
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sequential extraction of hemicelluloses by subcritical water improves saccharification of hybrid aspen wood grown in greenhouse and field conditions†
Pramod
Sivan‡
 a,
Emilia
Heinonen‡
ab,
Madhavi
Latha Gandla
c,
Amparo
Jiménez-Quero
a,
Hüsamettin Deniz
Özeren
a,
Leif J.
Jönsson
c,
Ewa J.
Mellerowicz
d and
Francisco
Vilaplana
*ab
a,
Emilia
Heinonen‡
ab,
Madhavi
Latha Gandla
c,
Amparo
Jiménez-Quero
a,
Hüsamettin Deniz
Özeren
a,
Leif J.
Jönsson
c,
Ewa J.
Mellerowicz
d and
Francisco
Vilaplana
*ab
aDivision of Glycoscience, Department of Chemistry, KTH Royal Institute of Technology, AlbaNova University Centre, 106 91 Stockholm, Sweden. E-mail: franvila@kth.se
bWallenberg Wood Science Centre, KTH Royal Institute of Technology, 100 44 Stockholm, Sweden
cDepartment of Chemistry, Umeå University, 901 87 Umeå, Sweden
dUmeå Plant Science Centre, Swedish University of Agricultural Sciences, Department of Forest Genetics and Plant Physiology, 901 83 Umeå, Sweden
First published on 23rd June 2023
Abstract
Fast growing hardwoods are one of the major renewable resources available to produce bio-based materials, platform chemicals and biofuels. However, the industrial processing of lignocellulosic biomass is hindered by the complex molecular structure of the cell wall components and their supramolecular organization. This highlights the necessity of improving green processing strategies to enhance biomass conversion to valuable products from industrial wood production species. In the present study, we implemented a hydrothermal step by sequential subcritical water (SW) in aspen wood prior to saccharification and validated the process for trees grown in greenhouse and field conditions. Subcritical water enables extraction of non-cellulosic cell wall polysaccharides in native polymeric form. A major part of the pectic fraction was easily extracted within the first 10 min, while acetylated xylan was enriched in the subsequent extracts after 20- and 30-min rounds. Prolonged extraction (above 60 min) resulted in partial deacetylation and a reduction of the molar mass of xylan. The analysis of the residues enriched with cellulose and lignin showed several micromorphological changes caused by subcritical water treatment, such as an increased porosity, a loosening of the fibre matrix and a decrease in the macrofibrillar dimensions. These morphological and molecular changes in the organization of cell wall polymers after SW treatment significantly enhanced saccharification yields compared to those of non-treated aspen wood chips from both field and greenhouse conditions. Our study demonstrates that SW can be implemented as pretreatment prior to saccharification reducing the requirements for chemical acid pretreatments. This process enables the extraction of native non-cellulosic cell wall polymers for potential material applications and promotes the subsequent biochemical conversion of the residual biomass into fermentable sugars and platform chemicals in future biorefineries.
Introduction
Lignocellulosic biomass represents the most abundant renewable resource on Earth for the sustainable production of eco-friendly materials,1,2 platform chemicals, and energy with almost CO2-neutral balance to slow down the global trend of CO2 increase.3,4 Lignocellulose is a heterogenous biocomposite composed of cellulose, hemicelluloses, pectins and lignin, tightly linked to each other through covalent linkages and non-covalent interactions. Cellulose has been traditionally used in the paper and pulp sectors, and through its nanotechnological modification it has the potential to substitute fossil-based structural polymers in all imaginable sectors, including electronics, energy storage and biomedical applications.5–7 Lignin is the major bio-based source of aromatics through thermal and catalytic valorization,8,9 and can be largely utilized as source of platform chemicals, polymers,10 and for the synthesis of lignin-based nanoparticles.11 Traditionally, hemicelluloses and pectins have been underexploited compared to the other lignocellulosic components, as they are largely degraded in the harsh conditions of the traditional pulp and paper processes. However, these polymers have large potential to be used for production of many high value products such as surface modifiers and texturing agents,6 packaging materials7,12 and precursors of bio-based plastics.13Lignocellulose recalcitrance has been the major challenge that hinders fractionation efficiency and conversion of biomass into derived bioproducts.14,15 Recalcitrance is conferred by multiple factors such as interactions among cell wall components, cellulose fibrillar characteristics such as crystallinity, microfibril diameter, and coalescence into macrofibrillar aggregates.2,14,15 Hemicelluloses and pectins contribute to lignocellulose recalcitrance due to their interactions with both cellulose microfibrils and lignin, acting as a transition phase between these components and as a physical barrier for the enzymatic hydrolysis of cellulose fibres.16,17 These important biological functions of hemicelluloses and pectins are regulated during biosynthesis and attributed to molecular features such as degree of polymerization, substitution pattern and crosslinking initiation sites.14,18 The morphological features of cellulose microfibrils (e.g. their diameter) and their aggregation are believed to be modulated through supramolecular interactions (both hydrogen bonding and van der Waals interactions) with hemicellulosic xylans and glucomannans at the microfibril surfaces; therefore, disruption of these aggregated forms constitutes a key target for increasing efficiency of biochemical conversion processes.14 This suggests the potential benefits of implementing hemicellulose-first approaches in biorefinery settings to increase the efficacy of biochemical conversion of lignocellulose.
Recent developments in the extraction of hemicellulose and lignin using sequential subcritical water extraction (SWE) with a buffered extraction solvent has provided novel insights on the native structure, composition and interactive domains of hemicelluloses and their lignin complexes (LCCs) from spruce (softwood) and birch (hardwood).2,19 SWE is a green extraction method based on pressurized water as a solvent under increased temperature, which facilitates the isolation of different pectin and hemicellulose populations depending on their recalcitrance in the lignocellulose network. SWE (also referred to as pressurized hot-water extraction or hydrothermal extraction) can be scaled up and run in both batch and semi-continuous mode.20,21 The different extractability of pectin and hemicellulose populations and the control of extraction times enable the targeted isolation of mannan and xylan populations with controlled acetylation and (in case of xylan) glucuronation.2,19 Moreover, the use of controlled buffered pH conditions during SWE preserves the acetylation in the extracted hemicelluloses, preventing its release into the aqueous phase and the occurrence of acid-induced autohydrolysis,2,22 as opposed to other studies where unbuffered conditions resulted in drastic depolymerization of the hemicelluloses and release of sigar monomers and oligomers.20 Hardwoods represent one of the most abundant resources for lignocellulose based biorefineries and biobased materials. Populus sp. trees including hybrid aspens and poplars are high yielding hardwood tree species with glucan content ranging from 40–62%, xylan content from 14–24% and lignin content 15–29% of the total dry weight and considered to be one of the most promising tree crops for production of bioenergy feedstock in short-rotation plantations.23 Being a model plant for hardwood species, Populus is extensively used for genetic engineering approaches to improve lignocellulose properties.24 The genetically engineered Populus trees are usually tested first under greenhouse conditions and then selected genotypes are evaluated under field conditions. In the present study, we have compared the potential of SWE for yielding high molecular weight hemicelluloses and pectins from the wood of hybrid aspen (Populus tremula L. X tremuloides Michx) trees. The structural changes in the extracted fractions have been monitored to identify links between the molecular structure and extractability of polymers. The potential of SWE process as an integrated hemicellulose-first approach prior to saccharification in biorefinery applications has been evaluated and validated with aspen trees grown in greenhouse and field conditions.
