
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrobial electrosynthesis with Clostridium ljungdahlii benefits from hydrogen electron mediation and permits a greater variety of products†
Santiago T.
Boto
 ab,
Bettina
Bardl
a,
Falk
Harnisch
c and
Miriam A.
Rosenbaum
*ab
ab,
Bettina
Bardl
a,
Falk
Harnisch
c and
Miriam A.
Rosenbaum
*ab
aLeibniz Institute for Natural Product Research and Infection Biology – Hans Knöll Institute (Leibniz-HKI), Jena, Germany. E-mail: miriam.rosenbaum@leibniz-hki.de
bFaculty of Biological Sciences, Friedrich Schiller University, Jena, Germany
cUFZ – Helmholtz-Centre for Environmental Research GmbH, Department of Environmental Microbiology, Permoserstraße, 15, 04318 Leipzig, Germany
First published on 17th May 2023
Abstract
Microbial electrosynthesis (MES) is a very promising technology addressing the challenge of carbon dioxide recycling into organic compounds, which might serve as building blocks for the (bio)chemical industry. However, poor process control and understanding of fundamental aspects such as the microbial extracellular electron transfer (EET) currently limit further developments. In the model acetogen Clostridium ljungdahlii, both direct and indirect electron consumption via hydrogen have been proposed. However, without clarification neither targeted development of the microbial catalyst nor process engineering of MES are possible. In this study, cathodic hydrogen is demonstrated to be the dominating electron source for C. ljungdahlii at electroautotrophic MES allowing for superior growth and biosynthesis, compared to previously reported MES using pure cultures. Hydrogen availability distinctly controlled an either planktonic- or biofilm-dominated lifestyle of C. ljungdahlii. The most robust operation yielded higher planktonic cell densities in a hydrogen mediated process, which demonstrated the uncoupling of growth and biofilm formation. This coincided with an increase of metabolic activity, acetate titers, and production rates (up to 6.06 g L−1 at 0.11 g L−1 d−1). For the first time, MES using C. ljungdahlii was also revealed to deliver other products than acetate in significant amounts: here up to 0.39 g L−1 glycine or 0.14 g L−1 ethanolamine. Hence, a deeper comprehension of the electrophysiology of C. ljungdahlii was shown to be key for designing and improving bioprocess strategies in MES research.
Introduction
The important commitment towards decarbonization made during the 26th United Nations Climate Change Conference of the Parties (COP26) represents a turning point. Countries must undertake political and economic changes to reduce greenhouse gas emissions, and double their efforts to mitigate climate change and its effects. From this situation an opportunity has emerged to create and improve key technologies enabling the transition to a bio-based and circular economy.1–3 Thereby, the substitution of fossil hydrocarbons for chemical production is essential but remains a challenging task.Most of the current strategies to address the substitution of hydrocarbons in the chemical industry are based on the utilization of bio-derived feedstock that are transformed into commodity chemicals through chemical or biotechnological ways as well as combinations thereof.4–6 Increasing attention is paid to the direct transformation of carbon dioxide (CO2) into commodity chemicals,7 as advancing such technologies will be key to a sustainable bioeconomy.8,9 Microbial electrosynthesis (MES)10 offers a biotechnological approach based on using electricity to provide the electrons needed for microbially catalyzed CO2 reduction. MES from CO2 is biochemically related to C1-gas fermentation,11 but here reducing power is not delivered into the system as hydrogen gas (H2) but directly as electric current using negatively poised electrodes (i.e. cathodes) within a bioelectrochemical system (BES).
Different microbes – being termed electrotrophs – are capable of consuming electrons from a cathode. This ability was first proven with Cupriavidus necator,12 by electrolytically generating H2 and oxygen, and since then many other electrotrophs have been found, as nicely reviewed here.13,14 Some electrotrophs are also capable of using these electrons to reduce CO2 and grow chemolithoautotrophically on CO2 and current as the sole sources of carbon and energy. These can be denominated as electroautotrophs. The first reported electroautotrophic microbe was Sporomusa ovata.15 In recent years, research on MES with electroautotrophs (mainly acetogens like Sporomusa spp. and Clostridium spp.) has shown the potential to produce commodity chemicals from CO2 and electricity, but it has also highlighted the technological and biological limitations.16 Drawbacks such as low product titers and coulombic efficiencies (CE), poor selectivity, challenging scale-up, and a very limited and economically unattractive product portfolio currently keep the field at an impasse.17 Key to overcoming this overall lack of performance, is a much deeper understanding of the physiology of electroautotrophs. Very often the focus of developments of MES from CO2 is on the technical side, developing new electrode materials, reactor configurations, or operational conditions.18,19 Meanwhile, the biological side is often treated as a “black box” where undefined microbial communities (microbial mixed cultures or reactor microbiomes) are preferred, because they are easier to handle, more robust, and the associated costs are lower.20 On the other hand, the use of pure cultures, although more challenging from the biological side, increases product specificity, reproducibility, and could potentially expand the range of products beyond the current limited portfolio.21
Although there is also great interest in the production of methane (CH4),22–24 the main products obtained by MES from CO2 are short-chain fatty acids and their corresponding alcohols (acetate/ethanol, butyrate/butanol, caproate/hexanol).25–28 The conversion of CO2 to acetyl-CoA, which then leads to the production of the aforementioned compounds, proceeds most often through the Wood–Ljungdahl pathway. Acetogens, as the main users of this pathway, utilize carbon monoxide (CO) or H2 as the electron donor for reducing CO2 during gas fermentation.29 However, during MES, it is still not experimentally verified if the cathodic electrons are channeled into the cell via membrane-associated redox components like cytochromes,30 nanowires31,32 or unknown means of direct extracellular electron transfer (DET), or by using diffusible electron shuttles like H2,33 flavins34 or quinones35 in mediated extracellular electron transfer (MET).
In our study, we clarify the electrophysiology of Clostridium ljungdahlii, as a model acetogen, at electroautotrophic conditions. This bacterium was initially reported to be able to perform DET25 but later studies rather suggested H2 mediation17 as the predominant mechanism of extracellular electron transfer (EET). More insight about the contribution of different EET mechanisms, as well as its influence in bacterial metabolism and growth, was essential to achieve a deeper comprehension about the physiology of this model microbe for MES from CO2. Ultimately, this allowed for higher throughput, better control over the biological aspects of the process and a broader range of products.
Experimental
Bacterial strains and culture conditions
All chemicals used in this study were of analytical grade, purchased from Thermo Fisher Scientific (Waltham, USA), Merck (Darmstadt, Germany) and Carl Roth (Karlsruhe, Germany). Clostridium ljungdahlii DSM 13528 (DSMZ) was cultured heterotrophically in RCM (10 g L−1 peptone, 10 g L−1 meat extract, 3 g L−1 yeast extract, 5 g L−1 NaCl, 5 g L−1 fructose, 1 g L−1 soluble starch, 5 g L−1 Na-acetate·3H2O, 0.5 g L−1L-cysteine·HCl, 1 mg L−1 resazurin) at 37 °C. Autotrophic and electroautotrophic growth were achieved in modified PETC medium (20 g L−1 MES, 2 g L−1 NH4Cl, 0.2 g L−1 KCl, 0.4 g L−1 MgSO4·7H2O, 0.2 g L−1 NaCl, 0.2 g L−1 KH2PO4, 80 mg L−1 CaCl2·2H2O, 2 mg L−1 CoCl2·6H2O, 8 mg L−1 (NH4)2Fe(SO4)2, 10 mg L−1 MnSO4, 20 mg L−1 nitrilotriacetic acid, 2 mg L−1 Na2MoO4, 0.2 mg L−1 Na2SeO4, 0.2 mg L−1 Na2WO4, 2 mg L−1 ZnSO4, 0.02 mg L−1 biotin, 0.05 mg L−1 pantothenic acid, 0.05 mg L−1 thiamine·HCl, 1 g L−1 yeast extract, 0.3 g L−1L-cysteine·HCl, 1 mg L−1 resazurin) adjusted to pH 5.7. Autotrophic pre-cultures for electroautotrophic growth were made in horizontally incubated 250 mL serum bottles with a filling volume of 30–50 mL. The headspace was filled with CO2 and H2 (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80; Air Liquide, Paris, France) at an overpressure of 1.5 bar. All the work performed with C. ljungdahlii was performed under strict anaerobic conditions. Escherichia coli DH5α (New England BioLabs, Ipswich, USA) and E. coli pANA1 (ref. 36) were cultured in LB at 37 °C. E. coli DH5α was used for the construction and propagation of clostridial shuttle plasmids, and E. coli pANA1 was used for plasmid methylation as previously described.36 The cloning strategy and construction of the C. ljungdahlii pFAST(thl) strain are detailed in the ESI (ESI Materials and methods: plasmid construction and genetic manipulation and Table S1†).
:80; Air Liquide, Paris, France) at an overpressure of 1.5 bar. All the work performed with C. ljungdahlii was performed under strict anaerobic conditions. Escherichia coli DH5α (New England BioLabs, Ipswich, USA) and E. coli pANA1 (ref. 36) were cultured in LB at 37 °C. E. coli DH5α was used for the construction and propagation of clostridial shuttle plasmids, and E. coli pANA1 was used for plasmid methylation as previously described.36 The cloning strategy and construction of the C. ljungdahlii pFAST(thl) strain are detailed in the ESI (ESI Materials and methods: plasmid construction and genetic manipulation and Table S1†).
Setup of bioelectrochemical systems
H-type glass reactors (Laborglas Lammek, Moers, Germany; Fig. S1†) were used for the bioelectrochemical experiments. Both half-cells were separated with a CMI-7000S cation exchange membrane (Membranes International, Ringwood, USA). Each half-cell had a nominal volume of 650 mL and a filling volume of 400 mL. All gas connections were made with oxygen-tight Tygon® tubing (VWR, Radnor, USA), and protected from contamination by using 0.2 μm PTFE vent filters (Merck, Darmstadt, Germany). All ports had butyl rubber septa (Reichelt Chemietechnik, Heidelberg, Germany) and were further sealed by using oxygen-tight PTFE tape (Würth, Künzelsau, Germany). Working and counter electrodes were made of graphite EDM-3 blocks (Novotec, Reuver, Netherlands). The standard electrode surface area was 26.59 cm2 (surface-area-to-reactor-volume ratio: 66.48 cm2 L−1). In certain experiments, as indicated in the respective sections, bigger cathodes of 153.05 cm2 (surface-area-to-reactor-volume ratio: 382.63 cm2 L−1) were used. In-house made Ag/AgClsat KCl electrodes were used as reference electrodes and reactors were operated using a potentiostat (VMP3; BioLogic, Seyssinet-Pariset, France). The off-gas stream of the reactors was analyzed using BlueVary analyzers (BlueSens, Herten, Germany) equipped with cartridges for H2 and CO2 quantification. The MES were set up in a temperature-controlled room at 37 °C. A scheme of the general setup is presented in Fig. 1A.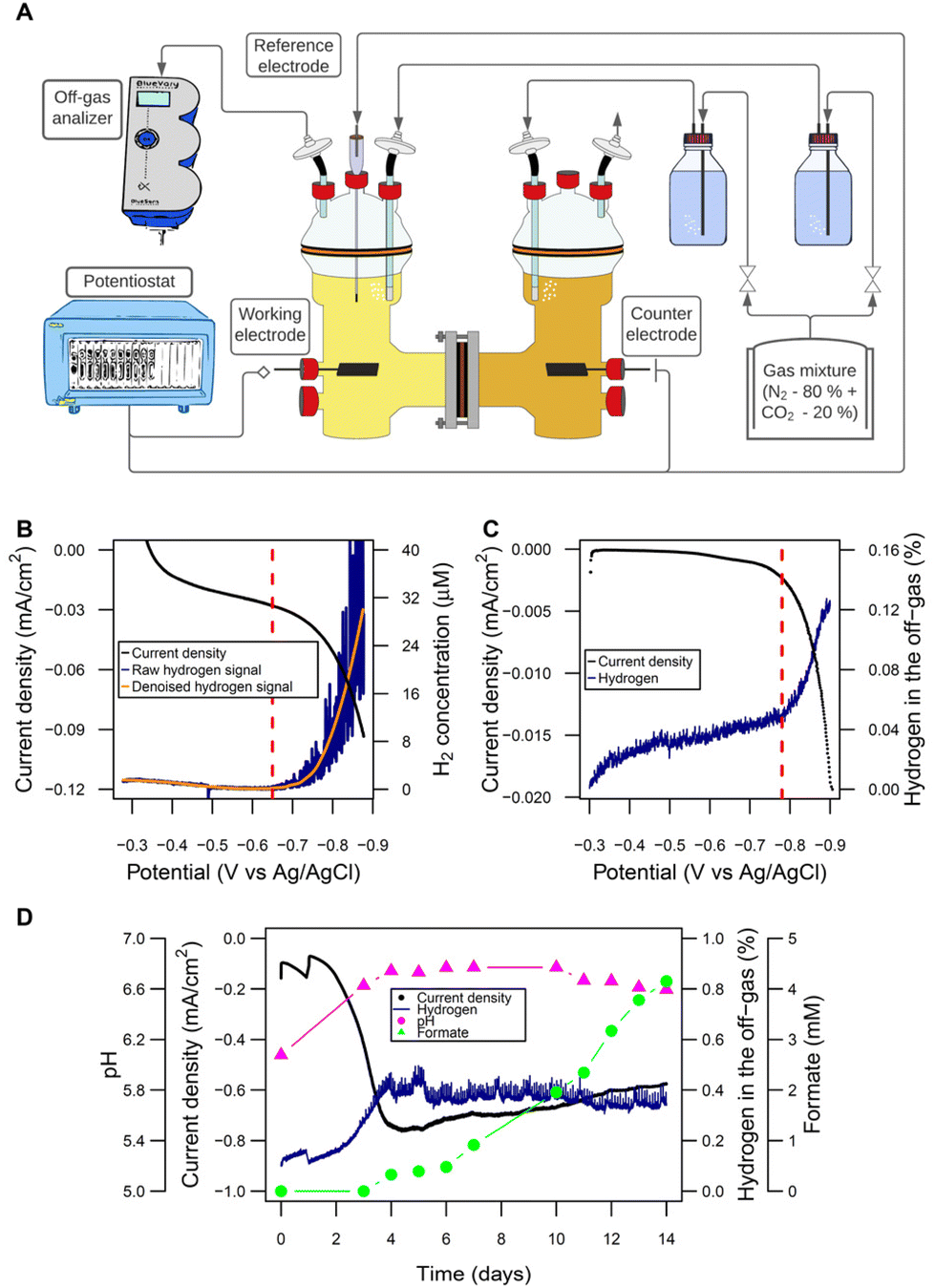 | ||
| Fig. 1 Microbial electrosynthesis cells: configuration and abiotic electrochemical performance. (A) Scheme of the H-type reactors and the general set-up used for the experiments (picture in Fig. S1†). (B) Linear sweep voltammetry (0.1 mV s−1) to determine the threshold of H2 evolution at pH 7 by the dissolved H2 microsensor (threshold indicated with a red vertical line: −720 mV). (C) Linear sweep voltammetry (0.1 mV s−1) to determine the threshold of H2 evolution at pH 7 by the off-gas analyzer (threshold indicated with a red vertical line: −780 mV). (D) Abiotic performance: H2 concentration in the off-gas and pH remained stable after a period of adaptation at a cathode potential of −900 mV. Formate is produced after about 4 days. Electrode surface area was 26.59 cm2 with a surface-area-to-reactor-volume ratio of 66.48 cm2 L−1. | ||
Operation of bioelectrochemical systems
If not stated otherwise, all provided potentials refer to the Ag/AgClsat KCl reference electrode, being +185 mV vs. standard hydrogen electrode (SHE) at 37 °C. Reactor electrolytes were constantly sparged with humidified and sterilized nitrogen and CO2 (80:20, Air Liquide, Paris, France) at an approximate volume flow of 0.1 L min−1. The cathodic chamber was stirred at 200 rpm. Potentiostat initialization and reactor inoculation (only for biotic experiments) were performed 24 and 48 h after the gassing started, respectively. Chronoamperometry (CA) was applied throughout the operation (the applied potential varied with the experiment), supplemented with cyclic voltammetry (CV) analysis at specific time points. CV was performed between −1 V and +0.2 V, with a scan rate of 1 mV s−1. Linear sweep voltammetry (LSV) in the abiotic experiments was performed from −0.3 V to −0.9 V with a scan rate of 0.1 mV s−1. Before performing LSV, overnight oxidation of the electrolyte was performed at −0.3 V to assure H2 depletion. C. ljungdahlii was inoculated in the MES from pre-grown autotrophic cultures using different inoculation densities: 3.4 × 107 ± 6.0 × 106 cells (further referred to as low inoculation density) and 1.0 × 108 ± 1.8 × 107 cells (further referred to as high inoculation density).
Microsensor measurements
A 1D motorized microprofiling system (Unisense, Aarhus, Denmark) equipped with a long H2 needle sensor (H2-NP; Unisense, Aarhus, Denmark), or with a long pH mini electrode (PH-500; Unisense, Aarhus, Denmark) in combination with a robust Ag/AgClsat KCl reference electrode (REF-RM; Unisense, Aarhus, Denmark), was used to measure dissolved H2 and pH gradients in close proximity to the cathode inside the catholyte. In order to keep anaerobic conditions, while the reactor lid was opened for the measurements, a customized anaerobic box was built (Fig. S2†).Electrochemical abiotic formate production
250 mL bottles (Schott, Mainz, Germany) with butyl septa were assembled with new graphite EDM-3 (Novotec, Reuver, Netherlands) electrodes. Modified PETC medium was used as electrolyte (filling volume: 200 mL). Cathode potential of −0.9 V and open circuit voltage (OCV) were applied in triplicate set-ups. 200 mM KCl electrolyte was used as negative control for formate production. Electrolyte samples were taken at times: 0, 1 and 2 weeks. In the case of KCl, 1 week was enough to obtain stable currents. The electrodes were dried and stored at −80 °C.ICP-OES analysis
Elemental concentration of the electrolytes of the bottles used for abiotic formate production was determined using ICP-OES (Spectro-Acros ICP OES, Spectro, Ameteck, Germany). Samples were diluted without further pretreatment 1:10 with 1% HNO3 and calibration was performed using TraceCERT® (Merck KG, Germany).
EDX-microscopy
To detect presumable CO2-to-formate reduction catalysts on the cathode surface, scanning electron microscopy (SEM) imaging coupled with energy dispersive X-ray spectroscopy (EDX) was used. A Zeiss Merlin VP compact field-emitting scanning electron microscope (Carl Zeiss Microscopy, Oberkochen, Germany) with the software package SmartSEM was used to acquire six to eight micrographs of different magnifications (100×, 300×, 1000×) on at least two fields-of-view per electrode. For that, the electron landing energy was set to 19.5 kV and secondary electron detection was used. The distribution of chemical elements, in particular metals, on the electrode surface was analyzed by EDX using a Bruker Quantax FlatQuad spectrometer (Bruker Nanoanalytics, Berlin, Germany) with the software Esprit. The electron landing energy amounted to 19.5 kV and the electron beam current was about 250 pA.Fluorescence microscopy
Microscopic images were taken with an Axio Observer Z1 fluorescence microscope (Carl Zeiss Microscopy, Oberkochen, Germany) and processed using the ZEN Pro software. The Filmtracer LIVE/DEAD biofilm viability kit (Thermo Fisher Scientific, Waltham, USA) was used to stain wild type cells attached to the cathode, following the manufacturer's instructions. The TFCoral fluorogen (Twinkle Bioscience, Paris, France) was used to stain C. ljungdahlii pFAST(thl) cells attached to the cathode, following the manufacturer's instructions.Analysis of the culture broths
The concentrations of organic acids and alcohols in the culture supernatant were determined using an HPLC system (Jasco, Tokio, Japan) equipped with PDA, RI and UV (210 nm) detectors. The column Aminex HPX-87H, 300 mm × 7.8 mm, 9 μm (Bio-Rad Laboratories, Hercules, USA) was used in combination with the precolumn Kromasil 100 C18, 40 mm × 4 mm, 5 μm (Dr Maisch, Ammerbuch, Germany). The samples were diluted 1:10 with the mobile phase (0.005 M H2SO4), and 50 μL were used for injection. The column was maintained at 50 °C and an isocratic flow rate of 0.5 mL min−1 was applied. The autosampler was flushed with water.
The concentrations of amino compounds in the culture supernatant were determined using an HPLC system (Jasco, Tokio, Japan) equipped with UV (338 nm) and fluorescence (excitation: 340 nm; emission: 455 nm) detectors. The column Kinetex XB C18, 75 mm × 2.1 mm, 2.6 μm (Phenomenex, Torrance, USA) was used in combination with the cartridge SecurityGuard Ultra Cartridge C18, 2 mm × 2.1 mm (Phenomenex Torrance, USA). The samples were derivatized by adding 96 μL of the derivatizing agent (phthaldialdehyde reagent) to 96 μL of the sample, with a reaction time of 2 min. 1 μL of the derivatized sample was used for injection. The column was maintained at 25 °C and a gradient using two mobile phases was applied as indicated in Table S2† with a flow rate of 0.25 mL min−1. The mobile phase A was 20 mM potassium phosphate buffer pH 7.2 and the mobile phase B was acetonitrile:methanol (50:50; v/v). The autosampler was flushed with acetonitrile:methanol:water (25:25:50; v/v/v).
Optical density at 600 nm was measured to follow bacterial growth in suspension with a BioPhotometer (Eppendorf, Hamburg, Germany). Culture pH was determined with a pH meter for small volumes (Mettler Toledo, Columbus, USA).
Data integration and calculations
Coulombic efficiencies (CE) were calculated as previously described,37 according to the following equation: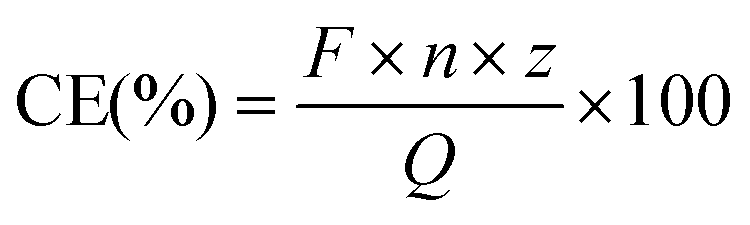
With F = Faraday constant (96485 C mol−1); n = moles of product (mol); z = number of electrons and Q = delivered charge (C). The moles of product were chromatographically measured as described above. The specific number of electrons was 2 (formate and H2), 6 (glycine and ethanolamine) or 8 (acetate). Delivered charge was calculated using EC-Lab V11.31 (BioLogic, Seyssinet-Pariset, France) or by integration with 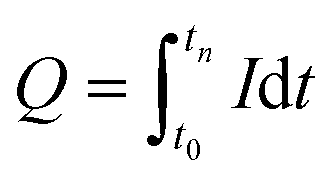 . The empirical biomass formula used was CH1.75O0.5N0.25.38 Biomass was approximated from total cell counts which, were estimated using the traditional Neubauer chamber cell counting method for the planktonic-dominant phenotype, and from fluorescence images of the cathode in the biofilm-dominant phenotype, using ImageJ.39 Potential corrections to a certain pH and predicted H2 evolution reaction (HER) potentials were calculated in accordance to the Nernst equation.37 All calculation and plots were performed in R/RStudio.40 Flux balance analysis (detailed information in the ESI†) was performed in Python,41 in combination with the tools CobraPy42 and Escher.43 The genome-scale metabolic model of C. ljungdahlii was updated and modified from Nagarajan et al.44
. The empirical biomass formula used was CH1.75O0.5N0.25.38 Biomass was approximated from total cell counts which, were estimated using the traditional Neubauer chamber cell counting method for the planktonic-dominant phenotype, and from fluorescence images of the cathode in the biofilm-dominant phenotype, using ImageJ.39 Potential corrections to a certain pH and predicted H2 evolution reaction (HER) potentials were calculated in accordance to the Nernst equation.37 All calculation and plots were performed in R/RStudio.40 Flux balance analysis (detailed information in the ESI†) was performed in Python,41 in combination with the tools CobraPy42 and Escher.43 The genome-scale metabolic model of C. ljungdahlii was updated and modified from Nagarajan et al.44
Results
Abiotic electrochemical performance
As a starting point, the establishment of a valid and reliable cathode potential was required. The goal was to set the potential low enough to allow for sufficient H2 evolution to support robust growth and product titers in the gram per liter range, which would be reached at a H2 production rate of >0.25 mL L−1 min−1.Using the Nernst equation, the formal potential of the hydrogen evolution reaction (HER) for the specific conditions given in the MES (Fig. S3†) was calculated as −548 mV. In a practical system overpotentials as well as other losses are in place. Therefore, we analyzed the amounts of H2 formed in the electrolyte solution close to the cathode but also in the headspace. Dissolved H2 started being detected by a H2 microsensor close to the cathode surface (Fig. 1B) at −650 mV when using LSV with 0.1 mV s−1. At −780 mV, the off-gas analyzer detected H2 in the headspace in the same test (potentials corrected to pH 5.7) (Fig. 1C). This “delay” of approximately 120 mV for the measurements in the off-gas relative to the solution was expected, since off-gas H2 is only detected once the solution was saturated. Based on these experiments and to give a high enough rate of reaction, −900 mV was chosen as cathode potential.
The abiotic performance at this potential was then evaluated in detail and over longer time (Fig. 1D). Interestingly, during the first days an increasing current was observed, from around −0.10 mA cm−2 during the first 2 days to −0.75 mA cm−2 at day 4. We speculated that it most likely resulted from the reduction of medium components. This led to an increase of the pH by 0.6 units and a 4-fold increase of H2 evolution, which then remained stable for the rest of the run. H2 production rate was estimated from delivered charge after reaching steady state as 0.31 mL L−1 min−1. Very surprisingly after this adaptation period of 3 days, formate started being produced abiotically at a rate of 0.014 g L−1 d−1. In additional experiments this was confirmed with plain PETC medium, where formate was produced at a rate of 0.038 ± 0.026 g L−1 d−1 (Fig. S4†). The electrochemical reduction of CO2 (CO2RR) to formate is not possible at plain graphite electrodes as used here.45–47
Furthermore, formate was not generated in abiotic electrochemical cell with the same carbon cathodes when 200 mM KCl was used as the electrolyte solution (Fig. S4†). We hypothesized that an initial reductive process changed the electrode surface composition by either deposition of catalytically active media components onto the cathode surface (e.g., metals) or structural changes of the electrode surface that gave access to trace metals within the cathode material, which subsequently catalyzed CO2RR to formate as well as the increased H2-evolution reaction. To test this hypothesis, the elemental composition of the electrolyte solution was analyzed via ICP-OES (Fig. S4†) showing enrichment of some elements over time, like calcium and magnesium in PETC and vanadium in the KCl control, indicating a release of these elements from the carbon electrode. In contrast, the concentration of other elements of the PETC medium like iron, manganese and molybdenum decreased over time indicating possible electrodeposition onto the cathode (Fig. S4†).
To confirm this, the elemental composition of the cathode surface was analyzed by EDX. Iron was found at higher levels on the electrode surface after the experiment than at unused control electrodes, while cobalt, aluminum, vanadium, nickel, and tungsten were only detected on treated electrodes but not at all on unused electrodes (Fig. 2A and B), indicating an electrodeposition or a change in the surface structure so they became accessible. Electron microscopy revealed that the morphology of the electrode surface also changed, exhibiting an eroded appearance (Fig. 2C and D). It can be concluded that changes in the surface metal composition of the cathode upon electrochemical application make catalytic effects for CO2RR to formate likely. However, this needs to be further confirmed.
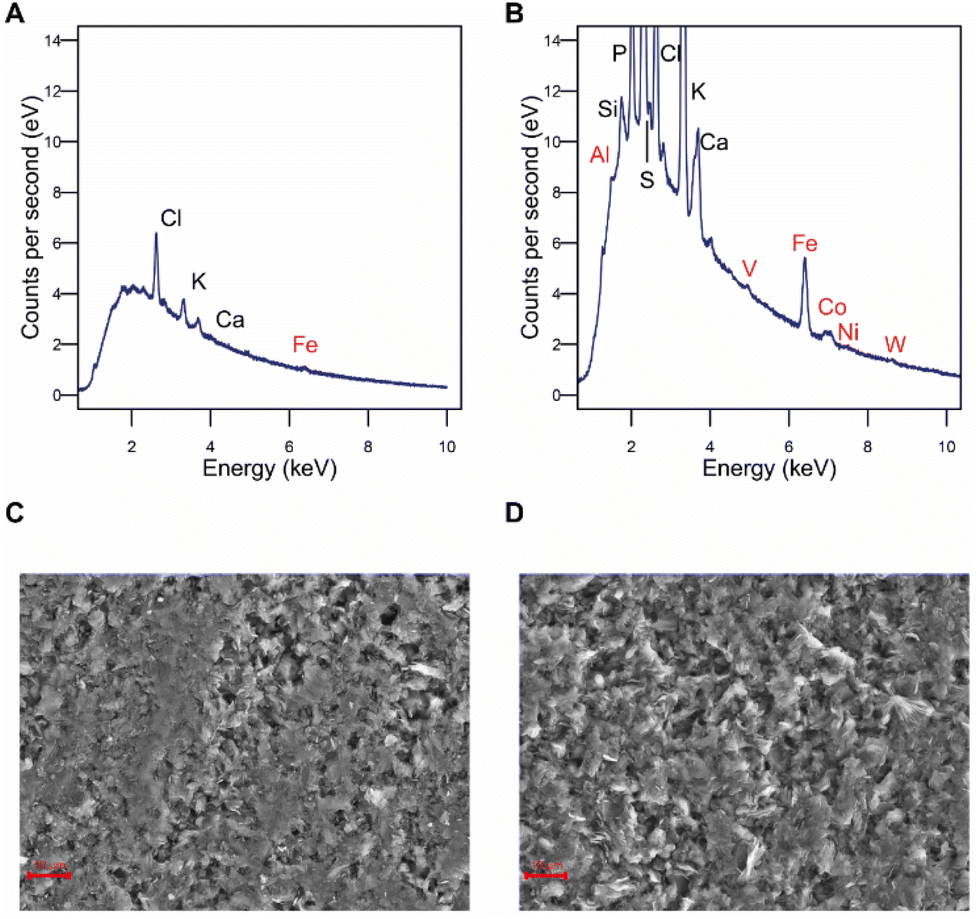 | ||
| Fig. 2 Evaluation of abiotic formate production (A) EDX spectrum of an unused electrode. (B) EDX spectrum of an electrode after 2 weeks poised at −900 mV in PETC medium. The spectrum shown belongs to a representative sample (n = 3). Metals for CO2RR to formate are marked in red. (C) SEM image of a new electrode. (D) SEM image of an electrode after 2 weeks poised at −900 mV in PETC medium, showing the erosion of the electrode surface. Images are representative of the electrode surface (n = 3). | ||
Controlling C. ljungdahlii planktonic or biofilm morphology in microbial electrosynthesis
When establishing MES with C. ljungdahlii, we noticed that certain inoculation densities led to distinctive growth phenotypes. With a high inoculation density (i.e., 1.0 × 108 ± 1.8 × 107 cells), a continuous increase of the optical density (OD600) was achieved. In contrast OD600 did not increase when using a low inoculation density (i.e., 3.4 × 107 ± 6.0 × 106 cells). However, both conditions showed MES activity. These differences between phenotypes were consistent and evident in all experiments and resulted in distinct differences in performance (see next section). Thus, we reached control over the MES operation by either using high inoculation density leading to planktonic-dominant conditions or low inoculation density leading to biofilm-dominant conditions. To confirm this, the surfaces of the cathodes were assessed by fluorescence microscopy in combination with the Filmtracer LIVE/DEAD staining. These showed no living cells attached to the cathode when planktonic growth was promoted (Fig. 3A). In contrast, alive cells and biofilm-like aggregates were present for the BES at biofilm-dominant conditions (Fig. 3B). This was confirmed independently in a different experiment with a C. ljungdahlii strain heterologously expressing the anaerobic fluorescent protein FAST (C. ljungdahlii pFAST(thl)). The fluorescence microscopy revealed equally: the high density inoculated BES showed no cell attachments at the cathode and grew well planktonically (Fig. 3C), while the low density inoculated BES showed promoted biofilm formation on the cathode, but exhibited no planktonic growth (Fig. 3D). This ability to grow in defined phenotypes depending on external factors, in this case the inoculation density, allowed us to evaluate C. ljungdahlii and its MES performance in both conditions: planktonic-dominant and biofilm-dominant.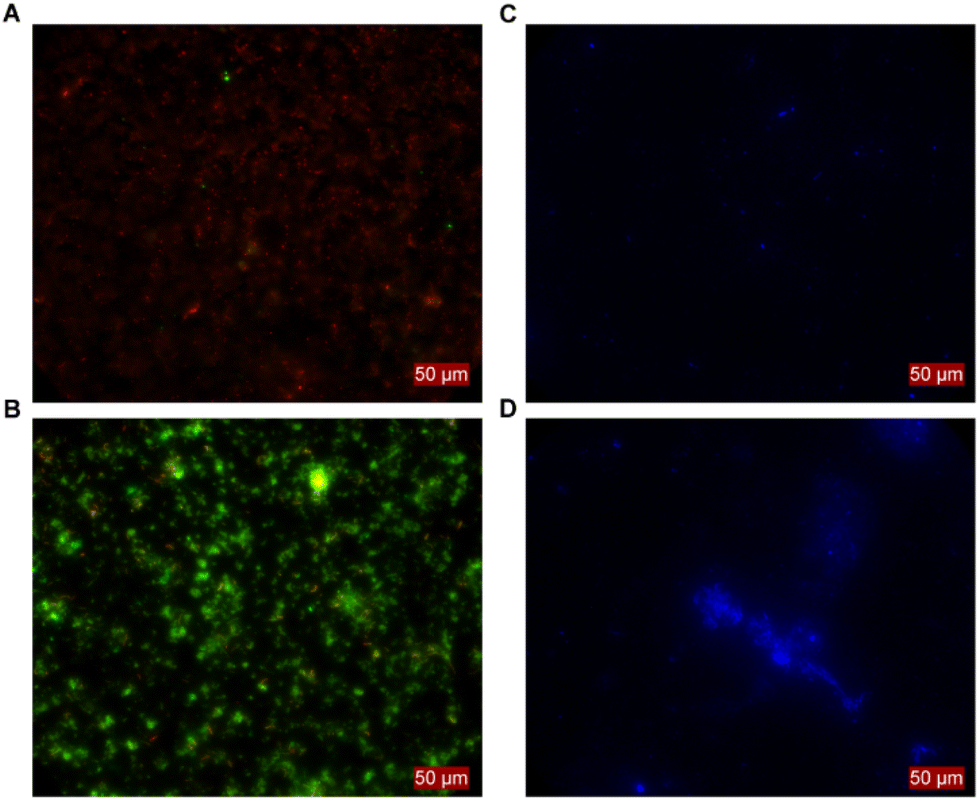 | ||
| Fig. 3 Confirmation of the phenotypic differences between biofilm-dominant and planktonic-dominant growth of Clostridium ljungdahlii in microbial electrosynthesis. Fluorescence microscopic image of cathodes in planktonic-dominant condition (A and C) and in biofilm-dominant condition (B and D). Filmtracer™ LIVE/DEAD™ biofilm viability kit (Invitrogen) was used for staining the wild type strain, in (A) and (B). The fluorogen TFCoral (Twinkle Bioscience) was used for staining metabolically active engineered strains expressing the anaerobic fluorescent protein FAST, in (C) and (D). No living cells are present on the cathode surface in planktonic-dominant condition (A and C), while the situation is the opposite for biofilm-dominant condition (B and D). | ||
H2 mediation is the likely mode of electron uptake for C. ljungdahlii
Interestingly microprofiling showed higher H2 concentrations and better diffusion into the electrolyte solution in the planktonic-dominant conditions compared to the abiotic and biofilm-dominant conditions (Fig. S5†). In order to clarify the role and importance of H2 more in detail, BES with planktonic-dominant as well as biofilm-dominant conditions were operated electroautotrophically for more than 25 days at a constant cathode potential of −900 mV to establish steady state.Peak current densities (1-day window from the maximum) during this period were −0.45 mA cm−2 and −0.67 mA cm−2 for the planktonic- and biofilm-dominant BES, respectively (two independent biological replicates of both phenotypes over 20 days are provided in Fig. S6 and S7†). Estimated H2 production rates from transferred charge were 0.21 mL L−1 min−1 for the planktonic- and 0.31 mL L−1 min−1 for the biofilm-dominant condition. Then, the potential was increased slowly in steps of 20 mV per 3–4 days, allowing the bacterial activity to reach a new steady state after each potential step (details see Fig. S8†). The goal was to progressively decrease H2 evolution beyond its thermodynamic limit and follow the metabolic activity of the cells during this process. With each step, the cathodic current density decreased (Fig. 4A), as well as the H2 in the headspace (i.e., the fraction of electron equivalents that was not consumed by the bacteria) (Fig. 4B). Nevertheless, the pH decline and the acetate increase proceeded at the same rate as before (Fig. 4A and C), indicating that the H2 availability was not limiting the bacterial activity. When reaching around −620 to −680 mV, H2 completely stopped being detected in the headspace, which directly resulted in a decline of the OD600 (only relevant for the planktonic-dominant condition) and the cease of acetate production in both systems. Most likely as a result of now metabolically inactive cells, formate accumulation became evident after H2 depletion, especially in biofilm-dominant conditions. The origin of formate is likely not only abiotic CO2RR reduction to formate in this case, but also other biotic processes. This can be concluded, since the CE for formate production between day 60 and 80 would have amounted to unrealistic 265%. After 80 days, the potential reached −400 mV and the current density was almost zero. A potential decrease back to −900 mV resulted in increases in OD600 and acetate concentrations only for the planktonic-dominant condition, showing that these cells were able to restore their metabolic activity. In contrast, for the biofilm-dominant conditions the cathodic currents remained much lower than before, H2 quickly accumulated in the headspace, no formate was reassimilated and no acetate was produced, indicating a more detrimental effect of the starvation period on the cells. Manual pH corrections were made to facilitate activity restoration, which was only feasible in the planktonic-dominant condition, indicating that the biofilm-dominant condition was irreversibly inactivated or alternatively, more time would be required to re-activate the biofilm-dominant BES.
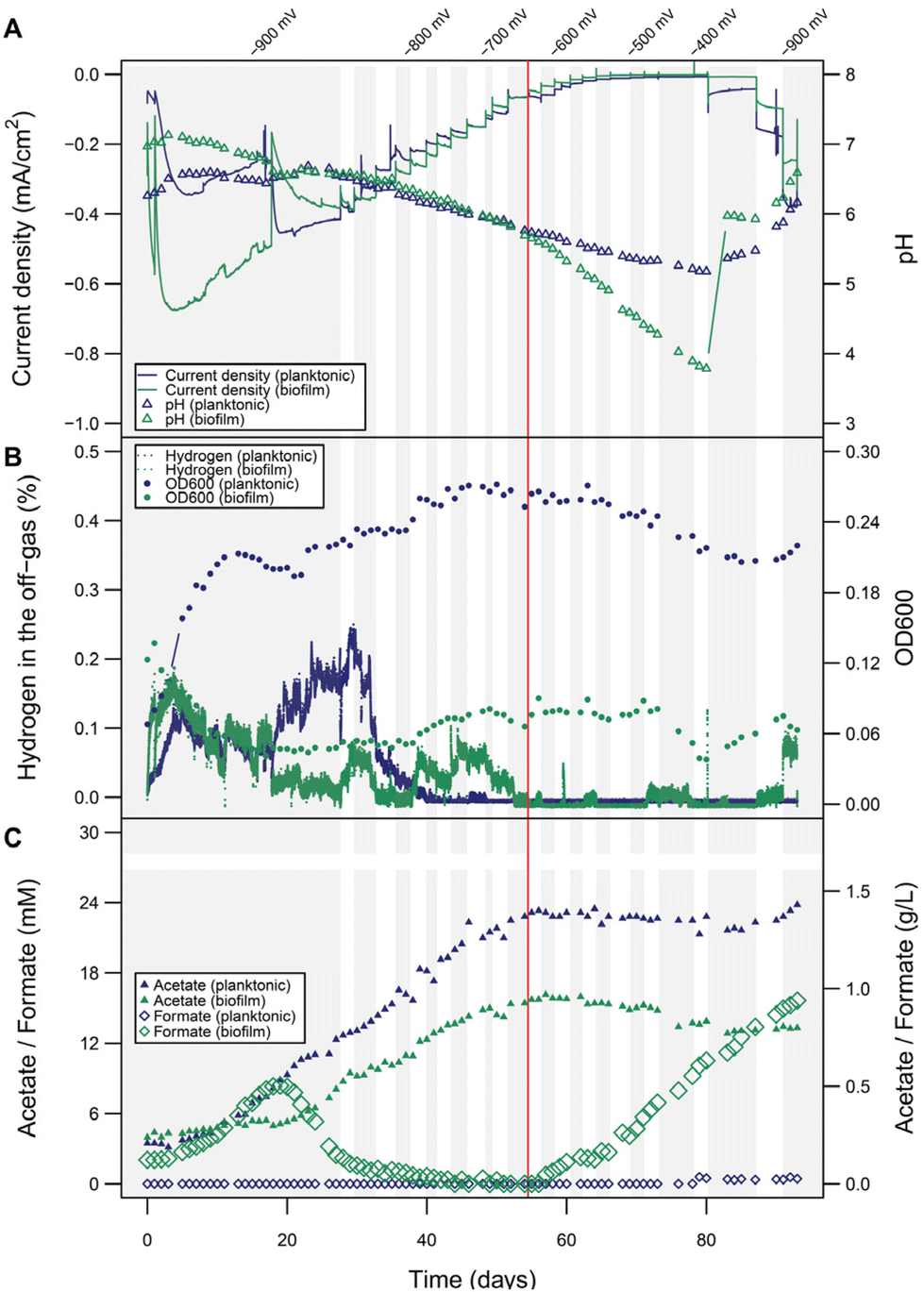 | ||
| Fig. 4 Microbial electrosynthesis using C. ljungdahlii with increasing potentials. A range of potentials was applied to two reactors after reaching a stable electrosynthetic performance at −900 mV (stepwise increase −900 to −400 mV and jump back to −900 mV), which were inoculated with low cell density (biofilm-dominant, green traces) and high cell density (planktonic-dominant, blue traces) cultures, respectively. (A) Current density and pH development. (B) H2 detected in the off-gas and OD600. (C) Acetate and formate titers. The changes in shaded areas indicate step changes in the applied potential. At around −650 mV (marked with a vertical red line), H2 stopped being detected in the headspace. Electrode surface area was 26.59 cm2 with a surface-area-to-reactor-volume ratio of 66.48 cm2 L−1. | ||
While the overall electrophysiological response of C. ljungdahlii to the potential change was similar for the two morphological conditions, the detailed performance was very different. The acetate titers obtained in the planktonic-dominant conditions (1.43 g L−1) were much higher than with the biofilm (0.8 g L−1) at similar or even higher electron equivalents respectively current densities for the biofilm-dominant conditions. The planktonic-dominant condition was found to be more productive overall (3.41 g m−2 d−1 acetate) and per cell (9.09 × 10−12 g per cell per d acetate; 9.99 × 108 cells) when comparing to the biofilm-dominant condition (1.91 g m−2 d−1 acetate and 5.01 × 10−12 g per cell per d acetate; 1.01 × 109 cells). CE (Fig. S9†) calculated for acetate formation between days 10 and 40 (no H2 limitation) were 18.2% in the planktonic-dominant conditions and 8.8% in the biofilm-dominant conditions. In contrast, the efficiency of H2 utilization seemed to be higher in the biofilm during the initial −900 mV growth phase, since less H2 was detected by the off-gas analyzers after the first few days. This also correlates to the H2 and pH measurements at the electrode using microsensors for both conditions after 15 days of operation (additional results in ESI: Fig. S5†). Surprisingly, this seemingly higher H2 (or electron equivalent) consumption did not translate into more products (see product distribution in Fig. S9†). Biomass formation is a possible sink of reducing equivalents, however, a reliable determination of the biomass amount for the biofilm was not possible despite attempts via gravimetry and image analysis. Nevertheless, values estimated from cell counts suggested a very low contribution (below 1.5%) of biomass formation to the CE. Formate accumulating in the starvation phases of the biofilm-dominant MES confirmed low or no metabolic activity since formate can be assimilated as carbon source using reduced ferredoxin (for which H2 is required), but it cannot be used as the electron source by C. ljungdahlii. CV analysis to screen for redox signatures was only applied sparsely during this study, since detrimental effects of redox cycling have been observed on the stability of the biotic systems by us and others.48 CVs from both planktonic- and biofilm-dominant reactors, performed at different time points, indicated no redox signatures in the relevant potential range that would point to the involvement of DET or the use of electron shuttles (Fig. S10 and 11†). It could be speculated on a likely irrelevant redox center with a midpoint potential close to −40 mV in the biofilm-dominant condition (Fig. S11†).
Overall, we conclude from these empirical observations that H2 availability is crucial for the metabolic activity and high productivities of C. ljungdahlii in MES. This is especially evident for the planktonic-dominant reactors, which also gave overall the better performance. For the biofilm-dominant system, the cease of activity is also highly inversely correlated to the evolution of H2, but we cannot exclude the involvement of other unknown electron transfer processes. In any case, if there are other processes of electron uptake, they seem less relevant than H2-promoted MES.
H2 availability determines process efficiency
Considering the H2 availability as limiting factor, especially in the planktonic-dominant condition where peak H2 production rate was below our objective of 0.25 mL L−1 min−1, and the achieved low CE, follow-up experiments were conducted at a cathode potential of −900 mV but with electrodes providing a 6-times larger electrode surface area-to-reactor volume ratio to provide more H2 in the reactor (Fig. 5). Another alternative could have been the use of more negative potentials, but linear increase of activity was not observed when potentials were lowered stepwise (data not shown). In the planktonic-dominant reactor, the acetate production was very high achieving a titer of 6.06 g L−1 with a productivity of 2.93 g m−2 d−1, and acetate-based CE of 87.4%. These are among the highest performance values reported for MES from CO2, both with pure culture acetogens as well as with undefined mixed cultures (Table 1). These high acetate titers might already have imposed some product inhibition or toxicity on C. ljungdahlii since a decrease in the rate of acetate formation after 40 days coincides with a pH drop below 5.5, a drop in OD600, and an increase in unused exhaust H2. The peak current density (1-day window from the maximum) was lower than in previous planktonic-dominant reactors: −0.10 mA cm−2 (compared to −0.33 ± 0.09 mA cm−2), but the peak of absolute current of −15.38 mA (compared to −8.66 ± 2.39 mA), and thus the estimated H2 estimated evolution rate (0.27 mL L−1 min−1), was higher. Electrodes were made from the same type of high purity graphite, but the increase in surface area did not linearly scale with current which can be especially assigned to changes in the mass-transfer regime for different surface area-to-volume ratios. Noteworthy, the biofilm-dominant reactor had a similar performance in terms of acetate titer (1.01 g L−1) as in previous experiments with smaller electrodes, at similar peak currents (−3.98 mA; compared to −6.38 ± 7.54 mA). The CE for acetate reached 25.9% (Fig. S9†). Overall, this showed that a planktonic morphology clearly provided a more robust, scalable and efficient performance of MES than a biofilm.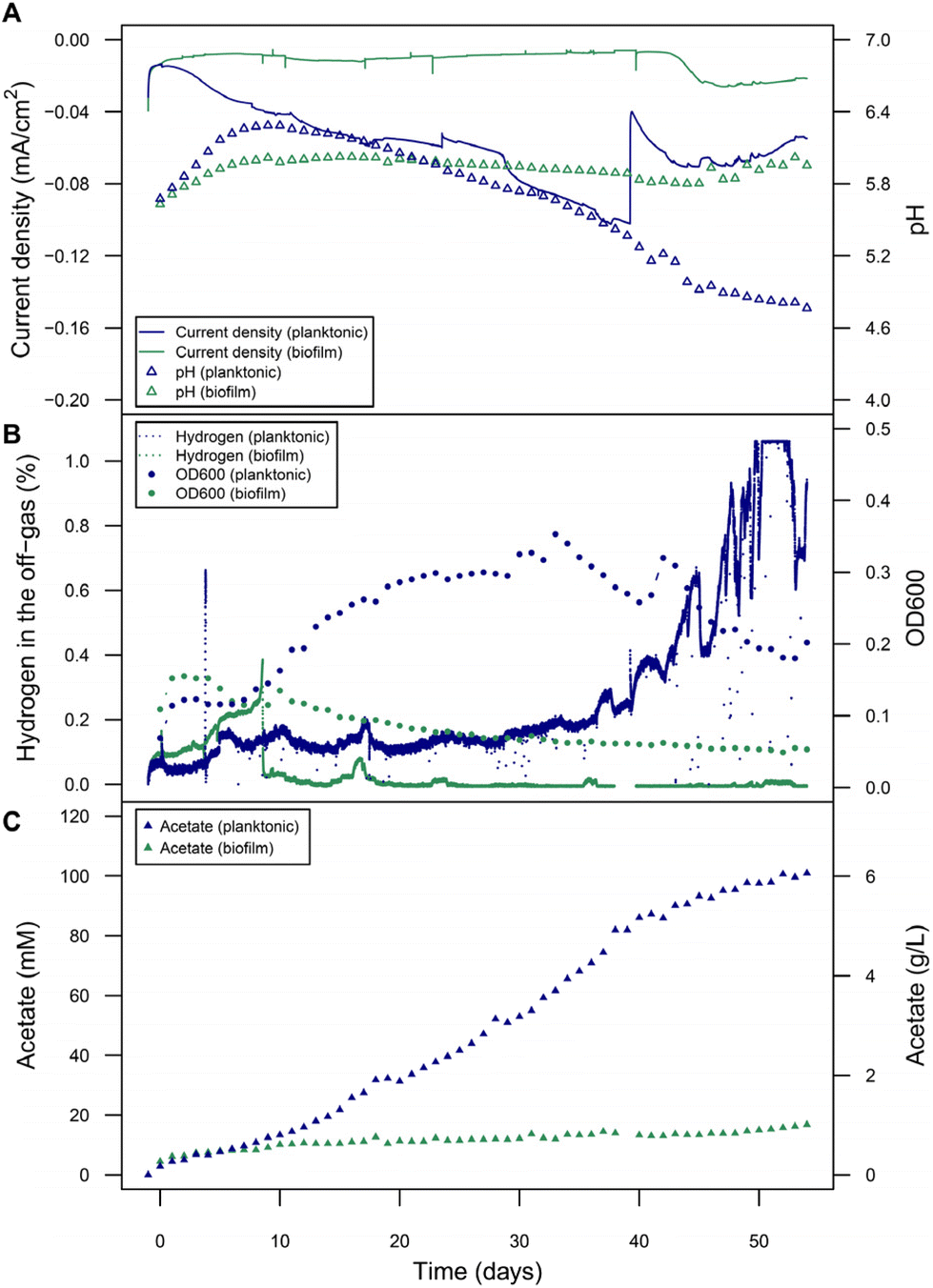 | ||
| Fig. 5 Microbial electrosynthesis with C. ljungdahlii at a fixed potential (−900 mV) using a 153.05 cm2 cathode to increase H2 supply. Two reactors were inoculated with low cell density (biofilm-dominant, green traces) and high cell density (planktonic-dominant, blue traces) cultures, respectively. (A) Current density and pH development. (B) H2 detected in the off-gas and OD600. (C) Acetate titers. Surface-area-to-reactor-volume ratio: 382.63 cm2 L−1. | ||
| Cathode type | Bacteria | Max. conc. (g L−1) | Rate (g L−1 d−1) | Claimed EET mechanism | CE (%) | E vs. SHE (mV) | Ref. |
|---|---|---|---|---|---|---|---|
| NA – not applicable. | |||||||
| Graphite block | C. ljungdahlii | 6.06 | 0.11 | H2 | 87 | −715 | This study |
| Cobalt phthalocyanine | C. ljungdahlii | 5.1 | 2.00 | Syngas | 17 | −1600 | 49 |
| Nickel phosphide modified carbon felt | Engineered C. ljungdahlii | 1.18 | 0.17 | H2 | 82 | −850 | 50 |
| Graphite rod | C. ljungdahlii | 1.14 | 0.14 | H2 | 42 | −1000 | 51 |
| Graphite felt with stainless steel | C. ljungdahlii | 0.6 | 0.086 | H2 | 40 | −700 | 52 |
| Graphite block | C. ljungdahlii | 0.37 | 0.06 | H2 | 18 | −800 | 53 |
| Graphite rod | C. ljungdahlii | 0.005 | 0.0008 | DET | 88 | −400 | 54 |
| Carbon felt | C. scatologenes | 0.44 | 0.016 | DET and H2 | 38 | −1000 | 55 |
| Carbon cloth | S. ovata | 3.60 | 0.68 | H2 | 63 | −600 | 56 |
| Ni–Mo-coated graphite rod | T. kivui | 6.0 | 3.36 | H2 | 95 | NA | 57 |
| EPD-3D | Mixed culture | 11.0 | 0.36 | NA | 94 | −850 | 58 |
| Graphite granules | Mixed culture | 10.5 | 1.04 | NA | 69 | −590 | 59 |
Redox imbalance promotes diversification of metabolic fluxes
The improved MES performance of the planktonic-dominated reactors suggests the existence of metabolic shifts caused by H2 availability. As detailed in the following we see indications that difference in metabolic states result in a redox imbalance, which might lead to a change of metabolic fluxes and hence MES performance. The main product of C. ljungdahlii growing electroautotrophically is acetate. However, additionally, other central metabolites of C. ljungdahlii were found at unusually high titers during MES: the two amino compounds, glycine and ethanolamine (Fig. 6). These amino compounds could putatively be produced in C. ljungdahlii through the glycine synthase-reductase pathway (GSRP) or the reductive glycine pathway (RGP), alternative pathways for carbon fixation to the Wood–Ljungdahl pathway, recently revealed for Clostridium drakei.60 Glycine was produced following a similar temporal pattern as acetate and reached titers up to 0.39 g L−1, which was higher in the biofilm-dominant reactors compared to planktonic-dominant conditions. Conversely, ethanolamine reached its peak during highest rate of acetate production, i.e., the peak in metabolic activity (titers up to 0.14 g L−1), and decayed afterwards. Ethanolamine production is possibly linked to the RGP, as the decarboxylation product of serine, which is part of the pathway. Other amino compounds did not change significantly during the experiments and their titers were negligible compared to glycine or ethanolamine. For MES using larger electrodes and hence increased H2 availability, lower titers of glycine and ethanolamine were detected in the planktonic-dominated reactor (0.15 g L−1 and 0.13 g L−1, respectively), with CE of 1.5% for glycine and 1.7% for ethanolamine (Fig. S9†). In the biofilm-dominant reactor, very low titers were reached (0.045 g L−1 glycine and 0.011 g L−1 ethanolamine) but with similar CE (1.2% for glycine and 0.5% for ethanolamine). Thus, the increase in H2 availability leads to an increased metabolic flux towards acetate production. On the other hand, it seemed that the production of glycine and ethanolamine were linked to some kind of metabolic stress. Both are not known to be produced in gas fermentation and were much lower in planktonic-dominant BES, which performs more similar to gas fermentation. This effect was further investigated in silico in a genome-scale metabolic model (Fig. S12† and respective code). Here, the simulation of an artificial surplus of NADPH resulted in the increase of metabolic flux through the putative pathways of carbon fixation via glycine, going from 1.13% to 100% of total carbon fixation flux (Fig. S12†). Flux diversification away from the Wood–Ljungdahl pathway seems to be an attempt of balancing the redox homeostasis, when it is disrupted by stress factors i.e., a surplus of NADPH, which resulted in the production of glycine and ethanolamine.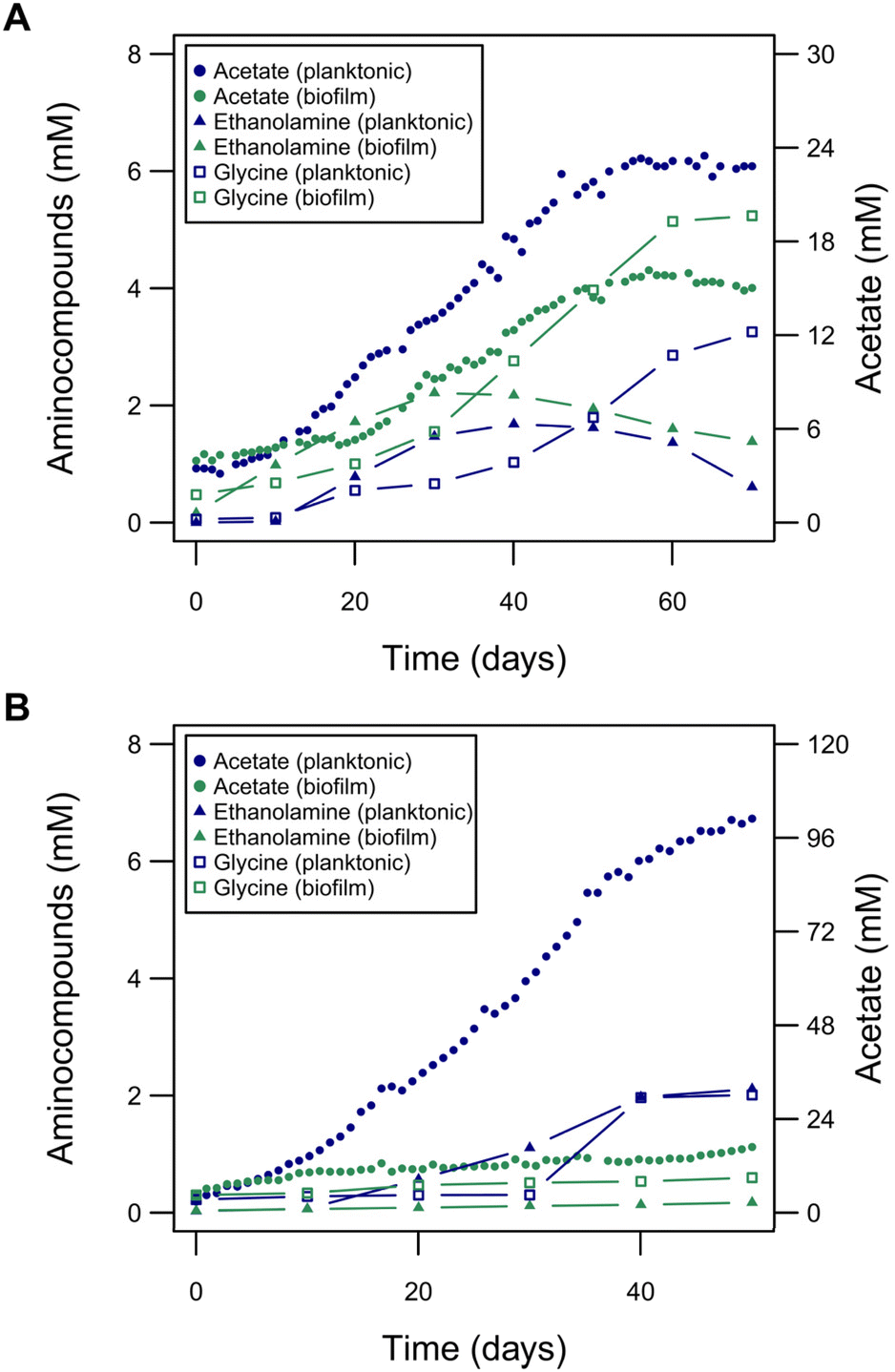 | ||
| Fig. 6 The role of amino compounds in electroautotrophic growth of C. ljungdahlii. (A) Glycine and ethanolamine produced during the experiment shown in Fig. 4, overlaid again with the acetate production. (B) Glycine and ethanolamine produced during the experiment shown in Fig. 5, overlaid again with the acetate production. Low cell density (biofilm-dominant, green traces) and high cell density (planktonic-dominant, blue traces) reactors are presented. | ||
Discussion
We established a robust and controllable microbial electrosynthesis converting CO2 and electrons to acetate ultimately outperforming previous studies (Table 1). The maximum acetate titers (6.06 g L−1) were the highest obtained in MES using pure cultures, surpassing the best Clostridium spp.49 and Sporomusa spp.56 results, although it was around half of the best titers achieved with mixed cultures.58,59 Noteworthy, this was achieved without any electrode modification or reactor optimization and only optimizing the biological part of the process. Although techno-economical evaluations of MES showed that its technology readiness level (TLR) corresponds to lab scale yet,18,28,61,62 efforts are currently being made to increase the TLR of this technology by academia as well as the private sector. Framed in that effort, our research set the foundations for a better control and understanding of the biocatalysts used in MES, which combined with the available technological advances could reduce the costs and improve the performance of the technology.Nevertheless, the main goal of the study was to set the basis for the understanding of electroautotrophic metabolism of C. ljungdahlii, and related bacteria. Constant flushing with inert gas of the electrolyte solution avoided H2 accumulation, which would have led to a process being similar to gas fermentation, and allowed the study of the electroautotrophic metabolism. The control of the microbial phenotypes of C. ljungdahlii by means of the cell density of the inoculum was the key to unveil the role of H2 as the mediator for extracellular electron transfer, and to de-couple electroautotrophic growth from biofilm formation in MES. The phenotype control was highly robust being barely affected by stochastic factors, such as the electrode characteristics and high variance of currents and current densities, partially induced by random differences in the growth level of the inoculum. Acetate was produced electroautotrophically by C. ljungdahlii reaching a maximum titer of 6.06 g L−1, with a productivity of 0.11 g L−1 d−1–2.93 g m−2 d−1. Acetate titers were significantly higher in the planktonic – than in the biofilm-dominant condition (Fig. S13†). Ethanol was likely also produced, but flushed out of the systems due to its volatility. Test experiments lowering the gas flow through the system allowed for ethanol accumulation in the liquid phase and prevented H2 loss (data not shown), which could further increase the efficiency. In previous publications, 2-oxobutyrate was detected in low concentrations,25 probably as a consequence of punctual redox imbalances linked to the activity of the glycine synthase.63 But this compound was never detected in our experiments (<1 mg L−1). Glycine and ethanolamine were produced at higher titers in the biofilm-dominant condition (up to 0.39 g L−1 and 0.14 g L−1, respectively), possibly due to a redox imbalance caused by a surplus of NADPH. RGP and GSRP are known to require more NADPH than reduced ferredoxin compared to the Wood–Ljungdahl pathway, making them less efficient but also more resilient under low energy conditions.64 Our genome-scale modelling points to a redox imbalance between NADPH and reduced ferredoxin as the mechanism behind the observed metabolic shift (Fig. S12†). It seems similar to the metabolic shift during solventogenesis, in which ethanol production is promoted by NADH accumulation.65 Formate was produced abiotically via CO2RR, which was found to proceed via in situ formed metal electrocatalysts by electroreduction of media components. Formate could be consumed in C. ljungdahlii only with further H2 reducing equivalents bypassing the formate dehydrogenase and saving reduced ferredoxin, presumably leading to increasing performance. However, the rate of formate accumulation in the biofilm-dominant condition during starvation cannot only be explained by abiotic CO2RR, since the concentration exceeded the provided electrochemical reducing equivalents by 2.5-fold. Another biotic source for formate release to the solution must be involved here. Furthermore, formate accumulation in this case could be another sign of H2 starvation, since C. ljungdahlii cannot grow solely on formate,66 unlike other acetogens.67
Combining different analytical approaches, H2 was shown to be required for efficient MES from CO2 using C. ljungdahlii. The growth of planktonic cells is physically incompatible with DET and the observed cease of activity when the potential was too positive for H2 evolution, strongly indicates that H2 mediates electron transfer in C. ljungdahlii, or at least plays a dominant role. In the case of biofilm-dominant conditions, activity followed the same pattern and also ceased at a similar cathode potential as in the planktonic-dominant reactor. Further, no indications pointing to other redox processes being relevant for EET were detected in CV analysis (Fig. S10 and 11†). Nevertheless, H2 in the off-gas and dissolved in the catholyte was still always detected albeit at lower shares in the biofilm-dominant conditions compared to the planktonic-dominant conditions. This suggests that H2 was still formed but was immediately consumed by the biofilm. An inconspicuous H2 mediation by the activity of uncharacterized hydrogenases68 able to efficiently channel H2 into the cell immediately after being generated at the cathode or even to promote transient H2 evolution by keeping a low H2 partial pressure in immediate electrode proximity69 are possible reasons for this observation. Yet, this immediate H2 consumption should translate into higher CE for the biofilm-dominant conditions, since the H2 would be metabolically utilized instead of “wasted”. However, this is not the case, as CE were always higher for the planktonic – than for the biofilm-dominant reactors. Besides, the higher H2 production observed in the planktonic-dominated reactors compared to the abiotic ones indicates a more active promotion of H2 evolution by the planktonic cells. A direct reduction of ferredoxin (with a similar redox potential to HER) is not possible, since ferredoxin is located in the cytoplasm. Unknown cellular components could potentially form a dedicated extracellular electron transfer machinery to ferredoxin, but there is no evidence for such a process. A DET process would likely result in a specific redox signature in the potential range more negative than the formal potential of ferredoxin in the CV (E′0 = −420 mV vs. SHE),70 for which there is no indication. Other putative electron mediators like flavins or quinones71 were examined but not found in the culture supernatants (data not shown). The electrochemical activity of media components such as resazurin,72 cysteine73 and yeast extract74 have been reported, but involving more oxidative potentials than the ones used in this study, which is supported by our CV data. Nevertheless, there could be a minor influence of yeast extract in bacterial growth, while cysteine could be converted to acetate even if it cannot support growth.75 Overall, the possible effect of these component was found negligible as evidenced in control runs at OCV (Fig. S14†).
Conclusions
The deeper understanding of the morphological state provides control over the MES performance of C. ljungdahlii. This is a key for further engineering and improvement in performance and to expand the range of products. In this study, glycine and ethanolamine were produced for the first time in MES from CO2, and by this bacterium. This highlights the importance of controlling and understanding electroautotrophic physiology not only as a production platform itself, but also as an exploratory tool to evaluate and extend the metabolic capacities of microbes capable of gas fermentation.76 This study provides the first experimental indication of the RGP and the GSRP activity in C. ljungdahlii. Further biochemical investigations are now required to detail the metabolic source of these amino compounds and to explore, if a tailored as well as enhanced production of these metabolites is feasible. Additionally, the presented advances in understanding the drivers of microbial catalytic activity can now be combined with more traditional efforts for process improvement like reactor and electrode engineering such as the use of chemically or structurally modified electrodes77–81 in various reactor scales and configurations.82–86 Hence, the innovation cycle can be closed and detailed biological investigations, which are only possible in a well-controlled pure-culture BES, can pave the road for future process as well as reactor engineering.Author contributions
Santiago T. Boto: methodology, software, formal analysis, investigation, writing – original draft and visualization. Bettina Bardl: methodology, investigation. Falk Harnisch: methodology, resources and writing – review & editing. Miriam A. Rosenbaum: methodology, formal analysis, conceptualization, writing – review & editing, supervision, project administration and funding acquisition.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors thank Michael Meyer from Leibniz-HKI for support with metabolite analysis, and the Max Plank Institute for Chemical Ecology (Jena, Germany) for technical assistance. Further, the authors thank Matthias Schmidt and the ProVIS – Centre for Chemical Microscopy of the UFZ for the micro-analysis with scanning electron microscope and energy-dispersive X-ray spectroscopy as well as the Department of Analytical Chemistry of the UFZ for ICP-OES-measurements. ProVIS is supported by the European Regional Development Funds (EFRE – Europe funds Saxony) and the Helmholtz Association. The authors gratefully acknowledge funding from the Deutsche Forschungsgemeinschaft (priority program SPP2240, project no. 445388719). Further, MAR and the work are supported by the European Research Council under the European Union’s Horizon 2020 research and innovation programme (Grant agreement No. 864669).References
- N. Nakicenovic and P. D. Lund, Nature, 2021, 596, 486 CrossRef CAS PubMed.
- Science Advice for Policy by European Academies (SAPEA), A systemic approach to the energy transition in Europe, 2021 Search PubMed.
- Y. Wang, Y. Liu and B. Gu, Adv. Atmos. Sci., 2022, 39, 1209–1216 CrossRef PubMed.
- J. M. Clomburg, A. M. Crumbley and R. Gonzalez, Science, 2017, 355 Search PubMed.
- Z. J. Schiffer and K. Manthiram, Joule, 2017, 1, 10–14 CrossRef.
- S. Y. Lee, H. U. Kim, T. U. Chae, J. S. Cho, J. W. Kim, J. H. Shin, D. I. Kim, Y. S. Ko, W. D. Jang and Y. S. Jang, Nat. Catal., 2019, 2, 18–33 CrossRef CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- P. Gabrielli, M. Gazzani and M. Mazzotti, Ind. Eng. Chem. Res., 2020, 59, 7033–7045 CrossRef CAS.
- F. Harnisch and C. Urban, Angew. Chem., Int. Ed., 2018, 57, 10016–10023 CrossRef CAS PubMed.
- U. Schröder, F. Harnisch and L. T. Angenent, Energy Environ. Sci., 2015, 8, 513–519 RSC.
- P. Dürre and B. J. Eikmanns, Curr. Opin. Biotechnol., 2015, 35, 63–72 CrossRef PubMed.
- H. G. Schlegel and R. Lafferty, Nature, 1965, 205, 308–309 CrossRef CAS.
- B. E. Logan, R. Rossi, A. Ragab and P. E. Saikaly, Nat. Rev. Microbiol., 2019, 17, 307–319 CrossRef CAS PubMed.
- C. Koch and F. Harnisch, ChemElectroChem, 2016, 3, 1282–1295 CrossRef CAS.
- K. P. Nevin, T. L. Woodard, A. E. Franks, Z. M. Summers and D. R. Lovley, mBio, 2010, 1(2), e00103–10 CrossRef PubMed.
- K. Rabaey and R. A. Rozendal, Nat. Rev. Microbiol., 2010, 8, 706–716 CrossRef CAS PubMed.
- A. Prévoteau, J. M. Carvajal-Arroyo, R. Ganigué and K. Rabaey, Curr. Opin. Biotechnol., 2020, 62, 48–57 CrossRef PubMed.
- L. Jourdin, J. Sousa, N. van Stralen and D. P. B. T. B. Strik, Appl. Energy, 2020, 279, 115775 CrossRef CAS.
- L. Jourdin and T. Burdyny, Trends Biotechnol., 2021, 39, 359–369 CrossRef CAS PubMed.
- A. ter Heijne, F. Geppert, T. H. J. A. Sleutels, P. Batlle-Vilanova, D. Liu and S. Puig, in Advances in Biochemical Engineering/Biotechnology, Springer Science and Business Media Deutschland GmbH, 2019, vol. 167, pp. 203–229 Search PubMed.
- M. A. Rosenbaum, C. Berger, S. Schmitz and R. Uhlig, in Advances in Biochemical Engineering/Biotechnology, Springer Science and Business Media Deutschland GmbH, 2019, vol. 167, pp. 181–202 Search PubMed.
- S. Cheng, D. Xing, D. F. Call and B. E. Logan, Environ. Sci. Technol., 2009, 43, 3953–3958 CrossRef CAS PubMed.
- R. Blasco-Gómez, P. Batlle-Vilanova, M. Villano, M. D. Balaguer, J. Colprim and S. Puig, Int. J. Mol. Sci., 2017, 18 Search PubMed.
- F. Mayer, F. Enzmann, A. M. Lopez and D. Holtmann, Bioresour. Technol., 2019, 289, 121706 CrossRef CAS PubMed.
- K. P. Nevin, S. A. Hensley, A. E. Franks, Z. M. Summers, J. Ou, T. L. Woodard, O. L. Snoeyenbos-West and D. R. Lovley, Appl. Environ. Microbiol., 2011, 77, 2882–2886 CrossRef CAS PubMed.
- R. Ganigué, S. Puig, P. Batlle-Vilanova, M. D. Balaguer and J. Colprim, Chem. Commun., 2015, 51, 3235–3238 RSC.
- I. Vassilev, P. A. Hernandez, P. Batlle-Vilanova, S. Freguia, J. O. Krömer, J. Keller, P. Ledezma and B. Virdis, ACS Sustainable Chem. Eng., 2018, 6, 8485–8493 CrossRef CAS.
- P. Dessì, L. Rovira-Alsina, C. Sánchez, G. K. Dinesh, W. Tong, P. Chatterjee, M. Tedesco, P. Farràs, H. M. V. Hamelers and S. Puig, Biotechnol. Adv., 2021, 46, 107675 CrossRef PubMed.
- S. W. Ragsdale and E. Pierce, Biochim. Biophys. Acta, Proteins Proteomics, 2008, 1784, 1873–1898 CrossRef CAS PubMed.
- M. A. Teravest, T. J. Zajdel and C. M. Ajo-Franklin, ChemElectroChem, 2014, 1, 1874–1879 CrossRef CAS.
- F. Wang, Y. Gu, J. P. O'Brien, S. M. Yi, S. E. Yalcin, V. Srikanth, C. Shen, D. Vu, N. L. Ing, A. I. Hochbaum, E. H. Egelman and N. S. Malvankar, Cell, 2019, 177, 361–369 CrossRef CAS PubMed.
- Y. Gu, V. Srikanth, A. I. Salazar-Morales, R. Jain, J. P. O'Brien, S. M. Yi, R. K. Soni, F. A. Samatey, S. E. Yalcin and N. S. Malvankar, Nature, 2021, 597, 430–434 CrossRef CAS PubMed.
- L. Jourdin, Y. Lu, V. Flexer, J. Keller and S. Freguia, ChemElectroChem, 2016, 3, 581–591 CrossRef CAS.
- E. Marsili, D. B. Baron, I. D. Shikhare, D. Coursolle, J. A. Gralnick and D. R. Bond, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3968–3973 CrossRef CAS PubMed.
- D. K. Newman and R. Kolter, Nature, 2000, 405, 94–97 CrossRef CAS PubMed.
- B. Molitor, K. Kirchner, A. W. Henrich, S. Schmitz and M. A. Rosenbaum, Sci. Rep., 2016, 6, 31518 CrossRef CAS PubMed.
- B. E. Logan, B. Hamelers, R. Rozendal, U. Schröder, J. Keller, S. Freguia, P. Aelterman, W. Verstraete and K. Rabaey, Environ. Sci. Technol., 2006, 40, 5181–5192 CrossRef CAS PubMed.
- E. Guedon, S. Payot, M. Desvaux and H. Petitdemange, J. Bacteriol., 1999, 181, 3262–3269 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- RStudio Team, 2020.
- G. Van Rossum and F. L. Drake, Python 3 Reference Manual, CreateSpace, Scotts Valley, CA, 2009 Search PubMed.
- A. Ebrahim, J. A. Lerman, B. O. Palsson and D. R. Hyduke, BMC Syst. Biol., 2013, 7, 1–6 CrossRef PubMed.
- Z. A. King, A. Dräger, A. Ebrahim, N. Sonnenschein, N. E. Lewis and B. O. Palsson, PLoS Comput. Biol., 2015, 11, e1004321 CrossRef PubMed.
- H. Nagarajan, M. Sahin, J. Nogales, H. Latif, D. R. Lovley, A. Ebrahim and K. Zengler, Microb. Cell Fact., 2013, 12, 118 CrossRef PubMed.
- P. Izadi and F. Harnisch, Joule, 2022, 6, 935–940 CrossRef.
- D. Li, H. Zhang, H. Xiang, S. Rasul, J. M. Fontmorin, P. Izadi, A. Roldan, R. Taylor, Y. Feng, L. Banerji, A. Cowan, E. H. Yu and J. Xuan, Sustainable Energy Fuels, 2021, 5, 5893–5914 RSC.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- S. M. de Smit, C. J. N. Buisman, J. H. Bitter and D. P. B. T. B. Strik, ChemElectroChem, 2021, 8, 3384–3396 CrossRef CAS.
- X. Zhu, J. Jack, A. Leininger, M. Yang, Y. Bian, J. Lo, W. Xiong, N. Tsesmetzis and Z. J. Ren, Resour., Conserv. Recycl., 2022, 184, 106395 CrossRef CAS.
- G. Wang, Q. Huang, T. S. Song and J. Xie, Energy Fuels, 2020, 34, 8666–8675 CrossRef CAS.
- M. Roy, R. Yadav, P. Chiranjeevi and S. A. Patil, Bioresour. Technol., 2021, 320, 124289 CrossRef CAS PubMed.
- S. Bajracharya, A. ter Heijne, X. Dominguez Benetton, K. Vanbroekhoven, C. J. N. Buisman, D. P. B. T. B. Strik and D. Pant, Bioresour. Technol., 2015, 195, 14–24 CrossRef CAS PubMed.
- C. Im, K. Valgepea, O. Modin and Y. Nygård, Bioresour. Technol. Rep., 2022, 19, 101156 CrossRef CAS.
- K. P. Nevin, S. A. Hensley, A. E. Franks, Z. M. Summers, J. Ou, T. L. Woodard, O. L. Snoeyenbos-West and D. R. Lovley, Appl. Environ. Microbiol., 2011, 77, 2882 CrossRef CAS PubMed.
- H. Liu, T. Song, K. Fei, H. Wang and J. Xie, Bioresour. Bioprocess., 2018, 5, 1–10 CrossRef CAS.
- A. Krige, U. Rova and P. Christakopoulos, J. Environ. Chem. Eng., 2021, 9, 106189 CrossRef CAS.
- J. S. Deutzmann, F. Kracke, W. Gu and A. M. Spormann, Environ. Sci. Technol., 2022, 56, 16073–16081 CrossRef CAS PubMed.
- L. Jourdin, T. Grieger, J. Monetti, V. Flexer, S. Freguia, Y. Lu, J. Chen, M. Romano, G. G. Wallace and J. Keller, Environ. Sci. Technol., 2015, 49, 13566–13574 CrossRef CAS PubMed.
- C. W. Marshall, D. E. Ross, E. B. Fichot, R. S. Norman and H. D. May, Environ. Sci. Technol., 2013, 47, 6023–6029 CrossRef CAS PubMed.
- Y. Song, J. S. Lee, J. Shin, G. M. Lee, S. Jin, S. Kang, J. K. Lee, D. R. Kim, E. Y. Lee, S. C. Kim, S. Cho, D. Kim and B. K. Cho, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 7516–7523 CrossRef CAS PubMed.
- H. M. Fruehauf, F. Enzmann, F. Harnisch, R. Ulber and D. Holtmann, Biotechnol. J., 2020, 15, 2000066 CrossRef CAS PubMed.
- J. C. Wood, J. Grové, E. Marcellin, J. K. Heffernan, S. Hu, Z. Yuan and B. Virdis, Water Res., 2021, 201, 117306 CrossRef CAS PubMed.
- Y. Hong, P. Arbter, W. Wang, L. N. Rojas and A. P. Zeng, Biotechnol. Bioeng., 2021, 118, 1366–1380 CrossRef CAS PubMed.
- A. Bar-Even, E. Noor and R. Milo, J. Exp. Bot., 2012, 63, 2325–2342 CrossRef CAS PubMed.
- H. Richter, B. Molitor, H. Wei, W. Chen, L. Aristilde and L. T. Angenent, Energy Environ. Sci., 2016, 9, 2392–2399 RSC.
- R. S. Tanner, L. M. Miller and D. Yang, Int. J. Syst. Bacteriol., 1993, 43, 232–236 CrossRef CAS PubMed.
- J. Moon, J. Dönig, S. Kramer, A. Poehlein, R. Daniel and V. Müller, Environ. Microbiol., 2021, 23, 4214–4227 CrossRef CAS PubMed.
- J. S. Deutzmann, M. Sahin and A. M. Spormann, mBio, 2015, 6, 1–8 CrossRef CAS PubMed.
- J. Philips, Front. Microbiol., 2020, 10, 2997 CrossRef PubMed.
- W. Buckel and R. K. Thauer, Front. Microbiol., 2018, 9, 401 CrossRef PubMed.
- C. M. Paquete, Comput. Struct. Biotechnol. J., 2020, 18, 3796–3802 CrossRef CAS PubMed.
- S. Khazalpour and D. Nematollahi, RSC Adv., 2014, 4, 8431–8438 RSC.
- M. Zhou, J. Ding, L. P. Guo and Q. K. Shang, Anal. Chem., 2007, 79, 5328–5335 CrossRef CAS PubMed.
- J. S. Lee and B. J. Little, Corrosion, 2015, 71, 1434–1440 CrossRef CAS PubMed.
- L. Göbbels, A. Poehlein, A. Dumnitch, R. Egelkamp, C. Kröger, J. Haerdter, T. Hackl, A. Feld, H. Weller, R. Daniel, W. R. Streit and M. C. Schoelmerich, Sci. Rep., 2021, 11, 1–15 CrossRef PubMed.
- F. M. Liew, M. E. Martin, R. C. Tappel, B. D. Heijstra, C. Mihalcea and M. Köpke, Front. Microbiol., 2016, 7, 694 Search PubMed.
- L. Jourdin, S. Freguia, B. C. Donose, J. Chen, G. G. Wallace, J. Keller and V. Flexer, J. Mater. Chem. A, 2014, 2, 13093–13102 RSC.
- J. M. Fontmorin, P. Izadi, D. Li, S. S. Lim, S. Farooq, S. S. Bilal, S. Cheng and E. H. Yu, Electrochim. Acta, 2021, 372, 137853 CrossRef CAS.
- N. Aryal, A. Halder, P. L. Tremblay, Q. Chi and T. Zhang, Electrochim. Acta, 2016, 217, 117–122 CrossRef CAS.
- T. Zhang, H. Nie, T. S. Bain, H. Lu, M. Cui, O. L. Snoeyenbos-West, A. E. Franks, K. P. Nevin, T. P. Russell and D. R. Lovley, Energy Environ. Sci., 2012, 6, 217–224 RSC.
- V. Flexer and L. Jourdin, Acc. Chem. Res., 2020, 53, 311–321 CrossRef CAS PubMed.
- M. Abdollahi, S. Al Sbei, M. A. Rosenbaum and F. Harnisch, Front. Microbiol., 2022, 13, 947550 CrossRef PubMed.
- C. G. S. Giddings, K. P. Nevin, T. Woodward, D. R. Lovley and C. S. Butler, Front. Microbiol., 2015, 6, 468 Search PubMed.
- S. Bajracharya, A. Krige, L. Matsakas, U. Rova and P. Christakopoulos, Bioresour. Technol., 2022, 354, 127178 CrossRef CAS PubMed.
- W. Cai, K. Cui, Z. Liu, X. Jin, Q. Chen, K. Guo and Y. Wang, Chem. Eng. J., 2022, 428, 132093 CrossRef CAS.
- A. Ayol, L. Peixoto, T. Keskin and H. N. Abubackar, Int. J. Environ. Res. Public Health, 2021, 18, 11683 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00471f |
| This journal is © The Royal Society of Chemistry 2023 |