
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biomass-derived functional materials as carriers for enzymes: towards sustainable and robust biocatalysts
Meena
Bisht†
*ab,
Sarath Kumar
Thayallath†
c,
Pranav
Bharadwaj
c,
Gregory
Franklin
 a and
Dibyendu
Mondal
*ac
a and
Dibyendu
Mondal
*ac
aInstitute of Plant Genetics (IPG), Polish Academy of Sciences, Strzeszyńska 34, 60-479 Poznań, Poland. E-mail: meenabisht56@gmail.com; meena@svc.ac.in; dmtapu@gmail.com; dmon@igr.poznan.pl
bDepartment of Chemistry, Sri Venkateswara College, Delhi University, Dhaula Kuan, New Delhi, 110021, India
cCentre for Nano and Material Sciences, JAIN (Deemed-to-be University), Jain Global Campus, Bangalore 562112, India
First published on 31st May 2023
Abstract
The unique catalytic properties of enzymes have led to the production of useful medicinal intermediates, foods, and biofuels from sustainable sources. However, the instability of soluble/free enzymes under several challenging conditions (e.g., pH, proteolysis, temperature, ionic potential, chemical denaturants) restricts the use of enzyme-based biocatalysts. Encapsulation of enzymes on suitable carriers would mitigate the instability issues faced in robust biocatalysis. An “ideal” carrier material employed for protein immobilization should be nontoxic, scalable, biocompatible, and should not compromise the biological activity and structure of proteins/enzymes. Thus, biodegradable and renewable biomass-derived functional materials (BDFMs) are envisaged as promising carriers for enzymes. BDFMs have in-built chemical functionalities and desirable physicochemical properties that enable their use in enzyme catalysis at the industrial scale. Numerous BDFMs have been used as immobilization matrices to improve the biocatalytic activity and stability of various enzymes. These solid materials are renewable and environmentally friendly compared with synthetic polymers. This review highlights the advancements, challenges and prospects in the emerging field of BDFMs (cellulose, silk protein, chitin, chitosan, lignocellulose, and a combination of biopolymers such as chitin/lignin and chitosan/alginate) for immobilization of enzymes (e.g., α-chymotrypsin, cytochrome c, carbonic anhydrase, glucose oxidase, ribonuclease, cholesterol oxidase, alkaline phosphatase, β-glucosidase, lipase, horseradish peroxidase, catalase, tyrosinase, acetylcholinesterase, amylase, invertase, protease, laccase, β-galactosidase, and several others) for biocatalytic processes. This review also describes the relationship between the structural properties and functionality of several enzymes immobilized in BDFMs, and profiles the impact of pH, temperature, reusability, stability of storage, and the activity of these enzymes. Future perspectives in this promising field, as well as potential difficulties, are discussed. This review will help in refining biocatalysis technologies whereby biomass-derived, environmentally friendly materials are employed as enzyme supports.
 Meena Bisht | Dr Meena Bisht received a doctorate in chemistry from Department of Chemistry, University of Delhi (Delhi, India). She is an Assistant Professor in the Department of Chemistry at Sri Venkateswara College (University of Delhi). She was a postdoctoral fellow at CICECO (University of Aveiro, Aveiro, Portugal) and Institute of Plant Genetics (Poznan, Poland). She has carried out substantial and innovative research in protein stabilization using a wide variety of “green” solvents (e.g., ionic liquids and deep eutectic solvents). She has developed a green, innovative, and cost-effective technology for the enhanced dissolution and extraction of bioactive compounds from sustainable biomass. |
 Sarath Kumar Thayallath | Mr Sarath Kumar T. is working as a Research Associate in a Research and Development department at Syngene International Limited (Bangalore, India). He completed a postgraduate degree in chemistry in 2020 from the Centre for Nano Materials and Sciences (CNMS) in Jain University (Bangalore, India). Immediately after completing a Master's degree, he worked for 1 year as a Junior Research Fellow at CNMS. His research interests are the synthesis, characterization, and purification of oligonucleotides. |
 Pranav Bharadwaj | Mr Pranav Bharadwaj holds a Master's degree in chemistry from the Centre for Nano Materials and Sciences (CNMS) in Jain University (Bangalore, India), which he earned in 2022 after completing a Bachelor's degree in chemistry in 2020 from Karnataka University (Dharwad, India). His interests lie in protein packaging and green chemistry. He works as a Junior Research Fellow at CNMS with a focus on developing robust solvent-manipulation strategies for biocatalytic applications. Pranav has received numerous accolades, including a Gold Medal for his excellent academic performance during his Master's degree and a Young Achiever Award in 2020 for his overall accomplishments. |
 Gregory Franklin | Dr Gregory Franklin is a professor at the Institute of Plant Genetics within the Polish Academy of Sciences (Poznan, Poland). He received a doctorate in plant biotechnology from the University of Madras (Chennai, India) in 2001. He held various research positions in the USA, Saudi Arabia, and Portugal before moving to Poland in 2015. He is an interdisciplinary scientist whose research focuses on plant nanotechnology and materials science. Understanding the fundamental mechanisms of green chemistry and the interaction between plants and nanomaterials is the main focus of his research team. |
 Dibyendu Mondal | Dr Dibyendu Mondal is the ERA chair holder in the NANOPLANT project at the Institute of Plant Genetics within the Polish Academy of Sciences (Poznan, Poland) since September 2021. He received a doctorate in chemical science from the Central Salt And Marine Chemical Research Institute (Bhavnagar, India) in 2015. From 2015 to 2017 he was a post-doctoral research assistant at CICECO within the University of Aveiro (Aveiro, Portugal) and then he joined as an Assistant Professor at the Centre for Nano Materials and Sciences in Jain University (Bangalore, India) in 2017. His main research areas focus on (i) value addition of bioresources using neoteric solvents; (ii) protein engineering and biocatalysis; (iii) bio-inspired and active nano-constructs; (iv) nanotechnology for sustainable agriculture. |
1. Introduction
Society has high dependence on non-renewable resources, which is unsustainable in the long run. Increases in population sizes and environmental pollution have prompted strategies for “green” chemistry, sustainable resources for fuels, chemicals, and materials, and reducing waste.1,2 In this scenario, the development of flexible and integrated “biorefineries” to produce biofuels and bioproducts from renewable biomass sources can aid transition to a novel “bioeconomy” for more efficient and sustainable global development. The “circular economy” concept is taking a central role in sustainable development and, for this reason, deserves attention.3–6 Renewable denotes sustainable and abundantly available feedstocks for the production of biofuel and biochemicals via suitable bioconversion. “Biocatalysis” is a charming technology used in various industrial applications. An enzyme or biocatalyst is essentially a non-hazardous, non-toxic material that is easily obtained from widely available renewable resources.7 Enzymes are Nature's sustainable catalysts which drive a variety of reactions. In accordance with the theory of green chemistry, biocatalytic processes are more sustainable, environmentally friendly, and cost-effective than traditional chemical processes. In the context of green chemistry and sustainable development, the utilization of enzymes as efficient catalysts offers several interesting characteristics.1,2 Enzymes are biodegradable, reusable biocatalysts which do not create by-products. Enzymes are made up of several amino acids that are linked together by a peptide bond (–CONH–).8,9 The native state of a protein is a result of complex interactions (e.g., hydrogen bonds, van der Waals interactions, ionic interactions, hydrophobic interactions) which ultimately provide stability to proteins and prevent ruinous conformational changes.10 Each enzyme exhibits a great degree of chemo-, regio- and stereospecificity towards a substrate because of the binding properties at the active site. The native or tertiary structure of some common enzymes that have been modified with biomass-derived functional materials (BDFMs) is shown in Fig. 1.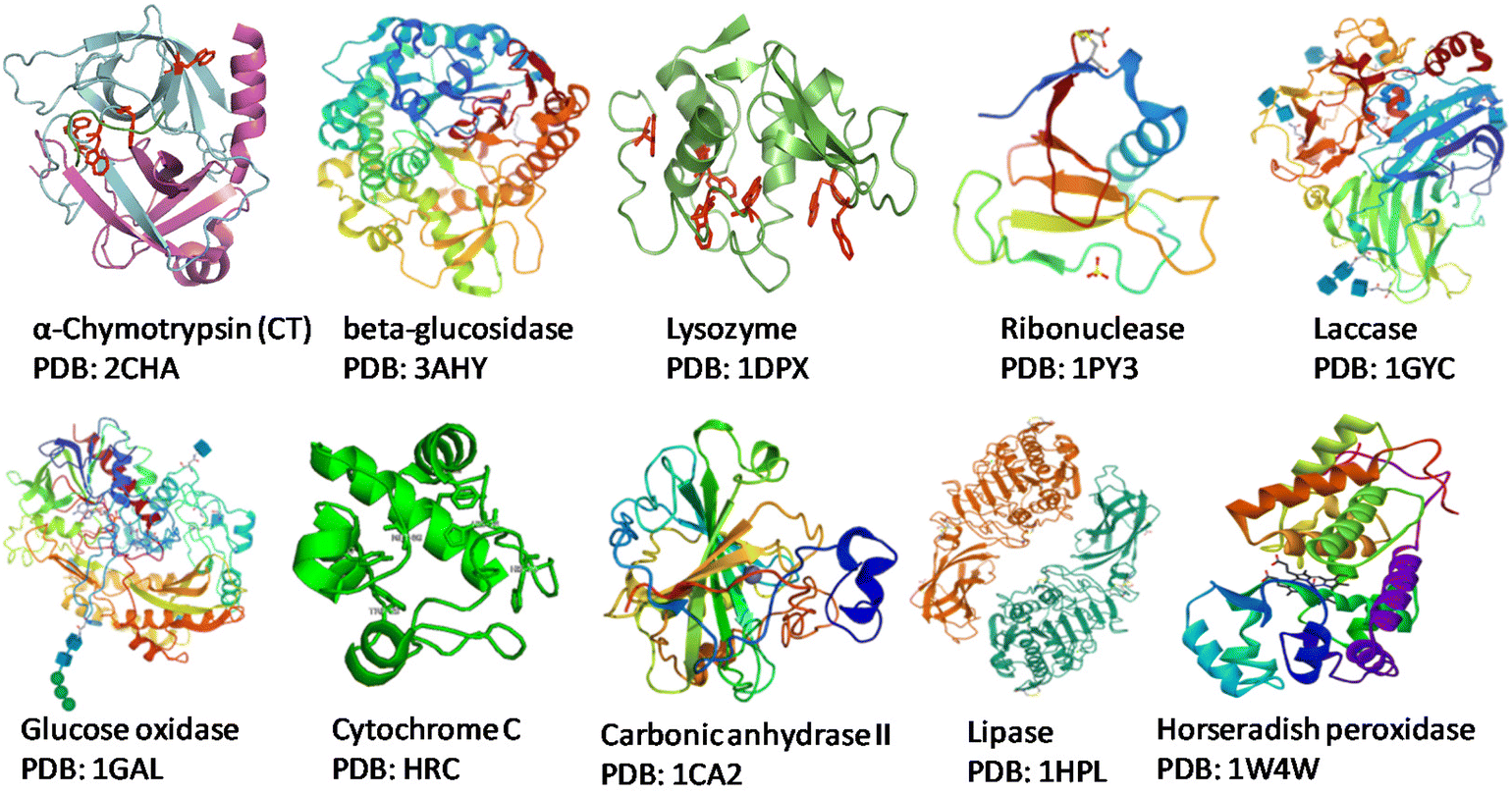 | ||
| Fig. 1 Structures of commonly used enzymes. Source: Protein Data Bank. | ||
In the synthesis of complex pharmaceuticals, enzymatic biocatalytic approaches are more environmentally friendly and sustainable than chemical methods. Due to their capacity to catalyse reactions with high efficiency and specificity, they have emerged as preferred tools in green chemistry and are being used more frequently in industrial processes.11Fig. 2 shows various factors that have an impact on the stability and activity of enzymes. In recent years, biocatalysis has emerged as an imperative technology for meeting the growing demands for green and sustainable chemical manufacturing. However, enzymes are moderately stable in terms of their structural and chemical stabilities, and enzymatic processes are conducted under physiological conditions in a buffer, with high rates and selectivities.1 Harsh processing conditions, such as the presence of volatile organic solvents, extreme pH, and temperature, are the main barriers to the effective use of enzymes in therapeutics and as biocatalysts. Moreover, a lack of long-term activity, stability, reusability, and a challenging recovery rate can hinder the industrial application of enzymes.1,2 To overcome these problems, numerous solutions have been postulated by researchers, such as methods for protein engineering, chemical modifications, excipient addition, and immobilization.12
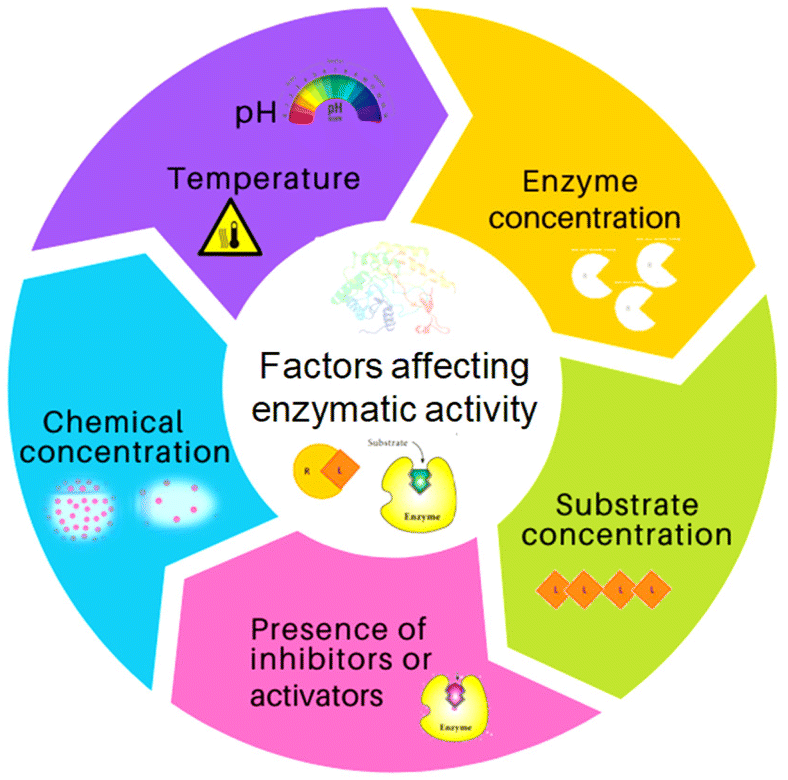 | ||
| Fig. 2 Various factors that influence the activity and stability of enzymes/proteins. | ||
The progress of effective immobilization strategies has paved the way for enhancing the recovery, recycling, operational stability, and storage of enzymes.13 Immobilization is also very important for effective biocatalysis. Enzyme immobilization is the stable fixation of the protein within a solid support/matrix by physical or chemical interactions (or both) so that the enzyme remains fully functional (or at least retains most of its catalytic activity). There are various methods for enzyme immobilization: adsorption, entrapment, covalent binding, and crosslinking.13–16 During immobilization, the integrity of the structure and the function of an enzyme are at great risk, and the biocatalytic performance may be compromised. In addition, the co-immobilization of two or more enzymes can afford multifunctional solid biocatalysts capable of catalysing processes in biocatalytic cascades.14 Therefore, immobilization of enzymes is an essential method that has a significant effect on the stability and effectiveness of enzymes. Several strategies for enzyme immobilization have been categorized: adsorption binding, entrapment, covalent binding, and crosslinking with/without a support.15,16 Immobilized proteins have been widely use in industrial applications (e.g., analytical, pharmaceutical, commodity chemicals, food, and cosmetic industries) as well as energy production and biomedicine.15,16 Therefore, immobilization technologies must be improved and diversified to support the development of fresh formats, better economies, and higher performance.13,14 Supports can be made of hybrid materials, inorganic materials, metal–organic frameworks (MOFs), nanoflowers, hydrogels, polymers, or biomass-based materials.15,17,18–21 BDFMs are exceptional supports for immobilising enzymes to enhance the activity and selectivity of enzymes.21,22 BDFMs have attracted much interest due to their abundance, environmental friendliness, sustainability, and unique structures.1 Biomass-based matrices have great microbiological resistance, biocompatibility, robust mechanical properties, acceptable stability, and controllable biodegradability, which might offer a favourable microenvironment for enzyme stability. BDFMs with superior properties and precise morphology for improving facile biocatalysis is a research “hotspot” (Fig. 3). BDFMs are created by transforming the natural resources (plants, food, animals, microorganisms) and their wastes into high-performance materials using physical or chemical processes.2 BDFMs promote the stabilization of entrained molecules, such as therapeutic proteins or enzymes, without compromising their activity.23 Furthermore, because BDFMs have been approved by the US Food and Drug Administration, these supports/matrices are ingestible and may have useful applications in vivo.23 The immobilization of industrial enzymes would enable the reuse of expensive enzymes for enhanced utility in industrial processes, whereas the immobilization of bioactive enzymes might be incorporated into diagnostic and therapeutic applications.24 In this review, we evaluate available data regarding the potential of BDFMs as sustainable carriers for enzyme stabilization.
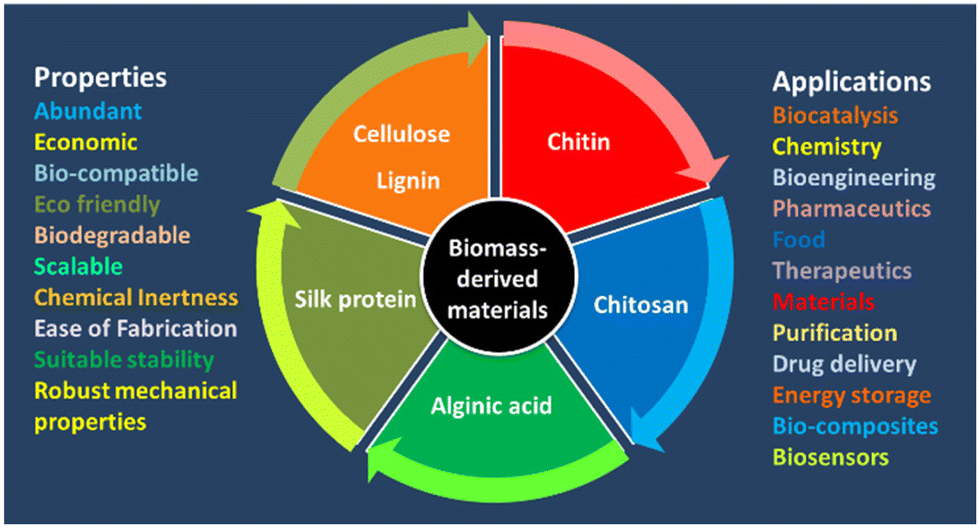 | ||
| Fig. 3 Potential applications of BDFMs in biocatalytic processes. | ||
Precursors for BDFMs are readily available, inexpensive, and primarily produced from plants (cellulose, lignin, alginate), animals (silk, chitin, chitosan), and microorganisms (bacterial cellulose). BDFMs possess numerous desirable inherent qualities that point to their potential use as an enzyme-stabilisation matrices to protect them from different stresses. This review focuses on the main categories of biomass-based support materials for different enzymes, or biocatalyst designs/functionalization for improving the stability and activity of enzymes and maximizing reaction efficiencies.
Fig. 4 depicts an overlay analysis which focuses on evaluating the qualities and academic importance describing BDFM use to increase enzymatic efficiency. An extensive evaluation of the academic and scientific literature accessible in this field of research was carried out using the Web of Science (WOS). In the current review, VOS viewer was used to display the co-occurrence of keywords, and an overlay map was developed. The circles in the overlay map depict the occurrence of the keywords in the chosen dataset (Fig. 4). The keyword co-occurrence map was created by choosing keywords that appeared ≥10 times. As a result, 315 out of 7211 keywords met the criteria and were further classified into eight major clusters. As shown in Fig. 4, since 2000, there have been sporadic reports on enzyme immobilizations using biomass-derived materials. In the years between 2008 and 2018, biomass-related research has experienced explosive growth. The important discoveries made possible by the use of biomass-derived materials as carriers for long-term stability and enhancing the catalytic activity of enzymes and proteins are summarised in this overview. Recent biocatalytic contributions of biomass-based supports are discussed in the next sections.
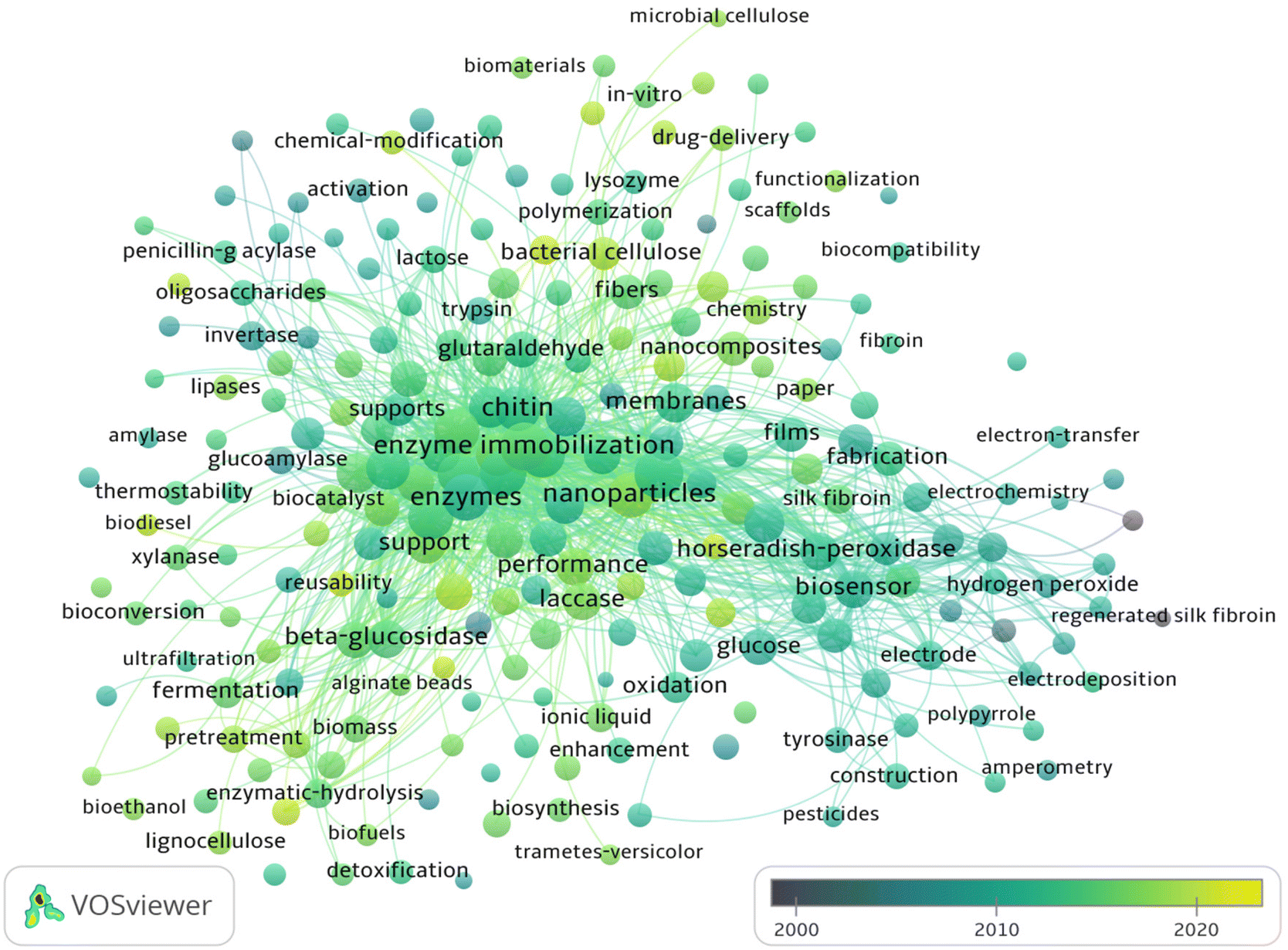 | ||
| Fig. 4 Overlay map of co-occurrence of keywords concerning BDFMs as carriers for enzymes. | ||
2. Lignocellulosic-based supports (carriers) for enzyme immobilization
Lignocellulose and cellulose-based materials are attractive support materials for enzyme immobilization because they have high porosity, strong mechanical properties, are highly resistant to microorganisms, as well as having tunable biodegradability, excellent stability, and dense network of hydrogen bond crosslinks.25–27 Being the most abundant polysaccharide, cellulose is made up of linear chains of D-glucose units connected through β(1 → 4)-fashioned glycosidic linkages, as well as intra and intermolecular H-bonding (Fig. 5). This results in the formation of cellulose microfibrils that are firm and strong. To create β(1 → 4) glycosidic bonds, every alternate glucose molecule in cellulose is inverted. Cellulose-functionalized materials can be made from cellulosic sources such as cotton and bacterial cellulose wherein the hydrolysis of cellulose generates nano-sized crystalline material.26–28 Several such forms of cellulose, such as cellulose hydrogel microspheres (CHMs), cellulose nanocrystal (CNCs), cellulose nanowhiskers (CNWs), and nanofibrillated cellulose (NFC) have been studied as possible supports for enzyme immobilization. They are characterized by high enzyme loading, enhanced stability, improved activity, and increased mass transfer rate.29–47 Various immobilization techniques can be applied to load enzymes onto the matrices of lignocellulosic biomass/modified cellulose-based materials for the conjugation and stabilization of enzymes and proteins. Recently, Ren et al.45 reported the superior potential of delignified bamboo over the immobilization of β-glucuronidase (BGU) with a significantly higher loading capacity (∼8 mg m−2·g−1), recyclability (13 cycles), and longer shelf life (≤7 weeks). Cellulose matrices were employed to enhance the robustness of the enzyme in analytical and synthetic applications for hierarchical flow-based bioreactors.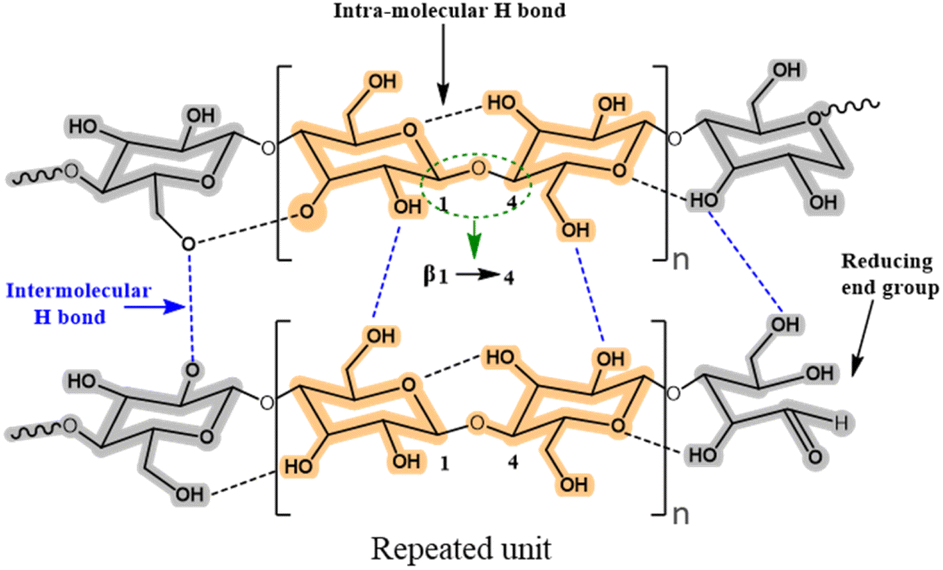 | ||
| Fig. 5 Intra- and inter-molecular hydrogen bonding and possible reaction sites in cellulose. | ||
Uth and co-workers47 used a modular approach for site-directed, biorthogonal protein immobilization. A general two-step method was created that took advantage of extremely effective oxime ligation along with enzyme-mediated protein coupling onto the surface of peptide-modified crystalline nanocellulose. Conjugate analyses revealed that, after being grafted onto CNC, the immobilized glucose oxidase (GOx) maintained its activity (Fig. 6). To improve the salt and thermal tolerance of cellulase enzyme, Montamedi and colleagues46 immobilized its cloned form (PersiCeL3) over a carboxymethyl cellulose (CMC) hydrogel matrix. With halotolerant behaviour >65% and almost two-folds higher activity than the native enzyme, immobilized PersiCel3 showed superb performance which could be used in lignocellulosic biomass industries to boost the production of value-added products in hot and salty environments.
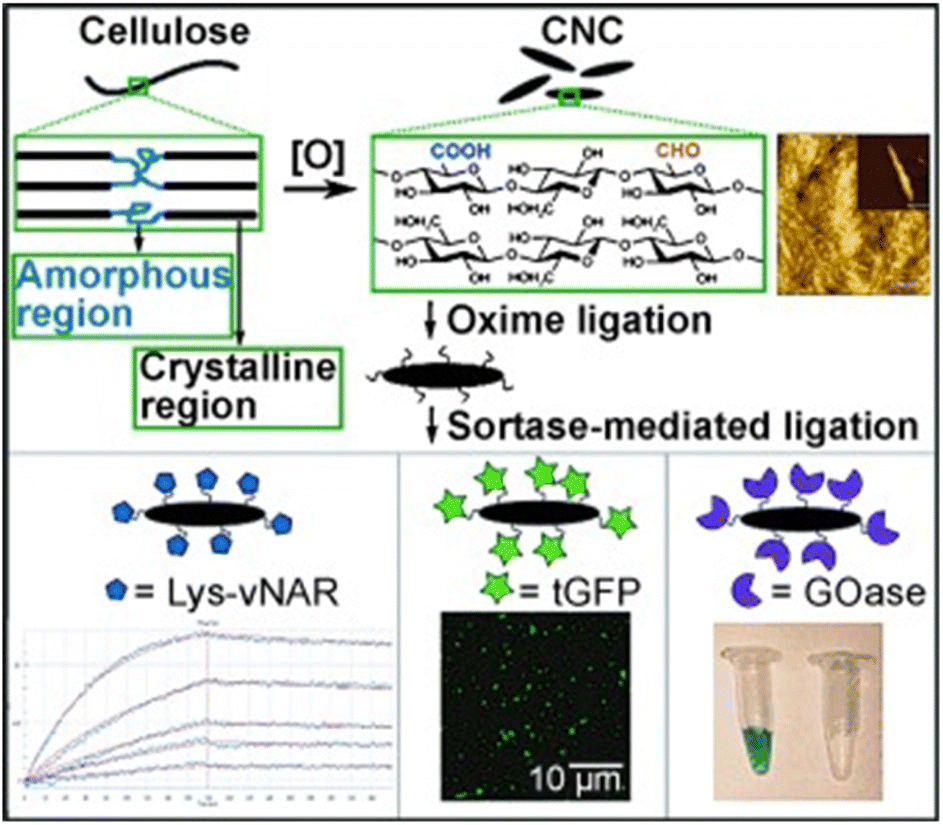 | ||
| Fig. 6 Postulated chemoenzymatic approach to protein immobilization onto crystalline cellulose nanoscaffolds (schematic). Reproduced from ref. 47 with permission from Wiley-VCH, copyright 2014. | ||
Authors transformed the primary alcohols on the surface of CNC to the corresponding aldehyde functionality, which enabled immobilization of bioactive modules via extremely effective oxime ligation. Immobilization of the enzyme on the CMC-hydrogel matrix could be used to improve the robustness of enzymes with strong resistance towards high temperatures and heavy salt concentrations.7,46 Suo et al.7 prepared ionic liquid (IL)-modified magnetic CMC nanoparticles on which porcine pancreatic lipase (PPL) was immobilized covalently (Fig. 7). The immobilized lipase (PPL-IL-MCMC) demonstrated specific activities that were 1.43- and 2.81-fold greater than those of free PPL and PPL-MCMC, respectively, with simple and efficient recoverability.
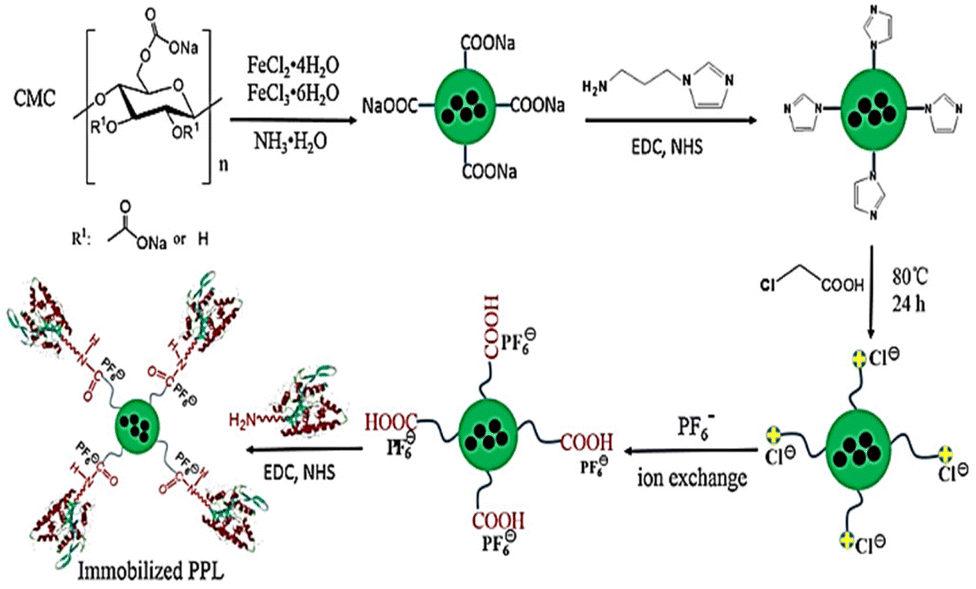 | ||
| Fig. 7 A common method for preparation of IL-modified BDFMs and enzyme immobilization upon it. Reproduced from ref. 7 with permission from Elsevier, copyright 2020. | ||
Extending the work further, the authors studied the effect of urea (Fig. 8a) and thermal stresses (Fig. 8b) over the activity and stability of PPL. The conjugate retained >68% activity but the native enzyme could barely maintain its functions. The immobilized enzymes PPL-MCMC and PLL-IL-MCMC could maintain 40.8% and 70.9% of their original activity, respectively, at a urea concentration of 6 mol L−1, whereas the native enzymes retained only 15% of their activity. A similar trend was reported for thermal stress for a long time (Fig. 8b). The advantage of combining two stabilizing systems can be clearly envisioned by recyclability and Michaelis–Menten parameters (Fig. 8c and d). This is due to ion interactions, covalent bonds, H-bonds, and π–π stacking between the carrier and enzyme. This could also support maintenance of the stiffness and integrity of the enzyme structure under harsh conditions, thereby stopping the enzymes from becoming partially inactivated.7 Utilizing halloysite nanotubes (HNTs) as a template, Sillu et al.,48 described the synthesis of a nanobiocatalyst that immobilized the enzyme that catalysed the breakdown of cellulose into glucose: cellulase.
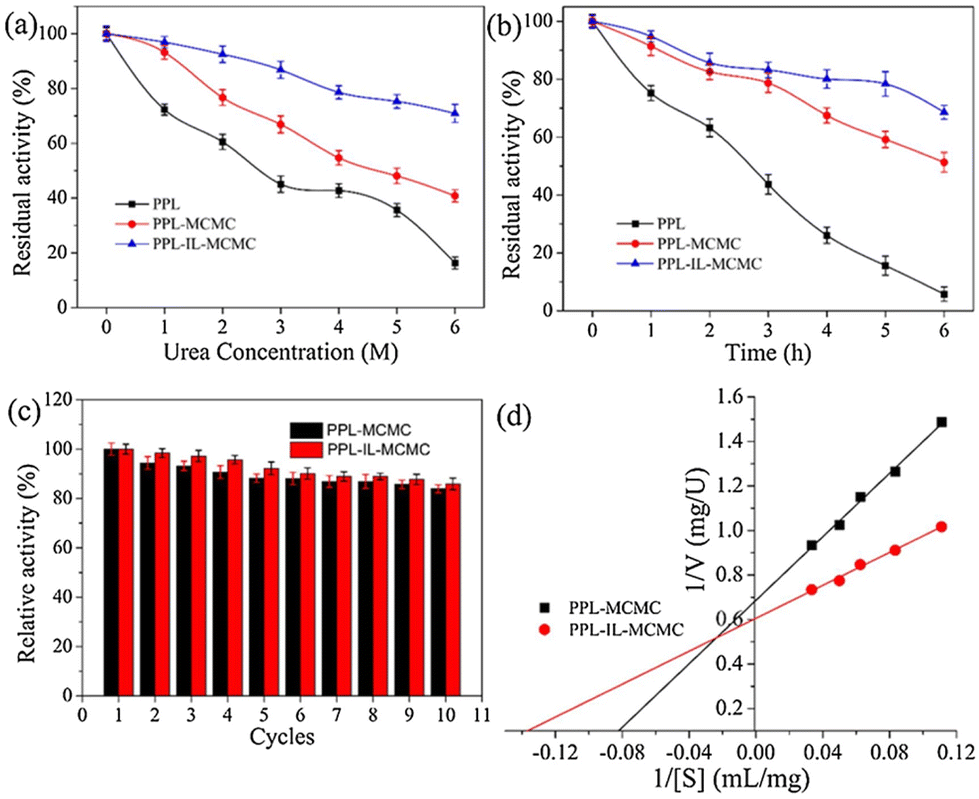 | ||
| Fig. 8 Effect of the CMC-hydrogel matrix on the stability (a–c) and kinetic parameters (d) of lipases. Reproduced from ref. 7 with permission from Elsevier, copyright 2020. | ||
As compared with free enzyme, immobilized cellulase showed greater stability at high temperatures (≥60 °C) and storage capacity and activity. Luo and co-workers49 created porous magnetic cellulose microspheres (MCMs) activated by epoxy chloropropane to boost the covalent immobilization of penicillin G acylase (PGA) to increase the effectiveness of catalyst recovery. The immobilized PGA displayed strong catalytic activity, improved pH tolerance, and enhanced thermal stability. The catalysts were recovered by separating the enzyme-loaded matrix from the reaction solution. In the quest of producing protein-friendly biomaterial for biocatalysis and biotechnological applications, our research team50 synthesized tendril-like functional carbon helices (TLFCHs) from lignocellulosic biomass via a “green” solvothermal technique utilising deep eutectic solvent (DES) as a soft template and catalyst. Exploiting the benefits of helicity and in-built chemical functionalities, we immobilized cytochrome c (Cyt c) on the surface of TLFCHs (Fig. 9). At optimized conditions, the specific activity, pH stability, and peroxidase activity of Cyt c were increased without impacting the structural integrity of the protein. Furthermore, lignocellulose-based hydrogels possess the advantages of low cost, biocompatibility, biodegradability, hydrophilicity, non-toxicity and, most importantly, controllable properties.17,51–62 Hydrogels based on lignocellulose have revealed promising applications in biocatalytic, biomedical, and bioelectronic areas.51,52 Jo et al.51 generated cellulose hydrogel microspheres by sol–gel transition employing a 1-ethyl-3-methylimidazolium acetate ([Emim][Ac])-in-oil emulsion for immobilization of lipase. Immobilized lipase showed greater efficiency as compared with the native enzyme. In contrast to lipase immobilized on MCC or mm-sized hydrogel beads, lipase immobilized on cellulose microspheres demonstrated a much greater loading efficiency, immobilization yield, and specificity constant.51 In another study,52 cellulose and lignin were co-dissolved in an IL to produce cellulose/lignin composite hydrogel beads. The activity, protein loading, and specific activity of the lipase immobilized on the cellulose/lignin beads were 2.6-, 2.2-, and 1.2-times higher than those of the lipase immobilized on cellulose beads, respectively.52 Kim et al.53 extended this work by conjugating cellulose with other bio-polymers, such as chitosan, carrageenan, agarose and agar. Candida rugose lipase (CRL) immobilized over these hydrogels in the presence of [Emim][Ac] demonstrated greater immobilization yield compared with that of cellulose beads.53 In addition to the cellulose materials mentioned above, chemically altered nanoscale cellulose materials make excellent matrices for enzyme immobilization. Arola and co-workers17 immobilized proteins over NFC through amine, epoxy, and carboxylic-acid functionalization.17 The NFC structure is beneficial for the stability and catalytic activity of proteins.17 Organophosphorus hydrolase (OPH) from Flavobacterium ATCC 27551 was immobilized on powdered plant cellulose treated with epoxy (Fig. 10) by Sharifi et al.54 The immobilized OPH demonstrated better storage, temperature, pH, and reusability properties than the free enzyme. Apart from epoxy groups, introducing aldehyde and carboxyl groups on cellulose matrices is an effective approach to attach them with the amino groups of enzymes.56,57
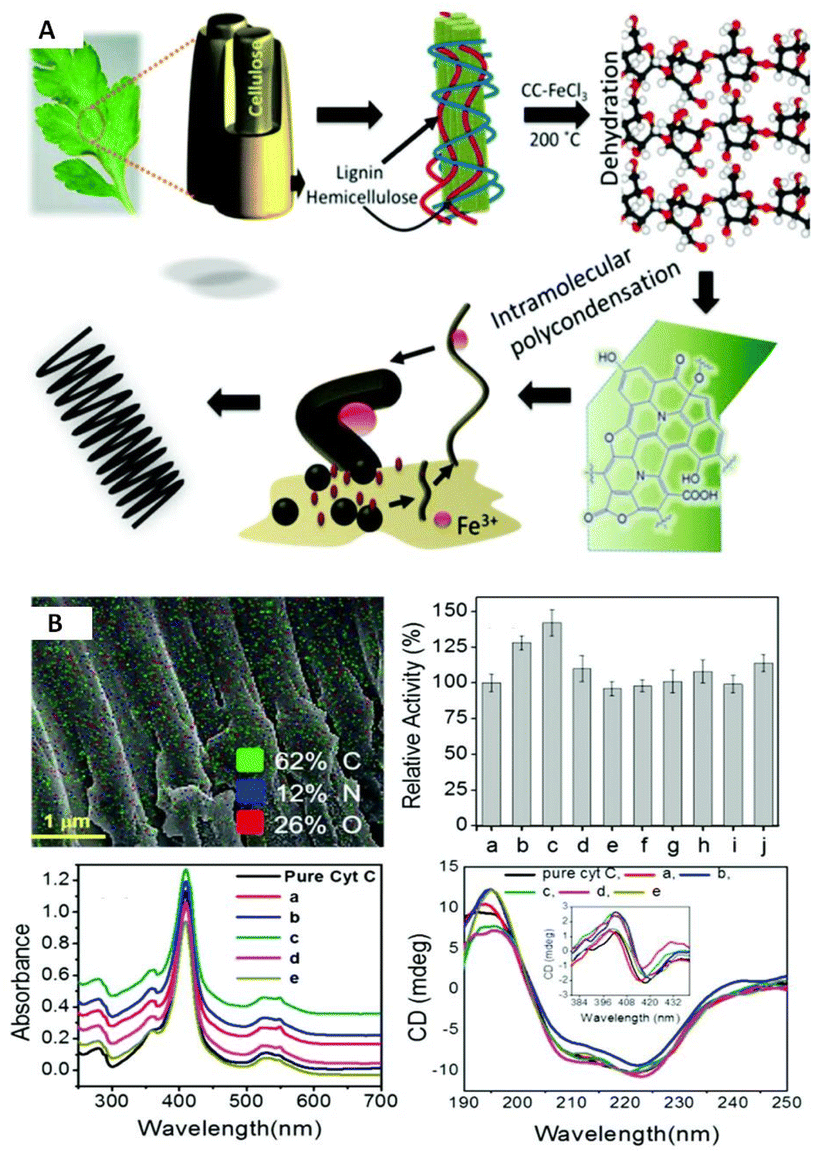 | ||
Fig. 9 Schematic depictions of (A) a plausible mechanism of the sequential growth of TLFCHs from Parthenium biomass during a solvothermal process in the presence of a DES (CC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :FeCl3, 1:2 mol ratio) (B) Cyt C immobilized on TLECH and showing activity and stability results (ref. 50). :FeCl3, 1:2 mol ratio) (B) Cyt C immobilized on TLECH and showing activity and stability results (ref. 50). | ||
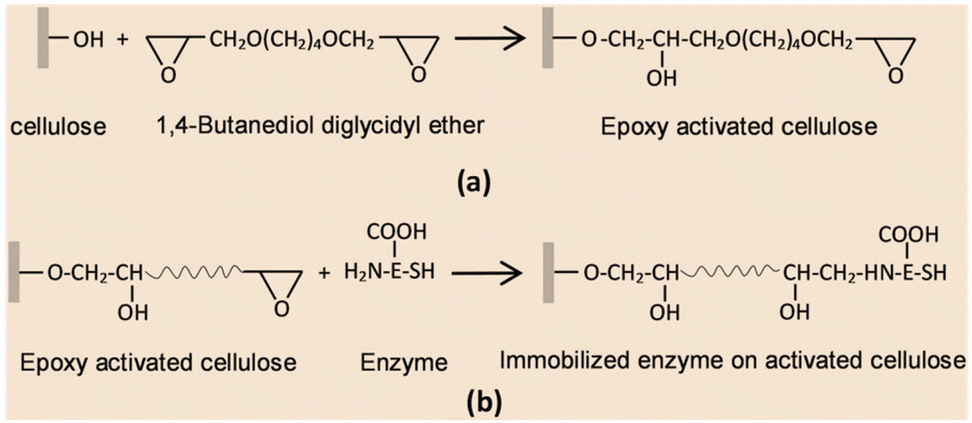 | ||
| Fig. 10 Plausible reaction mechanism of enzyme immobilization through the epoxy method. (a) Surface reaction of a cellulose matrix with 1,4-butanediol diglycidyl ether (BTDE). (b) Covalent coupling of enzyme onto the surface of activated cellulose. Reproduced from ref. 54 with permission from Elsevier, copyright 2018. | ||
Different chemical processes are used to oxidise the hydroxyl groups in cellulose to produce aldehyde or carboxyl groups. For example, Hao et al.56 used sodium periodate (NaIO4) for oxidizing cotton fibres to develop aldehyde groups in them.56 A similar study was reported for oxidation of cotton yarn by NaIO4 to introduce aldehyde groups and further used for immobilization of trypsin.57 The highest concentration of immobilized trypsin was 6.1 mg g−1 of dry cotton yarn. Over 60 days of storage, the activity of immobilized trypsin increased, and revealed >90% and >70% of the initial activity at 4 °C and 25 °C, respectively.57
In another study by the Gong group,58 NaIO4 first oxidised the cellulose in a loofah sponge to produce aldehyde groups. Then, the oxidised loofah sponge was used as a carrier for the covalent immobilization of lipase. The immobilized lipase provided better thermal stability, storage stability, and reusability. Besides periodate, 2,2,6,6-tetramethylpiperidine-1-oxyl radical (TEMPO) was used by Qian and co-authors to control the distribution of carboxylic groups over CNF macrogels, over which phospholipase from Thermomyces lanuginosus was immobilized and showed great reusability.43
To cutdown the time frame of purification and immobilization processes, Gennari et al.,59 engineered a single-step methodology by altering the surface of MCC with CBD. β-Galactosidase from Kluveromyces sp. was simultaneously purified and immobilized over this carrier and showed high stability at up to 40 cycles for the hydrolysis of milk lactose. Various other types of cellulose matrices have been used as carriers for enzyme immobilization (Table 1).63–85 To improve the tolerability of enzymes in enzymatic membrane bioreactors (EMBRs), it is crucial to immobilize enzymes on supports while retaining their structure and activity. In a recent study, Liu60 described a unique technique utilizing dicarboxylic acid halides as spacers for surface-initiated, covalent immobilization of enzymes onto cellulose microporous membranes. Sebacoyl chlorides, dicarboxylic acid halides, exhibit excellent reactivity with hydroxyl groups and amino groups, which are easily introduced onto membrane surfaces (Fig. 11). Trypsin and lipase were immobilized on the membrane surface while maintaining their molecular structure and activity substantially. The corresponding membrane demonstrated outstanding specific activity with strong activity retention (>25%) in a soaking mode at the optimal surface density. Moreover, the immobilized enzyme exhibited outstanding enhancement in reusability, thermal resistance, and continuous-operation capability.
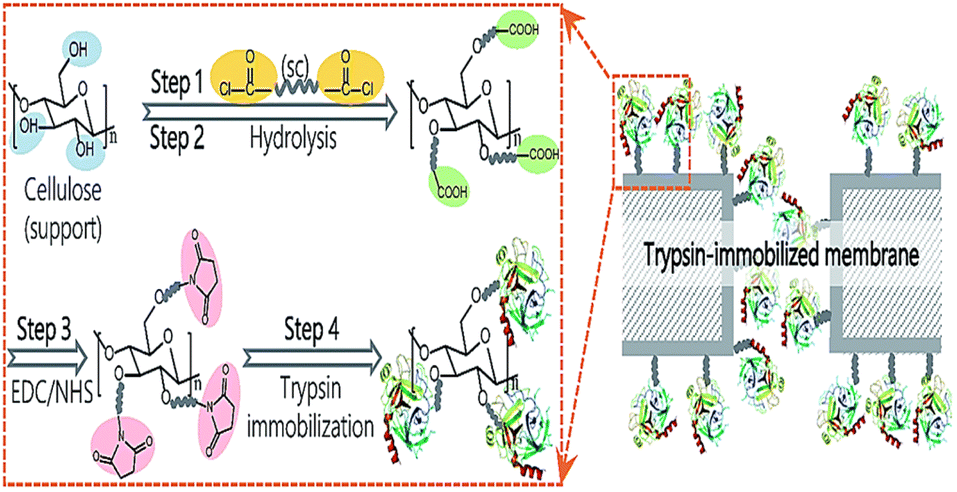 | ||
| Fig. 11 Proposed pathways for trypsin immobilization on regenerated cellulose membranes. Reproduced from ref. 60 with permission from RSC, copyright 2017. | ||
| Biomass | Biomass-derived material | Used enzyme | Immobilization method | Result/performance study | Ref. |
|---|---|---|---|---|---|
| Cellulose | Spin coating of protein NFC conjugates | Alkaline phosphatase | Conjugation | No loss in activity at 21 °C and >20% remaining activity after a week of incubation at 37 °C | 17 |
| Lignocellulosic | Tendril-like carbon helices | Cytochrome c | Enhanced structural stability and >150% higher activity than native Cyt c | 50 | |
| Cellulose | CHM | Lipase | Physical adsorption | 1.4 times higher specific activity, 41 folds half-life (at 45 °C), enhanced thermal and pH stabilities | 51 |
| Lignocellulosic | Cellulose/lignin hydrogel beads | Lipase | Physical adsorption | 2.6, 1.2 folds higher relative and specific activity than lipase-cellulose bead system. >3 times higher thermal stability and against lower pH (i.e. 3) | 52 |
| Wood mimetic | Cellulose–biopolymer composite hydrogel beads | Lipase | Entrapment | Improved reusability with >80% activity retention | 53 |
| Cellulose | Cellulose based membranes entrapping enzymes over the electrode | GOx | Entrapment | The enzyme electrode was stable at for least 6 months. Protected the leakage of enzyme. Glucose detection up to 10 μM with response time of ∼10 s | 55 |
| Cotton knit fabrics (cellulose) | Cotton fabrics oxidized by periodate | Cellulase | (1) Enhanced binding capacity of cellulase (>10.3 mg g−1). (2) Enhanced cotton hydrolysis and cellulase potential for adsorption | 56 | |
| Cotton yarn (cellulose) | Cotton yarn oxidised with sodium periodate | Trypsin | Covalent binding | Retained the initial activity after 60 days of storage in physiological solution | 57 |
| Cellulose | Cellulose-CBD | β-Galactosidase | Reusability for milk sugar hydrolysis till 40 cycles with 64% activity retention | 59 | |
| Paper wastes (PW) | Fe3O4 and chitosan functionalised αCFs | Laccase | Covalent bonding | Great loading capacity of >73 mg g−1; ∼92% activity recovery and excellent reusability capability with ∼74% activity retention at the end of 11th cycle | 61 |
| Waste newspaper | Dialdehyde-modified cellulose nanocrystals (DMC) | Laccase | Covalent bonding | 64.94% yield, excellent stability, and reusability | 62 |
| Cellulose | Magnetic dialdehyde cellulose nanoparticles (MDC) | Rhizopus lipase | Cross linking | Enhanced long term stability with recovery rate >50% after 30 days | 63 |
| Sugarcane bagasse | CNC | Lipase | Covalent bonding | With optimum pH shifted to 8.25, found best suited to catalyse a triglyceride lipolysis reaction from palm oil | 64 |
| Nanocellulose from almond shell | Nanocellulose (NC) extracted using p-toluenesulfonic acid (PTSA) and sulfuric acid (ASS) with sugar-based natural deep eutectic solvent (NADES1a) as a biocatalyst system | CRL | Significant improvement in half-life i.e., 14 days greater than native enzyme | 65 | |
| Cellulose | Cellulose beads | Glucose oxidase (GOx) | Effective antimicrobial action against Pseudomonas aeruginosa, Escherichia coli and two of the methicillin-resistant Staphylococcus species | 66 | |
| Cellulose | Nonwoven cotton fabric | Lysozyme | Cross linking | Enhanced antimicrobial activity and storage stability | 67 |
| Cellulose | CMC-silver nanoparticle (AgNp)-silica hybrids | Amylase | Adsorption | Activity increased and maintained the activity at room temperature (40 °C) for 15 days | 68 |
| Cellulose | Cellulose powder, cotton buds, disc make-up remover pads, cotton and linen tissues | Lipase | Covalent bonding | Increased effectiveness in removing aged linseed oil layers in 45 min at pH 6 and 40 °C | 69 |
| Cellulose from coffee filter | Cellulose nanofibers (CNFs) | α-Chymotrypsin (α-CT) | Enzyme precipitate coating (EPC) | Magnetic separation technique proved to be the best with >38 times higher activity than other methods. For long term, precipitate coating method proved to be superior with >70% activity retention even after 1 month of incubation | 70 |
| Cellulose | TEMPO-oxidized cellulose fibres used to build cellulose-based microspheres | Phospholipase | Covalent bonding | Showed a greater thermal stability and resistance to pH, as well as easy recovery and reusability | 71 |
| Cellulose | Dialdehyde cellulose | α-Amylase | Covalent binding | Lys142 has been found to be involved in α-amylase immobilization to dialdehyde cellulose | 72 |
| Cellulose | Magnetic dialdehyde cellulose (MDAC) | Bacterial laccase | Cross linking | (1) With >210 mg g−1 loading capacity and >34 emu g−1 of magnetization, the material offers 10 cycles of reusability with >70%. (2) Efficient against crystal violet decolouration | 73 |
| Cellulose | Microcrystalline cellulose (MCC) magnetic support | β-Galactosidase | >90% immobilization efficiency, 1.2 folds higher substrate affinity, 7 times higher thermal stability and recyclability up to 15 cycles | 74 | |
| Nanocellulose (NC) from an agro-waste of quinoa husks (QS) | Nanocellulose nano-carrier | Laccase enzyme (PersiLac1) | Adsorption | Having ∼98% and ∼60% dye degradation capacity pertaining to Malachite green and Congo red respectively, biocatalyst can be reused with >83% initial activity after 18 h | 75 |
| Cellulose from Balsa wood | Balsa wood-derived cellulose scaffold crosslinked with glutaraldehyde | Horseradish peroxidase (HRP), glucose oxidase (GOD), and catalase (CAT) | Adsorption | >90% phenol degradation rate and 95% sodium gluconate yield were achieved in the reactor | 76 |
Gajanan et al.61 used paper wastes (PWs) to extract multifunctional α-cellulose fibers (αCFs), which were further tuned with magnetic Fe3O4 and chitosan. This system was successfully employed for immobilization of laccase (Lac) with loading capacity of >70 mg g−1 and >90% of activity recovery. Due to excellent pH, storage and temperature stabilities, authors claimed that the catalytic system could be applied for the degradation of DR28 (xenobiotic benzidine-based azo dye).61 Overall, the unique microstructure of cellulose helps in improving the long-term stability and prevents back-diffusion of enzymes, which makes it an appropriate immobilization matrix for enzymes (Table 1).
3. Lignin-based supports (carriers) for enzyme immobilization
Lignin is a valuable and less explored natural resource. It is an intriguing class of material that has become a popular alternative in the search for more affordable and environmentally acceptable materials for enzyme immobilization. It possesses high strength and stability at extremes of pH, temperature, or pressures (Table 2).86–94 Weihua et al.86 demonstrated the use of lignin particles from bamboo shoot shells (BSSs) as novel carriers for immobilization of α-amylase from Bacillus subtilis with a maximum protein load of 19.0 mg g−1 in 20 min.86 The immobilized enzyme was superior compared with the free enzyme in terms of overall catalytic efficiency, storage stability, and recovery. Bebić and co-workers87 revealed amino-modified microspheres of kraft-lignin as potential supports for the immobilization of β-galactosidase and Lac enzymes. The former was used for the selective synthesis of galacto-oligosaccharides, whereas the latter was employed for the degradation of a pesticide (lindane). Lac immobilized onto modified kraft-lignin showed 3-fold greater efficiency in degrading the pesticide than amino-modified silica nanoparticles.87 Furthermore, multifunctional lignin/Fe3O4 nanoparticles were synthesized using green coconut fibres, which were used to remove colours from the effluents of the textile industry and to achieve a high stability for β-glucosidase.88 Immobilized β-glucosidase revealed significant adsorption capacity for the elimination of methylene blue (203.66 mg g−1), remazol red (96.46 mg g−1), and cibacron blue (112.36 mg g−1). It also showed excellent digestion performance towards crystalline cellulose compared with that of the native enzyme.88 Therefore, the proposed approach proved successful in obtaining lignins isolated from lignocellulosic residues.88 Łukasz et al.89 designed a hybrid TiO2/lignin support for the covalent immobilization of Aspergillus oryzae α-amylase. The immobilized enzyme displayed improvement in chemical and thermal stabilities with 10 cycles of reusability, and after 1 month of storage its initial activity was retained at >80%. Considering sustainable and green synthetic protocols, Capecchi and colleagues90 described a method of preparing sustainable bio-ink ingredient from poly(diallyldimethylammonium chloride) (PDDA) or chitosan-functionalized organosolv lignin nanocapsules. Pigments were catalysed by Lac and tyrosinase immobilized over functionalized lignin nanocapsules via a layer-by-layer (LBL) immobilization strategy.| Biomass | Biomass-derived material | Used enzyme | Immobilization method | Result/performance study | Ref. |
|---|---|---|---|---|---|
| Dendrocalamus Latiforu | BSS lignin as a support material | α-Amylase | Adsorption | With 19 mg g−1 loading capacity, immobilized enzyme showed >2-folds higher activity with >53% activity retention after 14 recycles | 86 |
| Bio-waste of kraft pulp lignin | Amino-modified microspheres (A-LMS) | β-Galactosidase | Electrostatic interaction | 1.5 times higher rate of galacto-oligosaccharide production and efficient in degradation of pesticide – lindane | 87 |
| Green coconut fiber (GCF) | Lignin/Fe3O4 nanoparticles synthesized by organosolv pre-treatment | β-Glucosidase | Covalent bonding | (1) Improved digestibility performance (>21 g L−1 of cellulose and >6 g L−1 of green coconut fibre) and stability. (2) Good synergism with cellulases during enzymatic hydrolysis. (3) High adsorption capacity of nanoparticles towards Methylene blue (203.66 mg g−1), Cibacron blue (112.36 mg g−1) and Remazol red (96.46 mg g−1) dyes | 88 |
| Kraft lignin | Novel hybrid support made of titanium and lignin | α-Amylase from Aspergillus oryzae | Covalent bonding | Improved pH range of 3–7 and best suited for low temperature catalytic applications (5–10 °C) | 89 |
| Lignin | Lignin nanocapsules | Tyrosinase and laccase | Used for the synthesis of bioinks with different shades. The nanocarriers found to prevent alkaline and UV degradation of synthesized melanin | 90 | |
| Organosolv lignin (OL) | LNPs | Enzymatic cascade of lipase-tyrosinase | Layer-by-layer method | Successfully synthesised high yield lipophilic hydroxytyrosol esters | 91 |
| Kraft lignin | Chitosan-coated LNPs | Glucose oxidase (GOx) | Adsorption | Enhanced the enzyme activity up to 70° C and display of self-scavenger property towards H2O2 | 92 |
| Lignin | Lignin-based spherical particle | Lipase | Electrostatic interaction | Retained 75% and 81% relative activity at higher temperature (60 °C) and after 10 reuses respectively | 94 |
The lignin nanocapsules were employed as multifunctional devices acting as scaffolds, activators for enzymes to catalyse pigment synthesis and, finally, being the vehicle or binder for pigments. L-Tyrosine, epicatechin, 3,4-dihydroxyphenyl-L-alanine (L-DOPA), and several other derivatives of phenol were used as reactants giving out melanin pigment with high alkaline and UV tolerance due to interaction with nanocapsules. Similarly, Tomaino et al.91 synthesized one-pot esterification of lipophilic hydroxytyrosol esters by a lignin nanoparticles (LNPs)-supported enzymatic cascade. Tyrosinase and lipase M were immobilized on LNPs by an LBL method using chitosan as a positive-charged polyelectrolyte and concanavalin A acting as molecular spacer to regulate the optimal separation between the active sites of the two enzymes. Interestingly, compared with the native counterparts, both immobilized enzymes on LNPs showed greater kinetic parameters (maximum reaction speed (Vmax) and Michaelis constant (Km)). By further extending the LBL strategy, Moreno and colleagues92 prepared a multifunctional particulate emulsifier where GOx was immobilized over chitosan-coated LNPs (Fig. 12).
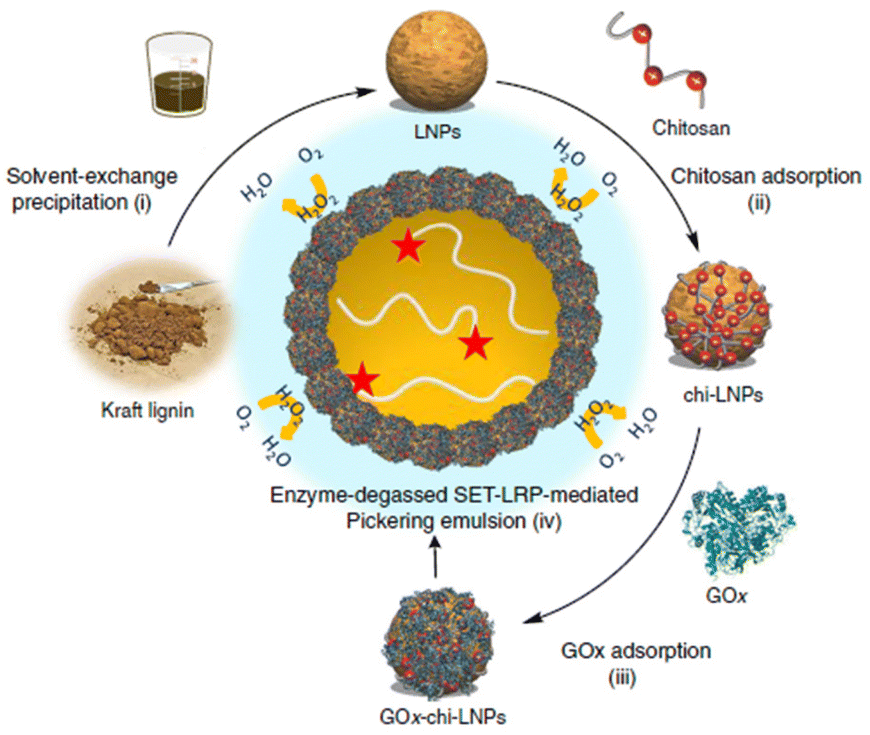 | ||
| Fig. 12 Preparation of GOx-chi-LNPs and application in enzyme-degassed SET-LRP-mediated Pickering emulsion (Schematic). (i) Preparation of LNPs via solvent-exchange precipitation. (ii) Adsorption of chitosan on LNPs to yield chi-LNPs. (iii) Adsorption of GOx on chi-LNPs to yield Gox-chi-LNPs. (iv) Application of GOx-chi-LNPs as biocatalytic degassing stabilizers for SET-LRP in Pickering emulsion media. Reproduced from ref. 92 with permission from Nature Portfolio, copyright 2020. | ||
This approach proved to be efficient in stabilizing the Pickering emulsions and in situ enzymatic degassing of single electron transfer-living radical polymerization (SET-LRP). Because of the auto-degradation of H2O2, GOx is protected against the oxidative stress caused in situ, leading it to withstand temperatures higher than the optimum. This rational design of the process is a breakthrough in avoiding the use of additives for SET-LRP and in opening new avenues for the green synthesis of block co-polymers having complex architectures. For the first time, applications of functionalized lignin as a matrix for immobilizing a wide range of enzymes (transaminase, carboxylase, dehydrogenase) using various binding chemistries were carried out by Benítez and co-workers.93 As a proof of concept, ω-transaminase was incorporated into a packed bead reactor and reversibly immobilized on polyethyleneimine-lignin, and its endurance was investigated in continuous-flow deamination reactions maintaining the same conversion for 100 cycles. All of these results demonstrated that fully closed-loop sustainable flow-biocatalytic systems based on bio-waste and natural lignin material to be effective substitutes for typical immobilization supports.
4. Silk fibroin-based supports (carriers) for enzyme immobilization
Silk fibroin (SF) is a well-known and widely used natural protein procured from silkworm cocoons. Large numbers of serine, glycine and alanine residues are found in this macromolecular protein (Fig. 13),24,95,96 along with readily reacting chemical groups (e.g., imidazole, tyrosyl/phenol and sulfhydryl).97 Due of its low cost, abundance, nontoxicity, and biodegradability, SF makes an appealing material for enzyme entrapment. Its peculiar amino-acid sequence and structure result in the formation of discrete nanoscale pockets and ensure that a sufficient number of water molecules is retained for protein interaction and stabilization, which make it a promising substrate for enzyme immobilization.16 The distinctive structure of SF protein (α-helices, β-sheets and random coils) synchronize to create a propitious space for stabilizing entrapped enzymes. SF can be transformed into fibres, powdery nanoparticles or microspheres, membranes, gels, films, hydrogels, or scaffolds under mild, ambient, or aqueous conditions.90 Numerous studies have shown that SF can serve as a matrix for immobilization, and thereby to impart stability to a variety of enzymes.87–101 Recently, various works have been published addressing the use of silk biomaterials as excellent supports for the covalent and noncovalent immobilization of enzymes with high retention of catalytic activity and stability against various stresses (Table 3).100–114 Silk nanofiber (SNF) is an excellent candidate for enzyme immobilization due to superior stiffness, larger specific surface area, higher interconnectivity and porosity with high capacity for loading enzymes. In this regard, Lee et al.91 prepared SF nanofibers by electrospinning and applied it for immobilization of α-CT.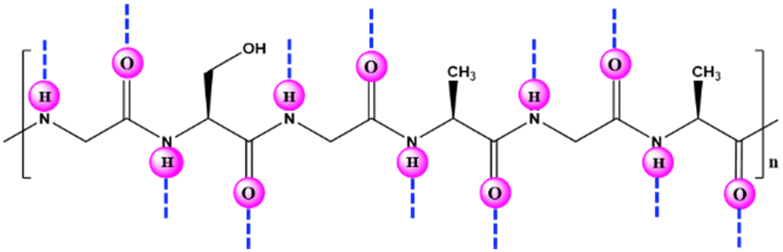 | ||
| Fig. 13 Chemical structure and illustration of the possible hydrogen bonding (––) sites in silk fibroin. | ||
| Biomass | Biomass-derived support material | Used enzyme | Immobilization method | Result/performance study | Ref. |
|---|---|---|---|---|---|
| Bombyx mori silkworm cocoons | Silk fibroin film | GOx | Entrapment | Activity maintained more than 10 months of preservation up to 37 °C of temperature | 23 |
| Bombyx mori silkworm cocoons | Silk fibroin film | HRP | Entrapment | Even 2 months later, 90% of activity retained at room temperature | 23 |
| Silkworm cocoons | Silk Fibroin nanofibers | α-CT | High loading capacity of 5.6% with 8 times higher activity than the one immobilized on silk fiber and >90% activity retention after 24 h incubation | 99 | |
| Woven Muga Silk | Glutaraldehyde – crosslinked silk fibers | Lipase | Cross linking | 12% lower activity than native enzyme but significant conversion rates in the emulsion phase with 3 cycles of recycling | 100 |
| Bombyx mori silkworm cocoons | Hydrophobic silk fiber treated with polydimethylsiloxane (PDMS) | Lipase | Adsorption | 2-Fold increase in the activity and improved thermal stability | 101 |
| Bombyx mori silk | Silk fibroin crosslinked with tyrosinase using glutaraldehyde | Tyrosinase | Cross linking | (1) A higher degree of stability. (2) 80% of initial activity retained even after multiple times reuses | 102 |
| Woven Muga silk | Silk mat using N-ethyl-N′-(3-dimethylaminopropyl carbodiimide) EDC-NHS (N-hydroxysuccinimide) linkage | ChOx | Covalent bonding | Greater catalytic activity and structural stability sustaining for 13 months and 25 individual assays | 103 |
| Bombyx mori silk | Silk fibroin-based hydrogel | CA | (1) ∼100% immobilization efficiency. (2) Stable against pH (pH-3) denaturation | 104 | |
| Bombyx mori cocoon | Silk film by entrapment | β-Glucosidase | Entrapment | Enhanced stability against various stresses such as heat, electrodialysis, and protease | 105 |
| Bombyx mori cocoons | Silk film entrapment | OPH | Entrapment | (1) Excellent long term stability with >60% activity even after 17 weeks of incubation. (2) Higher stability against UV radiation and extreme temperature | 106 |
| Bombyx mori silk cocoons | Silk powder by entrapment | Invertase | Entrapment | Enhanced thermal stability | 107 |
| Bombyx mori silk cocoons | Powder of Silk-ASNase bioconjugates by glutaraldehyde | ASNase | Bioconjugation | Improved half life (63 h), 6 times higher substrate affinity, resistance to trypsin digestion and good thermostability | 108 |
| Bombyx mori silk cocoon and lignin | Hydrogels composite made of silk fibroin and alkaline lignin (SF-AL) | Laccase (Lac) | Cross-linking | Enhanced working range of pH from 4.5 to 9 | 109 |
| Bombyx mori silk cocoons | SF nano-cocoons | GOx and HRP | Encapsulation | Improved the stability and efficiency of the catalytic reaction, as well as the temperature stability and storage stability | 111 |
| Bombyx mori silk cocoons | Silk fibroin hydrogel | Microalgae | Entrapment | Good mechanical strength and stability of material with 6 times higher oxygen generation compared to controlled algal culture | 112 |
| Bombyx mori silk cocoons | SF | GOx | Encapsulation | Biocompatible and biodegradable sensor with improved current response of 0.2 μA mM−1 | 113 |
| Bombyx mori silk cocoons | SF | Protease | Good extraction efficiency | 114 | |
| Silk | A film made of sericin, polyvinyl alcohol, and cassava starch | Botryosphaeria ribis EC-01 lipase | Adsorption | >9.8 folds higher activity, better yield, and recyclability up to 7 cycles | 115 |
| Bombyx mori silkworm cocoons | Sensor-based on regenerated silk fibroin film | HRP | Entrapment | Improved detection limit of 100 nM with <40 s response time | 116 |
| Bombyx mori silkworm cocoons | Porous membranes of Bombyx mori silk fibroin | GOx | Entrapment | Improved catalytic activity and thermal stability with >20 times increased permeability of NaCl and glucose | 117 |
| Bombyx mori and Philosamia cynthia ricini silk | SF membrane | GOx | Entrapment | Improved shelf life with lower extent of leakage (0.05%), and improved thermal stability at 60 °C | 118 |
| Woven silk | Immobilization on silk Fibre using the diazo method | Ribonuclease | Adsorption | 63% of activity retained for 7.2 months | 119 |
| Bombyx mori silk cocoons | Silk powder by entrapment | Phenylalanine ammonia-lyase | Entrapment | Improved resistance against chymotrypsin and trypsin | 120 |
| Bombyx mori silk cocoons | Entrapment with silk blend membrane and polyvinyl alcohol (PVA) | GOx | Entrapment | Improved catalytic activity | 121 |
| Bombyx mori silk cocoons | Graphene silk fiber nanocomposite decorated by platinum nanosphere | GOx | Cross linking | Stable conductivity of ∼57 S m−1 with an improved sensitivity of 150 μA mM−1 cm−2 and detection limit of 1 μM | 122 |
The activity of immobilized α-CT on SF nanofibers was shown to be eight-times more active than that of bulk material, and the activity was indirectly proportional to fibre diameter.99 Many reaction sites (e.g., –NH2, –COOH, imidazole and phenol) are provided by the diverse amino-acid constituents of silk fiber. Lipase was immobilized onto SF using a chemical method.100 This lipase was then used for the hydrolysis of Helianthus annuus (sunflower) oil for producing fatty acids.100 Chen and co-workers101 designed an inexpensive woven fabric support made from SF for the immobilization of Canadida sp. lipase. The stability and activity of immobilized lipases on two types of silk fabrics were examined, and it was shown that the lipase immobilized over the hydrophobic fabric had 2-fold higher esterification and hydrolysis activity than the lipase immobilized on native silk fibres. In comparison with free lipase, lipase after immobilization demonstrated stability over a larger pH range and a change in its optimum temperature. The method described in this work indicates that woven SF might be an ideal material for use as an immobilization matrix in industrial preparations.101 Similar to silk fibers, silk films offer handy and extremely efficient supports for the longstanding stability of enzymes that are confined. Lu and co-workers23 investigated the stabilization of horseradish peroxidase (HRP), lipase, and GOx entrapped in self-assembled SF films with higher enzyme loading. Among the three enzymes studied, GOx retained significant activity over 10 months of shelf storage at a wider temperatures range until 37 °C. The extent of molecular interaction between enzyme molecules and SF chains, the susceptibility of the enzyme to undergo oxidation, and hydrophobic–hydrophilic interfaces have been postulated to influence the relative stabilizing outcomes among the three enzymes used in that study.23 Acharya et al.102 explored an alternative and efficient method for the biosynthesis of L-DOPA from tyrosine using tyrosinase immobilized onto a novel silk protein: fibroin.102 Saxena and co-workers103 further demonstrated silk mat produced by weaving silk fibres of Antheraea assamensis as a promising biocompatible matrix for cholesterol oxidase (ChOx).103 The porous and fibrous structural morphology of the silk mat provided an ambient microenvironment for immobilizing ChOx with loading efficiency of >70%, which resulted in a good analytical display with higher sensitivity, stability, reproducibility and much greater selectivity for the substrate (cholesterol).103 On the other hand, protecting enzymes and proteins from undergoing inactivation induced by pH is a significantly difficult task. Taking up the challenge, Han and co-authors demonstrated that bombyx mori SF-based hydrogel with Ru(II)-mediated photo-chemical crosslinking served as an efficacious carrier for protecting an immobilized enzyme, carbonic anhydrase (CA), from pH denaturation.104 The free enzyme lost its activity entirely at pH 3, whereas the immobilized CA maintained >20% of its initial activity. These results implied that CA was stabilized to a greater extent in SF-based hydrogels than the non-immobilized counterpart. Furthermore, the authors applied the same method for xylanase and lysozyme. They found that the immobilized enzymes achieved comparatively higher activity even at an unfavourable basic pH of 9.104 Silk membranes have also been found to impart stability to enzymes against heat. Miyairi and colleagues105 used a fibroin membrane as a support to immobilize β-glucosidase. Apart from heat tolerance, the immobilized biocatalyst could counter the effects of electrodialysis and protease treatments as well.105 In addition, Dennis et al. demonstrated the value of SF entrapment for the preservation of OPH activity under a wide range of environmental circumstances, including high temperature, exposure to ultra violet light and to denaturing conditions imposed by organic reagents.106 An enhancement in thermal stability similar to this work has also been reported by Yoshimizu107 for SF powder over the enzyme invertase. In parallel, for treating acute lymphoblastic leukaemia, L-asparaginase (ASNase) is employed as a drug. However, to overcome its shortcomings in triggering an allergic response and a very short half-life, SF has been found to be suitable candidate for bioconjugation.108 In this regard, to lower the immunogenic responses and the innate property of antigenicity of the enzyme, Zhang and colleagues108 bioconjugated ASNase with polar functionalities of fibroin protein. The manipulated enzyme not only increased its stability against protease digestion (by trypsin), heat and storage, it also increased its half-life from 33 h to 63 h under laboratory conditions.108 SF protein also offers a promising strategy to exploit the tremendous capabilities of Lac.109 The adoption of gentle, aqueous processing techniques that were made possible by the specific chemistry, structure, and assembling of silk into nanodomains with low water content may be responsible for preservation of bioactivity during the manufacture of silk devices (Fig. 14).110
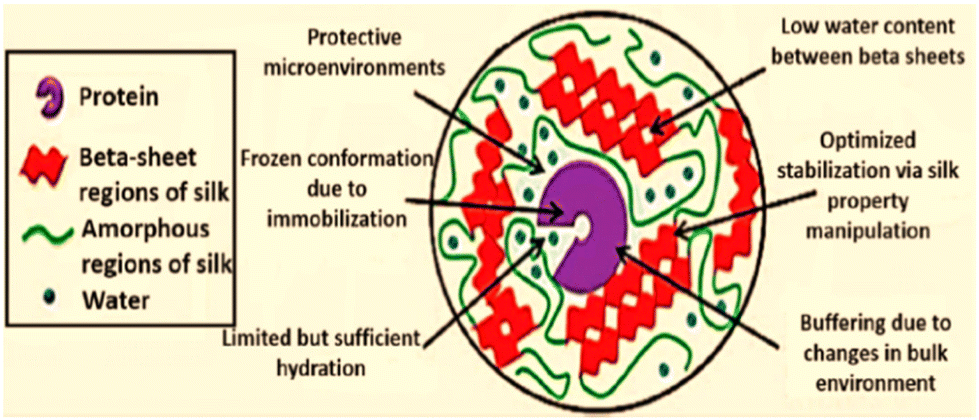 | ||
| Fig. 14 Summary of the effects of stabilization of silk fibroin on immobilized compounds. Reproduced from ref. 110 with permission from Wiley-VCH, copyright 2012. | ||
5. Chitin-based supports (carriers) for enzyme immobilization
Chitin is a biopolymer with potential applications in the biomedical sector.123,124 After cellulose, chitin is the second most abundant natural polysaccharide in the world.124–126 It is made up of β-(1–4) connected repeating units of N-acetyl-D-glucosamine (Fig. 15a) that can be found in the outer skeleton of arthropods, insects, marine invertebrates, and cell walls of some fungi.125–127 This biopolymer has many potential applications in medicine, food, cosmetic, agriculture, and immobilization processes due to its low toxicity and bioactivity, as well as high biocompatibility, biodegradability, and antibacterial properties.128–136 The supports based on chitin and its derivatives provide enzymes with good stability and affordable bioprocessing, and they can be used in various applications, ranging from agriculture to drug delivery. The enzymes bound to the chitin matrix also have greater thermal activity than that of free enzymes. Table 4130–150 displays examples of recent research that used chitin-based supports to enhance the stability and activity of enzymes.
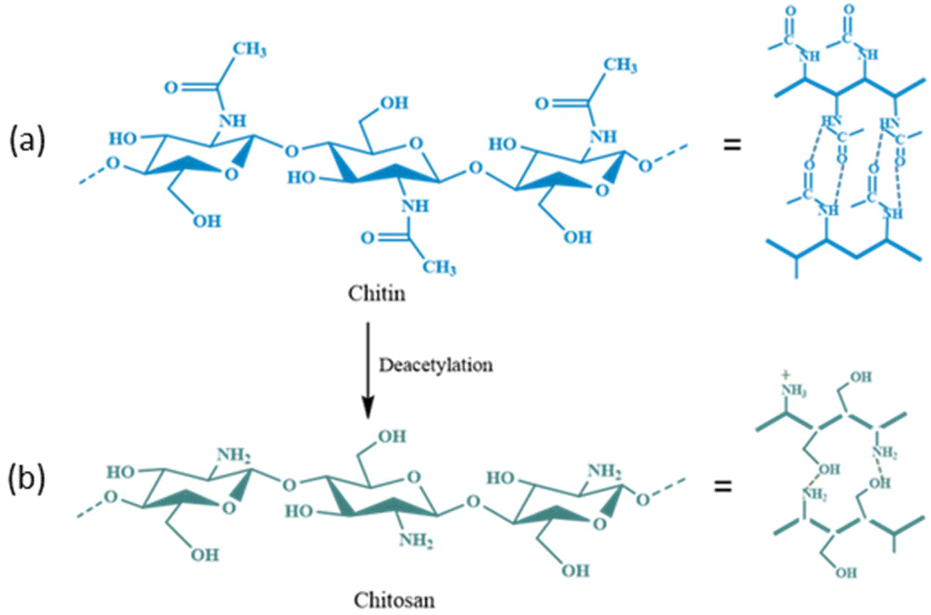 | ||
| Fig. 15 Chemical structure and possible intermolecular hydrogen bonding (––) in (a) chitin and (b) chitosan. | ||
| Chitin source | Chitin derived material | Used enzyme | Immobilization method | Result/performance study | Ref. |
|---|---|---|---|---|---|
| Chitin | Glutaraldehyde-activated chitin flakes | Amylase | Covalent bonding and crosslinking | Superior operational stability with ∼90% activity after 10 recycles and shelf life of >30 days with no loss in activity | 130 |
| Chitin from Krill | Krill chitin | Invertase and amyloglucosidase | Cross linking | Improved stability and activity | 131 |
| Chitin from squid pen | β-Crystalline chitin membrane | GOx | Entrapment | The enzyme-immobilized membrane is used for biosensors with wider linearity range of 0.125–2 mM | 133 |
| Chitin from crab shells | Hexamethylenediamine functionalized the chitin, and activated it with glutaraldehyde | CRL | Covalent bonding | Improved half-life (>425 h), excellent thermal stability, great activity yield and protein retention | 135 |
| Chitin from lobster shells | Sodium alginate-coated chitin support | Saccharomyces cerevisiae invertase | Electrostatic interactions | 97% initial activity retained and good thermal stability | 136 |
| Chitin from lobster shells | Hyaluronic-acid-coated chitin support | Baker yeast invertase | Polyelectrostatic interactions | Enhanced thermal stability, storage, and operational stability. 69% of enzyme activity retained at 37 °C after 50 days of storage | 137 |
| Chitin | Poly dopamine coated magnetic chitin (DMCT) particles with glutaraldehyde | α-Amylase | Cross linking | Immobilized α-amylase showed higher durability, magnetic recovery with 6 times reusability with >70% activity retained | 138 |
| Chitin | Immobilization of lysine decarboxylase (CadA) through fusion of a chitin-binding domain (ChBD) | Lysine decarboxylase (CadA) | Fusion of a chitin-binding domain | Better pH stability with >73% activity at pH8 and high molar yield of cadaverine (∼97%) | 139 |
| Chitin from lobster shells | Chitosan-coated chitin support with glutaraldehyde | Pectinase | Adsorption | Greater stability and after nine consecutive uses activity retained 100% | 140 |
| shells of shrimps and crabs | CHNW | Lysozyme | Adsorption | (1) Enhanced antimicrobial activity against Escherichia, Staphylococcus and Bacillus species. (2) Improved enzymatic activity (>1.5 folds) | 141 |
| Chitin | Chitin and starch linked as a support | Lipase and protease from sunflower seeds | Covalent binding | Enhanced the efficiency of detergents stain removal | 142 |
| Chitin powder | AuNPs immobilized on CNFs | GOx | Not mentioned | Enhanced colorimetric glucose detection with an LOD of 94.5 nM | 143 |
| Chitin | Aerogel beads made of chitin/graphene oxide (Ch/GO) composite crosslinked with glutaraldehyde | CRL | Adsorption followed by crosslinking | With immobilization capacity of >140 mg g−1, the biocatalyst can be reused for over 5 cycles with retention of 90% initial activity | 144 |
| Shrimp shells chitin | Chitin | Dextranase | Adsorption and covalent binding | A maximum of 88% immobilization yield achieved with superior 22 folds higher stability at 80 °C | 145 |
| Chitin | Magnetic chitin nanofiber composite (MCNC) | Chymotrypsin (CT) | Cross linking | Improved the loading capacity by 6.3 times after cross-linking, increased thermal stability and retained initial activity | 146 |
| Chitin | Chitin powder | α-Amylase from Bacillus subtilis ITBCCB148 | Adsorption | Immobilized α-amylase can be used up to five times and has an optimum temperature of 75 °C, increased the residual activity | 147 |
| Chitin | 3D fibrous chitinous scaffolds and silica nanopowder | HRP | Adsorption | Showed reusability of nano-SiO2 (HRP)-chitin (HRP) scaffolds and high efficiency towards removal of 17-α-ethinylestradiol (EE2) | 148 |
| Chitin | Chitin powder | Aspergillus Fumigatus α-Amylase | Adsorption | Improved thermal stability with >39% activity retained at 80 °C and 1.5 times higher activity at ambient conditions | 149 |
| Chitin | Chitin-Bentonite Hybrid matrix | α-Amylase from Aspergillus fumigatus | Physical adsorption | Enhanced stability with ∼4 folds higher t1/2 compared to native enzyme | 150 |
Important factors for choosing an appropriate enzyme support include the degree of chitin deproteinization, the availability of amino groups, the presence of minerals, mesh size, surface structure, and its conformation.131,132 The activity of invertase and amyloglucosidase has been improved by immobilizing them on chitin supports.131 Xavier et al.132 compared the characteristics of glucoamylase and α-amylase on two distinct types of support: ceramic and chitin. The initial activities of the glucoamylase complexes immobilized on chitin were five-times higher than those on ceramics, and the proportion of activity loss was two-times lower.132 The physical properties of chitin are also dependent upon its source. For example, due to the β-type crystalline structure, chitin from squid pens possesses superior physical qualities than chitin from crabs.133 Chitin from squid pens was converted into a thin membrane that was immobilized with GOx. The membrane had the potential to entrap enzyme molecules as well as sufficient glucose and oxygen permeabilities to be beneficial for biosensors. The immobilized GOx on the chitin membrane was combined with an oxygen electrode to create a glucose sensor that had good stability and a wide linearity range (0.125–20 mM).133 Immobilization methods can improved the properties of chitin further. To bind CRL, Gomes and co-workers135 functionalized chitin with hexamethylenediamine followed by activation of glutaraldehyde. The activity profile as a function of pH, temperature, and thermal stability was determined for free and immobilized lipases. Authors revealed that the immobilized CRL had higher thermal stability than free CRL.135 Leissy et al.136 described a novel technique for immobilizing enzymes that relied on creation of polyelectrolyte complexes with supports coated in polymers with opposite charge. They used chitosan to chemically modify Saccharomyces cerevisiae invertase, which was immobilized further on a chitin support coated with sodium alginate. The protein-immobilized yield and activity was 85% and 97%, respectively. After immobilization, the optimal temperature and thermostability of invertase were increased by about 9–10 °C. The immobilized enzyme was four-times more thermally resistant than its native counterpart at 65 °C and stable against incubation in solutions of high ionic strength.136 The authors suggested that the electrostatic immobilization technique they described might enhance the operational and functional features of the enzyme.136 In another study, the same authors137 described the immobilization of chitosan–invertase derivative on hyaluronic-acid-coated chitin. The immobilized enzyme was six-times more robust to thermal treatment at 65 °C than the native enzyme, and was stable against incubation in a solution of high ionic strength. Eighty percent of the initial activity of invertase was retained in the immobilized enzyme. The optimum temperature and thermal stability improved upon immobilization. The prepared biocatalyst showed exceptional temperature, storage, and operational-stability characteristics.137 The reusability and recovery of enzymes can be enhanced further by using magnetic-chitin (MCT) matrices,138 wherein magnetic granules were synthesized using chitin as a protective and dispersive matrix. The easily synthesised MCT particles with an average size of ∼1.5 μm were modified with dopamine to provide a useful matrix for immobilizing enzymes. Dopamine was self-polymerized and coated onto MCT to provide an enzyme-adhesive surface. An enzyme involved in starch hydrolysis, α-amylase, was immobilized on the polydopamine-coated MCT (DMCT). The effectiveness of enzyme immobilization on DMCT particles was boosted by glutaraldehyde treatment.138 Over a longer range of pH and temperature, the surface-immobilized α-amylase showed comparatively greater activity than that of the free enzyme. Also, the glutaraldehyde-treated polydopamine functionalized MCT microparticles with immobilized α-amylase had excellent durability, magnetic recovery and reusability.138 Zhou et al.139 used chitin as the carrier support for efficient immobilization of CadA through fusion with a chitin-binding domain (ChBD) for the synthesis of cadaverine from L-lysine.139 Furthermore, compared with wild CadA, the fusion protein (ChBD-CadA) had greater pH stability and maintained >73% activity at pH 8.139 In a batch conversion, the immobilized ChBD-CadA (I-ChBD-CadA) converted L-lysine (200.0 g L−1) to cadaverine (135.6 g L−1), obtaining a 97% molar yield of the substrate (L-lysine). Furthermore, I-ChBD-CadA was reusable at high L-lysine concentrations and retained >57% of its initial activity after four cycles of usage without the addition of acid to maintain pH (Fig. 16). These results indicate that immobilizing CadA with a chitin-binding domain could be used for the industrial synthesis of cadaverine.141 Ramirez and co-authors140 developed a novel bioconjugate for immobilization of pectinase on a chitosan-coated chitin support. As a result of immobilization, heat and temperature resistance were improved and the enzyme remained stable with 100% of activity retained after nine consecutive uses, and ∼70% of initial activity after 15 cycles of reuse.140
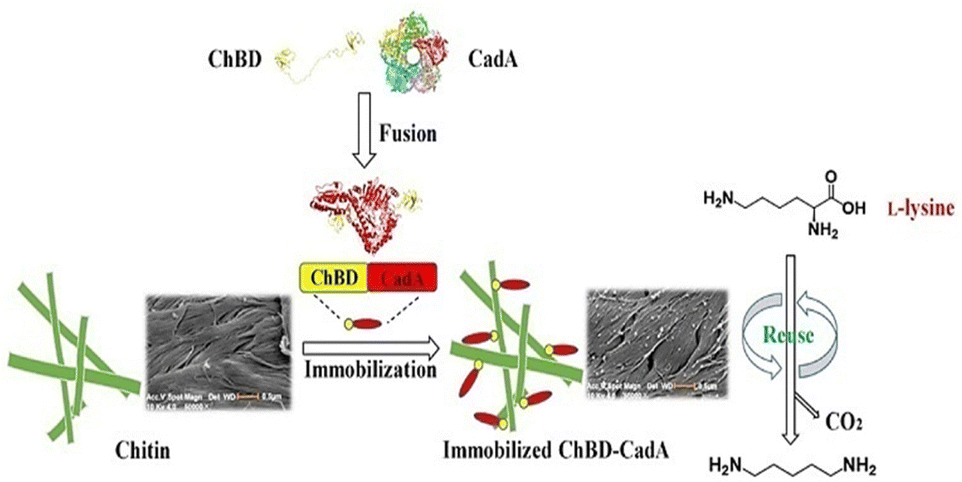 | ||
| Fig. 16 Immobilization of lysine decarboxylase for converting L-lysine to cadaverine via fusion of chitin-binding domains. Reproduced from ref. 139 with permission from Frontiers, copyright 2020. | ||
Chitin-derived supports can also be used to enhance the antibacterial activity of various enzymes. Suisui et al.141 investigated how chitin nanowhiskers (CHNWs) could enhance the antimicrobial and enzymatic activities of lysozyme adsorbed on CHNWs. Enzyme assays displayed that lysozyme-CHNW had 1.5-times higher enzymatic activity and greater antibacterial activity against Bacillus subtilis, Escherichia coli, and Staphylococcus aureus compared with those of the native lysozyme. These results revealed that lysozyme-CHNW could be used as a powerful antibacterial agent in food and medical fields.141 Mehdi and co-workers142 developed a unique method for immobilizing protease and lipase on chitin-starch material as an enzyme biocatalyst matrix. Compared with native enzymes, immobilized enzymes had better pH, thermal, reusability, and storage stability.142 Enzyme-immobilized chitin-coated nanoparticles are effective biocatalysts due to their surface area and mechanical strength.143,144 Yao et al.143 presented a simple green method for the heterogeneous synthesis of stable, size-controllable gold nanoparticles (AuNPs) immobilized on biocompatible chitin nanofibrils (CNFs). CNF-AuNPs showed peroxidase-mimic behaviour and catalysed the oxidation reaction of 3,3′,5,5′-tetramethylbenzidine (TMB) by H2O2. When coupled with GOx, with a limit of 94.5 nM, CNF-AuNPs could detect glucose sensitively. This method had excellent specificity and efficiency, was affordable, and suffered negligible contamination, which showed its importance for the diagnosis of diabetes mellitus.143 Chen et al.144 fabricated chitin/graphene oxide (Ch/GO) composite aerogel beads for lipase immobilization using chitin as a great blending material. The synthesised beads had a three-dimensional porous structure, and GO was attached tightly to chitin with enhanced mechanical strength and surface area. Under ideal conditions, CRL was immobilized on Ch/GO aerogel beads. The immobilized CRL demonstrated excellent thermal stability compared with that of the native enzyme and, even after recycling five times, the immobilized lipase maintained initial activity of >90%.144 Shahid et al.145 improved the functionality of dextranase (isolated from a thermophilic bacteria Bacillus megaterium KIBGE-IB31) with the help of its immobilization on chitin by various protocols. Adsorption and covalent binding strategies were used to immobilize the isolated dextranase on chitin. Compared with the enzyme immobilized by an adsorption approach, dextranase immobilized by covalent cross-linking demonstrated maximum stability at high temperatures combined with improved recycling efficiency. Thus, chitin appears to be an inexpensive and convenient matrix for immobilizing a variety of enzymes to increase the stability and reusability of enzymes at an industrial scale.
6. Chitosan-based supports (carriers) for enzyme immobilization
Chitosan is a linear polysaccharide made up of N-acetyl-D-glucosamine (acetylated unit) and randomly arranged β-(1 → 4)-linked D-glucosamine (deacetylated unit) (Fig. 15b).151,152 Chitosan can be used in medical applications due to its antibacterial, wound-healing, biodegradable, biocompatible, and non-toxic features.153–155 The chitosan matrix can be researched as a support for enzyme immobilization. The storage, thermal, operational, and pH stabilities can be greatly improved for enzymes that are immobilized on the surface of chitosan (Table 5).156–214 Chitosan-based materials can be produced in different geometrical configurations (e.g., microcapsules, membranes, beads/microspheres, coatings, fibers, gels, and sponges), which have shown good storage, thermal/operational stability, and reusability properties. Owing to these flexible features, their capacity as carriers for a wide range of enzymes could be applied practically. Immobilizing xylanase in xanthan/chitosan matrices has revealed higher thermal stability and activity than those of native enzymes.164 Çetinus et al.167 demonstrated that catalase (CAT) immobilized into chitosan beads had enhanced resistance against denaturation due to pH or heat. Immobilized CAT had a Km value that was two-times greater than that of free CAT and its Vmax decreased from 32000 μmol to 122 μmol (min mg protein)−1. However, immobilization enhanced the thermal, operational, and storage stabilities of CAT.167 Yang et al.168 immobilized GOx on a novel porous chitosan-SiO2 gel that was synthesized by coupling chitosan with tetramethoxysilane (TMOS) using a sol–gel method. With a profound immobilization yield of 97% and GOD activity of 1585 U g−1, even after 10 days and 15 days, the immobilized enzyme remained stable and functional, with activity of 86% and 56% at 30 °C, respectively, whereas most of the free enzyme became inactive after 5 days in a similar condition.168 Mao and co-workers developed chitosan microspheres and sponges in smaller sizes to immobilize cellulase.172 With rapid adsorption of cellulase over microspheres (<25 min), the immobilized cellulase displayed higher stability with respect to pH, higher Km, thermal stability, reuse, and storage stability than those of the native enzyme. Hence, this system could be employed in various industries demanding an enzyme with good performance at extreme physiochemical conditions. Dinçer et al.177 enhanced the stability of acid cellulase at neutral pH. Chitosan beads were coated with modified polyanionic PVA, and cellulase was immobilized on modified PVA-coated chitosan beads. Compared with the free enzyme, cellulase immobilized on chitosan beads demonstrated improved pH stability. The optimal pH of the enzyme moved from 4.0 to 7.0, signifying its ability for catalysis over a wide range of pH conditions, and also displayed excellent storage and operational stabilities.169
| Biomass | Biomass derived material | Used enzyme | Immobilization method | Result/performance study | Ref. |
|---|---|---|---|---|---|
| Chitosan | Magnetic gel microspheres made of chitosan, cellulose, and Fe3O4 | GOx | Cross linking | Higher conversion yield (>91%) and great recyclability with >84% activity retention even after 15 cycles | 79 |
| Chitosan | Mesoporous silica/titania with a chitosan coating | β-D-Galactosidase | Covalent binding | High activity (1223 U g−1), excellent efficiency (74%) and >90% activity retained after 15 cycles of reuses | 153 |
| Chitosan | Chitosan beads activated with glutaraldehyde | Lipase | Not mentioned | 46.5% of initial activity was improved | 155 |
| Chitosan | Glutaraldehyde cross-linked chitosan beads | Pleurotus nebrodensis WC 850 laccase | Cross linking | Great decolourization efficiencies for various reactive and disperse dyes (>83–90%) and >84% retained activity after 8 cycles of reuse | 156 |
| Chitosan | Manganese-ferrite nanoparticles coated with chitosan | Laccase | Covalent bonding | Best suited for pharmaceutical waste degradation with 80% removal rate for diclofenac | 157 |
| Chitosan | Genipin-activated chitosan support | β-Galactosidase | Cross linking | Increased thermal and storage stabilities with potential application in food industries harbouring lactose hydrolysis | 158 |
| Chitosan | Chitosan matrix | Bromelain, Papain and Cysteine proteases | Adsorption | (1) Intact optimum pH and temperatures. (2) ∼5.8 times and ∼7.6 enhanced stability for Bromelain and Papain respectively | 159 |
| Chitosan | Chitosan/organic rectorite composites | Polyphenol oxidase (PPO) | Physical adsorption and covalent binding | (1) Higher enzyme activity ∼8920–16370 U g−1. (2) 89–93% phenol derivative removal within 2 h with 10 cycles of recyclability | 161 |
| Chitosan | Chitosan nanoparticles | L-ASNase | Encapsulation | Significant decrease in the minimum inhibitory concentration (MIC) against microbes (5.26 mg mL−1) | 162 |
| Chitosan | Chitosan microbeads by microencapsulation | α-Amylase and invertase | Microencapsulation | Enhanced the stability and activity | 163 |
| Chitosan | Chitosan-xanthan hydrogels | Endo-1,4-β-xylanase | Noncovalent link | Enzymes show 60–70% higher activity than free enzymes | 164 |
| Chitosan | Copolymer of chitosan and poly-glycidyl methacrylate (GMA) | Urease | Covalent bonding | Enhanced enzyme specific activity, temperature stability, pH stability, and storage stability | 165 |
| Chitosan | flakes and porous chitosan beads (PCB) | Lipase | Physical adsorption | Immobilized enzyme showed better thermal stability at pH-6 and 40 °C | 166 |
| Chitosan | chitosan beads crosslinked with glutaraldehyde | Bovine liver catalase (CAT) | Cross linking | Improved enzyme thermal, operational, and storage stabilities | 167 |
| Chitosan | The chitosan and TMSO were coupled using the sol–gel method to create porous gels of chitosan-SiO2 | GOx | Entrapment | Enhanced stability | 168 |
| Chitosan from dried uttlebone cartilage | Chitosan and glutaraldehyde-crosslinked activated clay beads in equal weights | α-Amylase, glucoamylase and β-amylase | Cross linking | Superior operational stability with 81% retention of activity after 50 cycles of reuse | 169 |
| Chitosan | Magnetic chitosan microspheres cross-linked with glutaraldehyde | Laccase | Adsorption and cross-linking | Increased pH and temperature stability | 170 |
| Chitosan | Chitosan beads activated with glutaraldehyde | PGA | Covalent attachment | Good thermal and alkaline pH stabilities | 171 |
| Chitosan powder | Chitosan microspheres and sponges | Cellulase | Covalent method | Higher protein loading (∼145 mg g−1), better storage stability and reusability | 172 |
| Chitosan | Nanoparticles of chitosan made by the ionisation gelation process | Neutral proteinase | Adsorption | Enhanced operational, storage, and thermal stability | 173 |
| Chitosan | Nanoparticles of chitosan made by the ionisation gelation process | Neutral lipase | Adsorption | Increased the enzyme's efficiency by 13.17% compared to free lipase | 174 |
| Chitosan | Chitosan beads (Ch-bead) were attached to cibacron blue F3GA dye (CB-Ch-bead) | CAT | Adsorption | Better thermal, operational and storage stabilities | 175 |
| Chitosan | Chitosan beads | Pepsin | Cross linking | Increased thermal stability with shift in optimum temperature from 40 to 50 °C | 176 |
| Chitosan | Polyvinyl alcohol (PVA) coated chitosan beads | Cellulase | Adsorption | Immobilized cellulase showed better yield, pH stability (at pH 7), good storage and operational stability | 177 |
| Chitosan | Chitosan nanofibrous membrane | Lipase | Entrapment | Improved storage stability with ∼92% activity after 48 days of incubation | 178 |
| Antarctic krill shells | Glutaraldehyde-pretreated chitosan membranes | Urease | Entrapment | Improved thermal stability with raise in optimum temperature from 65 to 75 °C | 179 |
| Cuttlebone chitosan | Chitosan–clay composite bead cross-linked with glutaraldehyde | β-Glucosidase | Cross linking | Enhanced storage stability and activity in dried composite | 180 |
| Powdered chitosan | Chitosan beads | Chymotrypsin | Covalent multipoint attachment | At 65 °C, the immobilised enzyme's half-life increased from 0.57 hours at 55 °C to 7.8 hours | 181 |
| Powdered chitosan | Chitosan and agarose with glutaraldehyde | Candida Antarctica lipase type B (CALB) | Multipoint covalent attachment | Enhanced the thermal stability and activity | 183 |
| Chitosan | Nanocapsules made of CKGM-CS | L-ASNase | Entrapment | Prevent the leaking. Better stability and activity for broad pH range | 184 |
| Chitosan | Chitosan crosslinked with glutaraldehyde used for adsorption of Cu(II) | CAT | Cross linking | Though Cu-Ch-CAT had 3 folds lesser substrate affinity, but imparted greater thermal and storage stabilities compared to Ch-CAT | 186 |
| Chitosan | Magnetic chitosan nanoparticles | Laccase | Entrapment | Increased storage stability and operation stability | 187 |
| Pink shrimp | Modified chitosan beads | HRP | Covalent method | Enhanced the thermal stability and operational stability | 188 |
| Chitosan | Chitosan beads | CA | Physical adsorption | Half-life increased by 24 h and 48 h compared to native enzyme at −20 °C and R.T. respectively | 189 |
| Chitosan from crab shells | Chitosan-coated magnetite nanoparticles (Fe3O4-CS) | Laccase | Adsorption | Enhanced storage stability with >70% activity after 30 batches of use | 190 |
| Chitosan from shrimp shells | Chitosan macroparticles and nanoparticles | β-Galactosidase | Adsorption | 75–83% activity was retained even after 50 cycles of reuse | 191 |
| Chitosan | Glutaraldehyde crosslinked chitosan-clay composite beads | Tyrosinase | Cross linking | Optimum temperature, loading effectiveness, and activity were all increased | 192 |
| Chitosan powder | N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide (EDC) and N-hydroxysuccinimide (NHS) are utilised as coupling agents to covalently immobilise chitosan-coated Fe3O4 nanoparticles | Lipase | Covalent method | The stability remains same after 13 days of storage at 25 °C, the immobilised lipase exhibits enhanced operational stability, including wider temperature and pH ranges | 193 |
| Chitosan from shrimp shells | Chitosan macrobeads by covalent crosslinking using glutaraldehyde | Trypsin | Covalent cross-linking | Improved pH and temperature stability | 194 |
| Chitosan | Chitosan–halloysite hybrid-nanotubes cross-linking with glutaraldehyde | HRP | Cross-linking | The immobilised HRP did not lose any activity after 35 days of storage | 195 |
| Chitosan | Magnetic chitosan carriers | Lipase and β galactosidase | Entrapment | Demonstrated excellent long-term stability without enzyme leaking from the support | 196 |
| Chitosan | Glutaraldehyde-hardened alginate-chitosan beads | Inulinase | Cross linking | At 50 °C gel beads showed excellent activity. In six days at room temperature, the immobilised enzyme retained 76% of its activity | 197 |
| Chitosan | Fe3O4 magnetic nanoparticles with chitosan coating and glutaraldehyde coupling agent | Cellulase | Covalent method | Greater operating stability throughout a wider range of temperatures and pHs, as well as good activity and reusability during magnetic separation recovery | 198 |
| Chitosan | Hydrogel beads made of a bacterial cellulose (BC) and chitosan composite | Lipase | Physical adsorption and covalent cross-linking | BC-chitosan hydrogel beads immobilized showed higher catalytic activity (1.9 times), and greater enzyme stability than microcrystalline cellulose MCC-chitosan hydrogel beads (22.7 times higher t1/2) | 199 |
| Chitosan | Dextran polyaldehyde is used as a macromolecular cross-linking agent to create chitosan magnetic nanoparticles (CMNPs) | Pectinase | Cross-linking | 2 folds improved thermal stability (55–75 °C) excellent stability and durability reusability | 200 |
| Chitosan | Chitosan-blended cellulose monoacetate nanofibers | Protease | Physical adsorption | Higher operational stability, activity and reusability | 201 |
| Chitosan | Chitosan-montmorillonite nanocomposite beads with glutaraldehyde | α-Amylase | Covalent bonding | 95% activity retained after 40 days of incubation at 4 °C (∼2.6 times higher than native enzyme) | 202 |
| Chitosan | Chitosan nanoparticles (CS-NPs) | GOx | Covalent attachment, cross linking | Improved the stability and activity with ∼26 folds higher activity (with precipitation method) compared to covalent attachment | 203 |
| Shellfish derived chitosan (CS) powder | Clay/chitosan biocomposite systems | Protease | Covalent method | Increased the immobilization yield | 204 |
| Chitosan | Magnetic chitosan nanoparticles (CS-Fe3O4) modified with imidazole based ionic liquid (IL-CS-Fe3O4) | Porcine pancreatic lipase (PPL) | Physical adsorption | Having 1.93 folds higher specific activity with 382% activity recovery, immobilized PPL showed 84% remaining activity after 10 cycles of reuse | 205 |
| Chitosan | Chitosan-based nanoparticles | Glucoamylase | Cross linking | Greatly increased loading capacity and storage stability | 206 |
| Chitosan | Silica/chitosan composite | Laccase | Covalent method | Good residual activity after 7 months of storage | 207 |
| Chitosan from crab shell | biocompatible glutaraldehyde (GA) cross-linked chitosan beads as a matrix | Chitinase | Cross linking | Immobilized enzyme was found to be highly stable up to 1 month of storage and exhibited wider range of pH tolerance | 208 |
| Cuttle-fish waste | Highly swollen chitosan beads cross-linked with glutaraldehyde | Acid phosphatase and β-glucosidase | Cross linking | Enzymes were stable for long term | 209 |
| Chitosan | Fe2O3/chitosan coated superparamagnetic nanoparticles was cross-linked with glutaraldehyde | Lipase | Cross linking | (1) Broader working temperature range from 27–85 °C and pH tolerance in between 5.5–9.5. (2) Achieved 70.2% of microwave assisted biodiesel recovery | 210 |
| Chitosan | Chitosan functionalized with Concanavalin A (ConA) | α-Galactosidase | Adsorption, cross linking | Enhanced reusability and activity yield | 211 |
| Chitosan | Chitosan matrix | Papain | Adsorption | Enhanced stability against UV irradiation (up to 6040 J m−2) | 212 |
| Chitosan | Chitosan coated polylactic acid (PLA) based nanofiber (CCN) matrix | α-Amylase | Higher substrate conversion ratio of 0.85–0.99 obtained with 40 min of residence time and lower rate of dilution | 213 | |
| Chitosan | Chitosan gel activated with glutaraldehyde | Cellulases | Cross linking | Better hydrolysis performance with Avicel and sugarcane bagasse leading to glucose concentrations of 10–13 g L−1 in the hydrolysate | 214 |
To further explore the ability of a chitosan matrix to stabilize enzymes at different pH values, Tang et al.166 prepared chitosan nanoparticles via ionization gelation, which yielded spherical nanoparticles of diameter 100 nm. Immobilized lipase showed improved activity, thermal, storage, and operational stabilities. The immobilized neutral lipase had a higher Km (0.37 × 102 g l−1) than that of free neutral lipase (1.01 × 102 g l−1). Moreover, immobilization improved the stability of neutral lipase in the acidic range. The stability features of the immobilized enzyme were enhanced noticeably with respect to temperature, reuse, and storage duration.166
To solve the problem of the separation and reusability of a biocatalyst, Liu and group79 used the sol–gel transition method to develop magnetic Fe3O4-cellulose-chitosan hybrid gel microspheres by employing ILs as the solvent for the dissolution and regeneration of cellulose and chitosan. Glutaraldehyde was used to further immobilize GOx on hybrid gel microspheres (Fig. 17), which had a broader pH range, greater thermal stability, and enhanced storage stability than those of free GOx. The pH optimum of immobilized GOx was in the range 4–7, whereas that of free GOx exhibited maximal activity at pH 6. After 28 h of heating at 50 °C at pH 7, the immobilized enzyme exhibited 63% of its initial activity. However, under identical conditions, the free enzyme displayed only 22% of its initial activity.79 Similar to the report stated above, Lac was immobilized on chitosan-coated magnetite (Fe3O4) nanoparticles. Kinetic analyses revealed that the immobilized Lac system had identical catalytic efficiencies. However, at optimal conditions, after 30 batches of use, immobilized systems continued to exhibit >71% of their initial activity.190 With regard to other closure studies,188,191–196 lipase, β-galactosidase, cellulose, pectinase, and glucoamylase were immobilized onto chitosan-coated Fe3O4 NPs.191–196 These immobilized enzymes on magnetic chitosan carriers demonstrated prolonged stability with minimal leaching. They also conveyed better operational stability, including a wider thermal range, pH, reusability and storage stabilities, than those of the free enzyme. Hence, Fe3O4-chitosan nanoparticles could be used as efficient supports for lipase immobilization, particularly in applications involving the need for recycling.188 Huang et al.178 synthesised a chitosan nanofibrous membrane, and CRL was immobilized on the nanofibrous membrane using glutaraldehyde as a coupling reagent. Lipase loading on the nanofibrous membrane attained 63.6 mg g−1, and immobilized lipase activity was 49.8%. After immobilization on the chitosan nanofibrous membrane, the stability towards temperature, pH, reuse, and storage of immobilized lipase was enhanced. With this good immobilization fraction of loading capacity and improved stability, the membrane of chitosan nanofibers is an excellent biocompatible and potential support for enzyme immobilization.178 In the case pf porcine pepsin, immobilization onto chitosan beads shifted the optimum temperature by 10 °C, which was higher than that of the native version, revealing the resistance of the immobilized enzyme towards heat-induced denaturation along with a longer shelf-life. Observing these properties, Altun and colleagues176 demonstrated the applicability of this system in industries based on the clotting and processing of milk. Adriano and co-workers181 revealed a very noticeable improvement in the thermal stability of α-CT after covalent bonding of the enzyme on a chitosan-based hybrid gel. The half-life of immobilized chymotrypsin was enhanced from 0.57 h at 55 °C to 7.8 h at 65 °C.181 To improve the enzyme-holding capacity and to prevent leakage, Wang et al.184 prepared carboxymethyl konjac glucomannan–chitosan (CKGM–CS) nanocapsules as a new biocompatible matrix system for the immobilization L-ASNase. The semi-permeability of the matrix retained L-ASNase in the matrix and inhibited leakage while allowing the substrate and product to pass through. The immobilized enzyme system demonstrated noticeably greater stability and activity in comparison with those of free L-ASNase. These investigations could offer a brand-new material for the immobilization of pH- and temperature-sensitive enzymes.184 Pertaining to the challenge of reusability, Flores et al.182 discovered a technique to activate chitosan beads using genipin, over which β-galactosidase from Aspergillus oryzae was immobilized. During batch processing, the immobilized enzyme maintained 100% of relative initial activity up to 40 cycles without much compromise in thermal stability, thereby showing its potential applicability. Similarly, a system of inulin over chitosan beads showed a recycling capacity up to 14 cycles after engineering by Singh et al.185 Monier and co-workers188 developed a method to immobilize HRP on modified chitosan beads by graft copolymerization of polyethylacrylate with potassium persulphate and Mohr's salt as redox initiators (Fig. 18). Even after immobilization, the optimum temperature remained at 45 °C with higher relative activity than that of the native enzyme.188 Wanjari et al.189 immobilized CA on chitosan beads and its activity was evaluated using the p-nitrophenol assay (Table 5). The storage stability was profiled for up to 20 days and the half-life of immobilized CA increased up to 48 h. The authors suggested that this system could be applied for carbon-capture and higher efficiency for converting CO2 to CaCO3.189 Zhai and colleagues195 developed chitosan–halloysite hybrid-nanotubes (CTS-HNTs) through the assembly of chitosan onto halloysite (natural nanotubular aluminosilicate). HRP was covalently immobilized over nanotubes through crosslinking (Fig. 19). Even 35 days later, the immobilized HRP maintained its full activity whereas the free enzyme could maintain only 27% of its initial activity. The authors claimed that the CTS-HNT-immobilized HRP system could be employed for wastewater rejuvenation, particularly for the destruction of phenolic chemicals, because of its strong catalytic activity.195
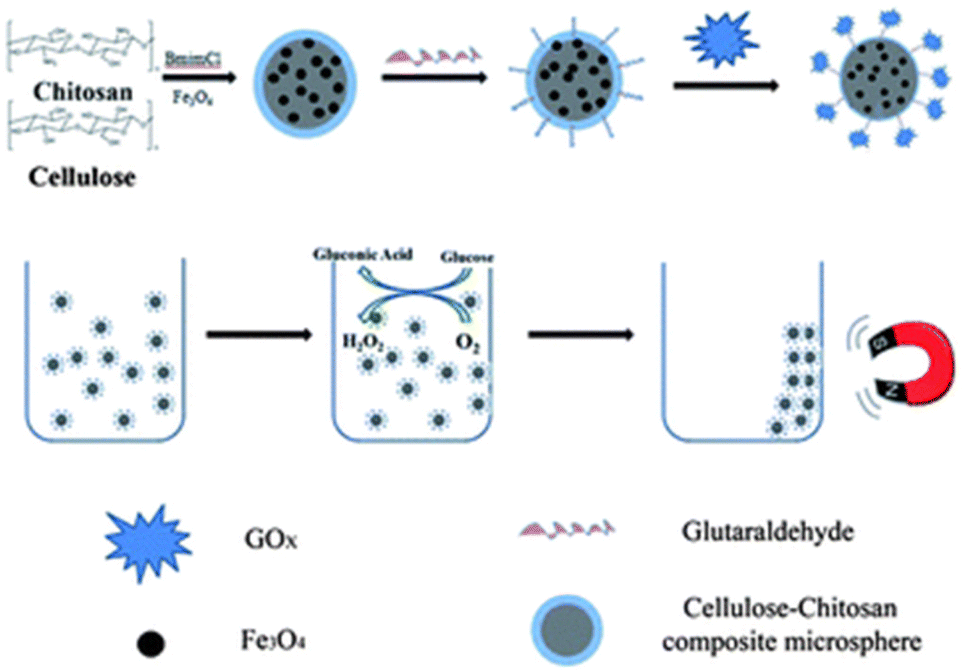 | ||
| Fig. 17 Preparation of magnetic Fe3O4–cellulose–chitosan microspheres after enzyme immobilization onto magnetic cellulose–chitosan (schematic). Reproduced from ref. 79 with permission from RSC, copyright 2012. | ||
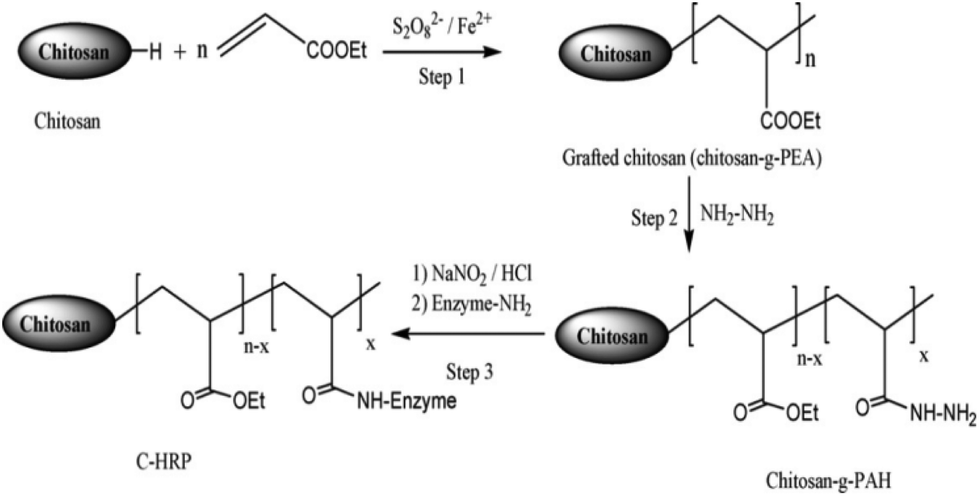 | ||
| Fig. 18 HRP immobilization onto modified chitosan beads (schematic). Reproduced from ref. 188 with permission from Elsevier, copyright 2010. | ||
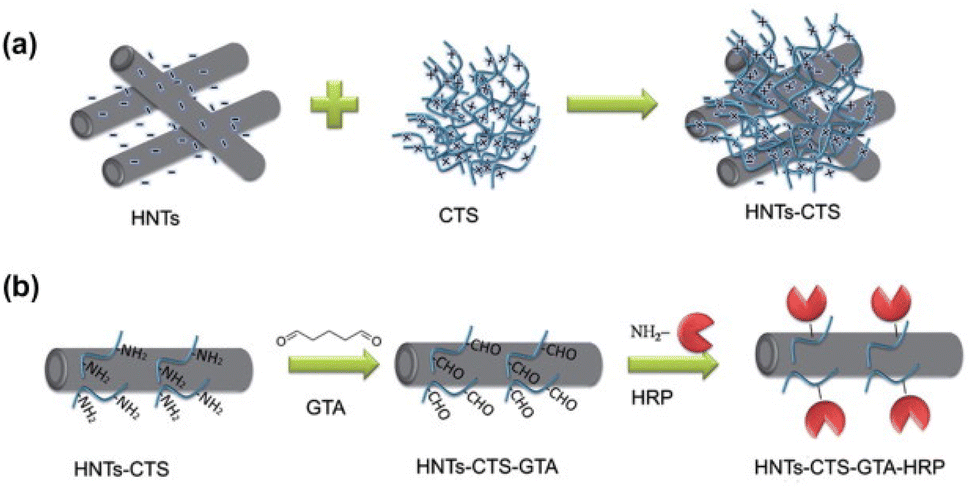 | ||
| Fig. 19 (a) Stepwise preparation and (b) immobilization of HRP on a chitosan (CTS) matrix. Reproduced from ref. 195 with permission from Elsevier, copyright 2013. | ||
7. Alginate-based supports (carriers) for enzyme immobilization
Alginic acid is a naturally occurring polysaccharide and has a negative charge. It is extracted from algal cell walls, in particular brown algae or seaweeds. Alginate is a liner copolymer that consists of two residues: β-D-mannuronate (termed “M block”) and α-L-guluronate (termed “G block”). Both residues are C5-epimers (Fig. 20).215 Depending on the source, the blocks M and G can be arranged in a consecutive manner (e.g., MMMM followed by GGGG or vice versa), alternating patterns (MGMGMGM), or random.215 The properties of alginate (e.g., interaction with cationic metal species, solubility, and viscosity,) are directly attributed to the molecular weight of mannuronic acid/guluronic acid.215,216 Alginate is stable, biocompatible, biodegradable, nontoxic, has chelating ability, and relatively inexpensive. Thus, this natural polysaccharide and its derivatives have a multitude of industrial uses.216 Alginate-based products have been used for drug delivery, regenerative engineering, environmental clean-up, wound dressing, biosensors, and transfection of genetic material.217,218 An attractive approach to increase the stability of enzymatic processes and economic feasibility in terms of reusability can be achieved with the use of alginate-based supports for enzyme immobilization.219–226 The mechanical characteristics and robustness of alginate-based supports can be enhanced further using polymer blending (much like cellulose and chitosan). These alginate forms have been demonstrated to increase thermal stability, enzyme activity, and reusability.227–236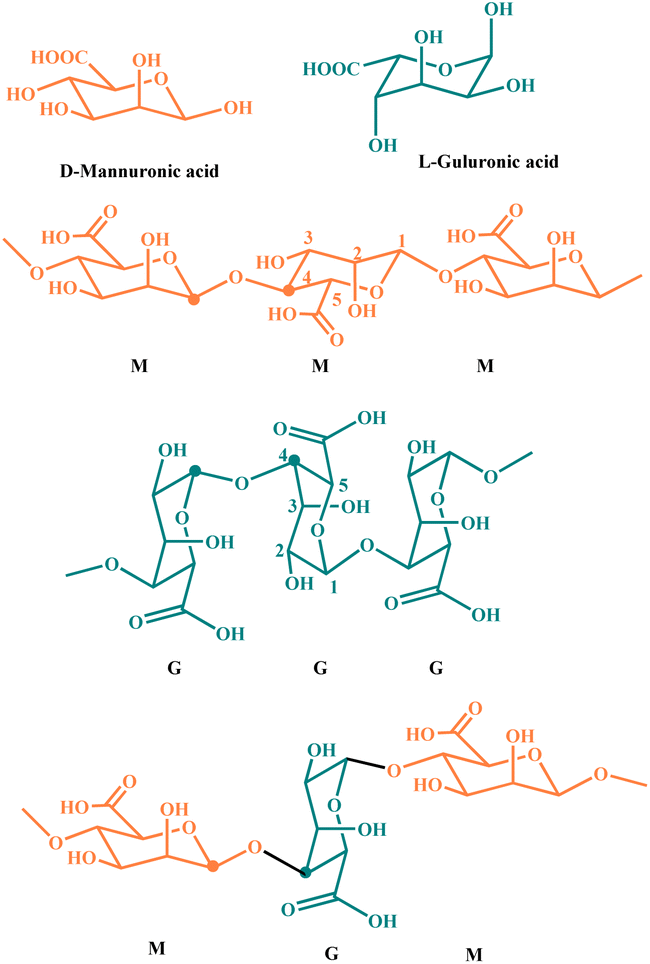 | ||
| Fig. 20 Chemical structure of alginate. Reproduced from ref. 215 with permission from ACS, copyright 2005. | ||
Palmieri et al.230 developed a novel procedure for enzyme immobilization by entrapment in copper alginate gel, which was applied for immobilizing fungal phenol oxidase. The immobilized enzyme boosted the rate of oxidation of various aromatic substrates, was active over a large pH range, and had a low temperature for optimal activity. Thus, a system for detoxifying wastewater contaminated with phenolic derivatives from industrial or agricultural sources could be developed using an enzyme that has been immobilized in this way.230 By first combining polysaccharides with Ca2+ and then cross-linking glutaraldehyde with the amine groups of the gelatine found in the initial mixture, Elçin and colleagues231 created urease-containing xanthan-alginate spheres. Higher enzymatic activity was shown by encapsulated urease even with variations in pH or temperature. Even after 20-times of its use in optimal conditions, xanthan-alginate spheres continued to have 75% of the maximum urease activity.231 More recently, a multifunctional alginate-derived carbon (AAC) material having variable oxygen functionalities was created by our research team232 for the immobilization of Cyt c. Without affecting the structural stability if Cyt C, the enzyme activity was increased significantly in comparison with that of the native protein (Fig. 21).
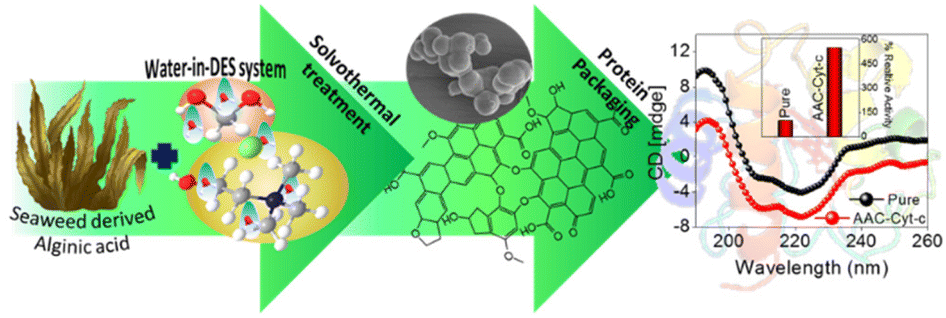 | ||
| Fig. 21 Preparation of functionalised solvothermal carbon derived from alginate using “water-in-deep eutectic solvents” for enhancing enzyme activity (schematic). From ref. 232. | ||
For the entrapment of different enzymes, calcium alginate (Ca-ALG) use seems to be economical and biocompatible. Blandino et al.233 systematically studied the activity of encapsulated GOx within Ca-ALG gel capsules. Comparison of the apparent kinetic characteristics of immobilized and free GOx revealed that the immobilized GOx had a higher Km and more pronounced differences in Vmax.233 Tanriseven and co-authors235 immobilized Saccharomyces cerevisiae invertase in alginate capsules, which achieved 87% relative activity along with enzyme stabilization at high pH and temperatures.235 Alginate-based gels have also enhanced the thermal and storage stabilities of various proteins.236 Vu and colleagues236 investigated the biochemical characteristics of invertase entrapped within alginate gels. The Km of immobilized invertase (139.19 mM) was higher than that of free invertase (93.19 mM), but its Vmax was smaller. Nevertheless, the support induced greater thermal stability to invertase (as manifested by a longer half-life) and endowed significant long-term stability (up to 40 days).236 To study the normalized effect of alginate matrices, Fadnavis et al.237 employed lipase from porcine pancreas, Pseudomonas cepacia, and Candida rugosa. Hydrogels prepared by crosslinking a blend of sodium alginate (5%) and gelatin (3%) with glutaraldehyde were used to immobilize these lipases, which had excellent efficiency and greater stability and reclaimability for ∼10 recycles without much loss of enzyme activity. Then, using a straightforward procedure of protein binding at pH 5 and release at pH 8.5, these functionalized beads were employed to purify crude porcine pancreatic lipase 7.4-times, showing their feasibility for biomedical applications.237 Thanks to advances in core–shell chemistry within nanoscience, Taqieddin and co-workers238 designed alginate-chitosan core–shell microcapsules to create biocompatible carriers for enzyme immobilization. These shells offered permeability control over substrates and end products, whereas the protein was maintained in a liquid or solid core. To create a liquid or solid core for the microcapsule, sodium alginate was crosslinked with calcium or barium ions, respectively. o-Nitrophenyl-β-D-galactopyranoside (ONPG) was used to assess the catalytic activity of a model enzyme, β-galactosidase, which was immobilized in the alginate core. Due to the extra layer required for the influx of the substrate and outflow of the product, Vmax for Ca2+ and Ba2+ alginate-chitosan core–shell microcapsules was much lower than that of the free enzyme.238 Ultrathin alginate/protamine/silica (APSi) composite membranes were constructed by Wang et al.239 using a co-extrusion minifluidic and biosilicification methodology for Lac immobilization. The prepared hybrid capsules were highly monodisperse and dispensed Lac with significantly high thermal, storage, and pH stabilities. With ∼100% immobilization yield, Lac displayed an activity of 61.8 mmol g−1 min−1, along with withholding 67% activity after 20 days and residual relative activity in APSi capsules remaining at 45% after 10 cycles.239 Alginate-based hybrid composites have become an innovative class of materials, providing a larger range of uses for enzyme encapsulation encompassing biocatalysis, biomedicine, bioseparation, and biosensing.240–245 To immobilize Lac, Lu et al. created alginate-chitosan microcapsules using emulsification-internal gelation.240 In addition to stability, a higher loading efficiency and immobilized yields were obtained under ideal immobilization conditions (0.3% chitosan, 2% sodium alginate, 2% CaCl2, and a 1:8 ratio by volume of enzyme:alginate). However, the recyclability was only three cycles, so finding a remedy for this problem is an issue.240 A multidimensional approach using biomass and nanomaterials to prevent leakage of yeast alcohol dehydrogenase (YADH) from Saccharomyces cerevisiae source was availed by Xu et al.241 By adding silica nanotubes (SiNTs) to the alginate (ALG) gel and then crosslinking it with calcium, they designed an alginate-silica nanotube (ALG-SiNT) composite, which was used to encapsulate YADH. Enzyme leakage from the carrier was reduced significantly (∼50%) compared with the alginate matrix composite only. These results showed that an ALG-SiNT carrier furnished stronger affinity with the enzyme compared with YADH immobilized over simple ALG support and led to longer storage, increased activity retention, and a stable operation.241
We applied a new and straightforward approach to prepare stable and versatile alginate-based white light-emitting hydrogels (WLEs) for protein packaging.242 Using the notion of complementary colors, a WLE composite was constructed by engineering the surface of an orange light-emitting ZnS quantum dot (QD) doped with Mn2+ using a blue light-emitting IL-choline-tosylate. The WLE QD-IL composite was combined with an alginate bio-polymer to fabricate the WLE hydrogel. After that, Cyt c was confined within the WLE hydrogel matrix, which led to higher relative activity of ∼180% compared with that of the free enzyme (Fig. 22). The authors counteracted the harsh effects of chemical denaturants (e.g., urea) and extreme temperature over the stability of the metalloprotein Cyt c. Extending this concept further, Ma et al.243 discovered an efficient carrier, double walled carbon nanotube-doped alginate gel (DWCNT-ALG) for immobilization of lactate dehydrogenase (LDH). Leakage of LDH from the LDH-DWCNT-ALG biocomposite was decreased significantly, and LDH that had been immobilized displayed increased activity. The authors demonstrated the practical use of this type of carrier for lactate bioconversion in industry, as well as its usage for the immobilization of additional enzymes and microorganisms (including LDH and ADH).243 Enzyme entrapment in ALG beads has been shown to be relatively inexpensive, safe, and straightforward technique for separation of the product and enzyme at the industrial scale.244 Bhushan et al.244 prepared ALG beads in an aqueous mixture containing sodium alginate, lipase from Arthrobacter sp. (ABL), and CaCl2 to empower its reusability and all-round enzyme stability. In comparison with the free enzyme, the entrapped enzyme was much more stable throughout greater temperature domains, pH, and storage times.244–248 Viet and colleagues246 immobilized cellulase in Ca-ALG beads by entrapment and its activity was assessed using CMC. With several cycles of use, the immobilized enzyme exhibited greater stability in response to changes in pH and temperature. The activity of immobilized cellulase remained 69.2% after five recycles and 20.3% after eight recycles.246 Manganese peroxidase (MnP), generated from an indigenous variant of Ganoderma lucidum IBL-05, was immobilized on Ca-ALG beads by Bilal and co-workers247 using a promising and environmentally friendly entrapment method. Ca-ALG-bound MnP was catalytically more vigorous and could be used eventually to decolorize dyes and detoxify industrial effluents more effectively.247 Nunes and co-workers248 developed conjugated polyvinyl alcohol-alginate (PVA-ALG) beads with excellent thermochemical and mechanical stabilities at high temperatures (>80 °C). Naringinase, an enzyme derived from Penicillium decumbens, was immobilized in ALG (0.2–1.0%)-PVA (10%) beads featuring three sizes (1–3 mm). At pH 4.0 and 70 °C, immobilized naringinase bestowed 80% activity and displayed >90% retention of initial activity after 6 weeks of incubation in acetate buffer. The importance of this immobilization approach for the system under consideration was illustrated by these promising outcomes, which also implied that it could be used to entrap other biocatalysts.248 Bonine et al.249 reported immobilization of a novel lipase isolated from the seeds of Pachira aquatica (PAL) using Ca-ALG beads and PVA. Similar results were obtained with immobilization of PAL in ALG and ALG/PVA beads, with improvement in their stability against temperature than the free enzyme.249 Ortega et al.250 covalently immobilized neutrase on ALG-glutaraldehyde beads. Under optimized conditions (pH 6.2, 2% ALG, 6.2% glutaraldehyde, and interaction for 60 min), the immobilization yield of encompassed neutrase (61.84 U mL−1) was ∼50% and the confined enzyme exhibited maximum activity at 10 °C higher than that of the native enzyme, which clearly showed the stability induced by the support. Immobilization caused the estimated activation energy to drop from 47.7 kJ mol−1 to 22.0 kJ mol−1, which suggested the role of the support in favouring of the forward reaction at the thermodynamic level.250 Eldin et al.251 extended the covalent immobilization technique towards glucoamylase to form a novel affinity with p-benzoquinone-activated ALG beads. The covalently mounted enzyme sustained its activity after 30 successive runs and shelf space of 36 days.251 To improve the stability and catalytic rate of GOx, Wang and co-workers252 designed a protocol for undertaking emulsification-internal gelation followed by GOx adsorption and chitosan deposition to encapsulate the enzyme in CaALG-chitosan composite microspheres (CACMs). Before grafting with chitosan, the Ca-ALG matrix was made highly porous with the release of CO2, which helped to enhance the adsorption efficiency of the carrier. The concentration and molecular weight of chitosan, incubation duration, and pH seemed to have an impact on the GOx loading, encapsulation efficiency, and activity of CACM-GOx. Exhibiting the maximum enzymatic activity and encapsulation efficiency at an isoelectric point of 4, immobilized GOx retained >70% activity, which was tenfold higher than the activity retention by the free enzyme.252 With some modification of this work, Zhao and colleagues253 reported a unique and feasible approach for non-covalent GOx immobilization in chemically reduced graphene oxide (RGO)/ALG hybrid gel beads. The enzyme contained in the hybrid microbeads demonstrated high environmental tolerance and could maintain optimal activity over a wide range of conditions (45–60 °C and pH 4–6). To produce a continuous fixed-bed enzyme catalytic process, the microbeads could also be recycled simply through conventional filtering and reintroduced to a column.253 Both of these works revealed the great potential to further explore CACM-GOx and ALG/RGO for developments in glucose bio-sensing. Abd El-Ghaffar et al.254 used a grafting-encapsulation approach for α-CT immobilization. First, chitosan grafted with polymethyl methacrylate (PMMA-g-CS) through free-radical polymerization was used as a support for α-CT. Ca-ALG beads were fabricated for sheathing PMMA-g-CS-CT to produce composite beads. For immobilized α CT, greater retention of activity (∼97.7%) was attained at pH 9 for 24 h. Immobilized α-CT retained 75% of its original activity after 60 days of storage at 25 °C and continued to work very well even after 25 reuses. Such results suggested that the recycling ability of ALG composite carriers could be used for continuous catalytic reactions in the industrial sector.254 Muthuvelu and colleagues255 designed a tri-enzyme biocatalyst by co-immobilizing Lac, cellulase, and β-glucosidase with ALG for evaluating bioethanol. The co-immobilized enzyme system had long shelf-life and thermal stability, offering an effective one-pot pre-treatment protocol for the production of bioethanol from lignocellulosic biomass, of which the maximum ethanol conversion was obtained with Ipomoea carnea (Fig. 23).
 | ||
| Fig. 22 Alginate-based hydrogels for protein packaging with enhanced activity and stability of Cyt c. (A) Schematic of the interaction of Cyt C with hydrogel composites. Percentage relative activity of Cyt C at room temperature (B), at various temperatures (C), and in presence of 8 M urea (D). (E) UV–vis spectra of Cyt C in presence of WLE and OLE hydrogels. (F) Fluorescence spectra of only WLE hydrogel and Cyt C + WLE hydrogels. Reproduced from ref. 242 with permission from Wiley-VCH, copyright 2020. | ||
 | ||
| Fig. 23 Immobilization of co-immobilized laccase, cellulase, and β-glucosidase for ethanol production. Reproduced from ref. 255 with permission from Elsevier, copyright 2018. | ||
Electrospun nanofibers comprising ALG-supporting immobilized lipase showed high enzyme loading capacity, catalytic activity, and stability. Doğaç et al.256 produced PVA/ALG and polyethylene oxide/alginate (PEO/ALG) nanofibres using electrospinning, and lipase was adsorbed and crosslinked further with glutaraldehyde. Free lipase lost all of its activity at high temperatures after 40–60 min, but lipase-immobilized nanofibers sustained approximately 65–70% activity during the same timeframe. Furthermore, PVA/ALG and PEO/ALG composite nanofibers immobilized with lipase exhibited 50% of their original activity after 14 and seven cycles, respectively.256 Zhao et al.257 synthesized novel nanoflower/ALG microbeads and immobilized α-acetolactate decarboxylase (ALDC) using a facile method. First, ALDC was co-precipitated with Ca3(PO4)2 to form enzyme-inorganic composite nanoflowers (ALDC-Ca3(PO4)2). Later, these nanoflowers were encapsulated in ALG gel beads (ALDC-Ca3(PO4)2 + ALG).256 The microbeads demonstrated outstanding stability and environmental endurance in contrast to free ALDC and ALDC-Ca3(PO4)2 nanoflowers, displaying optimal activity over a wide range of temperature (45–70 °C) and pH (3.5–7), but retained >98% of activity in comparison with unbound ALDC. Furthermore, the immobilized enzyme was employed in a 300 L beer fermenter to stop diacetyl generation. This resolved the issue of off-flavor in beer and reduced the time it takes for beer to mature, thereby demonstrating enormous scope for application in breweries.257 Xylanase has several uses in the food industry. Kumar et al.258 purified xylanase from Bacillus licheniformis Alk-1, and entrapped it within crosslinked Ca-ALG beads, and undertook activation through glutaraldehyde. In comparison with the free form, immobilized xylanase demonstrated improved overall chemical characteristics, recycling, and reusability efficiency. Hence, xylanase-ALG beads with higher xylanolytic activity and stability could be prepared, thereby aiding the design of efficient bioreactors for various applications (e.g., formulations of poultry feeds).258 Using citric acid as a nontoxic crosslinker, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) as activators, Bedade and co-authors259 mounted acrylamidase from Cupriavidus oxa laticus ICTDB921 on chitosan-coated Ca-ALG beads. After four cycles, the immobilized acrylamidase maintained 80% of its activity while exhibiting increased pH, temperature, and shelf stability.259 Pectic compounds cause turbidity in juices. The aesthetics and storage stability of such products can be enhanced by removing these substances with the help of pectinase.
Mohammadi and co-workers260 immobilized Aspergillus aculeatus-originated pectinase on ALG-montmorillonite beads. At extremes of pH, immobilized pectinase showed higher efficacy than that of the free enzyme. After six successive cycles, immobilized pectinase exhibited >53% of its initial activity, demonstrating improved stability and renewability for pectin hydrolysis. Pineapple juice was clarified using this catalytic system, demonstrating the potential of this enzyme-carrier model for use in fruit juices.260Table 6 summarizes the immobilization of other enzymes on the ALG matrix due to abundant functional groups.261–268 For instance, Kamaci et al.264 observed enhanced catalytic activity of immobilized phytase into polyvinyl alcohol-sodium alginate (PVA-SA) electrospun nanofibers. Nanofibers were fabricated by mixing PVA and SA at a 80:20 ratio, voltage of 23 kV, and distance of 14 cm via electrospinning. Sharma et al.269 prepared bio-based and low-cost hybrid alginate-protein cryogel beads as novel adsorbent materials for the purification of immunoglobulin (Ig)G from human serum (Fig. 24). Due to soft interaction between the protein and IgG, the stability and integrity of the antibody were retained after the desorption step. Similarly, upcycling of marine chitin was improved with immobilized chitinolytic enzymes.270
 | ||
| Fig. 24 Use of hybrid alginate-protein cryogel beads to purify IgG (schematic). From ref. 269. | ||
| Biomass | Biomass derived material | Used enzyme | Immobilization method | Result/performance study | Ref. |
|---|---|---|---|---|---|
| Alginate | Polyacrylamide alginate cryogel (PAG) functionalized with glycidyl methacrylate (GMA) | Laccase | Covalent immobilization | >70% removal of phenolics from olive mill wastewater, >55% dye removal from textile wastewater and 93–99% decolouration of few other dyes were achieved | 219 |
| Alginate | Carrageenan-alginate beads | CAT | Covalent immobilization | Enhanced pH tolerance towards alkaline media and 6 folds higher Vmax achieved | 220 |
| Alginate | sodium alginate (SA)-polyethylene glycol (PEG) | Cellulase | Cross linking | With only 22.68% immobilization yield, 133% of yield (reducing sugar) obtained | 221 |
| Alginate | Polypyrrole/silver nanocomposite coated with calcium alginate | Polygalacturonase (PG) | Excellent immobilization efficiency (>84%) and long term stability (83% of initial activity after 2 months) | 222 | |
| Alginate | Silver-alginate nanoparticle matrix | Lipase | Conjugation | Application in high-fat meat processing is demonstrated with loss of 40% initial weight after 2 weeks | 223 |
| Alginate | Dopamine-alginate beads | Laccase from Streptomyces cyaneus | Amido Black 10B, Reactive Black 5, Evans Blue, and Remazol Brilliant Blue were all completely decoloured with 100% efficiency | 224 | |
| Alginate | Alginate micron and submicron beads | Alcalase | Covalent immobilization | Improved performance and utilised in 7 subsequent cycles of soy protein hydrolysis with a slight decrease in activity | 225 |
| Alginate | Sodium alginate (SA)-polyethylene glycol (PEG)-Chitosan (CS) | Cellulase | Entrapment | Increased stability and recyclability with 22.68% activity higher than native enzyme after 5 cycles | 226 |
| Alginate | Oxidized tyramine-alginates micro beads | HRP | Encapsulation | With 20 mol% of tyramine-alginate, 96% of the phenol was eliminated from the solution. Increased reusability | 227 |
| Alginate | Carrageenan-alginate beads with chitosan-glutaraldehyde activation | β-D-Galactosidase | Covalent immobilization | Reusability was improved, and 79.74% of the activity from the first round maintained till the tenth reusability round | 228 |
| Alginate | Calcium alginate hydrogels with liposomes | Bovine Carbonic Anhydrase (BCA) | Entrapment | Entrapment of BCA in alginate hydrogels is stable and effectiveness for using to catalytic reactions in bioreactors | 229 |
| Alginate | Entrapment in copper-alginate beads | Fungal phenol oxidase | Entrapment | Enhanced thermal activity and stability. Wider pH ranges allowed the enzyme to remain active, and storage at 4 °C considerably enhanced | 230 |
| Alginate | The gelatin component contained in the gel solution was cross-linked with glutaraldehyde (GA) and encapsulated in xanthan-alginate (XA) spheres | Urease | Encapsulation | Increased temperature and pH stability. Even after 20 times of use, the xanthan-alginate spheres still had 75% of their initial activity | 231 |
| Alginate | Calcium alginate gel capsule encapsulation | GOx | Encapsulation | Studied gelation conditions affected on capsule factors like capsule thickness, enzyme leakage rate, and encapsulation effectiveness | 233 |
| Alginic acid (AA) | AAC | Cyt c | Enhanced enzymatic activity up to 5.5-fold and improved the thermal stability | 232 | |
| Alginate | Calcium alginate gel capsule encapsulation | GOx | Encapsulation | Studied gelation conditions affected on capsule factors like capsule thickness, enzyme leakage rate, and encapsulation effectiveness | 233 |
| Alginate | Encapsulation in microcapsules alginate coated with alternating multiple layers membrane of poly N-vinylamine and polyacrylic acid | Cyt c | Encapsulation | Increased the encapsulation yield, activity as well as stability | 234 |
| Alginate | Ca-ALG gel capsules | Saccharomyces cerevisiae invertase | Entrapment | Relative activity was found 87% and active for 36 days. More stable at high pH and temperatures | 235 |
| Alginate from Sargassum | Entrapped in alginate gel | Invertase | Entrapment | 3.5 times higher t1/2 than native enzyme at 60 °C | 236 |
| Alginate | Hydrogels produced by combining glutaraldehyde-stabilized gelatin and natural polysaccharide alginate | Lipase | Cross linking | Stable and can be recycled for 10 and 20 times in aqueous and micellar media respectively without noticeably losing enzyme activity | 237 |
| Alginate | Microcapsules with an alginate-chitosan shell | β-Galactosidase | Entrapment | (1) Ca2+ alginate has a lesser loading efficiency than Ba2+ alginate. (2) Compared to liquid core Ca2+ alginate microcapsules and the unbound enzyme, solid core Ba2+ alginate microcapsules increased the stability of the enzyme at 37 °C | 238 |
| Alginate | Ultrathin alginate/protamine/silica (APSi) hybrid membranes in the core–shell capsules | Laccase | Entrapment | After 20 days, the stability of the encapsulated laccase increased by 67%. Improved storage, pH, and thermal stabilities | 239 |
| Alginate | Alginate-chitosan microcapsules made using an internal gelation and emulsification process to create alginate beads | Laccase | Not mentioned | Increased loading effectiveness and stability. In the Alizarin Red dye decolorization test, the free and immobilised laccase alone both had very low decolorization efficiency | 240 |
| Alginate gel | Alginate-silica nanotubes (ALG-SiNTs) composite was created by incorporating silica nanotubes (SiNTs) into the alginate (ALG) gel and then encapsulating them with Ca2+ through cross-linking | Yeast alcohol dehydrogenase (YADH) | Encapsulation | Improved storage and operational stability with ALG-SiNT composite showing ∼50% lesser leaching compared to only alginate matrix | 241 |
| Alginate | Biomaterials made from alginate gel doped with double-walled carbon nanotubes (DWCNT-ALG) | LDH | Adsorption, encapsulation and cross linking | Improved enzyme adherence with >61% reduction in leaching and 25 folds higher shelf-life than native enzyme | 243 |
| Alginate | Ca-alginate beads by entrapment | Lipase | Entrapment | 40% higher activity than the free enzyme with 10 cycles of reusability without any loss of activity | 244 |
| Alginate | Calcium alginate beads | Protease | Entrapment | Activity retained for a longer period of time and reused for 3 cycles. Up to 10 days of storage stability at 4 °C was observed | 245 |
| Alginate | Calcium alginate gel by entrapment method | Cellulase | Entrapment | Increased stability with 5 °C raise in optimum temperature (55 to 60 °C) and pH tolerance in acidic regime | 246 |
| Alginate | Calcium-alginate beads made using the entrapment technique | MnP | Entrapment | (1) >83% immobilization yield; tremendous shift in the optimum temperature from 35 to 60 °C and pH (towards acidic range). (2) >82–95% decolouration capacity with Sandal-fix dyes | 247 |
| Sodium salt of alginic acid from brown algae | PVA-ALG beads | Naringinase | Entrapment | Residual activity retained 70% after 8 successive batches | 248 |
| Alginate | PVA with calcium alginate (Alg) beads | Lipase | Entrapment | Improved the thermal stability and reusability | 249 |
| Alginate | Alginate-glutaraldehyde beads made through covalent bonding | Neutrase | Covalent bonding | Immobilization yield was ∼50% and bestowed 10 °C higher thermal tolerance than free enzyme (50 °C) | 250 |
| Alginate | ρ-Benzoquinone-activated alginate beads | Glucoamylase | Entrapment | No change in optimum pH, temperature and activity over 36 days and 30 cycles of recycling | 251 |
| Alginate | Calcium alginate-chitosan microspheres (CACM) | GOx | Encapsulation and adsorption method | Increased the enzymatic activity and storage stability was increased up to 2 month and retained activity of CACM-GOX | 252 |
| Alginate | Hybrid gel beads made of chemically reduced graphene oxide (CRGO) and alginate | GOx | Non-covalent adsorption–entrapment method | Good recyclability, mechanical properties, and enhanced stability in a wide pH (4–6) and temperature (45–60 °C) range | 253 |
| Alginate | Polymethyl methacrylate (PMMA-g-CS) nanoparticles were grafted onto chitosan encapsulated calcium alginate beads | α-CT | Covalent bonding | Higher retained activity at pH 9 for 24 h | 254 |
| Alginate | polyvinyl alcohol/alginate and polyethylene oxide/alginate nanofibers by electrospinning method | Lipase | Adsorption and cross linking method | Enhanced the stability. Maintained 60% of their activities after 14 and 7 reuses and increased the stability | 256 |
| Alginate | Nanoflower/alginate microbeads | ALDC | Entrapment | Restored 98% of activity with improving stability and recyclability | 257 |
| Alginate | Calcium alginate beads with glutaraldehyde activation | Xylanase | Entrapment and cross linking | After 30 days at 4 °C, the enzyme still exhibits 80% of its initial activity and maintains recycling efficiency up to five reaction cycles with 37% retention activity | 258 |
| Alginate | Functionalized calcium alginate beads with chitosan coating | Acrylamidase | Cross linking and covalent immobilization | Enhanced pH, thermal, shelf, and mechanical stability, and maintained 80% activity after four cycles | 259 |
| Alginate | Activated alginate-montmorillonite(MMT) beads | Pectinase | Covalent binding | Showed greater activity, however the optimal pH dropped from 5.5to 5.0. The immobilised enzyme's initial activity was maintained at around 53% after 6 cycles of reuse | 260 |
| Alginate | Glutaraldehyde is utilized to harden hybrid alginate-chitosan beads | Saccharomyces cerevisiae alcohol dehydrogenase (SCAD) | Cross linking | Improved thermal stability with enhanced optimum temperature (from 30 to 40 °C) | 261 |
| Alginate | Boehmite/alginate hybrid beads | YADH | Encapsulation | After 67 hours of incubation, encapsulated YADH can practically approach “zero leaching” and retain 86.6% of its activity after 12 cycles | 262 |
| Alginate | Entrapped BG crosslinked glutaraldehyde in calcium alginate particles | β-Glucosidase | Entrapment and crosslinking | Under the optimum conditions, more than 60% of enzyme activity restored and cross linkage of glutaraldehyde reduced leakage of BG from the calcium alginate particles | 263 |
| Alginate | Polyvinyl alcohol-sodium alginate (PVA-SA) nanofibers by electrospun nanofiber | Phytase | Cross linking | Enhanced the catalytic activity with optimum pH shifting towards higher pH (from 5 to 6) and temperature from 45 to 55 °C | 264 |
| Alginate | Entrapment of alginate beads | Lipase | Entrapment | High lipase activity in the acidic pH of 3 and 40 °C temperature | 265 |
| Alginate | 3D transition metal cation-crosslinked alginate nanogels (Mn2+, Fe3+, and Co2+) | Urease | Encapsulation | Fe3+-alginate crosslinked nanogels demonstrated high enzyme activity, efficient enzyme loading, and zeta potential compared to Co2+ and Mn2+ | 266 |
| Alginate | Cu-alginate beads | Trametes Versicolor laccase | Entrapment | Enhanced activity with >96% degradation capacity towards bisphenol at pH 5 and 30 °C in 60 min | 268 |
8. Conclusions and future prospects
The use of enzyme-based biocatalysts at the industrial scale has garnered interest in recent decades. However, the use of enzymes is limited due to their low thermal stability, complicated reusability, self-cleavage, and aggregation under extreme processing conditions. Immobilization of enzymes on a biocompatible and renewable carrier is a promising method to overcome such drawbacks. BDFMs are promising carriers for enzyme immobilization due to their availability, biodegradability, biocompatibility, and low risk of environmental contamination. In this review, the main types of BDFMs were presented as promising carriers for enzymes to improve their stability and reusability. In addition, the immobilization of enzymes on various biomass-derived carriers (e.g., cellulose, lignin, SF, chitin, chitosan, and ALG) was surveyed. Although various approaches have been developed for BDFM fabrication, studies on the practical applications of these materials are in the early stages. Immobilization/adsorption/entrapment of various enzymes on BDFMs can improve the stability, activity, reusability, and recyclability of biocatalysts compared with those of their native state. In many cases, impressive results have been obtained whereas, in several other cases, the biocatalytic activity must be improved for commercial application. For selection of the best support materials and preparation of an efficient biocatalyst, all reaction conditions must be optimized in terms of monodispersity and surface chemistry. Moreover, the literature is based mainly on enzymes of a single type, which may not be suitable for industrial needs. Therefore, studies with multiple enzyme “cocktails” should be undertaken to lower the cost and increase the utility of biocatalysts.Fig. 25 provides an overview of the analysis of the strength, weakness, opportunities and threats (SWOT) of BDFMs as carriers for enzymes. These challenges should be considered when selecting the biomass matrix and immobilization methods for direct industrial application. Cost-effective sources (e.g., non-commercial biomass, waste biomass, non-wood forest products, and biomass from unconventional resources) as support materials as well as controlled immobilization methods with soft interactions between enzymes and BDFMs must be selected. This review also provides an opportunity to understand the availability of different types of biomass as catalyst supports to develop efficient biocatalytic systems. The recyclability and reusability of enzymes is another crucial parameter that ensures the practicality of a biocatalytic process. We believe that this review will aid use of BDFMs for further studies with different enzymes under extreme conditions (e.g., high temperature, pH, denaturing substances). We attempted to collect all the information on the immobilization of enzymes on macro- or microscopic biomass-derived materials as carriers to improve the enzyme activity, stability, and recovery of biocatalysts. Therefore, this review aims to create economic value from various biomass and biocatalytic processes to strengthen the economy, with high scientific, technological, and societal impacts. This review also aids the use of biomaterials as supports for the development of immobilized biocatalysts for applications based on energy, environmental issues, and chemical syntheses. From the perspective of green and sustainable chemistry, this review offers a promising approach for effective evaluation of biomass.
 | ||
| Fig. 25 SWOT analysis of BDFMs for biocatalysis. | ||
Abbreviations
| BDFM | Biomass-derived functional material |
| AA | Alginic acid |
| MOF | Metal–organic framework |
| WOS | Web of science |
| BGU | β-Glucuronidase |
| CHM | Cellulose hydrogel microsphere |
| CNC | Cellulose nanocrystal |
| CNW | Cellulose nanowhisker |
| NFC | Nanofibrillated cellulose |
| MCC | Microcrystalline cellulose |
| CBD | Cellulose-binding domain |
| GOx | Glucose oxidase |
| CMC | Carboxymethyl cellulose |
| IL | Ionic liquid |
| PPL | Porcine pancreatic lipase |
| HNT | Halloyite nanotube |
| MCM | Cellulose porous microsphere |
| PGA | Penicillin G acylase |
| TLFCH | Tendril-like functional carbon helix |
| DES | Deep eutectic solvent |
| Cyt c | Cytochrome c |
| [Emim][Ac] | 1-Ethyl-3-methylimidazolium acetate |
| CRL | Candida rugosa lipase |
| OPH | Organophosphorus hydrolase |
| BTDE | 1,4-Butanediol diglycidyl ether |
| EMBR | Enzymatic membrane bioreactor |
| αCF | α-Cellulose fiber |
| α-CT | α-Chymotrypsin |
| PDDA | Poly(diallyldimethylammonium chloride) |
| L-DOPA | 3,4-Dihydroxyphenyl-L-alanine |
| LNP | Lignin nanoparticle |
| SET-LRP | Single electron transfer-living radical polymerization |
| SF | Silk fibroin |
| HRP | Horseradish peroxidase |
| ChOx | Cholesterol oxidase |
| CA | Carbonic anhydrase |
| ASNase | L-Asparaginase |
| CadA | Lysine decarboxylase |
| ChBD | Chitin-binding domain |
| CHNW | Chitin nanowhisker |
| AuNP | Gold nanoparticle |
| AgNp | Silver nanoparticle |
| CNF | Chitin nanofibril |
| TMB | 3,3′,5,5′-Tetramethylbenzidine |
| GO | Graphene oxide |
| RGO | Reduced graphene oxide |
| BSS | Bamboo shoot shell |
| LBL | Layer-by-layer |
| SGP | Seal gastric protease |
| CAT | Catalase |
| NaIO4 | Sodium periodate |
| TEMPO | 2,2,6,6-Tetramethylpiperidine-1-oxyl radical |
| SNF | Silk nanofiber |
| ONPG | o-Nitrophenyl-β-D-galactopyranoside |
| DWCNT | Double-walled carbon nanotube |
| LDH | Lactate dehydrogenase |
| PDMS | Polydimethylsiloxane |
| PEO | Polyethylene oxide |
| ALDC | α-Acetolactate decarboxylase |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride |
| NHS | N-Hydroxysuccinimide |
| t 1/2 | Half life |
| PEG | Polyethylene glycol |
| Ca-ALG | Calcium-alginate |
| AAC | Alginate-derived carbon |
| SiNT | Silica nanotube |
| TMOS | Tetramethoxysilane |
| BCA | Bovine carbonic anhydrase |
| SA | Sodium alginate |
| XA | Xanthan-alginate |
| PVA-SA | Polyvinyl alcohol-sodium alginate |
| MMT | Montmorillonite |
| DMC | Dialdehyde-modified cellulose nanocrystal |
| MDC | Magnetic dialdehyde cellulose nanoparticle |
| MCNC | Magnetic chitin nanofiber composite |
| CS-Nps | Chitosan nanoparticle |
| ALG-SiNT | Alginate-silica nanotube |
| CMNP | Chitosan magnetic nanoparticle |
| MnP | Manganese peroxidase |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre (UMO-2021/43/D/ST4/00699). D. M. and G. F. acknowledge the NANOPLANT project, which received funding from the European Union's Horizon 2020 Research and Innovation Program (856961).References
- Y. Liu, J. Chen, B. Cui, P. Yin and C. Zhang, C, 2018, 4, 53 CAS.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- P. Manzanares, Acta Innov., 2020, 47–56 Search PubMed.
- P. Morone, Acta Innov., 2020, 5–16 Search PubMed.
- D. M. Kochepka, L. P. Dill, D. H. Fockink and R. M. Łukasik, Acta Innov., 2020, 35, 29–56 Search PubMed.
- L. P. Ramos, M. J. Suota, D. H. Fockink, G. Pavaneli, T. A. da Silva and R. M. Łukasik, Recent Advances in Bioconversion of Lignocellulose to Biofuels and Value-Added Chemicals within the Biorefinery Concept, Elsevier, 2020, pp. 61–100 Search PubMed.
- H. Suo, L. Xu, Y. Xue, X. Qiu, H. Huang and Y. Hu, Carbohydr. Polym., 2020, 234, 115914 CrossRef CAS PubMed.
- S. Doonan, E. W. Abel, D. Phillips, J. D. Woollins and A. G. Davies, Peptides and Proteins, Royal Society of Chemistry, 2002, vol. 15, 10.1039/9781847551634m3-X001.
- C. I. Branden and J. Tooze, Introduction to protein structure, Garland Science, 2012 Search PubMed.
- S. Ohnishi and K. Takano, Cell. Mol. Life Sci., 2004, 61, 511–524 CrossRef CAS PubMed.
- A. Lesk, Introduction to protein science: architecture, function, and genomics, Oxford university press, 2010 Search PubMed.
- A. Sindhu, S. Kumar and P. Venkatesu, ACS Sustainable Chem. Eng., 2022, 10, 4323–4344 CrossRef CAS.
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307 CrossRef CAS.
- S. van Pelt, F. van Rantwijk and R. A. Sheldon, Adv. Synth. Catal., 2009, 351, 397–404 CrossRef CAS.
- M. L. Verma, S. Kumar, A. Das, J. S. Randhawa and M. Chamundeeswari, Environ. Chem. Lett., 2020, 18, 315–323 CrossRef CAS.
- H. T. Imam, P. C. Marr and A. C. Marr, Green Chem., 2021, 23, 4980–5005 RSC.
- S. Arola, T. Tammelin, H. Setälä, A. Tullila and M. B. Linder, Biomacromolecules, 2012, 13, 594–603 CrossRef CAS PubMed.
- K. Xu, B. Appiah, B.-W. Zhang, Z.-H. Yang and C. Quan, J. Catal., 2023, 418, 31–39 CrossRef CAS.
- J. Meyer, L. E. Meyer and S. Kara, Eng. Life Sci., 2022, 22, 165–177 CrossRef CAS PubMed.
- V. C. Badgujar and B. M. Bhanage, Curr. Opin. Green Sustain. Chem., 2022, 100621 CrossRef.
- U. T. Bornscheuer, in Biotechnology for the Future, ed. J. Nielsen, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 181–203, DOI:10.1007/b136413.
- R. S. Khan, A. H. Rather, T. U. Wani, S. u. Rather, T. Amna, M. S. Hassan and F. A. Sheikh, Biotechnol. Bioeng., 2023, 120, 22–40 CrossRef CAS PubMed.
- S. Lu, X. Wang, Q. Lu, X. Hu, N. Uppal, F. G. Omenetto and D. L. Kaplan, Biomacromolecules, 2009, 10, 1032–1042 CrossRef CAS PubMed.
- S. Lv, Molecules, 2020, 25, 4929 CrossRef CAS PubMed.
- Y. A. Rodríguez-Restrepo and C. E. Orrego, Environ. Chem. Lett., 2020, 18, 787–806 CrossRef.
- H. J. Kim, S. Park, S. H. Kim, J. H. Kim, H. Yu, H. J. Kim, Y.-H. Yang, E. Kan, Y. H. Kim and S. H. Lee, J. Mol. Catal. B: Enzym., 2015, 122, 170–178 CrossRef CAS.
- R. S. Varma, ACS Sustainable Chem. Eng., 2019, 7, 6458–6470 CrossRef CAS.
- V. Incani, C. Danumah and Y. Boluk, Cellulose, 2013, 20, 191–200 CrossRef CAS.
- H. Liu, A. D. Štiglic, T. Mohan, R. Kargl, K. S. Kleinschek and B. Nidetzky, Int. J. Biol. Macromol., 2022, 222, 217–227 CrossRef CAS PubMed.
- Y. Liu and J. Y. Chen, J. Bioact. Compat. Polym., 2016, 31, 553–567 CrossRef CAS.
- W. Sheng-Chi and L. Ying-Ke, J. Mol. Catal. B: Enzym., 2008, 54, 103–108 CrossRef.
- A. Gennari, A. J. Führ, G. Volpato and C. F. V. de Souza, Carbohydr. Polym., 2020, 246, 116646 CrossRef CAS PubMed.
- M. P. Klein, C. W. Scheeren, A. S. G. Lorenzoni, J. Dupont, J. Frazzon and P. F. Hertz, Process Biochem., 2011, 46, 1375–1379 CrossRef CAS.
- S. Sulaiman, M. N. Mokhtar, M. N. Naim, A. S. Baharuddin and A. Sulaiman, Appl. Biochem. Biotechnol., 2015, 175, 1817–1842 CrossRef CAS PubMed.
- I. B. V. Erramuspe, E. Fazeli, T. Näreoja, J. Trygg, P. Hänninen, T. Heinze and P. Fardim, Biomacromolecules, 2016, 17, 3188–3197 CrossRef PubMed.
- M. Bagheri, H. Rodríguez, R. P. Swatloski, S. K. Spear, D. T. Daly and R. D. Rogers, Biomacromolecules, 2008, 9, 381–387 CrossRef CAS PubMed.
- N. F. Vasconcelos, F. K. Andrade, L. d. A. P. Vieira, R. S. Vieira, J. M. Vaz, P. Chevallier, D. Mantovani, M. d. F. Borges and M. d. F. Rosa, Cellulose, 2020, 27, 3055–3083 CrossRef CAS.
- H. Chen and Y.-L. Hsieh, Biotechnol. Bioeng., 2005, 90, 405–413 CrossRef CAS PubMed.
- E. Mascheroni, G. Capretti, M. Marengo, S. Iametti, L. Mora, L. Piergiovanni and F. Bonomi, Packag. Technol. Sci., 2010, 23, 47–57 CrossRef CAS.
- S. Martins de Oliveira, S. Velasco-Lozano, A. H. Orrego, J. Rocha-Martín, S. Moreno-Pérez, J. M. Fraile, F. López Gallego and J. M. Guisán, Biomacromolecules, 2021, 22, 927–937 CrossRef CAS PubMed.
- K. A. Mahmoud, E. Lam, S. Hrapovic and J. H. T. Luong, ACS Appl. Mater. Interfaces, 2013, 5, 4978–4985 CrossRef CAS PubMed.
- C. Goldhahn, I. Burgert and M. Chanana, Adv. Mater. Interfaces, 2019, 6, 1900437 CrossRef CAS.
- Q. Luan, H. Zhang, Y. Lei, Y. Cai, Y. Bao, Y. Li, H. Tang and X. Li, Cellulose, 2021, 28, 5735–5744 CrossRef CAS.
- S.-L. Cao, X.-H. Li, W.-Y. Lou and M.-H. Zong, J. Mater. Chem. B, 2014, 2, 5522–5530 RSC.
- Y. Ren, L. Zhang, T. Sun, Y. Yin and Q. Wang, ACS Sustainable Chem. Eng., 2022, 10, 6244–6254 CrossRef CAS.
- E. Motamedi, S. F. S. Motahar, M. Maleki, K. Kavousi, S. Ariaeenejad, A. A. Moosavi-Movahedi and G. H. Salekdeh, Cellulose, 2021, 28, 3485–3503 CrossRef CAS.
- C. Uth, S. Zielonka, S. Hörner, N. Rasche, A. Plog, H. Orelma, O. Avrutina, K. Zhang and H. Kolmar, Angew. Chem., Int. Ed., 2014, 53, 12618–12623 CAS.
- D. Sillu and S. Agnihotri, ACS Sustainable Chem. Eng., 2020, 8, 900–913 CrossRef CAS.
- X. Luo and L. Zhang, Biomacromolecules, 2010, 11, 2896–2903 CrossRef CAS PubMed.
- K. Aruchamy, M. Bisht, P. Venkatesu, D. Kalpana, N. R. Maalige, N. Singh, D. Ghosh, D. Mondal and S. K. Nataraj, Green Chem., 2018, 20, 3711–3716 RSC.
- S. Jo, S. Park, Y. Oh, J. Hong, H. J. Kim, K. J. Kim, K. K. Oh and S. H. Lee, Biotechnol. Bioprocess Eng., 2019, 24, 145–154 CrossRef CAS.
- S. Park, S. H. Kim, J. H. Kim, H. Yu, H. J. Kim, Y.-H. Yang, H. Kim, Y. H. Kim, S. H. Ha and S. H. Lee, J. Mol. Catal. B: Enzym., 2015, 119, 33–39 CrossRef CAS.
- M. H. Kim, S. An, K. Won, H. J. Kim and S. H. Lee, J. Mol. Catal. B: Enzym., 2012, 75, 68–72 CrossRef CAS.
- M. Sharifi, S.-M. Robatjazi, M. Sadri and J. M. Mosaabadi, React. Funct. Polym., 2018, 124, 162–170 CrossRef CAS.
- S. Yabuki, Y. Hirata, Y. Sato and S. Iijima, Anal. Sci., 2012, 28, 373–377 CrossRef CAS PubMed.
- L. Hao, R. Wang, Y. Zhao, K. Fang and Y. Cai, Cellulose, 2018, 25, 6759–6769 CrossRef CAS.
- N. Tanja, K. Mirjana, P. Jovana, P. Biljana, P. Zivomir and S. Petar, Carbohydr. Polym., 2010, 82, 976–981 CrossRef.
- R. Gong, J. Zhang, J. Zhu, J. Wang, Q. Lai and B. Jiang, Korean J. Chem. Eng., 2013, 30, 1620–1625 CrossRef CAS.
- A. Gennari, R. Simon, N. D. de Moura Sperotto, C. V. Bizarro, L. A. Basso, P. Machado, E. V. Benvenutti, G. Renard, J. M. Chies, G. Volpato and C. F. De Souza, Int. J. Biol. Macromol., 2022, 199, 307–317 CrossRef CAS PubMed.
- C. Liu, D. Saeki and H. Matsuyama, RSC Adv., 2017, 7, 48199–48207 RSC.
- S. G. Gajanan, Y. Jiwook, S. S. Surendra, M. M. Bhupendra, K. Dae-Young, S. Jung-Suk and A. K. Avinash, Bioresour. Technol., 2018, 261, 420–427 CrossRef PubMed.
- X. Xing, Y. Han, Q. Jiang, Y. Sun, X. Wang, G. Qu, G. Sun and Y. Li, Cellulose, 2021, 28, 4793–4805 CrossRef CAS.
- G. Hui, L. Bingshuang, Y. Jianwei, C. Yunfei and Q. Junqing, Int. J. Biol. Macromol., 2021, 185, 287–296 CrossRef PubMed.
- E. Restiawaty, F. A. Yatasya, N. T. U. Culsum and Y. W. Budhi, InIOP Conference Series: Materials Science and Engineering, IOP Publishing, 2021, vol. 1143, p. 012009.
- S. S. S. Putra, W. J. Basirun, A. A. M. Elgharbawy, A. Hayyan, M. Hayyan and M. A. Mohammed, Mol. Catal., 2022, 528, 112422 CrossRef CAS.
- D. Califano, B. L. Patenall, M. A. S. Kadowaki, D. Mattia, J. L. Scott and K. J. Edler, Biomacromolecules, 2021, 22, 754–762 CrossRef CAS PubMed.
- J. V. Edwards, N. T. Prevost, B. Condon and A. French, Cellulose, 2011, 18, 1239–1249 CrossRef CAS.
- V. Singh and S. Ahmad, Cellulose, 2012, 19, 1759–1769 CrossRef CAS.
- A. Girelli, L. Salvagni and A. Tarola, J. Braz. Chem. Soc., 2012, 23, 585–592 CAS.
- H. H. Je, S. Noh, S.-G. Hong, Y. Ju, J. Kim and D. S. Hwang, Chem. Eng. J., 2017, 323, 425–433 CrossRef CAS.
- Z. Hao, L. Qian, L. Yan, W. Jiahui, B. Yuping, T. Hu and H. Fenghong, Int. J. Biol. Macromol., 2022, 199, 61–68 CrossRef PubMed.
- V. N. Kumar and R. Neera, Int. J. Biol. Macromol., 2022, 200, 618–625 CrossRef PubMed.
- Q. Weichuan, Z. Ziyan, Q. Yi, X. Lijie and G. He, Colloids Surf., A, 2022, 632, 127818 CrossRef.
- A. Gennari, R. Simon, N. D. de Moura Sperotto, C. V. Bizarro, L. A. Basso, P. Machado, E. V. Benvenutti, A. D. Viegas, S. Nicolodi, G. Renard and J. M. Chies, Bioresour. Technol., 2022, 345, 126497 CrossRef CAS PubMed.
- S. Ariaeenejad, E. Motamedi and G. H. Salekdeh, Bioresour. Technol., 2022, 349, 126833 CrossRef CAS PubMed.
- Z. Zhihong, J. Mengchen, C. Guiru, L. Jiandu, W. Luying and G. Jun, Green Chem. Eng., 2023, 4(1), 39–48 CrossRef.
- K. A. Mahmoud, K. B. Male, S. Hrapovic and J. H. T. Luong, ACS Appl. Mater. Interfaces, 2009, 1, 1383–1386 CrossRef CAS PubMed.
- N. E. Kotel'nikova, S. A. Mikhailova and E. N. Vlasova, Russ. J. Appl. Chem., 2007, 80, 322–329 CrossRef.
- Z. Liu, H. Wang, B. Li, C. Liu, Y. Jiang, G. Yu and X. Mu, J. Mater. Chem., 2012, 22, 15085–15091 RSC.
- H. Xiao-Jun, C. Peng-Cheng, H. Fu, O. Yang, C. Ming-Rui and X. Zhi-Kang, J. Mol. Catal. B: Enzym., 2011, 70, 95–100 CrossRef.
- B. Muhammad and M. N. I. Hafiz, Int. J. Biol. Macromol., 2019, 130, 462–482 CrossRef PubMed.
- P. Bayazidi, H. Almasi and A. K. Asl, Int. J. Biol. Macromol., 2018, 107, 2544–2551 CrossRef CAS PubMed.
- S. P. de Souza, I. I. Junior, G. M. A. Silva, L. S. M. Miranda, M. F. Santiago, F. L.-Y. Lam, A. Dawood, U. T. Bornscheuer and R. O. M. A. de Souza, RSC Adv., 2016, 6, 6665–6671 RSC.
- S. A. Fatma, M. Monier and H. E. Nadia, Int. J. Biol. Macromol., 2018, 114, 1018–1025 CrossRef PubMed.
- C. J. R. Frazão, N. H. C. Silva, C. S. R. Freire, A. J. D. Silvestre, A. M. R. B. Xavier and A. P. M. Tavares, Eng. Life Sci., 2014, 14, 500–508 CrossRef.
- G. Weihua, R. Zhanxiang, Y. Fayin and Z. Guohua, Food Chem., 2017, 228, 455–462 CrossRef PubMed.
- J. Bebić, K. Banjanac, J. Rusmirović, M. Ćorović, A. Milivojević, M. Simović, A. Marinković and D. Bezbradica, RSC Adv., 2020, 10, 21495–21508 RSC.
- C. E. de A. Padilha, C. da C. Nogueira, D. F. de S. Souza, J. A. de Oliveira and E. S. dos Santos, Ind. Crops Prod., 2020, 146, 112167 CrossRef.
- K. Łukasz, Z. Jakub and J. Teofil, Colloids Surf., B, 2018, 162, 90–97 CrossRef PubMed.
- E. Capecchi, D. Piccinino, B. M. Bizzarri, D. Avitabile, C. Pelosi, C. Colantonio, G. Calabrò and R. Saladino, Biomacromolecules, 2019, 20, 1975–1988 CrossRef CAS PubMed.
- E. Tomaino, E. Capecchi, D. Piccinino and R. Saladino, ChemCatChem, 2022, 14, e202200380 CrossRef CAS.
- A. Moreno and M. H. Sipponen, Nat. Commun., 2020, 11, 5599 CrossRef CAS PubMed.
- A. I. Benítez-Mateos, S. Bertella, J. Behaghel de Bueren, J. S. Luterbacher and F. Paradisi, ChemSusChem, 2021, 14, 3198–3207 CrossRef PubMed.
- M. Stanisz, K. Bachosz, K. Siwińska-Ciesielczyk, Ł. Klapiszewski, J. Zdarta and T. Jesionowski, Catalysts, 2022, 12, 1031 CrossRef CAS.
- Y. Takahashi, M. Gehoh and K. Yuzuriha, J. Polym. Sci., Part B: Polym. Phys., 1991, 29, 889–891 CrossRef CAS.
- S. Kaushik, P. D. Thungon and P. Goswami, ACS Biomater. Sci. Eng., 2020, 6, 4337–4355 CrossRef CAS PubMed.
- L. Grasset, D. Cordier and A. Ville, Biotechnol. Bioeng., 1977, 19, 611–618 CrossRef CAS PubMed.
- M. A. Tomeh, R. Hadianamrei and X. Zhao, Pharmaceutics, 2019, 11, 494 CrossRef CAS PubMed.
- K. H. Lee, C. S. Ki, D. H. Baek, G. D. Kang, D.-W. Ihm and Y. H. Park, Fibers Polym., 2005, 6, 181–185 CrossRef CAS.
- S. Chatterjee, L. Barbora, S. S. Cameotra, P. Mahanta and P. Goswami, Appl. Biochem. Biotechnol., 2009, 157, 593–600 CrossRef CAS PubMed.
- B. Chen, C. Yin, Y. Cheng, W. Li, Z. Cao and T. Tan, Biomass Bioenergy, 2012, 39, 59–66 CrossRef CAS.
- C. Acharya, V. Kumar, R. Sen and S. C. Kundu, Biotechnol. J., 2008, 3, 226–233 CrossRef CAS PubMed.
- U. Saxena and P. Goswami, Appl. Biochem. Biotechnol., 2010, 162, 1122–1131 CrossRef CAS PubMed.
- Y. Han, S. Yu, L. Liu, S. Zhao, T. Yang, Y. Yang, Y. Fang and S. Lv, Mol. Catal., 2018, 457, 24–32 CrossRef CAS.
- S. Miyairi, M. Sugiura and S. Fukui, Agric. Biol. Chem., 1978, 42, 1661–1667 CAS.
- P. B. Dennis, A. Y. Walker, M. B. Dickerson, D. L. Kaplan and R. R. Naik, Biomacromolecules, 2012, 13, 2037–2045 CrossRef CAS PubMed.
- H. Yoshimizu and T. Asakura, J. Appl. Polym. Sci., 1990, 40, 127–134 CrossRef CAS.
- Y.-Q. Zhang, W.-L. Zhou, W.-D. Shen, Y.-H. Chen, X.-M. Zha, K. Shirai and K. Kiguchi, J. Biotechnol., 2005, 120, 315–326 CrossRef CAS PubMed.
- L. Lichao, H. Chengzhi, X. Menglin, A. Zeyu and L. Shanshan, Appl. Catal., A, 2020, 597, 117541 CrossRef.
- E. M. Pritchard, P. B. Dennis, F. Omenetto, R. R. Naik and D. L. Kaplan, Biopolymers, 2012, 97, 479–498 CrossRef CAS PubMed.
- Z. Chen, F. Hu, Z. Lin, J. Hu, R. Shen, Y. Lin and X. Y. Liu, Small Sci., 2021, 1, 2000049 CrossRef CAS.
- Y. Fu, X. Xie, Y. Wang, J. Liu, Z. Zheng, D. L. Kaplan and X. Wang, ACS Biomater. Sci. Eng., 2021, 7, 2734–2744 CrossRef CAS PubMed.
- M. Denise, D. Aleksander, I. Heiko, K. Nadja, Z. Max, S. Ralf, K. Marius, K. Alexander and J. S. Michael, Biosens. Bioelectron., 2021, 183, 113204 CrossRef PubMed.
- C. Bungthong, C. Wrigley, T. Sonteera and S. Siriamornpun, Molecules, 2021, 26, 3455 CrossRef CAS PubMed.
- M. d. C. de Castro, P. S. Garcia, M. M. Andrade, M. V. E. Grossmann, B. M. Simões, R. B. Samulewski and A. M. Baron, Biotechnol. Appl. Biochem., 2022, 69, 660–667 CrossRef CAS PubMed.
- Q. Jianghong, L. Yongcheng, L. Haiying, Y. Tongyin and D. Jiaqi, Anal. Biochem., 1996, 236, 208–214 CrossRef PubMed.
- D. Makoto and A. Tetsuo, J. Membr. Sci., 1991, 59, 39–52 CrossRef.
- K. Akio, A. Tetsuo, T. Ryuzo and M. Tadashi, J. Biotechnol., 1987, 5, 199–207 Search PubMed.
- D. Cordier, R. Couturier, L. Grasset and A. Ville, Enzyme Microb. Technol., 1982, 4, 249–255 CrossRef CAS.
- S. Inoue, Y. Matsunaga, H. Iwane, M. Sotomura and T. Nose, Biochem. Biophys. Res. Commun., 1986, 141, 165–170 CrossRef CAS PubMed.
- Y. Liu, X. Chen, J. Qian, H. Liu, Z. Shao, J. Deng and T. Yu, Appl. Biochem. Biotechnol., 1997, 62, 105–117 CrossRef CAS PubMed.
- B. Liang, L. Fang, Y. Hu, G. Yang, Q. Zhu and X. Ye, Nanoscale, 2014, 6, 4264–4274 RSC.
- A. Einbu, S. N. Naess, A. Elgsaeter and K. M. Vårum, Biomacromolecules, 2004, 5, 2048–2054 CrossRef CAS PubMed.
- R. Marguerite, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef.
- E. Cabib, in Plant Carbohydrates II: Extracellular Carbohydrates, ed. W. Tanner and F. A. Loewus, Springer Berlin Heidelberg, Berlin, Heidelberg, 1981, pp. 395–415. DOI:10.1007/978-3-642-68234-6_16.
- V. Farkaš, Acta Biotechnol., 1990, 10, 225–238 CrossRef.
- D. Knorr, Food Technol., 1984, 38, 92–97 Search PubMed.
- B. Amit and S. Mika, Adv. Colloid Interface Sci., 2009, 152, 26–38 CrossRef PubMed.
- A. A. M. Riccardo, Carbohydr. Polym., 2009, 76, 167–182 CrossRef.
- T. N. Nwagu, B. Okolo and H. Aoyagi, Bioresour. Technol. Rep., 2021, 13, 100645 CrossRef CAS.
- J. Synowiecki, Z. E. Sikorski and M. Naczk, Biotechnol. Bioeng., 1981, 23, 2211–2215 CrossRef CAS.
- M. T. Xavier, V. F. Soares, D. G. Freire, C. P. Moreira, M. F. Mendes and E. Bon, Biomass, 1987, 13, 25–32 CrossRef CAS.
- O. Eiji and K. Tsuyoshi, Anal. Chim. Acta, 1992, 262, 19–25 CrossRef.
- H. Xiao-Qing and S. Fereidoon, Food Chem., 1995, 52, 71–76 CrossRef.
- F. M. Gomes, E. B. Pereira and H. F. de Castro, Biomacromolecules, 2004, 5, 17–23 CrossRef CAS PubMed.
- G. Leissy, L. R. Hector, L. V. Maria, H. Junior and V. Reynaldo, Enzyme Microb. Technol., 2006, 38, 22–27 CrossRef.
- L. Gomez, H. L. Ramirez, G. Cabrera, B. K. Simpson and R. Villalonga, J. Food Biochem., 2008, 32, 264–277 CrossRef CAS.
- S. Manthiriyappan and L. Cheng-Kang, Carbohydr. Polym., 2011, 84, 775–780 CrossRef.
- N. Zhou, A. Zhang, G. Wei, S. Yang, S. Xu, K. Chen and P. Ouyang, Front. Bioeng. Biotechnol., 2020, 8, 103 CrossRef PubMed.
- H. L. Ramirez, L. G. Brizuela, J. Ú. Iranzo, M. Arevalo-Villena and A. I. B. Pérez, J. Food Process Eng., 2016, 39, 97–104 CrossRef CAS.
- J. Suisui, Q. Yang, Y. Jie, L. Man, X. Liu and S. Qingjie, Food Chem., 2017, 221, 1507–1513 CrossRef PubMed.
- W. A. Mehdi, A. A. Mehde, M. Özacar and Z. Özacar, Int. J. Biol. Macromol., 2018, 117, 947–958 CrossRef CAS PubMed.
- H. Yao, F. Yan, C. Lingyun, L. Ang and Z. Lina, Chem. Eng. J., 2017, 315, 573–582 CrossRef.
- M. Chen, H. Wu, Z. Li, K. Wu, Y. Jiao and C. Zhou, J. Porous Mater., 2020, 27, 549–554 CrossRef CAS.
- F. Shahid, A. Ansari, A. Aman and S. A. U. Qader, Catal. Lett., 2020, 150, 613–622 CrossRef CAS.
- W.-C. Huang, W. Wang, C. Xue and X. Mao, ACS Sustainable Chem. Eng., 2018, 6, 8118–8124 CrossRef CAS.
- Y. Yandri, T. Suhartati, H. Satria, S. Karlinasari, S. D. Yuwono and S. Hadi, J. Adv. Pharm. Educ. Res., 2021, 11, 63–69 CrossRef CAS.
- T. Machałowski, K. Jankowska, K. Bachosz, W. Smułek, H. Ehrlich, E. Kaczorek, J. Zdarta and T. Jesionowski, Molecules, 2022, 27, 1354 CrossRef PubMed.
- Y. Yandri, E. R. Tiarsa, T. Suhartati, B. Irawan and S. Hadi, Emerg. Sci. J., 2022, 7, 77–89 CrossRef.
- E. R. Tiarsa, Y. Yandri, T. Suhartati, H. Satria, B. Irawan and S. Hadi, Biochem. Res. Int., 2022, 2022, 5692438 Search PubMed.
- H. Ibrahim and E. El-Zairy, Concepts, compounds and the alternatives of antibacterials, 2015, vol. 1, pp. 81–101 Search PubMed.
- S. K. Arya, M. Manohar, G. Singh and W. A. Siddiqui, Chitosan: Derivatives, Composites and Applications, 2017, pp. 151–165 Search PubMed.
- N. C. Ricardi, L. T. Arenas, E. V. Benvenutti, R. Hinrichs, E. E. E. Flores, P. F. Hertz and T. M. H. Costa, Food Chem., 2021, 359, 129890 CrossRef CAS PubMed.
- D. R. Baidamshina, V. A. Koroleva, S. S. Olshannikova, E. Y. Trizna, M. I. Bogachev, V. G. Artyukhov, M. G. Holyavka and A. R. Kayumov, Mar. Drugs, 2021, 19, 197 CrossRef CAS PubMed.
- T. A. Costa-Silva, A. K. F. Carvalho, C. R. F. Souza, H. F. De Castro, L. Bachmann, S. Said and W. P. Oliveira, Appl. Catal., A, 2021, 622, 118217 CrossRef CAS.
- A. Sadia, A. Muhammad, K. N. Ahmad and B. Muhammad, J. Water Process. Eng., 2021, 40, 101971 CrossRef.
- A. Apriceno, I. Silvestro, A. Girelli, I. Francolini, L. Pietrelli and A. Piozzi, Polymers, 2021, 13, 1453 CrossRef CAS PubMed.
- P. C. Lima, I. Gazoni, A. M. G. de Carvalho, D. Bresolin, D. Cavalheiro, D. de Oliveira and E. Rigo, Food Chem., 2021, 349, 129050 CrossRef CAS PubMed.
- H. Marina, F. Dzhigangir, K. Victoria, O. Svetlana, Z. Nataliya, Z. Yuriy, K. Maxim, Z. Ekaterina and A. Valeriy, Int. J. Biol. Macromol., 2021, 180, 161–176 CrossRef PubMed.
- K. Belho and P. Ambasht, J. Sci. Res., 2021, 65, 111–119 Search PubMed.
- L. Zhong, J. Li, D. Tian, J. Cai, H. Wang and Q. Ma, Water Sci. Technol., 2021, 83, 906–921 CrossRef CAS PubMed.
- V. Archana and K. Awanish, J. Drug Delivery Sci. Technol., 2021, 61, 102231 CrossRef.
- M. I. G. Siso, E. Lang, B. Carrenõ-Gómez, M. Becerra, F. O. Espinar and J. B. Méndez, Process Biochem., 1997, 32, 211–216 CrossRef CAS.
- S. Dumitriu and E. Chornet, Biotechnol. Prog., 1997, 13, 539–545 CrossRef CAS.
- M. Chellapandian and M. R. V. Krishnan, Process Biochem., 1998, 33, 595–600 CrossRef CAS.
- E. Pereira, G. Zanin and H. Castro, Braz. J. Chem. Eng., 2003, 20, 343–355 CrossRef CAS.
- Ş. A. Çetinus and H. N. Öztop, Enzyme Microb. Technol., 2003, 32, 889–894 CrossRef.
- Y. M. Yang, J. W. Wang and R. X. Tan, Enzyme Microb. Technol., 2004, 34, 126–131 CrossRef CAS.
- C. Min-Yun and J. Ruey-Shin, Enzyme Microb. Technol., 2005, 36, 75–82 CrossRef.
- J. De-Sheng, L. Sheng-Ya, H. Jun, X. Hai-Yan and Z. Ju-Ying, Biochem. Eng. J., 2005, 25, 15–23 CrossRef.
- W. Adriano, J. Silva, R. Giordano and L. Gonçalves, Braz. J. Chem. Eng., 2005, 22, 529–538 CrossRef CAS.
- X. Mao, G. Guo, J. Huang, Z. Du, Z. Huang, L. Ma, P. Li and L. Gu, J. Chem. Technol. Biotechnol., 2006, 81, 189–195 CrossRef CAS.
- T. Zhen-Xing, Q. Jun-Qing and E. S. Lu, Process Biochem., 2006, 41, 1193–1197 CrossRef.
- Z. X. Tang, J. Q. Qian and L. E. Shi, Mater. Lett., 2007, 61, 37–40 CrossRef CAS.
- Ş. A. Çetinus, H. N. Öztop and D. Saraydin, Enzyme Microb. Technol., 2007, 41, 447–454 CrossRef.
- G. D. Altun and S. A. Cetinus, Food Chem., 2007, 100, 964–971 CrossRef CAS.
- A. Dinçer and A. Telefoncu, J. Mol. Catal. B: Enzym., 2007, 45, 10–14 CrossRef.
- X.-J. Huang, D. Ge and Z.-K. Xu, Eur. Polym. J., 2007, 43, 3710–3718 CrossRef CAS.
- B. Krajewska, M. Leszko and W. Zaborska, J. Chem. Technol. Biotechnol., 1990, 48, 337–350 CrossRef CAS PubMed.
- C. Min-Yun and J. Ruey-Shin, Biochem. Eng. J., 2007, 35, 93–98 CrossRef.
- W. S. Adriano, D. B. Mendonça, D. S. Rodrigues, E. J. Mammarella and R. L. C. Giordano, Biomacromolecules, 2008, 9, 2170–2179 CrossRef CAS PubMed.
- E. E. E. Flores, F. D. Cardoso, L. B. Siqueira, N. C. Ricardi, T. H. Costa, R. C. Rodrigues, M. P. Klein and P. F. Hertz, Process Biochem., 2019, 84, 73–80 CrossRef CAS.
- S. R. Dasciana, A. M. Adriano, S. A. Wellington, R. B. G. Luciana and L. C. G. Raquel, J. Mol. Catal. B: Enzym., 2008, 51, 100–109 CrossRef.
- R. Wang, B. Xia, B.-J. Li, S.-L. Peng, L.-S. Ding and S. Zhang, Int. J. Pharm., 2008, 364, 102–107 CrossRef CAS PubMed.
- R. S. Singh, R. P. Singh and J. F. Kennedy, Int. J. Biol. Macromol., 2017, 95, 87–93 CrossRef CAS PubMed.
- Ş. A. Çetinus, E. Şahin and D. Saraydin, Food Chem., 2009, 114, 962–969 CrossRef.
- H. Fang, J. Huang, L. Ding, M. Li and Z. Chen, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2009, 24, 42–47 CrossRef CAS.
- M. Monier, D. M. Ayad, Y. Wei and A. A. Sarhan, Int. J. Biol. Macromol., 2010, 46, 324–330 CrossRef CAS PubMed.
- S. Wanjari, C. Prabhu, R. Yadav, T. Satyanarayana, N. Labhsetwar and S. Rayalu, Process Biochem., 2011, 46, 1010–1018 CrossRef CAS.
- N. A. Kalkan, S. Aksoy, E. A. Aksoy and N. Hasirci, J. Appl. Polym. Sci., 2012, 123, 707–716 CrossRef CAS.
- M. P. Klein, M. R. Nunes, R. C. Rodrigues, E. V. Benvenutti, T. M. H. Costa, P. F. Hertz and J. L. Ninow, Biomacromolecules, 2012, 13, 2456–2464 CrossRef CAS PubMed.
- D. Ayşe, B. Seda and A. Tülin, Int. J. Biol. Macromol., 2012, 50, 815–820 CrossRef PubMed.
- K. Chia-Hung, L. Yung-Chuan, J. C. Chieh-Ming, C. Jiann-Hwa, C. Cheng and S. Chwen-Jen, Carbohydr. Polym., 2012, 87, 2538–2545 CrossRef.
- M. Kamburov and I. Lalov, Biotechnol. Biotechnol. Equip., 2012, 26, 156–163 CrossRef.
- R. Zhai, B. Zhang, Y. Wan, C. Li, J. Wang and J. Liu, Chem. Eng. J., 2013, 214, 304–309 CrossRef CAS.
- P. Kristyna and S. Ivo, Carbohydr. Polym., 2013, 96, 545–548 CrossRef PubMed.
- J. Missau, A. J. Scheid, E. L. Foletto, S. L. Jahn, M. A. Mazutti and R. C. Kuhn, Sustainable Chem. Processes, 2014, 2, 13 CrossRef.
- L. Zang, J. Qiu, X. Wu, W. Zhang, E. Sakai and Y. Wei, Ind. Eng. Chem. Res., 2014, 53, 3448–3454 CrossRef CAS.
- H. J. Kim, J. N. Jin, E. Kan, K. J. Kim and S. H. Lee, Biotechnol. Bioprocess Eng., 2017, 22, 89–94 CrossRef CAS.
- V. S. Uttam, S. N. Shamraja and K. R. Virendra, Carbohydr. Polym., 2017, 157, 677–685 CrossRef PubMed.
- E. Demirkan, T. Avci and Y. Aykut, J. Ind. Text., 2018, 47, 2092–2111 CrossRef CAS.
- T. Mardani, M. S. Khiabani, R. R. Mokarram and H. Hamishehkar, Int. J. Biol. Macromol., 2018, 120, 354–360 CrossRef CAS PubMed.
- K.-M. Yeon, J. You, M. D. Adhikari, S.-G. Hong, I. Lee, H. S. Kim, L. N. Kim, J. Nam, S.-J. Kwon, M. I. Kim, W. Sajomsang, J. S. Dordick and J. Kim, Biomacromolecules, 2019, 20, 2477–2485 CrossRef CAS PubMed.
- C. Ilaria, L. Claudio, B. Ilaria and E. Marco, J. Mater. Res. Technol., 2019, 8, 3644–3652 CrossRef.
- S. Hongbo, G. Zhen, X. Lili, X. Chao, Y. Dinghua, X. Xinran, H. He and H. Yi, Mater. Sci. Eng., C, 2019, 96, 356–364 CrossRef PubMed.
- W. Deqiang and J. Weifeng, Int. J. Biol. Macromol., 2019, 126, 1125–1132 CrossRef PubMed.
- A. M. Girelli, L. Quattrocchi and F. R. Scuto, J. Biotechnol., 2020, 318, 45–50 CrossRef CAS PubMed.
- M. Prasad and P. Palanivelu, Biotechnol. Appl. Biochem., 2015, 62, 523–529 CrossRef CAS PubMed.
- J. Ruey-Shin, W. Feng-Chin and T. Ru-Ling, Adv. Environ. Res., 2002, 6, 171–177 CrossRef.
- F. Maryam and I. Ani, Renewable Energy, 2022, 185, 1362–1375 CrossRef.
- Ç. Esin and Ö. Seçil, React. Funct. Polym., 2022, 172, 105181 CrossRef.
- M. G. Holyavka, S. M. Pankova, Y. M. Vyshkvorkina, A. N. Lukin, M. S. Kondratyev and V. G. Artyukhov, Biophysics, 2022, 67, 365–373 CrossRef CAS.
- K. K. Gali, N. Soundararajan, V. Katiyar and S. Sivaprakasam, J. Mater. Res. Technol., 2021, 13, 686–699 CrossRef CAS.
- M. B. Pereira, B. L. Nogueira, I. D. C. Montano, D. D. S. Rodrigues and C. A. G. Suarez, Cellul. Chem. Technol., 2021, 55, 829–837 CAS.
- K. Matsushima, H. Minoshima, H. Kawanami, Y. Ikushima, M. Nishizawa, A. Kawamukai and K. Hara, Ind. Eng. Chem. Res., 2005, 44, 9626–9630 CrossRef CAS.
- H. Zimmermann, F. Ehrhart, D. Zimmermann, K. Müller, A. Katsen-Globa, M. Behringer, P. J. Feilen, P. Gessner, G. Zimmermann, S. G. Shirley, M. M. Weber, J. Metze and U. Zimmermann, Appl. Phys. A, 2007, 89, 909–922 CrossRef CAS.
- I. P. S. Fernando, W. Lee, E. J. Han and G. Ahn, Chem. Eng. J., 2020, 391, 123823 CrossRef CAS.
- C. H. Goh, P. W. S. Heng and L. W. Chan, Carbohydr. Polym., 2012, 88, 1–12 CrossRef CAS.
- R. Yavaşer and A. A. Karagözler, Process Biochem., 2021, 101, 137–146 CrossRef.
- A. O. Ali, M. S. Abdalla, Y. E. Shahein, A. Shokeer, H. M. Sharada and K. A. Ali, 3 Biotech., 2021, 11, 341 CrossRef PubMed.
- R. Guo, X. Zheng, Y. Wang, Y. Yang, Y. Ma, D. Zou and Y. Liu, Appl. Biochem. Biotechnol., 2021, 193, 2043–2060 CrossRef CAS PubMed.
- Y. Q. Almulaiky and S. A. Al-Harbi, Catal. Lett., 2022, 152, 28–42 CrossRef CAS.
- A. B. A. Mohammed, A. E. Hegazy and A. Salah, Appl. Nanosci., 2023, 13(1), 641–649 CrossRef CAS.
- N. Popović, D. Pržulj, M. Mladenović, O. Prodanović, S. Ece, K. I. Đurđić, R. Ostafe, R. Fischer and R. Prodanović, Int. J. Biol. Macromol., 2021, 181, 1072–1080 CrossRef PubMed.
- M. Jonović, M. Žuža, V. Đorđević, N. Šekuljica, M. Milivojević, B. Jugović, B. Bugarski and Z. Knežević-Jugović, Catalysts, 2021, 11, 305 CrossRef.
- W. Yang, F. Chenyu, G. Rongxin, M. Yifang, Y. Yu and L. Yanping, Process Biochem., 2021, 107, 38–47 CrossRef.
- N. Pantić, R. Prodanović, K. I. Đurđić, N. Polović, M. Spasojević and O. Prodanović, Environ. Technol. Innovation, 2021, 21, 101211 CrossRef.
- I. W. Marwa, E. H. Mohamed and A. A. Korany, Biocatal. Biotransform., 2021, 39, 138–151 CrossRef.
- J. Moriyama and M. Yoshimoto, ACS Omega, 2021, 6, 6368–6378 CrossRef CAS PubMed.
- G. Palmieri, P. Giardina, B. Desiderio, L. Marzullo, M. Giamberini and G. Sannia, Enzyme Microb. Technol., 1994, 16, 151–158 CrossRef CAS PubMed.
- Y. M. Elçin, Biomaterials, 1995, 16, 1157–1161 CrossRef PubMed.
- N. Yadav, M. H. Mruthunjayappa, M. Bisht, S. K. Nataraj, P. Venkatesu and D. Mondal, Chem. Commun., 2020, 56, 9659–9662 RSC.
- A. Blandino, M. Macías and D. Cantero, Process Biochem., 2001, 36, 601–606 CrossRef CAS.
- P. Rilling, T. Walter, R. Pommersheim and W. Vogt, J. Membr. Sci., 1997, 129, 283–287 CrossRef CAS.
- A. Tanriseven and Ş. Doğan, Process Biochem., 2001, 36, 1081–1083 CrossRef CAS.
- T. Vu and V. Le, ASEAN Food J., 2008, 15, 73–78 CAS.
- N. W. Fadnavis, G. Sheelu, B. M. Kumar, M. U. Bhalerao and A. A. Deshpande, Biotechnol. Prog., 2003, 19, 557–564 CrossRef CAS PubMed.
- E. Taqieddin and M. Amiji, Biomaterials, 2004, 25, 1937–1945 CrossRef CAS PubMed.
- J.-Y. Wang, H.-R. Yu, R. Xie, X.-J. Ju, Y.-L. Yu, L.-Y. Chu and Z. Zhang, AIChE J., 2013, 59, 380–389 CrossRef CAS.
- L. Lu, M. Zhao and Y. Wang, World J. Microbiol. Biotechnol., 2007, 23, 159–166 CrossRef CAS.
- S. Xu, Y. Lu, Z. Jiang and H. Wu, J. Mol. Catal. B: Enzym., 2006, 43, 68–73 CrossRef CAS.
- S. M. Shet, M. Bisht, S. Pramanik, S. Roy, S. K. Thayallath, S. K. Nataraj, D. Mondal and S. Bhandari, Adv. Opt. Mater., 2020, 8, 1902022 CrossRef CAS.
- L. Ma, J. Wen, W. Lu, Q. Caiyin and Y. Liang, Enzyme Microb. Technol., 2008, 42, 235–241 CrossRef CAS.
- I. Bhushan, R. Parshad, G. N. Qazi and V. K. Gupta, J. Bioact. Compat. Polym., 2008, 23, 552–562 CrossRef CAS.
- A. Anwar, S. A. U. Qader, A. Raiz, S. Iqbal and A. Azhar, World Appl. Sci. J., 2009, 7, 1281–1286 CAS.
- T. Q. Viet, N. P. Minh and D. T. A. Dao, Am. J. Res. Commun., 2013, 1, 254–267 Search PubMed.
- M. Bilal and M. Asgher, BMC Biotechnol., 2015, 15, 1–14 CrossRef PubMed.
- M. A. P. Nunes, H. Vila-Real, P. C. B. Fernandes and M. H. L. Ribeiro, Appl. Biochem. Biotechnol., 2010, 160, 2129–2147 CrossRef CAS PubMed.
- B. M. Bonine, P. P. Polizelli and G. O. Bonilla-Rodriguez, Enzyme Res., 2014, 2014, 738739 Search PubMed.
- N. Ortega, M. Perez-Mateos, M. C. Pilar and M. D. Busto, J. Agric. Food Chem., 2009, 57, 109–115 CrossRef CAS PubMed.
- M. S. M. Eldin, E. I. Seuror, M. A. Nasr and H. A. Tieama, Appl. Biochem. Biotechnol., 2011, 164, 45–57 CrossRef CAS PubMed.
- X. Wang, K.-X. Zhu and H.-M. Zhou, Int. J. Mol. Sci., 2011, 12, 3042–3054 CrossRef CAS PubMed.
- F. Zhao, H. Li, X. Wang, L. Wu, T. Hou, J. Guan, Y. Jiang, H. Xu and X. Mu, J. Mater. Chem. B, 2015, 3, 9315–9322 RSC.
- M. A. Abd El-Ghaffar and M. S. Hashem, Carbohydr. Polym., 2013, 92, 2095–2102 CrossRef CAS PubMed.
- K. S. Muthuvelu, R. Rajarathinam, N. K. Manickam and S. Uthandi, Bioresour. Technol., 2018, 269, 227–236 CrossRef PubMed.
- Y. İ. Doğaç, I. Deveci, B. Mercimek and M. Teke, Int. J. Biol. Macromol., 2017, 96, 302–311 CrossRef PubMed.
- F. Zhao, Q. Wang, J. Dong, M. Xian, J. Yu, H. Yin, Z. Chang, X. Mu, T. Hou and J. Wang, Process Biochem., 2017, 57, 87–94 CrossRef CAS.
- S. Kumar, I. Haq, J. Prakash and A. Raj, Int. J. Biol. Macromol., 2017, 98, 24–33 CrossRef CAS PubMed.
- D. K. Bedade, Y. B. Sutar and R. S. Singhal, Food Chem., 2019, 275, 95–104 CrossRef CAS PubMed.
- M. Mohammadi, M. K. Heshmati, K. Sarabandi, M. Fathi, L.-T. Lim and H. Hamishehkar, Int. J. Biol. Macromol., 2019, 137, 253–260 CrossRef CAS PubMed.
- Z. Zhi-de, L. Gui-yin and L. Yuan-jian, Int. J. Biol. Macromol., 2010, 47, 21–26 CrossRef PubMed.
- Q. Ai, D. Yang, Y. Zhu and Z. Jiang, Ind. Eng. Chem. Res., 2013, 52, 14898–14905 CrossRef CAS.
- C.-T. Tsai and A. S. Meyer, Molecules, 2014, 19, 19390–19406 CrossRef PubMed.
- U. D. Kamaci and A. Peksel, Catal. Lett., 2021, 151, 821–831 CrossRef.
- C. Benjamas, J. Prawit and H. K. Aran, J. Mol. Catal. B: Enzym., 2009, 59, 206–211 CrossRef.
- A. Saxena, S. Sharda, S. Kumar, B. Kumar, S. Shirodkar, P. Dahiya and R. Sahney, Polymers, 2022, 14, 1277 CrossRef CAS PubMed.
- A. Omar and Q. A. Yaaser, Biocatal. Biotransform., 2022, 1–10 Search PubMed.
- L. Abdul, M. Ahsan, S. Kai and S. Youbin, J. Environ. Chem. Eng., 2022, 10, 107089 CrossRef.
- M. Sharma, A. P. M. Tavares, J. C. F. Nunes, N. Singh, D. Mondal, M. C. Neves, K. Prasad and M. G. Freire, Green Chem., 2020, 22, 2225–2233 RSC.
- A. Charoenpol, D. Crespy, A. Schulte and W. Suginta, Green Chem., 2023, 25, 467–489 RSC.
Footnote |
| † This authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |