
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClosed-loop recycling of microparticle-based polymers†
Takumi
Watanabe‡
 a,
Haruka
Minato‡
a,
Yuma
Sasaki
a,
Seina
Hiroshige
a,
Hayato
Suzuki
a,
Nahomi
Matsuki
a,
Koki
Sano
a,
Takeshi
Wakiya
c,
Yuichiro
Nishizawa
d,
Takayuki
Uchihashi
de,
Takuma
Kureha
f,
Mitsuhiro
Shibayama
g,
Toshikazu
Takata
hi and
Daisuke
Suzuki
*ab
a,
Haruka
Minato‡
a,
Yuma
Sasaki
a,
Seina
Hiroshige
a,
Hayato
Suzuki
a,
Nahomi
Matsuki
a,
Koki
Sano
a,
Takeshi
Wakiya
c,
Yuichiro
Nishizawa
d,
Takayuki
Uchihashi
de,
Takuma
Kureha
f,
Mitsuhiro
Shibayama
g,
Toshikazu
Takata
hi and
Daisuke
Suzuki
*ab
aGraduate School of Textile Science & Technology, Shinshu University, 3-15-1 Tokida Ueda, Nagano 386-8567, Japan. E-mail: d_suzuki@shinshu-u.ac.jp
bResearch Initiative for Supra-Materials, Interdisciplinary Cluster for Cutting Edge Research, Shinshu University, 3-15-1 Tokida Ueda, Nagano 386-8567, Japan
cResearch & Development Institute, Sekisui Chemical Co., Ltd., 2-1 Hyakuyama, Shimamoto, Mishima, Osaka, Japan
dDepartment of Physics and Structural Biology Research Center, Graduate School of Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8602, Japan
eDepartment of Creative Research, Exploratory Research Center on Life and Living Systems, National Institutes of Natural Sciences, Okazaki, Aichi 444-8787, Japan
fDepartment of Frontier Materials Chemistry, Graduate School of Science and Technology, Hirosaki University, Bunkyo-cho 3, Hirosaki, Aomori 036-8561, Japan
gComprehensive Research Organization for Science and Society Neutron Science and Technology Center, 162-1 Shirakata, Tokai, Naka, Ibaraki 319-1106, Japan
hSchool of Materials and Chemical Technology, Tokyo Institute of Technology, O-okayama, Meguro, Tokyo 152-8552, Japan
iGraduate School of Advanced Science and Engineering, Hiroshima University, 1-4-1 Kagamiyama, Higashi-Hiroshima, Hiroshima 739-8527, Japan
First published on 21st March 2023
Abstract
Contemporary polymer science is shifting toward the development of recycling systems to curb global resource depletion and environmental contamination. However, most methods of polymer recycling require cleavage of chemical bonds, which diminishes the quality of the polymers during recycling. Here, we propose a recycling strategy for tough polymers based on microparticles, which allows materials recycling without loss of their properties (‘closed-loop’ recycling). The polymer microparticles can be used to generate tough polymer films by controlling the interparticle physical cross-linking, and subsequently recycled on demand by disassembling into individual microparticles without chemical reactions. Our “microparticle-based concept” for polymer recycling circumvents the infamous trade-off between mechanical stability and degradability of polymers and be expected to open new avenues for closed-loop recycling of polymer materials.
Introduction
If materials could be easily decomposed into individual resources after use and recycled without deterioration of their properties, human society could maintain its quality of life while being one step closer to a truly sustainable society.1–6 However, huge amounts of materials are currently incinerated or deposited in landfills, which contributes to resource depletion and environmental contamination on a global scale.7 Especially in the case of polymer materials, commercially available and recyclable products that can be recycled without impairment of their properties, such as polyethylene terephthalate (PET) bottles, are limited.4,8 Therefore, much attention has recently been focused on fundamental research that would allow polymers to be recycled without loss of their properties (‘closed-loop’ recycling). Although several methods to convert polymers to monomers, such as thermal decomposition and the use of dynamic covalent bonding, have been proposed,2,9–11 these methods still require custom-designed monomers, complicated purification processes, and the manufacturing costs are often much higher than those of conventional polymers.6,7 Clearly, several innovations are necessary to improve the practical feasibility of fully recyclable polymers.Polymer microparticles are colloids, whose size ranges from tens of nanometers to several micrometers, that are typically suspended in water. They are usually synthesized in an aqueous system, and thus, they are generally regarded as environmentally friendly materials.12–18 Such polymer microparticles can be transformed into films through solvent evaporation and are of crucial importance in the polymer industry in e.g., adhesives, paints, and paper processing, evident from their worldwide production, which was on the multimillion-ton scale as of 2020.19,20 In general, the mechanical properties of polymer films composed of microparticles are low due to the presence of fracture regions such as particle interfaces linked by non-covalent bonds.21–25 Thus, various additives including plasticizers, as well as post-polymerization modifications for creating chemically cross-linking networks, are required,26–28 resulting in materials that are mechanically stable but cannot be decomposed after use. It has been believed that it is difficult to obtain mechanically stable microparticulate films without chemical bonding at the interface.
Under such background, in this study, we discovered that tough polymers are formed via simple water evaporation from particle dispersions without chemical crosslinking between microparticles, and microparticle-based recycling of the tough polymer materials can be achieved without loss of mechanical properties of polymers (Fig. 1). Our experimental results indicate that mechanically stable polymer films composed entirely of polyacrylate-based microparticles can be prepared without chemical bond at the particle interfaces and be disassembled into individual microparticles by reversibly controlling the interparticle physical cross-linking, which enables the resource circulation not only for polymers, but also for other valuable resources (Fig. 1).
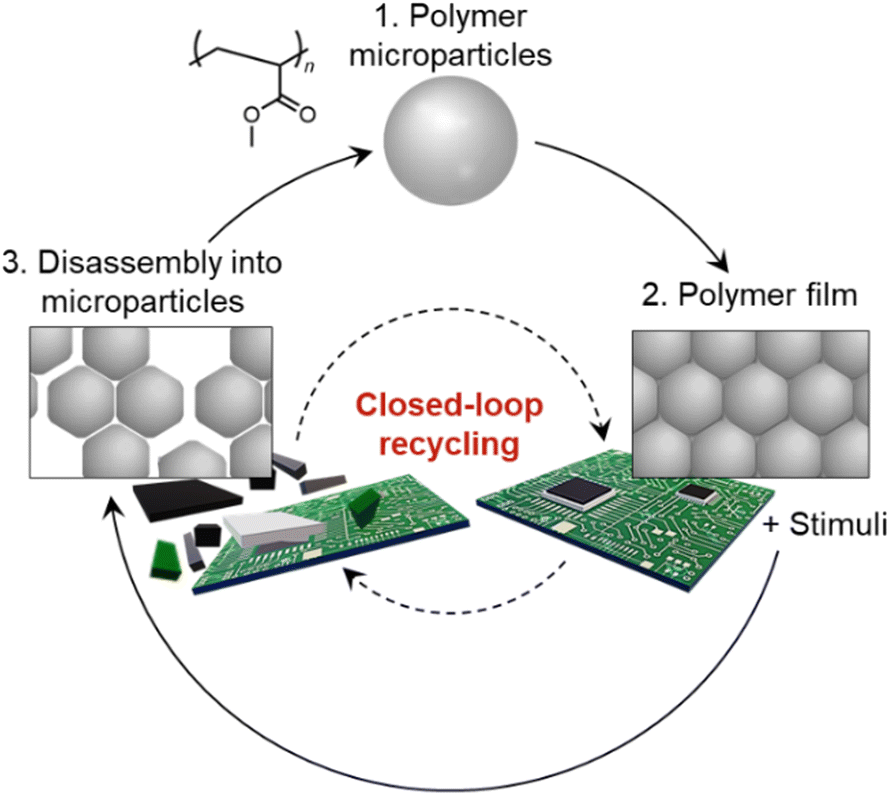 | ||
| Fig. 1 Recycling of microparticle-based polymers. Schematic illustration of the closed-loop recycling process of a polymer film composed of polyacrylate-based microparticles, which can be disassembled into individual microparticles. The recycling of microparticle-based polymers could potentially be applied to the circulation of other resources in advanced heterogeneous (composite) materials. | ||
Results and discussion
The polymer microparticles used to realize this concept here were prepared via aqueous emulsion polymerization12,13 from methyl acrylate (MA), which is the simplest acrylate monomer (Fig. 2a and Table S1†). The formation of poly-MA microparticles proceeded via the controlled aggregation of polymer chains in water, which afforded uniform spherical microparticles (Fig. S1†). After exhaustive purification by centrifugation and dialysis, poly-MA microparticles maintain their nanostructures due to their intraparticle chemical cross-linking and are colloidally stable in water for at least a year. The aqueous suspensions that contained these poly-MA microparticles were dried at ambient temperature to form thin polymer films (Fig. 2a). The resulting poly-MA-microparticle films exhibited unexpectedly high mechanical stability against external forces such as pulling and piercing (Fig. 2b). In more detail, the quantitative stress–strain relationship evaluated using a tensile test revealed that the fracture energy, which is an indicator of the toughness of a material, of the poly-MA-microparticle films was comparable or superior to those of conventional poly-MA bulk films (Fig. 2c). The fracture energy of these films is moreover no longer inferior to the strength of natural-rubber-latex films29–31 and other tough latex films, whose particle interfaces are chemically crosslinked.32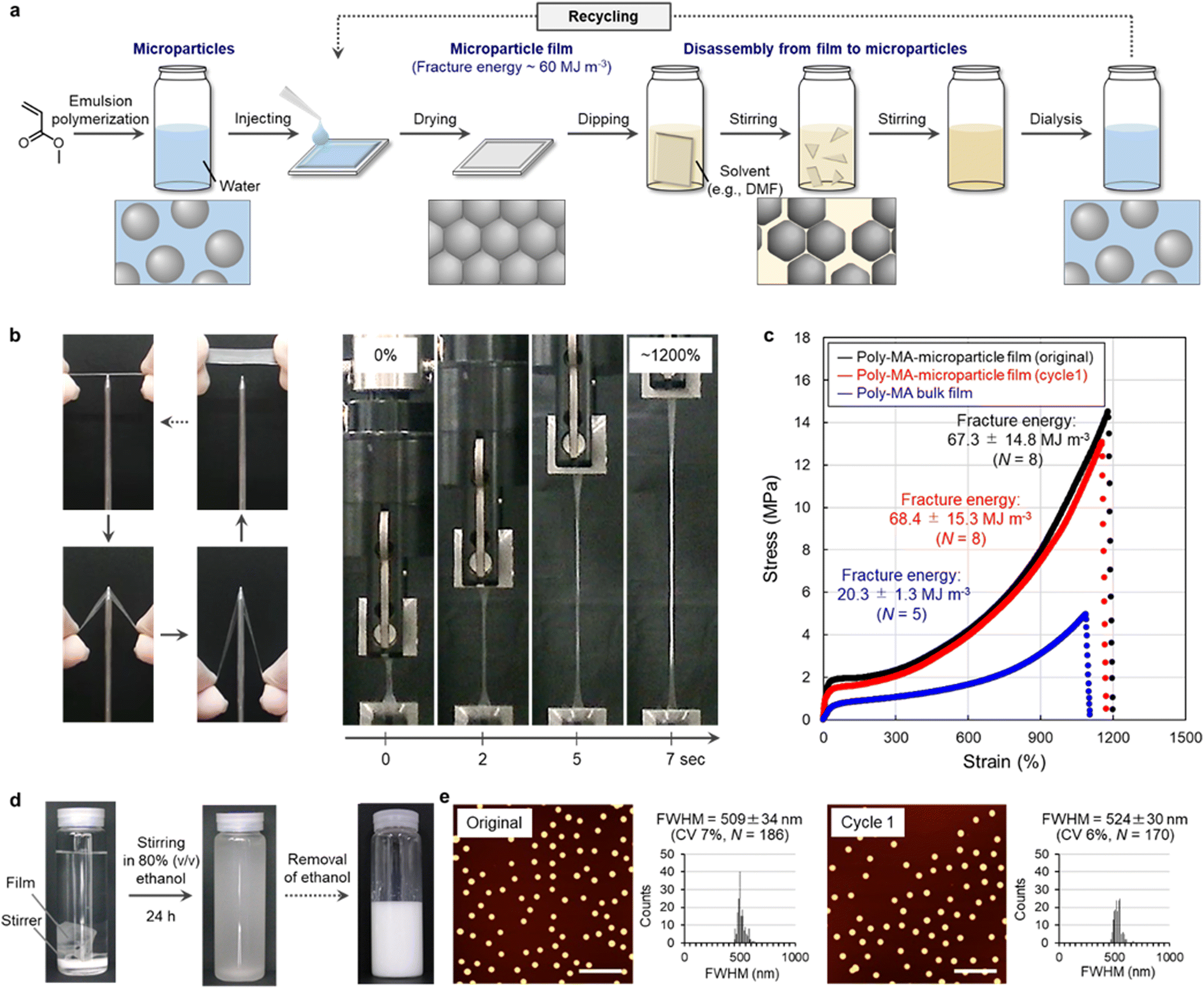 | ||
| Fig. 2 Polymer recycling using poly-MA microparticles. (a) Schematic illustration of the closed-loop-recycling process of a polymer film. (b) Representative photographs of tough poly-MA-microparticle(0.1 mol%_122 nm) films during mechanical testing. (c) Representative mechanical properties of poly-MA-microparticle(0.1 mol%_497 nm) films (original and cycle1), which exhibit higher mechanical properties than a poly-MA bulk film. (d) Degradation in an aqueous solution of 80% (v/v) ethanol and resuspension in water of poly-MA-microparticle(0.1 mol%_497 nm) films. (e) AFM height images and determined size (i.e., full width at half maximum (FWHM)) of poly-MA-microparticles(0.1 mol%_497 nm) before and after the recycling process; scale bar: 5 μm. | ||
Conversely, we confirmed that the tough poly-MA-microparticle films could be readily disassembled by immersing the films in a good solvent such as an aqueous solution of 80% (v/v) ethanol (Fig. S2†). The poly-MA-microparticle films disassembled into individual particles (Fig. 2d). When the poly-MA microparticles were not completely disassembled into individual particles, the corresponding recycling yield was low due to the presence of large microparticle aggregates (Fig. S2†). The maximum recycling yield (∼99%) was achieved for a degradation time of 24 h, at which point the films were already disassembled into individual microparticles but did not yet show deterioration (Fig. 2d, e, and Fig. S3, S4†). Furthermore, stable poly-MA-microparticle films could be reformed by drying the aqueous suspension that contain the recycled microparticles (Fig. 2c, and Fig. S3†). Importantly, the recycled poly-MA-microparticle films were also tough, and their fracture energy was close to that of the original films. Based on these results, we concluded that closed-loop recycling of polymer materials can be realized using microparticles (e.g., based on poly-MA) as the key. It should also be noted here that, in principle, this recycling system can be scaled-up to the industrial level (tons), considering that e.g., ton-scale centrifuges for the isolation and purification of the microparticles are available. Closed-loop recycling based on polymer microparticles has potential as a versatile approach, given that the degradation conditions for the polymer materials are mild and straightforward, i.e., simple immersion in a good solvent. The recyclable and tough polymer shown in Fig. 2 is particularly fascinating because facile degradability and good stability are the ultimate trade-off in polymer recycling. That is, the mechanical properties of polymer films that consist of microparticles are generally poor, given the high degree of interfaces between the microparticles in the films.21–25 Moreover, other types of tough films that consist of microparticles cross-linked by rotaxanes16 can also be disassembled into single particles by good solvents (Fig. S5†), suggesting that it is feasible to expect that this microparticle-based recycling strategy can be improved and extended to other functional polymers, including hard microparticles and soft microgels,33–39 considering that living organs are mechanically stable even though they are composed of organized ultrasoft microparticles (cells).
In order to clarify the origin of the desirable mechanical properties and high degradability of the polymer films of poly-MA microparticles, we subsequently examined the morphology and nanostructure of pristine and recycled poly-MA-microparticle films (Fig. 3a). High-speed atomic force microscopy (HS-AFM),40–44 which allows imaging with high spatial resolution, revealed that the original spherical shape of poly-MA microparticles was retained in the films and that the poly-MA microparticles are ordered on/in the films regardless of the cross-linking density of the microparticles (Fig. 3b, Fig. S6, and Movies S1, S2†). Moreover, a clear characteristic in the nanostructures of the microparticle films was confirmed via small-angle X-ray scattering (Fig. 3c). Initially, for the films that are degradable but fragile, (i.e., highly cross-linked poly-MA microparticles), the Bragg peaks derived from face-centered cubic (fcc) colloidal crystals were observed in the low-scattering vectors (Fig. 3c, black), which is consistent with conventional colloidal films.45 Conversely, the scattering profiles of degradable and tough poly-MA-microparticle films (i.e., sparsely cross-linked poly-MA microparticles) do not exhibit these characteristic peaks (Fig. 3c, yellow, blue, green); similar profiles were observed when the films were prepared from sparsely cross-linked poly-MA microparticles of different sizes (Fig. S7†), even though the poly-MA microparticles were ordered for fcc colloidal crystals on/in the films (Fig. 3b, Fig. S6, and Movies S1, S2†). Moreover, the Bragg peaks observed for highly cross-linked poly-MA-microparticle films was attenuated by thermal annealing (Fig. 3c, red). These results indicate that the disappearance of the Bragg peaks is most likely due to the high deformation of the poly-MA microparticles and their fusion to each other, while the poly-MA microparticles are indeed ordered in the films. The scattering profiles of the poly-MA-microparticle films did not change significantly upon recycling, suggesting that the nanostructures of the microparticles and the films were preserved without deterioration (Fig. S7†). In more detail, the interfacial thickness between microparticles, tint, which correlates well with the fracture energy of the microparticle films, is affected by the degree of cross-linking in the microparticles and the thermal annealing time (Fig. 3d and Fig. S8†). It is thus plausible that the recyclable poly-MA-microparticle films exhibit tough mechanical properties comparable to bulk materials because the polymer chains on the microparticle surfaces are so deeply interpenetrated with each other in the films that the presence of a particle interface becomes negligible.
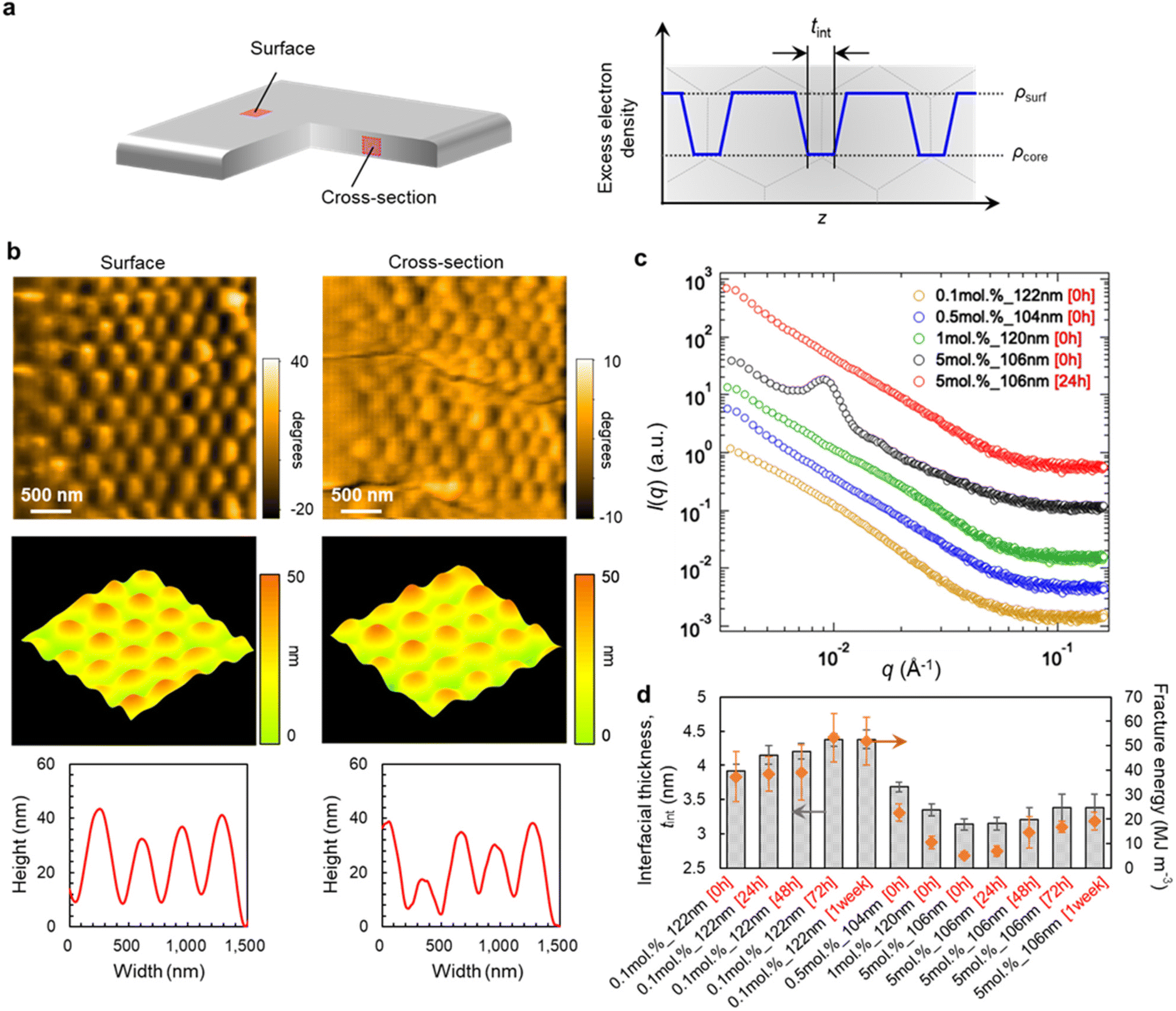 | ||
| Fig. 3 Structural characterization of tough poly-MA-microparticle films. (a) Schematic illustration of a poly-MA-microparticle film. (b) AFM images of films composed of poly-MA microparticles (0.1 mol%_497 nm), phase image, and 3D image reconstructed from the height image, as well as cross-sectional height profile for the surface and cross-section of the film. (c) Small-angle X-ray scattering (SAXS) intensities of a series of poly-MA-microparticle films. The scattering intensities are vertically offset for clarity. (d) Relationship between interfacial thickness (tint) and fracture energy of the poly-MA-microparticle films. The key to realizing mechanically stable microparticle film is the deep interpenetration of an interface between the microparticles. For the annealing process, the films were heated in a convection oven (70 °C; 24 h, 48 h, 72 h, or 1 week). Henceforth, the annealed poly-MA-microparticle films will be denoted as (X mol.%_Y nm [Z]) film, whereby Z represents the annealing time (0 h, 24 h, 48 h, 72 h, or 1 week). | ||
Finally, to improve the properties of the recyclable polymer films, the concept was expanded to mixtures with other nanomaterials (Fig. 4a). Initially, tougher microparticle films were obtained by drying mixtures of poly-MA microparticles and a silica nanofiller (Fig. 4b and Fig. S9†). The resulting films were disassembled into individual microparticles by immersion in a good solvent, i.e., an aqueous solution of 80% (v/v) ethanol, and the resultant mixtures were successfully separated via a short centrifugation into polymer microparticles and silica nanofiller. Thus, toughness (i.e., fracture energy) of the composite poly-MA-microparticle film could be controlled repeatedly with silica nanofiller (Fig. 4b and Fig. S10†). Importantly, such mixing/separation cycles can be continued four times without significant loss of the mechanical properties of the poly-MA-microparticle film (Fig. 4b and Fig. S10†). In addition to the mechanical properties, the optical properties of such poly-MA-microparticle films can be tuned by adding a pigment (Fig. 4c and Fig. S11†). For instance, blue microparticle films were obtained by drying mixtures of poly-MA microparticles and a blue pigment (Fig. 4d and e). The resulting film did not exhibit any color irregularities. Subsequently, the colored films were disassembled into individual microparticles by immersion in a good solvent, and the resultant mixtures were successfully separated via a short centrifugation into polymer microparticles and inorganic pigments (Fig. 4f). Since the inorganic pigments were completely separated from the polymer microparticles as confirmed by the reflection spectra of microparticle films (Fig. 4c), the recycled aqueous suspension of poly-MA microparticles afforded colorless films after drying (Fig. 4g), and the fracture energy of the films was close to that of the original films (Fig. S11b, d†). It is worth noting here that the pigment can be recycled without deterioration of their optical properties (Fig. S11c†). After disassembling the colorless films, differently colored microparticle films were successfully obtained using the recycled poly-MA microparticles and a red pigment (Fig. 4h and i). The color of the microparticle films could subsequently be changed to yellow, with high efficiency and without deterioration of mechanical properties of poly-MA-microparticle films (Fig. 4j, k and Fig. S11†). These investigations regarding the functionalization with inorganic nanoparticles support our concept (Fig. 1) that closed-loop recycling of polymer materials is possible and expandable using polymer microparticles as the key.
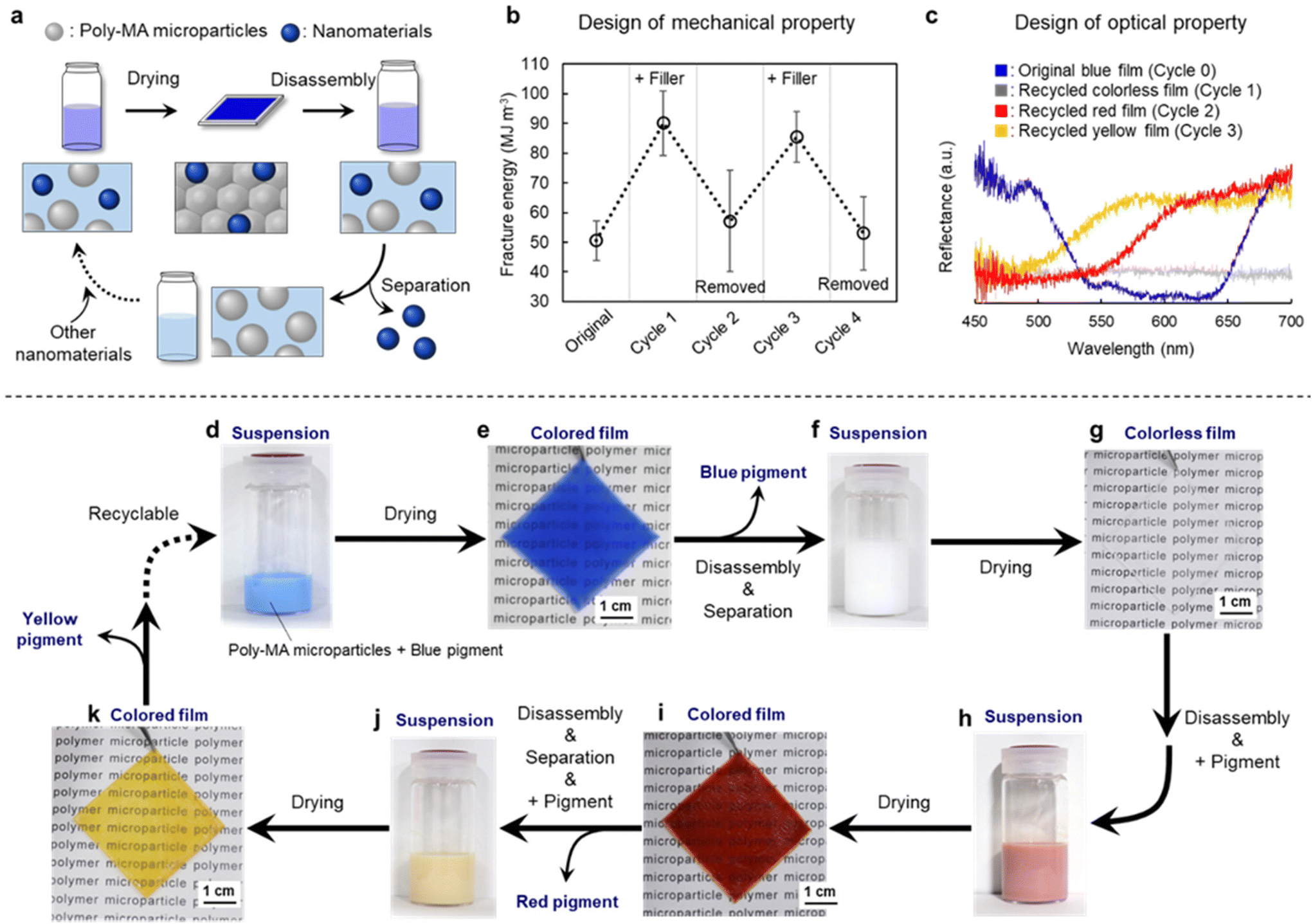 | ||
| Fig. 4 Recycling of poly-MA-microparticle films that contain other nanomaterials. (a) Schematic illustration of closed-loop recycling of poly-MA-microparticle films with other nanomaterials. (b) Fracture energy of poly-MA-microparticle(0.1 mol%_80 nm) films containing silica nanofiller at different number of recycling. (c) The reflection spectra of poly-MA-microparticle(0.1 mol%_122 nm) films with and without inorganic pigments. (d, f, h, j) Photographs of the poly-MA-microparticle(0.1 mol%_122 nm) suspension with and without inorganic pigments. (e, g, i, k) Photographs of colored and pigment-free poly-MA-microparticle(0.1 mol%_122 nm) films after different numbers of recycling processes: (e) cycle 0, (g) cycle 1, (i) cycle 2, and (k) cycle 3. | ||
Conclusion
In summary, polymer microparticles promise outstanding potential as microconstituents of recyclable polymer materials, given that polymer-microparticle films can be readily disassembled into individual microparticles with the aid of a suitable solvent under mild conditions. The key to realizing this concept is the design of an appropriate interface between the microparticles, which results in mechanically stable polymer materials despite their large interface area. This work constitutes only the proof of concept, albeit that we are convinced that it represents a significant step toward a more sustainable human society, since vast amounts of polymer microparticles have already been commercially produced, and these are crucially initially produced via environmentally friendly synthetic methods in water.Author contributions
D. S. conceived the concept of this study. Taku. W., Y. S., H. M., H. S., S. H., and N. M. performed the experiments (recycling of polymer-microparticles and their films). Y. N. and T. U. performed the AFM experiments. T. K. and M. S. performed the SAXS experiments. Taku. W., S. H., and H. M. investigated the scientific background. Taku. W., H. M., and D. S. designed the experiments. Taku. W., and H. M. wrote the original manuscript with input from D. S., T. T., K. S., and Take. W. D. S. reviewed and edited the entire manuscript. D. S. directed, supervised, and managed the entire project. All authors discussed the results and commented on the manuscript.Conflicts of interest
No competing interests to declare.Acknowledgements
This work was supported by a CREST Grant-in-Aid (JPMJCR21L2) from the Japan Science and Technology Agency (JST). The X-ray diffraction analysis and preliminary experiments were performed on the BL03XU beamline at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (Proposal 2022A7221).References
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- D. J. Fortman, J. P. Brutman, G. X. De Hoe, R. L. Snyder, W. R. Dichtel and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2018, 6, 11145–11159 CrossRef CAS.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. Harmelen, P. Wild, G. P. Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed.
- Z. O. G. Schyns and M. P. Shaver, Macromol. Rapid Commun., 2021, 42, 2000415 CrossRef CAS PubMed.
- J. G. Rosenboom, R. Langer and G. Traverso, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef PubMed.
- V. G. Zuin and K. Kümmerer, Nat. Rev. Mater., 2022, 7, 76–78 CrossRef PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- K. Ghosal and C. Nayak, Mater. Adv., 2022, 3, 1974–1992 RSC.
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442–448 CrossRef CAS PubMed.
- M. Häußler, M. Eck, D. Rothauer and S. Mecking, Nature, 2021, 590, 423–427 CrossRef PubMed.
- Z. Huang, M. Shanmugam, Z. Liu, A. Brookfield, E. L. Bennett, R. Guan, D. E. V. Herrera, J. A. Lopez-Sanchez, A. G. Slater, E. J. L. Mclnnes, X. Qi and J. Xiao, J. Am. Chem. Soc., 2022, 144, 6532–6542 CrossRef CAS PubMed.
- W. D. Harkins, J. Am. Chem. Soc., 1947, 69, 1428–1444 CrossRef CAS PubMed.
- W. V. Smith and R. H. Ewart, J. Chem. Phys., 1948, 16, 592–599 CrossRef CAS.
- H. Kawaguchi, Prog. Polym. Sci., 2000, 25, 1171–1210 CrossRef CAS.
- D. Suzuki, K. Horigome, T. Kureha, S. Matsui and T. Watanabe, Polym. J., 2017, 49, 695–702 CrossRef CAS.
- S. Hiroshige, T. Kureha, D. Aoki, J. Sawada, D. Aoki, T. Takata and D. Suzuki, Chem. – Eur. J., 2017, 23, 8405–8408 CrossRef CAS PubMed.
- M. Karg, A. Pich, T. Hellweg, T. Hoare, L. A. Lyon, J. J. Crassous, D. Suzuki, R. A. Gumerov, S. Schneider, I. I. Potemkin and W. Richtering, Langmuir, 2019, 35, 6231–6255 CrossRef CAS PubMed.
- S. Hiroshige, H. Minato, Y. Nishizawa, Y. Sasaki, T. Kureha, M. Shibayama, K. Uenishi, T. Takata and D. Suzuki, Polym. J., 2021, 53, 345–353 CrossRef CAS.
- S. Jiang, A. V. Dyk, A. Maurice, J. Bohling, D. Fasano and S. Brownell, Chem. Soc. Rev., 2017, 46, 3792–3807 RSC.
- D. D. Harrier, P. J. A. Kenis and D. Guironnet, Macromolecules, 2020, 53, 7767–7773 CrossRef CAS.
- M. A. Winnik, J. Coat. Technol., 2002, 74, 49–63 CrossRef.
- E. Limousin, N. Ballard and J. M. Asua, J. Appl. Polym. Sci., 2019, 136, 47608 CrossRef.
- J. L. Haye, I. Martin-Fabiani, M. Schulz, J. L. Keddie, F. D’Agosto and M. Lansalot, Macromolecules, 2017, 50, 9315–9328 CrossRef.
- E. Limousin, N. Ballard and J. M. Asua, Prog. Org. Coat., 2019, 129, 69–76 CrossRef CAS.
- N. Jiménez, N. Ballard and J. M. Asua, Macromolecules, 2019, 52, 9724–9734 CrossRef.
- H. H. Pham and M. A. Winnik, Macromolecules, 2006, 39, 1425–1435 CrossRef CAS.
- H. Bakhshi, M. Z. Zohuriaan-Mehr, H. Bouhendi and K. Kabiri, J. Mater. Sci., 2011, 46, 2771–2777 CrossRef CAS.
- J. Hu, K. Peng, J. Guo, D. Shan, G. B. Kim, Q. Li, E. Gerhard, L. Zhu, W. Tu, W. Lv, M. A. Hickner and J. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 17499–17510 CrossRef CAS PubMed.
- S. Amnuaypornsri, J. Sakdapipanich and Y. Tanaka, J. Appl. Polym. Sci., 2009, 111, 2127–2133 CrossRef CAS.
- M. Sriring, A. Nimpaiboon, S. Kumarn, K. Higaki, Y. Higaki, K. Kojio, A. Takahara, C. C. Ho and J. Sakdapipanich, Colloids Surf., A, 2020, 592, 124571 CrossRef CAS.
- Y. C. Wei, G. X. Liu, L. Zhang, W. Z. Xu, S. Liao and M. C. Luo, ACS Appl. Mater. Interfaces, 2020, 12, 14468–14475 CrossRef CAS PubMed.
- Y. Wu, Y. Wang, X. Wan, C. Gao and Y. Liu, Prog. Org. Coat., 2022, 164, 106705 CrossRef CAS.
- D. Suzuki, T. Kobayashi, R. Yoshida and T. Hirai, Soft Matter, 2012, 8, 11447–11449 RSC.
- H. Jiang, Y. Sheng and T. Ngai, Curr. Opin. Colloid Interface Sci., 2020, 49, 1–15 CrossRef CAS PubMed.
- J. Oberdisse and T. Hellweg, Colloid Polym. Sci., 2020, 298, 921–935 CrossRef CAS.
- A. Scotti, M. F. Schulte, C. G. Lopez, J. J. Crassous, S. Bochenek and W. Richtering, Chem. Rev., 2022, 122, 11675–11700 CrossRef CAS PubMed.
- F. Xu, C. Dawson, M. Lamb, E. Mueller, E. Stefanek, M. Akbari and T. Hoare, Front. Bioeng. Biotechnol., 2022, 10, 849831 CrossRef PubMed.
- T. Kawaguchi, M. Isshiki, M. Takeda and M. Shibayama, Polymer, 2001, 42, 3875–3881 CrossRef.
- G. Mahadevan and S. Valiyaveettil, Sci. Rep., 2021, 11, 1–15 CrossRef PubMed.
- T. Ando, T. Uchihashi and T. Fukuma, Prog. Surf. Sci., 2008, 83, 337–437 CrossRef CAS.
- T. Ando, T. Uchihashi and S. Scheuring, Chem. Rev., 2014, 114, 3120–3188 CrossRef CAS PubMed.
- S. Matsui, T. Kureha, S. Hiroshige, M. Shibata, T. Uchihashi and D. Suzuki, Angew. Chem., Int. Ed., 2017, 56, 12146–12149 CrossRef CAS PubMed.
- Y. Nishizawa, S. Matsui, K. Urayama, T. Kureha, M. Shibayama, T. Uchihashi and D. Suzuki, Angew. Chem., Int. Ed., 2019, 58, 8809–8813 CrossRef CAS PubMed.
- Y. Nishizawa, K. Honda and D. Suzuki, Chem. Lett., 2021, 50, 1226–1235 CrossRef CAS.
- T. Kureha, S. Hiroshige, D. Suzuki, J. Sawada, D. Aoki, T. Takata and M. Shibayama, Langmuir, 2020, 36, 4855–4862 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00090g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |