
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recycling of a thermoresponsive “catalyst pill”: separation of a molecular catalyst in solid ethylene carbonate in various reactions†
Jeroen T.
Vossen
ab,
Noah
Hülsken
b,
Andreas J.
Vorholt
 *a and
Walter
Leitner
ab
*a and
Walter
Leitner
ab
aMax Planck Institute for Chemical Energy Conversion, Stiftstraße 34-36, 45470 Mülheim an der Ruhr, Germany. E-mail: andreas-j.vorholt@cec.mpg.de
bInstitute for Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany
First published on 9th March 2023
Abstract
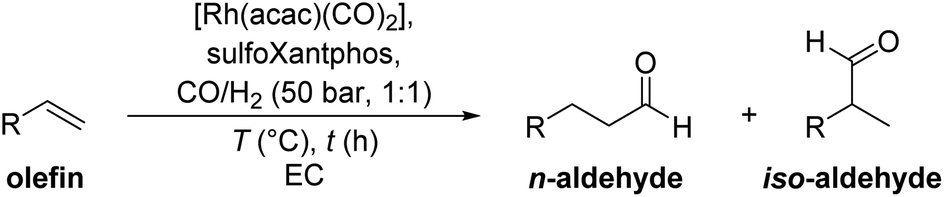
One approach to recycle homogeneous catalysts is through multiphase catalysis. Multiphase catalysis is not only limited to liquid–liquid multiphase systems but also includes for example solid and liquid phases. In this work, we present a catalyst recycling system based on the crystallization of the entire catalyst phase after the reaction at ambient temperature. Using the green and polar solvent ethylene carbonate, the polar Rh/sulfoXantphos catalyst is trapped in the crystallized ethylene carbonate phase. The product can be decanted under air as the catalyst is stabilized in the solid phase and the entire solid phase including the solvent is recycled. Several reactions such as the hydroformylation of hexene, octene and decene, and the hydrogenation of C14 aldol products were conducted with this system. A TTON of 8627 could be achieved in the hydroformylation of 1-octene with initial turnover frequencies up to 1460 h−1. In addition, the catalyst “pill” was switched between different reactions to show the flexibility of the system.
1 Introduction
Catalysts are key agents for realising sustainable and efficient processes; however, they need to be removed from the products after the reaction due to possible interference in further processes and consumer safety in the final product.1,2 Catalyst recycling is one of the major challenges when using homogeneous catalysis.3 Methods to remove and recycle catalysts from the reaction solution are for example precipitation, distillation, extraction and membrane filtration. However, these methods may not be suitable for continuous processes or could result in decomposition of the catalyst.3,4 Multiphase catalysis in contrast relies on in situ extraction of the reaction product from the separate catalyst phase. These systems usually consist of two or more immiscible solvents or a self-separating product phase.5A different method to recover the catalyst is based on crystallization, such as crystallization of the products after the reaction.6–10 An example of industrial application is the crystallization of the pharmaceutical L-DOPA which is selectively crystallized from the racemic mixture after the reaction.11,12 Another method is the crystallization or precipitation of the catalyst from the reaction mixture to recycle the catalyst.13–16 Using ionic liquid modified polyoxometalate catalysts (POMs), systems with a self-separation of the catalyst have been developed by multiple groups. These systems are based on a change of the phase behaviour during the reaction and by the consumption/formation of substrates and products.17,18
A novel method which has rarely been used for recycling to the best of our knowledge is the crystallization of the entire catalyst phase in a multiphase system. This thermoresponsive method allows for easy separation of the catalyst phase (including the catalyst phase solvent) by decanting the product phase. In addition, the trapping in the solid phase improves the stability of otherwise air sensitive catalysts. Suitable solvents for this methodology are for example higher molecular weight polyethylene glycols (PEG) such as PEG-1000 and higher. They are viscous liquids under reaction conditions at higher temperatures and turn highly viscous or solid when cooled to room temperature, trapping and stabilizing the catalyst.19–23 Extending the idea of using oligomers and polymers as solvents even further, the capture of a palladium catalyst in a solidified polyethylene phase after the substitution and ring-closing metathesis reaction has been reported. The catalyst could be easily separated and handled as a solid after cooling the reaction mixture.20 Another group of solvents, which may have a melting point above room temperature, is ionic liquids (ILs). Their use as solvents for the catalyst phase and solidification when cooling the mixture after the reaction was reported by Andersen et al. in 1998 in the hydroformylation of hexene.24 Leitner et al. influenced the melting point of ILs using super critical CO2 as a solvent in the hydroformylation of 2-vinyl-naphthalene. In the presence of super critical CO2, the melting point of the IL is decreased and it turns solid again after relieving the pressure from the reactor, resulting in the trapping of the catalyst in the solid IL.25 In 2012, Yang et al. used a Rh/TPPTS catalyst in guanidinium-based ILs to capture the catalyst after the reaction, showing its long-term recyclability in the hydroformylation of long chain olefins from C8 to C14. The TOF of this system is rather low, requiring long reaction times for high conversions.26 A different system developed by Jun et al. relies on the self-assembly through hydrogen bonding of the catalyst complex and a pyridinium-based linker. Upon cooling, the linker and the catalyst complex assemble and form a solid catalyst phase which can easily be recycled.27
A class of solvents which can be used for the catalyst phase are (cyclic) carbonates and specifically ethylene carbonate (EC). Currently, EC is mainly used as a battery electrolyte and has found few applications in chemical reactions.28,29 It is a green and polar solvent which is a solid at ambient temperature with a melting point of 36 °C and a high boiling point of 248 °C. In molten form, it is a low viscosity liquid in comparison with PEG and ILs and dissolves polar ligands and catalysts well.28,30,31 The solid state of EC at ambient temperature will be used as a feature for catalyst recycling through crystallization of the entire EC-based catalyst phase (Fig. 1). This solid catalyst phase or “catalyst pill” is easy to handle, store and recycle in different reactions. The system and the long-term stability of the catalyst will be studied in recycling experiments in different reactions.
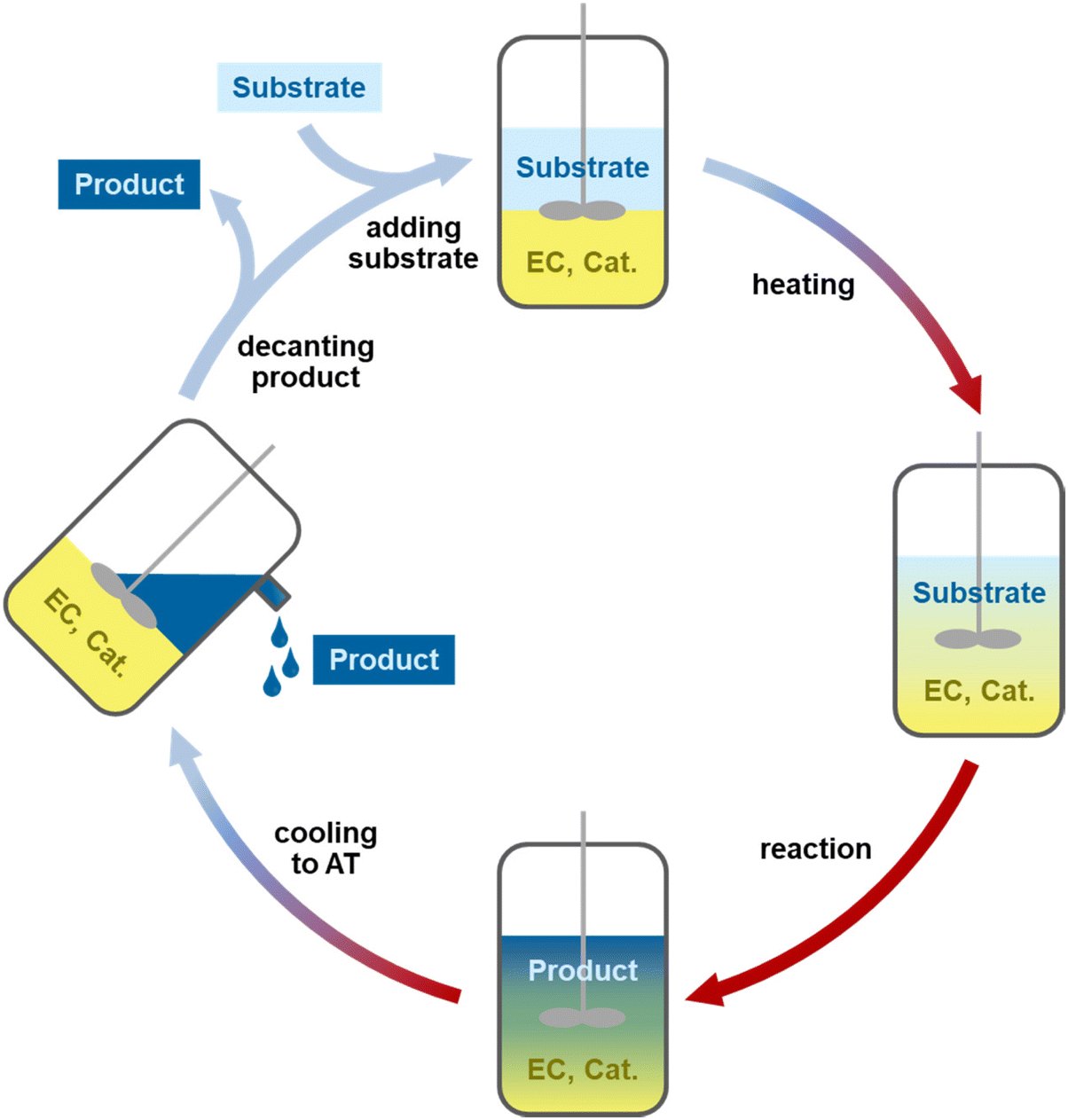 | ||
| Fig. 1 Graphical representation of the recycling process, starting with a solid catalyst phase and substrate, which turns liquid upon heating and reacts with the product. After cooling the reactor to ambient temperature (AT), the catalyst phase crystallizes, the product phase is decanted and the fresh substrate can be resupplied. | ||
2 Results and discussion
The hydroformylation of 1-octene was the first reaction studied using the catalyst “pill” reaction and the recycling strategy with ethylene carbonate (EC). When EC is liquified at 40 °C, it dissolves the rhodium and sulfoXantphos-based catalyst well and forms a biphasic mixture with octene and nonanal (Fig. 2). After the reaction, the liquid, colourless organic phase was decanted from the bottom, yellow catalyst phase under a normal atmosphere and could be analysed. After initial screening experiments, variation of the reaction temperature showed a decent activity even at a low temperature of 80 °C with a maximum aldehyde yield at 120 °C of 80% (Table 1, entries 1–4). The conversion and yield are significantly lower at 100 °C and 110 °C due to the lower reaction rate. At 130 °C, the conversion and aldehyde yield are similar to those obtained at 120 °C. The entire temperature screening can be found in the ESI (ESI, Fig. S1†). Increasing the reaction time to obtain higher yields even at 110 °C resulted in nearly full conversions but the aldehyde yields remained around 70% (entries 5 and 6). The remaining substrate was converted to octane and to internal olefins via double bond isomerization with a ratio of approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The hydrogenation of nonanal to nonanol was only observed in traces. Decreasing the catalyst concentration still resulted in a decent reaction rate; however, the n:iso ratio decreased with lower catalyst concentrations due to the decrease in the absolute ligand concentration in the catalyst phase and due to the relatively low Rh-to-P ratio of 1:4 (entry 7). Doubling the catalyst concentration only showed little improvement over 0.1 mol% of the catalyst except for a slight increase in regioselectivity (entry 8). The full catalyst concentration screening can be found in the ESI (Fig. S2†). Small catalyst concentrations of 0.007 mol% could be used, reaching a turnover number (TON, moloct molcat) of 9770 in batch experiments (entry 9). The catalyst-to-organic phase ratio of 1:2 was chosen based on the literature results of aqueous hydroformylation systems.32 A variation of the ratio showed lower yields with smaller amounts of ethylene carbonate which is a result of the increasing absolute ligand concentration in EC inhibiting the rhodium catalyst (ESI, Fig. S3†). A larger phase ratio did not provide any improvements. In comparison with reactions with an aqueous catalyst phase conducted under the same reaction conditions, the system with EC showed a much higher turnover frequency (TOF, moloct molcat−1 h−1) with an initial turnover frequency (TOFini) of approximately 1460 h−1 at 110 °C. Throughout the reactions, a high selectivity was maintained with n:iso ratios usually in the range of 97:3.
:1. The hydrogenation of nonanal to nonanol was only observed in traces. Decreasing the catalyst concentration still resulted in a decent reaction rate; however, the n:iso ratio decreased with lower catalyst concentrations due to the decrease in the absolute ligand concentration in the catalyst phase and due to the relatively low Rh-to-P ratio of 1:4 (entry 7). Doubling the catalyst concentration only showed little improvement over 0.1 mol% of the catalyst except for a slight increase in regioselectivity (entry 8). The full catalyst concentration screening can be found in the ESI (Fig. S2†). Small catalyst concentrations of 0.007 mol% could be used, reaching a turnover number (TON, moloct molcat) of 9770 in batch experiments (entry 9). The catalyst-to-organic phase ratio of 1:2 was chosen based on the literature results of aqueous hydroformylation systems.32 A variation of the ratio showed lower yields with smaller amounts of ethylene carbonate which is a result of the increasing absolute ligand concentration in EC inhibiting the rhodium catalyst (ESI, Fig. S3†). A larger phase ratio did not provide any improvements. In comparison with reactions with an aqueous catalyst phase conducted under the same reaction conditions, the system with EC showed a much higher turnover frequency (TOF, moloct molcat−1 h−1) with an initial turnover frequency (TOFini) of approximately 1460 h−1 at 110 °C. Throughout the reactions, a high selectivity was maintained with n:iso ratios usually in the range of 97:3.
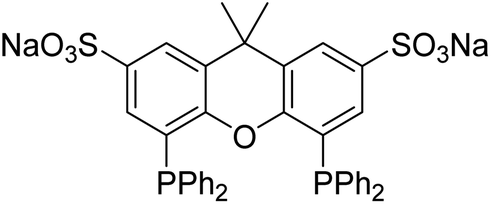 | ||
| Fig. 2 Polar sulfoXantphos ligand (due to the sodium sulfonate groups) used in these investigations. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | T (°C) | t (h) | c Rh (mol%) | X 1-octene (%) | Y aldehyde (%) |
n:iso |
| Conditions: 1-octene (2.0 mL, with 5 vol% mesitylene as the internal standard) in EC (1.0 mL), [Rh(acac)(CO)2]/sulfoXantphos catalyst (mol% relative to 1-octene, Rh:P = 1:4) at various temperatures, 50 bar CO/H2 (1:1) in 20 mL autoclave reactors and 1000 rpm stirring speed for various reaction times. |
||||||
| 1 | 100 | 2 | 0.1 | 43 | 33 | 94:6 |
| 2 | 110 | 2 | 0.1 | 71 | 62 | 97:3 |
| 3 | 120 | 2 | 0.1 | 90 | 80 | 97:3 |
| 4 | 130 | 2 | 0.1 | 89 | 76 | 97:3 |
| 5 | 110 | 4 | 0.1 | 92 | 71 | 96:4 |
| 6 | 110 | 16 | 0.1 | 99 | 72 | 94:6 |
| 7 | 110 | 2 | 0.05 | 55 | 37 | 92:8 |
| 8 | 110 | 2 | 0.2 | 73 | 68 | 98:2 |
| 9 | 110 | 16 | 0.007 | 71 | 57 | 97:3 |
Based on these results, 0.1 mol% Rh/sulfoXantphos at 120 °C for a reaction time of 2 h was used for the recycling experiments; as not all substrates were fully converted, a potential catalyst deactivation or leaching could still be observed. The melting point of 36 °C and fast crystallization of EC allowed for a method in which the catalyst is captured in the crystallized EC phase after the reaction (Fig. 1). The organic solution was removed under a normal atmosphere by decanting or using a syringe without a significant effect on the catalyst. The solid catalyst phase was handled under air for about an hour during work up without showing any change in colour. 12 runs were conducted using this system without a significant drop in the yield (Fig. 3). During the recycling experiments, a total of 0.54% of the starting amount of rhodium and 0.10% of phosphorus leached into the organic phase (detailed leaching per run can be found in the ESI Table S1†). Besides the catalyst, ethylene carbonate also leached into the organic phase due to its solubility in nonanal. The amount of leached EC per run increased with the increase of the aldehyde yield and was determined by GC FID. After the next run, the leached EC was resupplied to avoid a diminishing catalyst phase, which would cause a decrease in the reaction rate. A total of 0.79 g of EC leached across all runs (60% of the initial amount and 39% of the total amount including resupply). The EC leaching into the organic phase can be reduced if the mixture is crystallized for a longer time period.10 During the recycling, a total TON of 8627 was achieved with the results comparable to the batch experiments of the hydroformylation of 1-octene under the same conditions (Table 1, entry 3).
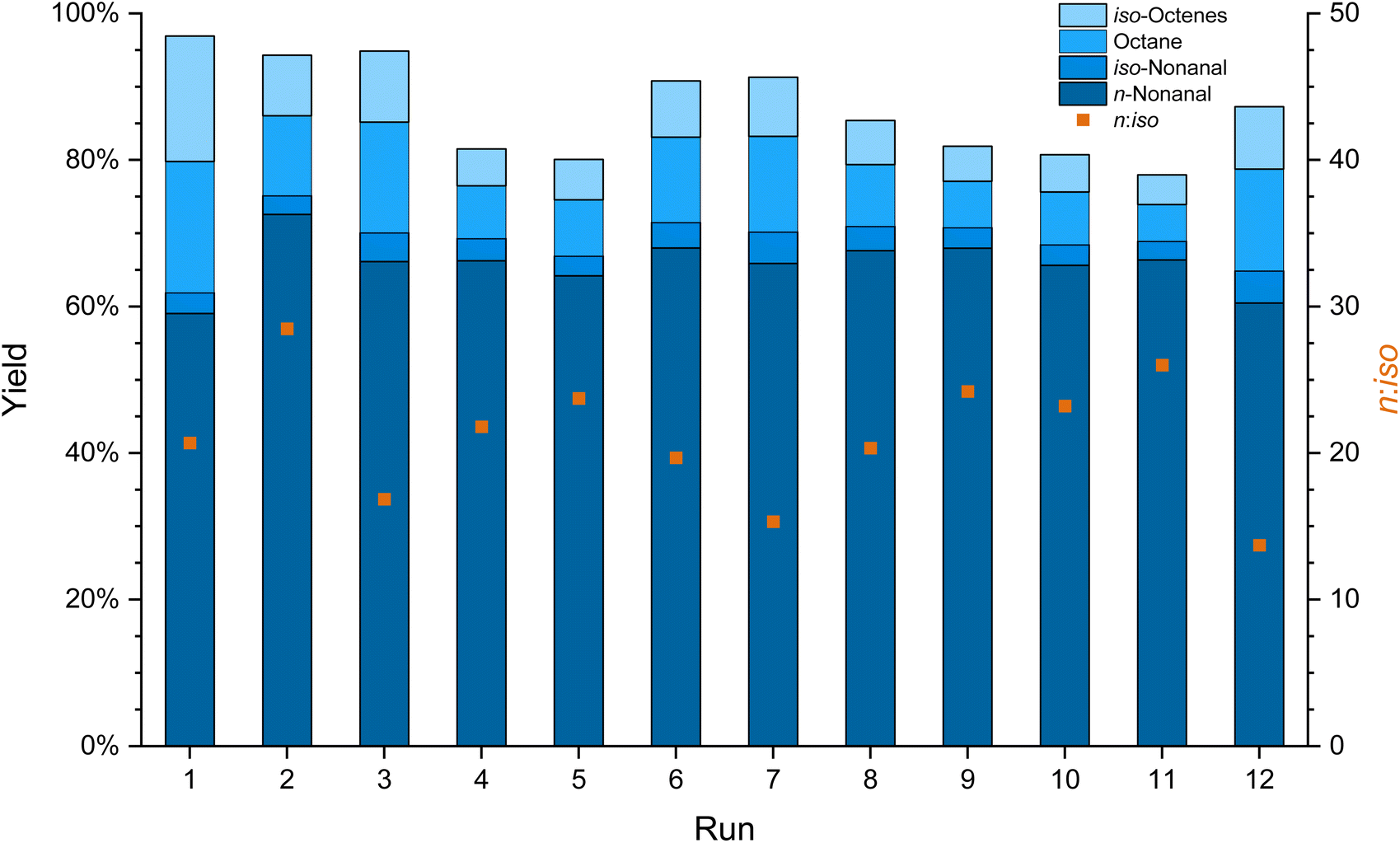 | ||
| Fig. 3 Yield, conversion and n:iso ratios of the catalyst recycling experiment in the hydroformylation of 1-octene (2.0 mL, with 5 vol% mesitylene as the internal standard) in ethylene carbonate (1.0 mL) over 12 runs with [Rh(acac)(CO)2] and sulfoXantphos as a catalyst (0.1 mol% Rh relative to 1-octene, Rh:P = 1:4) at 120 °C, 50 bar CO/H2 (1:1) in 20 mL autoclave reactors and with 1000 rpm stirring speed for 2 h reaction time. Total Rh leaching of 0.54% and P leaching of 0.10% across all 12 runs. | ||
2.1 Hydroformylation of other substrates using this system
Besides 1-octene, other substrates such as 1-hexene and 1-decene were tested in the hydroformylation to their corresponding aldehydes, heptanal and undecanal to examine the flexibility of the recycling strategy. The reaction of 1-hexene is significantly faster and reaches higher yields than 1-octene under the same reaction conditions (Table 2, entries 1 and 2). Hexene isomers lead to the main side products (16% in entry 1) with only traces of hydrogenated hexene (2% in entry 1). However, the reaction mixture does not form a well separated catalyst and product phase but rather a dispersion of yellow and colourless solids and liquids. The catalyst phase does not properly separate it from the organic phase due to the higher polarity of heptanal in comparison with nonanal in the conversion of octene. This system would not be suitable for catalyst recycling.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | T (°C) | t (h) | X 1-olefin (%) | Y aldehyde (%) |
n:iso |
| Conditions: 1-hexene or 1-decene (2.0 mL, with 5 vol% mesitylene as the internal standard) in EC (1.0 mL), [Rh(acac)(CO)2]/sulfoXantphos catalyst (0.1 mol% relative to the olefin, Rh:P = 1:4) at various temperatures, 50 bar CO/H2 (1:1) in 20 mL autoclave reactors and 1000 rpm stirring speed for various reaction times.a 1 mL of 1-hexene:1 mL of 1-decene. |
||||||
| 1 | Hexene | 110 | 2 | 94 | 76 | 97:3 |
| 2 | Hexene | 120 | 2 | 97 | 89 | 98:2 |
| 3 | Decene | 120 | 2 | 62 | 47 | 97:3 |
| 4 | Decene | 120 | 8 | 98 | 72 | 95:5 |
| 5 | Hexene/decenea | 120 | 4 | 98/98 | 84/81 | 96:4 |
1-Decene on the other hand provides a well separated catalyst and product phase after the reaction. The conversion and yield under the same conditions are lower and longer reaction times are required to reach full conversion (Table 2, entries 3 and 4). Isomerization products and hydrogenation products of the substrate are formed approximately in a 1:1 ratio (13% decene isomers, 14% decane in entry 4).
To recycle the catalyst phase while still converting hexenes to aldehydes, a 1:1 volumetric mixture of 1-hexene and 1-decene has been applied (Table 2, entry 5). Using this mixture, the phases were well separated and the catalyst phase could be recycled. In addition, the mixing of the substrates showed a positive influence on the aldehyde yield and conversion of 1-decene. The amount of the hydrogenated (9%) and isomerized substrate (9%) was reduced while the aldehyde yield increased.
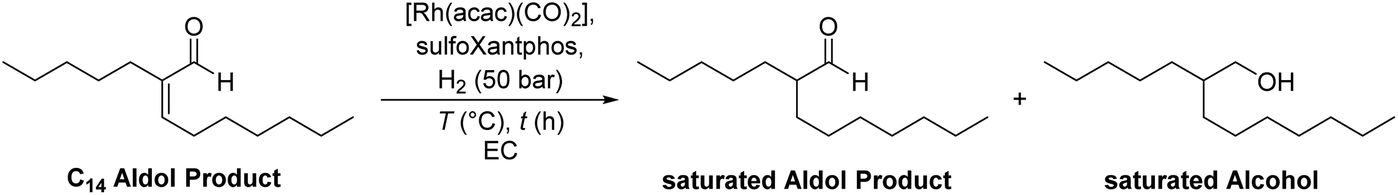
2.2 Hydrogenation of C14 aldol products
Besides the hydroformylation, the hydrogenation of C14 aldol products was tested, to investigate the reaction scope of this recycling method. 2-Pentylnon-2-enal (C14 aldol product) was synthesized from heptanal according to a literature procedure reported by Vorholt et al.33 During the hydrogenation, a saturated aldol product in which only the double bond is hydrogenated (2-pentylnonanal) and the saturated alcohol after a subsequent hydrogenation of the aldehyde (2-pentylnonanol) are formed (reaction scheme, Table 3). This group of hydrogenated aldol products can be used in the production of surfactants, as plasticizer alcohols or in further chemical processes.34 After a reaction time of 2 h, full conversion is reached with much of the substrate being converted to the saturated aldehyde intermediate (Table 3, entry 1). Only after a much longer reaction time of 16 h or at a higher reaction temperature, the main product saturated alcohol is obtained (Table 3, entries 2 and 3).|
|
|||||
|---|---|---|---|---|---|
| Entry | T (°C) | t (h) | X aldol (%) | Y sat. aldol (%) | Y sat. alcohol (%) |
| Conditions: C14 aldol product (2.0 mL, with 5 vol% mesitylene as the internal standard) in EC (1.0 mL), [Rh(acac)(CO)2]/sulfoXantphos catalyst (0.15 mol% relative to the substrate, Rh:P = 1:4) at various temperatures, 50 bar H2 in 20 mL autoclave reactors and 1000 rpm stirring speed for various reaction times. |
|||||
| 1 | 120 | 2 | 98 | 62 | 36 |
| 2 | 130 | 2 | 95 | 26 | 68 |
| 3 | 120 | 16 | >99 | 10 | 90 |
2.3 Switch recycling systems
The concept of the recycling of a solid catalyst phase in the form of a catalyst “pill” can be taken one step further by using the same catalyst “pill” in various reactions. This has been investigated by performing the so-called switch recycling experiments where the same catalyst phase was used for the hydroformylation of 1-octene and 1-decene always switching the substrate after every run. The same reaction conditions of 120 °C, 2 h reaction time and 50 bar synthesis gas have been used for both reaction steps (Fig. 4).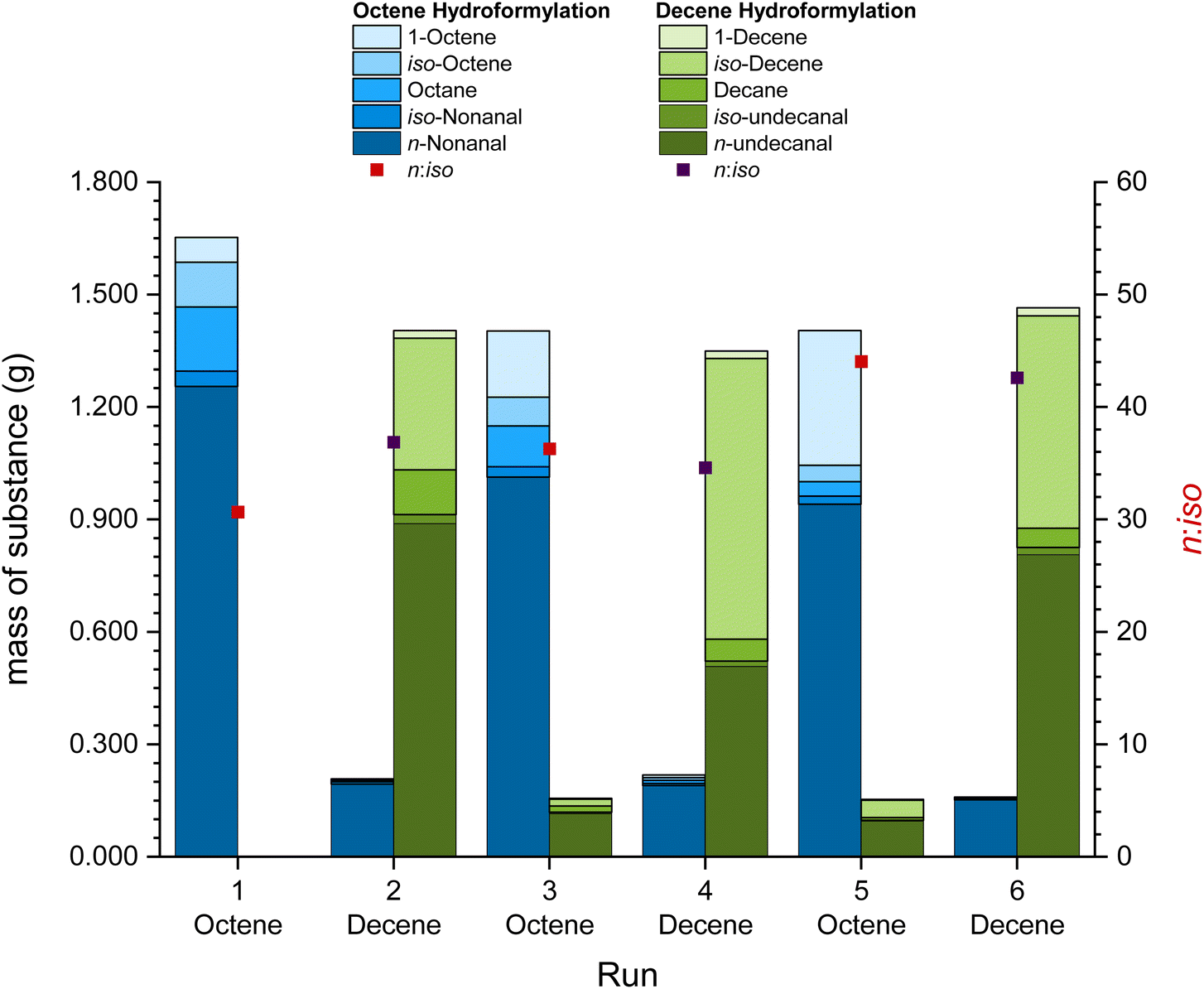 | ||
| Fig. 4 Mass of the substance and n:iso ratios of the switch catalyst recycling experiment in the hydroformylation of 1-octene and 1-decene (each 2.0 mL, with 5 vol% mesitylene as the internal standard) in ethylene carbonate (1.0 mL) over 6 runs (3 runs each) with [Rh(acac)(CO)2] and sulfoXantphos as a catalyst (0.1 mol% Rh relative to 1-octene, Rh:P = 1:4) at 120 °C, 50 bar CO/H2 (1:1) in 20 mL autoclave reactors and with 1000 rpm stirring speed for 2 h reaction time. Total Rh leaching of 0.47% and P leaching of 0.08% across all 6 runs. | ||
When switching between two different substrates, the retention of the substances in the previous run in the catalyst phase becomes obvious. About 8% to 15% of the substrates and products remain enclosed in the catalyst phase during each run. These substances, however, have the possibility of improving the overall reaction. As it has been the case for the reaction with a mixture of hexene and decene (Table 2, entry 5), the presence of nonanal in the reaction mixture improves the undecanal formation in the hydroformylation of decene, resulting in better results in comparison with batch reactions (Table 2, entry 3). In addition, the first run showed the highest level of isomerization and hydrogenation of the substrate which decreased for all further runs. During the 6 runs in the switch-recycling system, a total of 0.47% of the initial amount of rhodium, 0.08% of the initial amount of phosphorus and 0.177 g of EC leached into the organic phase (detailed leaching in the ESI, Table S2†). A TTON of 3702 was obtained with a higher TON during the reactions with 1-octene, as the reaction with shorter carbon chains is faster than that with longer carbon chains (see Tables 1 and 2).
Besides the recycling of the catalyst “pill” in the same reaction with different substrates, a recycling of the catalyst across different reactions can be tested. The switch catalyst recycling with 1-octene and the C14 aldol product was investigated (Fig. 5). Hydroformylation was conducted at 120 °C for 2 h and with 50 bar synthesis gas, while hydrogenation was conducted at 120 °C, 2 h and with 50 bar of hydrogen. The inhibiting effect of CO on the hydrogenation was observed in the first aldol hydrogenation run, which was far below the full conversion reported for batch experiments in Table 3 (entry 1). This effect was observed by Vorholt et al. previously.33 By applying vacuum to the catalyst phase at 60 °C after the third and fifth run, the CO was (partially) removed and the hydrogenation showed significantly better conversions increasing from 10% in run 1 to 63% in run 6. Additionally, the remaining nonanal from the previous run was also partly hydrogenated to nonanol during the reaction. Similar to the switch recycling with 1-decene, the hydrogenation and isomerization of 1-octene decreased and the selectivity increased after the first run. 1.04% of the initial amount of rhodium, 0.15% of phosphorus and 0.347 g of EC leached into the organic phase during the reactions (detailed leaching in the ESI, Table S3†). Leaching was significantly higher in reactions with the aldol product, likely as the formation of the alcohols in the hydrogenation enhances the leaching in comparison with the formation of aldehydes in the hydroformylation. A TTON of 3105 was achieved with a low TON for initial aldol hydrogenation runs of 204, but it increased up to 568 as the product yield improved after applying vacuum during the work-up.
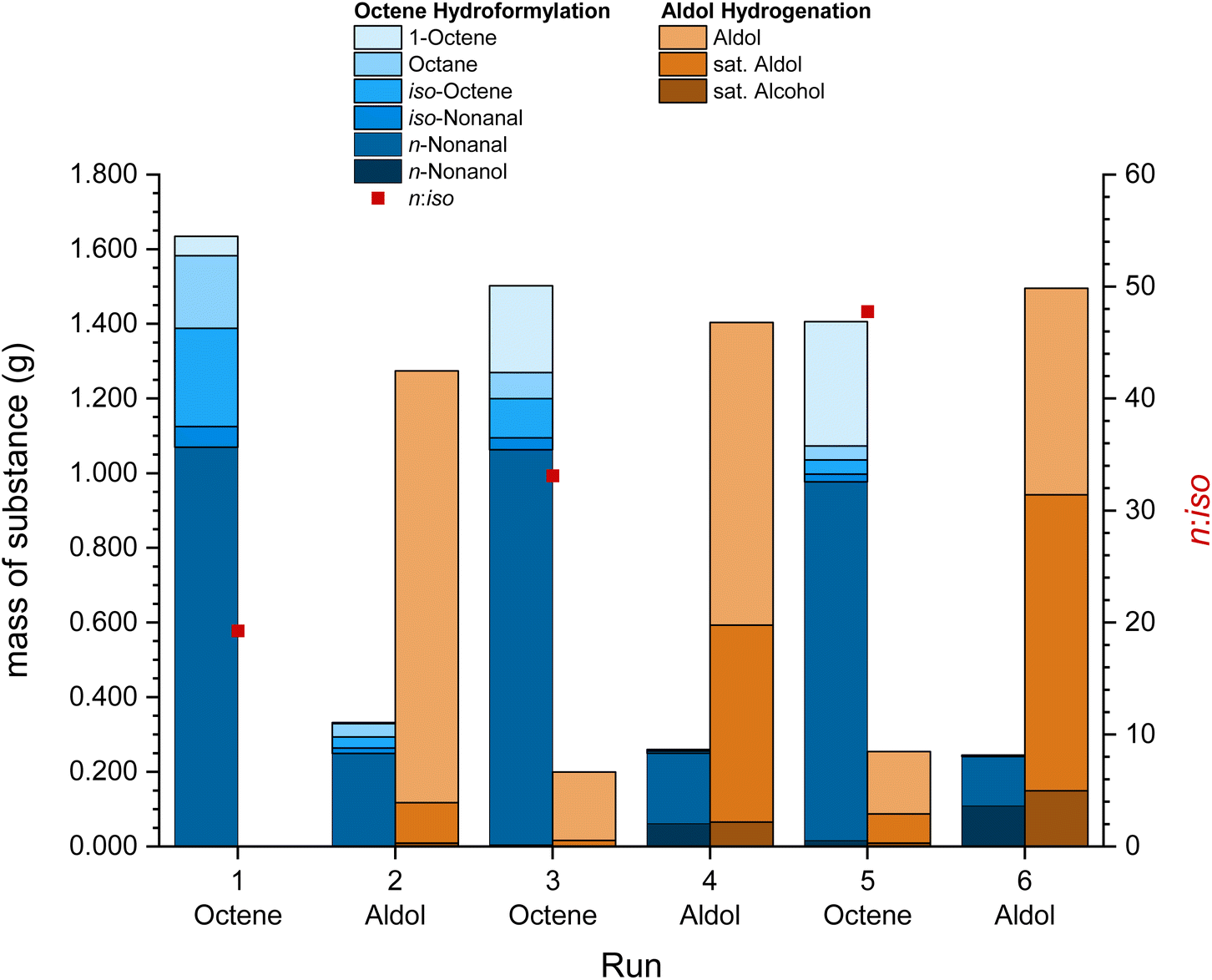 | ||
| Fig. 5 Mass of the substance and n:iso ratios of the switch catalyst recycling experiment in the hydroformylation of 1-octene and the hydrogenation of the C14 aldol product (each 2.0 mL, with 5 vol% mesitylene as the internal standard) in ethylene carbonate (1.0 mL) over 6 runs (3 runs each) with [Rh(acac)(CO)2] and sulfoXantphos as a catalyst (0.1 mol% Rh relative to 1-octene, Rh:P = 1:4) at 120 °C, 50 bar CO/H2 (1:1) for the hydroformylation and 50 bar H2 in 20 mL autoclave reactors and with 1000 rpm stirring speed for 2 h reaction time. Total Rh leaching of 1.04% and P leaching of 0.15% across all 6 runs. Vacuum applied to the catalyst phase at 60 °C between run 3 and 4 and between 5 and 6 to remove CO traces from the catalyst phase. | ||
3 Conclusion
Ethylene carbonate has been used as a solvent for multiphase catalyst recycling and solidification of the entire catalyst phase after the reaction at ambient temperature, forming a solid catalyst “pill” which could be easily stored and handled. This method was used in the multiphase hydroformylation of 1-hexene, 1-octene and 1-decene. High TOFs up to 1460 h−1 and n:iso ratios up to 98:2 were achieved using this system. The catalyst was recycled over 12 runs without a major loss in activity in the conversion of 1-octene, resulting in a TTON of 8627 and a total rhodium leaching of 0.54%. Systems involving 1-hexene as a substrate only provided a well separated catalyst “pill” when converted in a mixture with longer chain substrates such as 1-decene. Besides the hydroformylation, the hydrogenation of the C14 aldol product was investigated, showing high conversions to the saturated aldehyde and alcohol. The cross-recyclability of the catalyst “pill” in different reactions and with different substrates was shown by switch catalyst recycling in the hydroformylation of 1-octene and 1-decene, as well as in the hydroformylation of 1-octene and the hydrogenation of the C14 aldol product in combination. Both systems were well recyclable with a total Rh leaching of 0.47% and 1.04%. These experiments also provided an idea of substrates retained by the catalyst phase during the recycling. The cross-contamination between switch-recycling runs of different substrates can be investigated in future research. Methods such as washing the EC phase with a non-polar solvent or removing trace substances in a vacuum could overcome this issue. In addition, EC traces in the product mixture could be reduced by further investigating the crystallization behaviour of the catalyst phase and of EC.
This system offers significant benefits over aqueous multiphase systems, in particular for long chain olefins, due to their slower rate of reaction in aqueous systems. The phase behaviour of this reaction system and the optimization of this system for the hydroformylation of long chain olefin substrates can be studied in detail in future projects. Long-term catalyst stability studies can be carried out.
4 Experimental
4.1 General considerations
All reactions were conducted under an argon inert gas atmosphere unless stated otherwise with argon supplied by AirLiquide (99.9999 purity). Air sensitive chemicals were stored in a glovebox and standard Schlenk techniques were applied. Mesitylene was obtained from Alfa Aesar, n-decane was supplied by TCI and 1-octene was obtained from Across Organics, Sigma Aldrich and abcr. All other chemicals were obtained from Sigma Aldrich. All chemicals were used as obtained from the vendors and degassed before application in catalytic reactions. Deionized water was obtained from a MilliQ IQ 7000 device purchased from Merck Millipore. Carbon monoxide (99.999% purity) and hydrogen (99.999% purity) for catalytic reactions were obtained from AirLiquide.GC measurements were conducted on a Nexis GC-2030 purchased from Shimadzu with a flame ionization detector. A GCMS-QP2020 purchased from Shimadzu was used to conduct GC with in-line mass spectrometry measurements. For both apparatus Rtx-1 columns of the company Restek with a length of 30 m, an inner diameter of 0.25 mm and a particle size of 0.5 μm were installed and helium was used as a carrier gas. A GC FID of the same model with a CP Wax 52 CB column with a length of 60 nm, an inner diameter of 0.25 mm and a particle size of 0.25 μm was used for measuring the product solution after the aldol reaction and hydrogenation. The Sternberg method was applied to approximate GC factors for non-calibrated substances.35
ICP-MS measurements were conducted on an ICPMS-2030 from Shimadzu with sample preparation being carried out using a Mars 6 microwave from CEM and Mikroanalytisches Laboratorium Kolbe and on an ICP-OES Acros Spectro from the company Spectro with the sample preparation being carried out on a Mars 6 microwave from CEM. The samples were analyzed for rhodium and phosphorus contents.
4.2 General procedure for hydroformylation autoclave reactions
Hydroformylation experiments were conducted in 20 mL (total volume) autoclave reactors (details in the ESI†). [Rh(acac)(CO)2], sulfoXantphos and EC were weighed into a glass inlet in a glovebox. The inlet was inserted into the autoclave reactor outside the glovebox, which was then closed and placed under inert gas at the Schlenk line. The substrate stock solution was filled into the reactor. Most reactions were conducted with a total reaction volume of 3 mL (1 mL EC and 2 mL organic phase). The autoclave was placed in a pre-heated cone after pressurization with CO and H2 to the desired pressure at ambient temperature and stirring was started 10 min after to ensure that the EC phase was fully liquified. After the reaction, the reactor was placed in an ice bath for cooling, the remaining gas was vented and the autoclave was opened. The product solution was decanted from the solid catalyst phase and a sample of the organic phase was taken. Mesitylene was used as internal standard in all hydroformylation reactions. The samples were analyzed using GC FID.4.3 General procedure for hydroformylation recycling experiments
Recycling reactions were carried out as described for the hydroformylation reactions above. The organic phase was removed under air after the reaction and the inlet with the solid catalyst phase was placed back into the autoclave. Vacuum was applied briefly and the autoclave was placed under inert gas. A fresh substrate stock solution was added, the reactor was pressurized and the next run was started according to the same procedure as for batch reactions. The organic phase of each run was analyzed by GC FID. The amount of leached EC was determined by GC FID analysis and ICP-MS measurements of the organic phase were conducted to determine the amount of leached rhodium and phosphorus. Leached EC was resupplied one run later (for example the EC leached in the first run was resupplied before the start of the third run). Recycling reactions were conducted in 20 mL autoclave reactors.4.4 Synthesis of the C14 aldol product from heptanal
The synthesis of the C14 aldol product was carried out according to the literature of Vorholt et al.334.5 General procedure for the hydrogenation of the C14 aldol product
The hydrogenation experiments of the aldol product were carried out in 20 mL stainless steel autoclave reactors in a similar manner to the hydroformylation reactions. The reactors were pressurized with H2 only. The reaction products were analyzed by GC FID using an Rtx-1 and a CpWax column as mentioned above.4.6 General procedure for switch recycling experiments
The switch catalyst recycling experiments were carried out in a similar manner to the hydroformylation recycling experiments. Runs with the aldol product as a substrate were carried out using only H2 gas according to the procedure for batch hydrogenation reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Verband der Chemischen Industrie e.V. for the stipend of Jeroen T. Vossen and for financing this research. In addition, we would like to thank Nina Bäumer for her assistance in the lab concerning this work. We acknowledge Open Access funding provided by the Max Planck Society.References
- K. S. Egorova and V. P. Ananikov, Organometallics, 2017, 36, 4071–4090 CrossRef CAS.
- A. Behr, Angewandte Homogene Katayse, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 1st edn, 2008 Search PubMed.
- D. J. Cole-Hamilton and R. P. Tooze, in Catalyst Separation, Recovery and Recycling: Chemistry and Process Design, Springer Netherlands, Dordrecht, 2006, pp. 1–8 Search PubMed.
- W. Keim, Green Chem., 2003, 5, 105–111 RSC.
- B. Cornils, W. A. Herrmann, I. T. Horváth, W. Leitner, S. Mecking, H. Olivier-Bourbigou and D. Vogt, Multiphase Homogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005, vol. 1, pp. 1–2 Search PubMed.
- C. Wibowo and K. M. Ng, Conceptual Design of Crystallization Processes, De Gruyter, Berlin/Boston, 2020 Search PubMed.
- J. Vondran, A. I. Seifert, K. Schäfer, A. Laudanski, T. Deysenroth, K. Wohlgemuth and T. Seidensticker, Ind. Eng. Chem. Res., 2022, 61, 9621–9631 CrossRef CAS.
- M. A. McDonald, H. Salami, P. R. Harris, C. E. Lagerman, X. Yang, A. S. Bommarius, M. A. Grover and R. W. Rousseau, React. Chem. Eng., 2021, 6, 364–400 RSC.
- J. Vondran, R. Moeschke, T. Deysenroth and T. Seidensticker, Eur. J. Lipid Sci. Technol., 2022, 2200126 CrossRef.
- T. Seidensticker, H. Busch, C. Diederichs, J. J. von Dincklage and A. J. Vorholt, ChemCatChem, 2016, 8, 2890–2893 CrossRef CAS.
- W. S. Knowles, J. Chem. Educ., 1986, 63, 222 CrossRef CAS.
- D. F. Reinhold, R. A. Firestone, W. A. Gaines, J. M. Chemerda and M. Sletzinger, J. Org. Chem., 1968, 33, 1209–1213 CrossRef CAS PubMed.
- Q. Chen, L. Lv, M. Yu, Y. Shi, Y. Li, G. Pang and C. Cao, RSC Adv., 2013, 3, 18359–18366 RSC.
- W. Wang, L. Cui, P. Sun, L. Shi, C. Yue and F. Li, Chem. Rev., 2018, 118, 9843–9929 CrossRef CAS PubMed.
- Á. Molnár and A. Papp, Coord. Chem. Rev., 2017, 349, 1–65 CrossRef.
- S. Han, J. Yang and H. Huang, Fuel, 2022, 315, 122815 CrossRef CAS.
- Y. Zhou, G. Chen, Z. Long and J. Wang, RSC Adv., 2014, 4, 42092–42113 RSC.
- P. Zhao, J. Wang, G. Chen, Y. Zhou and J. Huang, Catal. Sci. Technol., 2013, 3, 1394–1404 RSC.
- V. S. Shende, V. B. Saptal and B. M. Bhanage, Chem. Rec., 2019, 19, 2022–2043 CrossRef CAS PubMed.
- Y. Yang, N. Priyadarshani, T. Khamatnurova, J. Suriboot and D. E. Bergbreiter, J. Am. Chem. Soc., 2012, 134, 14714–14717 CrossRef CAS PubMed.
- X.-Y. Dou, J.-Q. Wang, Y. Du, E. Wang and L.-N. He, Synlett, 2007, 3058–3062 CAS.
- D. J. Heldebrant, H. N. Witt, S. M. Walsh, T. Ellis, J. Rauscher and P. G. Jessop, Green Chem., 2006, 8, 807–815 RSC.
- Y. Du, J.-Q. Wang, J.-Y. Chen, F. Cai, J.-S. Tian, D.-L. Kong and L.-N. He, Tetrahedron Lett., 2006, 47, 1271–1275 CrossRef CAS.
- N. Karodia, S. Guise, C. Newlands and J.-A. Andersen, Chem. Commun., 1998, 2341–2342 RSC.
- A. M. Scurto and W. Leitner, Chem. Commun., 2006, 3681–3683 RSC.
- X. Jin, D. Yang, X. Xu and Z. Yang, Chem. Commun., 2012, 48, 9017–9019 RSC.
- J. H. Yoon, Y. J. Park, J. H. Lee, J. Yoo and C.-H. Jun, Org. Lett., 2005, 7, 2889–2892 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- M. I. Chaudhari, A. Muralidharan, L. R. Pratt and S. B. Rempe, in Modeling Electrochemical Energy Storage at the Atomic Scale, ed. M. Korth, Springer International Publishing, Cham, 2018, pp. 53–77 Search PubMed.
- R. Naejus, D. Lemordant, R. Coudert and P. Willmann, J. Chem. Thermodyn., 1997, 29, 1503–1515 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press Inc., Boca Raton, FL, 79th edn, 1998– 1999 Search PubMed.
- H. Warmeling, D. Hafki, T. von Söhnen and A. J. Vorholt, Chem. Eng. J., 2017, 326, 298–307 CrossRef CAS.
- M. Strohmann, A. J. Vorholt and W. Leitner, Chem. – Eur. J, 2022, 28, e202202081 CrossRef CAS PubMed.
- H. An, D. Wang, S. Miao, Q. Yang, X. Zhao and Y. Wang, RSC Adv., 2020, 10, 28100–28105 RSC.
- S. Decker, L. Schubert, F. Schroter, M. Klaas and F. Roessner, Sorption Chromatogr. Processes, 2013, 13, 785–793 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional graphics, detailed experimental procedures and analytical data. See DOI: https://doi.org/10.1039/d2gc04822a |
| This journal is © The Royal Society of Chemistry 2023 |