
Graphitic carbon nitride materials in dual metallo-photocatalysis: a promising concept in organic synthesis
Binoyargha
Dam
,
Bubul
Das
and
Bhisma K.
Patel
 *
*
Department of Chemistry, Indian Institute of Technology Guwahati, Guwahati-781039, India. E-mail: patel@iitg.ac.in
First published on 5th April 2023
Abstract
In recent years, dual metallo-photocatalysis has grown rapidly, to emerge as one of the most powerful tools in functionalizing organic molecules. In contrast to conventional catalysis, photocatalysis exploits the formation of reactive intermediates by photo-induced atom, electron, or energy transfer processes. An interesting synergy evolved when photocatalysts were pooled together with a non-photochemical second catalytic system comprising mostly metals. In this strategy, the photocatalyst and the metallic system in the presence of light work in synergy with one another, sensitize the organic substrates, and manipulate the reactivity of photogenerated species. Graphitic carbon nitrides encompass a class of transition metal-free photocatalysts, which possess several merits like high chemical stability, low cost, recyclability, and high absorption coefficient (>105 cm−1). Blending this heterogeneous photocatalyst with Earth-abundant metal-based catalytic systems has contributed in abundance towards sustainable and effective synthetic transformations. Most significantly, this rather new branch of catalysis has stimulated an interdisciplinary branch of research that resonates from inorganic and organic chemistry to materials sciences, thereby establishing itself as one of the trending fields in contemporary organic synthesis. The objective of this review is to highlight and illustrate (with mechanisms) the milestones in organic synthesis, where dual metallo-photocatalytic strategies using graphitic carbon nitride have been used. Finally, forward-looking opportunities in this emerging field of research have also been discussed.
 Binoyargha Dam | Binoyargha Dam was born in the year 1989. He received his B.Sc (Hons.) degree from St. Edmund's College, Shillong in the year 2010 and M.Sc (Chemistry) degree from North-Eastern Hill University, Shillong in 2012. He joined the same institute for his doctoral studies in the year 2012 and completed his Ph.D in 2018. Then he joined Professor B. K. Patel's group in the Department of Chemistry, IIT Guwahati for his post-doctoral research. His research interests include photoredox catalysis, green synthetic protocols, synthetic organic chemistry, and heterogeneous catalysis. |
 Bubul Das | Bubul Das was born in 1996 in Assam, India. He received his BSc. degree from Nalbari College, Gauhati University in 2017 and his M.Sc. degree from Delhi University in 2019. He qualified for the CSIR National Eligibility Test (2018) & GATE (2019) and is currently pursuing his doctoral research under the supervision of Prof. Bhisma K. Patel at Indian Institute of Technology Guwahati (India). His research primarily focuses on transition metal-catalyzed C–H bond activation and annulation reactions. |
 Bhisma K. Patel | Bhisma Kumar Patel (born in August 1965) received his B.Sc (Hons) and M.Sc degrees from Sambalpur University, Odisha, India. He joined IIT Kanpur for his PhD in the research group of Prof. S. Ranganathan (FNA) (1988–1994). After three years of post-doctoral tenure with Prof. Dr Fritz Eckstein at the Max Planck Institute for Experimental Medicine (1994–1997), he joined the Department of Chemistry, Indian Institute of Technology Guwahati as an Assistant Professor in April 1997, where he was elevated to the post of Full Professor in August 2005 and HAG Professor in September 2011 and continued as the Head of the Department. His current research interests include photochemical and electrochemical synthesis, green chemistry, C–H activation, cross dehydrogenative coupling, metal catalyzed/metal-free oxidative functionalization, and hypervalent iodine-mediated organic transformations. Altogether, 27 students have been awarded a PhD degree under his supervision and his research work has resulted in the publication of 180 research papers along with 15 book chapters and three patents. |
1. Introduction
Developing environmentally benign strategies is one of the prime challenges faced by chemical researchers in order to diminish the detrimental effect of environmental pollution. The philosophy with which modern-day chemical researchers work in order to develop eco-friendly procedures, is known as green chemistry. Following the principles of green chemistry, emerging strategies involving the formation and cleavage of chemical bonds symbolize the central topic in modern-day chemical research.1–9 At the heart of the catalysis field, heterogeneous catalysts have occupied a supreme position because of their high rate of activity, product selectivity, and high conversion efficiency. The recyclability of heterogeneous catalysts allows for sustainable and green synthesis and avoids unwanted pollution.10–13 Over the years, photocatalysis has played a marvellous role in assisting the growth of new reactions.14–16 An astonishingly varied range of structures including both metals and organocatalysts have been developed over the years to enhance the rate and practicability of the reaction processes. The interaction of small organic molecules with light is generally weak and electronically excited states are available only under ultra-violet (UV) irradiation. But, because of the uncontrolled photodecomposition caused by high-energy photons, the broad utility of photochemical organic synthesis is hindered. In contrast to organic molecules, photocatalysts absorb light with greater efficiency and convert the energy of absorbed photons into chemical potential which can be applied for transforming organic substrates.17Photochemically, a wide range of complex synthetic conversions have been developed in recent years.18–24 In contrast to conventional catalysis, photocatalysis exploits the formation of reactive intermediates by photo-induced electron, atom, or energy transfer processes.25,26 Commonly used photocatalysts (PCs) are mainly conjugated systems that consist of aromatic chromophores, inorganic clusters, organic dyes, and transition metal complexes. Amongst all the metal complexes, the application of ruthenium (Ru) and iridium (Ir) salts has been extensively studied.27 More specifically, in these cases the photocatalytic properties of metal salts are tuned by varying the electronic nature of organic ligands. When a metal's electron density increases, the oxidative power of the PC decreases and the reductive power increases. This is the basic concept that is used while designing a PC. Hence, it must be highlighted that the synthesis and selection of both the ligand and metal complexes should be done very cautiously. Moreover, keeping the greener and sustainable approach in mind, the application of these metal complexes has declined considerably in recent years owing to their high cost, toxicity and non-availability.27,28 Substitution of these metal-based PCs is of prime importance and because of this, cheaper, abundant and less harmful metal-free catalysts are coming into the picture. Molecular organic dyes are one of those candidates which have emerged as metal-free PCs.28–36 They are cheaper, easily available and most importantly their photocatalytic properties are easily tunable. But, the application of these metal-free organic dyes is hindered because of their non-recyclability, instability under certain conditions, and application on a high loading scale in some cases (20 mol%).24,37 To overcome these limitations, the development of proficient recyclable semiconductor-based heterogeneous photocatalytic systems is in high demand and desirable.38,39 When a semiconductor material absorbs energy of an appropriate wavelength, its electron gets excited from the valence band (VB) to the conduction band (CB), thereby creating a hole and an electron pair in the VB and CB respectively (Fig. 1). If the absolute values of the energy levels of the VB and CB are higher than the redox potentials of donors and acceptors, then they will be thermodynamically competent to oxidize donors and to reduce acceptors via single electron transfer (SET).38,40 In previous years, various heterogeneous semiconductor materials have been used as photocatalysts.38,40–44 Among others, graphitic carbon nitride (g-C3N4) materials are a class of heterogeneous semiconductor photocatalysts which absorb light in the visible region (with a band gap of 2.7 eV; λmax ∼ 460 nm) (Fig. 1).29,45–49 Currently, the application of g-C3N4 materials is in high demand as evidenced from their publication rate, which is enhancing exponentially. At the time of writing, Web of Science records approximately 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 937 reports with carbon nitride or C3N4 in the title, from 2013 to early 2023 (Fig. 2).
937 reports with carbon nitride or C3N4 in the title, from 2013 to early 2023 (Fig. 2).
 | ||
| Fig. 1 General catalytic mechanism of the semiconductor photocatalyst (carbon nitride). | ||
 | ||
| Fig. 2 Number of publications per year containing “carbon nitride” or “C3N4” in searched titles from 2013 until March 2023. | ||
Graphitic carbon nitrides are those materials that consist of carbon, nitrogen and a substantial amount of hydrogen in them. They are mainly of 2 types, i.e., CxNy and CxNyHz. In a classical series of investigations, Liebig described the formation and properties of various CxNyHz compounds that were given names such as “melem” (C6N10H6), “melam” (C6N11H9), and “melamine” (C3N6H6).50–52 Liebig first applied the term “melon”50 to an amorphous yellow residue. The limiting stoichiometry of this compound was found to lie near C2N3H (or C6N9H3). Modern approaches to form g-CN materials related to Liebig's melon involve thermolytic condensation of molecular precursors including melamine (C3N3(NH2)3), cyanamide and its dimer (dicyandiamide, C2N4H4, DCDA), as well as N-rich molecules such as urea (NH2CONH2).29,53–56 McMillan and co-workers in their review discussed various types of carbon nitride materials as shown in Fig. 3.57 A series of compounds containing planar carbon nitride layers formed by polytriazine imide-linked (PTI) units that provide hosts for intercalated ions including Li+, Br−, Cl−, and H+ have also been reported.58–61 It is interesting to note that the PTI layers have the composition C2N3H, equivalent to that of Liebig's melon. Finally, the expected end result from continued elimination of NH3 would cause the formation of fully graphitic g-C3N4 layers based on linked polyheptazine units. McMillan and his group suggested that the specific term “g-C3N4” should be reserved for those materials that are determined to have a composition that closely matches the ideal stoichiometry, with minimal incorporation of heteroatoms such as H and O, whose reports are extremely limited. The Dontsova and Lotsch groups simultaneously reported the synthesis of poly(heptazine imide) graphitic carbon nitride materials.62,63 There are also several other classes of materials that have been described as “graphitic carbon nitrides”. These include N-doped graphites or graphenes, which usually contain up to only a few percent nitrogen distributed randomly over the sp2-bonded sites (Fig. 3).64,65 These materials are typically metallic to semi-metallic and this distinguishes them from the semiconducting g-C3N4 compounds.
 | ||
| Fig. 3 Various classes of carbon nitrides, the dashed box indicates materials often designated as graphitic carbon nitride. | ||
Savateev and co-workers in their review described that, depending on the structure, carbon nitride materials can be categorized into two types: (i) covalent and (ii) ionic carbon nitrides.40 Covalent carbon nitrides contain only covalent C–N bonds or contain an equal number of anions or cations intercalated between the N–H functionality and π-conjugated layers. In contrast to this, ionic carbon nitrides possess negatively charged N-atoms bonded to metal cations (Fig. 4). Powder XRD, SEM and TEM are considered as important tools to identify these types of carbon nitrides. Graphitic carbon nitrides (g-CN), mesoporous graphitic carbon nitride (mpg-CN), and poly(triazine imide) intercalated with LiCl (PTI/Li+Cl−) are represented as covalent carbon nitrides, whereas poly(heptazine imide)s (PHIs) and cyanamide surface-functionalized melon-type carbon nitrides (NCNCNx) which consist of heptazine units as building blocks are ionic carbon nitrides. Structures and powder XRD characterization details of these various kinds of carbon nitride materials are shown in Fig. 4.
 | ||
| Fig. 4 Structures of various classes of carbon nitrides. | ||
Currently, an interesting synergy is evolving which involves the pooling of these metal-free heterogeneous photocatalysts with non-photochemical second catalytic systems comprising mostly metals to meet more challenging chemical conversions.66,67 Metal-catalyzed reactions have always been fine tools for organic synthesis and the entire rate of the reaction gets enhanced when the metal catalyst is merged with a photosensitizer via a dual photocatalytic approach (Fig. 5).68,69 This dual catalytic approach can be mostly classified into two types. Firstly, a semi-heterogeneous combination is possible, which involves the application of a metal salt with a heterogeneous photocatalyst and secondly, the metal can be deposited into the heptazine units of carbon nitride (due to the presence of various nitrogens), thereby giving an integrated single entity-based fully heterogeneous photocatalytic system (Fig. 6).70 These single-entity catalysts are the best representative alternatives and complement the drawbacks of the former type, which include catalyst deactivation and non-recyclability of metal salts. This rather new branch of catalysis has emerged as a trending field in contemporary organic synthesis, stimulating a link between inorganic and organic chemistry to materials sciences. The purpose of this review is to highlight and illustrate the milestones in organic synthesis, where dual metallo-photocatalytic materials comprising graphitic carbon nitride have been used. It will be useful to researchers working in the field of sustainable photocatalytic organic transformation and will assist them in adopting detailed mechanistic investigations during catalytic applications.
 | ||
| Fig. 5 Metal catalysis vs. dual metallo-photocatalysis involving carbon nitride. | ||
 | ||
| Fig. 6 Two types of dual metallo-photocatalytic approaches involving carbon nitride. | ||
2. Semi-heterogeneous dual carbon nitride metallo-photoredox catalysis
As discussed previously, in a dual catalytic approach, where a metal salt is added separately to the heterogeneous carbon nitride photocatalyst, a semi-heterogeneous combination is formed. In the past few years, several works have been reported in this field, which has led to the construction of unprecedented molecular complexities. Pieber and co-workers in the year 2019, reported the application of carbon nitride materials accompanied by homogeneous nickel catalysts.71 They carried out visible light-mediated cross-coupling of aryl halides with alcohols and thiols under mild conditions. Carbon nitride materials (CN-OA-m) were synthesized via the polymerization of urea and oxamide. Application of this CN-OA-m (3.33 mg mL−1) material, under oxygen-free conditions, white light irradiation at 40 °C, in combination with a catalytic amount of NiBr2·3H2O (10 mol%), di-tert-butylbipyridyl (dtbbpy) (10 mol%) and N-tert-butylisopropylamine (BIPA) (5.0 equiv.), gave a diverse range of ethers and thioethers (Scheme 1). Various aryl halides (1, 1.2 mmol), when treated with alcohols (2, 4–4.8 mmol) under the standardized reaction conditions, in the presence of acetonitrile (6 mL) showed positive outcomes. Aryl bromides bearing electron-withdrawing groups (EWGs) worked well under the reaction conditions (4a, 87%; 4b, 83%). The reaction also proceeded smoothly with heteroaryl bromides (4c, 39%). However, in the absence of EWGs, the desired product was isolated in lower yield. Methyl-4-bromobenzoate under the standard reaction conditions coupled with methanol and gave the desired product (4d, 80%) within 24 h. Deuterium-labeled anisoles were also prepared by this methodology in excellent yield (4e, 92%). Primary alcohol when coupled with an organic halide gave the desired product with high selectivity (4f, 92%). Simple secondary alcohols reacted efficiently (4g, 84%; 4h, 82%), whereas sterically encumbered secondary (4i, 8%) and tertiary (4j, 2%) alcohols gave a lower yield of the desired product after 48 h of stirring. This semi-heterogeneous strategy was also applied for the synthesis of pharmaceutically important intermediates (4k, 61%). The same catalytic system was further employed for the coupling of thiols (3) with aryl iodides and a diverse range of thioethers (5a, 33%; 5b, 79%) were synthesized. At the later stage, the reaction was also carried out in the presence of blue LEDs which increased the reaction rate significantly. This intensified condition was used for studying the recyclability of heterogeneous graphitic carbon nitride material. CN-OA-m was recycled for six consecutive runs without much decrease in the catalytic activities, thereby establishing its high potential for sustainable photocatalysis. The recycled catalyst was also further analyzed and it showed the presence of Ni deposited on it, however, this deposited Ni was not catalytically active, as evident from the reaction showing no positive result when carried out only with recycled catalyst without the application of additional Ni(II) salts. | ||
| Scheme 1 Semi-heterogeneous dual nickel/photocatalytic (thio)etherification. | ||
The same research group also demonstrated a combination of carbon nitrides and nickel catalysis to induce C–O cross-coupling of aryl halides (6) and carboxylic acids (7).72 After carrying out a detailed study for the optimization of reaction parameters, the authors reported that when a cocktail of CN-OA-m (3.33 mg mL−1), NiCl2·glyme or Ni(OAC)2·4H2O (10 mol%), 4,4-di-tert-butyl-2,2-dipyridyl (dtbbpy) (10 mol%), and N-tert-butylisopropylamine (BIPA) (3/5 equiv.) in 3 mL of dimethyl sulfoxide (DMSO) was irradiated in the presence of white light, the reaction gave the best result. Reactions went smoothly in the presence of aryl iodides bearing EWGs [8a–d, 78–92% with NiCl2·glyme; 56–80% with Ni(OAC)2·4H2O], whereas substrates lacking EWGs showed lower reactivity (8e, <15%). 1-Bromo-4-iodobenzene gave the anticipated product [8f, 84% with NiCl2·glyme; 62% with Ni(OAC)2·4H2O] without the formation of any 1,4-diester. The scope of various carboxylic acids was also investigated and aliphatic [8g, 91% with NiCl2,glyme; 77% with Ni(OAC)2·4H2O], olefinic [8h, 89% with NiCl2,glyme; 59% with Ni(OAC)2·4H2O], benzylic [8i, 65% with NiCl2,glyme; 45% with Ni(OAC)2·4H2O] and benzoic acid derivatives [8j, 78% with NiCl2,glyme; 68% with Ni(OAC)2·4H2O] were found to be compatible under the optimized reaction conditions. Esterification of biotin [8k, 93% with NiCl2·glyme; 77% with Ni(OAC)2·4H2O] showed the applicability of the protocol for conjugation purposes. No formation of the decarboxylative C–C coupling product (9, 0%) was detected, thereby indicating selective photosensitization. The organic semiconductor was able to harvest a broad range of visible light spectra as monitored by in situ FT-IR analysis. After the completion of the reaction, the carbon nitride materials were recycled and reused for 5 consecutive runs by adding fresh Ni salt and dbppy. The reused catalyst was also characterized by various analytical techniques (Scheme 2).
 | ||
| Scheme 2 Esterification of carboxylic acids with aryl halides by using Ni/carbon nitride photocatalysis. | ||
Cross-coupling reactions are well known for C–C bond formation, therefore, König et al. in 2020, carried out a C(sp2)–C(sp3) cross-coupling by using a synergistic combination of mpg-CN and the homogeneous nickel catalyst.73 The protocol in the presence of visible light irradiation at room temperature afforded cross-coupling between aryl halides and potassium alkyl trifluoroborates via single electron transmetallation. This form of transmetallation is different from traditional two-electron transmetallation which proceeds via three-step catalytic cycle i.e., oxidative addition, transmetallation and reductive elimination. Although this strategy is highly effective for C(sp2)–C(sp2) coupling but faces challenges when extended to C(sp3) centers because of lower rates of oxidative addition, transmetallation, and tendency of alkyl metallic intermediates to undergo facile β-hydride elimination. This mode of transmetallation also requires high activation energy, elevated temperature, and stoichiometric application of the base and the rate of transmetallation is C(sp) > C(sp2) > C(sp3). In contrast to this, in single electron transmetallation, a series of SET occurs, enabling cross-coupling at room temperature. In lieu of high activation energy, here the alkyl transfer is affected through radical combination with the transition metal catalyst, thereby making it a nearly barrier less energetic process. It requires no application of base and exciting the photocatalyst supplies virtually all of the energy required for the cross-coupling to occur, diminishing the need for heat in the reaction. Under these conditions, minimal side products are formed and the yield of the desired product enhances. The rate of electron transfer in single electron transmetallation follows the trend C(sp3) > C(sp2) > C(sp).74–76
Initially, the optimization of reaction parameters was performed by the research group and it was observed that when 4-bromobenzoate (10) (0.2 mmol) and potassium benzyl trifluoro borate (11) (1.5 equiv.) were irradiated for 24 h under nitrogen in the presence of blue LEDs (455 nm) using NiBr2·glyme (2.5 mol%) as the catalyst, neocuproine (2.5 mol%) as a ligand, 2,6-lutidine (1.25 equiv.) as an additive and mpg-CN (10 mg) as a heterogeneous photocatalyst, the reaction gave the best result in DMF solvent. Following that, the scope of various substrates was explored. Aryl bromides possessing both electron-withdrawing and donating groups, worked effectively when coupled with potassium benzyl trifluoroborate (Scheme 3). The corresponding C(sp2)–C(sp3) coupled products were isolated in good yields (13a–c, 55–96%). The reaction was also compatible with heteroaryl bromides (13d–e, 63–91%) and worked well under gram scale (g·s) conditions (13e, 81%). Several borate salts were effective as a source of the C(sp3) radical, thereby yielding desired products in good yields (13f–g, 42–83%). The reaction was also compatible with heteroaryl chlorides (13h, 55%). This protocol also allowed the installation of allyl groups onto (hetero)arenes (14a–b, 70–73%).
 | ||
| Scheme 3 Ni–carbon nitride photocatalyzed C(sp2)–C(sp3) cross-coupling. | ||
The authors proposed two plausible mechanistic pathways as shown in (Scheme 4). According to the first pathway, Ni(0) species (A1) undergoes oxidative addition to aryl halides (10) to give Ni(II) intermediate (B1). Mesoporous g-CN gets excited in the presence of a light source and generates an electron and a hole pair. The photogenerated hole acts as an effective site for oxidative generation of benzyl radical (C1), which eventually get trapped by Ni(II) species and generates Ni(III) organometallic adduct (D1). Alternatively, (D1) could have formed from Ni(0) species A1, through radical trapping followed by oxidative addition. This intermediate (D1) eventually undergoes reductive elimination and gives the desired coupling products (13/14). Finally, Ni(I) species (E1) undergoes reduction from Ni(I) to Ni(0) using electrons at the semiconductor surface, to regenerate the catalytic cycle. Further identification of bibenzyl via radical coupling reactions was confirmed by gas chromatography (GC) and GS-MS, which supports the generation of a benzyl radical (C1, under photoredox conditions) from potassium benzyl trifluoroborate (11). After completion of the reaction, mpg-CN was recycled by simple filtration or centrifugation and reused six times without significant loss of photocatalytic activity. But, the application of the recovered catalyst, without renewed addition of Ni catalyst gave no desired products, indicating precipitation of nickel on the surface of the heterogeneous photocatalyst.
 | ||
| Scheme 4 Plausible mechanism for the dual metallophotocatalytic transformation. | ||
The same research group very recently reported another application of mpg-CN and nickel dual-catalyzed cross-coupling between aryl (15) or alkenyl bromides (16) and alkyl bis (catecholato)silicates (17) as radical precursors.77 Firstly, optimizations of reaction parameters were evaluated, following which scopes of various substrates were investigated. This synergistic dual catalysis between heterogeneous mpg-CN and homogeneous Ni-based catalyst gave cross-coupling products (18a–h, 44–98%) under sustainable conditions (Scheme 5).
 | ||
| Scheme 5 Dual-metallophotocatalytic arylation of alkylbis(catecholato)silicates. | ||
Mechanistically, the success of the current protocol lies in the generation of electron and hole pairs in photoexcited mpg-CN (Scheme 6). It possesses an oxidation potential of (+1.2 V vs. SCE) and a reduction potential of (−1.5 V vs. SCE) on the material's surface. The former potential is sufficient to oxidize alkyl bis(catecholato)silicates (17) (<0.9 V vs. SCE) and form alkyl radicals (B2), whereas the latter potential reduces Ni(II) to Ni(I) and subsequently to Ni(0) (E2), which readily undergoes oxidative addition with aryl bromides to give the Ni(II) complex (A2). This complex serves as a radical trap for formerly generated alkyl radical (B2) to give intermediate (C2), which after reductive elimination gave the cross-coupling product (18). The SET-mediated reduction of the resulting Ni(I) complex (D2) by mpg-CN closes the catalytic cycle. Differently substituted aryl (15) and alkenyl bromides (16) gave the expected C(sp2)–C(sp3) cross-coupling products under dual synergistic catalytic conditions (18a–e, 44–71%). Finally, scopes of several silicate radical precursors were explored and both primary (18f, 98%) and secondary silicates (18g–h, 77–78%) worked well under the current photochemical protocol. A mixture of β-bromostyrene (E/Z = 90:10) gave 18d (E/Z = 92:8) in 52% yield, whereas (Z)-β-bromostyrene (E/Z = 2:98) gave anticipated 18e (E/Z = 22:78) in 44% yield. Recycling experiments of mpg-CN were also carried out. Up to three consecutive runs, the yield of the anticipated product was almost constant (76%), but decreased thereafter to around 43%. The authors described that this suddenly reduced activity might have occurred because of the formation of bis(catecholato)silane side products, which get separated along with the catalyst during recycling. The accumulation of this side product after each cycle obstructs the substrate's access to mpg-CN's surface, which inhibits the redox process. Time-correlated single-photon counting time-resolved emission spectroscopy (TCSPC-TRES) and electron paramagnetic resonance (EPR) spectroscopy experiments were performed for both the freshly prepared and the recycled catalysts. Assessment of TCSPC-TRES data indicated a decrease in the fluorescence lifetime which was consistent with the results of the EPR signal, which decreased upon the recovered sample.
 | ||
| Scheme 6 Plausible mechanism for dual-metallophotocatalytic arylation of alkyl bis(catecholato)silicates. | ||
König and co-workers again in the year 2021, described an mpg-CN and nickel-mediated arylation of C(sp3)–H bonds.78 The reaction was selective for C–H activations of α to amide groups. Kinetic studies and mechanistic investigations suggested that the reaction proceeded via energy transfer from the semiconductor-based catalyst to nickel metal, thereby differentiating this protocol from the archetypal SET process. This synthetic protocol possesses broad scope and more than 70 derivatives were synthesized along with late-stage functionalizations of agrochemicals and drugs. In a model reaction 4-bromobenzonitrile (19) was used toward the C(sp3)–H bond arylation of N,N-dimethyl acetamide (DMA) (20) as both solvent and C–H precursor. At first the optimization of various reaction parameters was carried out and after that, a library of compounds was synthesized. A multitude of aryl halides bearing electron-withdrawing as well as electron-donating groups underwent C–H arylation with DMA (Scheme 7). The reaction was also compatible with aryl chlorides (21b–c; 84–89%). Pleasingly, the developed conditions were also used for a gram scale reaction (21a, 83%) and a noticeable erosion in the yield was observed. The photochemical protocol was used effectively on aryl moieties having more than one reactive site and on polycyclic aryl bromides (21e–f, 67–82%). Following that, amenability towards heteroaryl halides was also tested (21g–h, 81–83%). Compatibility of the procedure was also assessed with an unprotected polar functional group (21i, 85%), which is vital for the synthesis of bioactive molecules and drug discovery. Further, the scope of the procedure was also expanded to other precursors as a substitute to DMA (21j–k, 43–59%). Finally, late-stage functionalizations of various bioactive molecules were also carried out by following this protocol and it gave the desired product in good yield (21l, 72%).
 | ||
| Scheme 7 Nickel mediated aryaltion of C(sp3)–H bonds α to amide groups. | ||
After completion of the reaction, the catalyst was recovered and reused for five runs. The recovered material was characterized by a series of techniques and showed stability and durability of the semiconductor photocatalyst. Based on prior mechanistic explorations two mechanistic pathways were proposed as shown in Scheme 8. The first pathway proceeds via conventional electron and hole formation by mpg-CN which mediate the formation of the product by a SET pathway. In the second pathway, mpg-CN absorbs light and undergoes an energy-transfer process (EnT) to produce an electronically excited intermediate (H3), which is a Ni(II) species. Breakage of Ni(II) halogen bonds and hydrogen atom transfer, followed by the fusion of consequential carbon-centered radical and intermediate (I3) generates intermediate J3. Intermediate J3 undergoes EnT in the presence of mpg-CN to generate intermediate K3, which undergoes reductive elimination to provide the desired product (21) and regenerates Ni(0) species, thereby completing the catalytic cycle. However, after a few control experiments and kinetic studies, it was concluded that the latter mechanistic pathway is more favorable.
 | ||
| Scheme 8 Plausible mechanism for dual nickel–carbon nitride mediated arylation of C(sp3)–H bonds α to amide groups. | ||
Further, König et al. in the year 2021 also reported the synthesis of 1,4-dicarbonyl compounds (25) and substituted alkenes (26) by the Mizoroki–Heck-type coupling (Scheme 9).79 They used mesoporous graphitic carbon nitride as a semiconductor photocatalyst and Ni(II) as a Lewis acid catalyst. Secondary and tertiary alkyl halides (24) along with vinyl acetates (22) or styrenes (23) were used as the starting materials for this protocol. First the reactivity of mpg-CN was explored for the synthesis of 1,4-dicarbonyls (25a–c, 69–83%). Optimization of reaction parameters was carried out and the reaction gave the best results when starting materials were reacted in the presence of mpg-CN (10 mg), NiBr2·glyme (1.25 mol%), 2,6-lutidine (1.25 equiv.), DMF (1 mL) and stirred for 24 h at 25 °C under blue light emission under a nitrogen atmosphere. Even though the reaction was working with other metal salts like Cu(OTf)2, In(OTf)3, Sc(OTf)3, Zn(OTf)2, AlCl3, and Y(OTf)3, the best result was obtained in the presence of NiBr2·glyme (1.25 mol%). One of the commendable features of this protocol was the synthesis of carbonyl compounds bearing all-carbon quaternary stereocentres when bromo-compounds possessing cyclobutane or cyclohexane rings were used (25b–c, 69–73%). After synthesizing a library of 1,4-dicarbonyls, the present protocol was used for the addition of alkyl radicals to styrene derivatives which led to the formation of substituted alkene derivatives (26a–c, 63–77%). In this case, all the reaction conditions remained the same except the presence of the base. The reaction was also effective when carried out on the gram scale (26b–c, 63–64%). The heterogeneous nature and stability of mpg-CN allowed the recyclability of the catalyst and after washing with different solvents and water, it was reused for six runs. Detailed characterization of the reused catalyst was also carried out.
 | ||
| Scheme 9 Dual-metallo-photocatalytic synthesis of 1,4-dicarbonyl compounds and substituted alkenes. | ||
Based on the experimental results, a working hypothesis was elucidated by the authors (Scheme 10). Even though the accurate role of Ni(II) salt was not clear, the authors hypothesized that the nickel salt acted as the Lewis acid to coordinate with the ester group of alkyl bromide (24), which reduced its reduction potential. Alternatively, it may activate the double bond of the olefin by coordinating with it. However, a typical cross-coupling mechanism was ruled out as reactions of non-activated alkyl bromides showed no positive outcome. Based on these results, the authors believed that mpg-CN under photo-irradiation generated redox centers (electron and hole pair). Alkyl bromide (24) gets effectively reduced by the photogenerated electron and generates alkyl radical A4. This alkyl radical A4 then gets added to vinyl acetate's double bond, thereby giving intermediate B4. A photogenerated hole oxidizes B4 delivering carbocation C4. Consecutive loss of the acetyl group, most likely mediated by nucleophiles (bromide anions or solvent) present in the reaction medium gave corresponding 1,4-dicarbonyl compounds (25), whereas in other cases de-protonation of the cation intermediate leads to the formation of a Mizoroki–Heck type cross-coupling product (26).
 | ||
| Scheme 10 Plausible mechanism for dual-metallo-photocatalytic synthesis of 1,4-dicarbonyl compounds and substituted alkenes. | ||
A Ni-catalyzed aryl amination and etherification using surface-modified carbon nitride (NCNCNx) was reported by the Nocera group in the year 2020.80 Cyanamide-modified carbon nitride (NCNCNx) was found to be a better photocatalyst for its long-lived electrons in the conduction band than the other surveyed photocatalysts such as SiC, carbon nitride (NH2CNx), ZnSe, GaP and CdTe. These long-lived electrons serve as the continuous source of the redox equivalents and thus reduce the Ni(II) complexes and drive the productive Ni(I/III) dark cycle. The importance of this catalytic system is that, it can harness an extended spectral range of sunlight for the photoredox cross-coupling reactions. The authors verified this by allowing sunlight irradiation in the reaction through a filter. The reaction was found to remain active in broadband sunlight (from 345 nm to 590 nm) for both the cross-couplings. The added advantages of the NCNCNx heterogeneous photocatalyst are facile recyclability and no loss of catalytic activity even after 5 cycles. The photocatalyst can be easily recycled by subjecting it to three rounds of washing, and centrifugation followed by drying at 130 °C. Various aryl bromides (27) were subjected to this solar-driven nickel-catalyzed cross-coupling reaction with different nucleophiles (28 or 30) and anticipated products (29 or 31) were isolated in appreciable yields (72–99%) (Scheme 11).
 | ||
| Scheme 11 Photoredox Ni and NCNCNx dual catalyzed C–N and C–O couplings. | ||
Another efficient hydroxylation reaction for the synthesis of phenols (33) from alkyl halides (32) was reported by Zhang and co-workers using a carbon nitride and Ni complex cooperative catalyst. The protocol was well tolerant to a wide variety of aryl halides (32) as shown in Scheme 12.81 To get the mechanistic insight, a set of trapping experiments were performed using scavengers for hydroxyl radicals, superoxide radicals, electrons and holes. The results of these trapping experiments showed that in the presence of an electron scavenger (AgNO3), the photocatalytic activity of the catalyst decreased drastically. This confirmed that electrons play a crucial role in this hydroxylation protocol. A probable mechanism proposed by the authors is elucidated in Scheme 13. Electron and hole pairs are generated by visible light illumination of g-C3N4. The photogenerated hole carries out the oxidation of Et3N which gives cationic radical A5. Then, radical A5 undergoes a hydrogen atom transfer process with water to give the hydroxyl radical. Concurrently, oxidative addition of aromatic halides (32) to Ni(0) species delivered Ni(II) species, which were trapped by the hydroxyl radical to form a Ni(III) organometallic adduct. Thereafter, reductive elimination gave the desired phenol product (33) and Ni(I) species. Finally, the photogenerated electrons by semiconductor excitation was utilized for the reduction of the Ni(I) species to Ni(0) species via a single electron transfer. Concomitant oxidative addition of aromatic halides to Ni(0) species delivered Ni(II) species to complete the Ni catalytic cycle. Carbon nitride was recycled and reused for five runs.
 | ||
| Scheme 12 Hydroxylation reaction for the synthesis of phenols from alkyl halides by using carbon nitride and Ni complex. | ||
 | ||
| Scheme 13 A plausible mechanism for the synthesis of phenols from alkyl halides by using carbon nitride and Ni complex catalysis. | ||
In the year 2022, Su et al. demonstrated a semi-heterogeneous dual metallo-photoredox catalysis for various coupling reactions using polymeric carbon-nitrides (PCN).82 PCN is eco-friendly, cost-effective and scalable. The photoexcited electrons of PCN with strong reducing ability make it an effective semi-heterogeneous photocatalyst for many unique bond-forming reactions. A merger of Cu and PCN dual catalytic system has been utilized to effectively enable C–S and C–Se cross-couplings (chalcogeneation), and C–N cross-coupling of various N-heterocycles (Scheme 14). Initially, C–H arylation of benzothiazole (35) was explored using iodobenzene (34) as a coupling partner in the presence of the PCN photocatalyst, CuI catalyst, and LiOtBu base under irradiation of 20 W blue LEDs. Various control experiments suggested that light, PCN, CuI and base are essential for the successful C–H arylation. Aryl iodides with electron-donating or withdrawing substituents at para or meta positions effectively delivered the anticipated cross-coupling products (38b–c, 65–70%). However, with the ortho-substituted aryl iodides, the efficiency of delivering the anticipated product was lower (38d, 44%). A wide array of heterocycles including benzothiazole (35), benzoxazoles (36), and benzimidazole (37) were efficiently coupled with aryl iodides (34). Under similar conditions, the C–H bond functionalization of heterocycles with aryl iodides and elemental chalcogen (S and Se) was evaluated. A variety of 2-arylsulfanylbenzothiazoles (39a, 65%) and 2-arylselanylbenzothiazoles (39b, 64%) were synthesized using the said protocol. The authors further extended this photo-Cu-dual photocatalytic system for effective C–N cross-coupling in N-containing heterocycles (40) (Scheme 14). Different C–N cross-coupled products were isolated in acceptable yields (41a–c, 73–87%). Analyses such as SEM, XPES, and UV-visible spectroscopy of PCN indicated that the morphology of the catalyst had no significant changes even after six catalytic cycles.
 | ||
| Scheme 14 Various cross-coupling using the photo-Cu-dual catalytic system with PCN. | ||
The control experiments and the literature studies revealed that the reaction proceeded via a photocatalytic radical pathway (Scheme 15). Under blue light irradiation, the heterogeneous photocatalyst, PCN resulted in charge separation, thus furnishing redox surfaces. The SET reaction of aryl halides (34) with a reductive electron from PCN eventually generates an aryl radical and a hole (h+). These holes in the presence of a base are effective for the oxidation of Cu complex A6 to another key Cu(II) intermediate B6. This intermediate then traps aryl radical to produce unstable Cu(III) complex C6. Finally, reductive elimination of the intermediate C6 furnishes the desired product (38) and completes the catalytic cycle. The authors also proposed a similar energy transfer pathway for these coupling reactions.
 | ||
| Scheme 15 Proposed SET mechanism for various coupling involving copper and PCN dual catalysis. | ||
3. Single entity-based fully heterogeneous dual metallophotocatalysis
Since the semi-heterogeneous approaches suffer from certain drawbacks like catalyst deactivation and non-recyclability of metal salts, therefore, the development of single entity-based, metal-integrated carbon nitride photocatalysts was initiated to complement the drawbacks of the former type. In the year 2020, the Song group reported a light-driven dual metal/photoredox catalytic procedure for cross-coupling of alkyl halides with alcohols using the Ni-coordinated graphitic nitride catalyst (C3N4–Ni).83 Herein, taking advantage of graphitic nitride as a light-harvesting material as well as an adaptive ligand, the C3N4–Ni platform was designed and synthesized. C3N4–Ni was synthesized by stirring a mixture of NiCl2·6H2O (0.2–1 mM) and carbon nitride (2 g L−1) suspension in N,N-dimethylacetamide (DMAc) for 6 h under dark conditions. This allowed the appropriate interaction of Ni(II) with vacant nitrogen sites of the carbon nitride. The reaction mixture was then centrifuged and washed with DMAc (2 times) to remove uncoordinated Ni(II), followed by drying under vacuum at 80 °C for 12 h. This synthesized catalyst was characterized by various analytical techniques like HR-TEM, HAADF-STEM, XPS, etc. The actual coordination of Ni onto carbon nitride was analyzed by DFT calculations. After successful characterization of the catalyst, it was employed for cross-coupling of 4-bromoacetophenone (42) (0.1 mmol, 1 equiv.) and methanol (43) (6 mmol). Firstly, optimization of various reaction parameters was performed. A combination of C3N4–Ni (2 g L−1, 0.19 mol% Ni), imidazole (0.2 equiv.), quinuclidine (1.8 equiv.) in 5 mL of DMAc under irradiation with light of wavelength above 400 nm at room temperature under an Ar atmosphere provided 4-methoxyactophenone (44a) in 91% isolated yield. A gram scale reaction was also performed and it provided 86% yield of the desired product after 15 h of visible light irradiation. In this reaction, imidazole was considered as an auxiliary ligand for Ni; other potential ligands like pyrimidine or pyrrole were also investigated but provided inferior yields. Following that, appropriate investigation was carried out to check the efficacy of the current protocol. At first, the scope of several substituted aryl bromides and iodides was investigated and it was found that methanol when coupled with aryl bromides and iodides with EWGs in the para-position produced desired products (44a–c, 81–91%) in good yields. Further, other alcohols as coupling partners were also investigated. It was observed that under the optimized reaction conditions, various alcohols afforded anticipated products in good yields (44d–e, 91%). Notably, reaction with water gave 4-hydroxyacetophenone (44f) in 75% isolated yield (Scheme 16). | ||
| Scheme 16 Scope of C–O coupling reactions by using the Ni-coordinated graphitic nitride catalyst (C3N4–Ni). | ||
Mechanistically, upon excitation, the transfer of the hole to quinuclidine and electrons to Ni generates a quinuclidine radical cation (A7) and Ni(I) species (B7) respectively. B7 initiates oxidative addition, alcohol co-ordination and reductive elimination via Ni(I) and Ni(III) states to yield the desired coupling product (44). Even though the Ni(I)/Ni(III) catalytic pathway is a self-sustained, the off-pathway process (designated by dotted arrows) which includes re-oxidation of Ni(I) by a quinuclidine radical cation and reduction of Ni(III) intermediates by accumulated electrons would have disrupted the overall conversion. Therefore, continuous irradiation is necessary to sustain catalysis. After completion of the photoreaction, the heterogeneous C3N4–Ni was recollected, rinsed and dried under vacuum for recycling. The catalyst was reused for five consecutive runs, however, extended irradiation time was required to reach 90% yield from the third run onwards (Scheme 17).
 | ||
| Scheme 17 Proposed mechanism for light-driven C–O coupling, catalyzed by C3N4–Ni. | ||
In the year 2021, Reisner and collaborators reported the synthesis of an integrated single entity, which consisted of mesoporous graphitic-CNx with Ni deposited onto it (Ni-mpg-CNx).84 The catalyst was applied for performing C–O coupling between alcohols and aryl halides under visible light irradiation (Scheme 18). Application of Ni-mpg-CNx overcomes the requirement of additives, organic ligands, and expensive photocatalysts without the deactivation of the catalyst. Firstly mpg-CNx was synthesized by heating cyanamide in the air at 550 °C and then NiCl2 was added in the presence of acetonitrile and triethyl amine at 80 °C to get the desired catalyst. It was then characterized by various analytical techniques like SEM, TEM, ICP-OES, EDX, XPS, etc. The XPS analysis confirmed the presence of Ni(II) sites on the mpg-CNx surface. After careful investigation of the reaction parameters, scopes of various substrates were explored for C–O coupling. Several aryl and heteroaryl halides (45) worked smoothly under the optimum reaction conditions. para-Substituted electron-withdrawing bromoarenes provided coupling products in good yields (47a–b, 80–81%). Ortho and meta acetyl substituents were also tolerable but reacted slowly (47c, 60%; 47d, 84%). The reaction was also compatible with electron-rich aryl bromides but required a longer reaction time to form 47f (42%). Deuterated aryl ether (47g, 78%) was also prepared by using this protocol. The scope of this protocol was also further investigated by coupling methanol with iodo and chlorobenzene (47b, I = 84%, Cl = 30%). Aryl iodides reacted faster to give the coupling products, followed by aryl bromides and aryl chlorides. The catalyst was easily recyclable (up to 94% recovered material per run) by simple centrifugation and could be reused effectively for three subsequent cycles. The lower yield after three catalytic runs may be due to the leaching of Ni from Ni-mpg-CNx.
 | ||
| Scheme 18 Nickel deposited mpg-CNx-catalyzed visible-light promoted C–O bond formation. | ||
Based on computational calculations, experimental evidence and previously reported results, a plausible mechanism was proposed. Initially, mpg-CNx on irradiation with visible light generates an electron–hole pair. Potentials of mpg-CNx are +1.7 V vs. SHE (VB) and −1.0 V vs. SHE (CB). Therefore, alcohol substrates with Eox = +1.54 V vs. SHE, quench the photogenerated hole. Concurrently, the photogenerated electron is transferred to Ni(II) to give Ni(I) (A8) as supported by DFT calculations. Following that, oxidative addition and ligand exchange occur during which Ni(III) intermediates (B8 and C8) were generated and finally reductive elimination delivers the product (47) and completes the catalytic cycle (Scheme 19).
 | ||
| Scheme 19 Proposed mechanism for light-driven C–O bond formation, catalyzed by Ni-mpg-CNx. | ||
In organic synthesis, the preparation of primary anilines by a sustainable process remains a challenge. Therefore, Reisner and co-workers very recently reported a procedure for the selective synthesis of primary anilines by cross-coupling of sodium azides (49) and aryl halides (48).85 They used Ni(II) deposited on mesoporous carbon nitride (Ni-mpg-CNx) as a light-absorbing photocatalyst which had a ‘built in’ solid-state ligand for the catalytic Ni site (Scheme 19). This reported protocol holds potential to surmount the need for complex ligands in homogeneous catalysis, expensive metals and soaring pressure/temperature. To start the work, the amination protocol was first performed by using 4-bromobenzonitrile and sodium azide as model substrates. Reaction was set up under visible light irradiation in the presence of the Ni-mpg-CNx catalyst. Diverse bases, solvents, temperature and concentrations of the photocatalyst and substrates were checked and optimized reaction parameters were identified. The desired aniline product was attained in 84% isolated yield (50a) after irradiating (LED, λ = 447 nm, 2.4 W) 0.4 mmol of 4-bromobenzonitrile (48) and 2 mmol of NaN3 (49) for 24 h using a methanol:water (5:3) mixture containing triethyl amine as a base/electron donor under a nitrogen atmosphere at 60 °C. With the optimized reaction conditions in hand, the scope of this amination protocol was investigated (Scheme 20). The reaction was effective with various electron-withdrawing (50a–b; 84%, 83%), neutral (50c; 56%) and electron-donating (50d, 26%) alkyl halides. The reaction showed excellent reactivity with bromo heteroarenes (50e–f; 56%, 87%). A few biologically active compounds including benzocaine (50g, 83%) and sulfanilamide (50h, 85%) were also synthesized by this procedure. After completion of the reaction, the catalyst was recycled by simple centrifugation and reused for photocatalytic runs. A gradual decrease in the yield of the desired product was observed after each run and this may be attributed to the leaching of the Ni (17.6% after the 4th cycle) component from the catalyst. This also leads to the formation of darker materials, which may be due to the formation of small Ni aggregates as observed by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM).
 | ||
| Scheme 20 Ni deposited mpg-CNx-catalyzed visible-light promoted amination of aryl halides using sodium azide. | ||
Next, based on reactivity, DFT and kinetic studies of the plausible mechanism was proposed as shown in Scheme 21. Initially, on photo-irradiation, electron and hole pairs are generated in the VB and CB of Ni-mpg-CNx respectively. Et3N quenches the photogenerated hole generating F8 and at the same time, the photoexcited electrons are transferred from the mpg-CNx matrix to the deposited Ni(II) center, which generates a Ni(I) species (A8). According to DFT calculations, consecutive reduction of Ni(I) to Ni(0) by mpg-CNx is kinetically and thermodynamically less favorable. Then oxidative addition of aryl halide (48) generates a Ni(III) species (B8). Subsequently, transmetallation leads to the Ni(III)-azide intermediate (D8) which via reductive elimination generates Ni(I) intermediate (E8) and eventually on photoreduction yields anticipated aniline derivatives (50) (Scheme 21).
 | ||
| Scheme 21 Proposed mechanism for light-promoted amination catalyzed by Ni-mpg-CNx. | ||
Yoo et al. in 2022 introduced a single Ni atom supported (SAC) on carbon nitride (NiSAC/CN) as a heterogeneous catalytic system.86 They performed visible-light-driven C–N coupling through a photoredox catalytic cycle using this NiSAC/CN catalytic system. Carbon nitride is preferred in such SACs as they have abundant anchoring sites for a single atom, low production cost, polymeric ring structures and an appropriate band gap and band position to replace various prized metal-based photocatalysts such as Ru or Ir complexes. This NiSAC/CN shows no aggregation of Ni atoms under blue light irradiation and a high yield was achieved compared to a semi-heterogeneous system. The catalytic system was synthesized by the wet impregnation method followed by calcination under an Ar/H2 atmosphere. Different analyses including XRD, EDS, STEM, EXAFS confirmed the homogeneous distribution of Ni atoms throughout the CN surface without agglomeration. Inductively coupled plasma optical emission spectroscopy (ICP-OES) measured the Ni content in NiSAC/CN and it was found to be 0.53%. XPS and XANES studies suggested that both Ni(0) and Ni(II) species can be stabilized on the surface of CN, this implies the redox versatility one expects in a catalyst for efficient photoredox reactions. Similar UV-vis spectra for both CN and NiSAC/CN suggested that stabilization of the Ni atoms does not significantly improve the absorption properties of CN. With the optimized conditions, CN coupling of aryl halides (51) with amines (52) was conducted using the NiSAC/CN heterogeneous catalyst and quinuclidine as a base under blue light irradiation. The scope of the reaction was tested with various aryl halides (51) which gave the C–N coupled products (53). Various aryl bromides having electron-withdrawing groups such as cyano, ester, trifluoromethyl at para (53a, 99%; 53b, 79%; 53c, 97%) and ortho (53d, 82%) positions deliver the corresponding product with excellent to moderate coupling efficiency. The C–N coupling of 1,4-dibromobenzene gives the mono amino product (53e, 67%). However, electron-rich aryl halides gave the anticipated aminated product in a lower yield (53f, 29%). Heteroaryl bromides also delivered the corresponding product when coupled with amine (53g, 63%). Finally, the scope of various secondary amines containing a six-member ring (53h, 92%) and acyclic amine (53i, 99%) was investigated (Scheme 22).
 | ||
| Scheme 22 NiSAC/CN catalyzed blue-light promoted C–N coupling reaction. | ||
A plausible dual catalytic mechanistic pathway with synergistic interactions between single Ni atoms and CN was proposed after various comprehensive experiments including EPR, photoluminescence and fluorescence quenching studies. EPR experiments suggested that Ni atoms possess some dynamic redox activity during the photoredox C–N coupling. Control experiments demonstrated that electron transfer from the CN to the single Ni atoms is a prerequisite for the C–N coupling. Then, blue-light irradiation generates Ni(0) species (A9) which on oxidative addition of aryl halides (51) generated Ni(II) intermediate B9. The photogenerated holes are quenched by pyrrolidine and form pyrrolidine radical cation C9 which serves as a proton donor to quinuclidine forming another radical D9. This radical then coordinates with intermediate B9 to give Ni(III) intermediate E9. Reductive elimination of E9 leads to the formation of the desired product (53) and subsequent electron transfer from CN regenerates Ni(0) species and completes the catalytic cycle (Scheme 23).
 | ||
| Scheme 23 Plausible mechanism for NiSAC/CN catalyzed blue-light promoted C–N coupling reaction. | ||
In 2022, Song and co-workers developed a cost-effective and robust Ni/C3N4 photocatalytic system for the highly selective semi-hydrogenation of alkynes.87 Polymeric carbon nitride (C3N4) scaffolds with an incorporated Ni metal catalyst facilitate the interaction of substrates, which eventually helps in the selectivity of the light-driven semi-hydrogenation reaction. The Ni/C3N4 catalyst was prepared by the NaBH4 reduction method. Initially, 100 mg of C3N4 was dispersed in 20 mL Ni(NO3)2·6H2O and allowed to stir for 30 minutes. Then, NaBH4 was added dropwise to reduce Ni(II) to metallic Ni. Following this reduction, the reaction mixture was centrifuged, washed, and vacuum-dried at 60 °C overnight. Ni/C3N4 (0.32% Ni content) was then used under the optimized semi-hydrogenation reaction conditions. Various spectroscopic studies revealed the structure of the synthesized catalyst as homogeneously distributed Ni nanoparticles. Photocatalytic semi-hydrogenation was examined by taking phenylacetylene (54) as the model substrate in the presence of Ni/C3N4, TEA in methanol solvent under 420 nm LED irradiation. The reaction exclusively yielded the kinetically favored styrene (55) without over-hydrogenation and H2 evolution. The interesting feature of the reaction is that the incorporated hydrogen in alkene was derived from the hydroxy group of methanol solvent via in situ formed metallic Ni-mediated proton reduction by photogenerated electrons. Following this strategy, they have synthesized different deuterated substituted alkenes from readily available CH3OD or D2O and unlabelled alkyne precursors. In terms of substrate scope, this photocatalytic protocol was successful with various terminal and internal alkyne and even well compatible with various functionalities that are prone to hydrogenation. Although, the reaction needs an extended exposure time in the case of internal alkynes, but proceeded well with high alkene yields and good stereo-selectivity towards synthetically valuable cis-isomers. The heterogeneous nature of the catalyst is supported by easy catalyst separation, reuse and recyclability although to a lesser extent due to partial Ni leaching in each cycle (Scheme 24).
 | ||
| Scheme 24 Semi-hydrogenation of alkyne using the Ni/C3N4via photocatalytic pathway. | ||
In 2020, Liu et al. disclosed a dual platinum nanocluster/graphitic carbon nitride solar light catalysis for the oxidation of alcohols to ketones.88 Desired products were obtained in good yields, at room temperature from various functionalized alcohols including unactivated alcohols. They started the investigation of reaction parameters by taking benzhydrol (56) as a model substrate under a nitrogen atmosphere. Initial investigation suggested that Pt/g-C3N4 and Ru/g-C3N4 were the best catalytic material and are essential for the desired oxidation to deliver the anticipated ketone (57a). Control experiments indicated that light is necessary for the desired transformation and reduction of the yield of the product in the presence of radical scavenger (BHT) hints towards the radical intermediacy in the reaction. To further identify these radicals AgNO3, benzoquinone and tert-butyl alcohol were used as radical scavengers, respectively, for electrons, superoxide (˙O2−) radicals and hydroxyl radicals (˙OH). The reaction went smoothly and the synthesis of 57a was unaffected. Following this, application of ammonium oxalate as a hole scavenger was carried out and both selectivity and conversion decreased under those conditions, indicating a direct hole oxidation pathway in the reaction. 1,1,2,2-Tetraphenylethane-1,2-diol (58), a by-product, was detected during hole scavenging experiments both with ammonium oxalate and NaHCO3. These revealed that the reaction proceeds via the formation of an alcohol radical intermediate. With optimum reaction conditions in hand, the scope of various alcohols was explored under the current photochemical conditions. Aromatic alcohols with several electronic effects worked smoothly to give the desired product in good yield and high selectivity. Effects of substituent on para-, meta- and ortho-positions were also investigated and it was observed that conversion decreases gradually with a change in the position of the substituent (57a–d, 69–97%). This may be because of the high steric resistance of the substituent which affects the reaction kinetics. Heterocyclic (57e, 61%), primary (57b, 89%) and aliphatic alcohols (57f, 88%; 57g, 80%) also showed attractive results for this photocatalytic anaerobic dehydrogenation protocol (Scheme 25).
 | ||
| Scheme 25 Pt/g-C3N4 catalyzed sunlight-promoted oxidation of alcohols. | ||
To further divulge the involvement of Pt-mediated hole oxidation, the authors also calculated electronic structure of Pt/g-C3N4 by using the Pt atom and the heptazine unit for DFT calculations. Based on the result obtained from this calculation and related reports, authors proposed a plausible mechanism. Firstly, hole and electron pairs were generated on irradiation with sunlight. Following this, Pt-mediated holes extracted hydrogen from alcohol (56) and releases α-alcohol radicals (A10). Subsequently, this radical intermediate oxidizes to afford the desired product (57). Concurrently, electrons in the conduction band were transferred to Pt nanoclusters to drive the formation of the hydrogen molecule. The generation of this hydrogen molecule was detected by GC analysis (Scheme 26). Reusability of the Pt/g-C3N4 composite was also checked after the completion of the reaction. The catalyst was easily recycled and reused for five consecutive runs. IR and XPS analyses of the reused catalyst were also performed and the results showed a stable microstructure, interestingly only the binding energy altered as evident from XPS data, indicating charge redistribution in the catalyst during the photochemical process.
 | ||
| Scheme 26 Proposed mechanism for the Pt/g-C3N4-catalyzed oxidation process. | ||
Teixeira and co-workers in 2022, used an ion exchange strategy for the preparation of an iron(III) based single-atom catalyst from economical and easily available first-row transition metals.89 In this approach, the melamine precursor was used for the preparation of sodium-poly(heptazine imides) (Na-PHI), and subsequently, by using different concentrations of FeCl3·6(H2O), Fe-PHI materials were obtained by cation exchange methods. The authors characterized the synthesized catalyst by various techniques and confirmed that the oxidation state of Fe in the catalyst is Fe(III), and this metal center is strongly coordinated to the PHI scaffold. This was then investigated for photo-oxidation of ethylbenzene (59) to furnish acetophenone (60a) using sulphuric acid and molecular oxygen as green oxidants under blue light irradiation. Even though the reaction scope was restricted to a few substrates, the synthesized graphitic carbon nitride based single atom catalyst with 0.1 wt% of Fe(III) was effective enough to carry out the oxidation of a few more hydrocarbons (Scheme 27) to their ketones in considerable yields. The research group claimed that the active Fe(III) single-atom sites generate strongly oxidative Fe(IV) and Fe(V) species. This oxidation of the metal site is encouraged by a light-driven pathway, because of the highly positive position of the VB of the PHI scaffold, where holes possess enough oxidation power to oxidize Fe(III) to Fe(IV)/Fe(V) species. Electrons stay delocalized in the π-conjugated structure to compensate for the positive charge of Fe(V) species. C–H bonds of benzene are then activated through the oxygen rebound mechanism.
 | ||
| Scheme 27 Fe-PHI catalyzed photo-oxidation of alcohols. | ||
J. Liu and co-workers in the year 2022, synthesized a hybrid catalyst by grafting a cobalt complex on mesoporous carbon nitride through the amide bond linkage, and this catalyst proved to be efficient and selective for aerobic oxidation of organic molecules under photocatalytic conditions.90 The molecular hybrid catalytic system Co(tBusalophen)@mpg-C3N4, was synthesized by stirring a mixture of 1-ethyl-3-(3-(dimethylaminopropyl)carbodiimide (EDC, 2.0 mmol), triethylamine (TEA, 2.0 mmol) and a suspension of Co(tBusalophen)-COOH (759 mg, 1 mmol) in DMF (30 mL) for 20 minutes at room temperature. Following that, a solution of 1-hydroxybenzotriazole (HOBt, 2 mmol) in 5 mL DMF was added to it and stirred for another 1 h. Finally, a suspension of mpg-CN (920 mg) in 5 mL of DMF was added to the mixture and stirred for 7 more days. The final catalyst was filtered from the suspension, washed with solvents, dried and characterized by various analytical techniques. After the characterization of the catalyst, its photocatalytic activity in aerobic oxidation was scrutinized. A variety of alkenes (61) (0.5 mmol), iPrCHO (1.5 mmol) as an additive in 2 mL of MeCN, and 10 mg (0.2 wt% Co) of the synthesized catalyst under air were irradiated in the presence of a 25 W 450 nm LED lamp. Reaction mixtures were stirred at room temperature for a time period of 9–48 h (Scheme 28). A diverse range of substrates with varied substituents yielded the desired product in good yields (62a–c; 70–87%). Inspired by the positive result, epoxidation of chalcone derivatives was also investigated (63a–b; 33–45%). In addition to that, a few substituted cyclic carbonates 64 (67%) and 65 (65%) were readily accessed in a one-pot process, when this aerobic epoxidation was combined with a CO2 cycloaddition reaction. This protocol was also employed for the aerobic oxidation of other substrates like benzylamine (66, 80%), thioanisole (67, 67%), β-ketoesters (68, 70%), etc.
 | ||
| Scheme 28 Co(tBusalophen)@mpg-C3N4-catalyzed photo-aerobic oxidation of alkenes. | ||
In the year 2020, H. Kaur and their research group reported the use of copper nanoparticles supported on graphitic carbon nitride (Cu2O NPs@ g-C3N4) as a photocatalyst for ynone synthesis under eco-friendly conditions.91 This reaction progressed efficiently under visible light at room temperature. A large number of substrates were prepared (73a–c, 70–99%) as shown in Scheme 29. The versatility of Cu2O NPs@ g-C3N4 was also tested for the synthesis of indolizines and quinolones (74a–d, 97–99%). Green chemistry metrics like atom economy, E-factor, etc. were also calculated for the protocol. A comparative study of the protocol was also carried out with other known methods and the results were found to be superior. Turnover number (TON) and turnover frequency (TOF) values were calculated and recyclability of the catalyst was also performed (Scheme 29).
 | ||
| Scheme 29 Cu2O NPs@g-C3N4 photocatalyzed synthesis of ynones, indolizines and quinolones. | ||
Cai et al. in 2021 developed an efficient and eco-friendly heterogeneous photocatalytic system for the fluoroalkylation-distal heteroaryl/imine migration.92 They used K-modified carbon nitride (K-CNs) as a recyclable and non-toxic photocatalyst for the fluoroalkyl radical mediated functionalization of alkenes (75) via intramolecular functional group migrations. Potassium intercalated CN (CN–K) was prepared by thermal polymerization of melamine in the presence of NH4Cl and KCl salts at 823 K (melamine/NH4Cl/KCl = 1/3/10 wt%). Different techniques like FTIR, XPS, XRD, SEM and TEM confirmed that the structure of CN–K consisted of a K-intercalated poly(heptazine) motif. Again, compared to traditional bulk carbon nitrides (g-C3N4) which are amorphous aggregates on the microscale, CN–K consisted of lamellar nanocrystalites. A weaker emission peak in the photoluminescence spectra, a short average lifetime in fluorescence decay spectra and a stronger transient photocurrent response imply that CN–K is more effective in promoting the generation and separation of photogenerated electron–holes. This proves that potassium ion-incorporated carbon nitride has more photocatalytic activity than simple carbon nitride. Incorporated K however, plays no role in the mechanistic pathway. The higher photocatalytic activity of CN–K is also evident from the band gap of 2.72 eV with a CB potential of −0.88 eV vs. NHE and this thermodynamically favors single electron reduction of O2 {(E(O2/˙O2−) = −0.16 vs. NHE} to the superoxide radical anion ˙O2−. The effectiveness of the engineered CN–K heterogeneous catalyst for the fluoroalkylaton-distal functionalization of unactivated alkenes was established by synthesizing a library of heteroaryl migrated products (Scheme 30). Optimization studies revealed that irradiation with blue light and the application of air as oxidants were very much essential for the smooth functioning of this protocol. Benzothiazole, benzoxazolyl, thiazolyls, etc. were a few other compatible heteroaryl migratory groups in this fluoroalkylation-distal functionalization strategy (77a–e, 20–82%). A sterically hindered as well as non-terminal alkene also delivered multi-substituted products (77b, 50%; 77c, 20%) although the yield is low with the latter one. Formyl or imino groups also underwent migration in this protocol delivering corresponding CF3 functionalized aldehydes or imines (77e–f, 70–87%). Different fluoroalkylating sources (76) also worked well under standard conditions (77g, 67%). The practicability of the said protocol was illustrated by performing the same reaction with the CN–K-based catalyst recycled at least 5 times without much loss of catalytic activity (Scheme 30).
 | ||
| Scheme 30 CN–K catalyzed blue light promoted distal functionalization of unactivated alkenes with RFSO2Na. | ||
Various radical scavenger experiments, EPR analysis, and the light on–off experiments suggested that the reaction proceeded via a radical pathway. The generation of the oxygen superoxide radical anion (˙O2−) and light irradiation were crucial for the reaction. Based on all this experimental evidence, a plausible reaction mechanism was proposed (Scheme 31). The electronic properties of CN–K with a band gap of 2.72 eV help in the efficient separation of electron–hole pairs under blue light irradiation (460 nm). As explained earlier, the photoelectrons can reduce O2 to ˙O2−. Simultaneously, the photogenerated hole of CN–K via SET with RFSO2Na (76) generated the RF radical, which initiates the reaction with alkene (75) and generates the radical A11. The intramolecular radical addition of A11 to the migratory groups gives cyclic species B11 which subsequently yields another radical intermediate C11via C–C bond cleavage. Intermediate C11 then undergoes oxidation by ˙O2−, followed by deprotonation, to deliver the corresponding product (77) and completes the catalytic cycle (Scheme 31).
 | ||
| Scheme 31 A plausible mechanism for CN–K catalyzed blue light promoted distal functionalization of unactivated alkenes with RFSO2Na. | ||
The same research group also employed the abovementioned catalyst for both decarboxylative addition and reductive dimerization of para-quinone methides (p-QMs) (78) (Scheme 32).93 Mild reaction conditions, heterogeneous nature of the catalyst and its admirable recyclability, broad substrate scope and scale-up in a continuous flow are a few added advantages of this protocol.
 | ||
| Scheme 32 CN–K-catalyzed decarboxylative addition and reductive dimerization of para-quinone methides. | ||
According to the proposed mechanism by authors (Scheme 33), for the reductive dimerization reaction, initially, holes and electron pairs are created by light-absorbed CN–K. Photo-electrons then reduce p-QMs to the diaryl methyl radical (A12) via a proton-coupled electron transfer (PCET) process. Following that, a 1,6-radical addition of A12 generated radical B12. HCOONa underwent oxidation to HCOO˙, by the photo-induced hole of CN–K (+0.84 eV) which after hydrogen atom transfer to B12 yielded the desired dimerized product (82). For, the decarboxylative 1,6-addition reaction, holes of CN–K underwent SET with carboxylic acid anions to give corresponding alkyl or acyl radicals (C12 or D12). The direct coupling of radical A12 with radical C12 or D12 gave the anticipated 1,6-addition adducts (81).
 | ||
| Scheme 33 Proposed mechanism for CN–K catalyzed decarboxylative addition and reductive dimerization. | ||
Very recently, a synergistic dual catalyst consisting of g-C3N4 and few-layered black phosphorus (FLBP/g-CN) was developed and applied for C–H arylation reactions under visible light irradiation (Scheme 34). Metin and co-workers in their work investigated a variety of heteroarenes like furan, thiophene and N-Boc-pyrrole (83) with substituted diazonium salts bearing both electron-donating as well as withdrawing groups (84) to give corresponding C–H arylated products (85).94
 | ||
| Scheme 34 FLBP/g-CN-catalyzed C–H arylation reactions under visible light irradiation. | ||
From the literature survey and control experiments, a plausible mechanism was proposed (Scheme 35). According to this, after photoexcitation, the generated electrons of FLBP/g-CN get transferred to the diazonium salt (84a) to generate a radical intermediate (A13). This radical intermediate then gets captured by heteroarene (83) to form another radical intermediate B13, which ultimately undergoes oxidation to cation intermediate C13via transferring an electron to the photogenerated holes. Finally, deprotonation of this cation intermediate leads to the formation of the anticipated product (85a) (Scheme 35).
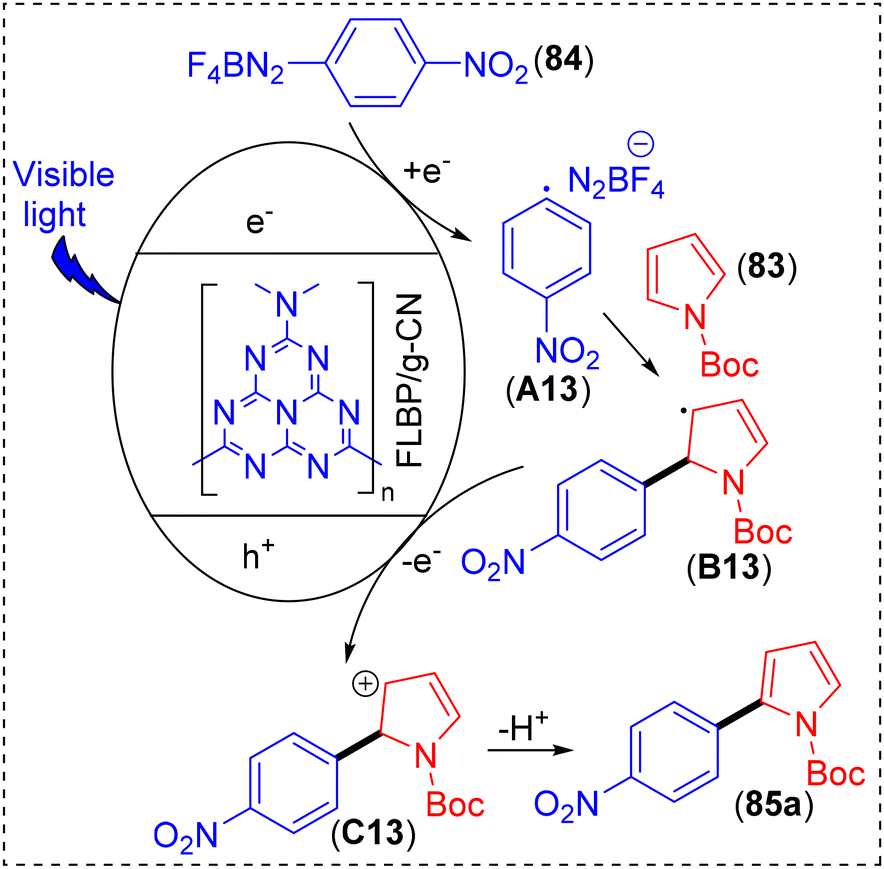 | ||
| Scheme 35 Proposed mechanism for C–H arylation reaction catalyzed by FLBP/g-CN. | ||
In the study of graphitic carbon nitride's action in the photocatalytic cycle, hole (or electron) scavengers are often added to remove one charge carrier effectively from the VB (or CB) of the light absorber to facilitate studies of the complementary oxidation/reduction process. However, due to strong kinetic asymmetry, while removing charge carriers from the light absorber, accumulation of one type of charge occurred. This build-up of charge has been shown to accelerate charge recombination which inherently decreases the photocatalytic activity efficiency. Unfortunately, graphitic carbon-nitride materials tend to aggregate in solution and therefore form an optically inhomogeneous suspension, limiting the understanding of the carrier dynamics for their photocatalytic performance. Therefore, engineering a carbon nitride photocatalyst to improve its catalytic activity is an emerging topic while dealing with graphitic carbon nitride. Fine-tuning of CNx to overcome charge recombination or rate asynchrony will increase its scope and acceptability as a photo agent. A long-lived triplet excited-state is found to be optimal to allow for diffusive excited-state quenching and avoid rate asynchrony between redox processes.95,96
In 2021, Nocera et al. utilized long-lived triplet excited states of the heterogeneous cyanamide modified carbon nitride (NCNCNx) photocatalyst in the hydroamidation reaction (Scheme 36).97 Product (87) was isolated in substantial yield. The authors synthesized heterogeneous cyanamide modified carbon nitride (NCNCNx) via thermal KSCN treatment of NCNCNx. However, the major concern associated with these CNx materials is the excess build-up of trapped electrons due to inconsistency in oxidation and reduction rates. This build-up of trapped electrons has been found to accelerate the rate of charge recombination and thus reduces the efficiency of CNx as a photoreagent. To avoid this rate asynchrony, the authors further reduced the particle size of NCNCNxvia liquid-assisted grinding in a planetary ball-mill. Two types of CNx materials with Z-average particle sizes, as measured by dynamic light scattering (DLS), of 304 nm (NCNCNx304) and 148 nm (NCNCNx148) were obtained, as compared to the 607 nm of the parent material (NCNCNx607). Again, no major changes were noticed after ball-milling and NCN modification was preserved in the photocatalyst and the same is confirmed by IR stretching frequency, XPS and PXRD studies. The reduction of the particle size of NCNCNx helps in the formation of homogeneous suspension in an organic solvent which assists in achieving a long-lived emissive state. A slightly red-shifted long-lived emissive rate with τphos = 1.77 μs was observed with this reduced particle sized NCNCNx. This is because of the phosphorescent triplet state and the same is indicated by the fact that exposing the solution to air, quenches this long-lived emission and cooling the suspension of NCNCNx148 increased the intensity to a long-time scale of 4.8 μs. Furthermore, they utilized this particle size-dependent long-lived excited state for hydroamidation of olefins under blue light irradiation in the presence of phenyl disulphide (PhSSPh) and tributylmethylammoniumdibutylphosphate (PiBu2). Ball-milling treatment and reduction of the particle size of NCNCNx substantially accelerated the hydroamidation rate compared to NCNCNx, KClCNx, mpgCNx and unmodified NH2CNx. Again, the intramolecular hydroamidation product obtained from NCNCNx148 and NCNCNx304 was significantly better compared to the parent NCNCNx607.
 | ||
| Scheme 36 Synthesis of NCNCNx and its application in intramolecular hydroamidation. | ||
4. Conclusion and outlook
Dual-metallo-photocatalysis by using carbon nitride materials as photocatalysts is a highly fascinating concept. Passionate interest from the synthetic community in this approach is reflected in the literature summarized in this review. As the literature precedents demonstrate, the interaction of a non-metallic recyclable photocatalyst with a metal-based co-catalyst comes up with a bag full of advantages. These include (i) the application of a cheap, recyclable carbon nitride photocatalyst which makes the catalytic design more economical and sustainable, (ii) enhancing the rate of existing reactions, and (iii) carrying out effective transformations that were impossible or difficult to achieve by using a single catalyst. The possibility of synthesizing a ‘fully heterogeneous’ dual catalyst by inserting single atoms into the photocatalyst surface has also been discussed. This is believed to be the frontier in dual catalytic organic transformations, because, here the catalyst is a single material and apart from recyclability and characterization simplicities, it can help in making the mechanistic understanding easier. In this review, literature reports have been properly documented with the synthetic procedures of dual catalytic systems, their characterization, reusability, and applications in organic synthesis. Various mechanistic pathways involved therein have also been discussed. Even though a plethora of reactions have been reported by using this strategy, further advancement in certain fields is highly anticipated, like (i) extension of this approach to other metals, which have been centered mainly on nickel, (ii) application of fully recyclable dual photocatalytic systems for the synthesis of optically active compounds and (iii) employment of this strategy for cross-coupling of the most abundant yet precluded aryl chlorides in the formation of C–C bonds. Apart from some scattered reports, the C–Cl bond activation using graphitic carbon nitride is still elusive. The key challenge associated with C–Cl bonds in cross-electrophile coupling is the requirement of higher reactivity without sacrificing the selectivity. So, broadening this strategy towards the activation of strong C–Cl bonds using emerging g-CN-based catalysts will open up a new avenue. We hope that the review on this rather new branch of catalysis which resonates from inorganic and organic chemistry to materials science will help researchers from both academia and industry to determine the future direction of organic synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. K. P. acknowledges the support of this research by SERB (SCP/2022/000195, CRG/2022/004023). B. Dam acknowledges CSIR 09/731(0176)/2020-EMR-I for the fellowship, and B. D. thanks MHRD for fellowships.References
- P. T. Anastas and J. C. Warner, Green chemistry: theory and practice, Oxford University Press, Oxford, First publ. new as paperback, 2000 Search PubMed.
- P. Spargo, Org. Process Res. Dev., 2004, 8, 299–300 CrossRef CAS.
- Handbook of green chemistry and technology, ed. J. H. Clark and D. J. Macquarrie, Blackwell Science, Oxford [England], Malden, MA, 2002 Search PubMed.
- M. Poliakoff, J. M. Fitzpatrick, T. R. Farren and P. T. Anastas, Science, 2002, 297, 807–810 CrossRef CAS PubMed.
- A. S. Matlack, Introduction to green chemistry, M. Dekker, New York, 2001 Search PubMed.
- G. Albano, A. Punzi, M. A. M. Capozzi and G. M. Farinola, Green Chem., 2022, 24, 1809–1894 RSC.
- C.-M. Zhu, R.-B. Liang, Y. Xiao, W. Zhou, Q.-X. Tong and J.-J. Zhong, Green Chem., 2023, 25, 960–965 RSC.
- A. N. V. Satyanarayana, N. Mukherjee and T. Chatterjee, Green Chem., 2023, 25, 779–788 RSC.
- V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J.-M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS PubMed.
- P. Choudhary, A. Kumar and V. Krishnan, Chem. Eng. J., 2022, 431, 133695 CrossRef CAS.
- K. Kumari, P. Choudhary, D. Sharma and V. Krishnan, Ind. Eng. Chem. Res., 2023, 62, 158–168 CrossRef CAS.
- P. Choudhary, K. Kumari, D. Sharma, S. Kumar and V. Krishnan, ACS Appl. Mater. Interfaces, 2023, 15, 9412–9420 CrossRef CAS PubMed.
- N. Kumari, T. Chhabra, S. Kumar and V. Krishnan, Catal. Commun., 2022, 168, 106467 CrossRef CAS.
- X. Zhu, Y. Lin, Y. Sun, M. C. Beard and Y. Yan, J. Am. Chem. Soc., 2019, 141, 733–738 CrossRef CAS PubMed.
- K. Gadde, P. Mampuys, A. Guidetti, H. Y. V. Ching, W. A. Herrebout, S. Van Doorslaer, K. A. Tehrani and B. U. W. Maes, ACS Catal., 2020, 10, 8765–8779 CrossRef CAS.
- N. Chakraborty, K. K. Rajbongshi, A. Dahiya, B. Das, A. Vaishnani and B. K. Patel, Chem. Commun., 2023, 59, 2779–2782 RSC.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef PubMed.
- M. Parasram and V. Gevorgyan, Chem. Soc. Rev., 2017, 46, 6227–6240 RSC.
- H. N. Dhara, A. Rakshit, T. Alam, A. K. Sahoo and B. K. Patel, Org. Lett., 2023, 25, 471–476 CrossRef CAS PubMed.
- C. Rosso, G. Filippini, A. Criado, M. Melchionna, P. Fornasiero and M. Prato, ACS Nano, 2021, 15, 3621–3630 CrossRef CAS PubMed.
- J. D. Bell and J. A. Murphy, Chem. Soc. Rev., 2021, 50, 9540–9685 RSC.
- S. P. Pitre and L. E. Overman, Chem. Rev., 2022, 122, 1717–1751 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- B. König, Chemical photocatalysis, Walter de Gruyter GmbH & Company KG, 2013 Search PubMed.
- M. Marchi, G. Gentile, C. Rosso, M. Melchionna, P. Fornasiero, G. Filippini and M. Prato, ChemSusChem, 2022, 15, e202201094 CrossRef CAS PubMed.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- F.-L. Zeng, H.-L. Zhu, X.-L. Chen, L.-B. Qu and B. Yu, Green Chem., 2021, 23, 3677–3682 RSC.
- B. Dam, A. K. Sahoo and B. K. Patel, Green Chem., 2022, 24, 7122–7130 RSC.
- D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355–360 CrossRef CAS.
- A. K. Sahoo, A. Rakshit, A. Pan, H. N. Dhara and B. K. Patel, Org. Biomol. Chem., 2023, 21, 1680–1691 RSC.
- D.-M. Yan, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 378–380 CrossRef CAS PubMed.
- L. Xia, M. Jin, Y. Jiao and S. Yu, Org. Lett., 2022, 24, 364–368 CrossRef CAS PubMed.
- B. Nagar and B. B. Dhar, J. Org. Chem., 2022, 87, 3195–3201 CrossRef CAS PubMed.
- C. Bo, Q. Bu, J. Liu, B. Dai and N. Liu, ACS Sustainable Chem. Eng., 2022, 10, 1822–1828 CrossRef CAS.
- A. Das and K. R. J. Thomas, Green Chem., 2022, 24, 4952–4957 RSC.
- S. G. E. Amos, M. Garreau, L. Buzzetti and J. Waser, Beilstein J. Org. Chem., 2020, 16, 1163–1187 CrossRef CAS PubMed.
- S. Gisbertz and B. Pieber, ChemPhotoChem, 2020, 4, 456–475 CrossRef CAS.
- A. Savateev and M. Antonietti, ACS Catal., 2018, 8, 9790–9808 CrossRef CAS.
- S. Mazzanti and A. Savateev, ChemPlusChem, 2020, 85, 2499–2517 CrossRef CAS PubMed.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC.
- S. Reischauer and B. Pieber, iScience, 2021, 24, 102209 CrossRef CAS PubMed.
- J. A. Caputo, L. C. Frenette, N. Zhao, K. L. Sowers, T. D. Krauss and D. J. Weix, J. Am. Chem. Soc., 2017, 139, 4250–4253 CrossRef CAS.
- Y.-Y. Liu, D. Liang, L.-Q. Lu and W.-J. Xiao, Chem. Commun., 2019, 55, 4853–4856 RSC.
- J. Tian, L. Zhao, C. Yang, C. Yang, L. Guo and W. Xia, ACS Catal., 2023, 13, 866–876 CrossRef CAS.
- S. K. Verma, R. Verma, Y. R. Girish, F. Xue, L. Yan, S. Verma, M. Singh, Y. Vaishnav, A. B. Shaik, R. R. Bhandare, K. P. Rakesh, K. S. S. Kumar and K. S. Rangappa, Green Chem., 2022, 24, 438–479 RSC.
- P. P. Singh and V. Srivastava, RSC Adv., 2022, 12, 18245–18265 RSC.
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS.
- T. Chhabra, A. Bahuguna, S. S. Dhankhar, C. M. Nagaraja and V. Krishnan, Green Chem., 2019, 21, 6012–6026 RSC.
- J. Liebig, Ann. Pharm., 1834, 10, 1–47 CrossRef.
- J. Liebig, Ann. Chem. Pharm., 1844, 50, 294–295 CrossRef.
- J. Liebig, Ann. Chem. Pharm., 1855, 95, 257–282 CrossRef.
- E. Kroke, Coord. Chem. Rev., 2004, 248, 493–532 CrossRef CAS.
- E. Kroke, M. Schwarz, E. Horath-Bordon, P. Kroll, B. Noll and A. D. Norman, New J. Chem., 2002, 26, 508–512 RSC.
- I. Ghosh, J. Khamrai, A. Savateev, N. Shlapakov, M. Antonietti and B. König, Science, 2019, 365, 360–366 CrossRef CAS PubMed.
- P. Geng, Y. Tang, G. Pan, W. Wang, J. Hu and Y. Cai, Green Chem., 2019, 21, 6116–6122 RSC.
- T. S. Miller, A. B. Jorge, T. M. Suter, A. Sella, F. Corà and P. F. McMillan, Phys. Chem. Chem. Phys., 2017, 19, 15613–15638 RSC.
- Z. Zhang, K. Leinenweber, M. Bauer, L. A. J. Garvie, P. F. McMillan and G. H. Wolf, J. Am. Chem. Soc., 2001, 123, 7788–7796 CrossRef CAS PubMed.
- E. Wirnhier, M. Döblinger, D. Gunzelmann, J. Senker, B. V. Lotsch and W. Schnick, Chem. – Eur. J., 2011, 17, 3213–3221 CrossRef CAS PubMed.
- M. B. Mesch, K. Bärwinkel, Y. Krysiak, C. Martineau, F. Taulelle, R. B. Neder, U. Kolb and J. Senker, Chem. – Eur. J., 2016, 22, 16878–16890 CrossRef CAS PubMed.
- M. J. Bojdys, J.-O. Müller, M. Antonietti and A. Thomas, Chem. – Eur. J., 2008, 14, 8177–8182 CrossRef CAS PubMed.
- Z. Chen, A. Savateev, S. Pronkin, V. Papaefthimiou, C. Wolff, M. G. Willinger, E. Willinger, D. Neher, M. Antonietti and D. Dontsova, Adv. Mater., 2017, 29, 1700555 CrossRef PubMed.
- H. Schlomberg, J. Kröger, G. Savasci, M. W. Terban, S. Bette, I. Moudrakovski, V. Duppel, F. Podjaski, R. Siegel, J. Senker, R. E. Dinnebier, C. Ochsenfeld and B. V. Lotsch, Chem. Mater., 2019, 31, 7478–7486 CrossRef CAS PubMed.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- Y. Deng, Y. Xie, K. Zou and X. Ji, J. Mater. Chem. A, 2016, 4, 1144–1173 RSC.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- U. B. Kim, D. J. Jung, H. J. Jeon, K. Rathwell and S. Lee, Chem. Rev., 2020, 120, 13382–13433 CrossRef CAS PubMed.
- B. Das, A. Dahiya, A. K. Sahoo and B. K. Patel, J. Org. Chem., 2022, 87, 13383–13388 CrossRef CAS PubMed.
- M. M. Mastandrea and M. A. Pericàs, Eur. J. Inorg. Chem., 2021, 2021, 3421–3431 CrossRef CAS.
- Z. Chen, S. Mitchell, E. Vorobyeva, R. K. Leary, R. Hauert, T. Furnival, Q. M. Ramasse, J. M. Thomas, P. A. Midgley, D. Dontsova, M. Antonietti, S. Pogodin, N. López and J. Pérez-Ramírez, Adv. Funct. Mater., 2017, 27, 1605785 CrossRef.
- C. Cavedon, A. Madani, P. H. Seeberger and B. Pieber, Org. Lett., 2019, 21, 5331–5334 CrossRef CAS PubMed.
- B. Pieber, J. A. Malik, C. Cavedon, S. Gisbertz, A. Savateev, D. Cruz, T. Heil, G. Zhang and P. H. Seeberger, Angew. Chem., Int. Ed., 2019, 58, 9575–9580 CrossRef CAS PubMed.
- J. Khamrai, I. Ghosh, A. Savateev, M. Antonietti and B. König, ACS Catal., 2020, 10, 3526–3532 CrossRef CAS.
- J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433–436 CrossRef CAS PubMed.
- J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef CAS PubMed.
- D. N. Primer, I. Karakaya, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2015, 137, 2195–2198 CrossRef CAS PubMed.
- T. E. Schirmer, M. Abdellaoui, A. Savateev, C. Ollivier, M. Antonietti, L. Fensterbank and B. König, Org. Lett., 2022, 24, 2483–2487 CrossRef CAS PubMed.
- S. Das, K. Murugesan, G. J. V. Rodríguez, J. Kaur, J. P. Barham, A. Savateev, M. Antonietti and B. König, ACS Catal., 2021, 11, 1593–1603 CrossRef CAS.
- J. Khamrai, S. Das, A. Savateev, M. Antonietti and B. König, Green Chem., 2021, 23, 2017–2024 RSC.
- Y. Qin, B. C. Martindale, R. Sun, A. J. Rieth and D. G. Nocera, Chem. Sci., 2020, 11, 7456–7461 RSC.
- K. Wang, M. Tong, Y. Yang, B. Zhang, H. Liu, H. Li and F. Zhang, Green Chem., 2020, 22, 7417–7423 RSC.
- Z. Zhang, Y. Xu, Q. Zhang, S. Fang, H. Sun, W. Ou and C. Su, Sci. Bull., 2022, 67, 71–78 CrossRef CAS PubMed.
- X. Zhao, C. Deng, D. Meng, H. Ji, C. Chen, W. Song and J. Zhao, ACS Catal., 2020, 10, 15178–15185 CrossRef CAS.
- A. Vijeta, C. Casadevall, S. Roy and E. Reisner, Angew. Chem., Int. Ed., 2021, 60, 8494–8499 CrossRef CAS PubMed.
- A. Vijeta, C. Casadevall and E. Reisner, Angew. Chem., Int. Ed., 2022, 61, e202203176 CrossRef CAS PubMed.
- M. Kwak, J. Bok, B.-H. Lee, J. Kim, Y. Seo, S. Kim, H. Choi, W. Ko, W. H. Antink, C. W. Lee, G. H. Yim, H. Seung, C. Park, K.-S. Lee, D.-H. Kim, T. Hyeon and D. Yoo, Chem. Sci., 2022, 13, 8536–8542 RSC.
- T. Jia, D. Meng, H. Ji, H. Sheng, C. Chen, W. Song and J. Zhao, Appl. Catal., B, 2022, 304, 121004 CrossRef CAS.
- D. Sun, P. Li, X. Wang, Y. Wang, J. Wang, Y. Wang, Y. Lu, L. Duan, S. Sarina, H. Zhu and J. Liu, Chem. Commun., 2020, 56, 11847–11850 RSC.
- M. A. R. da Silva, I. F. Silva, Q. Xue, B. T. W. Lo, N. V. Tarakina, B. N. Nunes, P. Adler, S. K. Sahoo, D. W. Bahnemann, N. López-Salas, A. Savateev, C. Ribeiro, T. D. Kühne, M. Antonietti and I. F. Teixeira, Appl. Catal., B, 2022, 304, 120965 CrossRef CAS.
- W. Jiang, H. Deng and J. Liu, Catal. Commun., 2022, 170, 106498 CrossRef CAS.
- A. S. Sharma, V. S. Sharma and H. Kaur, ACS Appl. Nano Mater., 2020, 3, 1191–1202 CrossRef CAS.
- Y. He, X. Dan, Y. Tang, Q. Yang, W. Wang and Y. Cai, Green Chem., 2021, 23, 9577–9582 RSC.
- Q. Yang, G. Pan, J. Wei, W. Wang, Y. Tang and Y. Cai, ACS Sustainable Chem. Eng., 2021, 9, 2367–2377 CrossRef CAS.
- Z. Eroglu, M. S. Ozer, T. Kubanaliev, H. Kilic and Ö. Metin, Catal. Sci. Technol., 2022, 12, 5379–5389 RSC.
- W. Yang, R. Godin, H. Kasap, B. Moss, Y. Dong, S. A. J. Hillman, L. Steier, E. Reisner and J. R. Durrant, J. Am. Chem. Soc., 2019, 141, 11219–11229 CrossRef CAS PubMed.
- V. W. Lau, D. Klose, H. Kasap, F. Podjaski, M.-C. Pignié, E. Reisner, G. Jeschke and B. V. Lotsch, Angew. Chem., Int. Ed., 2017, 56, 510–514 CrossRef CAS PubMed.
- A. J. Rieth, Y. Qin, B. C. M. Martindale and D. G. Nocera, J. Am. Chem. Soc., 2021, 143, 4646–4652 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |