, 2, 1250–1257 RSC
- F. Mastronardi, B. Gutmann and C. O. Kappe, Org. Lett., 2013, 15, 5590–5593 CrossRef CAS PubMed
- M. Brzozowski, M. O'Brien, S. V. Ley and A. Polyzos, Acc. Chem. Res., 2015, 48, 349–362 CrossRef CAS PubMed
- D. Dallinger, V. D. Pinho, B. Gutmann and C. O. Kappe, J. Org. Chem., 2016, 81, 5814–5823 CrossRef CAS PubMed
- G. Wu, E. Cao, P. Ellis, A. Constantinou, S. Kuhn and A. Gavriilidis, Chem. Eng. J., 2019, 377, 120086 CrossRef CAS
- P. T. Anastas, M. M. Kirchhoff and T. C. Williamson, Appl. Catal., A, 2001, 221, 3–13 CrossRef CAS
- N. Zotova, K. Hellgardt, G. H. Kelsall, A. S. Jessiman and K. K. Hii, Green Chem., 2010, 12, 2157–2163 RSC
- D. Obermayer, A. M. Balu, A. A. Romero, W. Goessler, R. Luque and C. O. Kappe, Green Chem., 2013, 15, 1530–1537 RSC
- D. V. Bavykin, A. A. Lapkin, S. T. Kolaczkowski and P. K. Plucinski, Appl. Catal., A, 2005, 288, 175–184 CrossRef CAS
- Y. Mo, J. Imbrogno, H. Zhang and K. F. Jensen, Green Chem., 2018, 20, 3867–3874 RSC
- R. V. Chaudhari and P. A. Ramachandran, AIChE J., 1980, 26, 177–201 CrossRef CAS
- V. G. Pangarkar, Chem. Eng. Process., 2017, 120, 1–8 CrossRef CAS
- I. K. Stamatiou and F. L. Muller, AIChE J., 2017, 63, 273–282 CrossRef CAS
- B. Venezia, M. Douthwaite, G. Wu, M. Sankar, P. Ellis, G. J. Hutchings and A. Gavriilidis, Chem. Eng. J., 2019, 378, 122250 CrossRef CAS
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362 CrossRef CAS PubMed
- M. Schmitz, A. Steiff and P.-M. Weinspach, Chem. Eng. Technol., 1987, 10, 204–215 CrossRef CAS
- A. Heyouni, M. Roustan and Z. Do-Quang, Chem. Eng. Sci., 2002, 57, 3325–3333 CrossRef CAS
- N. N. Dutta and K. V. Raghavan, Chem. Eng. J., 1987, 36, 111–121 CrossRef CAS
- J. J. Heijnen and K. Van't Riet, Chem. Eng. J., 1984, 28, B21–B42 CrossRef CAS
- S. H. Kang, S. G. Lee, W. M. Jung, M. C. Kim, W.-S. Kim, C. K. Choi and R. S. Feigelson, J. Cryst. Growth, 2003, 254, 196–205 CrossRef CAS
- M. W. Losey, M. A. Schmidt and K. F. Jensen, Ind. Eng. Chem. Res., 2001, 40, 2555–2562 CrossRef CAS
- F. Galvanin, M. Sankar, S. Cattaneo, D. Bethell, V. Dua, G. J. Hutchings and A. Gavriilidis, Chem. Eng. J., 2018, 342, 196–210 CrossRef CAS
- G. Wu, G. L. Brett, E. Cao, A. Constantinou, P. Ellis, S. Kuhn, G. J. Hutchings, D. Bethell and A. Gavriilidis, Catal. Sci. Technol., 2016, 6, 4749–4758 RSC
- D. I. Enache, D. W. Knight and G. J. Hutchings, Catal. Lett., 2005, 103, 43–52 CrossRef CAS
- F. Li, Q. Zhang and Y. Wang, Appl. Catal., A, 2008, 334, 217–226 CrossRef CAS
- V. Pascanu, A. Bermejo Gómez, C. Ayats, A. E. Platero-Prats, F. Carson, J. Su, Q. Yao, M. À. Pericàs, X. Zou and B. Martín-Matute, ACS Catal., 2015, 5, 472–479 CrossRef CAS
- C. Bäcktorp, L. Hagvall, A. Börje, A.-T. Karlberg, P.-O. Norrby and G. Nyman, J. Chem. Theory Comput., 2008, 4, 101–106 CrossRef PubMed
- S. E. Dapurkar, H. Kawanami, M. Chatterjee, C. V. Rode, T. Yokoyama and Y. Ikushima, Appl. Catal., A, 2011, 394, 209–214 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01242e |
| This journal is © The Royal Society of Chemistry 2023 |