Materials and methods
Plant material
Hybrid aspen (Populus tremula L. X tremuloides Michx.), clone T89, was used for all experiments. Greenhouse grown trees were harvested after 13 weeks of cultivation in conditions previously described.20 Field grown trees were harvested in July 2014 after five growth seasons in the field as described previously.25 The wood from greenhouse-grown trees was dissected and freeze-dried20 whereas the wood from the field-grown trees was dissected and dried at 60 °C.25Chemicals
All chemicals, analytical standards and reagents were from Sigma-Aldrich (Stockholm, Sweden) unless otherwise stated.Implementation of the extraction and saccharification process
Characterization of the extracts
The starting wood powder material and the SWE residues were subjected to a two-step sulfuric hydrolysis for total sugar compositional analysis considering cellulose and non-cellulosic polysaccharides.30 In brief, 1 mg of starting wood biomass or residue was incubated with 125 μL of 72% H2SO4 at room temperature for 3 h, then diluted with 1375 μL of deionized water and subsequently incubated at 100 °C for 3 hours. Hydrolysates were diluted with MilliQ water and further filtered through 0.2 mm syringe filter (Chromacol 17-SF-02-N) into HPAEC-PAD vials and analyzed as above. Quantification of monosaccharide composition (monomeric form) was performed by standard calibration of ten monosaccharides (Ara, Rha, Fuc, Xyl, Man, Gal, Glc, GalA, 4-O-MeGlcA and GlcA) with concentrations between 0.005–0.1 g L−1. All experiments were conducted in triplicates.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Å (Polymer Standard Services, Mainz, Germany) at a flow rate of 0.5 mL min−1 and 60 °C. The columns were calibrated using pullulan standards between 345 and 708000 Da (Polymer Standard Services, Mainz, Germany).
000 Å (Polymer Standard Services, Mainz, Germany) at a flow rate of 0.5 mL min−1 and 60 °C. The columns were calibrated using pullulan standards between 345 and 708000 Da (Polymer Standard Services, Mainz, Germany).
Characterization of the solid residues after subcritical water treatment
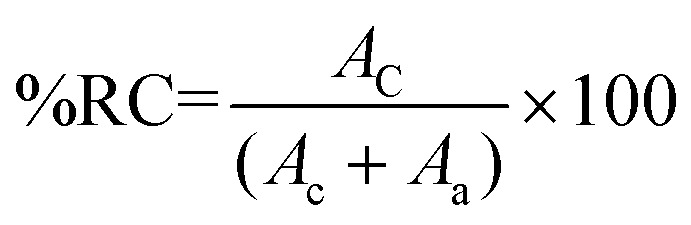 | (1) |
Results and discussion
Hemicellulose-first fractionation of aspen wood: validation in greenhouse and field conditions
Aspen has high industrial relevance as a promising biorefinery feedstock due to its fast growth and the desirable structural and chemical features of its woody biomass.41 As a model plant for tree biotechnology, aspen trees are usually grown in controlled greenhouse conditions for approximately three months. However, trees grown in field conditions are exposed to both biotic and abiotic stresses leading to variation in phenotype and cell wall chemistry and are usually harvested after several growing seasons (Fig. 1A). Here we used the two types of hybrid aspen wood to test the applicability of SWE as an alternative pretreatment method that provides benefit of extracting its matrix cell wall polysaccharides for biorefinery applications. The sequential SWE process was implemented under buffered conditions on hybrid aspen wood without any pretreatment for delignification to yield maximum hemicellulose population with minimum deacetylation and autohydrolysis. The present study aimed to evaluate the efficiency of a hemicellulose-first hydrothermal process using SWE to promote saccharification of the biomass, compared to the traditional acid pretreatment (Fig. 1B).27 This hemicellulose-first approach has the benefit to obtain a polymeric population of hemicelluloses that could be exploited in structural applications, as opposed to the degradative effect of acidic treatments. Aspen hemicelluloses include acetylated glucuronoxylan as the main component, with minor content of acetylated glucomannan (Fig. 1C). | ||
| Fig. 1 Hemicellulose-first processing prior to enzymatic saccharification of hybrid aspen grown in greenhouse and field conditions. (A) Comparison of hybrid aspen grown under greenhouse (G) and field (F) conditions. (B) Subcritical water extraction (SW) of hemicelluloses from aspen wood. Comparison to the untreated starting material (SM) and to conventional acid pretreatment (AT) prior to enzymatic saccharification (EH). (C) Molecular structures of the main hemicelluloses in hybrid aspen. Acetylated glucuronoxylan consists of a backbone of β-(1 → 4)-xylose (Xyl) units decorated with α-(1 → 2)-4-O-methyl glucuronic acid (mGlcA) units and acetylated in the O-2 and/or O-3 positions. Acetylated glucomannan consists of a backbone of β-(1 → 4)-mannose (Man) and β-(1 → 4)-glucose (Glc) units decorated with acetylations in the O-2 and/or O-3 positions. | ||
Since growth conditions and growth duration are expected to influence the chemical composition of the lignocellulosic plant cell wall material, the SWE process and saccharification efficiency were validated in industrially relevant aspen wood grown in both greenhouse and field conditions. The cell wall chemistry analysis of both starting materials (Tables 1 and 2), reveals that aspen wood from the field has relatively higher lignin content, lower syringyl to guaiacyl (S/G) ratio, lower pectin and mannan content compared to that of greenhouse grown aspen (statistically significant at P ≤ 0.05, ESI Table S3†). These differences between greenhouse and field grown trees can be attributed to changes in the wood cell wall structure and composition during wood juvenility-maturity transition42,43 and to differences in stress exposure.44 A similar higher guaiacyl (G) lignin content and lower S/G ratio in the field grown trees compared to greenhouse grown trees were reported previously.20
| SM | A1 | A2 | A3 | A4 | ΣA | R | |
|---|---|---|---|---|---|---|---|
| a Yields determined gravimetrically and referred to the starting aspen wood chips. b Determined from the complete monosaccharide composition (ESI Table S1†). c Determined by starch gelatinization and enzymatic degradation. d Determined from acetyl bromide lignin. e Determined from pyrolysis GC-MS. f Determined after saponification and HPLC-UV analysis. g Determined by SEC-DRI. A1–A4 indicate sequential extraction time: A1 (10 min), A2 (20 min), A3 (30 min), A4 (60 min). n.a. not applicable; n.d. not detected; S/G Syringyl–Guaiacyl ratio; C/L Carbohydrate–Lignin ratio. | |||||||
| Extraction time (min) | n.a | 10 | 20 | 30 | 60 | 120 | n.a |
| Total yielda (%) | 100.0 | 2.9 | 2.2 | 2.7 | 3.3 | 11.1 | 88.9 |
| Xylan yieldb (%) | 100.0 | 2.5 | 4.4 | 7.7 | 9.8 | 24.5 | 68.0 |
| Lignin yieldb (%) | 100.0 | 2.1 | 1.7 | 2.0 | 2.4 | 8.3 | 27.7 |
| Carbohydrate contentb (mg/g) | 647.0 | 706.5 | 670.8 | 773.7 | 791.1 | 679.7 | |
| Cellulloseb (%) | 61.4 | n.d. | n.d. | n.d. | n.d. | 74.7 | |
| Xylanb (%) | 31.4 | 32.8 | 72.2 | 88.0 | 92.0 | 21.5 | |
| Mannanb (%) | 3.9 | 16.7 | 6.8 | 2.1 | 0.7 | 2.9 | |
| Pectinb (%) | 3.3 | 50.5 | 21.0 | 9.9 | 7.3 | 0.9 | |
| MeGlcA:Xylb |
n.d | 0.32 | 0.18 | 0.20 | 0.21 | n.d | |
| Starch contentc | 0.16 | 0.4 | 0.2 | 0.2 | 0.2 | ||
| Lignin contentd (ABL) (mg/g) | 258.4 | 234.8 | 248.8 | 231.8 | 231.2 | 229.4 | |
| Lignin contente (PyGCMS)(%) | 34.9 | 20.0 | 17.2 | 16.3 | 13.2 | ||
| Guaiacyle (%) | 8.6 | 3.8 | 4.3 | 2.8 | 2.0 | ||
| Syringyle (%) | 25.4 | 12.1 | 7.8 | 10.6 | 8.9 | ||
| p-Hydroxyphenyle (%) | 0.8 | 3.9 | 5.0 | 2.8 | 2.2 | ||
| Phenolicse (%) | 0.05 | 0.2 | 0.1 | 0.1 | 0.0 | ||
| S/Ge | 3.0 | 3.2 | 1.8 | 3.9 | 4.5 | ||
| C/Le | 1.7 | 3.9 | 4.7 | 5.0 | 6.4 | ||
| Acetyl contentf (%) | 2.5 | 4.9 | 6.1 | 2.9 | n.a | ||
| M n (kDa)g | 2.3 | 3.2 | 4.2 | 3.7 | n.a | ||
| M w (kDa)g | 12.4 | 12.7 | 12.9 | 9.3 | n.a | ||
| SM | A1 | A2 | A3 | A4 | ΣA | R | |
|---|---|---|---|---|---|---|---|
| a Yields determined gravimetrically and referred to the starting aspen wood chips. b Determined from the complete monosaccharide composition (ESI Table S2†). c Determined by starch gelatinization and enzymatic degradation. d Determined from acetyl bromide lignin. e Determined from pyrolysis GC-MS. f Determined after saponification and HPLC-UV analysis. g Determined by SEC-DRI. A1–A4 indicate sequential extraction time: A1 (10 min), A2 (20 min), A3 (30 min), A4 (60 min). n.a. not applicable; n.d. not detected; S/G Syringyl–Guaiacyl ratio; C/L Carbohydrate–Lignin ratio | |||||||
| Extraction time (min) | n.a | 10 | 20 | 30 | 60 | 120 | n.a |
| Total yielda (%) | 100.0 | 2.2 | 2.1 | 3.5 | 4,1 | 11.9 | 88.1 |
| Xylan yieldb (%) | 100.0 | 1.8 | 4.7 | 10.0 | 13,1 | 29.6 | 67.9 |
| Lignin yieldb (%) | 100.0 | 1.9 | 1.9 | 2.9 | 3,5 | 10.2 | 27.3 |
| Carbohydrate contentb (mg g−1) | 673.5 | 629.9 | 719.3 | 826.0 | 921,7 | n.a. | 691.4 |
| Cellulloseb (%) | 61.7 | n.d. | n.d. | n.d. | n.d. | 75.5 | |
| Xylanb (%) | 32,5 | 38.1 | 80.6 | 91.5 | 93.2 | 21.4 | |
| Mannanb (%) | 3.1 | 21.2 | 5.1 | 1.4 | 0.8 | 2.2 | |
| Pectinb (%) | 2.7 | 40.6 | 14.4 | 7.2 | 6.0 | 0.9 | |
| MeGlcA:Xylb |
n.d | 0.31 | 0.20 | 0.21 | 0.24 | n.d | |
| Starch contentc | 0.45 | 6.6 | 1.6 | 0.5 | 0.2 | ||
| Lignin contentd (ABL) (mg g−1) | 285.7 | 258.4 | 263.4 | 242.2 | 249.0 | 242.6 | |
| Lignin contente (PyGCMS) (%) | 36.9 | 29.2 | 19.8 | 14.5 | 15.3 | ||
| Guaiacyle (%) | 10.4 | 6.9 | 3.1 | 1.8 | 1.8 | ||
| Syringyle (%) | 23.2 | 17.4 | 14.0 | 10.7 | 11.8 | ||
| p-Hydroxyphenyle (%) | 3.2 | 4.9 | 2.6 | 1.9 | 1.6 | ||
| Phenolicse (%) | 0.04 | 0.1 | 0.1 | 0.1 | 0.0 | ||
| S/Ge | 2.2 | 2.5 | 4.6 | 5.8 | 6.7 | ||
| C/Le | 1.8 | 2.4 | 4.0 | 5.8 | 5.4 | ||
| Acetyl contentf (%) | 2.8 | 4.6 | 6.8 | 2.9 | |||
| M n (kDa) | 1.7 | 3.2 | 4.6 | 3.3 | |||
| M w (kDa) | 7.8 | 12.1 | 13.3 | 7.9 | |||
Subcritical water extraction of aspen wood: mass balances, composition, and molar mass
The sequential subcritical water extraction process was evaluated for aspen wood grown in greenhouse and field conditions (Tables 1 and 2, respectively), based on the yields, composition and average molar mass of the sequential extracts (A1–A4) and the residue (R). Complete mass balances show that between 11–12% of the total solids were extracted. The monosaccharide composition of the sequential extracts confirms that non-cellulosic polysaccharides (pectins, glucomannan and mainly glucuronoxylan) were primarily extracted in the SWE process, while cellulose was mainly left in the residue (Fig. 2A). The A1 and A2 extracts were rich in pectin, as revealed by the high content of diagnostic rhamnose (Rha), arabinose (Ara) galactose (Gal) and galacturonic acid (GalA) monosaccharides. As expected, these extracts from the greenhouse grown trees showed a higher pectin content compared to field grown trees (P ≤ 0.05, t-test). The higher pectin content may be related to the reduced amount of the secondary wall relative to the compound middle lamella in the more juvenile wood of greenhouse grown trees.43,45–48 A relatively high glucan content was observed in the 10 min extracts (A1) for both greenhouse and field samples, which could be attributed to residual starch, primary cell wall xyloglucan, and/or callose. Interestingly, the glucan content in the field extract at 10 min was more than double than for greenhouse (P ≤ 0.05, t-test), which could be related to the higher occurrence of starch (Tables 1 and 2, P ≤ 0.05, t-test) and probably also callose in the field due to overwintering and higher stress levels experienced by the trees. Indeed, both starch and callose are known to accumulate in responses to biotic and abiotic stresses.44 Mannan represents a minor part of hemicellulose composition in hardwoods.49 Aspen glucomannan is reported to have a mannose to glucose ratio of 1.3–2:1 and are attached with O-acetyl groups at C2 and C3 of mannopyranosyl residues.50 The SWE process showed a higher mannan population in the A1 extract compared with subsequent extracts suggesting preferential extraction of mannan along with pectic components during short extraction time (10 min). The high extractability of the mannan could be related to its spatial distribution in primary and secondary cell walls, and/or its interaction with cellulose and other cell wall components.51,52
 | ||
| Fig. 2 Composition of extracts obtained by SWE. (A) Monosaccharide composition of starting material (SM), SW extracts at 10, 20, 30 and 60 min, and residue (R) from greenhouse and field samples. (B) Molar mass distributions of field (F) and greenhouse (G) SW extracts. | ||
Xylan represents approximately between 31–32% of the aspen wood dry weight. About 25–30% of this xylan could be extracted in polymeric form with during the 2-hour SWE process (Tables 1, 2 and Fig. 2). However, 68% of the xylan in the aspen wood could not be fractionated and was left in the residue, whereas 3–7.5% of the total solid were lost during dialysis corresponding to low molar mass compounds. These yields are comparable to a 5-hour extraction from birch wood.2 The total xylan yield in the extracts was relatively higher in field grown aspen compared to that of greenhouse aspen which probably reflects the higher relative content of secondary walls in more mature wood from the field.45 As expected, the purity of xylan increased with extraction time, reaching about 93% of the carbohydrate composition for the A3 and A4 extracts. The molecular xylan structure in aspen was assessed by the glucuronosyl (mGlcA) and acetyl (Ac) substitution contents and patterns. The acetyl content increased gradually during extraction from A1 to A3 while it decreased significantly in A4, suggesting deacetylation of xylan during longer extraction periods. On the other hand, the MeGlcA:Xyl ratio gradually increased from A2 to A4 (Tables 1 and 2) with a significant difference between A3-A4 (P ≤ 0.05, ESI Table S4†), which indicate tighter glucuronation for the recalcitrant xylan fractions, as previously reported for birchwood.2
The molar mass profiles of the sequential SWE extracts (Fig. 2B) show similar trends between the greenhouse and the field starting materials. A1 extracts showed multimodal distributions, including a population of larger molar mass polymers (between 105–106 Da) that could be attributed to starch,2 and a population of small molar mass compounds (500 Da) that might be assigned to extractives. The xylan-rich extracts (A2–A4) show that the extracted compounds conserved their polymeric structure between 104 to 105 Da corresponding to hardwood xylan.2 The A2 extracts showed bimodal molar mass distributions, with a main population above 104 Da and a minor shoulder between 103–104 Da. These two distinct populations become more evident by comparing the signals from the refractive index (DRI) and ultraviolet (UV) detectors (ESI Fig. S1†). The high molar mass regions in the A2 extracts exhibited relatively weak UV signals that correlate with lower lignin content, whereas the low molar mass populations exhibited a prominent UV peak that indicates a larger aromatic abundance that could be assigned to lignin-carbohydrate complexes. This contrasts with previous observations on alkali-extracted xylan that can effectively remove the lignin moieties attached to the hemicelluloses.53 Finally, longer extraction times result in a shift of the molar mass distributions to lower values as evidenced in the A4 extracts, confirming the occurrence of hydrolytic events during longer exposure times. Indeed, Martínez-Abad et al. (2018)2 reported that the extraction in subcritical conditions is largely governed by mass transfer and diffusion kinetics of matrix cell wall polymers to the liquid phase, and that higher temperature and controlled mild acidic pH provide a compromise between molar mass, yield and purity. This explains the similar composition and molar mass in extracts A2–A3 and the drastic decrease in molar mass of A4 extract due to hydrolytic depolymerization (Fig. 2B).
The cell wall compositional analysis by pyrolysis GC-MS revealed that the lignin content and monolignol yield for G, S and H units decreased in successive extracts whereas the S/G ratio increased (Tables 1, 2 and P ≤ 0.05, ESI Table S5†). This suggests a high recalcitrance of guaiacyl units to water extraction process. The field grown trees showed higher S/G ratio in A2–A4 extracts than greenhouse grown trees, which is mainly contributed by their higher syringyl monolignol content. On the other hand, the carbohydrate to lignin (C/L) ratio increased over the time due to the high hemicellulose extraction yield. The different extractability of monolignol components between greenhouse and field grown aspen also suggests the possible change in the dynamics of molecular interaction between cell wall polymers during xylem juvenility-maturity transition.
The effect of SWE treatment on the morphology of the wood residue
The effect of the hydrothermal conditions on the morphology and ultrastructure of the cellulose-rich insoluble fractions after subcritical water treatment were evaluated by Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD) and scanning electron microscopy. SEM analysis of starting material showed the occurrence of cellulose fibrils embedded in matrix polymers, giving a smooth appearance to the inner surface layer (S3 layer) (Fig. 3A). Macrofibrils were readily observed in the compound middle lamellae (CML) region often stretched and fractured during the milling process (Fig. 3A). The effect of growth conditions on wood macrofibril dimensions was evaluated. The field grown trees showed larger macrofibril width compared to greenhouse grown trees (P ≤ 0.05, Table 3 and ESI Table S6†). In Pinus and Populus wood, macrofibril diameter is linearly related to lignin concentration.54 Lignin is assumed to infiltrate the cellulose macrofibril aggregates during lignification resulting in swelling of the aggregates.55 Moreover, the macrofibril size also depends on xylan content.56 More mature wood of field grown aspen might have higher lignin concentration and higher xylan content than that of greenhouse grown plants. SEM analysis was also used to evaluate the effect of SWE process on the structure and dimensions of cellulose macrofibrils in the residue. The residue from the greenhouse grown trees after SWE extraction showed loosening of macrofibrils in the form of a web-like organization (Fig. 3A). The field grown trees also showed swelling of the secondary wall and macrofibril loosening. Previous studies have reported that cellulose aggregation after pulping processes is related to the residual hemicellulose content, where cellulose fibrils aggregate and the macrofibril size increases with decreasing hemicelluloses content.57,58 However, in the present study, the width of macrofibrils showed a slight reduction between aspen wood and residue after hydrothermal treatment. This suggests that the lignin infiltrated within the macrofibril organization somehow preserves its dimension after hydrothermal treatment and prevents macrofibril aggregation, in contrast to cellulose fibrils observed in wood pulps that had been subjected to delignification processes. The changes in the cell wall architecture during SWE were further evident from the BET analysis of lignocellulose porosity. A significant increase in surface area was noticed in the residue compared to that of starting material (P ≤ 0.05, ESI Table S7† and Table 3), which is related to the preferential extraction of acetylated GX during SWE. The increase in porosity was twofold higher in greenhouse samples compared to field samples (P ≤ 0.05, ESI Table S7†). This could be attributed to the more juvenile and less compact wood cell wall structure in greenhouse samples compared to the field ones.59 | ||
| Fig. 3 Structural characteristics of starting material (SM) and residue (R) of aspen wood grown in Greenhouse (G) and field (F) conditions. (a) FTIR spectra, (b) X-ray diffraction pattern and (c) Scanning electron microscopy (SEM), SEM scale bar = 200 nm. | ||
| G-SM | F-SM | G-R | F-R | |
|---|---|---|---|---|
| Mean ± STDev of 3 technical replicates. Asterisk (*) indicates statistically significant difference for significant difference between SWE treated and SWE untreated samples of same growth condition determined by Student's t-test P ≤ 5% (*); P ≤ 1% (**). Indices calculated at TCI = α1364/2892; LOI = α1418/893; HBI = α3338/1334. | ||||
| Macrofibril width (nm) | 14 ± 0.6 | 17.1 ± 1.0 | 12.0 ± 0.4 | 15.0 ± 0.5 |
| BET surface area (m2 g−1) | 1.40 ± 0.04 | 1.86 ± 0.17 | 3.08 ± 0.20** | 2.2 ± 0.17* |
| Lateral order index | 0.80 ± 0.08 | 0.77 ± 0.10 | 0.91 ± 0.10 | 0.91 ± 0.20 |
| Hydrogen bond intensity | 0.99 ± 0.2 | 0.95 ± 0.3 | 0.91 ± 0.2 | 0.91 ± 0.2 |
| Total crystallinity intensity | 1.63 ± 0.01 | 1.56 ± 0.03 | 1.76 ± 0.02 | 1.58 ± 0.06 |
| Degree of crystallinity (%) | 42.8 ± 0.11 | 43.7 ± 0.05 | 48.6 ± 0.1* | 50.2 ± 0.6* |
The FTIR spectra from the starting aspen wood showed the chemical features attributed to the specific cell wall components (Fig. 3B), including C–H stretching at 2800–3000 cm−1, O–H stretching at 3300–3600 cm−1, and several peaks in the region between 700 and 1750 cm−1 that have contributions from both carbohydrates and lignin. The evidence of the preferential extraction of acetylated glucuronoxylan was evident from a decrease in the intensities of 1735 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ester) and 1230 cm−1 (–C–O– stretching band) in the FTIR spectra of the SWE residues, which are assigned for the ester stretching of the acylated hemicelluloses.38,39 On the other hand, the increase at 1590 cm−1 (lignin skeletal vibrations from the C–C,38,39 1080 cm−1 and 1055 cm−1 (C–O and C–O–C of cellulose38,39 further confirmed the relative enrichment of cellulose and lignin in the SWE residue (R). Moreover, ATR-FTIR was used to investigate the crystallinity and hydrogen bonding characteristics of the cellulose microfibrils.60 The Lateral order index (LOI, α1418/894), hydrogen bond intensity (HBI, α3336/1336), and total crystallinity intensity (TCI, α1364/2892), based on the ratio of absorbance bands at specific wavenumbers, were used to interpret qualitative changes in cellulose crystallinity.60 An increase of LOI and TCI were observed in the residues from SWE treatment from the field and the greenhouse compared to the starting material (Table 3), which suggest an increase in overall crystallinity of the residue due to selective removal of amorphous hemicellulose material.
O ester) and 1230 cm−1 (–C–O– stretching band) in the FTIR spectra of the SWE residues, which are assigned for the ester stretching of the acylated hemicelluloses.38,39 On the other hand, the increase at 1590 cm−1 (lignin skeletal vibrations from the C–C,38,39 1080 cm−1 and 1055 cm−1 (C–O and C–O–C of cellulose38,39 further confirmed the relative enrichment of cellulose and lignin in the SWE residue (R). Moreover, ATR-FTIR was used to investigate the crystallinity and hydrogen bonding characteristics of the cellulose microfibrils.60 The Lateral order index (LOI, α1418/894), hydrogen bond intensity (HBI, α3336/1336), and total crystallinity intensity (TCI, α1364/2892), based on the ratio of absorbance bands at specific wavenumbers, were used to interpret qualitative changes in cellulose crystallinity.60 An increase of LOI and TCI were observed in the residues from SWE treatment from the field and the greenhouse compared to the starting material (Table 3), which suggest an increase in overall crystallinity of the residue due to selective removal of amorphous hemicellulose material.
X-ray diffraction (XRD) was performed on the original solid material and after SWE treatment to obtain a deeper understanding of the crystalline arrangement of the aspen biomass (Fig. 3c). The XRD patterns from all the samples exhibit the typical crystalline peaks of cellulose I at 15–16° (100) and 22° (200), as previously reported61–63 The crystal peaks were investigated with the deconvolution method, in order to obtain the relative crystallinity of the different samples.64 The XRD results verified the preliminary assessment from FTIR, indicating that the residue after SWE have higher crystallinity after the extraction of hemicelluloses and result in more pronounced XRD crystalline peaks (Table 3 and ESI Table S8†). We have qualitatively normalized the overall degree of crystallinity to the cellulose content in the starting materials and residues after SWE, to assess whether the hydrothermal process had an impact on the specific crystallinity of the cellulose components. The comparison suggests that the hemicelluloses in the starting materials contribute to their overall crystallinity, and that the SWE treatment somehow influences the organization of the cellulose components reducing the specific crystallinity of the cellulose microfibrils. This different organization of the cellulose components will have an influence on the recalcitrance and saccharification potential of the aspen biomass, as will be discussed later.
Subcritical water enhances the saccharification potential of aspen wood
To understand the influence of hydrothermal extraction of matrix polymers on reducing lignocellulose recalcitrance of aspen wood, we evaluated the effect of SWE on enzymatic saccharification of the SW residue in comparison with the untreated starting material and conventional acid pretreatment as a reference. The effect of applying the conventional acid treatment on the subcritical water residue prior to saccharification was also evaluated. In both greenhouse and field grown aspen, subcritical water extraction clearly improved the saccharification efficiency of wood, as evidenced by the significantly higher glucose production rate (GPR) compared to the control sample without any pretreatment (Fig. 4A). Moreover, SWE treatment increased fourfold the total glucose and xylose yields after saccharification of the residue, compared to those of starting material without any pretreatment (Fig. 4B and ESI Tables S9, S10†). This can be correlated with the higher porosity, lower inherent crystallinity of the cellulose, and the increased fibrillation caused by the SWE treatment compared to the starting material, as revealed by the XRD, SEM and BET results (Fig. 3). The removal of hemicelluloses attached to cellulose microfibrils, the subsequent reduced acetyl content, and the increase in surface area in the residues caused by SWE seems to enhance enzymatic accessibility towards cellulose, leading to efficient conversion of glucose during saccharification without pretreatment. Indeed, a lower surface area of wood starting material compared to the SWE residue due to hemicellulose coating the cellulose surfaces and the presence of lignin, along with the high crystallinity of cellulose are major limiting factors for effective access of hydrolytic enzymes to cellulose during saccharification process.14,15 Interestingly, the starting materials from greenhouse grown trees showed relatively higher glucose production rate compared to that of field grown trees with and without SWE process (Fig. 4A and ESI Table S11†), suggesting that enzyme accessibility might differ in both cases due to composition (i.e. lower lignin content) and wood cell ultrastructure (i.e. smaller microfibril diameter and degree of crystallinity).23,65 | ||
| Fig. 4 Enzymatic saccharification of aspen lignocellulose biomass from greenhouse (G) and field (F) under four different treatment conditions as depicted in Fig. 1B: untreated starting material (NT), subcritical water (SW), acid pretreatment (AP) and subcritical water followed by acid pretreatment (SW/AP). (a) Glucose production rate (g L−1 h−1). (b) Monosaccharide yield for glucose (Glc), xylose (Xyl) and total sugars from the enzymatic saccharification. The complete released monosaccharides are presented in ESI Tables S9 and S10† for field and greenhouse aspen, respectively. PL refers to the liquid hydrolysate obtained after acid pretreatment (AP) containing fermentable sugars. The asterisks (*) indicate statistically significant differences between the enzymatic saccharification of the SW residue compared to the other treatments determined by Student's t-test (ESI Tables S12 and S13†): P ≤ 5% (*); P ≤ 1% (**); P ≤ 0.1% (***). n.s: not statistically significant. | ||
Comparing the conventional acid pretreatment (AP) with the subcritical water (SW) process, interesting conclusions can be drawn. The acid pretreatment causes a significant increase in the glucose production rate compared to the subcritical water process, related to the higher glucose content released in the acid hydrolysate (PL) and the subsequent enzymatic hydrolysis (EH). However, comparing the total sugars, the SW process enables similar release compared to the acid pretreatment, which can be attributed to the enzymatic release of xylose from the residual xylan that could not be extracted in the SW process. Indeed, most of the xylose is released during the acid pretreatment in the liquid phase (PL) and therefore is not available in the following enzymatic saccharification step. The fact that the xylose released in the saccharification step after SW extraction is similar to the xylose released in the acid hydrolysate (PL) suggests the occurrence of degradation, as it will be confirmed later in the full mass balances. Finally, performing an acid pretreatment step after subcritical water significantly increases the release of glucose release and overall total sugars compared to both the subcritical water and the acid pretreatment performed alone.
Mass balances were performed on the total polysaccharides and the xylan/xylose for the four different treatments evaluated in this study (Table 4). The acid pretreatment still offers higher total yields that the subcritical water process, all in the form of hydrolysed monosaccharides, although the SW process is able to preserve the polymeric structure of the xylan in the extraction step prior to saccharification. Interestingly, the combination of SW and AP can reach almost 85–90% conversion of the initial polysaccharides into fermentable sugars. When comparing the xylan yields alone, the SW process clearly outperforms the traditional acidnhydrolysis, which could be due to the degradation of the xylose into furan derivatives caused by the acid conditions. All in all, our results suggests that the hemicellulose first extraction by SWE process is an efficient process to reduce the requirement of acid pretreatment during the conversion of wood biomass for biorefinery applications, by on one hand preserving the polymeric structure of the extracted xylan that could be used for valuable material applications (i.e. as barrier or biomedical matrices) and enabling the subsequent conversion of the residual biomass into fermentable sugars by enzymatic saccharification.
| Field (F) | Greenhouse (G) | |||||||
|---|---|---|---|---|---|---|---|---|
| NT | SW | AP | SW/AP | NT | SW | AP | SW/AP | |
| SW (subcritical water extraction); PL (hydrolysate acid pre-treatment); EH (hydrolysate enzymatic saccharification). | ||||||||
| SWE polysaccharides (%) | — | 11.9 | — | 11.9 | — | 11.1 | — | 11.1 |
| Total sugars PL (%) | — | — | 17.8 | 18.6 | — | — | 23.5 | 17.8 |
| Total sugars EH (%) | 6.8 | 40.9 | 41.2 | 60.3 | 10.1 | 38.4 | 41.7 | 56.7 |
| Total mass balance (%) | 6.8 | 52.8 | 59.0 | 90.8 | 10.1 | 49.5 | 65.2 | 85.6 |
| SWE xylan (%) | — | 29.6 | — | 29.6 | — | 24.5 | — | 24.5 |
| Total xylose PL (%) | — | — | 30.9 | 31.2 | — | — | 43.5 | 33.0 |
| Total xylose EH (%) | 3.1 | 28.1 | 1.7 | 3.2 | 4.5 | 27.5 | 1.5 | 3.4 |
| Xylan/xylose balance (%) | 3.1 | 57.7 | 32.5 | 64.0 | 4.5 | 52.0 | 45.0 | 60.9 |
Conclusions
The present study demonstrates the efficacy of subcritical water extraction in the fractionation of non-cellulosic polysaccharides from aspen wood and validates the process for aspen trees grown in controlled greenhouse and real field conditions. Greenhouse conditions enable the control of the different environmental parameters during aspen growth, whereas field growth represent a more realistic case close to industrial application, where biotic and abiotic conditions also play a role during lignocellulose development. For both conditions, subcritical water induces morphological changes in the residual lignocellulosic biomass, reducing its native recalcitrance to enzymatic hydrolysis and favoring its subsequent saccharification. The sequential SWE process of aspen wood extracts pectic polysaccharides at short times (below 10 min), whereas longer extraction times (between 2–60 min) promote the extraction of hemicelluloses in polymeric form, mainly acetylated glucuronoxylan. Longer extraction times (>60 min) induce deacetylation of the isolated xylan and a reduction in the molecular weight, caused by hydrolytic processes under the subcritical water conditions. When comparing aspen grown in greenhouse and field conditions, the SWE shows very similar behaviour, with only slight differences in the pectin and glucan content in the short time extracts, which could be assigned to the different stages of wood development between growth conditions and the response to environmental stress (abiotic and biotic) in the field trees. The fractionation of cell wall matrix polysaccharides by SWE process induces structural changes in the lignocellulosic biomass, including higher porosity and lower fibril dimensions. These structural and chemical changes lead to significantly higher yields of glucose and xylan in enzymatic saccharification compared to yields from not treated aspen wood chips. We hypothesize that the saccharification potential was enhanced by the increased accessibility of hydrolytic enzymes into the residual biomass, caused by the removal of acetylated glucuronoxylan and associated changes in the surface morphology and cell wall porosity. Compared to the traditional acid pretreatment that hydrolyses the hemicellulose components, SW offers comparable yields of fermentable sugars with the advantage of providing an initial hemicellulose fraction with preserved macromolecular structure, which could be used as a biopolymeric matrix in barrier and structural applications. This study highlights the potential of SWE to extract native hemicellulose, and also as a green pretreatment strategy to reduce biomass recalcitrance contributed by non-cellulosic polysaccharides, thereby decreasing the requirement of acid pre-treatments during biochemical conversion of aspen wood.Conflicts of interest
Francisco Vilaplana reports a relationship with Oatly AB that includes employment.Acknowledgements
The authors acknowledge the financial support from the Swedish Research Council (Project Grant 2020-04720) and the Knut and Alice Wallenberg Foundation (KAW) to the Wallenberg Wood Science Centre. F. V. and P. S. acknowledge the BioUPGRADE project for the financial contribution. This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 964764. The content presented in this document represents the views of the authors, and the Commission is not responsible for any use that may be made of the information it contains. The authors are thankful for the access to the facilities and the technical assistance of the Umeå Centre for Electron Microscopy – National Microscopy Infrastructure (UCEM-NMI) and Bioanalytical Platform, Umea Plant Science Centre (UPSC), Umeå for access to Pyrolysis-GCMS analysis.References
- Y. M. Bar-On, R. Phillips and R. Milo, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6506–6511 CrossRef CAS PubMed.
- A. Martínez-Abad, N. Giummarella, M. Lawoko and F. Vilaplana, Green Chem., 2018, 20, 2534–2546 RSC.
- X. Yang, D. Liu, H. Lu, D. J. Weston, J.-G. Chen, W. Muchero, S. Martin, Y. Liu, M. M. Hassan, G. Yuan, U. C. Kalluri, T. J. Tschaplinski, J. C. Mitchell, S. D. Wullschleger and G. A. Tuskan, BioDesign Res., 2021, 9798714 CAS.
- D. Keegan, B. Kretschmer, B. Elbersen and C. Panoutsou, Biofuels, Bioprod. Biorefin., 2013, 7, 193–206 CrossRef CAS.
- B. Thomas, M. C. Raj, B. K. Athira, H. M. Rubiyah, J. Joy, A. Moores, G. L. Drisko and C. Sanchez, Chem. Rev., 2018, 118, 11575–11625 CrossRef CAS PubMed.
- E. Kontturi, P. Laaksonen, M. B. Linder, A. H. Nonappa, A. H. Gröschel, O. J. Rojas and O. Ikkala, Adv. Mater., 2018, 30, 1703779 CrossRef PubMed.
- T. Li, C. Chen, A. H. Brozena, J. Y. Zhu, L. Xu, C. Driemeier, J. Dai, O. J. Rojas, A. Isogai, L. Wågberg and L. Hu, Nature, 2021, 590, 47–56 CrossRef CAS PubMed.
- S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17, 1077–1087 RSC.
- W. J. Liu, H. Jiang and H. Q. Yu, Green Chem., 2015, 17, 4888–4907 RSC.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- W. Zhao, B. Simmons, S. Singh, A. Ragauskas and G. Cheng, Green Chem., 2016, 18, 5693–5700 RSC.
- A. Duval and M. Lawoko, React. Funct. Polym., 2014, 85, 78–96 CrossRef CAS.
- K. S. Mikkonen, Green Chem., 2020, 22, 1019–1037 RSC.
- M. C. McCann and N. C. Carpita, J. Exp. Bot., 2015, 66, 4109–4118 CrossRef CAS PubMed.
- M. E. Himmel, S. Y. Ding, D. K. Johnson, W. S. Adney, M. R. Nimlos, J. W. Brady and T. D. Foust, Science, 2007, 315, 804–807 CrossRef CAS PubMed.
- P. A. Penttilä, A. Várnai, J. Pere, T. Tammelin, L. Salmén, M. Siika-aho, L. Viikari and R. Serimaa, Bioresour. Technol., 2013, 129, 135–141 CrossRef PubMed.
- E. Heinonen, G. Henriksson, M. E. Lindström, F. Vilaplana and J. Wohlert, Carbohydr. Polym., 2022, 285, 119221 CrossRef CAS PubMed.
- J. Berglund, D. Mikkelsen, B. M. Flanagan, S. Dhital, S. Gaunitz, G. Henriksson, M. E. Lindström, G. E. Yakubov, M. J. Gidley and F. Vilaplana, Nat. Commun., 2020, 11, 4692 CrossRef CAS PubMed.
- A. Martínez-Abad, A. Jiménez-Quero, J. Wohlert and F. Vilaplana, Green Chem., 2020, 22, 3956–3970 RSC.
- G. Gallina, E. R. Alfageme, P. Biasi and J. García-Serna, Bioresour. Technol., 2018, 247, 980–991 CrossRef CAS PubMed.
- P. O. Kilpeläinen, S. S. Hautala, O. O. Byman, L. J. Tanner, R. I. Korpinen, M. K. J. Lillandt, A. V. Pranovich, V. H. Kitunen, S. M. Willför and H. S. Ilvesniemi, Green Chem., 2014, 16, 3186–3194 RSC.
- R. C. Rudjito, A. Jiménez-Quero, M. Hamzaoui, S. Kohnen and F. Vilaplana, Green Chem., 2020, 22, 8337–8352 RSC.
- S. Pramod, M. L. Gandla, M. Derba-Maceluch, L. J. Jönsson, E. J. Mellerowicz and S. Winestrand, Front. Plant Sci., 2021, 12, 704960 CrossRef PubMed.
- P. Sannigrahi, A. J. Ragauskas and G. A. Tuskan, Biofuels, Bioprod. Biorefin., 2010, 4, 209–226 CrossRef CAS.
- E. N. Donev, M. Derba-Maceluch, Z. Yassin, M. L. Gandla, P. Sivan, S. E. Heinonen, V. Kumar, G. Scheepers, F. Vilaplana, U. Johansson, M. Hertzberg, B. Sundberg, S. Winestrand, A. Hörnberg, B. Alriksson, L. J. Jönsson and E. J. Mellerowicz, Plant Biotechnol. J., 2023, 1005–1021 CrossRef CAS PubMed.
- M. L. Gandla, M. Derba-Maceluch, X. Liu, L. Gerber, E. R. Master, E. J. Mellerowicz and L. J. Jönsson, Phytochemistry, 2015, 112, 210–220 CrossRef PubMed.
- Z. Wang, S. Winestrand, T. Gillgren and L. J. Jönsson, Biomass Bioenergy, 2018, 109, 125–134 CrossRef CAS.
- F. Bertaud, A. Sundberg and B. Holmbom, Carbohydr. Polym., 2002, 48, 319–324 CrossRef CAS.
- L. S. McKee, H. Sunner, G. E. Anasontzis, G. Toriz, P. Gatenholm, V. Bulone, F. Vilaplana and L. Olsson, Biotechnol. Biofuels, 2016, 9, 2 CrossRef PubMed.
- J. F. Saeman, W. E. Moore, R. L. Mitchell and M. A. Millett, Tappi J., 1954, 37, 336–343 CAS.
- C. E. Foster, T. M. Martin and M. Pauly, J. Visualized Exp., 2010, 37, 1745 Search PubMed.
- M. Stitt, R. M. C. Lilley, R. Gerhardt and H. W. Heldt, Methods Enzymol., 1989, 174, 518–552 CAS.
- J. H. M. Hendriks, A. Kolbe, Y. Gibon, M. Stitt and P. Geigenberger, Plant Physiol., 2003, 133, 838–849 CrossRef CAS PubMed.
- A. M. Smith and S. C. Zeeman, Nat. Protoc., 2006, 1, 1342–1345 CrossRef CAS PubMed.
- L. Gerber, M. Eliasson, J. Trygg, T. Moritz and B. Sundberg, J. Anal. Appl. Pyrolysis, 2012, 95, 95–100 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- S. Lowell and J. E. Shields, in Powder Surface Area and Porosity, ed. S. Lowell and J. E. Shields, Springer Netherlands, Dordrecht, 1991, pp. 14–29. DOI:10.1007/978-94-015-7955-1_4.
- F. Nindiyasari, E. Griesshaber, T. Zimmermann, A. P. Manian, C. Randow, R. Zehbe, L. Fernandez-Diaz, A. Ziegler, C. Fleck and W. W. Schmahl, J. Compos. Mater., 2016, 50, 657–672 CrossRef CAS.
- M. Fasoli, R. Dell’Anna, S. Dal Santo, R. Balestrini, A. Sanson, M. Pezzotti, F. Monti and S. Zenoni, Plant Cell Physiol., 2016, 57, 1332–1349 CrossRef CAS PubMed.
- S. I. Andersen, J. O. Jensen and J. G. Speight, Energy Fuels, 2005, 19, 2371–2377 CrossRef CAS.
- C. Lars, in Economic Effects of Biofuel Production, ed. B. Marco Aurélio dos Santos, IntechOpen, Rijeka, 2011, ch. 8, DOI:10.5772/17198.
- S. Adamopoulos, E. Voulgaridis and C. Passialis, Holz Roh- Werkst., 2005, 63, 327–333 CrossRef CAS.
- U. Sahlberg, L. Salmén and A. Oscarsson, Wood Sci. Technol., 1997, 31, 77–86 CrossRef CAS.
- D. Ellinger and C. A. Voigt, Ann. Bot., 2014, 114, 1349–1358 CrossRef CAS PubMed.
- F. C. Bao, Z. H. Jiang, X. M. Jiang, X. X. Lu, X. Q. Luo and S. Y. Zhang, Wood Sci. Technol., 2001, 35, 363–375 CrossRef CAS.
- R. Funada, H. Abe, O. Furusawa, H. Imaizumi, K. Fukazawa and J. Ohtani, Plant Cell Physiol., 1997, 38, 210–212 CrossRef CAS.
- J. C. Mortimer, G. P. Miles, D. M. Brown, Z. Zhang, M. P. Segura, T. Weimar, X. Yu, K. A. Seffen, E. Stephens, S. R. Turner and P. Dupree, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17409–17414 CrossRef CAS PubMed.
- R. L. Silveira, S. R. Stoyanov, S. Gusarov, M. S. Skaf and A. Kovalenko, J. Am. Chem. Soc., 2013, 135, 206–211 Search PubMed.
- T. E. Timell, Wood Sci. Technol., 1967, 1, 45–70 CrossRef CAS.
- A. Teleman, M. Nordström, M. Tenkanen, A. Jacobs and O. Dahlman, Carbohydr. Res., 2003, 338, 525–534 CrossRef CAS PubMed.
- M. G. Handford, T. C. Baldwin, F. Goubet, T. A. Prime, J. Miles, X. Yu and P. Dupree, Planta, 2003, 218, 27–36 CrossRef CAS PubMed.
- J. S. Kim and G. Daniel, Planta, 2012, 236, 1275–1288 CrossRef CAS PubMed.
- P. M.-A. Pawar, M. Derba-Maceluch, S.-L. Chong, M. L. Gandla, S. S. Bashar, T. Sparrman, P. Ahvenainen, M. Hedenström, M. Özparpucu, M. Rüggeberg, R. Serimaa, M. Lawoko, M. Tenkanen, L. J. Jönsson and E. J. Mellerowicz, Biotechnol. Biofuels, 2017, 10, 98 CrossRef PubMed.
- L. Donaldson, Wood Sci. Technol., 2007, 41, 443–460 CrossRef CAS.
- L. A. Donaldson, Wood Sci. Technol., 1994, 28, 111–118 CrossRef CAS PubMed.
- J. J. Lyczakowski, M. Bourdon, O. M. Terrett, Y. Helariutta, R. Wightman and P. Dupree, Front. Plant Sci., 2019, 10, 1398 CrossRef PubMed.
- I. Duchesne, E. Hult, U. Molin, G. Daniel, T. Iversen and H. Lennholm, Cellulose, 2001, 8, 103–111 CrossRef CAS.
- E. L. Hult, P. T. Larsson and T. Iversen, Polymer, 2001, 42, 3309–3314 CrossRef CAS.
- J. Fromm, in Cellular Aspects of Wood Formation, ed. J. Fromm, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 3–39, DOI:10.1007/978-3-642-36491-4_1.
- J. Široký, R. S. Blackburn, T. Bechtold, J. Taylor and P. White, Cellulose, 2010, 17, 103–115 CrossRef.
- S. Collazo-Bigliardi, R. Ortega-Toro and A. Chiralt-Boix, Carbohydr. Polym., 2018, 191, 205–215 CrossRef CAS PubMed.
- R. Moriana, F. Vilaplana and M. Ek, Carbohydr. Polym., 2016, 139, 139–149 CrossRef CAS PubMed.
- R. Requena, A. Jiménez-Quero, M. Vargas, R. Moriana, A. Chiralt and F. Vilaplana, ACS Sustainable Chem. Eng., 2019, 7, 13167–13177 CrossRef.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels, 2010, 3, 10 CrossRef PubMed.
- M. Derba-Maceluch, F. Amini, E. N. Donev, P. M. A. Pawar, L. Michaud, U. Johansson, B. R. Albrectsen and E. J. Mellerowicz, Front. Plant Sci., 2020, 11, 651 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01020a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |