
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lignin-based bisguaiacol diisocyanate: a green route for the synthesis of biobased polyurethanes†
Sébastien
Lemouzy‡
 a,
Aliénor
Delavarde‡
a,
Frédéric
Lamaty
b,
Xavier
Bantreil
bc,
Julien
Pinaud
a and
Sylvain
Caillol
*a
a,
Aliénor
Delavarde‡
a,
Frédéric
Lamaty
b,
Xavier
Bantreil
bc,
Julien
Pinaud
a and
Sylvain
Caillol
*a
aICGM, Univ Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: sylvain.caillol@enscm.fr
bIBMM, Univ Montpellier, CNRS, ENSCM, Montpellier, France
cInstitut universitaire de France (IUF), France
First published on 1st June 2023
Abstract
The synthesis of a new biobased aromatic diisocyanate derived from lignocellulosic raw material, namely guaiacol and vanillyl alcohol, through phosgene-free route offers the prospect of greener approaches for isocyanate production and the polyurethane industry. Indeed, bisguaiacol F diisocyanate (BGI) was obtained via a three-step process from readily available bisguaiacol F (BGF), involving conversion of aromatic amine into aromatic isocyanate. The unusual transition-metal-free conversion of BGF to bisguaiacol F diamine (BGA) was performed by a two-step approach: (a) the Williamson-type alkylation of BGF and then (b) the base-promoted Smiles rearrangement of bis O-alkylated BGF. In order to improve the sustainability of this process, the first step was realized under solvent-free mechanochemical conditions, and the second step was performed using two different activating methods: thermal and microwave. The thermal process provided an isolated BGA yield of ca. 70%. Microwave activation proved to be an interesting alternative, although a lower yield (32%) of the desired BGA was achieved. Finally, the diisocyanate synthesis was performed via a phosgene-free/room temperature protocol using di-tert-butyl dicarbonate in the presence of a catalytic amount of 4-dimethylaminopyridine (DMAP). Two polyurethane thermosets were designed and synthesized using the aromatic diisocyanates, biobased BGI and petrochemical-based methylene diphenyl diisocyanate (MDI), by a solvent-free two-step polymerization. Their thermo-chemical properties as well as their reactivities for polymer synthesis were evaluated. These preliminary results suggest that BGI could be potentially used as a nearly fully biobased surrogate for MDI.
Introduction
Within a few decades, polyurethanes have emerged as a powerful class of organic polymers. These materials possess a wide range of industrial applications,1 ranging from foams to CASE (Coatings, Adhesives, Sealants and Elastomers), owing from the increasing array of polyfunctional alcohol and isocyanate monomers available. Until very recently, polyurethane materials were synthesized from petrochemical-based compounds with high toxicity. However, with the growing concerns regarding raw material origin and resource scarcity in the upcoming decades,2 the design of monomers based on renewable sources is becoming a need.Therefore, both academic and industrial research have focused on the enhancement of the sustainability in polyurethanes by developing a wide range of new bio-based polyols.3,4 In particular, polyurethanes can be easily prepared from renewable resources such as plant oils,5 as unsaturated fatty acids have proven to be efficient precursors of polyols6–8 and isocyanate9 monomers. However, bio-based poly-isocyanate monomers have seen less development,10 especially aromatic isocyanates, which have received very little attention.11 One of the main reasons for this lack of development is the need to use phosgene gas to obtain isocyanate molecules. Indeed, the acute toxicity of phosgene gas is a major hinder to the development of phosgene-based industrial processes. Since 1928, when the first accident related to phosgene occurred in Hamburg,12 numerous industrial incidents and accidents involving phosgene production and/or use have been reported. Especially, lethal industrial accidents have been reported by DuPont13 and BASF14 in the last decade. Henceforth, phosgene is always produced in situ where isocyanate synthesis occurs and drastic regulations (such as the Seveso directive in the European Union) have been implemented to make sure that the companies that use phosgene possess the appropriate technology and knowledge to avoid such severe accidents. Consequently, phosgene-free routes have been explored to design isocyanate compounds in a greener way. The triphosgenation of amine with bis(trichloromethyl) carbonate (triphosgene) can be explored to synthesize isocyanate compound. This one-step reaction is easy to implement but does not prevent phosgene production during the reaction. Thermolysis of urethanes15 and reductive carbonylation of nitro-compounds16 are both energy-consuming, requiring high reaction temperatures (>200 °C). Regarding the reductive carbonylation reaction, high-pressure of carbon monoxide (85 atm) is used, which severely impacts the sustainability of the process. On the other hand, since the thermolysis of urethane requires a reagent with urethane functions, the synthetic possibilities of this method are limited. Another promising approach is the preparation of isocyanate via the Curtius rearrangement, but the preparation and handling of acyl azide intermediates raises safety concerns, preventing this method from reaching industrial scale-up production. However, recent development in this chemistry using continuous-flow reaction allow for more promising environmental impact.17,18
In the 1990s, Knolker reported a very mild method for the preparation of isocyanates.19,20 This reaction operates in the presence of 4-dimethylaminopyridine (DMAP) as the catalyst (usually 10 mol%) using cheap and readily available di-tert-butyl dicarbonate (Boc2O) as the carbonyl source,21–23 in acetonitrile or dichloromethane at 25 °C. The main limitation of this method lies in the choice of the amine starting material, as only anilines and hindered alkyl amines are suitable for this reaction. More nucleophilic primary alkyl amines often yield ureas as the major side-product. We thought that this process could be viewed as a much greener alternative to the phosgene route (room temperature, much less hazardous reagents), especially since Boc2O can be synthesized directly from CO2.23 However, to our great surprise, it has hardly ever been used for the synthesis of isocyanate monomers for the preparation of polyurethanes materials.24 Thus, in our quest to design a greener alternative to methylene diphenyl diisocyanate (MDI), we decided to synthesize a structurally analogous molecule from lignin feedstock using the Boc2O route (Scheme 1B).
 | ||
| Scheme 1 Process for the preparation of MDI (A) and synthetic pathway of BGI (B). | ||
Methylene diphenyl diisocyanate (MDI) is a diaromatic diisocyanate with three common isomers (4,4′-MDI, 2,4′-MDI and 2,2′-MDI) produced by a 4 step synthesis starting from aniline.25,26 The process for producing MDI (Scheme 1A) involves the reaction of aniline with formaldehyde in the presence of an acid catalyst to produce a mixture of methylene (diphenyldiamines) (MDA) and polymeric aniline (pMDA).
Further reaction with phosgene provides the corresponding poly-isocyanates (step C). Finally, isolation of MDI isomers is performed through distillation (step D). Aniline, involved in the first step, is classified as highly toxic for human beings and environment, CMR and corrosive. Formaldehyde is fatal if inhaled and is also classified as highly corrosive, CMR and flammable. Therefore, raw materials for MDI synthesis are highly toxic compounds obtained from petro-based resources. Indeed, aniline is usually obtained from nitration of benzene followed by nitro to amine reduction. Product of the second step is a mixture of MDA isomers and polymeric aniline that is deemed as more toxic than MDI isomers. Finally, in the third step, phosgene gas is used to transform amine into isocyanate. Phosgene is classified by EU as highly toxic material. It has the highest NFPA 704 health rating of 4, “very short exposure could cause death or major residual injury”.27 Since MDI is the most produced diisocyanate with a total European production capacity of 770![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 metric tons in 2020, the design of an analogous bio-based aromatic isocyanate could be of high interest. In addition to use renewable feedstocks, a greener and safer synthetic pathway is proposed in order to avoid the use of lethal compounds such as aniline, formaldehyde and phosgene gas.
000 metric tons in 2020, the design of an analogous bio-based aromatic isocyanate could be of high interest. In addition to use renewable feedstocks, a greener and safer synthetic pathway is proposed in order to avoid the use of lethal compounds such as aniline, formaldehyde and phosgene gas.
Results and discussion
Our initial idea was to use bisguaiacol F (BGF) as the starting material. Indeed, BGF can be easily synthesized by acid-mediated Friedel–Crafts-type alkylation28,29 of guaiacol with vanillyl alcohol, which are both bio-based and readily available lignin-derived phenols. We selected the procedure of Periyasamy et al.28 (1:1 ratio on both bio-based phenol, sulfuric acid, EtOH, 100 °C) for multi-gram synthesis of BGF. Using these conditions, we were able to obtain 125 g of BGF in an approximately 3:1 mixture (p,p′-BGF: o,p′-BGF ratio) of isomers (Scheme 2).
 | ||
| Scheme 2 Synthesis of BGF according to literature procedure.28 | ||
We then turned our attention to the transformation of BGF 3 into the corresponding diamine 6 (BGA). However, the direct amination of phenol derivatives, although very attractive, is usually difficult and requires harsh conditions and expensive transition-metal catalysts to achieve effective dearomative “hydrogen-borrowing strategy” (Scheme 3A).30–35 Moreover, the substrate scope of such transformations is somewhat limited, especially when using ammonia as the nitrogen nucleophile.31,32 Alternatively, the amination of phenols via transition-metal-free Smiles rearrangement has also been described (Scheme 3B).36–39
 | ||
| Scheme 3 Strategies for the amination of phenol derivatives. | ||
The reaction operates via a three-step process, first a Williamson-type O-alkylation of the phenolic substrate, then the subsequent base-promoted Smiles rearrangement of the pendant amide moiety, followed by the base-promoted amide cleavage. Such a rearrangement is highly kinetically disfavoured, owing from the loss of aromaticity in the Meisenheimer-type intermediate (especially when using a phenol substituted with electron-donating groups), and therefore high temperatures (>100 °C) are usually required. In addition, it also seems that the formation of the Meisenheimer intermediate is accelerated by Thorpe–Ingold effect, as highlighted by the superior reactivity of secondary aryl ethers (R2 = Me or Ph) versus primary ones (R2 = H).37 Interestingly, this method features a broad scope of phenol derivatives, even for the formation of unsubstituted anilines.37 Another advantageous feature of the reaction is the possibility to perform the two-step sequence in one-pot36–38 as the conditions used for the O-alkylation are very close to the ones required for the Smiles rearrangement/amide cleavage. Therefore, we settled for a strategy based on the transition-metal-free Smiles rearrangement of bisguaiacol F. Initial attempts using bisguaiacol F 3 and 2-bromopropionamide 4 as the amination reagent in a one-pot process in DMSO under previously reported conditions31,40,41 resulted in very low yields (<10%) of bisguaiacol F diamine 6.
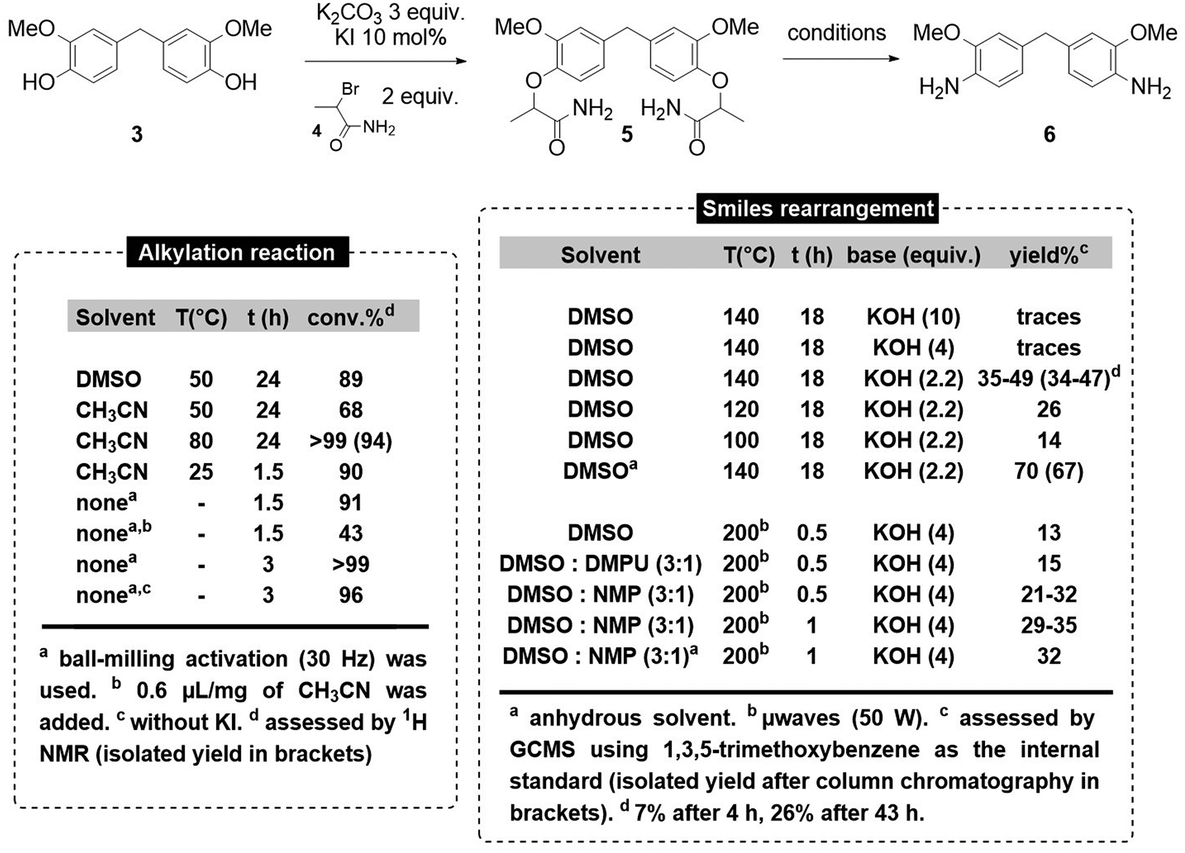
Thus, we decided to synthesize 6via a two-step sequence: (a) the Williamson-type alkylation of BGF 3 and (b) the base-promoted Smiles rearrangement/amide hydrolysis of bis O-alkylated BGF 5 (Scheme 4). The alkylation of BGF was first carried out in DMSO at 50 °C for 24 h using K2CO3 as a base and KI as a catalyst. Surprisingly, incomplete conversion (89%) was obtained. Moreover, the high solubility of 5 in water made its purification difficult, since a substantial amount of product (>50%) was lost when attempting to remove DMSO from the mixture via aqueous work-up. When switching the reaction solvent to acetonitrile, incomplete conversion was observed at 50 °C (68%), but full conversion of BGF was observed at 80 °C after 24 h, and pure compound 5 was isolated in nearly quantitative yield (94%) after evaporation of acetonitrile followed by filtration on silica gel. To further improve this synthetic step in which using a solvent was found to be problematic, we turned our attention to solvent-free ball-milling.42–44
 | ||
| Scheme 4 Optimization of the synthesis of BGA 6. | ||
Indeed, this approach already gave excellent results in our hands for monomer or polymer synthesis.45–47 Gratifyingly, using a 20 mL stainless steel milling jar with a 1 cm diameter stainless steel ball agitated at a frequency of 30 Hz, in a vibratory ball-mill, we obtained an excellent yield (90%) of 5 after only 90 minutes of milling. The yield of the transformation was even improved to nearly quantitative (>99%), when running the reaction for 3 h in the presence of potassium iodide catalyst.48 It is worth noting that under these mechanochemical conditions, the reaction can be carried out with similar success (96% conversion) in the absence of potassium iodide as catalyst. The purified product 5 was fully characterized by 1H NMR, 13C NMR, FTIR, LC(MS) and DSC analysis (see ESI†).
With these alkylation conditions in hand, we then moved to the optimization of the Smiles rearrangement of bis-amide 5 to yield the desired diamine 6. We first examined the feasibility of such a rearrangement under thermal activation (140 °C) in DMSO and we observed that decreasing the amount of potassium hydroxide from 10 to 2.2 equiv. resulted in the increase of the yield of 6. We attributed this result to the potential irreversible hydrolysis of the starting di-amide in 5 at high hydroxide anion concentrations. However, at 140 °C using 2.2 equiv. of KOH, 6 was isolated in yields ranging from 34 to 47%. Decreasing the reaction temperature resulted in lower yields of the desired diamine and increasing the reaction time at 140 °C allowed the degradation of the desired product. Concerned by the reproducibility issues at 140 °C, we decided to run the reaction using dry DMSO, hypothesizing that an inconsistent amount of water in the reaction mixture might be detrimental to the effectiveness of the Smiles rearrangement. Using dried DMSO, we obtained compound 6 in an improved 67% isolated yield, and more importantly, with an enhanced reproducibility, obtaining differences in yields within the range of experimental error. The purified product 6 was fully characterized by 1H NMR, 13C NMR, FTIR, LC(MS), GC(MS) and DSC analysis (see ESI†). Next, we examined the possibility of performing the Smiles rearrangement using microwaves activation. After prior optimization, we found that 4 equiv. of KOH at 200 °C for 30 minutes (50 W) was the optimal set of conditions. However, in DMSO, very low yield of 6 was obtained (13%). The use of polar co-solvents was found to be beneficial for the yield of rearrangement product in that case, with NMP being the best co-solvent (21–32%). The yield could be slightly increased when increasing the reaction time to 1 h (29–35%). Once again, reproducibility issues prompted us to explore the use of anhydrous solvents, but under microwave activation, these conditions did not improve the efficiency of the process (32%).
After having optimized the Smiles rearrangement conditions, we finally carried out the isocyanate synthesis using phosgene-free conditions (Scheme 5). Inspired by the work of Knolker, we synthesized the bisguaiacol F diisocyanate 7 from the corresponding diamine 6. We first tried the described set of conditions11 using catalytic amount of DMAP (10 mol%) along with a slight excess of di-tert-butyl dicarbonate (2.2 equiv.) in dry acetonitrile. Full conversion into the diisocyanate was observed by GCMS after 2 h, but the crude product was found to contain a substantial amount (ca. 15%) of Boc2O.
 | ||
| Scheme 5 Synthesis of BGI 7 from 6 under phosgene-free conditions. | ||
Therefore, we isolated BGF diisocyanate by flash column chromatography on silica gel. However, fast column chromatography with less polar solvents was found to be critical to obtain good yield of the diisocyanate, as 7 decomposes rapidly on silica gel. In these conditions, the desired BGF diisocyanate 7 was successfully isolated as a white solid with an isolated yield of 70%. A greener solvent, 1,3-dioxolane, was also used in substitution of acetonitrile and showed promising results (90%). Noteworthy, diisocyanate 7 possesses a calculated biomass degree of 88% against a biomass degree of 0% for MDI. Finally, the chemical structure of product 7 was ascertained by 1H NMR, 13C NMR, and FTIR, spectroscopy, while LC(MS) and GC(MS) confirmed the good purity of the isolated product. The latter possesses a melting point of 82.5 °C as determined by DSC analysis.
In order to prepare a polymeric system from our BGI monomer, we decided to synthesize a BGI-based polyurethane thermoset and to compare its properties with the MDI-based analogue. Since BGI and MDI are solid compounds, a two-step method was used to design these materials. This method consists in synthesizing an isocyanate-terminated prepolymer followed by the formation of a 3D network though the addition of a trifunctional chain extender (Scheme 6).
 | ||
| Scheme 6 Synthesis of isocyanate-terminated prepolymer. | ||
Thereby, the selected isocyanate was heated up above its melting point (∼90 °C) and the system was purged with nitrogen to avoid side reactions. Velvetol® H500, which is a fully biobased difunctional hydroxyl-terminated poly(1,3-propanediol) with an approximative molecular weight of 500 g mol−1, was slowly added to the system. An excess of diisocyanate (2.5 equiv.) was used to react with Velvetol® H500 (1 equiv.) to control the system stoichiometry and to form an isocyanate-terminated prepolymer. The prepolymer structure was confirmed by 1H NMR, 13C NMR and FTIR analyses (Fig. S22–S24 and S26–S28†). Finally, the obtained prepolymer was poured in a polypropylene flask and the appropriate amount of glycerol was added. The mixture was stirred and cured into a silicon mould at 90 °C for 24 h in an oven. In the case of BGI-based thermoset, an additional curing step has been done during 48 h to obtain a complete disappearance of isocyanate peaks. Fig. 1 shows a picture of both cured materials.
 | ||
| Fig. 1 Picture of MDI-based thermoset (left) and BGI-based thermoset (right). | ||
In addition to fully characterizing both materials by FTIR (ATR), TGA and DSC analysis, we monitored the curing reaction of both materials through the determination of the gelation time. We also followed the disappearance of NCO function by FTIR monitoring to monitor the degree of conversion. The determination of the crosslinked materials gelation time can be very useful to compare the reactivity of monomers. The gelation times were determined by rheological analyses using a multi-frequency method and a plate-plate geometry at 90 °C with a stress of 3 Pa. The cross-over between G′ and G′′ for each frequency was followed and used to determine the average gelation time of BGI- and MDI-based thermosets. G′ and G′′ curves are displayed in Fig. S32 and S37,† whereas Table 1 shows the gelation times values obtained at the three different frequencies and the average gelation times for each material.
| Frequency | Gelation times (s) | |
|---|---|---|
| MDI-based thermoset | BGI-based thermoset | |
| 0.8 Hz | 1802 | 14520 |
| 2.4 Hz | 1853 | 14500 |
| 7 Hz | 1950 | 14460 |
| Average | 1868 ± 75 | 14493 ± 30 |
MDI-based thermoset presents a gelation time around 31 min while BGI-based thermoset have a gelation time around 4 h. The lower reactivity of BGI-based thermoset can be attributed to a combination of steric and electronic effect of the neighbouring ortho-methoxy groups. To monitor the conversion curing at 90 °C, we used a Nicolet 210 Fourier transform infrared (FTIR) spectrometer equipped with a Specac golden gate attenuated total reflection (ATR) heating cell.
In line with the gelation time results, the conversion of BGI-based thermoset was slower than that of the MDI-based one as seen in Fig. 2 and Fig. S33, S38.† Indeed, MDI-based material exhibits 80% isocyanate conversion after 55 min of reaction while BGI-based material exhibits 80% isocyanate conversion after 13 h 35 min of reaction.
 | ||
Fig. 2 Evolution of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching peak of NCO function of MDI-based (A) and BGI-based (B) formulations over time at 90 °C. O stretching peak of NCO function of MDI-based (A) and BGI-based (B) formulations over time at 90 °C. | ||
Finally, the FTIR (ATR) spectra of cured materials made with BGI and MDI monomers are displayed in Fig. S29 and S34.† In both cases, a broad band between 3600 and 3300 cm−1, corresponding to the N–H bond of the urethane groups, is observed. A sharp peak around 1700 cm−1, corresponding to CO function of urethane groups, is also observed in both spectra. Moreover, the absence of a band around 2300–2200 cm−1 corresponding to isocyanate bond shows fully conversion of isocyanates functions for both thermosets.
The thermal stability of each cured materials was studied by TGA measurements under nitrogen atmosphere (Table 2). Both materials have quite similar degradation profiles. The BGI-based thermoset tends to be slightly less thermally resistant compared to the MDI-based thermoset. Moreover, the BGI-based material exhibits a lower Tg value (16 °C) compared to the MDI-based material (34 °C) (Table 2). This can partially result from the presence of a neighbouring methoxy group in the BGI-based material.49 Finally, gel contents were measured for both materials (Table 2) and indicate that the material synthesized with BGI possesses a lower crosslink density than the one synthesized with MDI. This can also be observed by comparing the swelling index of both materials since the MDI-based thermoset has a much lower swelling index (100%) than the BGI-based thermoset (290%). However, the formation of a crosslinked material was confirmed since the BGI-based thermoset features a 75% of insoluble fraction.
| Unit | T 5%d | T 50%d | T 85%d | T g | SI | GC |
|---|---|---|---|---|---|---|
| (°C) | % | |||||
| MDI-based thermoset | 287 | 380 | 487 | 34 | 100 | 99 |
| BGI-based thermoset | 287 | 391 | 428 | 16 | 286 | 77 |
Consequently, ortho-methoxy groups induce some differences regarding reactivity and properties of BGI-based materials compared to MDI-based ones. However, we proved that these differences do not preclude the use of BGI monomer for the synthesis of PUs.
Conclusions
In summary, we have developed an efficient synthetic strategy to produce a greener lignin-based surrogate of MDI with a biobased content of 88%. This approach involves a 4-step synthesis from lignin-based, readily available guaiacol and vanillic alcohol. Unlike the MDI synthesis, our strategy relies on a transition-metal-free and phosgene-free route to afford the biobased aromatic diisocyanate BGI, avoiding the use of very toxic and environmentally harmful compounds such as aniline, formaldehyde and phosgene. BGI was found to be a convincing alternative to MDI for the preparation of cross-linked polyurethanes, as similar thermal properties were obtained for both MDI- and BGI-based materials. Even though BGI exhibits a lower reactivity compared to MDI, as confirmed by rheological analyses and FTIR monitoring, BGI can be considered as a new biobased aromatic diisocyanate of great interest to replace MDI.Moreover, chemical companies such as BASF50 are actively working on the development of biobased MDI from biobased aniline, highlighting once more the urgent need for the design of more sustainable aromatic isocyanates monomers. In this study, we offer a possibility to avoid the risks associated with the conventional preparation of MDI and to valorise greenhouse gas through Boc2O synthesis.23 In particular, we believe that bypassing the use of phosgene is a significant advance in the design of greener isocyanate monomers, and further studies using phosgene-free isocyanate monomers are currently underway in our laboratory.
Conflicts of interest
There are no conflicts to declare.References
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- Developments and Forecasts of Aggravating Resource Scarcity | Knowledge for policy, https://knowledge4policy.ec.europa.eu/foresight/topic/aggravating-resource-scarcity/more-developments-relevant-aggravating-resource-scarcity_en#water, (accessed 14 February 2023).
- M. Desroches, M. Escouvois, R. Auvergne, S. Caillol and B. Boutevin, Polym. Rev., 2012, 52, 38–79 CrossRef CAS.
- P. Furtwengler and L. Avérous, Polym. Chem., 2018, 9, 4258–4287 RSC.
- M. A. R. Meier, Macromol. Rapid Commun., 2019, 40, 1800524 CrossRef PubMed.
- M. A. Sawpan, J. Polym. Res., 2018, 25, 184 CrossRef.
- G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, Biomacromolecules, 2010, 11, 2825–2835 CrossRef CAS PubMed.
- Z. Petrovic, Polym. Rev., 2008, 48, 109–155 CrossRef CAS.
- B. Schemmer, C. Kronenbitter and S. Mecking, Macromol. Mater. Eng., 2018, 303, 1700416 CrossRef.
- B. V. Tawade, R. D. Shingte, S. S. Kuhire, N. V. Sadavarte, K. Garg, D. M. Maher, A. B. Ichake, A. S. More and P. P. Wadgaonkar, PU Today, 2017, pp. 41–46 Search PubMed.
- S. S. Kuhire, S. S. Nagane and P. P. Wadgaonkar, Polym. Int., 2017, 66, 892–899 CrossRef CAS.
- M. Norton, Explosion of phosgene gas at Hamburg, ed. Sir Hosman Leslie, The National Archives, Berlin, 1928 Search PubMed.
- DuPont Corporation Toxic Chemical Releases | CSB, https://www.csb.gov/dupont-corporation-toxic-chemical-releases/, (accessed 14 February 2023).
- Two Dead in BASF Ludwigshafen Blast | CHEManager, https://www.chemanager-online.com/en/news/two-dead-basf-ludwigshafen-blast, (accessed 14 February 2023).
- J. Chambers, J. Jiricny and C. B. Reese, Fire Mater., 1981, 5, 133–141 CrossRef CAS.
- F. J. Weigert, J. Org. Chem., 1973, 38, 1316–1319 CrossRef CAS.
- T. A. P. Hai, L. J. S. De Backer, N. D. P. Cosford and M. D. Burkart, Org. Process Res. Dev., 2020, 24, 2342–2346 CrossRef.
- O. Kreye, H. Mutlu and M. A. R. Meier, Green Chem., 2013, 15, 1431–1455 RSC.
- H.-J. Knölker, T. Braxmeier and G. Schlechtingen, Angew. Chem., Int. Ed. Engl., 1995, 34, 2497–2500 CrossRef.
- H.-J. Knölker and T. Braxmeier, Synlett, 1997, 925–928 CrossRef.
- Y. Basel and A. Hassner, J. Org. Chem., 2000, 65, 6368–6380 CrossRef CAS PubMed.
- J. K. Augustine, Y. A. A. Naik, V. Vairaperumal and S. Narasimhan, Tetrahedron, 2009, 65, 134–138 CrossRef CAS.
- N. Hirano and F. Iwasaki, US6020514, Process for producing di-tert-butyl dicarbonate, 2000 Search PubMed.
- H. W. I. Peerlings and E. W. Meijer, Tetrahedron Lett., 1999, 40, 1021–1024 CrossRef CAS.
- M. Reif, B. Bruchmann, S. Pohl, C. Bettendorf, H. Plaumann and K. Thiele, DE10111337A1, Process for the production of MDI, in particular 2.4′-MDI, 2001 Search PubMed.
- G. Marlair, Rubrique 1158: MDI di-isocyanate de diphényl méthane, 2002 Search PubMed.
- Codes and Stardands of the NFPA, retrieved from https://www.nfpa.org.
- T. Periyasamy, S. P. Asrafali, S. Muthusamy and S. C. Kim, New J. Chem., 2016, 40, 9313–9319 RSC.
- S. F. Koelewijn, D. Ruijten, L. Trullemans, T. Renders, P. Van Puyvelde, H. Witters and B. F. Sels, Green Chem., 2019, 21, 6622–6633 RSC.
- Z. Chen, H. Zeng, S. A. Girard, F. Wang, N. Chen and C. J. Li, Angew. Chem., Int. Ed., 2015, 54, 14487–14491 CrossRef CAS PubMed.
- T. Cuypers, P. Tomkins and D. E. De Vos, Catal. Sci. Technol., 2018, 8, 2519–2523 RSC.
- Z. Qiu, L. Lv, J. Li, C.-C. Li and C.-J. Li, Chem. Sci., 2019, 10, 4775–4781 RSC.
- X. Chen, Z. Yang, X. Chen, W. Liang, Z. Zhu, F. Xie and Y. Li, J. Org. Chem., 2020, 85, 508–514 CrossRef CAS PubMed.
- W. Liang, F. Xie, Z. Yang, Z. Zeng, C. Xia, Y. Li, Z. Zhu and X. Chen, Org. Lett., 2020, 22, 8291–8295 CrossRef CAS PubMed.
- K. Chen, Q. K. Kang, Y. Li, W. Q. Wu, H. Zhu and H. Shi, J. Am. Chem. Soc., 2022, 144, 1144–1151 CrossRef CAS PubMed.
- J. Yu, Y. Wang, P. Zhang and J. Wu, Synlett, 2013, 1448–1454 CAS.
- J. Yu, P. Zhang, J. Wu and Z. Shang, Tetrahedron Lett., 2013, 54, 3167–3170 CrossRef CAS.
- X. Chang, Q. Zhang and C. Guo, Org. Lett., 2019, 21, 4915–4918 CrossRef CAS PubMed.
- S.-B. Lin, W.-W. Wang, J.-P. Meng, X.-W. Li, J. Wu and X.-L. Sun, Tetrahedron Lett., 2019, 60, 151309 CrossRef CAS.
- W. Liang, F. Xie, Z. Yang, Z. Zeng, C. Xia, Y. Li, Z. Zhu and X. Chen, Org. Lett., 2020, 22, 8291–8295 CrossRef CAS PubMed.
- Z. Qiu, L. Lv, J. Li, C.-C. Li and C.-J. Li, Chem. Sci., 2019, 10, 4775–4781 RSC.
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- A. Krusenbaum, S. Grätz, G. T. Tigineh, L. Borchardt and J. G. Kim, Chem. Soc. Rev., 2022, 51, 2873–2905 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- T. F. Burton, J. Pinaud, N. Pétry, F. Lamaty and O. Giani, ACS Macro Lett., 2021, 10, 1454–1459 CrossRef CAS PubMed.
- R. Tavernier, L. Granado, M. Tillard, L. van Renterghem, T.-X. Métro, F. Lamaty, L. Bonnaud, J.-M. Raquez, G. David and S. Caillol, Polym. Chem., 2022, 13, 5745–5756 RSC.
- D. Breilly, S. Fadlallah, V. Froidevaux, F. Lamaty, F. Allais and T.-X. Métro, Green Chem., 2022, 24, 7874–7882 RSC.
- G. A. Bowmaker, Chem. Commun., 2013, 49, 334–348 RSC.
- E. D. Hernandez, A. W. Bassett, J. M. Sadler, J. J. la Scala and J. F. Stanzione, ACS Sustainable Chem. Eng., 2016, 4, 4328–4339 CrossRef CAS.
- BASF expands portfolio of climate friendly products introducing the first isocyanate not carrying a CO2 backpack, https://www.basf.com/global/en/media/news-releases/2022/02/p-22-132.html, (accessed 19 April 2023).
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00704a |
| ‡ Both authors equally contributed to this study. |
| This journal is © The Royal Society of Chemistry 2023 |