
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1,3-Dioxolane compounds (DOXs) as biobased reaction media†
Massimo
Melchiorre
 ab,
Peter H. M.
Budzelaar
a,
Maria E.
Cucciolito
ac,
Roberto
Esposito
ac,
Emanuela
Santagata
a and
Francesco
Ruffo
*ac
ab,
Peter H. M.
Budzelaar
a,
Maria E.
Cucciolito
ac,
Roberto
Esposito
ac,
Emanuela
Santagata
a and
Francesco
Ruffo
*ac
aDipartimento di Scienze Chimiche, Università degli Studi di Napoli Federico II, Complesso Universitario di Monte S. Angelo, Via Cintia 21, 80126, Napoli, Italy. E-mail: ruffo@unina.it
bISusChem S.r.l., Piazza Carità 32, 80134 Napoli, Italy
cConsorzio Interuniversitario di Reattività Chimica e Catalisi (CIRCC), Via Celso Ulpiani 27, 70126, Bari, Italy
First published on 2nd March 2023
Abstract
Solvents constitute around 80% of the total volume of chemicals used in fine-chemical processes and contribute significantly to their environmental impact and hazard profile. Thus, there is a strong driving force towards the replacement of traditional fossil-based solvents by alternatives that are more benign in terms of their origin, availability, convenience of synthesis, handling, biodegradability and environmental impact. In the class of polar aprotic solvents, the most successful “green” replacement is γ-valerolactone (GVL). Here, we propose the use of a structurally related compound, 5-methyl-1,3-dioxolane-4-one (LA-H,H), as a reaction medium. It is easily prepared from lactic acid and formaldehyde and satisfies the criteria for a green solvent. It is stable under neutral or basic conditions. Despite the presence of a ketal functionality, it even survives mildly acidic conditions. Evaluation of the Kamlet–Taft and Hansen solvent parameters shows that indeed LA-H,H and GVL are closely similar, suggesting that LA-H,H is an effective new green entry in the class of polar aprotic solvents. We have tested its performance in Pd-catalyzed Heck arylation of methyl acrylate and in the Menschutkin reaction of N-methylimidazole with 1-iodobutane. LA-H,H is also the parent of a whole family of potential solvents easily prepared from two green precursors: α-hydroxy carboxylic acids (lactic, mandelic, and α-hydroxyisobutyric acids) and aldehydes/ketones (formaldehyde, acetaldehyde, and acetone); five such variations were briefly examined. Interestingly, LA-H,H has the unusual property of forming a three-phase system when combined with water and hexane, which may allow technological variations that are not possible with the more normal one- and two-phase systems. As a curiosity, a rare four-phase system is achievable combining LA-H,H with octane, water and perfluorodecaline.
Introduction
More than twenty years ago, Anastas and Warner introduced the “12 principles of Green Chemistry,” providing a basis for the rational design of safer production methods for chemicals and materials.1 These principles call for a series of actions, including the use of renewable feedstocks, the adoption of benign substances and catalytic methods, the elimination of auxiliaries, the prevention of waste, and the saving of energy. The choice of solvent used in a synthetic process is crucial, as traditional organic solvents usually come with environmental issues, including volatility, high flammability, and toxicity. Moreover, they are often obtained from fossil sources with a negative environmental impact on their production. The use of solvent-free processes would naturally appear as the best choice, but it is often hampered by the high viscosity or melting point of the reagents and by the required purification steps.Therefore, enormous scientific efforts and stricter regulations have been directed towards the search for improved solvents whose qualities include inertness, desired polarity and proticity, sustainable manufacture using renewable sources, low toxicity, low flammability, biodegradability, recyclability, ease of storage and transport.2
It is obvious that no single solvent can simultaneously meet all these requirements: a palette of solvents is needed to match diverse chemical process implementations, which may require specific ranges of viscosity, polarity or density.3 Alternative and more harmless solvents are slowly becoming available, and major companies have developed their own guidelines, e.g., GSK, Pfizer, Sanofi and Astra Zeneca.4,5
In light of the sustainable development goals, solvents that are bio-based or sourced from non-critical materials are preferred in chemical processes.6,7 Relevant examples include essential oils (limonene, p-cymene, and terpinene), lignocellulosic solvents (γ-valerolactone, Cyrene™, and 2-methyltetrahydrofuran), and vegetable oil derivatives (fatty acid esters and glycerol), which have already found applications in synthetic processes.8 For example, 2-methyltetrahydrofuran is suitable for use with Grignard reagents,9 and limonene10 has shown potential in the extraction of oils (replacing hexane). More recently, other valuable green alternative solvents have been proposed in the literature, such as 2,2,5,5-tetramethyltetrahydrofuran (TMTHF) and 2,5-diethyl-2,5-dimethyloxolane (DEDMO) as non-polar and non-peroxide forming ether solvents.11,12 3-Methoxybutan-2-one (MO) has been used instead of chlorinated solvents.13 γ-Valerolactone (GVL) is a polar aprotic solvent effective in supporting cross-coupling reactions14–20 and is (amongst) the best established bio-based alternatives to conventional dipolar aprotic solvents (e.g., DMF, NMP and DMSO). However, even GVL is not problem free: legal issues associated with the functioning of GVL as a pro-drug21 have led to the decision that its use should not be recommended in a Unified Solvent Selection Guide.22 Thus, there is still a need for additional options and for “families” of solvents that can be tuned to specific process needs, both in terms of chemical yield and regarding choices of process steps for isolation and purification. This applies in particular to the category of polar aprotic solvents. Polarity is often essential in promoting the formation of ionic intermediates or the cleavage of polar bonds. Protic solvents tend to interfere with process chemistry. As a result, polar aprotic fossil-based solvents such as DMF, NMP and DMSO have become almost ubiquitous, but it is clear that this must change.23,24 Ketones and esters, mostly in cyclic variations, are the obvious starting points for polar aprotic replacements. It occurred to us that ketals derived from α-hydroxycarboxylic acids (1,3-dioxolan-4-ones – DOXs) have useful solvent properties. Here, we propose the use of 5-methyl-1,3-dioxolan-4-one (Fig. 1, LA-H,H ketal derived from formaldehyde and lactic acid) as a replacement for polar aprotic solvents.
 | ||
| Fig. 1 Polar aprotic cyclic oxygenated solvents: GVL, LA-H,H and Cyrene™. | ||
More in general, the two components of DOX solvents (acids: lactic, mandelic, and α-hydroxyisobutyric; carbonyls: formaldehyde, acetaldehyde, and acetone) are individually bio-based and can be combined to produce the ketal in a single and simple catalytic process step (Scheme 1, left). Here, we focus on the parent LA-H,H but also present five additional members of this family of solvents (Scheme 1, bottom).
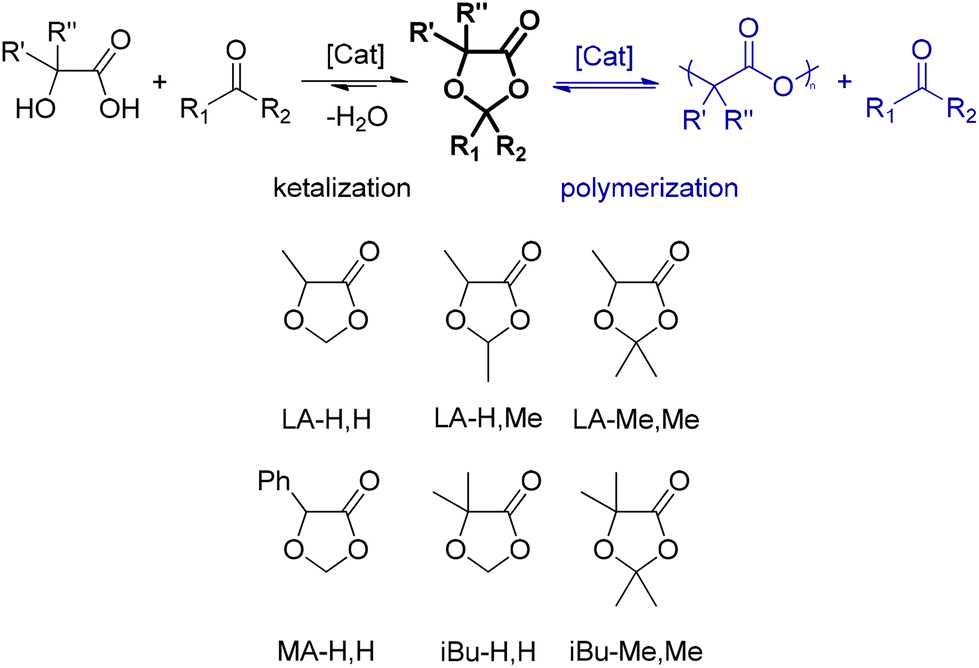 | ||
| Scheme 1 Formation of DOX through ketalization of α-hydroxyacids (left); unwanted polycondensation (right); potential solvents studied in this work (bottom). Compound labelling: the first two letters refer to the parent acid (lactic: LA-; mandelic: MA-; and α-hydroxyisobutyric: iBu-), and the following letters refer to the substituent groups at position 2 (-H and -Me). For example, in the case of 2,2,5-trimethyl-1,3-dioxolan-4-one, it is LA-Me,Me. | ||
They are not new compounds, but so far they have not been proposed as solvents. In fact, one might expect such use to be problematic due to the sensitivity of ketals to acid-catalyzed hydrolysis or to polycondensation to polylactides25,26 (Scheme 1, right). However, we have found that such side reactions require fairly forcing conditions and thus do not rule out the use of the ketals as solvents under mild conditions. In fact, some degree of degradation over time is important so that they do not become persistent contaminants.
We recently proposed27 the use of ester ketals in electrolytes for electrical double-layer capacitors, and LA-H,H turned out to be stable up to high potentials (2.6 V under operative conditions) and able to dissolve high amounts of tetraalkylammonium salt (1.0 M Et3NMeBF4) with good conductivity (8.5 mS cm−1). Precise tunability is particularly important in electrochemical applications.
Results and discussion
Synthesis and characterization of the solvents
All 6 solvents were prepared from the selected α-hydroxyacids (lactic, mandelic or α-hydroxyisobutyric acids)28–31 and carbonyl precursors (formaldehyde, acetaldehyde or acetone)32–34 obtainable from bio-based processes. The acid-catalyzed ketalization was performed with a Dean–Stark apparatus to remove water. Okada35 used Amberlite® as the acidic catalyst, but later authors (and the present authors) used p-toluenesulfonic acid with satisfactory results. The original methods and also more recent works mostly used azeotropic distillation with benzene25,35,36 for water removal (Miyagawa37 and Watanabe38 used petroleum ether); we used petroleum ether (bp 40–60 °C) or cyclohexane (also proposed by Xu26), which worked equally well. At the end of the reaction, the mixture is quenched with a base (to prevent DOX hydrolysis), and the product is isolated through vacuum distillation. Details and NMR data for the individual solvents are available in the ESI (Fig. S1–S6 and Table S1†).Solvent properties
Solvent replacement is in part a matter of trial and error: different solvents may promote different reaction paths, or determine unexpected physical phenomena (crystallization and azeotrope). But there are other aspects that help rational selection. Kamlet and Taft have developed numerical measures for hydrogen bond donor strength (α), hydrogen bond acceptor strength (β), and solvent dipolarity (π*).39,40 Later, Reichardt developed an indicator for solvent polarity (ENT) that allowed its application to ionic liquids, gas phase, reactive surfaces and other special cases.41 In a parallel development, Hansen42 proposed three solubility parameters that cover similar concepts: δd for dispersion, δp for polarity and δhb for hydrogen bonding. One advantage of the Hansen approach is that the three parameters are defined on a common energy scale, which allows comparison of two compounds in terms of a three-dimensional “distance” Ra between them. Kamlet–Taft parameters (KT) correlate to reaction rates and equilibria and so are helpful for linear free energy relationships, while Hansen solubility parameters on the other hand relate to solubility.It stands to reason that such parameters can be helpful in finding solvent “equivalencies”. To aid rational selection, we evaluated the Kamlet–Taft and Hansen parameters for LA-H,H and for their variations (Table 1). Kamlet–Taft parameters were obtained from solvatochromic effects on p-nitrophenol and p-nitroanisole (see the ESI for details of the procedure, Fig. S7a, b for calibration lines and Table S2† for DOX absorption frequencies). The DOXs are of course not hydrogen bond donors (HBDs), so α is consistently 0. The hydrogen bond acceptor character(HBA, β) varies from strong (0.6, for the least substituted examples LA-H,H) to medium (0.3, for the more substituted LA-Me,Me). The dipolarity π* in the series slightly decreases with increasing numbers of methyl groups. In addition, Table 1 includes the parameters of a few relevant polar aprotic solvents (GVL, Cyrene™, NMP, DMF, and DMSO), and Fig. 2 shows a KT plot positioning DOXs and classical solvents over KT parameter (β and π*) space. Inspection of the plot shows clearly that it is not easy to find a green solvent that matches the hydrogen bond acceptor strength of the highly polar amides (DMF and NMP) or DMSO using simple ketones or esters. But compared to other HBA examples, the present series looks fairly promising, with LA-H,H being close to GVL in both polarity and hydrogen bond acceptor characteristics. Hansen parameters were evaluated for DOXs with the method proposed by Stefanis43 (included in Table 1; details are shown in the ESI, Tables S3–8†), to allow a direct comparison with classical solvents in terms of the 3D Hansen distance Ra (Tables S9–11,†Fig. 3 and Fig. S8, 9†). Inspection of Table 2 shows that LA-H,H is fairly similar to NMP and GVL; however, the introduction of further substituents does not cause large changes in polarity. It should be noted that these are not the only possible solvent property measures. Several others have been proposed; we have evaluated them for our ketals, and compared them to other popular solvents in Table 1.
