
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biological conversion of cyclic ketones from catalytic fast pyrolysis with Pseudomonas putida KT2440†
Andrew J.
Borchert‡
 ,
A. Nolan
Wilson
,
William E.
Michener
,
Joseph
Roback§
,
William R.
Henson¶
,
Kelsey J.
Ramirez
and
Gregg T.
Beckham
*
,
A. Nolan
Wilson
,
William E.
Michener
,
Joseph
Roback§
,
William R.
Henson¶
,
Kelsey J.
Ramirez
and
Gregg T.
Beckham
*
Renewable Resources and Enabling Sciences Center, National Renewable Energy Laboratory, Golden, CO, USA. E-mail: gregg.beckham@nrel.gov
First published on 29th March 2023
Abstract
Catalytic fast pyrolysis (CFP) of lignocellulosic biomass is under intense investigation to produce sustainable biofuels. CFP produces a heterogenous bio-oil fraction rich in oxygenated species, including cyclic ketones. While the high oxygen content of CFP bio-oil renders it unsuitable for direct use as transportation fuel replacement or blendstock, many oxygenates can be separated and upgraded to value-added chemicals to offset biofuels’ production cost. In this work, we isolated fractions enriched in cyclic ketones from ex situ CFP bio-oil and metabolically engineered the robust soil bacterium Pseudomonas putida KT2440 to upgrade these ketones to hydroxy and dicarboxylic acids, which have a broad set of industrial applications, especially for use in polymers. P. putida was adapted to higher concentrations of the cyclic ketone substrate, 2-cyclopenten-1-one, to further improve the conversion process. Overall, this work demonstrates the valorization of an important class of compounds found in CFP bio-oil, further expanding the possibilities of producing valuable bioproducts in CFP processes, especially from substrates that are disadvantaged for biofuels production.
Introduction
Over the last several decades, much focus has been placed on the production of fuels, chemicals, and materials from lignocellulosic biomass as a potential route to reduce reliance upon fossil fuels and limit greenhouse gas emissions.1 Among many process options, one of the most well-developed approaches to date for biofuel production is catalytic fast pyrolysis (CFP).2 In particular, ex situ CFP, where the catalyst is applied separately from the pyrolysis reactor, allows separation of the catalyst from contaminants and enables optimization of pyrolysis and catalytic upgrading separately.3 CFP generates bio-oil and an aqueous phase, which are both heterogenous mixtures rich in oxygenated compounds.4 The high oxygen content of bio-oil decreases its heating value and chemical stability, making it unsuitable as a direct transportation fuel replacement or blendstock.5,6 Therefore, bio-oil oxygen content is typically reduced by fractionation and hydrotreating.7,8 However, the costs associated with fractionation and hydrotreating can increase the overall cost of CFP-based biofuel production9A common approach for reducing biofuel production costs is to separate valuable lower-molecular weight coproducts for sale alongside the biofuels.10–13 This practice is common throughout the petroleum refining industry, where 15% of crude oil dedicated to petrochemical production accounts for nearly 50% of the profits.14,15 Additionally, since many valuable petroleum-derived coproducts are produced via oxygen addition to hydrocarbon components, but biomass-derived coproducts already exist as oxygenates, atom efficiency can be improved by obtaining these chemicals from biomass-based sources instead of petroleum-based processes, decreasing the production costs associated with these commodities. Extraction of coproducts shows promise in a CFP context, where a recent techno-economic analysis (TEA), which was informed by bench-scale CFP and hydrotreating data, revealed that the production of valuable coproducts from CFP bio-oil could decrease the minimum fuel selling price of CFP-derived fuels by 12% and reduce greenhouse gas emissions by 39%, relative to a process devoid of co-product production.16 Additionally, distillation approaches have been successfully used to isolate valuable commodities such as olefins, aromatic hydrocarbons, and phenolics from CFP wastewater,12,17 and bio-insecticide fractions rich in alkylated phenols from CFP bio-oil.13 Further, CFP oxygenates that are not themselves industrially useful may also be used as precursors to produce valuable chemicals.18,19 These examples illustrate the potential economic and environmental value in fractioning CFP bio-oil for obtaining value-added chemical fractions able to offset the cost of biofuels produced.
Cyclic ketones, including cyclopentanone (CP), 2-cyclopenten-1-one (2CP), and methylated derivatives represent a promising class of compounds found in CFP bio-oil10,13 that can be used to generate a number of industrially relevant chemicals with applications in the production of bio-degradable polyesters and other important commodities.20–23 Therefore, low-cost separation and valorization of cyclic ketones from CFP bio-oil may represent an additional strategy for offsetting CFP biofuel costs. However, the heterogeneous nature of CFP-derived chemical streams may hinder typical catalytic strategies used for valorization of cyclic ketones.24,25 Generally, the use of engineered microorganisms naturally suited for consumption of heterogeneous feedstocks as biocatalysts for the precise funneling of substrates to desired products has gained in popularity over recent years.26–30 This biocatalytic approach has been previously applied to ex situ CFP aqueous wastewater valorization, where expression of heterologous catabolic pathways and overexpression of native toxicity tolerance machinery in Pseudomonas putida enabled utilization of 89% (w/w) of carbon present and production of (methyl)muconates, which have value as plasticizers.18,19
This work aimed to explore how fractionation of complex chemical streams, such as ex situ CFP bio-oil, could be combined with microbial biocatalysis to produce high purity, value-added chemicals. Specifically, distillation was used to separate a chemical fraction rich in cyclic ketones from CFP bio-oil, and subsequently, P. putida was employed as a biocatalyst to upgrade this cyclic ketone fraction into several value-added chemicals, including hydroxy acids and dicarboxylic acids, which find use in polymers and as plasticizers, among other applications.31,32 Additionally, many complex biomass-derived chemical streams contain toxic compounds that hinder microbial growth, and consequently, engineering stress tolerance against these chemicals can improve biocatalytic outcomes.27,33 Accordingly, this work shows that adaptive laboratory evolution (ALE) targeting improved metabolic stress tolerance represents an important tool for overcoming stress-based biocatalytic bottlenecks. Altogether, this work aims to underscore the value in using distillation of CFP bio-oil to obtain a chemical fraction enriched in compounds inappropriate for use in biofuels, but suitable for bio-catalysis to value-added coproducts, the sale of which could offset biofuels production cost.
Materials and Methods
Isolation of a cyclic ketone-rich stream from CFP bio-oil
2CP rich fractions were generated from bio-oil that was produced using platinum on titanium oxide (Pt/TiO2) to upgrade pyrolysis vapors generated from whole woody pine biomass.34 The resulting bio-oil (∼200 mL) was subjected to three sequential vacuum distillation runs using a spinning band distillation column (model 800-SB-A, BR Instruments) in batch mode. This distillation method was described previously by Wilson and colleagues.12 A Teflon band, simulating 30 theoretical trays, was used for distillation at 30 torr and with a reflux ratio of 10–60. The reflux ratio was controlled by a solenoid needle valve. In this setup, the reflux ratio is the total amount of time, in seconds, for one cycle. A reflux ratio of 10 indicates the valve was open for 1 s and closed for 9 s. All reported temperatures are atmospheric equivalent temperatures (AET) calculated using B/R AET Utility software 1.0. Fractions subject to separation by distillation were taken as follows: fraction 1, 90 °C–110 °C; fraction 2, 110 °C–130 °C; fraction 3, 130 °C–185 °C; fraction 4, 185 °C–230 °C; fraction 5, 230 °C–250 °C; F6, >250 °C.For catalytic hydrogenation, Fraction 3 from the third distillation of CFP bio-oil (∼6.5 g), 10 mL methanol, and 10% Pd/C (∼0.25 mol% Pd relative to 2CP content) were added to a 50 mL three-neck flask equipped with a magnetic stir bar and the headspace was purged using N2 and a H2 balloon was attached. The reaction was stirred at room temperature for 24 h, and the H2 balloon was refilled as needed (approximately every 8 h). After 24 h, the H2 balloon was removed, and the head space was once again purged with N2. The reaction mixture was filtered through a 0.2 μm syringe filter, and the reaction vessel and filter were rinsed with 2 × 10 mL methanol to capture residual product.
Excess methanol from the reduction of 2CP and 2-methyl-2-cyclopenten-1-one (2m-2CP) was removed via rotary evaporation. An aliquot of the post-reduction stream (∼4 g) was placed in a 25 mL pear-shaped vessel and a Buchi R-300 rotary evaporator was operated with a water bath set to 50 °C with 100 RPM and 205 mbar vacuum pressure to deplete the sample of methanol. The gas phase, composed of methanol, was condensed using crushed dry ice and evaporation was allowed to proceed until a drip rate > 1 drop per min was observed. This yielded 1 g of a methanol-reduced product stream. The methanol and cyclic ketone content of the sample prior to and following rotary evaporation was assessed by high-performance liquid chromatography (HPLC), as described below.
Assessing the chemical composition of separated CFP streams
Chemical identification and compositional determination of the distillate fractions were performed on an Agilent Technologies 7890A, 5975c gas chromatography-mass spectrometer (GC-MS), and gas chromatography-flame ionization detector (GC-FID) equipped with a DB-5 column with a Polyarc® microreactor upstream of the FID detector. Since the Polyarc converts all carbon to methane, moles of a species in the sample are proportional to the GC peak area divided by the number of carbons in the compound. All samples were prepared in methanol solvent. Peak integration employed Agilent's Enhanced Chemstation software with the Chemstation Integrator for area determination and NIST library v2.0A 2011 for identification. The resulting areas were analyzed with Python 3.6, using the chemspipy-1.0.5 and pyvalence-0.0.2 libraries.Quantitation of methanol
Quantitation of methanol was conducted using chromatographic conditions utilizing a Aminex HPX-87H column (Bio-Rad Laboratories), as described in Rorrer et al., 202235 with the addition of methanol as an analyte. A minimum of 6 calibration levels were used for quantification with a linear range 0.1 g L−1 to 10 g L−1.Quantitation of (methyl)cyclopentanones and (methyl)glutaric acids
CP, 2CP, 2-methylcyclopentanone (2-mCP), 2m-2CP, 3-methylcyclopentanone (3-mCP), 3-methyl-2-cyclopenten-1-one (3m-2CP), glutaric acid, 2-methylglutaric acid (2-mGA), and 3-methylglutaric acid (3-mGA) quantitation was performed on an Agilent 1290 Infinity II LC system (Agilent Technologies) equipped with a G7117 diode array detector (DAD) and a G7102A evaporative light scattering detector (ELSD). All samples and standards were injected at a volume of 8 μL onto a Phenomenex Luna C18(2)-HST column 100 Å, 25 μm, 2.0 × 100 mm column (Phenomenex). The column temperature was maintained at 45 °C and the buffers used to separate the analytes of interest were 0.16% formic acid in water (A)/acetonitrile (B). The following gradient program was used to separate the analytes of interest: (A) = 100% and (B) = 0% at t = 0; (A) = 100% and (B) = 0% at t = 1 min; (A) = 77.5% and (B) = 22.5% at t = 4 min; (A) = 60% and (B) = 40% at t = 10 min; (A) = 50% and (B) = 50% at t = 11 min; (A) = 100% and (B) = 0% at t = 11.01 min; (A) = 100% and (B) = 0% at t = 13 min. The flow rate was held constant at 0.50 mL min−1 resulting in a total run time of 13 min. Detection and quantification of each analyte was accomplished by using either ELSD or DAD detectors. Glutaric acid, 2-mGA, and 3-mGA analytes were quantitated by ELSD. The ELSD evaporator temperature was set to 45 °C, nebulizer temperature was set to 40 °C, gas flow rate was set to 1.6 standard L min−1 and data were acquired at 10 Hz. CP, 2CP, 2-mCP, 2m-2CP, 3-mCP, 3m-2CP analytes were quantitated by the DAD. A wavelength of 225 nm was used for 2CP, 2m-2CP, and 3m-2CP, and a wavelength of 280 nm was used for CP, 2-mCP, and 3-mCP. Unique standards for each compound were used to establish retention time of each analyte of interest. A minimum of 5 calibration levels were used for each analyte with an r2 ≥ 0.995. A calibration check standard was analyzed every 10 samples to ensure the integrity of the initial calibration. All HPLC quantitation measurement data presented in this work were compiled into ESI File 1.†Quantitation of (methyl)hydroxyvaleric acids, 5-hydroxyhexanoic acid and 5-oxohexanoic acid
5-hydroxyvaleric acid, 2-methyl-5-hydroxyvaleric acid (2m-5HVA), 3-methyl-5-hydroxyvaleric acid (3m-5HVA), 4-methyl-5-hydroxyvaleric acid (4m-5HVA), 5-hydroxyhexanoic acid (5-HHA), and 5-oxohexanoic acid quantitation was performed on an Agilent 1100 HPLC system (Agilent Technologies) equipped with an Agilent 6120 mass spectrometer (MS) detector, a G1362A refractive index detector (RID) and a G1315D DAD. Each sample and standard were injected at a volume of 20 μL on a BioRad Aminex HPX-87H 9 μm, 7.8 × 300 mm column (BioRad). The column and RID temperatures were maintained at 55 °C and the buffer used to separate the analytes of interest was 0.2% formic acid in water (A). Both RID and DAD were used to verify analyte detection for MS quantification. An isocratic program was used to separate the analytes of interest: (A) = 100% for t = 65 min. The flow rate was held constant at 0.60 mL min−1. The MS system was set in negative electrospray ionization mode with a gas temperature of 350 °C, drying gas at 12 L min−1, nebulizer pressure set to 35 psig, and a Vcap voltage of 3000 V. The MS was operated in both SIM and Scan modes. A scan range of 40–200 m/z with a fragmentor voltage of 70 V was used to detect all masses within that range. A total of four different masses in SIM mode from the MS detector were used to identify the analytes of interest. A mass of 131.20 m/z (M − H)− was used for quantitation of 2m-5HVA, 3m-5HVA, 4m-HVA, and 5-HHA, 129.10 m/z (M − H)− was used for 5-oxohexanoic acid, and 117.1 m/z (M − H)− was used for 5-hydroxyvaleric acid. All quantitation measurement data presented in this work were compiled into ESI File 1.†Bacterial strains, plasmids, and primers
Strains used in this work are provided in Table 1 and are all derivatives of the genome reduced Pseudomonas putida EM42 strain, which is derived from P. putida KT2440 (ATCC 47054).36 Gene disruptions and integrations were performed using the non-replicative pK18sB33 plasmid backbone via a selection (nptII, kanamycin)/counterselection (sacB, sucrose) approach, as described by Johnson and Beckham.37 A full description of strain construction details as well as a list of plasmids are provided in ESI Table S1.†| Strains | Genotype | Source |
|---|---|---|
| EM42 | Pseudomonas putida KT2440 Δprophage1 Δprophage4 Δprophage3 Δprophage2 ΔTn7 ΔendA-1 ΔendA-2 ΔhsdRMS Δflagellum ΔTn4652 | 36 |
| RH300 | EM42 PP_5042:Ptac:mekAB:cpnDE | This work |
| AJB87-AJB91 | RH300 isolates following growth in M9 + 10 mM CP | This work |
| AJB137 | EM42 PP_5042:Ptac:mekAB:cpnDE ΔgcdH:gcoT ΔcsiD | This work |
| AJB171-172, 175-176, 179-180 | AJB137 TALE isolates following growth in M9 + 10 mM D-glucose, 5.5 mM 2CP | This work |
| AJB185 | EM42 PP_5042:Ptac:mekAB:cpnDE ΔpaaYX | This work |
| AJB186 | EM42 ΔgcdH:gcoT ΔcsiD PP_5042:Ptac:mekAB:cpnDE ΔpaaYX | This work |
| AJB195 | EM42 ΔgcdH:gcoT ΔcsiD PP_5042:Ptac:mekAB:cpnDE ΔnemA | This work |
| AJB196 | EM42 PP_5042:Ptac:mekAB:cpnDE ΔpaaYX ΔpaaFGHIJKABCDE | This work |
| AJB197 | EM42 PP_5042:Ptac:mekAB:cpnDE ΔpaaYX ΔPP_3270:PP_3273 | This work |
| AJB217 | EM42 PP_5042:Ptac:mekAB:cpnDE ΔgcdH:gcoT ΔcsiD ΔPP_3273:actP-III:phaL:paaZ | This work |
The strain used for plasmid construction and storage was chemically competent NEB 5α F'IqE. coli (New England Biolabs). For gcdH:gcoT, csiD, and paaYX deletions, homology arms used for recombination encoded ∼800 bp upstream of the start codon and ∼800 bp downstream of the stop codon for each deleted gene. Heterologous expression of mekAB and cpnDE used a vector containing ∼800 bp directly upstream and downstream of an intergenic locus downstream of PP_5042.38 Ptac, an optimized RBS, mekAB, and cpnDE were all encoded between these homology fragments. Heterologous genes were codon optimized for P. putida using the online OPTIMIZER tool39 and plasmids were assembled using the New England Biolabs Hifi Gibson Mix. All plasmids were sequence verified by Genewiz®. Gene deletions were confirmed by PCR and correct integration of the PP_5042:Ptac:mekAB:cpnDE locus was confirmed by Sanger sequencing. All oligonucleotide sequences and sequences for Ptac:mekAB:cpnDE constructs used in this work are provided in ESI Tables S2 and S3.† AJB87-AJB91 were obtained when full density populations from two of the RH300 biological replicates grown for 168 h on 10 mM CP were streaked for isolation on LB agar plates and three isolated colonies from one and two from the other population were used to prepare glycerol stocks AJB87-AJB91. AJB171-AJB180 were obtained after AJB137 was subjected to TALE, where cultures were grown to full density in 10 mM D-glucose and 2.5 mM 2CP and sub-cultured into 10 mM D-glucose containing increased 2CP, up to5.5 mM (see method below). The resultant pools were streaked for isolation on LB agar plates and four isolated colonies from each population were used to prepare glycerol stocks AJB171-AJB180, though only two isolates from this study were analyzed further in this work.
Culture media and chemicals
Minimal medium was modified M9 (6.78 g L−1 Na2HPO4, 3.00 g L−1 K2HPO4, 0.50 g L−1 NaCl, 1.66 g L−1 NH4Cl, 0.24 g L−1 MgSO4, 0.01 g L−1 CaCl2, and 0.002 g L−1 FeSO4) supplemented with the indicated carbon source. Rich medium was Luria–Bertani (LB) medium (Lennox). Solid medium was prepared by the addition of 1.50% (wt/vol) agar. When needed, 50 μg mL−1 kanamycin was supplemented into the growth medium. Solid sucrose counterselection medium was 10 g L−1 yeast extract, 20 g L−1 tryptone and 250 g L−1 sucrose (YTS) medium, containing 3.67% (wt/vol) agar. All chemicals for cell culture were purchased from Sigma Aldrich. CP, 2CP, 2-mCP, 2m-2CP, 3-mCP, 3m-2CP, 5-HHA, 5-oxohexanoic acid, glutaric acid, 2-mGA, and 3-mGA were obtained from Sigma Aldrich. 2-mHVA, 3-mHVA, and 4-mHVA were all synthesized and ordered through ChemSpace US Inc.Growth analyses
All growth analyses were performed in biological triplicate, unless otherwise stated. For shake flask experiments, isolated colonies were inoculated into a 100 mm by 13 mm test tube containing 2 mL LB medium and allowed to grow at 30 °C shaking with 225 rpm overnight (∼16 h). Overnight cultures were centrifuged 1 min at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g to obtain cells pellets which were resuspended in 1× M9 salts and used to inoculate 10 mL of M9 minimal medium with the appropriate carbon source in 50 mL baffled shake flasks at an approximate starting optical density at 600 nm (OD600) of 0.2. Cultures were incubated at 30 °C shaking at 225 rpm and growth was periodically assessed as the change in OD600 over time. Samples used for quantification of metabolites were collected by harvesting 1 mL of cell culture and spinning down at 16000g for 2 minutes and supernatant was filtered through a 0.2 μL nylon filter and placed into a 1.5 mL glass HPLC vial for storage at −20 °C. All growth results were plotted using GraphPad Prism 8.4.2 to generate curves represented as the composite of the averages and standard deviation of the replicates. Absorbance measurements (OD600) for all growth data presented in this work were compiled into ESI File 1.†
000g to obtain cells pellets which were resuspended in 1× M9 salts and used to inoculate 10 mL of M9 minimal medium with the appropriate carbon source in 50 mL baffled shake flasks at an approximate starting optical density at 600 nm (OD600) of 0.2. Cultures were incubated at 30 °C shaking at 225 rpm and growth was periodically assessed as the change in OD600 over time. Samples used for quantification of metabolites were collected by harvesting 1 mL of cell culture and spinning down at 16000g for 2 minutes and supernatant was filtered through a 0.2 μL nylon filter and placed into a 1.5 mL glass HPLC vial for storage at −20 °C. All growth results were plotted using GraphPad Prism 8.4.2 to generate curves represented as the composite of the averages and standard deviation of the replicates. Absorbance measurements (OD600) for all growth data presented in this work were compiled into ESI File 1.†
Tolerance adaptive laboratory evolution (TALE)
For tolerance adaptive laboratory evolution against 2CP, three isolates of AJB137 from LB agar plates were inoculated into 100 mm by 13 mm test tubes containing 2 mL LB medium and allowed to grow overnight (∼16 h) at 30 °C with shaking (225 rpm). These cultures were used to inoculate (1:100) 50 mL baffled flasks containing 10 mL of M9 medium supplemented with 10 mM D-glucose and 2.0 mM 2-CP and allowed to grow at 30 °C shaking with 225 rpm. Cultures were observed daily to see whether they had reached full density (OD600–1.5) and if they had, 200 μL of culture was used to inoculate 10 mL fresh M9 medium supplemented with 10 mM D-glucose and a 0.5 mM increase in 2-CP. Following six passages (up to 5.5 mM 2CP in 11 days), the three independent TALE pools were stored as glycerol stocks. These pools were streaked onto LB agar plates, from which four colonies from each TALE pool were isolated and saved. Two of the isolates from each pool were selected for downstream phenotyping and whole genome sequencing.
Whole genome sequencing
Strains used for whole genome sequencing were grown overnight (∼16 h) at 30 °C with shaking (225 rpm) in 100 mm by 13 mm test tubes containing 3 mL of LB medium, and 2 mL cell pellets were collected by centrifugation (16000g, 1 min) before sending to Genewiz® for Illumina library preparation using the Nextera XT DNA Library Preparation platform and HiSeq 2 × 150 bp paired-end sequencing (AJB087-AJB091) or Illumina MiSeq 2 × 150 bp paired-end sequencing (AJB171-AJB180). Coverage for all strains was greater than 30×. Reads for each strain were assembled to derivatives of the P. putida KT2440 reference genome from GenBank (GCA_000007565.2, https://www.ncbi.nlm.nih.gov/assembly/GCA_000007565.2) using Geneious Prime (2020.0.5) with the built-in Geneious mapper set to medium-low sensitivity and 5 iterations. Variants were called inside and outside CDS regions using Geneious Prime software with default settings (minimum coverage: 5 reads, minimum variant frequency: 75%, maximum variant p value: 10−6, minimum strand-bias p value: 10−5 when exceeding 65% bias, using approximate p value calculations). The parental strain, RH300 was also sequenced, and any variants found in this background, relative to the predicted sequence derived from the NC_002947 reference sequence, were accounted for during the identification of mutations arising during growth selection (ESI Table S4†). Any additional mutations arising in the RH300 derivative used for the 2-CP TALE experiment (AJB137) were also identified (ESI Table S5†). Sequencing data (fastq files) were deposited at the NCBI Sequence Read Archive (SRA) and can be assessed under the accession number PRJNA783002.
Results
Separation of cyclic ketone-rich fractions from CFP bio-oil
Bio-oil was produced by CFP and separated into fractions to concentrate specific chemical classes, specifically cyclic ketones (Fig. 1A). A pine wood feedstock was pyrolyzed at 500 °C, and the vapors are then upgraded over a platinum on titanium oxide (Pt/TiO2) catalyst at 400 °C and near atmospheric pressure in a fixed bed reactor in the presence of H2, as described previously.34 Vapors derived from this treatment were then condensed into a bio-oil and subjected to distillation into six fractions using vacuum distillation, as previously described.12 The first five fractions were collected from the top of the distillation column, where the first fraction (5.1 wt% of feed CFP bio-oil) was primarily an aqueous stream, the second fraction (0.9 wt%) was rich in small (C2–C4) aliphatic acids, the third fraction (6.2 wt%) was the target cyclic ketone rich fraction used in this work, the fourth fraction (14.7 wt%) contained many simple phenols (phenols containing up to one <C3 alkyl substitution), and the fifth fraction (5.9 wt%) contained more complex phenols (phenols with more than one alkyl substitution or a single >C3 alkyl substitution) (Fig. 1B). | ||
| Fig. 1 Production of fractions rich in CP and CP derivatives from CFP bio-oil. (A) Process flow diagram showing the distillation approach for the separation of streams rich in mixed cyclic ketones. Percentages denote the wt% (g/g) composition of each fraction, compared to the CFP bio-oil input (ESI Table S6†). (B) wt% composition of chemicals found in each fraction following the first distillation (C) wt% composition of compounds in the third fraction (F3) following the second and third distillations (D2 & D3). The chemical class data for panels B and C are in ESI File 1.† Abbreviations are cyclopentanone (CP), 2-methylcyclopentanone (2-mCP), 3-methylcyclopentanone (3-mCP), 2-cyclopenten-1-one (2CP), 2-methyl-2-cyclopenten-1-one (2m-2CP), 3-methyl-2-cyclopenten-1-one (3m-2CP). Simple phenols are defined as phenols containing up to one <C3 alkyl substitution and complex phenols represent phenols with more than one alkyl substitution or a single >C3 alkyl substitution. | ||
A viscous liquid comprising 64.0 wt% of the bio-oil input remained in the column as the sixth fraction, the properties of which are described elsewhere.13 Minor losses (3.2 wt%) of the bio-oil input material were observed during the bench-scale batch distillation, as expected. Notably, the compounds found in fractions 4 and 5 can be used directly as bioinsecticides or valorized to value-added polymer building blocks.13,19 However, valorization of the compounds found in the third distillate fraction remains relatively unexplored. The cyclic ketone-rich fraction (F3) was subjected to two further rounds of distillation, until an F3 fraction containing 70.2 wt% (g/g) cyclic ketones (83.5 wt% of identified metabolites) was obtained (Fig. 1C and ESI File 1†). The cyclic ketone-rich fraction (F3) obtained from the second and third distillations represented 2.6 wt% and 0.5 wt% of the original CFP bio-oil input material, respectively (ESI Table S6†). 2CP is more toxic than CP, (ESI Fig. S1†), likely owing to the electrophilicity of the enone functional group.40 Therefore, conversion of a chemical stream rich in 2CP potentially requires a more dilute feed than conversion of a stream with an equal amount of CP in a batch process. To preclude 2CP toxicity from interfering with conversion outcomes, chemo-catalytic hydrogenation of the alkenes in the cyclic ketone-rich fraction was conducted to produce their alkane counterparts. Reduction of the 2CP and 2m-2CP found in the cyclic ketone-rich fraction from the third distillation to CP and 2-mCP, respectively, was achieved using a palladium on carbon (Pd/C) catalyst in methanol, as described in the Materials and Methods Section. Consistent with 100% hydrogenation of 2CP by the Pd/C catalyst, no 2CP was observed above the limit of detection (1 ppm) and CP was quantified to 13.4 wt% of the resultant chemical stream (Table 2). Assuming that all 2-mCP in the hydrogenated stream was derived from 2m-2CP, only about 39% of the input 2m-2CP was successfully reduced to 2-mCP (0.11 M 2m-2CP and 0.07 M 2-mCP in the hydrogenated sample). It is possible that the methyl group disrupted the efficiency of alkene bond hydrogenation for 2m-2CP. The methanol introduced during hydrogenation of 2CP and 2m-2CP was removed by rotary evaporation (Table 2).
| Pd/C Hydrogenated fraction | Concentration (M) | Weight% (g/g) |
|---|---|---|
| Methanol | 16.49 | 77.7 |
| 2-Cyclopenten-1-one | 0.00 | 0.0 |
| 2-Methyl-2-cyclopenten-1-one | 0.11 | 1.6 |
| Cyclopentanone | 1.08 | 13.4 |
| 2-Methylcyclopentanone | 0.07 | 1.0 |
| Other | — | 6.3 |
| Methanol-depleted Sample | Concentration (M) | Weight% (g/g) |
|---|---|---|
| Methanol | 5.99 | 23.4 |
| 2-Cyclopenten-1-one | 0.00 | 0.0 |
| 2-Methyl-2-cyclopenten-1-one | 0.55 | 6.4 |
| Cyclopentanone | 4.01 | 41.1 |
| 2-Methylcyclopentanone | 0.30 | 3.6 |
| Other | — | 25.5 |
Engineering cyclopentanone catabolism
As an initial step in the biocatalysis of cyclic ketones, P. putida EM42 was engineered to target CP utilization as a sole carbon and energy source. P. putida EM42 is a genome-reduced derivative of KT2440 (ATCC 47054) and was chosen as a biological chassis owing to its improved growth properties (lag times, growth rates, and biomass yield) and improved tolerance against oxidative stress, as compared to P. putida KT2440.36 CP can be oxidized by Baeyer–Villiger monooxygenases (BVMOs) to generate 5-valerolactone, which can be subsequently ring-opened to produce 5-hydroxyvaleric acid and oxidized to glutaric acid, a viable carbon and energy source for P. putida.41 To this end, an artificial operon encoding mekAB from Pseudomonas veronii MK700, whose products oxidize CP to 5-hydroxyvalerate,42 and cpnDE from Comamonas sp. NCIMB 9872, whose products catabolize 5-hydroxyvalerate to glutaric acid,43 were placed under the control of the constitutive tac promoter (Ptac)44 and chromosomally integrated into P. putida EM42 downstream of the PP_5042 locus, creating RH300 (EM42 PP_5042:Ptac:mekAB:cpnDE) (Fig. 2A). | ||
| Fig. 2 Consumption of cyclic ketones by P. putida RH300. (A) Pathway for conversion of CP to TCA cycle intermediates. Blue enzyme names indicate heterologous genes expressed in P. putida. (B) Growth of EM42 (orange) and RH300 (blue) at 30 °C in M9 medium supplemented with 10 mM CP (squares) or 2.5 mM 2CP (circles) as sole carbon and energy sources. (C) RH300 was grown in M9 medium containing 10 mM D-glucose and supplemented with the reduced and methanol-depleted cyclic ketone rich CFP fraction to an approximate initial CP concentration of ∼10 mM at 30 °C for 72 h. CP, 2-m-2CP, and 2-mCP concentrations were tracked for biological samples as well as an abiotic control. Growth of RH300 was measured using absorbance at 600 nm (OD600). The data represent the mean from three biological replicates (singlet abiotic control) and error bars denote the standard error of the mean. | ||
Expression of mekAB and cpnDE enabled growth using 10 mM CP as a sole carbon and energy source, following an extensive lag phase of ∼120 h (Fig. 2B and ESI File 1†). Interestingly, the addition of 2.5 mM 2CP, the oxidized form of CP, as the sole carbon and energy source did not allow for growth of RH300 within 216 h (Fig. 2B). To assess whether RH300 could successfully consume CP in the CFP-derived cyclic ketone-rich fraction described above, a shake flask experiment was performed using RH300 grown in minimal M9 medium containing 20 mM D-glucose and supplemented with the reduced and methanol-depleted cyclic ketone-rich fraction, diluted to an approximate concentration of 10 mM CP. The concentration of cyclic ketones was tracked over 72 h. RH300 reached a final OD600 of ∼3.0, during which all CP and 2-mCP was consumed by 48 h and all 2-m-2CP was consumed by 72 h (Fig. 2C and ESI File 1†). All residual methanol in this stream was also consumed by 72 h (ESI Fig. S2†).
Importantly, the abiotic control revealed that, owing to their volatility, a fraction of the methanol (24%), CP (13%), 2-mCP (46%), and 2-m-2CP (3%) evaporated over the course of the 72 h experiment. Nonetheless, consumption by RH300, and not evaporation, contributed to most of the observed conversion of these substrates from the growth medium (Fig. 2C and ESI Fig. S2†). Overall, these results demonstrate that RH300, which can catabolize CP, is also capable of consuming CP found in CFP-derived streams. Therefore, the other compounds found in this stream do not appear to prevent growth of P. putida at the concentrations used and do not seem to otherwise disrupt CP consumption. Additionally, these results showed that RH300 could consume the 2-mCP and 2-m-2CP found in this stream, suggesting that, in the presence of glucose, RH300 can at least partially catabolize oxidized and methylated forms of CP. Methanol was likely oxidized to CO2, providing reducing units in the form of NADH.45
Biological conversion of CP and 2CP to glutaric acid
Increasing interest has been placed upon the renewable synthesis of C5 diacids, specifically glutaric acid, which has broad applications as a platform chemical in consumer goods, textiles, and footwear industries.46–48 Therefore, this work aimed to accumulate glutaric acid produced from CP by eliminating the native genes encoding the glutaric acid catabolic pathways (gcdH:gcoT and csiD) from the RH300 background, generating AJB137 (EM42 PP_5042:Ptac:mekAB:cpnDE ΔgcdH:gcoT ΔcsiD) (Fig. 2A). To determine whether this construct could successfully generate glutaric acid from CP, AJB137 was grown in M9 medium containing 10 mM glucose and supplemented with 10 mM CP. Consistent with the disruption of glutaric acid catabolism perturbing utilization CP as a carbon source, AJB137 reached the same final density as EM42 (OD600 < 2.0), while the parental RH300 strain, which can assimilate CP-derived carbon, obtained an OD600 of ∼3.0 (Fig. 3A and ESI File 1†).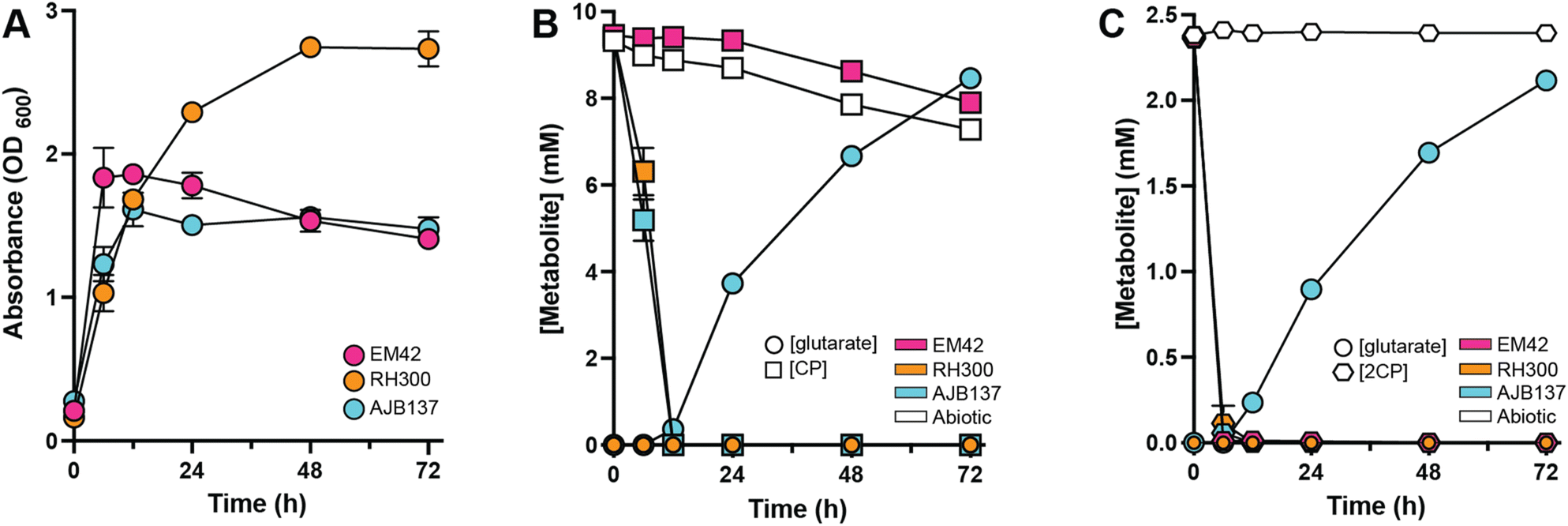 | ||
| Fig. 3 Production of glutaric acid from cyclopentanone and 2-cyclopenten-1-one. (A) Growth of EM42, RH300, and AJB137 at 30 °C in M9 medium supplemented with 10 mM D-glucose and 10 mM CP. (B) CP and glutaric acid concentrations were quantified over the course of the experiment from Fig. 3A for EM42, RH300, AJB137, and an abiotic control. (C) 2CP and glutaric acid concentrations were quantified for EM42, RH300, AJB137, and an abiotic control following during growth in M9 minimal medium containing 10 mM D-glucose and 2.5 mM 2CP. For all experiments, the data represent the mean from three biological replicates and error bars denote the standard error of the means. | ||
Additionally, while RH300 did not accumulate glutaric acid, AJB137 accumulated ∼8.5 mM glutaric acid following 72 h of growth, having consumed all CP by 12 h. Noting that the calculated initial concentration of CP was ∼9.4 mM, this corresponded to >90% glutaric acid yield (Fig. 3B and ESI File 1†). For the abiotic control, 4.7% of the initial CP was lost by 12 h, with ∼22% lost by 72 h, likely due to evaporation. Similar loss of CP was observed for the EM42 cultures, while RH300 consumed CP at the same rate as AJB137, indicating that P. putida did not consume any CP without the presence of the engineered mekAB:cpnDE construct. Despite finding that 2CP alone could not sustain growth of the CP-utilizing strain (RH300) (Fig. 2A), when AJB137 was grown in M9 glucose medium containing 2.5 mM 2CP, glutaric acid production was observed (Fig. 3C and ESI File 1†). The EM42 and RH300 backgrounds both consumed 2CP at the same rate as AJB137, indicating that P. putida can natively reduce 2CP to CP, but neither EM42 nor RH300 could accumulate glutaric acid from 2CP (Fig. 3C). Notably, only 2.5 mM 2CP was added to the growth medium since supplementation of 2CP at or above 5.0 mM hindered growth of AJB137 (ESI Fig. S1†). CP supplementation to 80 mM still permitted growth, indicating that 2CP is significantly more toxic than CP (ESI Fig. S1†).
Biological conversion of methyl-cyclopentanones to methylated 5-hydroxyvaleric acids
Compositional analysis of CFP-derived streams indicated that these streams also contain appreciable amounts of methylated CP and 2CP species (Table 2).10 Given that an RH300-dependent loss of 2-mCP and 2m-2CP was observed during growth in the presence of the cyclic ketone-rich CFP fraction (Fig. 2C), it was possible that the engineered MekAB/CpnDE pathway could allow production of methylated glutaric acid species from 2-mCP and 2m-2CP (Fig. 4A). | ||
| Fig. 4 Production of hydroxy acids from methylated cyclopentanone species. (A) Putative pathways for conversion of 2-mCP by MekAB and CpnDE. (B) Analysis of 2-mCP conversion by AJB137. 2-mCP and 5-hydroxyhexanoic (5-HHA) acid concentrations were quantified for AJB137 and an abiotic control following growth in M9 minimal medium containing 10 mM D-glucose and 10 mM 2-mCP. Note, since the 0 h 2-mCP values were quantitated at 70% of expected concentrations, reported 2-mCP values reflect measured values normalized to a 10 mM starting concentration. Both measured and normalized 2-mCP values are reported in ESI File 1† (C) Putative pathways for conversion of 3-mCP by MekAB and CpnDE. (D) Analysis of 3-mCP conversion by AJB137. 3-mCP, 3-methyl-5-hydroxyvaleric acid (3m-5HVA) and 4-methyl-5-hydroxyvaleric acid (4m-5HVA) concentrations were quantified for AJB137 and an abiotic control following during growth in M9 minimal medium containing 10 mM D-glucose and 10 mM 3-mCP. For all experiments, the data represent the mean from three biological replicates and error bars denote the standard error of the means. | ||
To test this hypothesis, AJB137 was grown in M9 medium containing 10 mM glucose as a carbon source and supplemented with 10 mM 2-mCP. AJB137 consumed all the 2-mCP substrate, but <0.05 mM 2-mGA accumulated in the growth media (ESI File 1†). Almost all 2-mCP substrate was converted to 5-HHA, indicating both that MekA has a strong preference for generating 5-methyl-5-valerolactone over 2-methyl-5-valerolactone, and that CpnD is unable to act upon 5-HHA substrate (Fig. 4B). Abiotic controls indicated that the base media contained minimal amounts of 5-HHA (0.03 mM) and 5-oxohexanoic acid (0.11 mM), likely from impurities in the 2-mCP substrate. Interestingly, over the course of the experiment, all 5-oxohexanoic acid was consumed by AJB137, while levels did not change in the abiotic control (ESI File 1†). Conversely, when AJB137 was grown in medium supplemented with 10 mM 3-mCP, both 3m-5HVA and 4m-5HVA were produced, with a slight preference for 4m-5HVA (Fig. 4D). No 2-mGA and only minimal (0.1 mM) 3-mGA was observed (ESI File 1†).
Overall, the data from 2-mCP and 3-mCP experiments indicate that MekA and MekB can accept methylated cyclopentanones and 5-valerolactones as substrates, respectively. Additionally, these data indicate that when acting upon methylated CP species, MekA prefers to insert the oxygen atom at positions between the carbonyl carbon and closest to the methylated carbon. Therefore, an important consequence of this study is that it expands upon prior work characterizing MekA substrate range against aliphatic, aromatic, and cyclic ketone chemical species.49 The data also indicate that 5-HHA and methylated 5-hydroxyvaleric acid species are poor substrates for CpnD, though, at very low frequency, 2-mCP and 3-mCP can be processed to 2-mGA and 3-mGA, respectively.
Genetic exploration of CP utilization
As stated previously, introduction of mekAB and cpnDE into P. putida EM42 only allowed growth using CP as a sole carbon source following a 120 h lag phase, and the conversion of CP to glutaric acid in AJB137 reached >90% only after 72 h growth (Fig. 2B and 3B). To identify mutants with the potential to improve CP utilization and conversion, individual isolates from a stationary phase culture of RH300 were obtained following growth in M9 minimal medium containing 10 mM CP. When these isolates were again grown using 10 mM CP as a sole carbon source, the lag phase was reduced by ∼50 h, compared to RH300 (ESI Fig. S3†). To explore whether the improved growth in CP containing medium stemmed from mutational changes, five isolates obtained following a single passage on M9 minimal medium containing 10 mM CP were sequenced. All isolates contained mutations predicted to disrupt paaX expression (Fig. 5A). | ||
| Fig. 5 Deletion of paaX influences CP utilization. (A) Table of mutations present at the paaX locus in isolates of RH300 taken following growth in M9 + 10 mM CP as the sole carbon source. (B) Growth of RH300 (EM42 PP_5042:Ptac:mekAB:cpnDE), AJB185 (RH300 ΔpaaYX), AJB196 (RH300 ΔpaaYX ΔpaaFGHIJKABCDE) and AJB197 (RH300 ΔpaaYX ΔPP_3270-PP_3273) at 30 °C in M9 medium supplemented with 10 mM CP as the sole carbon and energy source was measured using absorbance at 600 nm (OD600). The data represent the mean from three biological replicates and error bars denote the standard error of the mean. (C) AJB137 and AJB186 (AJB137 ΔpaaYX) were grown in M9 medium containing 10 mM D-glucose and supplemented with 10 mM CP at 30 °C for 72 h. CP and glutaric acid concentrations were tracked for biological samples and an abiotic control. The data represent the mean from three biological replicates (triplicate abiotic controls) and error bars denote the standard error of the mean. | ||
When a ΔpaaYX mutant of RH300 (AJB185) was grown in M9 containing 10 mM CP, it also exhibited a reduced lag phase (∼50 h), supporting the hypothesis that loss of paaX results in improved growth in CP-containing medium (Fig. 5B and ESI File 1†). PaaX is a transcriptional repressor that regulates expression of two operons involved in phenylacetic acid transport and catabolism.50 The smaller of the two operons, hereafter termed the paaZ operon, encodes a hypothetical membrane protein (PP_3273), an acetate permease (PP_3272, ActP-III), a phenylacetic acid porin (PP_3271, PhaK), and a bifunctional oxepin-CoA hydrolase/3-oxo-5,6-dehydrosuberyl-CoA semialdehyde dehydrogenase (PP_3270, PaaZ), while the larger operon (paaFGHIJKABCDE) encodes many enzymes involved in phenylacetic acid catabolism. Elimination of the paaZ operon from the AJB185 background (AJB197), but not the paaFGHIJKABCDE operon (AJB196), reversed the lag time phenotype contributed by paaYX deletion (Fig. 5B). Indeed, the elimination of the paaZ operon from the ΔpaaYX background led to worse growth outcomes than initially observed for the RH300 strain. These results are consistent with a model where induced expression of one or more of the paaZ operon genes improves the ability for RH300 to catabolize CP. However, since growth was not abolished for AJB197, the paaZ operon is not essential for use of CP as a sole carbon and energy source.
Since P. putida can use glutaric acid as a carbon and energy source,41 it is doubtful that the extended lag time associated with growth on CP is resultant of a metabolic bottleneck following glutaric acid production. Therefore, it is also unlikely that paaX deletion or increased expression of the paaZ operon influences CP catabolism downstream of glutaric acid. Additionally, since the elimination of the paaFGHIJKABCDE catabolic operon did not reverse the ΔpaaYX phenotype, promiscuous catabolism of glutaric acid by the paa system is unlikely. Consistent with this reasoning, while previous work showed that a ΔpaaYX mutant of P. putida has improved catabolism of longer dicarboxylic acids (C6–C10), no change in glutaric acid catabolism was observed.51 It is possible that the phenylacetic acid transport system encoded by the paaZ operon is involved in the promiscuous transport of CP into the cell. Unfortunately, elimination of paaYX from the glutaric acid production strain background, AJB137, did not improve either the rate of CP consumption or glutaric acid production (Fig. 5C and ESI File 1†). Altogether, these data suggest that paaX-mediated control of expression for the paaZ operon is inconsequential when glucose is also provided in the growth medium. This was supported by the observation that AJB217 (AJB137 ΔPP_3270-73) consumed CP and produced glutaric acid at the same rate as AJB137, during growth in medium containing CP and glucose (ESI Fig. S3†). Additionally, work in P. putida U revealed that expression of the paaZ operon is derepressed when a paaX mutant is grown in the presence of glucose,52 indicating that paaZ is still subject to transcriptional repression when glucose is present. Altogether, these results suggest that the paaZ operon may be dispensable for cyclopentanone uptake and catabolism when glucose is used as a carbon and energy source. It is possible that a higher concentration of CP or an experiment with greater sampling frequency might better resolve CP consumption or glutaric acid production discrepancies between AJB137 and AJB186/AJB217, but this was not pursued further for this work. Regardless of these findings, paaX deletion may still be a future strategy for improving CP valorization with different experimental setups or when using CP as a sole carbon source.
Genetic exploration of 2CP tolerance
The glutaric acid production strain, AJB137, also showed a significant sensitivity to 2CP at concentrations above 2.5 mM (ESI Fig. S1†). Tolerance Adaptive Laboratory Evolution (TALE) can be a useful technique for identifying genetic interventions that improve tolerance of P. putida against toxic chemicals found in biomass-derived chemical streams.53,54 Therefore, to identify mutants with increased tolerance to 2CP, a TALE experiment was performed, where AJB137 was subjected to sequential passaging in M9 + 20 mM D-glucose medium supplemented with 2CP increasing from 2.5 mM to 5.5 mM (Fig. 6A). | ||
| Fig. 6 TALE of AJB137 for increased 2CP tolerance. (A) Scheme for the tolerance adaptive laboratory evolution (TALE) experiment adapting AJB137 to increasing amounts of 2CP. AJB137 was grown in triplicate in M9 medium + 10 mM D-glucose and 2.5 mM 2CP at 30 °C and, upon reaching full density, was passaged (1:100) into fresh medium containing an incrementally higher amount (+0.5 mM) of 2CP. This was repeated up to 5.5 mM 2CP (six passages) and two isolates from each replicate were sequenced to identify mutations present. (B) Table of mutations present at the nemA locus in isolates of AJB137 taken following growth in M9 + 10 mM D-glucose and 5.5 mM 2CP. (C) Growth of AJB137 (EM42 PP_5042:Ptac:mekAB:cpnDE ΔgcdH:gcoT ΔcsiD), AJB171 (AJB137 TALE isolate), and AJB195 (AJB137 ΔnemA) at 30 °C in M9 medium supplemented with 10 mM D-glucose and 5 mM 2CP was monitored using absorbance at 600 nm (OD600). The data represent the mean from three biological replicates and error bars denote the standard error of the mean. (D) AJB137 (EM42 PP_5042:Ptac:mekAB:cpnDE ΔgcdH:gcoT ΔcsiD) and AJB195 (EM42 PP_5042:Ptac:mekAB:cpnDE ΔgcdH:gcoT ΔcsiD ΔnemA) were grown in M9 medium containing 10 mM D-glucose and supplemented with 2.5 mM 2CP at 30 °C for 72 h. 2CP and glutaric acid concentrations were tracked for biological samples as well as an abiotic control. The data represent the mean from three biological replicates (triplicate abiotic controls) and error bars denote the standard error of the mean. | ||
Six of the resultant isolates were sequenced to determine the genetic determinants for improved 2CP tolerance. Interestingly, comparison of these isolates against AJB137 revealed that 3/6 isolates contained mutations in their chromosome upstream of the nemA locus and 3/6 harbored mutations in the coding DNA sequence (CDS) of nemA, where two of the CDS mutations (AJB171 and AJB172) led to a predicted frame shift in the nemA gene (Fig. 6B). NemA is one of three native alkene reductases (along with XenA and XenB) and previous work has demonstrated that all three can reduce 2CP to CP.55 When a ΔnemA mutant of AJB137 (AJB195) was grown alongside the isolate from the TALE experiment expressing a truncated NemA (AJB171) and the AJB137 parental strain in M9 containing 10 mM D-glucose and 5 mM 2CP, both AJB195 and AJB171 showed similar growth behavior, reaching full density by 24 h, while AJB137 showed poor growth over the same time period (Fig. 6C and ESI File 1†). These data support the hypothesis that loss of nemA results in improved tolerance to 2CP. Improving 2CP tolerance via elimination of one of the three proteins responsible for 2CP reduction to CP is somewhat counterintuitive. It is possible that the three alkene reductases possess different preferences for NADH vs. NADPH, such that deletion of NemA serves a role in changing the balance of NADH/NADPH pools during 2CP consumption. Engineered increases in NADPH abundance has helped overcome metabolic bottlenecks in other systems.56–59 However, the influence of nemA deletion on NADH/NADPH pools was not investigated further. To see whether nemA deletion disrupted glutaric acid production from 2CP substrate, AJB137 and AJB195 were grown in M9 medium containing 10 mM glucose as a carbon source and supplemented with 2.5 mM 2CP and 2CP consumption and glutaric acid production was tracked over 72 h (Fig. 6D and ESI File 1†). Fortunately, elimination of nemA from the glutaric acid production strain background, AJB137, did not perturb the rate of 2CP consumption or glutaric acid production.
Discussion
Potential cyclic ketone valorization process improvements
While obtaining fractions more enriched in cyclic ketones is important for abiotic valorization approaches, where contaminant chemicals may interfere with processing reactions, biological systems are naturally well-suited for consumption of heterogeneous feedstocks.26 This work focused upon biocatalysis of a CFP bio-oil steam obtained following three distillation steps, but future work may investigate whether P. putida is capable of valorizing more complex streams that have not undergone multiple separation steps. Moreover, another strategy that has been used to separate oxygenated compounds from CFP bio-oil is fractional condensation.60 It is possible that the costs associated with thermal processing of CFP bio-oil could be reduced through incorporation of fractional condensation to target production of a fraction rich in cyclic ketones.The oxidized cyclopentanones present in the CFP-derived cyclic ketone-rich fraction were reduced using catalytic hydrogenation. Since the strains engineered to produce value-added chemicals from CP and its methylated intermediates were able to reduce and catabolize 2CP and its methylated intermediates, it is perhaps possible to improve the economics of bio-oil coproduct valorization through elimination of the catalytic reduction step prior to bioconversion. However, 2CP is significantly more toxic than CP (ESI Fig. S1†), likely owing to the electrophilicity of the enone group.40 Therefore, valorization of a chemical stream rich in 2CP requires significantly more dilution prior to valorization than the same stream where the 2CP is reduced to CP. As a note, P. putida was less sensitive to methylated forms of 2CP, likely owing to the reduced electrophilicity of these species (ESI Fig. S1†).40 This work describes an initial attempt at using TALE to identify mutants that overcome the 2CP toxicity bottleneck. This led to the discovery that a ΔnemA mutant is more tolerant to 2CP, and further investigation revealed that elimination of NemA-dependent alkene reductase activity did not prohibit the production of glutaric acid from 2CP. If it is the case that deletion of nemA serves to change the balance of NADH/NADPH pools during 2CP consumption, targeting rebalancing of these pools may be a strategy for further improving 2CP tolerance. Additionally, continued evolution of AJB195 against 2CP may lead to the isolation of mutants that can tolerate even higher concentrations of the cyclic ketone fraction from CFP bio-oil. This could further reduce the need to dilute chemical streams rich in 2CP and/or subject them to chemo-catalytic reduction prior to bioprocessing, a strategy that could improve process viability.
Diverting glucose for additional product yield
The native L-lysine degradation pathway in P. putida KT2440 catabolizes L-lysine to glutaric acid using the davBATD gene products.61 Additionally, recent work in Corynebacterium glutamicum describes expression of P. putida davBAT to synthesize 4-oxovalerate from L-lysine and introduction of one of several aldehyde reductases, like YahK or YqhD from Escherichia coli, to produce 5-hydroxyvalerate.20 Therefore, introduction of davBATD may represent a reasonable strategy for producing glutaric acid from glucose, by way of L-lysine. Then, subsequent expression of YqhD or another aldehyde reductase in a P. putida strain where glutaric acid production from 4-oxovalerate is blocked (ΔdavD) may allow for production of 5-hydroxyvalerate from glucose. Importantly, prior work has demonstrated glutaric acid overproduction in P. putida when lysine was supplemented into the growth medium, but targeted engineering of metabolic factors controlling lysine biosynthesis, such as overcoming feedback inhibition of lysine biosynthetic pathways, would be required to obtain appreciable amounts of glutaric acid or 5-hydroxyvalerate production from glucose.41 Additionally, E. coli was also used to engineer a biosynthetic route for 3-methyl-5-valerolactone from glucose.62,63 Expression of this pathway in the P. putida strains described in this work may offer a strategy for the production of 3-methyl-5-valerolactone or 3-methyl-5-hydroxyvaleric acid (when mekB is expressed) from the glucose supplemented into the growth medium.Direct production of methylated valerolactone species
Recent work has seen that thermoplastic poly(ester-amide)s (PEAs) prepared from 3-methyl-5-valerolactone display high toughness and ductility, compared to unbranched 5-valerolactone-derived counterparts.64 Polyesters have also been prepared from 2-methyl-5-valerolactone.65,66 Therefore, preparation of methylated 5-valerolactone chemical building blocks from methyl-cyclopentanones found in CFP bio-oil may also be desirable. Dehydration of the methyl-valeric acids produced by AJB137 from 2-mCP and 3-mCP could represent one strategy for producing methylated 5-valerolactone species. While not pursued in this work, another strategy may involve direct production of the methylated 5-valerolactones from 2-mCP and 3-mCP using a P. putida EM42 PP_5042:Ptac:mekA strain.Stability of engineered and adapted strains
When employing engineered or adapted microbial constructs within industrial biocatalytic processes, where seed train propagation or continuous production schemes can result in cultures containing individuals that are several generations removed from the original strain, it is important to consider the evolutionary durability of the desired mutations. Even if industrially advantageous, mutations may be selectively neutral or deleterious to microbial fitness, allowing for their eventual loss during periods of extended propagation. To combat this, several strategies have been employed to increase the genetic stability of various constructs including genomic integration of engineered constructs,67 tight control of gene regulation,68 and entangling engineered constructs with the expression of essential genetic features.69 As it relates to this work, the heterologous constructs employed were integrated into the P. putida chromosome for improving genetic stability. The relative toxicity of 2CP may also act as selective pressure for maintaining at least part of the bio-conversion pathway and the nemA mutation, so long as the feedstock contains 2CP. Further strategies, such as those described above, could be employed to further improve the genetic stability of this strain or its engineered progeny prior to industrial implementation.Conclusions
This work demonstrated an approach combining fractionation of ex situ CFP bio-oil with use of a microbial biocatalyst to upgrade oxygenates unsuitable for direct use in biofuels and found in CFP bio-oil into a suite of industrially relevant chemical coproducts. This approach represents an avenue for valorization of CFP-derived oxygenates as a mechanism to offset the costs associated with biofuel production. Overall, this work has successfully demonstrated (i) a distillation process for the separation of chemical fractions enriched in cyclic ketones away from chemicals better suited for use in biofuels, (ii) the engineering of P. putida for the bioconversion of cyclic ketones into value-added hydroxy acids and dicarboxylic acids, (iii) and the use of genetic screens and adaptive laboratory evolution for the characterization of genetic factors influencing cyclic ketone catabolism and tolerance, respectively. More generally, this work extends beyond CFP bio-oil and cyclic ketones to demonstrate the potential in coupling separation and bioconversion approaches for the renewable generation of valuable and industrially relevant coproducts from chemical streams produced during the manufacture of biofuels or from other heterogeneous chemical waste streams.Author contributions
G. T. B., A. N. W., W. R. H., and A. J. B. conceived the study, A. N. W. and J. R. performed distillations, J. R. performed hydrogenation reactions, A. N. W. analyzed different fraction compositions, A. J. B. and W. R. H. constructed strains, A. J. B. performed biological experiments, W. E. M. and K. J. R. performed analytics following biological valorization, A. J. B. wrote initial manuscript drafts, and all authors reviewed and approved the manuscript.Conflicts of interest
A. J. B., W. R. H., and G. T. B. have filed a patent application on the cyclic ketone bioconversion pathways described in this work.Acknowledgements
This work was authored by the Alliance for Sustainable Energy, LLC, the manager and operator of the National Renewable Energy Laboratory for the U.S. Department of Energy (DOE), under Contract No. DE-AC36-08GO28308, and in collaboration with the Chemical Catalysis for Bioenergy Consortium (ChemCatBio), a member of the Energy Materials Network (EMN). Funding provided by the U.S. DOE Energy Efficiency and Renewable Energy (EERE) Bioenergy Technologies Office (BETO). We thank Calvin Mukarakate, Kristiina Iisa, Scott Palmer, Kellene Orton, and Rick French for preparation of the CFP bio-oil and Lisa Kirchner for assistance with rotary evaporation.References
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- D. A. Ruddy, J. A. Schaidle, J. R. Ferrell III, J. Wang, L. Moens and J. E. Hensley, Green Chem., 2014, 16, 454–490 RSC.
- A. Dutta, A. H. Sahir, E. Tan, D. Humbird, L. J. Snowden-Swan, P. A. Meyer, J. Ross, D. Sexton, R. Yap and J. Lukas, Process design and economics for the conversion of lignocellulosic biomass to hydrocarbon fuels: thermochemical research pathways with in situ and ex situ upgrading of fast pyrolysis vapors, Report PNNL-23823, Pacific Northwest National Laboratory, United States, 2015 Search PubMed.
- S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 18, 590–598 CrossRef CAS.
- R. Doukeh, D. Bombos, M. Bombos, E.-E. Oprescu, G. Dumitrascu, G. Vasilievici and C. Calin, Sci. Rep., 2021, 11, 6176 CrossRef PubMed.
- H. Wang, P. A. Meyer, D. M. Santosa, C. Zhu, M. V. Olarte, S. B. Jones and A. H. Zacher, Catal., 2021, 365, 357–364 CAS.
- A. Dutta, M. K. Iisa, M. Talmadge, C. Mukarakate, M. B. Griffin, E. C. Tan, N. Wilson, M. M. Yung, M. R. Nimlos, J. A. Schaidle, H. Wang, M. Thorson, D. Hartley, J. Klinger and H. Cai, Ex situ catalytic fast pyrolysis of lignocellulosic biomass to hydrocarbon fuels: 2019 state of technology and future research, Report NREL/TP-5100-80291, National Renewable Energy Laboratory, United States, 2020 Search PubMed.
- K. Iisa, D. J. Robichaud, M. J. Watson, J. ten Dam, A. Dutta, C. Mukarakate, S. Kim, M. R. Nimlos and R. M. Baldwin, Green Chem., 2018, 20, 567–582 RSC.
- L. R. Lynd, Nat. Biotechnol., 2017, 35, 912–915 CrossRef CAS PubMed.
- B. A. Black, W. E. Michener, K. J. Ramirez, M. J. Biddy, B. C. Knott, M. W. Jarvis, J. Olstad, O. D. Mante, D. C. Dayton and G. T. Beckham, ACS Sustainable Chem. Eng., 2016, 4, 6815–6827 CrossRef CAS.
- M. J. Biddy, R. Davis, D. Humbird, L. Tao, N. Dowe, M. T. Guarnieri, J. G. Linger, E. M. Karp, D. Salvachúa, D. R. Vardon and G. T. Beckham, ACS Sustainable Chem. Eng., 2016, 4, 3196–3211 CrossRef CAS.
- A. N. Wilson, A. Dutta, B. A. Black, C. Mukarakate, K. Magrini, J. A. Schaidle, W. E. Michener, G. T. Beckham and M. R. Nimlos, Green Chem., 2019, 21, 4217–4230 RSC.
- A. N. Wilson, M. J. Grieshop, J. Roback, S. Dell’Orco, J. Huang, J. A. Perkins, S. Nicholson, D. Chiaramonti, M. Nimlos, E. Christensen, K. Iisa, K. Harris, A. Dutta, J. R. Dorgan and J. A. Schaidle, Green Chem., 2021, 23, 10145–10156 RSC.
- M. J. Biddy, C. Scarlata and C. Kinchin, Chemicals from biomass: A market assessment of bioproducts with near-term potential, Report NREL/TP-5100-65509, National Renewable Energy Laboratory, United States, 2016 Search PubMed.
- R. Rinaldi and F. Schüth, Energy Environ. Sci., 2009, 2, 610–626 RSC.
- A. Dutta, H. Cai, M. S. Talmadge, C. Mukarakate, K. Iisa, H. Wang, D. M. Santosa, L. Ou, D. S. Hartley, A. N. Wilson, J. A. Schaidle and M. B. Griffin, Chem. Eng. J., 2023, 451, 138485 CrossRef CAS.
- A. K. Starace, B. A. Black, D. D. Lee, E. C. Palmiotti, K. A. Orton, W. E. Michener, J. ten Dam, M. J. Watson, G. T. Beckham, K. A. Magrini and C. Mukarakate, ACS Sustainable Chem. Eng., 2017, 5, 11761–11769 CrossRef CAS.
- W. R. Henson, A. W. Meyers, L. N. Jayakody, A. DeCapite, B. A. Black, W. E. Michener, C. W. Johnson and G. T. Beckham, Metab. Eng., 2021, 68, 14–25 CrossRef CAS PubMed.
- W. R. Henson, N. A. Rorrer, A. W. Meyers, H. B. Hoyt, T. A. VanderWall, R. Katahira, J. J. Anderson, B. A. Black, W. E. Michener, L. N. Jayakody, D. Salvachúa, C. W. Johnson and G. T. Beckham, Green Chem., 2022, 24, 3677–3688 RSC.
- Y. J. Sohn, M. Kang, K.-A. Baritugo, J. Son, K. H. Kang, M.-H. Ryu, S. Lee, M. Sohn, Y. J. Jung, K. Park, S. J. Park, J. C. Joo and H. T. Kim, ACS Sustainable Chem. Eng., 2021, 9, 2523–2533 CrossRef CAS.
- K. Duale, M. Zięba, P. Chaber, D. J. Di Fouque, A. Memboeuf, C. Peptu, I. Radecka, M. Kowalczuk and G. Adamus, Molecules, 2018, 23, 2034 CrossRef PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2016, 49, 2419–2428 CrossRef CAS.
- G. M. S. R. O. Rocha, T. M. Santos and C. S. S. Bispo, Catal. Lett., 2011, 141, 100–110 CrossRef CAS.
- G. J. ten Brink, I. W. Arends and R. A. Sheldon, Chem. Rev., 2004, 104, 4105–4124 CrossRef PubMed.
- A. Cavarzan, A. Scarso, P. Sgarbossa, R. Michelin and G. Strukul, ChemCatChem, 2010, 2, 7 CrossRef.
- C. W. Johnson, D. Salvachúa, N. A. Rorrer, B. A. Black, D. R. Vardon, P. C. St. John, N. S. Cleveland, G. Dominick, J. R. Elmore, N. Grundl, P. Khanna, C. R. Martinez, W. E. Michener, D. J. Peterson, K. J. Ramirez, P. Singh, T. A. VanderWall, A. N. Wilson, X. Yi, M. J. Biddy, Y. J. Bomble, A. M. Guss and G. T. Beckham, Joule, 2019, 3, 1523–1537 CrossRef CAS.
- A. J. Borchert, W. R. Henson and G. T. Beckham, Curr. Opin. Biotechnol., 2022, 73, 1–13 CrossRef CAS PubMed.
- L. T. Cordova, B. C. Lad, S. A. Ali, A. J. Schmidt, J. M. Billing, K. Pomraning, B. Hofstad, M. S. Swita, J. R. Collett and H. S. Alper, Bioresour. Technol., 2020, 313, 123639 CrossRef CAS PubMed.
- J. Lange, F. Müller, K. Bernecker, N. Dahmen, R. Takors and B. Blombach, Biotechnol. Biofuels, 2017, 10, 277 CrossRef PubMed.
- J. G. Linger, D. R. Vardon, M. T. Guarnieri, E. M. Karp, G. B. Hunsinger, M. A. Franden, C. W. Johnson, G. Chupka, T. J. Strathmann, P. T. Pienkos and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12013–12018 CrossRef CAS PubMed.
- R. M. Cywar, N. A. Rorrer, C. B. Hoyt, G. T. Beckham and E. Y. X. Chen, Nat. Rev. Mat., 2022, 7, 83–103 CrossRef CAS.
- C. M. Rohles, L. Gläser, M. Kohlstedt, G. Gießelmann, S. Pearson, A. del Campo, J. Becker and C. Wittmann, Green Chem., 2018, 20, 4662–4674 RSC.
- L. N. Jayakody, C. W. Johnson, J. M. Whitham, R. J. Giannone, B. A. Black, N. S. Cleveland, D. M. Klingeman, W. E. Michener, J. L. Olstad, D. R. Vardon, R. C. Brown, S. D. Brown, R. L. Hettich, A. M. Guss and G. T. Beckham, Energy Environ. Sci., 2018, 11, 1625–1638 RSC.
- M. B. Griffin, K. Iisa, H. Wang, A. Dutta, K. A. Orton, R. J. French, D. M. Santosa, N. Wilson, E. Christensen, C. Nash, K. M. Van Allsburg, F. G. Baddour, D. A. Ruddy, E. C. D. Tan, H. Cai, C. Mukarakate and J. A. Schaidle, Energy Environ. Sci., 2018, 11, 2904–2918 RSC.
- N. A. Rorrer, S. F. Notonier, B. C. Knott, B. A. Black, A. Singh, S. R. Nicholson, C. P. Kinchin, G. P. Schmidt, A. C. Carpenter, K. J. Ramirez, C. W. Johnson, D. Salvachúa, M. F. Crowley and G. T. Beckham, Cell Rep. Phys. Sci., 2022, 3, 100840 CrossRef CAS.
- E. Martinez-Garcia, P. I. Nikel, T. Aparicio and V. de Lorenzo, Microb. Cell Fact., 2014, 13, 159 CrossRef PubMed.
- C. W. Johnson and G. T. Beckham, Metab. Eng., 2015, 28, 240–247 CrossRef CAS PubMed.
- J. E. Chaves, R. Wilton, Y. Gao, N. M. Munoz, M. C. Burnet, Z. Schmitz, J. Rowan, L. H. Burdick, J. Elmore, A. Guss, D. Close, J. K. Magnuson, K. E. Burnum-Johnson and J. K. Michener, Metab. Eng. Commun., 2020, 11, e00139 CrossRef PubMed.
- P. Puigbo, E. Guzman, A. Romeu and S. Garcia-Vallve, Nucleic Acids Res., 2007, 35, W126–W131 CrossRef PubMed.
- R. J. Mayer, P. W. A. Allihn, N. Hampel, P. Mayer, S. A. Sieber and A. R. Ofial, Chem. Sci., 2021, 12, 4850–4865 RSC.
- M. Zhang, C. Gao, X. Guo, S. Guo, Z. Kang, D. Xiao, J. Yan, F. Tao, W. Zhang, W. Dong, P. Liu, C. Yang, C. Ma and P. Xu, Nat. Commun., 2018, 9, 2114 CrossRef PubMed.
- C. Onaca, M. Kieninger, K. H. Engesser and J. Altenbuchner, J. Bacteriol., 2007, 189, 3759–3767 CrossRef CAS PubMed.
- H. Iwaki, Y. Hasegawa, S. Wang, M. M. Kayser and P. C. Lau, Appl. Environ. Microbiol., 2002, 68, 5671–5684 CrossRef CAS PubMed.
- H. A. de Boer, L. J. Comstock and M. Vasser, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 21–25 CrossRef CAS PubMed.
- S. B. Hintermayer and D. Weuster-Botz, Biotechnol. J., 2017, 12, 1600720 CrossRef PubMed.
- M. Zhao, G. Li and Y. Deng, Appl. Environ. Microbiol., 2018, 84, e00814–18 CrossRef CAS PubMed.
- G. Li, D. Huang, X. Sui, S. Li, B. Huang, X. Zhang, H. Wu and Y. Deng, React. Chem. Eng., 2020, 5, 221–238 RSC.
- T. Han, G. B. Kim and S. Y. Lee, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 30328–30334 CrossRef CAS PubMed.
- A. Volker, A. Kirschner, U. T. Bornscheuer and J. Altenbuchner, Appl. Microbiol. Biotechnol., 2008, 77, 1251–1260 CrossRef PubMed.
- C. Fernández, E. Díaz and J. L. García, Environ. Microbiol. Rep., 2014, 6, 239–250 CrossRef PubMed.
- Y. S. Ackermann, W.-J. Li, L. Op de Hipt, P.-J. Niehoff, W. Casey, T. Polen, S. Köbbing, H. Ballerstedt, B. Wynands, K. O'Connor, L. M. Blank and N. Wierckx, Metab. Eng., 2021, 67, 29–40 CrossRef CAS PubMed.
- E. R. Olivera, B. Minambres, B. Garcia, C. Muniz, M. A. Moreno, A. Ferrandez, E. Diaz, J. L. Garcia and J. M. Luengo, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6419–6424 CrossRef CAS PubMed.
- E. T. Mohamed, A. Z. Werner, D. Salvachúa, C. A. Singer, K. Szostkiewicz, M. R. Jiménez-Díaz, T. Eng, M. S. Radi, B. A. Simmons, A. Mukhopadhyay, M. J. Herrgård, S. W. Singer, G. T. Beckham and A. M. Feist, Metab. Eng. Commun., 2020, 11, e00143 CrossRef PubMed.
- H. G. Lim, B. Fong, G. Alarcon, H. D. Magurudeniya, T. Eng, R. Szubin, C. A. Olson, B. O. Palsson, J. M. Gladden, B. A. Simmons, A. Mukhopadhyay, S. W. Singer and A. M. Feist, Green Chem., 2020, 22, 5677–5690 RSC.
- C. Peters, R. Kölzsch, M. Kadow, L. Skalden, F. Rudroff, M. D. Mihovilovic and U. T. Bornscheuer, ChemCatChem, 2014, 6, 1021–1027 CrossRef CAS.
- E. Kuatsjah, C. W. Johnson, D. Salvachúa, A. Z. Werner, M. Zahn, C. J. Szostkiewicz, C. A. Singer, G. Dominick, I. Okekeogbu, S. J. Haugen, S. P. Woodworth, K. J. Ramirez, R. J. Giannone, R. L. Hettich, J. E. McGeehan and G. T. Beckham, Metab. Eng., 2022, 70, 31–42 CrossRef CAS PubMed.
- A. M. Sanchez, J. Andrews, I. Hussein, G. N. Bennett and K. Y. San, Biotechnol. Prog., 2006, 22, 420–425 CrossRef CAS PubMed.
- A. Kabus, T. Georgi, V. F. Wendisch and M. Bott, Appl. Microbiol. Biotechnol., 2007, 75, 47–53 CrossRef CAS PubMed.
- H. C. Lee, J. S. Kim, W. Jang and S. Y. Kim, J. Biotechnol., 2010, 149, 24–32 CrossRef CAS PubMed.
- B. Peterson, C. Engtrakul, A. N. Wilson, S. Dell'Orco, K. A. Orton, S. Deutch, M. M. Yung, A. K. Starace, Y. Parent, D. Chiaramonti and K. A. Magrini, ACS Sustainable Chem. Eng., 2019, 7, 14941–14952 CrossRef CAS.
- O. Revelles, M. Espinosa-Urgel, T. Fuhrer, U. Sauer and J. L. Ramos, J. Bacteriol., 2005, 187, 7500–7510 CrossRef CAS PubMed.
- M. Xiong, D. K. Schneiderman, F. S. Bates, M. A. Hillmyer and K. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8357–8362 CrossRef CAS PubMed.
- C. Zhang, D. K. Schneiderman, T. Cai, Y.-S. Tai, K. Fox and K. Zhang, ACS Sustainable Chem. Eng., 2016, 4, 4396–4402 CrossRef CAS.
- D. M. Guptill, B. S. Chinta, T. Kaicharla, S. Xu and T. R. Hoye, Polym. Chem., 2021, 12, 1310–1316 RSC.
- H. Kikuchi, H. Uyama and S. Kobayashi, Polym. J., 2002, 34, 835–840 CrossRef CAS.
- K. Küllmer, H. Kikuchi, H. Uyama and S. Kobayashi, Macromol. Rapid Commun., 1998, 19, 127–130 Search PubMed.
- C. N. Santos, D. D. Regitsky and Y. Yoshikuni, Nat. Commun., 2013, 4, 2503 CrossRef PubMed.
- S. Yang, S. C. Sleight and H. M. Sauro, Nucleic Acids Res., 2013, 41, e33 CrossRef CAS PubMed.
- T. Blazejewski, H. I. Ho and H. H. Wang, Science, 2019, 365, 595–598 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00084b |
| ‡ Current address: Crysalis Biosciences, Golden, CO, USA. |
| § Current address: Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO, USA. |
| ¶ Current address: Battelle, Columbus, OH, USA. |
| This journal is © The Royal Society of Chemistry 2023 |