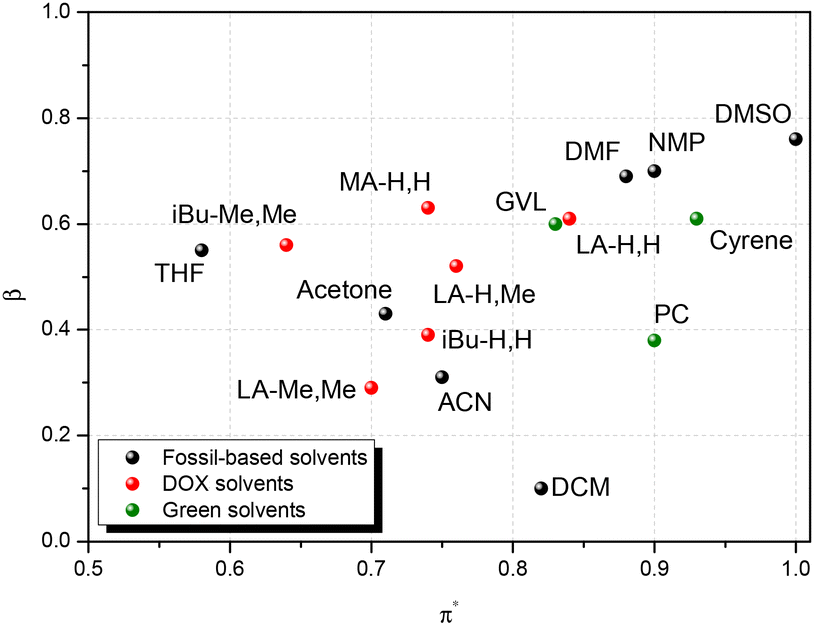 | ||
| Fig. 2 Kamlet–Taft plot for fossil-based solvents (black), green (green), and DOX solvents (red). | ||
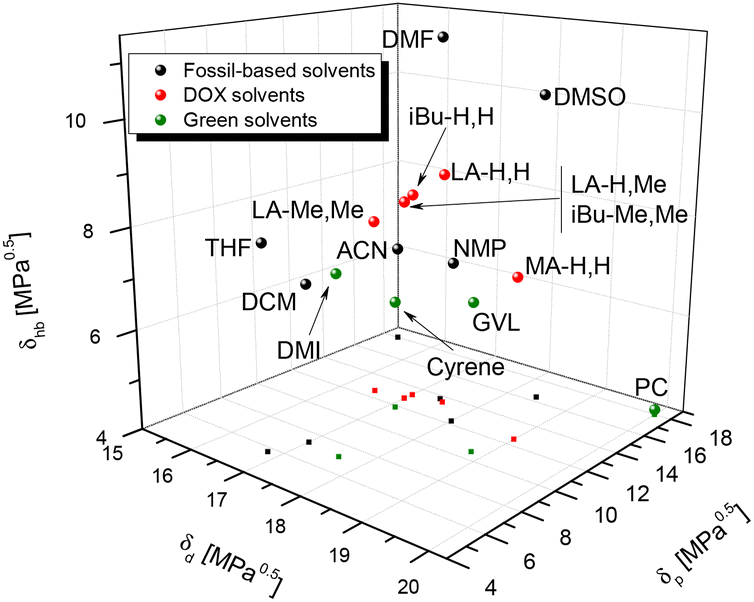 | ||
| Fig. 3 3D Hansen space for fossil-based (black), green (green), and DOX (red) solvents. LA-H,Me and iBu-Me,Me overlap. A magnification of this graph is available in Fig. S8.† | ||
| Solvent property | Solvents | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LA-H,H | LA-H,Mea | LA-Me,Me | MA-H,H | iBu-H,H | iBu-Me,Me | GVL | Cyrene | NMP | DMF | DMSO | ||
a Experimental results for 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 diastereomer mixture.
b Extrapolated from experimental value at reduced pressure in Table S1† (estimated at 1 atm by nomograph).
c At 1 atm.
d From ref. 44.
e From ref. 45.
f From ref. 46. :30 diastereomer mixture.
b Extrapolated from experimental value at reduced pressure in Table S1† (estimated at 1 atm by nomograph).
c At 1 atm.
d From ref. 44.
e From ref. 45.
f From ref. 46.
|
||||||||||||
| mp | [°C] | <−70 | <−70 | <−70 | ≈7–9 | −43 ± 2 | −43 ± 2 | −31 | −18 | −24 | −61 | 18 |
| bp | [°C] | (161–164)b | (163–165)b | (166–169)b | (125–127)b | (141–143)b | (150–153)b | 205c | 227c | 202c | 153c | 189c |
| fp | [°C] | 80.3 | 77.1 | 84 | 129.5 | 79.3 | 42.9 | 96 | 108 | 86 | 58 | 95 |
| ρ | [g mL−1] | 1.12 | 1.11 | 1.05 | 1.24 | 1.13 | 0.98 | 1.05 | 1.25 | 1.03 | 0.95 | 1.09 |
| n D | 1.4118 | 1.4062 | 1.4062 | 1.5282 | 1.4072 | 1.4038 | 1.4333 | 1.4732 | 1.4675 | 1.4305 | 1.479 | |
| [α]20D | [°] | +46.6°neat | — | +35.1°neat | +54.5°neat | — | — | — | — | — | — | — |
| β | 0.61 | 0.52 | 0.29 | 0.63 | 0.39 | 0.56 | 0.60d | 0.61d | 0.75e | 0.71e | 0.74e | |
| π* | 0.84 | 0.76 | 0.70 | 0.74 | 0.74 | 0.64 | 0.83d | 0.93d | 0.90e | 0.88e | 1.00e | |
| E NT | 0.306 | 0.275 | 0.251 | 0.267 | 0.267 | 0.227 | 0.301d | 0.339d | 0.355e | 0.386e | 0.444e | |
| δ d | [MPa1/2] | 17.5 | 17.0 | 15.2 | 19.0 | 15.7 | 18.5 | 17.1d | 18.8d | 18f | 17.4f | 18.4f |
| δ p | [MPa1/2] | 13.5 | 12.8 | 10.5 | 12.5 | 11.2 | 16.5 | 11.9d | 10.6d | 12.3f | 13.7f | 16.4f |
| δ hb | [MPa1/2] | 8.7 | 8.1 | 7.5 | 7.2 | 8.1 | 7.3 | 6.2d | 6.9d | 7.2f | 11.3f | 10.2f |
| δ t | [MPa1/2] | 23.7 | 22.7 | 19.9 | 23.8 | 20.9 | 25.8 | 21.7 | 22.7 | 23.0 | 24.9 | 26.7 |
| R a(LA-H,H) | R a(LA-H,Me) | R a(LA-Me,Me) | R a(iBu-Me,Me) | R a(iBu-H,H) | R a(MA-H,H) | R a(GVL) | |
|---|---|---|---|---|---|---|---|
| Acetone | 14.3 | 8.0 | 4.6 | 8.0 | 9.4 | 26.7 | 3.6 |
| ACN | 23.2 | 21.3 | 18.6 | 21.3 | 19.0 | 43.1 | 7.1 |
| DCM | 21.0 | 15.6 | 14.7 | 15.6 | 18.6 | 21.5 | 4.7 |
| DMF | 3.4 | 5.8 | 9.1 | 5.8 | 5.2 | 14.2 | 5.4 |
| DMSO | 6.9 | 12.6 | 17.8 | 12.6 | 10.7 | 12.8 | 6.6 |
| NMP | 2.3 | 2.5 | 4.6 | 2.5 | 3.0 | 2.0 | 2.1 |
| THF | 31.6 | 25.3 | 24.1 | 25.3 | 29.0 | 33.1 | 6.5 |
| PC | 33.2 | 39.5 | 45.2 | 39.5 | 37.5 | 21.9 | 8.7 |
| DMI | 21.2 | 17.1 | 17.6 | 17.1 | 20.2 | 18.5 | 5.1 |
| Cyrene™ | 9.2 | 9.6 | 12.8 | 9.6 | 11.0 | 1.9 | 3.7 |
| GVL | 4.7 | 2.2 | 1.9 | 2.2 | 3.0 | 7.9 | — |
The aliphatic DOX solvents have boiling points slightly lower than those of other proposed green solvents (Fig. S10†), which is good for lowered energy consumption (distillation) and does not substantially impact on fire safety (similar flash point, Fig. S11†). The boiling point of the aromatic MA-H,H is obviously much higher, but its properties are otherwise still close to those of the other five DOXs. Table 1 also provides the specific rotation for chiral solvents (LA-H,H; LA-Me,Me; MA-H,H). These represent a potential resource for under-explored solvent effects, such as asymmetric synthesis promoted or enhanced by the chirality of the solvent.47–50
Mizoroki–Heck reaction
The Mizoroki–Heck reaction (Scheme 2) is a very relevant cross-coupling reaction able to introduce an aryl group in an appropriate olefin. This reaction is strongly influenced by the choice of the reaction medium and usually requires a polar aprotic solvent such as DMF, which comes with a significant ecotoxicological profile. These features have led to the use of this reaction as a benchmark for evaluating catalysts and solvents.51,52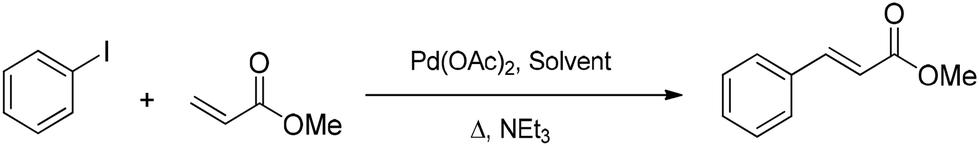 | ||
| Scheme 2 Mizoroki–Heck reaction scheme. | ||
A first screening of all six DOXs was performed under the conditions typically used in the literature, using palladium acetate as the catalyst precursor, triethylamine as the base, phenyl iodide as the aryl compound, and methyl acrylate as the olefin. An intermediate reaction time of 0.5 h at 100 °C was taken as the reference to avoid plateau effects due to high conversions; solvent stability was also qualitatively evaluated after 24 h (Table 3 and Fig. S12–S18†). As reported in Table 3, the coupling proved to be very effective in MA-H,H. Unfortunately, MA-H,H and LA-H,Me were not stable under the applied conditions: the 1H NMR spectra after 24 h clearly showed signals due to solvent degradation (Fig. S13 and S15†). Therefore, further optimization of the reaction conditions was only performed with LA-H,H:temperature (T); olefin:phenyl iodide molar ratio (MRO) and base:phenyl iodide molar ratio (MRB). Over the temperature range of 95–110 °C (Fig. 4a), the yield increases with temperature at a low reaction time (0.5 h), while at 2 and 24 hours, it increases until 105 °C and then decreases at 110 °C. Once the optimal temperature was established (105 °C), the MRO was screened (Fig. 4b) from a stoichiometric value (MRO=1.0) to 40 mol% excess (MRO=1.4). The best performance was achieved at an intermediate MRO of 1.2. This is reasonable considering that the olefin also acts as a neutral ligand: a moderate excess is functional to avoid catalyst deactivation, while a more pronounced excess may oppose the oxidative addition of phenyl iodide. The effect of the base was evaluated by varying the loading of triethylamine, testing the MRB (Fig. 4c) from the stoichiometric value (1.0) to 40 mol% excess (1.4). It is known that in the catalytic cycle, the amine has two roles: reducing the Pd(II) precursor to the Pd(0) active species and neutralizing the hydroiodic acid that is produced in the reductive elimination step. An excess of 20 mol% turned out to be much better than either stoichiometric amine or a large excess.
 | ||
| Fig. 4 Mizoroki–Heck reaction. (a) Temperature screening: phenyl iodide, 1 mmol; olefin, 1.2 mmol; and triethylamine, 1.2 mmol. (b) MRO screening: phenyl iodide, 1 mmol; triethylamine, 1.2 mmol; and olefin, 1 or 1.2 or 1.4 mmol. (c) MRB screening: phenyl iodide, 1 mmol; olefin, 1.2 mmol; and triethylamine, 1 or 1.2 or 1.4 mmol. Yield defined by 1H NMR, error ±2%. | ||
| Entry | Solvent [1 mL] | Methyl cinnamate | Is the solvent stable? |
|---|---|---|---|
| Yield [%] | [Y/N] | ||
| 1 | LA-H,H | 36 | Y |
| 2 | LA-H,Me | 20 | N |
| 3 | LA-Me,Me | 20 | Y |
| 4 | MA-H,H | 57 | N |
| 5 | iBu-H,H | 22 | Y |
| 6 | iBu-Me,Me | 27 | Y |
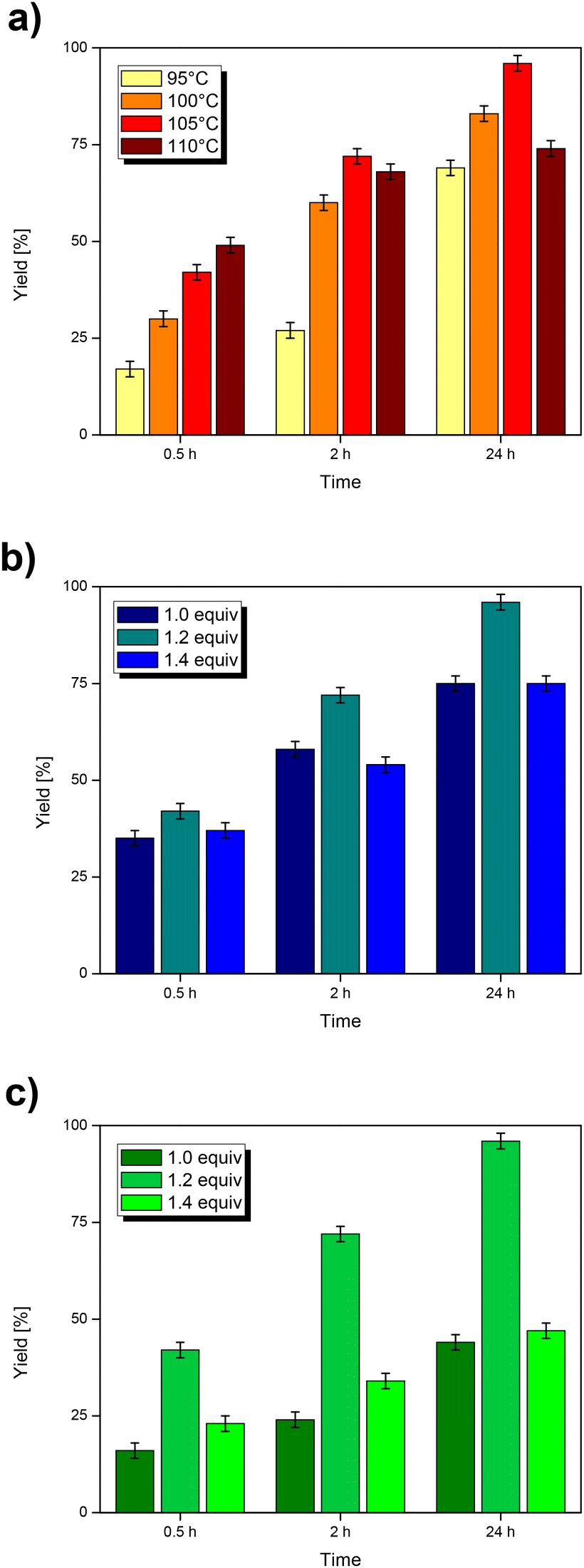
Finally, the reaction scope was explored by testing aryl sources with different functional groups. The results are collected in Table 4. The pattern of reactivities is similar to those observed with other polar solvents: the best/fastest conversions are obtained with substrates bearing electron-withdrawing substituents, while electron-rich aryl iodides react much more slowly. In addition, the reactivity with 1-bromo-4-iodobenzene (entry 4) is found to be most interesting, which leaves bromine unreacted and therefore suitable for further functionalization.
|
|
||||
|---|---|---|---|---|
| Entry | X– | Yield [%] | ||
| 0.5 h | 2 h | 24 h | ||
| 1 | H– | 71 | 88 | 99 |
| 2 | F– | 83 | 93 | >99 |
| 3 | Cl– | >99 | — | — |
| 4 | Br– | >99 | — | — |
| 5 | F3C– | 77 | 99 | >99 |
| 6 | NC– | >99 | — | — |
| 7 | CH3C(O)– | 99 | >99 | — |
| 8 | CH3OC(O)– | 86 | 96 | 97 |
| 9 | CH3– | 15 | 46 | 62 |
| 10 | CH3O– | 10 | 41 | 65 |
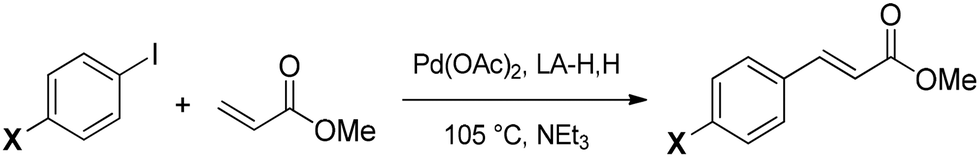
Menschutkin reaction
To validate its general applicability as a polar aprotic solvent, LA-H,H was used in the Menschutkin reaction (nucleophilic substitution) between 1-methylimidazole (MeIm) and 1-iodobutane (Scheme 3). The product is a room temperature ionic liquid, [BMIM+][I−], and the reaction kinetics correlate with the π* parameter.45,53 A parallel experiment was conducted in DMSO, and progress of the reactions was monitored through 1H NMR. We find that the reaction proceeds slightly slower in LA-H,H, but with a somewhat higher overall yield (Fig. 5). 1-Iodobutane undergoes nucleophilic attack by water present in DMSO, with the formation of n-butanol (triplet at 3.65 ppm that appears after 4 h, Fig. S19†), leading to some loss of the product. Probably, this side reaction is promoted by the presence of methyl imidazole, which acts as a base. In the case of LA-H,H, the product was isolated by simply keeping the mixture under vacuum at 40–45 °C (Fig. S20†). | ||
| Scheme 3 Menschutkin reaction conditions: 1 mmol MeIm, 1.1 mmol n-BuI, and 24 h at 50 °C. | ||
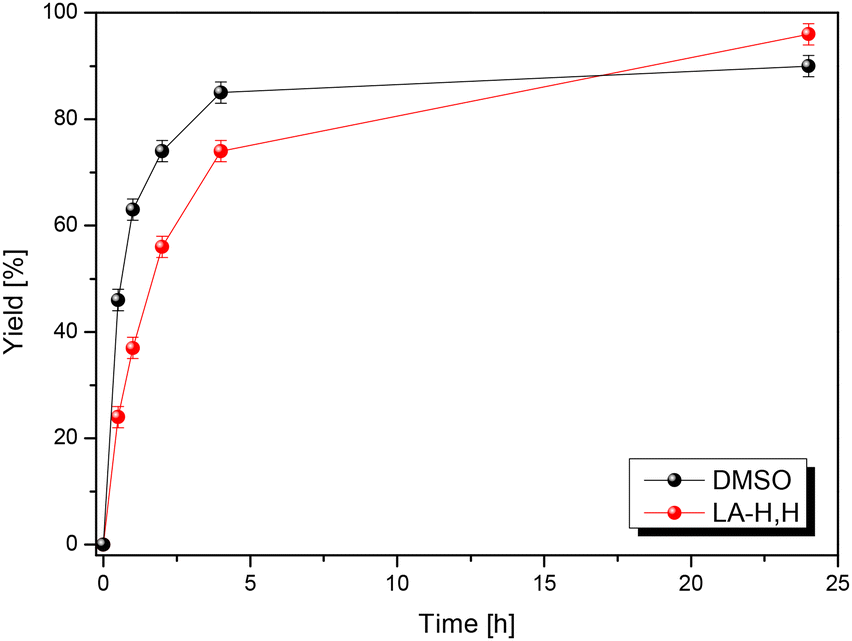 | ||
| Fig. 5 Menschutkin kinetic profile in DMSO (black) and in LA-H,H (red). | ||
Solvent stability
Ketals and acetals are usually stable in basic environments and are even used as protecting groups for aldehyde and ketone functionalities during conversions involving base. Therefore, it seems logical to contemplate the use of LA-H,H primarily under basic conditions. Stability tests were conducted by treating the solvent with a panel of common organic and inorganic bases at 25, 50 and 100 °C for 24 h. 1H NMR spectra recorded after 24 h at 100 °C (Fig. S20†) showed that LA-H,H has excellent stability. In all cases, no appreciable degradation was detected. Compared to Cyrene™, this result is remarkable since the latter is not compatible with most of the investigated bases even at room temperature.54Acetals tend to rapidly hydrolyse in the presence of aqueous acids, giving back the parent carbonyl compounds. In addition, the formation of lactides and/or polylactides is possible.25,55 Therefore, stability under acidic conditions was also tested with p-TsOH, aqueous hydrochloric acid and water buffer at pH 4.7 with acetic acid/acetate. LA-H,H was found to be stable for 24 h at 25 and 50 °C (Fig. S22†) in the presence of p-TsOH. Instead, at 100 °C, the solvent was mostly hydrolysed (more than 90%, estimated from 1H NMR), and signals related to lactic acid and its oligomers and polymer appeared56 along with those related to hydrated formaldehyde at 4.67 (HO(CH2O)2H) and 4.59 (CH2(OH)2) ppm (ref. 57) (Fig. 6, full spectra Fig. S23†).
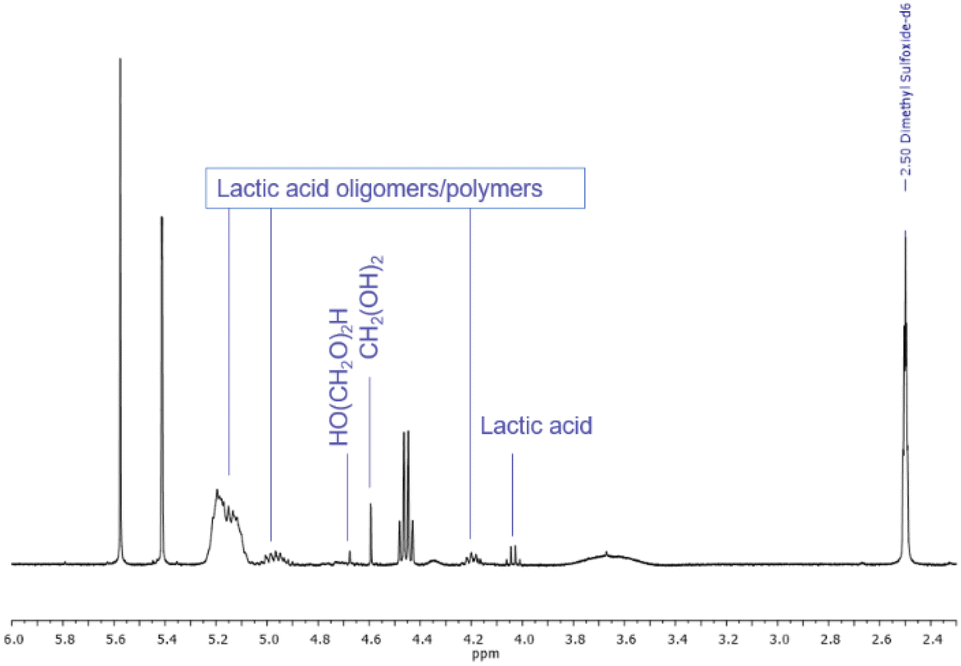 | ||
| Fig. 6 Relevant portion of 1H NMR of LA-H,H treated with p-TsOH for 24 h at 100 °C. | ||
Under mild conditions (25–50 °C), the hydrolysis is probably mitigated due to the low content of water in p-TsOH, which is however sufficient to start the hydrolysis reaction at high temperatures (100 °C).
In the presence of aqueous strong acids, such as HCl (Fig. S24,† trace 1), or a fully aqueous environment with mild acidity (Fig. S24,† trace 2), hydrolysis took place even at room temperature, reaching, respectively, 5 mol% and 9 mol% of free lactic acid after 24 hours. After two weeks in the aqueous buffer, the process reached almost 50% based on free lactic acid, and the NMR region related to hydrated formaldehyde became more complex (Fig. S24,† trace 3). Of course, the lability of the solvent under such conditions is a limitation of its use as a reaction medium, but it is an important aspect considering issues related to the persistence and bioaccumulation of chemicals in the environment.
Physical properties
Solubility values of LA-H,H in n-hexane, n-heptane, and cyclohexane were estimated by 1H NMR and resulted, respectively, in 1, 5 and 5 mol%. The corresponding solubility of aliphatic solvents in LA-H,H resulted in 3, 3, and 4 mol%, respectively, for n-hexane, n-heptane, and cyclohexane.Miscibility with classical solvents was qualitatively evaluated (Fig. S25†), and LA-H,H demonstrated an unusual behaviour compared to classical solvents, as it is immiscible with both water and aliphatic solvents (n-hexane, n-heptane, n-octane and cyclohexane) and can hence give rise to three-layer reaction mixtures on work-up. This is a relevant property that could be exploited for a liquid–liquid separation/purification step, or in biphasic catalysis as the reaction process. Notably, a rare example of a four-phase liquid mixture was obtained upon mixing LA-H,H with n-octane, water and perfluorodecaline (Fig. 7).
 | ||
| Fig. 7 Left: the four-phase system of perfluorodecaline (bottom), LA-H,H, water, n-octane (top). Right: the same system after the addition of Cu(NO3)2 and para red to make the phase separation more visible. | ||
Another notable aspect of this solvent is its electrochemical stability. As previously reported by our research group,27 LA-H,H has a large electrochemical stability window in combination with common tetraalkylammonium salts (from −1.95 to 2.55 V vs. Ag/Ag+, current cut-off=1 mA cm−2), which is comparable to that of other dipolar organic solvents (e.g., propylene carbonate). This aspect may be useful for application in electrocatalytic processes as well.
Toxicity and biodegradability considerations
A full experimental investigation of LA-H,H toxicity is beyond the scope of this work, but this aspect should be assessed before proposing its commercialisation. A limited computational approach was pursued to get preliminary indications about the general toxicity of this class of compounds.Three open-source software packages were used to predict their mutagenicity and carcinogenicity: VEGA58 (mutagenicity – CAESAR, SARpy, ISS and KNN models), Toxtree59 (mutagenicity – ISS model; carcinogenicity and mutagenicity – ISS model) and T.E.S.T.60 (mutagenicity – consensus method). The outputs provided by VEGA software were translated in scores following the approach reported by Noppawan.12 The results collected in Table S12† show how these compounds may have a non-toxic profile (average score <0.5 for all DOXs). However, due to the chemical nature of ketals and considering hydrolysis equilibria in biological environments, the toxicity of their parent AHA and carbonyl compounds also must be evaluated. The AHAs are hazardous on eye contact (MA – H318; LA – H315, H318; iBu – H315, H318, and H335), but they do not have any specific hazard statement for chronic or acute toxicity (GHS08 and GHS06). C1–C3 carbonyl compounds obtained with hydrolysis, especially formaldehyde and acetaldehyde, have several hazard statements. Despite their toxicity in cases of severe exposure, it should be pointed out that these compounds naturally occur in some foods and are endogenous to metabolic pathways (e.g., the human body produces about 50 grams of formaldehyde per day,61 and acetaldehyde is an intermediate of ethanol metabolism62). Both aldehydes have low half-lifes in human blood plasma (formaldehyde 1.5 min;63 acetaldehyde18–31 min (ref. 64)). Moreover, some commercial polymers based on formaldehyde (e.g., melamine resins) are used in kitchen utensils and drug delivery systems.65,66 In this context, it should be mentioned that GVL produces upon hydrolysis a bioactive compound, γ-hydroxyvaleric acid, which is considered potentially harmful.67
From the point of view of biodegradability and persistence of these compounds, a dedicated study should be performed. However, it is reasonable to consider the acetal moiety as the weak point subjected to hydrolysis under aqueous conditions. In this case, the parent synthons should be considered, and for LA-H,H, the half-life of formaldehyde is 30–50 min,68 while lactic acid is a readily biodegradable compound.69
Experimental
Dioxolane synthesis
The syntheses of DOXs are known in the literature and are briefly summarized here. In a round-bottom flask equipped with a Dean–Stark trap and an Allihn condenser, an appropriate amount of AHA is treated with the carbonyl compound under reflux in petroleum ether (bp 40–60 °C) or cyclohexane in the presence of an acid catalyst. After 24–36 h, the cooled mixture is treated with sodium carbonate and filtered. The volatile phase is removed using a rotary evaporator, and the crude mixture is purified by vacuum distillation. Details for each DOX are provided in the ESI.†Solvent characterization: solvent stability and solubility
The Kamlet–Taft and Hansen parameters were determined according to the method described in the ESI.† The experimental setup and procedure for determining the solvent stability and miscibility are also reported in the ESI.†Mizoroki–Heck reaction
At 0.5 h, 2 h and 24 h, an aliquot of few microliters of the reaction mixture was diluted in CDCl3, and a 1H NMR spectrum was recorded. The reaction yields were evaluated by the integration of suitable signals related to the unreacted phenyl iodide (2H at 7.10 ppm) and methyl cinnamate (2H at 7.55 ppm). Fig. S26† shows the relevant part of the spectrum and the equation employed.
In the case of low solubility of the catalyst in the chosen solvent, a stock solution in CHCl3 was prepared. The appropriate amount of catalyst solution was placed into the vial, and the organic solvent was evaporated under reduced pressure. The chosen solvent and the appropriate amounts of reagents were premixed in a different vial and added to the reaction vessel as a homogeneous solution.
Menschutkin reaction
To a 3 mL screw-cap vial were added 1.0 mmol of 1-methylimidazole (82 mg, 79 μL), 1 mL of the chosen solvent and 1.10 mmol of 1-iodobutane (202 mg, 125 μL). The reaction mixture was placed in a pre-heated oil bath at 50 °C and left under magnetic stirring for 24 h. The reaction progress was analyzed with 1H NMR spectroscopy at different times.Conclusion
This study contributes to the urgent implementation of greener production, to reduce the circulation of pollutants on the planet, and to achieve the ambitious zero waste goal of the circular economy. It has been demonstrated that LA-H,H, easily accessible from biobased starting materials, satisfies several conditions of sustainability and should be considered as a potential replacement for polar aprotic solvents. It has been validated as a reaction medium for the Mizoroki–Heck coupling between methyl acrylate and a series of substituted aryl iodides and in a nucleophilic substitution reaction. In both cases, the satisfactory outcome of the reactions is accompanied by clear advantages, such as the possibility of recovering the product by simple extraction, and higher selectivity with respect to a classical organic solvent such as DMSO or DMF. There might even be an incentive to replace “green” GVL by LA-H,H in view of the pro-drug issues of the former solvent. These benefits are added to excellent stability and predictable low toxicity and high biodegradability.The study, therefore, reinforces the awareness that it is possible to reduce the environmental impact of chemical processes and represents a step towards the full application of the principles of green chemistry and the circular economy.
Author contributions
Conceptualization, M. M. and F. R.; methodology, M. M., R. E. and E. S.; validation, M. M., P. H. M. B., M. E. C. and R. E.; investigation, M. M. and E. S.; data curation, M. M., and E. S; writing – original draft preparation, M. M., E. S. and F. R.; writing – review and editing, M. M., P. H. M. B., M. E. C., R. E. and F. R.; supervision, M. E. C. and F. R. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Miss Chiara Russo and Miss Ilaria Incoronato for their technical assistance.References
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford [England]; New York, 1998 Search PubMed.
- J. T. M. Jacobs, Improving the Identification, Evaluation, Adoption and Development of Safer Alternatives: Needs and Opportunities to Enhance Substitution Efforts within the Context of REACH, L. C. f. S. Production, University of Massachusetts Lowell, Lowell, 2016 Search PubMed.
- V. Hessel, N. N. Tran, M. R. Asrami, Q. D. Tran, N. Van Duc Long, M. Escribà-Gelonch, J. O. Tejada, S. Linke and K. Sundmacher, Green Chem., 2022, 24, 410–437 RSC.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- N. Winterton, Clean Technol. Environ. Policy, 2021, 23, 2499–2522 CrossRef PubMed.
- Y. Gu and F. Jérôme, Chem. Soc. Rev., 2013, 42, 9550–9570 RSC.
- E. J. Milton and M. L. Clarke, Green Chem., 2010, 12, 381–383 RSC.
- P. K. Mamidipally and S. X. Liu, Eur. J. Lipid Sci. Technol., 2004, 106, 122–125 CrossRef CAS.
- B. R. Trowse, F. P. Byrne, J. Sherwood, P. O'Brien, J. Murray and T. J. Farmer, ACS Sustainable Chem. Eng., 2021, 9, 17330–17337 CrossRef CAS.
- P. Noppawan, S. Sangon, N. Supanchaiyamat, J. Sherwood and A. J. Hunt, ACS Sustainable Chem. Eng., 2022, 10, 4486–4493 CrossRef CAS.
- S. Jin, F. P. Byrne, J. H. Clark, C. R. McElroy, A. Quinn, J. Sherwood and A. J. Hunt, RSC Adv., 2021, 11, 39412–39419 RSC.
- S. Sangon, N. Supanchaiyamat, J. Sherwood, C. R. McElroy and A. J. Hunt, React. Chem. Eng., 2020, 5, 1798–1804 RSC.
- X. Tian, F. Yang, D. Rasina, M. Bauer, S. Warratz, F. Ferlin, L. Vaccaro and L. Ackermann, Chem. Commun., 2016, 52, 9777–9780 RSC.
- D. Rasina, A. Kahler-Quesada, S. Ziarelli, S. Warratz, H. Cao, S. Santoro, L. Ackermann and L. Vaccaro, Green Chem., 2016, 18, 5025–5030 RSC.
- H. Mahmoudi, F. Valentini, F. Ferlin, L. A. Bivona, I. Anastasiou, L. Fusaro, C. Aprile, A. Marrocchi and L. Vaccaro, Green Chem., 2019, 21, 355–360 RSC.
- E. Ismalaj, G. Strappaveccia, E. Ballerini, F. Elisei, O. Piermatti, D. Gelman and L. Vaccaro, ACS Sustainable Chem. Eng., 2014, 2, 2461–2464 CrossRef CAS.
- G. Strappaveccia, L. Luciani, E. Bartollini, A. Marrocchi, F. Pizzo and L. Vaccaro, Green Chem., 2015, 17, 1071–1076 RSC.
- D. Fodor, T. Kégl, J. M. Tukacs, A. K. Horváth and L. T. Mika, ACS Sustainable Chem. Eng., 2020, 8, 9926–9936 CrossRef CAS.
- H. Andresen-Streichert, H. Jungen, A. Gehl, A. Müller and S. Iwersen-Bergmann, J. Anal. Toxicol., 2013, 37, 250–254 CrossRef CAS PubMed.
- A. Jordan, C. G. J. Hall, L. R. Thorp and H. F. Sneddon, Chem. Rev., 2022, 122, 6749–6794 CrossRef CAS PubMed.
- F. Gao, R. Bai, F. Ferlin, L. Vaccaro, M. Li and Y. Gu, Green Chem., 2020, 22, 6240–6257 RSC.
- L. Vaccaro, Eur. J. Org. Chem., 2020, 4273–4283 CrossRef CAS.
- S. A. Cairns, A. Schultheiss and M. P. Shaver, Polym. Chem., 2017, 8, 2990–2996 RSC.
- Y. Xu, M. R. Perry, S. A. Cairns and M. P. Shaver, Polym. Chem., 2019, 10, 3048–3054 RSC.
- M. Melchiorre, R. Esposito, M. Di Serio, G. Abbate, A. Lampasi, A. Balducci and F. Ruffo, Energies, 2021, 14, 4250 CrossRef CAS.
- T. Rohwerder and R. H. Müller, Microb. Cell Fact, 2010, 9, 13 CrossRef PubMed.
- S. K. Bhatia, R. K. Bhatia and Y.-H. Yang, Rev. Environ. Sci. Biotechnol., 2016, 15, 639–663 CrossRef CAS.
- R. Datta and M. Henry, J. Chem. Technol. Biotechnol., 2006, 81, 1119–1129 CrossRef CAS.
- A. Manandhar and A. Shah, Processes, 2020, 8, 199 CrossRef.
- L. E. Heim, H. Konnerth and M. H. G. Prechtl, Green Chem., 2017, 19, 2347–2355 RSC.
- A. Segawa, A. Nakashima, R. Nojima, N. Yoshida and M. Okamoto, Ind. Eng. Chem. Res., 2018, 57, 11852–11857 CrossRef CAS.
- T. H. Nguyen, I. Y. Sunwoo, C. H. Ra, G.-T. Jeong and S.-K. Kim, Bioprocess Biosyst. Eng., 2019, 42, 415–424 CrossRef CAS PubMed.
- M. Okada, H. Sumitomo and M. Atsumi, Macromolecules, 1984, 17, 1840–1843 CrossRef CAS.
- R. T. Martin, L. P. Camargo and S. A. Miller, Green Chem., 2014, 16, 1768–1773 RSC.
- T. Miyagawa, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1861–1865 CrossRef CAS.
- K. Watanabe, Y. Andou, Y. Shirai and H. Nishida, Chem. Lett., 2013, 42, 159–161 CrossRef CAS.
- M. J. Kamlet, J. L. Abboud and R. W. Taft, J. Am. Chem. Soc., 1977, 99, 6027–6038 CrossRef CAS.
- M. J. Kamlet and R. W. Taft, J. Am. Chem. Soc., 1976, 98, 377–383 CrossRef CAS.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- C. M. Hansen, Hansen Solubility Parameters – A User's Handbook, Boca Raton, 2nd edn, 2007 Search PubMed.
- E. Stefanis and C. Panayiotou, Int. J. Thermophys., 2008, 29, 568–585 CrossRef CAS.
- L. Germán, J. M. Cuevas, R. Cobos, L. Pérez-Alvarez and J. L. Vilas-Vilela, RSC Adv., 2021, 11, 19070–19075 RSC.
- J. Sherwood, H. L. Parker, K. Moonen, T. J. Farmer and A. J. Hunt, Green Chem., 2016, 18, 3990–3996 RSC.
- M. Díaz de los Ríos and E. Hernández Ramos, SN Appl. Sci., 2020, 2, 676 CrossRef.
- B. Pégot, G. Vo-Thanh, D. Gori and A. Loupy, Tetrahedron Lett., 2004, 45, 6425–6428 CrossRef.
- J. L. Greenfield, E. W. Evans, D. Di Nuzzo, M. Di Antonio, R. H. Friend and J. R. Nitschke, J. Am. Chem. Soc., 2018, 140, 10344–10353 CrossRef PubMed.
- Y. Nagata, R. Takeda and M. Suginome, ACS Cent. Sci., 2019, 5, 1235–1240 CrossRef CAS PubMed.
- R. Gausepohl, P. Buskens, J. Kleinen, A. Bruckmann, C. W. Lehmann, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2006, 45, 3689–3692 CrossRef CAS PubMed.
- J. Sherwood, J. H. Clark, I. J. S. Fairlamb and J. M. Slattery, Green Chem., 2019, 21, 2164–2213 RSC.
- H. L. Parker, J. Sherwood, A. J. Hunt and J. H. Clark, ACS Sustainable Chem. Eng., 2014, 2, 1739–1742 CrossRef CAS.
- J. Sherwood, M. De bruyn, A. Constantinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunt and J. H. Clark, Chem. Commun., 2014, 50, 9650–9652 RSC.
- K. L. Wilson, A. R. Kennedy, J. Murray, B. Greatrex, C. Jamieson and A. J. B. Watson, Beilstein J. Org. Chem., 2016, 12, 2005–2011 CrossRef CAS PubMed.
- K. Hyoi, A. Kanazawa and S. Aoshima, ACS Macro Lett., 2019, 8, 128–133 CrossRef CAS PubMed.
- J. L. Espartero, I. Rashkov, S. M. Li, N. Manolova and M. Vert, Macromolecules, 1996, 29, 3535–3539 CrossRef CAS.
- M. Rivlin, U. Eliav and G. Navon, J. Phys. Chem. B, 2015, 119, 4479–4487 CrossRef CAS PubMed.
- Laboratory of Environmental Chemistry and Toxicology – Milan (Italy) – V.E.G.A. Version 1.1.5., [https://vegahub.eu/download/-vega-qsar-download/], (accessed 17th September 2022).
- G. Patlewicz, N. Jeliazkova, R. J. Safford, A. P. Worth and B. Aleksiev, SAR QSAR Environ. Res., 2008, 19, 495–524 CrossRef CAS PubMed.
- US Enviromental Protection Agency T.E.S.T. (Toxicity Estimation Software Tool) Version 5.1.2.0, [https://www.epa.gov/ORD/NRMRL/std/cppb/qsar/index.html], (accessed 17th September 2022).
- B. A. Magnuson, G. A. Burdock, J. Doull, R. M. Kroes, G. M. Marsh, M. W. Pariza, P. S. Spencer, W. J. Waddell, R. Walker and G. M. Williams, Crit. Rev. Toxicol., 2007, 37, 629–727 CrossRef CAS PubMed.
- G. J. Lees and G. R. Jago, J. Dairy Sci., 1978, 61, 1205–1215 CrossRef CAS.
- World Health Organization, Regional Office for Europe, Air quality guidelines for Europe, World Health Organization. Regional Office for Europe, Copenhagen, 2nd edn, 2000 Search PubMed.
- A. W. Jones, J. Neiman and M. Hillbom, Br. J. Clin. Pharmacol., 1988, 25, 213–221 CrossRef CAS PubMed.
- E. Ordonez, L. L. Kendrick-Williams and E. Harth, Eur. Polym. J., 2021, 143, 110210 CrossRef CAS.
- K. H. Lund and J. H. Petersen, Food Addit. Contam., 2006, 23, 948–955 CrossRef CAS PubMed.
- L. P. Carter, W. Chen, H. Wu, A. K. Mehta, R. J. Hernandez, M. K. Ticku, A. Coop, W. Koek and C. P. France, Drug Alcohol Depend., 2005, 78, 91–99 CrossRef CAS PubMed.
- United States Environmental Protection Agency (U.S. EPA), Formaldehyde, Toxicity and Exposure Assessment for Children's Health, [https://archive.epa.gov/region5/teach/web/pdf/formaldehyde_summary.pdf], (accessed 25th June 2022).
- C. T. Bowmer, R. N. Hooftman, A. O. Hanstveit, P. W. M. Venderbosch and N. van der Hoeven, Chemosphere, 1998, 37, 1317–1333 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00227f |
| This journal is © The Royal Society of Chemistry 2023 |