
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe E factor at 30: a passion for pollution prevention
Roger Arthur
Sheldon
 ab
ab
aMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Wits 2050, Johannesburg, South Africa. E-mail: roger.sheldon@wits.ac.za
bBiocatalysis & Organic Chemistry, Delft University of Technology, 2628 BL Delft, Netherlands. E-mail: r.a.sheldon@.tudelft.nl
First published on 17th January 2023
Abstract
The introduction of the E Factor in 1992 focussed attention on the problem of waste generation, defined as everything but the desired product, in chemicals manufacture and gave rise to a paradigm shift in our concept of efficiency in chemical processes, from one based solely on chemical yield to one that assigns value to eliminating waste. Thirty years later, it has become clear that waste is the underlying cause of the major global environmental problems, from climate change to plastic pollution and that the solution to this ubiquitous waste problem is pollution prevention at source enabled by green and sustainable chemistry. The role played by (bio)catalysis, alternative solvents, the emergence of a carbon neutral circular economy based on renewable resources and the electrification of chemicals manufacture based on renewable energy in the drive towards pollution prevention and sustainable industries is delineated.
1. The ubiquity of waste: the source of global environmental problems
The daunting environmental problems – climate change, degradation of the natural habitat and its biodiversity – that humanity is currently facing on a global scale have a common denominator: WASTE. One could say that finding solutions to this global waste problem is one of the grand challenges of chemistry and chemical engineering for the foreseeable future.The major environmental concern thirty years ago was the damage caused by ozone depleting chemicals in the atmosphere, particularly chlorofluorocarbons (CFCs) and related halogen compounds. Incidentally, the same compounds are also potent greenhouse gases (GHGs). Now, thirty years later, the major environmental problem is global climate change, the root cause of which is GHG emissions, in particular carbon dioxide (the mother of all waste) methane and nitrous oxide (N2O). Another waste problem of global proportions is caused by discarded organic materials, in particular single use plastics, that are polluting our natural environment with disastrous consequences for flora and fauna. Recently, attention has focused on yet another major source of organic waste, most of which ends up as land-fill: food supply chain waste (FSCW)1 arising from the production, distribution and consumption of food. The ubiquity of organic waste is depicted in Scheme 1. The primary solution to pollution is to avoid the formation of waste in the first place. However, not all waste is avoidable; agricultural and forestry residues, for example, are largely unavoidable and valorisation of this renewable organic waste is the answer.
 | ||
| Scheme 1 The ubiquity of organic waste. | ||
The introduction, thirty years ago, of the E Factor (mass of waste/mass of product), usually expressed as (kgs/kg), constituted a paradigm shift in measuring the efficiencies of chemical processes, from being based mainly on chemical yield to an alternative that assigns value to waste elimination. It highlighted the amounts of waste generated in various segments of the chemicals industry (see section 2)2 and this struck a chord with the chemical industry, particularly in the pharmaceuticals and fine chemicals segments (Table 1).
| Industry segment | Product tonnage (p/a) | E-Factor (kgs waste/kg product) |
|---|---|---|
| Oil refining | 106–108 | <0.1 |
| Bulk chemicals | 104–106 | <1–5 |
| Fine chemicals | 102–104 | 5–50 |
| Pharmaceuticals | 10–103 | 25–>100 |
1.1 Enter green chemistry and sustainable development
In the 1990s, as a result of increasing environmental awareness, emphasis gradually switched from waste remediation through end-of-pipe solutions to waste prevention at source This not only eliminates waste treatment costs but also strengthens economic competitiveness through more efficient use of raw materials. It culminated, in the mid-1990s, in the introduction of the term Green Chemistry with the overall guiding element of ‘benign by design’ and the primary goal of pollution prevention.3Green chemistry efficiently utilises (preferably renewable) raw materials, eliminates waste and avoids the use of toxic and/or hazardous reagents and solvents in the manufacture and application of chemical products. The ultimate goal was zero waste production of chemicals.However, no economic component is implicit in green chemistry and industrial players prefer the concept of sustainable development, launched in the Brundtland report,4Our Common Future, published in 1987 by the World Commission on Environment and Development. It is defined as development that meets the needs of the present generation without compromising the ability of future generations to meet their own needs. A sustainable technology must fulfill two conditions: (i) natural resources must be used at rates that do not unacceptably deplete supplies over the long term and (ii) residues must be generated at rates no higher than can be readily assimilated by the natural environment.5 In reality, non-renewable fossil resources – oil, coal and natural gas – are being consumed at a much higher rate than that of their replacement by natural geological processes, a situation referred to6 as “ecological time-scale violation”, making their use unsustainable in the long term. At the same time, the use of fossil resources is generating carbon dioxide at rates that cannot be assimilated by the natural environment which causes devastating climate change.7
1.2 The role of catalysis in sustainable development
A major cause of waste in the production of chemicals, especially advanced pharmaceutical ingredients (APIs) and numerous other fine chemicals, was the widespread use of stoichiometric amounts of organic and inorganic reagents in organic synthesis. Prime examples include the use of metals and metal hydrides such as Na, Mg, Zn and NaBH4 and LiAlH4, respectively, as reducing agents, and chromium(VI) compounds and permanganate as oxidants, to mention but a few. Other major sources include mineral acids such as H2SO4 and Lewis acids, AlCl3, ZnCl2 and BF3 and bases such as NaOH and KOH used as stoichiometric reagents or in work-up. The key to sustainable manufacture of chemicals is clear: broad application of cleaner catalytic alternatives – heterogeneous, homogeneous, biocatalysis and organocatalysis in organic synthesis.8Why were catalytic technologies not widely used in the production of APIs and fine chemicals? A prime reason was that the much smaller volumes involved compared with those in commodity chemicals production made the necessity to minimise waste less acute. Moreover, the economics of commodity chemicals dictate the use of the least expensive reagents that are usually the most atom economical reagents, e.g. H2 for reductions, O2 or H2O2 for oxidations and CO for C–C bond formation. Furthermore, time-honoured, classical stoichiometric technologies generally have the broadest applicability and shortest development times. This resulted in the use of inferior stoichiometric technologies in order to meet stringent market deadlines. Subsequent process changes were generally prohibitive owing to problems associated with gaining regulatory approval.
Nonetheless, motivated by the pressing need to reduce waste, in the last thirty years more emphasis has been placed on the use of catalytic methods in (industrial) organic synthesis. This involved, for example, the application of catalytic technologies such as hydrogenation, carbonylation, hydroformylation and olefin metathesis that were already widely used in the production of commodity chemicals. Furthermore, catalytic technologies are also evolving. For example, there is a discernible trend, both in Industry and Academia, to replace the use of scarce precious metal catalysts, e.g. Pd. Pt, Rh and Ru, with non-precious metal catalysts (NPMCs),9 particularly Fe, Cu and Ni, otherwise known as Earth abundant metals (EAMs)10 as sustainable, cost-effective alternatives. It is worth noting, in this context, that these are the very same elements contained in the active sites of enzymes catalysing a variety of redox processes in nature.
The evolution of catalysis in industrial organic synthesis is being further stimulated by the ongoing decarbonisation of the energy sector and defossilisation of chemicals production. Renewable (green) electricity, generated using solar, wind and nuclear energy, is becoming increasingly important and has already led to a veritable renaissance in electro- and photo-catalysis (see section 6).
At the same time, the increasing importance of the circular bio-economy is stimulating the application of biocatalysis in chemicals manufacture. This will be manifest in integrated biorefineries involving the conversion of lignocellulose to monosaccharides as the key base chemicals in lieu of olefins in oil refineries.
1.3 The role of the solvent11
In 1992 we asserted2 that: “So many of the solvents favored by organic chemists are now on the black list that the whole question of solvents in organic synthesis requires rethinking. Not only do organic chemists generally use too much solvent, they very often choose the wrong ones. In the first place, is a solvent really necessary? If a solvent (diluent) is needed it should preferably be water”.Solvents account for 80–90% of the total mass of non-aqueous material used in pharmaceutical manufacture and are responsible for 75–80% of the waste.12 A survey of solvent usage in the period 1997–2012, led to the conclusion that there was considerable room for improvement throughout the global pharmaceutical industry13 and the last two decades have been devoted to rethinking the question of solvents in chemical processes.14 The best solvent is no solvent; reactions can be performed in neat liquid substrates as exemplified by biodiesel production from triglycerides.15
There is currently a marked trend away from the use of chlorinated solvents, low-boiling hydrocarbons and ethers that are classified as hazardous or highly hazardous based on flammability and/or toxicity issues. The list also includes certain polar, aprotic solvents, such as dimethylformamide (DMF) and N-methylpyrollidone (NMP) that are reproductive toxicity hazards and are classified as substances of very high concern (SVHCs). Many of these solvents are still being used, despite the availability of greener alternatives, which motivated pharmaceutical companies to develop Solvent Selection Guides (SSGs) to stimulate replacement of environmentally undesirable solvents, in particular chlorinated hydrocarbons.16 The overall trend is towards the use of lower alcohols, esters and, in some cases, ethers.
Bio-based esters such as isobutyl acetate, ethyl lactate (derived from two innocuous fermentation products), ethyl levulinate, diethyl carbonate, and γ-valerolactone (GVL)26 are environmentally attractive solvents. Similarly, bio-based ethers such as methyl-tetrahydrofuran (Me-THF),27 dimethyl tetrahydrofuran (diMeTHF), cyclopentyl methyl ether (CPME) and dimethylisosorbide, are useful solvents for, inter alia, biocatalysis (Scheme 2).28
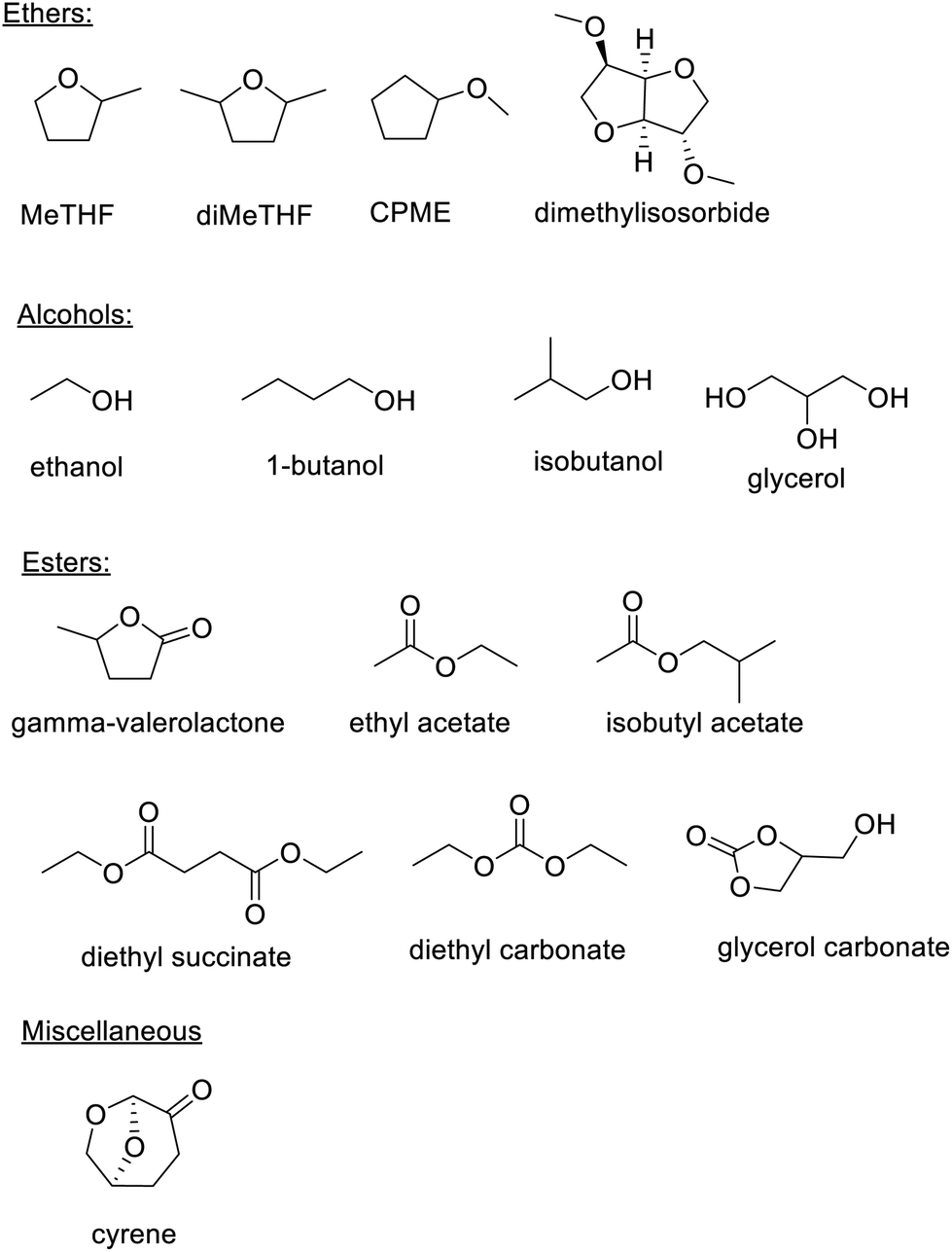 | ||
| Scheme 2 Examples of bio-based solvents. | ||
Dihydrolevoglucosenone, marketed as cyrene and produced by pyrolysis of (ligno)cellulose29 followed by hydrogenation of the resulting levoglucosenone, is a safe bio-based replacement for polar aprotic solvents with toxicity issues, such as DMF and NMP. It combines a good health and safety profile with excellent biodegradability.30,31 It has been successfully used as solvent, or co-solvent with water, in a wide variety of organic reactions,32 including biocatalytic transformations.33 Recently, the enzymatic reduction of levoglucosenone to cyrene, which has the advantage of affording a solvent free of traces of metal catalysts, was demonstrated.34
1.4 Aqueous–organic biphasic catalysis
Aqueous–organic biphasic catalysis45 is used to facilitate the separation and recycling of water soluble homogeneous catalysts from water insoluble products. The classic industrial example is the Ruhrchemie/Rhône Poulenc process for the hydroformylation of propylene employing a water-soluble Rh(I) catalyst.46 The process has an E Factor of 0.1 compared to 0.6–0.9 for conventional hydroformylation processes. Moreover, aqueous biphasic catalysis is readily adapted to continuous flow operation.Enzymes function optimally in water but if the product is only sparingly soluble in water it can be extracted into an environmentally acceptable solvent, such as ethyl acetate. Alternatively, extraction can be back-integrated into the reaction step by conducting the process as aqueous biphasic catalysis. The company Codexis, for example, developed a variety of cost-effective biocatalytic processes to pharmaceutical intermediates using highly engineered enzymes in this way.47
1.5 Micellar catalysis: aqueous micelles as nanoreactors
Another innovative approach, pioneered by Lipshutz and co-workers,48–50 involves the use of small amounts of environmentally benign designer amphiphiles (surfactants). The latter spontaneously self-assemble in water, at concentrations above their critical micelle concentrations (CMC) of ca. 10−4 M, to form nanomicelles. The lipophilic interiors of the latter act as nanoreactors for reactions involving water-insoluble substrates and, for example, noble metal complexes or metal nanoparticles.51 The choice of amphiphile is critical as it determines the size, shape and internal lipophilicity of the nanoreactors. Lipshutz and co-workers52,53 used small amounts (e.g. 2 wt%) of commercially available designer amphiphiles, such as TPGS-750-M (Scheme 3), to perform a variety of noble metal catalysed reactions at room temperature in organic solvent free water. The product was recovered by extraction with a minimum amount of a solvent that is environmentally acceptable and readily recyclable, such as ethyl acetate, while the amphiphile remains in the water phase. The E-Factors of reactions performed in water with 2–5% amphiphile were an order of magnitude less than the same reactions conducted in traditional organic solvents.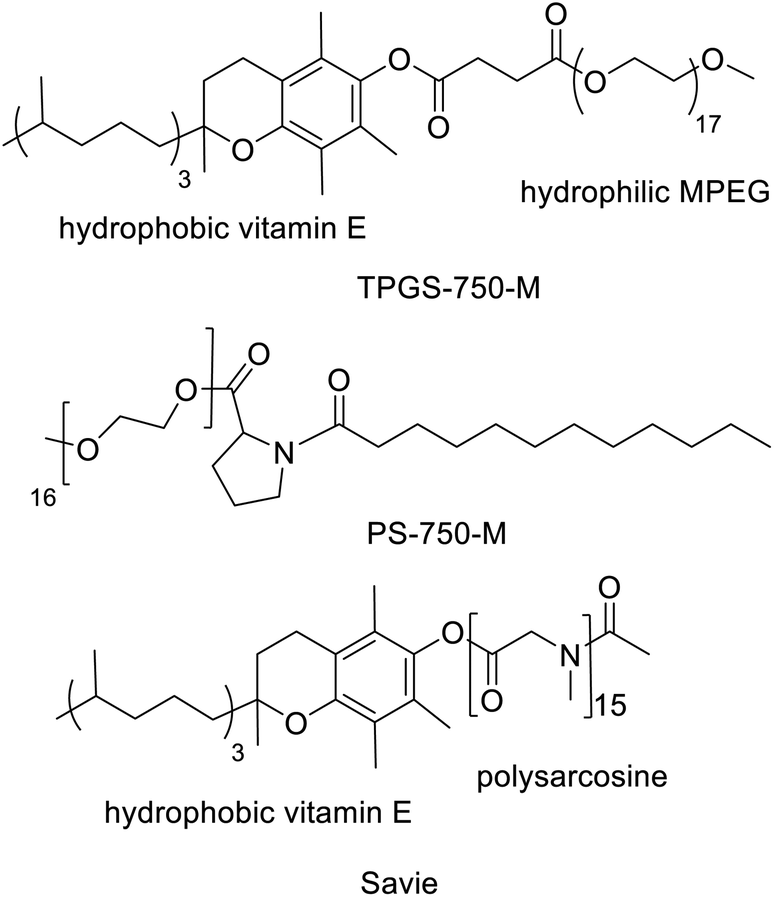 | ||
| Scheme 3 Structures of designer amphiphiles. | ||
Interestingly, Handa and co-workers54 recently designed a proline-based amphiphile, PS-750-M (Scheme 3), containing a tertiary amide function. Their goal was to replace polar aprotic solvents such as DMF and NMP through the formation of designer nanoreactors. PS-750-M instantly forms micelles containing a tertiary amide group in the hydrophobic core or at the interface, thus mimicking dipolar aprotic solvents. The micelles of PS-750-M were shown to mediate the formation of amides in water.55
Similarly, enzymatic reactions of hydrophobic substrates in water were enabled using designer amphiphiles, particularly TPGS-750-M.56 This enabled the combination of noble-metal catalysed synthesis of ketones with subsequent enantioselective KRED catalysed reduction to afford a one-pot production of chiral secondary alcohols in high enantioselectivities (Scheme 4). Interestingly, the activity of the KRED increased in the presence of the amphiphile compared to in buffer alone. This was attributed to a ‘reservoir’ effect in which the micelles control the supply of both substrate and product to the active site of the enzyme, thereby limiting substrate and product inhibition.
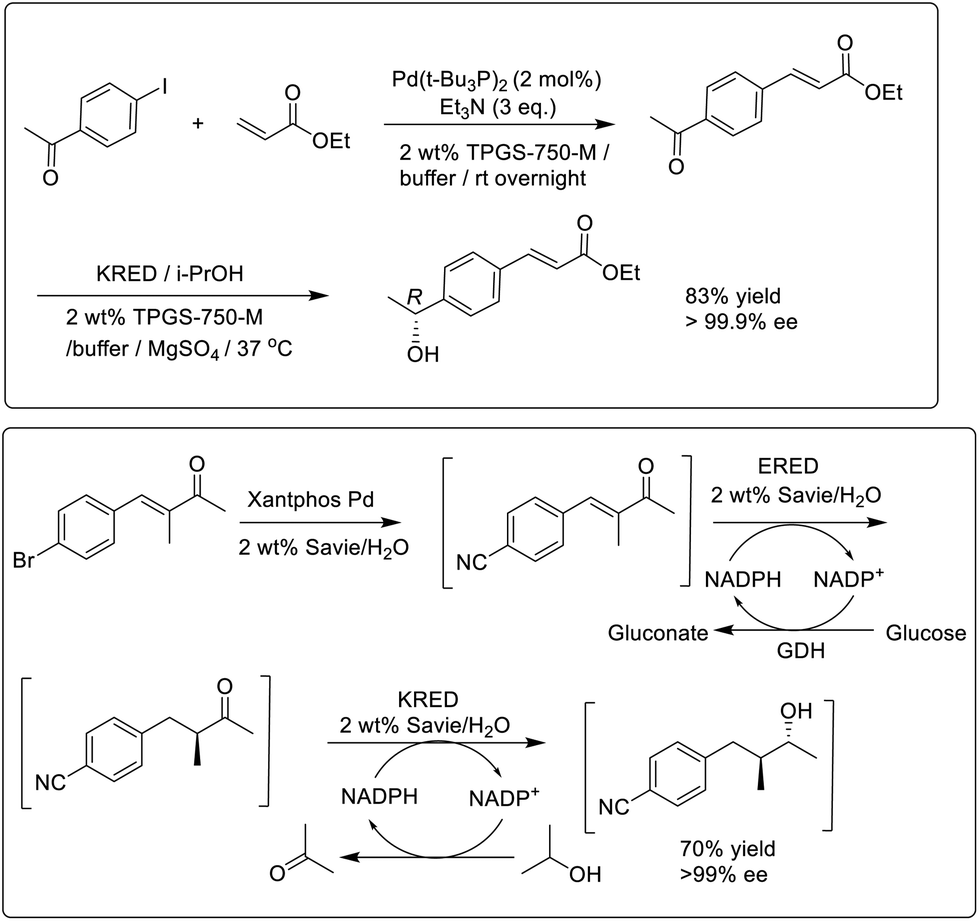 | ||
| Scheme 4 One-pot chemo-enzymatic conversions in water containing designer surfactants. | ||
Similarly, micellar catalysis with designer amphiphiles, such as TPGS-750-M, enabled a variety of one-pot cascade processes that combine chemo-catalytic and enzymatic steps.57–59 Product recovery involved extraction with a minimum amount of an environmentally acceptable solvent, such as ethyl acetate, or precipitation as an insoluble solid with the amphiphile remaining in the water phase. More recently, Lipshutz and coworkers reported60 the synthesis of a biodegradable amphiphile, Savie, and its use in one pot chemo-enzymatic cascade processes (see Scheme 4).
1.6 The role of circularity in waste minimisation and sustainable development
The increasing focus of attention on pollution prevention and resource efficiency is the major driver in the transition from a traditional linear flow of materials in a ‘take-make-use-dispose’ economy, to a sustainable, circular economy.61 The latter eliminates waste by design of products and processes that efficiently use resources and are recycled according to the underlying philosophy of the European Commission's “Roadmap to a resource efficient Europe”.62Circularity is not a new concept. Barry Commoner, the iconic industrial ecologist, already observed63 in the 1960s: “We have broken out of the circle of life, converting its endless cycles into man-made linear events: oil is taken from the ground, distilled into fuel, burned in an engine, converted thereby into noxious fumes which are emitted into the air”. Unfortunately, the transition from an unsustainable linear economy to a greener circular economy is seriously hampered by not conducting economic evaluations on a level playing field. The true costs of ‘take-make-use-dispose’ manufacturing chains must take resource efficiency and circularity into account. The currently externalised costs of resource depletion, waste management and environmental pollution must be internalised.
1.7 Waste valorisation in a circular bio-based economy
In the case of unavoidable but renewable organic waste, e.g. agricultural and forestry residues, valorisation is the answer. A sustainable circular bio-based economy will be largely based on conversion of waste lignocellulose into carbon neutral liquid fuels, bulk chemicals and novel materials in integrated biorefineries (see section 4).64 This concurs with the third element of green chemistry: utilisation of renewable resources.65 Hundreds of millions of tonnes of lignocellulosic waste are generated annually world-wide66,67 as agricultural residues such as sugar cane bagasse, corn stover, wheat straw and rice husks. The second generation bio-based economy, therefore, is based on the full utilisation of agricultural biomass. Moreover, substitution of existing products by safer bio-based alternatives, e.g. biocompatible and biodegradable plastics with reduced environmental footprints, is an added environmental benefit.681.8 The plastic pollution challenge
Industrial monomers constitute a large portion of the global production of commodity chemicals and in a circular economy the corresponding polymers must be recycled, as such or as the original monomers. The global production of plastics, for example, was 390 million tonnes in 2018, consisting of 174 million tonnes for packaging and 216 million tonnes for non-packaging applications. Virgin plastics represented 360 mio tonnes (92%) of the total and a mere 8% (30 mio tonnes) was recycled plastics. The total consumption of plastic products in the same period was 385 million tonnes resulting in the generation of 250 million tonnes of post-consumer plastic waste. 75 million tonnes of this were landfilled, 50 million tonnes burned for energy recovery, 75 million tonnes were improperly disposed in the environment, and 50 million tonnes were recycled.69 Packaging plastics consist mainly of polyolefins – polyethylene (PE), polypropylene (PP) and polystyrene (PS) – together with polyethylene terephthalate (PET). Production of the corresponding monomers accounts for ca. 4% of the global production of all petrochemicals.70 A paltry 9% of all the plastics ever produced have been recycled with the remainder being mostly discarded in the environment where they slowly fragment into microplastics and nanoplastics. The associated costs for society are profound, e.g. failure to recycle plastics costs the EU Euro 105 billion.71In 2020 a new species of Eurythenes amphipod was discovered at a depth of 6900 meters in the Mariana Trench of the North-West Pacific Ocean.72 It already had microplastic fibers in its gut, with 84% similarity to PET, and was ironically named Eurythenes plasticus.
Clearly, a linear take-make-use-dispose economy, that encourages production of virgin plastic over recycling, is not conducive to the goal of zero waste chemicals manufacture. In the post energy-transition era a resource efficient and environmentally benign circular economy, based on the principles of low waste by design, keeping products and materials in the loop, and regenerating natural systems73,74 will flourish. Particularly single use plastics will be based on renewable carbon and designed for efficient recycling.75,76
2. The metrics of waste
If we want to reduce the formation of waste in chemicals manufacture we must have metrics for measuring the mass efficiency of processes. Thirty years ago, two key mass efficiency metrics – atom economy (AE)77 and the E Factor2 – were introduced. AE is obtained by dividing the molecular weight of the product by the sum of the molecular weights of all the materials in the stoichiometric equation. It assumes the use of exactly stoichiometric amounts of starting materials and a 100% chemical yield. No experimental data is required and, hence, it is very useful for comparing different routes to a target molecule before performing any experiments.This contrasts with the E(nvironmental)-Factor which is the actual amount of waste formed, i.e. it is the environmental footprint of a process. By definition it is “everything but the desired product” produced per kg of product and includes both solvent losses and ancillary chemicals used. The ideal E Factor is zero in accordance with the first principle of Green Chemistry: “It is better to prevent waste than to treat or clean up waste after it is formed”. E Factors of multi-step processes are obtained simply by adding E Factors of individual steps.
That chemicals manufacture was responsible for a substantial environmental footprint, expressed as the E Factor, was illustrated with the now well-known Table of E Factors, based on actual data from mature commercial processes in various industry segments, from oil refining to pharmaceuticals. Publication of this table in 1992 laid down an important challenge for the industry, particularly the pharmaceuticals sector, to reduce its waste. The challenge was accepted and major pharmaceutical companies spent the next 2–3 decades cleaning up their manufacturing operations.78 However, these improvements are not always apparent in lower E Factors owing to the increasing molecular complexity and longer syntheses of APIs that is reflected in higher E Factors that partially neutralise the improvements.79 On the other hand these more recent APIs tend to be considerably more active than their predecessors and, hence, annual production volumes are substantially less.
The E Factor emphasizes the need for designing cleaner, waste-free processes. The ideal E-Factor of 0 clearly reflects the ultimate goal of zero waste manufacturing plants. In addition, lower E Factors translate to smaller quantities of materials consumed and are manifest in reduced manufacturing and waste disposal costs.80 The E Factor includes all reagents used and, if the actual amount is not known, 10% of the solvents used on the assumption 90% would be recovered and recycled. In hindsight, this was probably overoptimistic. Organic chemists generally optimise the solvent for each step in a multi-step synthesis, often leading to a different solvent for each step and cross-contamination that impedes solvent recycling. Water was excluded on the grounds that inclusion would produce skewed E Factors. However, disposal or recycling of process water generally involves some sort of pretreatment and we currently calculate E Factors both with and without water for comparison.81 This led to the designation of simple E Factors (sEF), that exclude both solvents and water, for initial route scouting, and complete E Factors (cEF) that include solvents and water with no recycling.5 The true E Factor falls between the sEF and cEF and can be calculated when reliable data are available for solvent losses on a production scale.
Inclusion of energy requirements in the E Factor was always implicit since we recognised that energy consumption generates carbon dioxide as waste. On the other hand, fine chemicals and pharmaceuticals production usually involves multi-purpose facilities where energy usage is often not allocated to individual product lines. In contrast, energy consumption is an important component of the production of bulk chemicals that is generally in dedicated plants. Consequently, the E+ Factor, comprising greenhouse gas emissions emanating from electricity used in cooling, heating, stirring and pumping, etc., was proposed.82
E Factors refer to chemical processes conducted at a manufacturing site, i.e. within gate-to-gate system boundaries, rather than cradle-to-grave.83 It is dependent on the starting point of the synthesis and E Factors of multi-step syntheses are substantially reduced overnight by purchasing early intermediates rather than producing them in-house. It is essential, therefore, to have a clear definition of what is considered as a starting material. In pharmaceuticals manufacture it has been defined as material that is readily available at a price of <$100 per kg from a reputable commercial supplier.84 Any intermediate costing >$100 per kg is referred to as an advanced starting material (ASM) and the E Factor for its production, the so-called intrinsic E Factor, must be included. Since E Factors are additive, the intrinsic E Factor for an ASM synthesis may be simply added to the main synthesis E Factor to obtain an E Factor for the complete synthetic pathway. For example, in conducting a meaningful comparison of routes to a key HIV protease inhibitor intermediate, it was essential to include intrinsic E Factors for the synthesis of ASMs in order to make comparisons on a level playing field.85
The E Factor's durability lies in its simplicity, in terms of concept and application, and broad familiarity. In the first two decades following its publication it was used to assess the extent of waste formation in the manufacture of pharmaceuticals, a broad range of fine chemicals, and a few bulk chemicals including industrial monomers such as caprolactam,86 in addition to being widely adopted in academic circles. In the last decade it has become increasingly applied in polymer synthesis. The major outlet of bulk chemicals is as monomers for the synthesis of a broad range of polymers and in recent years it has become increasingly apparent that the sustainability of polymeric materials is very much dependent on the amount of waste generated, both in the production of the monomer and in the process for its polymerization.87–89
In the drive towards a carbon-neutral bio-based economy there is increasing emphasis on the use of bio-based polymers derived from renewable raw materials, e.g. by modification of the most abundant biopolymer, cellulose (see also section 4).90 However, renewable is not enough: the percentage of bio-based feedstock in the polymer is relevant but the process for its manufacture must also have a low E Factor. For example, Fadlallah and coworkers91 employed the E Factor, the simple E Factor and atom economy as green metrics for comparing synthetic methods for the production of a wide variety of bio-based monomers from (waste) lignin.
Other mass-based metrics such as process mass intensity (PMI) and reaction mass efficiency (RME) have been proposed92 but have not attained the scope and broad acceptance of the E Factor. We recognised, however, that a limitation of the E Factor is that it didn't consider the environmental impact of the waste; all types of waste were assigned equal rating. Consequently, the E Factor must be considered in conjunction with other environmental metrics.93 We proposed the environmental quotient (EQ),94 where Q represents the nature of the waste, to assess environmental impact but then the problem becomes the quantification of Q. Eissen and Metzger95 introduced the user-friendly EATOS (Environmental Assessment Tool for Organic Synthesis) software for assessing the potential environmental impact of waste by assigning penalty points based on human and eco-toxicity. It was subsequently further refined by others.96
In our experience, the Green Motion penalty point system developed by Mane97 is very useful for rapid comparison of processes. It involves evaluation of seven fundamental process parameters – raw material, solvent, hazard and toxicity of reagents, reaction efficiency, process efficiency, hazard and toxicity of final product and waste generation using a questionnaire with simple yes/no answers. Penalty points are assigned and deducted from 100 to afford an overall score. The higher the score the more sustainable and the lower the environmental impact of the process.
In order to set meaningful goals for industrial research it is necessary to compare processes with an industry benchmark. The Green Aspiration Level (GAL™) is such a benchmark. It represents the average waste generated per kilogram API in 46 commercial manufacturing processes from nine large pharmaceutical companies.98 More recently, a further refinement, the innovative Green Aspiration Level 2.0 (iGAL 2.0) was published.99 However, we note that these tools are only relevant for process evaluations of multi-step syntheses of relatively complex APIs.
2.1 The C factor: the carbon footprint
There is currently an ongoing transition, referred to in the EU as the Green Deal,100 which has the goal of achieving climate neutrality by 2050. It is motivated by the pressing need for climate change mitigation, through replacing an economy based on fossil resources with a bio-based economy based on renewable energy and raw materials. It is manifest in the decarbonisation of the energy sector and defossilisation of chemicals manufacture. Christensen and co-workers101 proposed, already in 2008, the use of a (C)limate factor metric, defined as the total mass of CO2 emitted divided by the mass of product formed (kg CO2/kg product), to compare the CO2 footprints of different processes to a given product.| C Factor = kg CO2 emitted/kg product |
It is the sum total of kg CO2 emitted in the production of the raw material(s) and in the conversion of the raw materials to the product(s) and is useful for comparing processes based on renewable biomass vs. non-renewable fossil resources.102 Gallou and co-workers103 recently used it to assess the ecological footprints of APIs. We note, however, that system boundaries need to be defined for determining the C factor.
Summarising, the extent of waste formation is defined by the following mass-based metrics:
- Atom economy is the molecular weight of the product divided by the sum of the molecular weights of the raw materials in the stoichiometric equation expressed as a percentage. It is useful for a rough comparison of possible routes prior to experimentation.
- The E Factor is the total mass of waste formed per kg of product (kg waste/kg product) excluding water but including the solvent if the total amount leaving the plant is known. If not it is assumed that the solvent will be recycled with 10% lost as waste (in hindsight probably over-optimistic).
- The simple E Factor (sEF) excludes water and solvent used.
- The complete E Factor (cEF) includes water and solvent and assumes no recycling.
- E factors are based on gate-to-gate boundaries. If an advanced starting material (ASM) is used the E Factor for the synthesis of the ASM, the so-called intrinsic E Factor, must be included.
- Strictly speaking, the E Factor must always include waste, in CO2 equivalents, from the energy used to drive the process. In practice, however, this is generally not included but when it is it has been referred to as the E+ Factor.
- The C Factor is kg CO2 formed/kg product and is the carbon footprint of the process.
- The process mass intensity (PMI) is the total mass of materials used divided by the mass of product formed (kg/kg) and is E +1.
2.2 The ethanol equivalent
Horvath and coworkers104 proposed a relatively simple mass metric for assessing the sustainability of biomass-based routes to fuels and chemicals: the ethanol equivalent. It is defined as the “mass of ethanol required to deliver the equivalent amount of energy from a given feedstock using energy equivalency or produce the equivalent mass of a carbon-based chemical using molar equivalency”. Since ethanol is, inter alia, produced by fermentation, the required mass of biomass feedstock, the land area and even the volume of water can be readily calculated. The authors concluded, based on their calculations of ethanol equivalents, that production of the 387 × 106 tons of gasoline used in the US in 2008, using 1G corn-based bioethanol technology commercially practiced in the US is not even close to being a viable proposition. In contrast, the conversion of 2G waste lignocellulose to basic chemicals, such as ethylene, propylene and xylenes, via intermediate ethanol, could be a sustainable option in the near future.3. Biocatalysis is sustainable and green
Biocatalytic processes are eminently sustainable. They have definite economic and environmental benefits and conform to 10 of the twelve principles of green chemistry,105,106 the foremost of which is pollution (i.e. waste) prevention. Indeed, one could say that biocatalysis is the key to sustainable chemistry:107(i) The catalysts (enzymes) are derived from inexpensive, renewable resources with stable availability and costs in contrast with the fluctuating prices and availability of scarce precious metal catalysts. Moreover, enzyme production costs are steadily decreasing as a result of using more productive hosts coupled with improved protein expression and downstream enzyme recovery.
(ii) They avoid the need for costly removal of traces of noble metals, to an acceptable ppm level, from end products.
(iii) The catalysts (enzymes) are biocompatible, biodegradable, essentially non-toxic and non-hazardous.
(iv) The processes are conducted under mild conditions, in water at ambient pressures and temperatures. This enables relatively simple coupling of enzymatic steps in cost-effective, environmentally attractive cascade processes.108
(v) The processes are step economic.109 As a result of their exquisite functional group specificity the need for protection and deprotection steps is usually circumvented leading to less waste and more cost-effective processes.
(vi) Optimised enzymes exhibit superb chemo-, regio- and stereoselectivities, including near-perfect enantioselectivities (>99.99%), that are absolutely unparalleled.
(vii) Owing to the exquisite functional group specificity of enzymes, extra functional group protection and deprotection steps are avoided
(viii) Because of the ambient conditions enzymatic processes (but not fermentations) can be conducted in standard multi-purpose batch reactors, thus avoiding extra investments, e.g. in high-pressure equipment.
As if all this were not enough, procedures have been extensively optimised in the last decade by the application of immobilised enzymes in continuous processing (see below). Such advantages inevitably beg the question: if enzymatic processes are so good why haven't they been widely used until the last two decades? The answer is: lack of use was largely due to the limited commercial availability and poor stability of most enzymes under the harsh conditions – high substrate concentrations and elevated temperatures with substrates that are poorly soluble in water-characteristic of industrial processes severely limited their applications in industrial organic synthesis.
Two decades ago commercial availability of enzymes was rather limited to hydrolases of animal origin, e.g. proteases, lipases, esterases and glycosidases, used in food and beverages and detergents. This changed dramatically in the last two decades. Advances in high throughput genome sequencing in combination with bioinformatics have enabled the identification of an ever-increasing number of new and robust enzymes through metagenome mining.110,111 Subsequent synthesis of the targeted gene, ready for cloning into a host production organism, requires only two weeks at a cost of <$1000. Consequently, more enzymes are available and they are more cost-effective. Furthermore, developments in directed (in vitro) evolution techniques,112–114 with the aid of advanced machine learning techniques115 enable the re-engineering of enzymes to exhibit pre-defined properties with regard to substrate specificity, activity, selectivity, stability and optimum pH116,117 Two decades ago, the process had to be modified to fit the available enzyme, often resulting in a nightmare process. Today, the enzyme is evolved to fit a pre-defined optimum process that is benign by design. In the last decade this facilitated the broad integration of biocatalysis, as a mature sustainable technology, in mainstream (industrial) organic synthesis,118–124 particularly for the enantiospecific synthesis of APIs.125–130 It led to the introduction of the concept of biocatalytic retrosynthesis, with rules and guidelines to aid the identification of biocatalytic pathways to target molecules.131–133
The general discussion of the important role of the solvent in determining the overall greenness of a process, as discussed in section 1.3, is also applicable to biocatalytic processes. Using an organic solvent as such is not a problem. The preferred solvent for enzymatic processes is water but this can be problematic with hydrophobic substrates. In this case aqueous/organic biphasic systems are often used to enable recycling of the organic solvent. Alternatively, hydrophobic solvents, e.g. biobased ethers and esters, can sometimes be used, in the absence of a separate water phase, with immobilised enzymes.
3.1 The ever broadening scope of biocatalysis
Clearly catalysis with enzymes has successfully negotiated the long and winding road from laboratory to industrial process. Not only has the number of commercially available enzymes dramatically increased, the scope of biocatalysis has been rapidly widening.134–137Chiral secondary alcohols, for example, are key intermediates in the synthesis of a broad range of APIs. Thirty years ago they were produced either by stoichiometric ketone reduction with chiral hydride reagents or catalytic asymmetric hydrogenation.138 Thanks largely to vast improvements achieved through directed evolution, ketoreductase (KRED) mediated enantioselective reduction of prochiral ketones has essentially superseded asymmetric hydrogenation as the first choice method for industrial production of enantiomerically pure secondary alcohols (Scheme 5).139–142 The nicotinamide cofactors, NADH or NADPH, that supply the reducing equivalents are recycled in situ by adding a large excess of an alcohol co-substrate, such as isopropanol, or a second enzyme in combination with an inexpensive co-substrate, such as formate/formate dehydrogenase (FDH) or glucose/glucose dehydrogenase (GDH).
 | ||
| Scheme 5 Enzymatic production of chiral alcohols. | ||
Similarly, selective oxidation of primary and secondary alcohols to aldehydes and ketones, respectively, is a key transformation in organic synthesis. Historically, stoichiometric amounts of inorganic oxidants, such as hazardous chromium VI reagents, were used but there is a marked trend towards more sustainable chemocatalytic143 and biocatalytic methods.144 Alcohol oxidases (AOx) employ stoichiometric amounts of dioxygen as the oxidant together with catalase to rapidly degrade the H2O2 co-product. Since the reactions are performed in aqueous media, safety issues normally associated with use of dioxygen in combination with volatile organic compounds no longer apply. This is another virtue of enzymatic catalysis in aqua.
Typical examples are copper-dependent galactose oxidase (GOase) and flavin-dependent glucose oxidase (GOX) that catalyse substrate specific aerobic oxidations of carbohydrates. New flavin- and Cu-dependent AOxs, with improved stabilities and altered substrate specificities have been developed145 with the aid of genome mining and directed evolution. A choline oxidase variant, for example, is a broad spectrum primary alcohol oxidase146 and a GOase variant catalysed the ammoxidation of primary alcohols to nitriles.147
Enantiopure chiral primary amines are also key intermediates in the synthesis of a wide variety of pharmaceuticals and can be produced by a suite of biocatalytic methods148–150 using amine oxidases (AOs), amine dehydrogenases (AmDHs) and imine reductases (IREDs). A synthesis of Boceprevir, for example, involved an AO catalysed desymmetrisation as the key step.151 Reductive amination of prochiral ketones catalysed by AmDHs was unknown 10 years ago. In contrast, amino acid dehydrogenases (AADHs) were well-known. This inspired Bommarius and co-workers152,153 to use them as starting points for engineering highly active and enantioselective (>99.8% ee) AmDHs. Subsequently, AmDHs were used in conjunction with cofactor regeneration using glucose/GDH or ammonium formate/FDH (Scheme 7), for the reductive amination of a variety of ketones154–157 and a new family of native bacterial AmDHs was discovered.158
Another fairly recent development is the emergence of imine reductases (IREDs)159–165 and the closely related reductive aminases,166–169 that catalyse asymmetric reductive amination of ketones. For example, a synthesis of the JAK1 inhibitor, Abrocitinib, involved an enantioselective reductive amination as the key step (Scheme 6).170
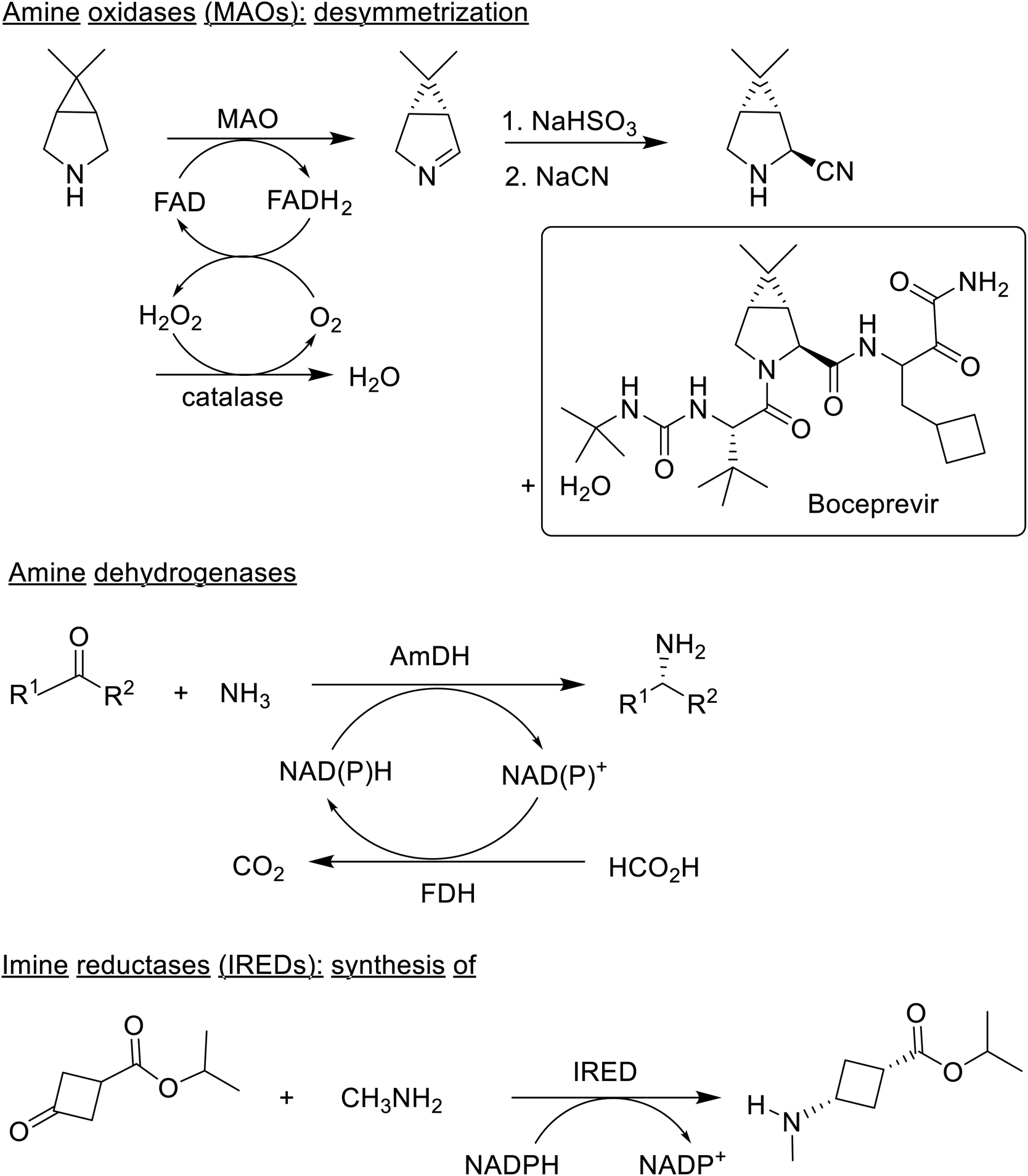 | ||
| Scheme 6 Enzymatic production of chiral amines. | ||
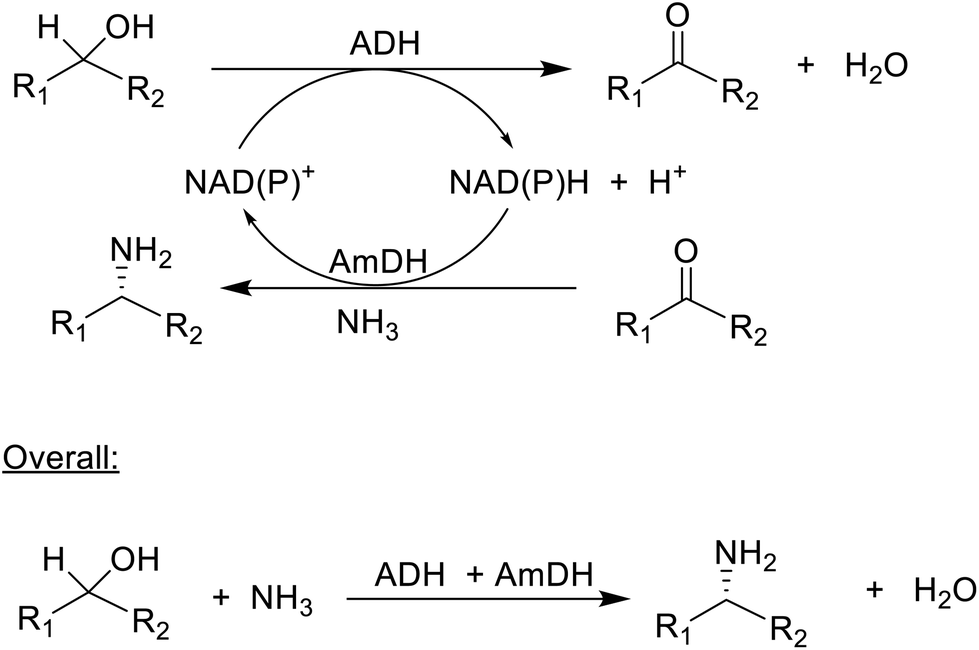 | ||
| Scheme 7 Enzymatic hydrogen borrowing. | ||
The synthesis of chiral amines by AmDH catalysed reductive amination of ketones was taken a step further by combining it with an ADH catalysed oxidation of an alcohol. This afforded a redox-neutral conversion of a racemic alcohol to a single enantiomer of the corresponding amine in a classic example of so-called hydrogen borrowing (Scheme 7).171 Overall the reaction involves nucleophilic substitution of a hydroxyl group in a secondary alcohol, a transformation on the list of 10 key research areas for greening the pharmaceutical industry.172 In this case OH is substituted by NH2, forming an equivalent of water as the sole co-product. Its success depends on the ADH catalysing the oxidation of both alcohol enantiomers,173 which is not trivial since ADHs are generally enantioselective. The overall efficiency was further improved by co-immobilisation of the ADH and AmDH.174
3.2 Chemomimetic biocatalysis: new to nature reactions
One way to develop new biocatalytic reactions is to use a chemomimetic approach,175 that is by imitating known chemo-catalytic reactions. Combining this concept with protein engineering using directed evolution afforded new-to-nature biocatalytic reactions (Scheme 8).176,177 For example, metalloporphyrins are known to catalyse cyclopropanations and aziridinations of olefins, by carbene and nitrene insertion, respectively, and C–H bond insertion reactions via high valent oxo-metal species. Arnold reasoned178,179 that heme-dependent enzymes, in particular cytochrome-P450-dependent mono-oxygenases, that catalyse olefin epoxidations via high-valent oxoiron intermediates should also be able to catalyse cyclopropanation and aziridination of olefins.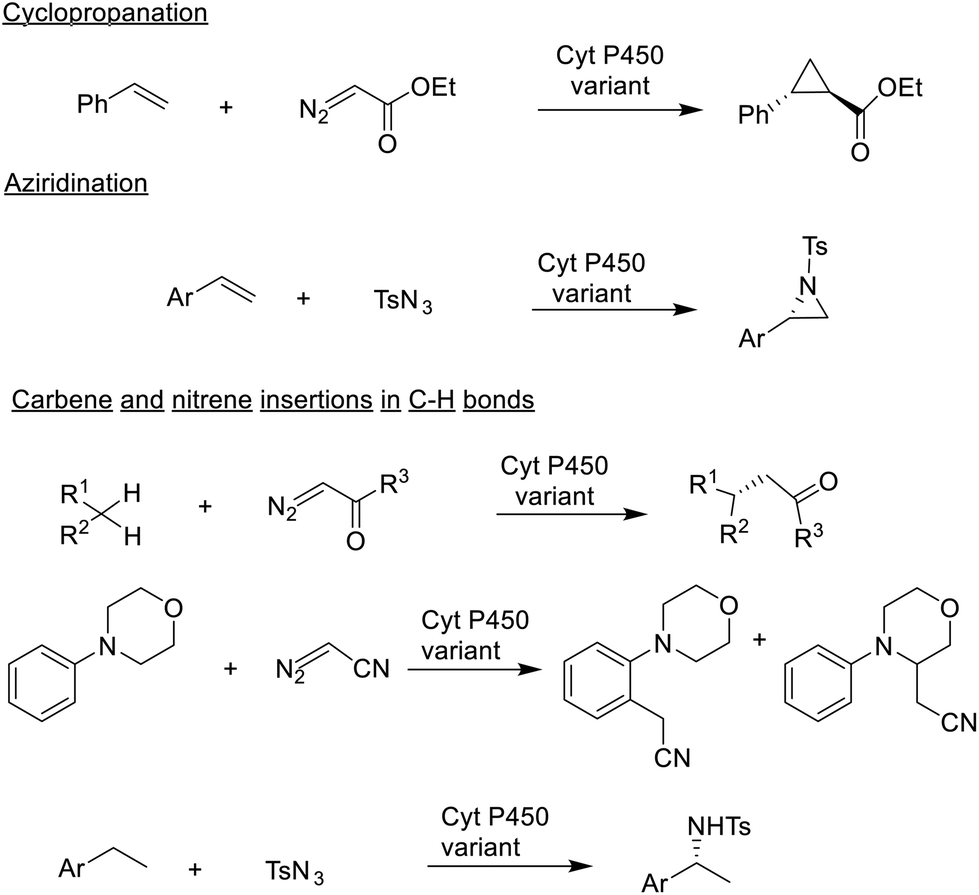 | ||
| Scheme 8 New to nature reactions. | ||
This proved to be the case: the P450BM3 from Bacillus megaterium catalysed olefin cyclopropanation and aziridination by reaction with ethyl diazoacetate and tosyl azide, respectively. More recently, Arnold and co-workers extended this to the cyanomethylation of sp3 and sp2 (arene) C–H bonds180 catalysed by two different P-450-based, carbene transferases, even in the same molecule, using diazoacetonitril as the stoichiometric reagent (see Scheme 8). New-to-nature reactions, developed with the help of directed evolution, can also be used for the synthesis of a wide variety of non-canonical amino acids that are structural motifs in many modern therapeutics.181
3.3 Enzyme immobilisation, biocatalysis in flow and cascade processes
Notwithstanding their many advantages enzymes have two shortcomings: (i) limited stability under the harsh conditions of industrial processes and (ii) limited recyclability. Enzymes are soluble in water and are typically used as homogeneous catalysts in an aqueous medium on a once-through, throw-away basis, which is not conducive to sustainability. This hurdle can be overcome by immobilising the enzyme as a solid heterogeneous catalyst that is recoverable by centrifugation or filtration and can be subsequently recycled.182 This is essentially a conditio sine qua non for developing sustainable processes with high catalyst productivities.183Enzymes are immobilised by attachment to an insoluble support (carrier), usually a natural or synthetic organic polymer or an inorganic solid such as silica or alumina or as insoluble cross-linked enzyme aggregates (CLEAs) formed by intermolecular cross-linking of enzyme molecules. Simple adsorption and ionic binding with carriers are widely used but have the drawback that they are susceptible to leaching of the enzyme under aqueous conditions, depending on the pH, ionic strength and temperature, especially under the harsh conditions of many industrial processes. Covalent bonding between the enzyme and the carrier has the advantage of stability towards leaching but the limitation that random covalent bonding at various positions in the enzyme can lead to loss of activity. There is a definite need, therefore, for less invasive methods for forming covalent bonds between the enzyme and the carrier, e.g. by employing bio-orthogonal chemistry to affect precision covalent bonding at preselected sites in the enzyme.184
Alternatively, affinity immobilisation makes use of polymers containing surface chelating functionalities, such as iminodiacetic acid, preloaded with metal ions – Ni2+, Zn2+, Fe2+, Co2+ and Cu2+ – that exhibit a strong affinity for the imidazole groups in surface histidine residues of the enzyme. It affords high activity recoveries through non-invasive binding. In addition, recombinant proteins are usually produced containing a so-called His-tag – a string of six to nine histidine residues – attached to the N- or C-terminus – to enable purification through affinity binding. This makes it possibile to combine enzyme isolation, purification and immobilisation into a single, cost-effective operation.185
Multi-step processes of APIs can often be significantly shortened through the use of biocatalytic reactions to circumvent the need for functional group protection and deprotection and functional group activation steps. Multi-step processes can be further shortened by combining steps in multi-enzyme or chemo-enzymatic cascade processes.186 Most enzymatic reactions are conducted under roughly the same conditions of temperature and pressure and, hence, it is relatively easy to integrate multiple steps into eco-efficient catalytic cascade processes.187–189 thus avoiding the separation, and purification of the products from individual steps characteristic of conventional processing. This also improves conversions by limiting product inhibition.190 It can involve the co-immobilisation of enzymes to afford a multi-functional biocatalysts, e.g. as genetically fused multi-enzymes191 or combi-CLEAs.192
The success story of biocatalysis in pharma is further underlined by the fact that more than 60% of all FDA-approved antiviral drugs were made accessible through biocatalytic processes,193 often involving multiple enzymes. Superb examples are provided by the synthesis of Islatravir, a nucleoside HIV inhibitor, in a three-step cascade involving nine enzymes194 and molnupiravir,195 the first SARS-coV-2 oral antiviral, in three steps and 69% overall yield from simple raw materials with isolation of only one intermediate (Scheme 9). The latter process replaced the original 10 step chemical synthesis that had an overall yield of 10%.
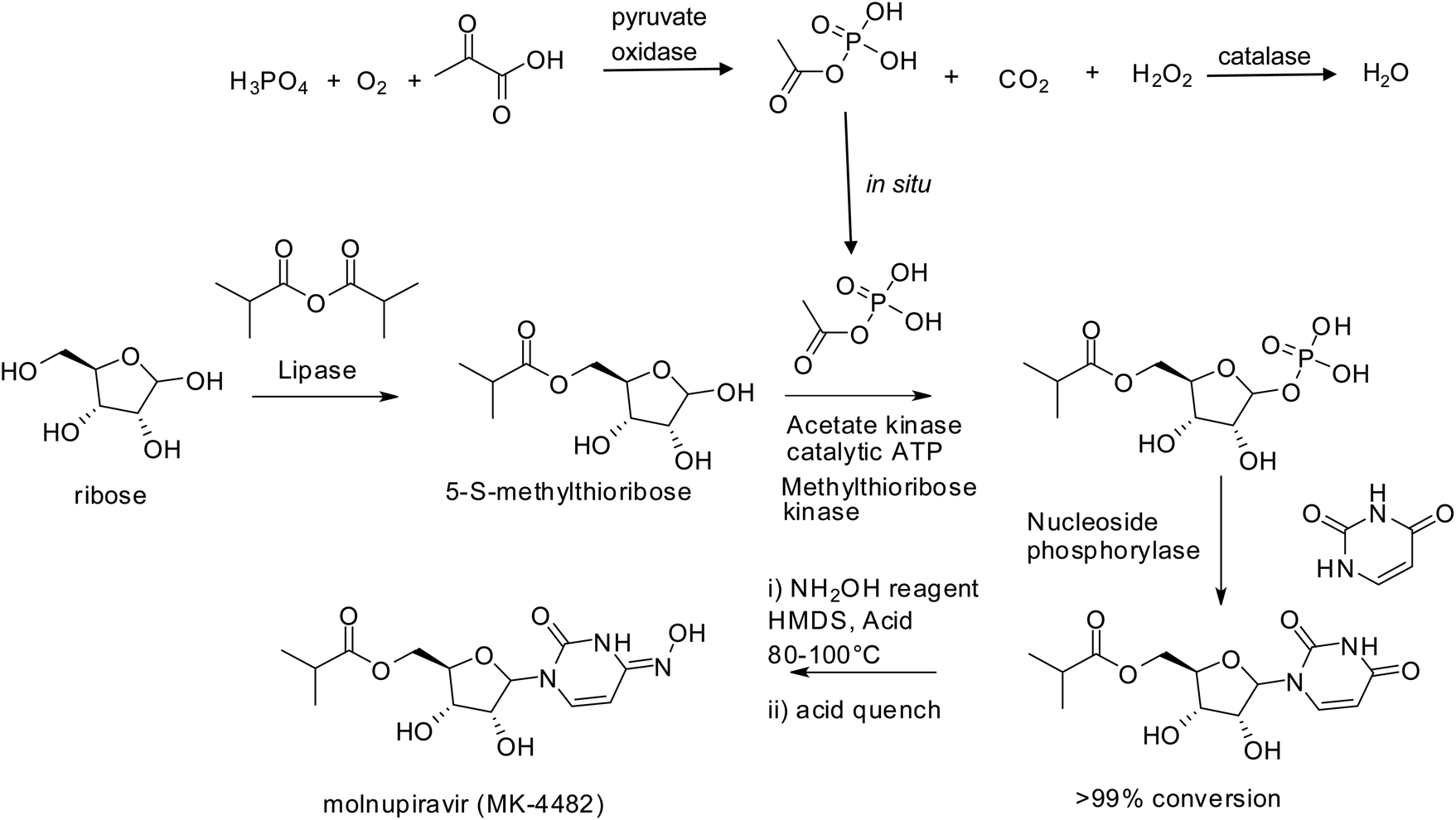 | ||
| Scheme 9 Molnupiravir synthesis. | ||
The pharmaceutical industry in general, and biocatalysis in particular,196–198 is rapidly evolving towards safer and more sustainable continuous processes199–201 that generate less waste and have lower E factors than conventional processes. Biocatalysis in flow facilitates process intensification202 and integration of multiple process steps in biocatalytic and chemo-enzymatic cascade processes.203,204 Conducting cascade processes in a continuous flow mode205 overcomes limitations resulting from reaction and reagent batch incompatibility.206,207 It was applied, for example, in transaminase catalysed continuous amination of ketones.208–210 In an interesting variation on this theme, Mutti and co-workers211 recently reported the use of 3D printing technology to facilitate the merging of biocatalysis and continuous processing.
4. The biobased economy: waste valorisation
Publication of the table of E Factors thirty years ago challenged the fine chemical and pharmaceutical industries to make the paradigm shift from a concept of process efficiency that was exclusively focused on chemical yield to one motivated by elimination of waste and conservation of resources. It played an undeniable role in the shaping of more sustainable chemicals manufacture by substitution of archaic wasteful technologies and toxic or hazardous solvents and reagents with more sustainable alternatives. This will continue to be an important driver in the future but will be underpinned by two additional developments. The first is the so-called defossilisation of chemicals manufacture by substituting fossil resources with renewable, bio-based feedstocks in the manufacture of commodity chemicals and materials. The second is the transition from an unsustainable linear economy to a circular alternative involving the deliberate design of products and processes with conservation of resources and elimination of waste in mind.In a bio-based economy unavoidable waste, such as agricultural and forestry residues, composed of lignocellulose, are the feedstocks in biorefineries. However, lignocellulose is more difficult to process than first generation feedstocks such as sucrose and starch. It consists of three polymeric components: lignin (ca. 20%), cellulose (ca. 40%), and hemicellulose (ca. 25%) that must be depolymerised and partially deoxygenated, using thermochemical or hydrolytic processes (Scheme 10).212
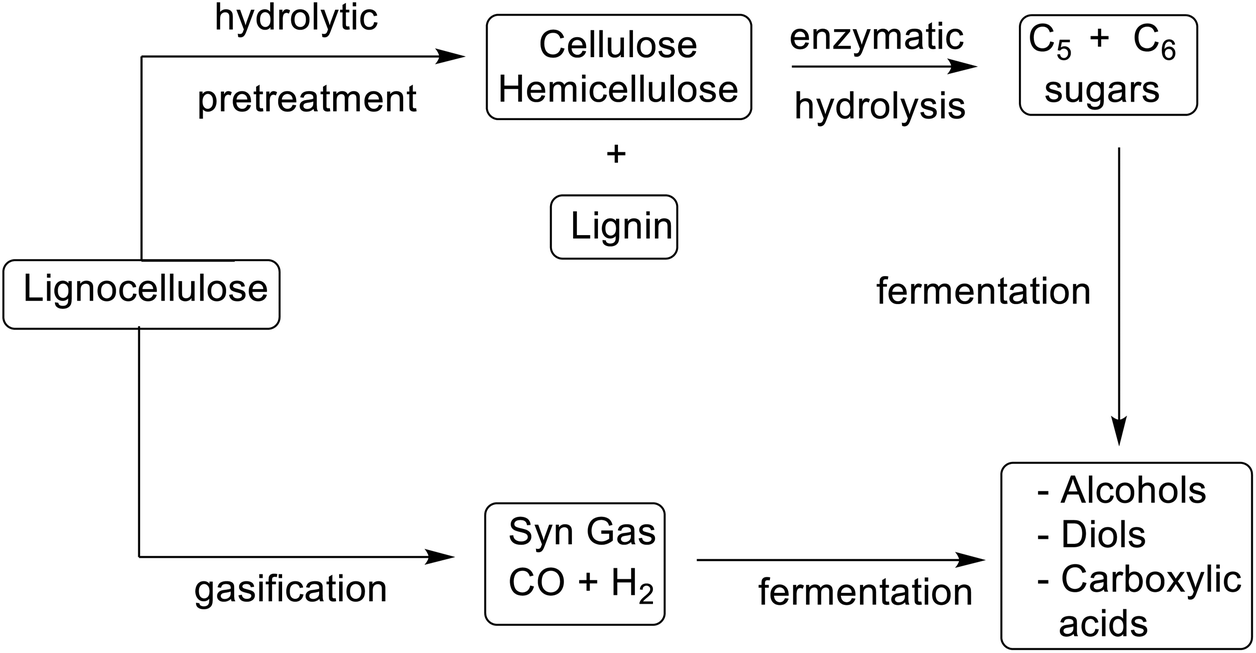 | ||
| Scheme 10 Conversion of lignocellulose to commodity chemicals. | ||
Gasification, for example, affords a mixture of carbon monoxide and hydrogen (syn gas) that is converted into biofuels and platform chemicals using established chemo-catalytic or fermentation technologies.213 Hydrolytic processes involve some form of pre-treatment, such as steam explosion, to open up the intractable lignocellulose, followed by hydrolysis of the cellulose and hemicellulose, catalysed by a cocktail of enzymes.214 This affords a mixture of hexoses and pentoses that are converted to platform chemicals by fermentation and/or chemo-catalysis.215 Traditionally the lignin was used to generate electricity but a commercially more attractive option is (bio)catalytic valorisation to commodity chemicals.
Although the cost contribution of the enzyme cocktail has decreased substantially over the last decade,216 the pretreatment and enzymatic hydrolysis contribute significantly to the overall costs. Further reductions may be achievable by enabling multiple recycling of the enzymes through suitable immobilisation, e.g. as cross-linked enzyme aggregates (CLEAs).217 An extra challenge in this context is the need to separate the immobilised enzyme cocktail from other insoluble solids present in the feedstock. This can be achieved using magnetic CLEAs in combination with magnetic separation equipment that is common in the mining industry.218
Currently, much attention is focused on the use of alternative reaction media such as ILs, and DESs, particularly if they are derived from renewable raw materials, to facilitate both saccharification of lignocellulose and subsequent conversion of the sugar building blocks to biofuels and commodity chemicals.219–221
4.1 Carbohydrates to commodity chemicals
Production of commodity chemicals from C6 and C5 sugars involves either (i) conversion to ‘drop-in’ petroleum hydrocarbons and further processing via established petrochemical technologies or (ii) direct, redox-economic conversion to oxygenates as platform chemicals.Various chemo- or biocatalytic methods are employed for converting hexoses and pentoses to petroleum hydrocarbons.222 For example, lower alcohols – ethanol and 1-butanol and isobutanol – are produced by fermentation and dehydrated to ethylene, 1-butene and isobutene, linking seamlessly with existing petrochemical supply chains and the optimum use of bioethanol could well be as a platform chemical rather than as a biofuel.223 Efficient production of isopropanol from ethanol by fermentation, using a metabolically engineered strain of E. coli, was recently reported.224 Subsequent dehydration of ethanol and isopropanol affords two of the top ten petrochemicals: ethylene and propylene. Similarly, 2,3-butane diol and 1,4-butane diol can be produced by fermentation and be subsequently dehydrated to butadiene. Alternatively, metabolic engineering can be used to re-engineer the isoprenoid pathway in bacteria or yeast, to directly produce hydrocarbons, such as isobutene and isoprene, by fermentation.225
Thanks to advances in metabolic engineering and synthetic biology in the last two decades, a wide variety of diols and mono- and di-carboxylic acids can be produced, in a redox economic and cost effective manner, by fermentation. Citric acid and lactic acid are examples of first generation, large volume commodity chemicals produced by fermentation. There are many examples of second generation (di) carboxylic acids, such as succinic acid and acrylic acid, and diols, such as 1,3-propane diol, 1,4-butane diol and 2,3-butane diol that are produced cost-effectively by fermentation (Scheme 11).
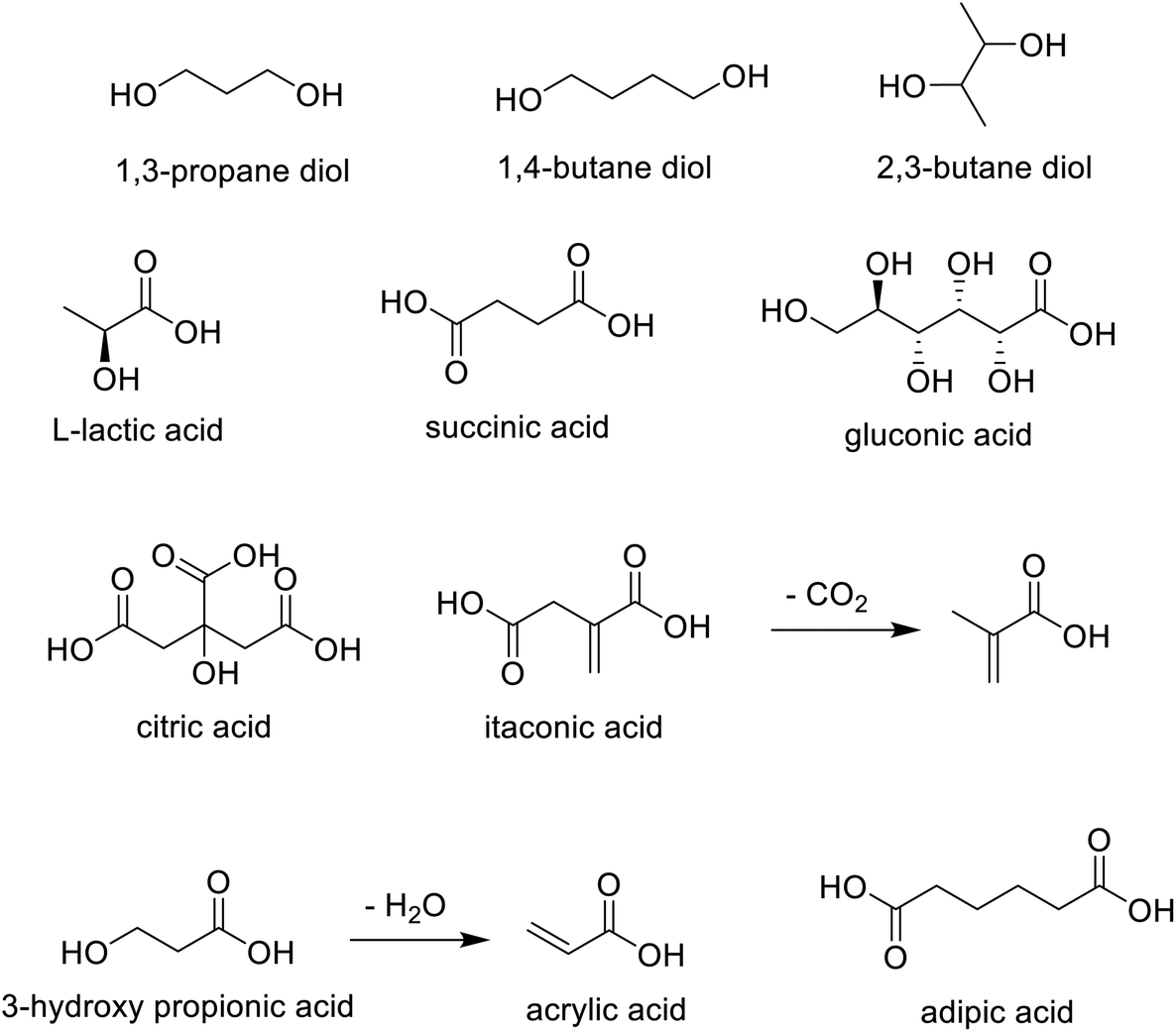 | ||
| Scheme 11 Carbohydrates to commodity chemicals. | ||
In yet another approach, so-called aqueous phase reforming (APR), over supported Pt or Pt–Re catalysts, pioneered by Dumesic and co-workers,226 is used to produce, inter alia, furfural, 5-hydroxymethyl furfural (HMF) and levulinic acid (LA), that are further hydrogenated in situ to a mixture of mainly C4–C6 alkanes.
Alternatively, direct conversion of hexoses and pentoses, derived from lignocellulose hydrolysis, to platform chemicals can be achieved using chemo- and/or bio-catalysis.227,228 For example, acid catalysed hydrolysis of pentoses and hexoses produces furfural229 and HMF, respectively. Furfural is an existing commodity chemical with broad applications and HMF has enormous potential230 as a feedstock for chemicals, polymers and biofuels. Aerobic oxidation of HMF affords 2,5-diformylfuran (DFF) that can be converted to the corresponding diol or diamine, potential polymer building blocks, by hydrogenation or reductive amination, respectively (Scheme 12). Alternatively, chemo-231,232 or biocatalytic oxidation233–237 produces furan-2,5-dicarboxylic acid (FDCA),238 the building block for polyethylene furanoate (PEF) (see section 5).
 | ||
| Scheme 12 Production of furanics from glucose. | ||
However, cost-effective dehydration of hexoses, in particular glucose, to HMF presents a significant challenge, mainly owing to the low stability of HMF under the acidic conditions where it decomposes to form humin, a polyfuranic resin.239 A possible option is to bypass HMF altogether by converting the hexose precursor directly, under mild conditions in high yield, to 5-chloromethyl furfural (CMF) by reaction with aq. HCl.240 CMF can be further converted to FDCA or to levulinic acid. The combination of good stability with lipophilicity for facile isolation from aqueous media make CMF an interesting platform chemical for production in 2G biorefineries.
5. The circular economy: waste-free by design
5.1 The plastic pollution challenge
Only 30% of all the plastics that have ever been produced are currently in use.241 Most of the rest, amounting to more than 6 billion tonnes, has accumulated in landfills or in our natural environment. Furthermore, the projected production of plastics in the period 2020–2050 will generate a further 12 billion tonnes of waste.There is clearly a pressing need to replace this linear take-make-use-dispose economy with a circular plastic economy that reduces waste and optimises resource utilisation through multiple rounds of recycling. The preferred option is closed-loop recycling to the original plastic by recovery of the corresponding monomers. The second option is open-loop recycling to afford lower value products by, for example, pyrolysis or energy recovery by incineration. Landfill is no longer a viable option.
Depolymerisation to the olefin monomers is the preferred option for recycling the largest volume plastics, i.e. polyethylene (PE) and polypropylene (PP). It is technically feasible with polystyrene (PS)242 but depolymerisation of PE and PP are highly endothermic processes. On the other hand, pyrolysis to a naphtha-like mixture of hydrocarbons and subsequent steam cracking to afford a mixture of lower olefins is feasible. More recently, a more selective approach to PE depolymerisation, involving catalytic transfer dehydrogenation, with a small amount of ethylene mediated by the Phillips ethylene polymerisation catalyst, followed by tandem catalytic isomerisation and olefin metathesis, afforded propylene in 80% yield (Scheme 13).243,244 The method is reminiscent of the Shell Higher Olefins Process (SHOP), started up in 1977, for the synthesis of higher olefins from ethylene,245 but operating in reverse.
 | ||
| Scheme 13 Catalytic conversion of PE to propylene. | ||
5.2 Biocatalytic recycling of plastics
The polyolefin recycling problem can be circumvented altogether by substituting polyolefin plastics with more readily recycled polyesters or polyamides. Polymers containing hydrolysable bonds, e.g. polyesters and polyamides, such as PET, polybutylene succinate and Nylon, can be converted to the original monomers by acid- or base-catalysed or enzyme-catalysed hydrolysis. The possibilities and limitations of biotechnological recycling of plastics was recently reviewed by Blank and Bornscheuer and co-workers.246,247In 2016, a bacterium, Ideonella sakaiensisin, was discovered248,249 in soil from an industrial waste PET-recycling facility, that catalysed the hydrolysis of PET. It secreted two different hydrolases: so-called IsPETase that catalyses hydrolysis of PET to mono-2-hydroxyethyl terephthalic acid (MHET) and a second enzyme that catalyses the hydrolysis of MHET (Scheme 14)
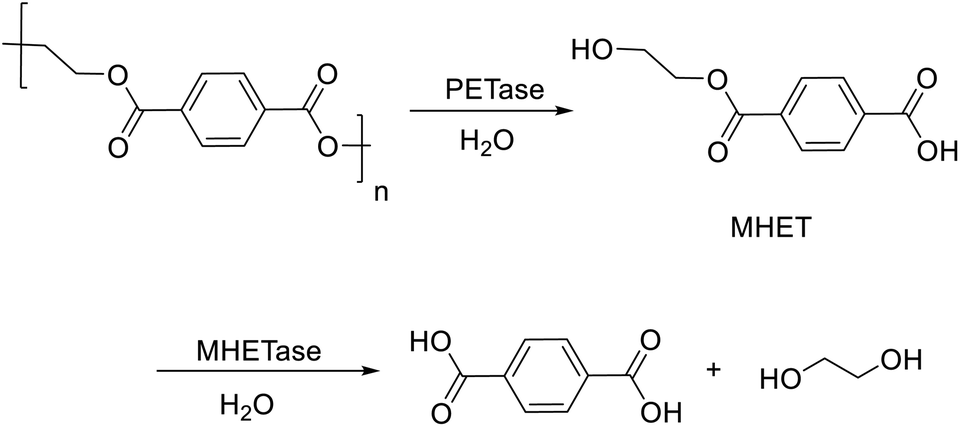 | ||
| Scheme 14 Enzymatic hydrolysis of PET. | ||
The IsPETase250–252 was subsequently shown to have structural features in common with both lipases and the plant cell-wall degrading cutinases253 that catalyse PET depolymerisation via hydrolysis.254,255 Kanaya and co-workers256 identified a cutinase, from leaf-branch compost (LCC) in a Japanese public park, which catalysed the hydrolysis of PET at 70 °C. Computer-aided protein engineering was subsequently used257 to produce a variant which catalysed the hydrolysis of PET to 90% conversion in less than 10 h at 72 °C using an enzyme loading of 3 wt%, and a Space Time Yield of 16.7 g l−1 h−1. The cost was estimated to be ca. 4% of the cost of virgin PET.
Similarly, an anaerobic thermophilic bacterium, Clostridium thermocellum was genetically engineered to enable high-level secretory expression of LCC.258 In addition to catalysing the depolymerisation of PET at 60 °C the microbial cells catalysed the efficient hydrolysis of cellulose, suggesting that they could be used to recycle textile waste containing both polymers.
The current state of research on PET-hyrolysing enzymes was recently reviewed by Kawai.259 They can be divided into mesophilic (e.g. IsPETase) and thermophilic cutinases. The former have the disadvantage that large scale hydrolysis requires a temperature close to the glass transition temperature of 65–70 °C in water. Economic viability is still a key issue for PET recycling but bio-recycling is expected to be the optimum solution as it is eco-friendly with low energy consumption.260 More recently, machine learning-aided engineering of PET hydrolases was used to produce a robust and active PETase that showed good performance in the degradation of thermoformed PET products.261
Clearly, (bio)catalytic depolymerisation of polyesters to the coresponding monomers will be industrially viable in the near future, particularly when costs of virgin vs. recycled plastic are compared on a level playing field.
5.3 Biodegradability of plastics
Biodegradability is widely viewed as a desirable property for plastics but technical and socio-economic arguments are not generally in favour of biodegradable plastics.71 Plastics that readily degrade remain an environmental hazard in a marine environment. Moreover, the biodegradable label will be interpreted by many as meaning that littering is not a problem. In contrast, plastics that are biodegradable-on-demand at their end-of-life (EoL), after multiple recycling, are attractive. For example, Carbios embedded an extremely thermostable esterase, that could withstand the extrusion temperature of 170 °C during its production, to afford polylactate (PLA) that is self-degrading at its EoL.2565.4 Bio-based plastics for sustainability
The sustainable macromolecular materials of the 21st century will have reduced environmental footprints and be based on the carbon neutral utilisation of polymers derived from renewable raw materials.262,263 Utilisation of renewable biomass is a conditio sine qua non for lower GHG emissions. PLA production from corn-starch, for example, affords a 27% reduction in GHG emissions compared with PE from fossil resources264 and bio-based PET is responsible for 25% less GHG emissions than PET from fossil resources.265,266 Indeed, the recent publication of several reviews on polymers from renewable feedstocks,267–271 and the current trends and challenges in their production,272 attests to its current importance.Bio-based plastics can be drop-in products, e.g. PE produced from ethylene derived from bioethanol. Alternatively, they can be totally new polymers e.g. PHAs (see later). An important advantage of drop-ins is that the product is already known in the marketplace and there is no need to change production equipment and processes. In contrast, if the bio-based resin is different, e.g. PLA to replace PET, different additives and processing technology may be required and the product will have different mechanical and diffusion barrier properties to e.g. water and oxygen. Such problems can sometimes be overcome by using polymer blends.273
The primary motive for switching to bio-based plastics is climate change mitigation through reductions in CO2 emissions but the envisaged facile recyclability and biodegradability will be welcome bonuses. However, the use of renewable feedstocks should not compete with food production or cause deforestation and/or loss of biodiversity. This will be avoided by utilising waste streams as the raw material in 2nd generation (2G) biorefineries, e.g. lignocellulosic waste from agricultural and forestry residues and food supply chain waste.274 Looking further into the future, they will be produced from 3rd generation (3G) polysaccharides from algae and photosynthetic bacteria, that have the advantage that arable land and fresh water are not necessary for their cultivation. Alginate and carrageenan from macroalgae (seaweed), for example, can function as feedstocks for bio-based plastics.275
In short, bio-based plastics are produced (i) directly from bio-based monomers, e.g. PLA from lactic acid produced by fermentation, (ii) directly by fermentation, or (iii) by conversion of natural biopolymers such as starch, cellulose and chitin.276 Examples of bio-based monomers – diols, dicarboxylic acids and diamines277 produced by fermentation or chemocatalytic conversion of glucose, are depicted in Scheme 15. In addition to using bio-based monomers there is also a growing trend towards the use of biocatalysis in the polymerisation process, e.g. in the synthesis of polyesters.278,279
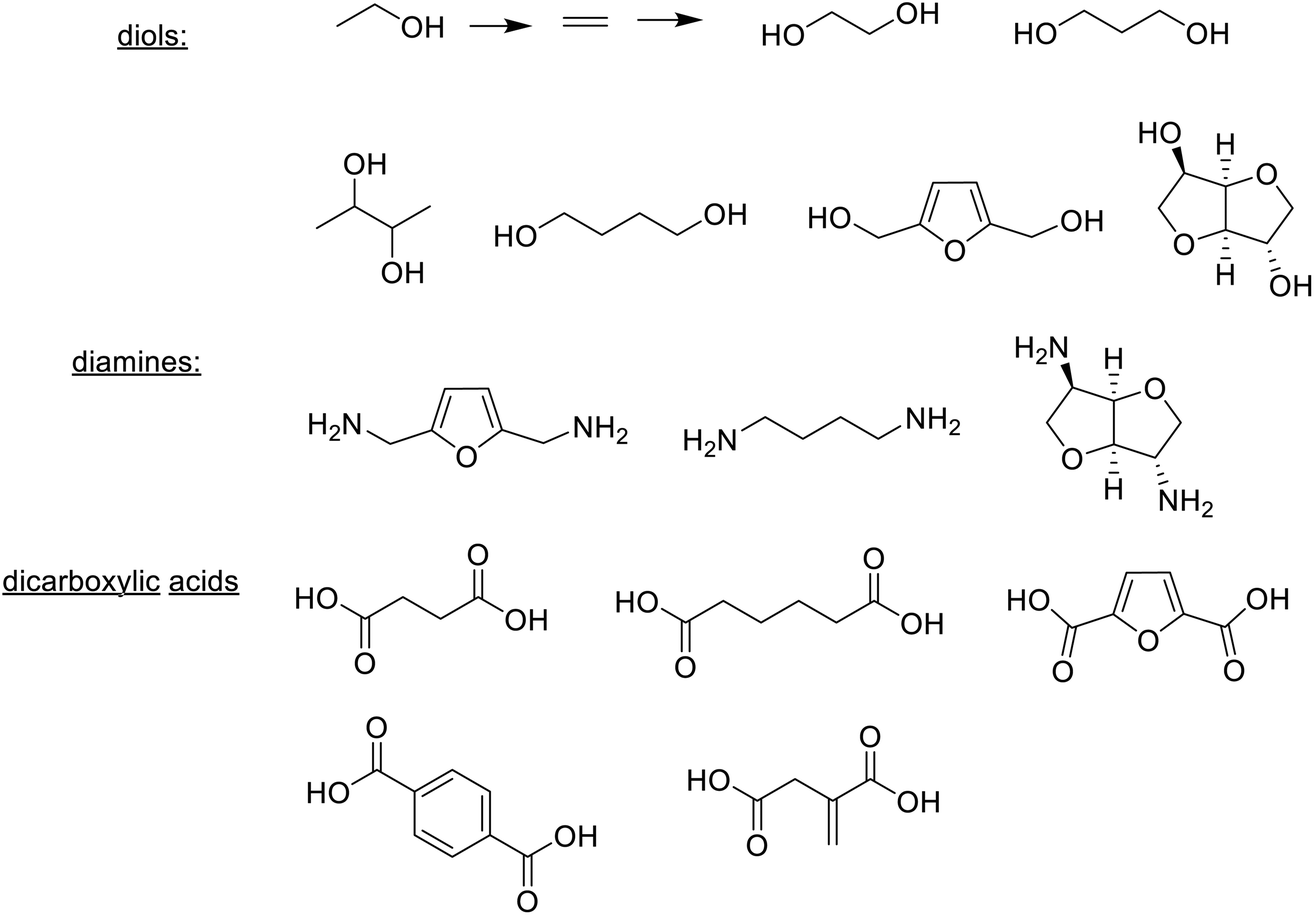 | ||
| Scheme 15 Bio-based monomers for thermoplastics. | ||
Drop-in bio-based PET, currently market leader, is 20% biobased with 2 carbons from bioethanol and 8 carbons from fossil-based p-xylene (Scheme 16). Considerable research effort is currently being devoted to developing an industrially viable route to bio-based terephthalic acid to enable production of 100% bio-based PET.280,281
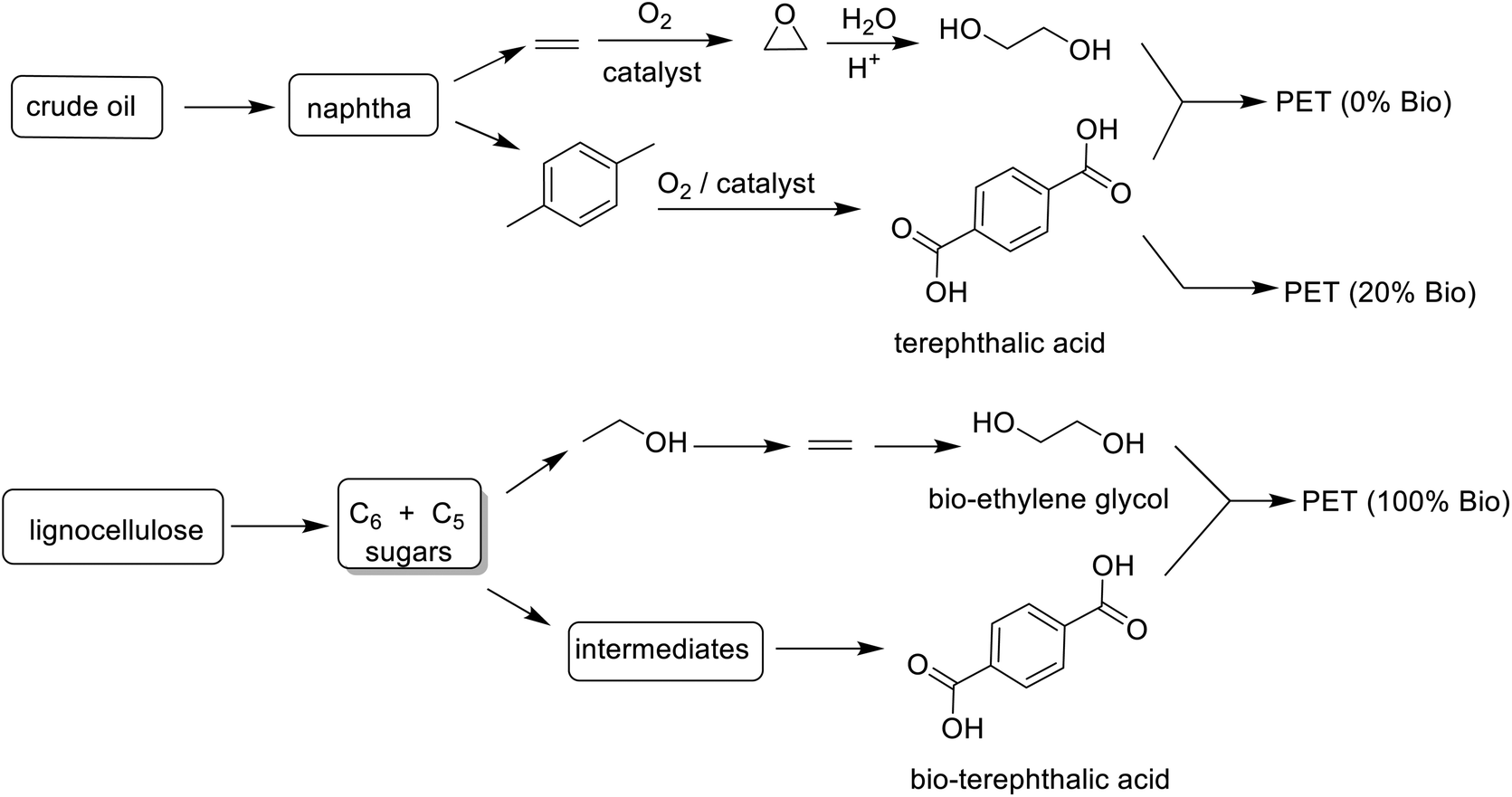 | ||
| Scheme 16 Production of fossil vs. biobased PET. | ||
Polyethylene furan 2,5-dicarboxylate (PEF), developed by Avantium,282 is 100% bio-based (Scheme 17), with a reduction in GHG emissions of up to 55%, and superior thermal, mechanical, and gas barrier properties, compared to PET.283,284 A 100% bio-based equivalent of polytrimethylene terephthalate (PTT) has been produced from FDCA and bio-based 1,3-propane diol.285
 | ||
| Scheme 17 Production of PEF. | ||
Polyhydroxyalkanoates (PHAs) function as a carbon and energy source for acetogenic bacteria and can represent up to 90% of the dry weight of the bacterium. They are particularly interesting plastics because they can be produced by fermentation of low-cost waste streams, e.g. municipal286,287 and paper mill288 waste water, even waste polystyrene,289 and they are biodegradable. Their physical properties are comparable with those of PE and PP, making them suitable for, inter alia, applications in single-use plastic packaging. Newlight Technologies produced a PHA by aerobic fermentation of methane or biogas from landfills and converted it to a thermoplastic, ‘AirCarbon’, that was licensed to IKEA for use in home furnishing products.290 Composites of PHAs with inexpensive natural fibres with superior physico-mechanical properties have also been described.291
According to a recent review of bio-based plastics in food packaging292 the total production of bio-based plastics has reached 7.5 million tonnes per annum, including bio-based polyurethanes, that is 2% of fossil-based plastics. It is worth noting, however, that the manufacture of fossil-based plastics has been optimised over a period of more than half a century and it is not surprising, therefore, that their bio-based cousins, such as PLA and PHAs, are currently more expensive.293 This gap is steadily decreasing albeit too slowly to seriously impact the plastic pollution challenge in the short term. This situation would change overnight, however, if EPR were implemented in the pricing of polyolefins, as was the case with the introduction of lead-free gasoline in the 1970s. A recent study of the environmental impact of bio-based plastics concluded that if two thirds of the global plastics were bio-based the annual generation of CO2 equivalents would be reduced by 241–316 million tonnes.294
Bio-based plastics are also produced by direct conversion of natural biopolymers, such as starch and cellulose. Less explored polysaccharides such as chitin,295 the second most abundant natural polymer after cellulose, are potential sources of packaging plastics. Chitin nanofibers, for example, afforded packaging materials that combine excellent gas barrier properties with flexibility and optical transparency.296 Similarly, polyamide and polyurethane plastics are derived from long-chain fatty acids present in plant oils (e.g. castor oil, sunflower oil)297 and waste proteins, such as the vast amounts of keratin from wool, hair and chicken feathers298 constitute another commercially viable source of novel polymer materials.
Finally, production of plastics directly from carbon dioxide will be facilitated by the envisaged future availability of sustainable green hydrogen from water electrolysis using renewable electricity.299 Carbon dioxide can be converted to syn gas as a source of monomers for plastics produced by existing petrochemical technologies or by fermentation.300 Alternatively, PHAs can be produced directly from light, CO2 and water.301
6. The energy transition and beyond: decarbonisation of energy and electrons versus reagents
The pressing need for reductions in GHG emissions to mitigate climate change has led to a so-called energy transition in which decarbonisation of the energy sector and defossilisation of chemicals manufacture are expected to play primary roles. Our reliance on finite fossil resources, for generating energy and materials, is clearly not sustainable in the long term and will be systematically reduced over the coming decades with the goal of completing the transition by the turn of the century. It will involve replacement of fossil resources by renewable, carbon neutral forms of energy: hydroelectric, geothermal, wind and solar energy. However, it is unlikely that this will generate enough energy to drive highly industrialised economies and sustainable nuclear energy302 will inevitably be part of the energy mix. Indeed, the anticipated replacement of classical uranium reactors by molten salt thorium reactors303,304 will make an important contribution to significantly reducing CO2 emissions and will afford serious reductions in long-lived radioactive waste, another waste problem of global proportions.In the envisaged scenario for energy decarbonisation, electricity will play a primary role as an energy source and off-peak electricity will be used for the production of renewable liquid fuels and chemicals, using electrons instead of ‘reagents’ to drive a so-called Power-to-X (P2X) industry305–308 in e-refineries. The importance of P2X is underlined by the rapidly growing number of demonstration plants worldwide (190 in 2019).309 It could involve, for example, the production of renewable hydrogen by electrolysis of water and its use in heating and the production of steel,310 transportation fuels and base chemicals. Alternatively, electrolysis of a mixture of carbon dioxide and water could be used to produce a mixture of CO and H2 (syn-gas) that is subsequently converted to hydrocarbons using the classical Fischer–Tropsch (FT) process or to oxygenates using chemo-catalysis or fermentation.311 The production of e-diesel in an electrically driven FT process will substantially reduce its global warming impact but the total environmental impacts of the two processes need to be compared for a complete evaluation (caveat emptor).312 Replacing energy intensive steam crackers, that use gas-fired furnaces and generate huge amounts of carbon dioxide, by electric cracking of hydrocarbons using renewable electricity is also a possibility.313
Electrolytic reduction of carbon dioxide can be utilised to produce methanol as a high-energy-density liquid fuel and raw material for commodity chemicals manufacture, in a methanol economy as envisaged by Olah and co-workers.314,315 Ammonia is also a serious contender for energy carrier in a global distribution of renewable energy.316 Ammonia is already, in a fossil resource-based industry, one of the top ten bulk chemicals with an annual production of 8 mio tonnes. In a sustainable economy it will be produced by hydrogenation of nitrogen with renewable hydrogen or by direct electrolytic reduction of nitrogen.317
The world-wide attention currently focused on P2X strategies for bulk chemicals production has also provoked a veritable renaissance in organic electrosynthesis.318–325 Moreover, microbial electrosynthesis (MES), involving coupling of renewable electricity generation with the metabolism of electroactive acetogenic and methanogenic microorganisms (electroautotrophs), in e-biorefineries, can drive the in vivo reduction of CO2 to acetic acid, ethanol, formate, methanol and methane.326,327
6.1 Electrobiocatalysis
Alternatively, electricity can drive reactions catalysed by isolated oxidoreductases through replacement of conventional co-factor regeneration systems, requiring a second enzyme and a co-substrate, with electrochemical regeneration. A broad range of NAD(P), FMN, FAD and PQQ-dependent metallo- and non-metallo-oxidoreductases, using both oxygen and hydrogen peroxide as terminal oxidants, has been studied.328 Enzymatic oxidations with hydrogen peroxide catalysed by peroxidases, for example, have the limitation that the enzymes undergo competing facile oxidative degradation by hydrogen peroxide. An advantage of in situ electrolytic generation is that the hydrogen peroxide concentration is maintained at a low level. Deactivation of chloroperoxidase from Caldariomyces fumago (CPO), that catalyses a variety of oxidations including enantioselective olefin epoxidation and sulfoxidations,329 for example, was reduced by using electrolytic in situ hydrogen peroxide generation.330In the innovative ‘electrochemical leaf’ concept331 an NADP(H)-dependent dehydrogenase and a small photosynthetic flavo-enzyme – ferredoxin-NADP+ reductase (FNR) – that supplies electrons to generate the NADPH cofactor are entrapped in a nanoporous metal oxide electrode. FNR is ubiquitous in nature where it channels electrons into biosynthesis.332 This concept can be used to drive a variety of reactions, including multi-enzyme cascade processes333,334 such as a one-pot deracemisation of chiral secondary alcohols (Scheme 18).335
 | ||
| Scheme 18 Electrobiocatalytic deracemisation of sec-alcohols. | ||
Alternatively, chemocatalytic oxidations can be integrated with biocatalytic steps, e.g. combination of TEMPO-catalysed aerobic oxidation of alcohols with enzymatic reductive amination of the resulting ketones to afford an enantioselective, environmentally attractive route to chiral amines from alcohols, with a molecule of water as the sole coproduct, in an aqueous medium (Scheme 19).336
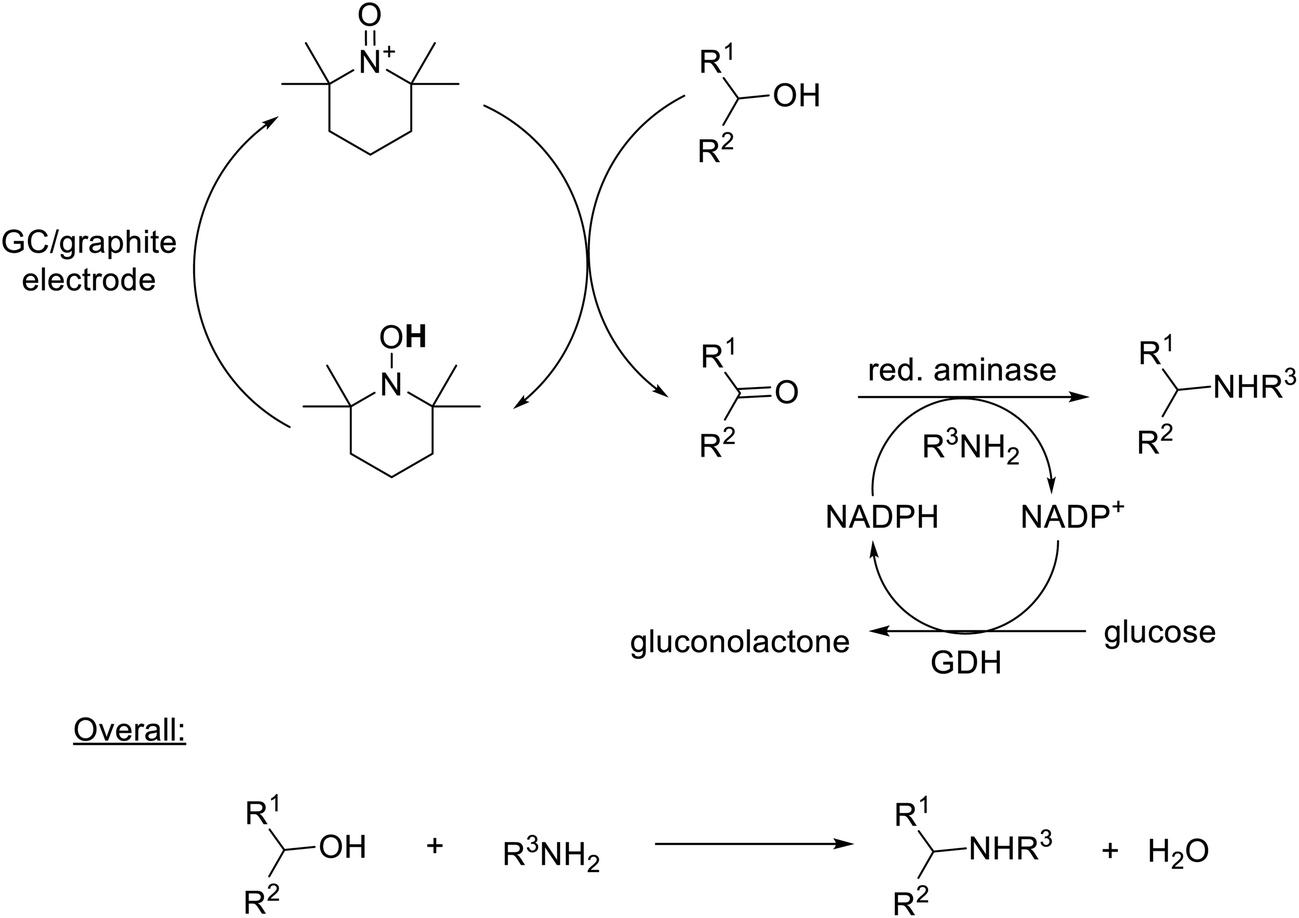 | ||
| Scheme 19 Electrochemoenzymatic conversion of sec-alcohols to chiral amines. | ||
7. Conclusions
Thirty years ago the E Factor drew attention to the problem of waste generation in chemicals manufacture and caused a paradigm shift in how the efficiency of chemical manufacturing processes is assessed. In the meantime, it has become increasingly clear that waste is the common denominator in the major environmental problems, such as climate change and pollution of our natural habitat. It is also clear that finding solutions to the ubiquitous waste problem, in its many facets, is one of the grand challenges for chemistry and chemical engineering.Hopefully, we have shown in this review that there is sufficient reason for optimism. The answer is not less chemistry but better more sustainable chemistry, based on a more efficient use of resources in a circular economy underpinned by advances in (bio)catalysis and coupled with extensive decarbonisation of the energy sector and defossilisation of chemicals manufacture.337 Technologies will be sustainable, and products recyclable, by design and the key words will be pollution prevention.
The organic chemist, A. W. von Hofmann, already said it almost 200 years ago:
“in an ideal chemical factory there is, strictly speaking, no waste but only products. The better a real factory makes use of its waste, the closer it gets to its ideal, the bigger is the profit”.
Conflicts of interest
There are no conflicts to declare.References
- L. A. Pfaltzgraff, M. De Bruyn, E. C. Cooper, V. Budarin and J. H. Clark, Green Chem., 2013, 15, 307–314 RSC
.
- R. A. Sheldon, Chem. Ind., 1992, 903–906 CAS
-
P. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed
-
C. G. Brundtland, Our Common Future, The World Commission on Environmental Development, Oxford University Press, Oxford, 1987 Search PubMed
-
Handbook of Green Chemistry and Technology, ed. T. E. Graedel, J. Clark and D. J. Macquarrie, Wiley, New York, 2002, pp. 56–61 Search PubMed
- J. M. Köhler, Green Process. Synth., 2014, 3, 33–45 Search PubMed
- https://www.ipcc.ch/report/sixth-assessment-report-working-group-ii/ .
-
R. A. Sheldon, I. W. C. E. Arends and U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH, Weinheim, 2007 Search PubMed
- H. Wu, B. Qu, T. Nguyen, J. C. Lorenz, F. Buono and N. Haddad, Org. Process Res. Dev., 2022, 26, 2281–2310 CrossRef CAS
- R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha and C. H. Hendon,
et al.
, Science, 2020, 369, 786 CrossRef PubMed
-
Biocatalysis in Green Solvents, ed. P. Lozano, Academic Press, Elsevier, London, 2022 Search PubMed
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133–137 CrossRef CAS
- C. Ashcroft, P. Dunn, J. Hayler and A. S. Wells, Org. Process Res. Dev., 2015, 19, 740–747 CrossRef CAS
- R. A. Sheldon, Curr. Opin. Green Sustainable Chem., 2019, 18, 13–19 CrossRef
- A. Canet, M. D. Benaiges and F. Valero, J. Am. Oil Chem. Soc., 2014, 91, 1499–1506 CrossRef CAS
- A. Jordan, P. Stof and H. F. Sneddon, Chem. Rev., 2021, 121, 1582–1622 CrossRef CAS PubMed
- C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed
- M. M. C. H. van Schie, J.-D. Spöring, M. Bocola, P. Dominguez de Maria and D. Rother, Green Chem., 2021, 23, 3191–3206 RSC
- A. G. Corrêa, M. W. Paixão and R. S. Schwab, Curr. Org. Synth., 2015, 12, 675–695 CrossRef
- J. H. Clark, T. J. Farmer, A. J. Hunt and J. Sherwood, Int. J. Mol. Sci., 2015, 16, 17101–17159 CrossRef CAS PubMed
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC
- K. Tekin, N. Hao, S. Karagoz and A. J. Ragauskas, ChemSusChem, 2018, 11, 3559–3575 CrossRef CAS PubMed
- A. E. Diaz-Alvarez, J. Francois, P. Crochet and V. Cadierno, Curr. Green Chem., 2014, 1, 51–65 CrossRef CAS
- G. Ou, B. He and Y. Yuan, Enzyme Microb. Technol., 2011, 49, 167–170 CrossRef CAS PubMed
- https://www.solvay.com/en/brands/augeo accessed 14 January 2023.
- F. Kerkel, M. Markiewicz, S. Stolte, E. Müller and W. Kunz, Green Chem., 2021, 23, 2962–2976 RSC
- V. Pace, P. Hoyos, L. Castoldi, P. Domínguez de María and A. R. Alcantara, ChemSusChem, 2012, 5, 1369–1379 CrossRef CAS PubMed
- C. Aranda and G. de Gonzalo, Molecules, 2020, 25, 3016 CrossRef CAS PubMed
- S. Kudo, X. Huang, S. Asano and J. Hayashi, Energy Fuels, 2021, 35, 9809–9824 CrossRef CAS
- J. Sherwood, M. de Bruyn, A. Constantinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunt and J. H. Clark, Chem. Commun., 2014, 50, 9650–9652 RSC
- A. A. C. Pacheco, J. Sherwood, A. Zhenova, C. R. McElroy and A. J. Hunt,
et al.
, ChemSusChem, 2016, 9, 3503–3512 CrossRef CAS PubMed
- D. Kong and A. V. Dolzhenko, Sustainable Chem. Pharm., 2022, 25, 100591 CrossRef CAS
- N. Guajardo and P. Domínguez de María, Mol. Catal., 2020, 485, 110813 CrossRef CAS
- L. M. M. Mouterd, F. Allais and J. D. Stewart, Green Chem., 2018, 20, 5528–5532 RSC
- P. Lozano, E. Alvarez, J. M. Bernal, S. Nieto, C. Gomez and G. Sanchez-Gomez, Curr. Green Chem., 2017, 4, 116–129 CAS
- T. Itoh, Chem. Rev., 2017, 117, 10567–10607 CrossRef CAS PubMed
- P. Xu, S. Liang, M.-H. Zong and W.-Y. Lou, Biotechnol. Adv., 2021, 29, 107702 CrossRef PubMed
- H. T. Imam, V. Krasnan, M. Rebros and A. C. Marr, Molecules, 2021, 26, 4791 CrossRef CAS PubMed
- M. Pätzold, S. Siebenhaller, S. Kara, A. Liese, C. Syldatk and D. Holtmann, Trends Biotechnol., 2019, 37, 943–959 CrossRef PubMed
- P. Xu, G.-W. Zheng, M.-H. Zong, N. Li and W.-Y. Lou, Bioresour. Bioprocess., 2017, 4, 34 CrossRef PubMed
- L. Cicco, G. Dilaurio, F. M. Perna, P. Vitali and V. Capriati, Org. Biomol. Chem., 2021, 19, 2558–2577 RSC
- L. Cicco, N. Rios-Lombardia, M. J. Rodriguez-Alvarez, F. Moris, F. M. Perna, V. Capriati, J. Garcia-Alvarez and J. Gonzalez-Sabin, Green Chem., 2018, 20, 3468–3475 RSC
- Y. Dai, J. Van Spronsen, G. J. Witkamp, R. Verpoorte and Y. H. Choi, Anal. Chim. Acta, 2013, 766, 61–68 CrossRef CAS PubMed
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS
-
Special Issue on Recent Advances in Catalysis in Green Aqueous Media, in Catal. Today, ed. G. Papadogianakis and R. A. Sheldon, 2015, vol. 247, pp. 1–190 Search PubMed
- C. W. Kohlpaintner, R. W. Fischer and B. Cornils, Appl. Catal., A, 2001, 221, 219–225 CrossRef CAS
- J. Lalonde, Curr. Opin. Biotechnol., 2016, 42, 152–158 CrossRef CAS PubMed
- B. H. Lipshutz, Curr. Opin. Green Sustainable Chem., 2018, 11, 1–8 CrossRef
- M. Cortes-Clerget, T. Yu, J. R. A. Kincaid, P. Walde, F. Gallou and B. H. Lipshutz, Chem. Sci., 2021, 12, 4237–4266 RSC
- B. H. Lipshutz, S. Ghorai and M. Cortes-Clerget, Chem. – Eur. J., 2018, 24, 6672–6695 CrossRef CAS PubMed
- For a recent review see: W. Bin, S. Miraghaee, S. Handa and F. Gallou, Curr. Opin. Green Sustainable Chem., 2022, 38, 100691 CrossRef
- B. H. Lipshutz, N. A. Isley, J. C. Fennewald and E. D. Slack, Angew. Chem., Int. Ed., 2013, 52, 10952–10958 CrossRef CAS PubMed
- B. H. Lipshutz and S. Ghorai, Green Chem., 2014, 16, 3660–3679 RSC
- G. Kaur, S. Kaur and S. Handa, Curr. Opin. Green Sustainable Chem., 2022, 38, 100690 CrossRef CAS
- S. Kaur, G. Kaur and S. Handa, Org. Process Res. Dev., 2021, 25, 1960–1965 CrossRef
- M. Cortes-Clerget, N. Akporji, J. Zhou, F. Gao, P. Guo, M. Parmentier, F. Gallou, J.-Y. Berthon and B. H. Lipshutz, Nat. Commun., 2019, 10, 2169 CrossRef PubMed
- V. Singhania, M. Cortes-Clerget, J. Dussart-Gautheret, B. Akkachairin, J. Yu, N. Akporji, F. Gallou and B. H. Lipshutz, Chem. Sci., 2022, 13, 1440–1445 RSC
- C. Tang and B. T. McInnes, Molecules, 2022, 27, 5611 CrossRef CAS PubMed
- C. J. Hastings, N. P. Adams, J. Bushi and S. J. Kolb, Green Chem., 2020, 22, 6187–6193 RSC
- J. R. A. Kincaid, M. J. Wong, N. Akporji, F. Gallou, D. M. Fialho and B. H. Lipshutz, DOI:10.26434/chemrxiv-2022-bhzns.
- J. H. Clark, T. J. Farmer, L. Herrero-Davila and J. Sherwood, Green Chem., 2016, 18, 3914–3934 RSC
- Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the Regions, 2011, Roadmap to a Resource Efficient Europe, COM/2011/0571 final.
-
B. Commoner, The Closing Circle: Nature, Man and Technology, Knopf, New York, 1971 Search PubMed
- E. F. Sousa-Aguiar, L. Gorenstin Appel, P. Costa Zonetti, A. do Couto Fraga, A. Azevedo Bicudo and I. Fonseca, Catal. Today, 2014, 234, 13–23 CrossRef CAS
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC
- C. O. Tuck, E. Perez, I. T. Horvath, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed
- J. K. Saini, R. Saini and L. Tewari, 3 Biotech, 2015, 5, 337–353 CrossRef PubMed
- E. de Jong, A. Higson, P. Walsh and M. Wellisch, Biofuels, Bioprod. Biorefin., 2012, 6, 606–624 CrossRef CAS
- https://www.carboliq.com/pdf/19_conversio_global_plastics_flow_2018_summary.pdf .
- D. R. Shonnard, E. Tipaldo, V. Thompson, J. M. Pearce, G. T. Caneba and R. Handler, Procedia CIRP, 2019, 80, 602–606 CrossRef
- The Environmental Impacts of Plastics and Micro-Plastics Use, Waste and Pollution: EU and National Measures—European Parliament Think Tank https://www.europarl.europa.eu/thinktank/en/document.html?reference=IPOL_STU(2020)658279.
- J. N. J. Weston, P. Carrillo-Barragan, T. D. Linley, W. D. K. Reid and A. J. Jamieson, Zootaxa, 2020, 4748(1), 163–181 CrossRef PubMed
- Ellen Macarthur Foundation 2013 Towards the Circular Economy https://www.ellenmacarthurfoundation.org/assets/downloads/publications/ellen-macarthurfoundation-towards-the-circular-economy-vol.1.pdf.
- R. A. Sheldon, Philos. Trans. R. Soc., A, 2020, 378, 20190274 CrossRef CAS PubMed
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310–6322 RSC
- J. P. Lange, ACS Sustainable Chem. Eng., 2021, 9, 15722–15738 CrossRef CAS
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed
- F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752–768 RSC
- F. Roschangar, J. Li, Y. Zhou, W. Aelterman, A. Borovika and J. Colberg, ACS Sustainable Chem. Eng., 2022, 10, 5148–5162 CrossRef CAS
- Y. Hayashi, Chem. Sci., 2015, 7, 866–880 RSC
- S. K. Ma, J. Gruber, C. Davis, L. Newman, D. Gray, A. Wang, J. Grate, G. W. Huisman and R. A. Sheldon, Green Chem., 2010, 12, 81–86 RSC
- F. Tieves, F. Tonin, E. Fernandez-Fueyo, J. M. Robbins, B. Bommarius, A. S. Bommarius, M. Alcalde and F. Hollmann, Tetrahedron, 2019, 75, 1311–1314 CrossRef CAS
- W. Klopffer, Int. J. Life Cycle Assess., 2005, 3, 173–177 Search PubMed
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS
- S. G. Akakios, M. L. Bode and R. A. Sheldon, Green Chem., 2021, 23, 3334–3347 RSC
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC
- Y. D. Y. L. Getzler and R. T. Mathers, Acc. Chem. Res., 2022, 55, 1869–1878 CrossRef CAS PubMed
- O. Coulombier, S. Moins and P. Dubois, Green Mater., 2013, 1, 203–208 CrossRef
- A. L. Holmberg, J. F. Stanzione, R. P. Wool and T. H. A. Epps, ACS Sustainable Chem. Eng., 2014, 2, 569–573 CrossRef CAS
- K. N. Onwukamike, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, ACS Sustainable Chem. Eng., 2019, 7, 1826–1840 CrossRef CAS
- S. Fadlallah, P. Sinha Roy, G. Garnier, K. Saito and F. Allais, Green Chem., 2021, 23, 1495–1535 RSC
- S. G. Koenig and B. Dillon, Curr. Opin. Green Sustainable Chem., 2017, 7, 56–59 CrossRef
- L. M. Tufvesson, P. Tufvesson, J. M. Woodley and P. J. Börjesson, Int. J. Life Cycle Assess., 2013, 18, 431–444 CrossRef CAS
- R. A. Sheldon, CHEMTECH, 1994, 38–47 CAS
- M. Eissen and J. O. Metzger, Chem. – Eur. J., 2002, 8, 3580–3585 CrossRef CAS PubMed
- J. Martinez, J. F. Cortes and R. Miranda, Processes, 2022, 10, 1274 CrossRef
- T. V. T. Phan, C. Gallardo and J. Mane, Green Chem., 2015, 17, 2846–2852 RSC
- F. Roschangar, J. Colberg, P. J. Dunn, F. Gallou, J. D. Hayler and S. G. Koenig,
et al.
, Green Chem., 2017, 19, 281–285 RSC
- F. Roschangar, J. Li, Y. Zhou, W. Aelterman, A. Borovika and J. Colberg,
et al.
, ACS Sustainable Chem. Eng., 2022, 10, 5148–5162 CrossRef CAS
- https://www.europeaninterest.eu/article/parliament-supports-european-green-deal/ accessed 19.09.2022.
- C. H. Christensen, J. Rass-Hansen, C. C. Marsden, E. Taarning and K. Egeblad, ChemSusChem, 2008, 1, 283–289 CrossRef CAS PubMed
- B. Voss, S. I. Andersen, E. Taarning and C. H. Christensen, ChemSusChem, 2009, 2, 1152–1162 CrossRef CAS PubMed
- U. Onken, A. Koettgen, H. Scheidat, P. Schueepp and F. Gallou, Chimia, 2019, 73, 730–736 CrossRef CAS PubMed
- E. Cséfalvay, G. R. Akien, L. Qi and I. T. Horvath, Catal. Today, 2015, 239, 50–55 CrossRef
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed
- J. M. Woodley, Front. Catal., 2022, 2, 883161 CrossRef
- A. R. Alcantara, P. Dominguez de Maria, J. A. Littlechild, M. Schürmann, R. A. Sheldon and R. Wohlgemuth, ChemSusChem, 2022, e202102709 CAS
- A. Bruggink, R. Schoevaart and T. Kieboom, Org. Process Res. Dev., 2003, 7, 622–640 CrossRef CAS
- P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 4049 CrossRef PubMed
-
A. Zaparucha, V. D. de Beradinis and C. Vaxelaire-Vergne, in Modern Biocatalysis: Advances Towards Synthetic Biological Systems, in G. Williams and M. Hall, Royal Society of Chemistry, 2018, ch. 1 Search PubMed
- J. A. Littlechild, Ind. Microbiol. Biotechnol., 2017, 44, 711–720 CrossRef CAS PubMed
- M. Ferrer, F. Martinez-Abarca and P. N. Golyishin, Curr. Opin. Biotechnol., 2005, 16, 588–593 CrossRef CAS PubMed
-
M. T. Reetz, Directed Evolution of Selective Enzymes: Catalysts for Organic Chemistry and Biotechnology, Wiley-VCH, Weinheim, 2016 Search PubMed
- M. S. Morrison, C. J. Podracky and D. R. Liu, Nat. Chem. Biol., 2020, 16, 610–619 CrossRef CAS PubMed
- G. Li, Y. Dong and M. T. Reetz, Adv. Synth. Catal., 2019, 361, 2377–2386 CAS
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed
- A. Illanes, A. Cauerhff, L. Wilson and G. R. Castro, Bioresour. Technol., 2012, 115, 48–57 CrossRef CAS PubMed
-
P. Sutton and J. Whitall, Applied Biocatalysis: the Chemist's EnzymeToolkit, Wiley-VCH, Weinheim, Germany, 2020 Search PubMed
-
Green Biocatalysis, ed. R. N. Patel, Wiley, Hoboken, New Jersey, 2016 Search PubMed
- R. A. Sheldon and D. Brady, ChemSusChem, 2019, 12, 2859–2881 CrossRef CAS PubMed
- B. Hauer, ACS Catal., 2020, 10, 8418–8427 CrossRef CAS
- U. Hanefeld, F. Hollmann and C. E. Paul, Chem. Soc. Rev., 2022, 51, 594–627 RSC
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed
- K. Hecht, H. P. Meyer, R. Wohlgemuth and R. Buller, Catalysts, 2020, 10, 1420 CrossRef CAS
- J. P. Adams, M. J. B. Brown, A. Diaz-Rodriguez, R. C. Lloyd and G.-D. Roiban, Adv. Synth. Catal., 2019, 361, 2421–2432 CAS
- P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem., 2018, 2, 409–421 CrossRef
- F. Romero, B. S. Jones, B. N. Hogg, A. Rué Casamajo, M. A. Hayes and S. L. Flitsch,
et al.
, Angew. Chem., Int. Ed., 2021, 60, 16824–16855 CrossRef PubMed
- G. de Gonzalo, A. R. Alcantara, P. Dominguez de Maria and J. M. Sanchez-Montero, Expert Opin. Drug Discovery, 2022, 10, 1159–1171 CrossRef PubMed
- A. Fryszkowska and P. N. Devine, Curr. Opin. Chem. Biol., 2020, 55, 151–160 CrossRef CAS PubMed
- A. M. Bezborodov and N. A. Zagustina, Appl. Biochem. Microbiol., 2016, 52, 237–249 CrossRef CAS
- N. J. Turner and E. O'Reilly, Nat. Chem. Biol., 2013, 9, 285–288 CrossRef CAS PubMed
-
N. J. Turner and L. Humphreys, Biocatalysis in Organic Synthesis: The Retrosynthesis Approach. Turner, Royal Society of Chemistry, Cambridge, 2018 Search PubMed
- R. O. M. A. de Souza, L. S. M. Miranda and U. T. Bornscheuer, Chem. – Eur. J., 2017, 23, 12040–12063 CrossRef CAS PubMed
- J. R. Marshall, J. Mangas-Sanchez and N. J. Turner, Tetrahedron, 2021, 82, 131926 CrossRef CAS
- R. A. Sheldon and D. Brady, ChemSusChem, 2019, 12, 2–25 CrossRef PubMed
- U. Hanefeld, F. Hollmann and C. E. Paul, Chem. Soc. Rev., 2022, 51, 594–627 RSC
- R. A. Sheldon, M. L. Bode and D. Brady, Chem. Sci., 2020, 11, 2587–2605 RSC
- G.-W. Zheng, Y.-Y. Liu, Q. Chen, L. Huang, L.-H. Yu, W.-Y. Lou, C.-X. Li, Y.-P. Bai, A.-T. Li and J.-H. Xu, ACS Catal., 2017, 7, 7174–7181 CrossRef CAS
- R. Zhang, Y. Xu and R. Xiao, Biotechnol. Adv., 2015, 33, 1671–1684 CrossRef CAS PubMed
- G. W. Huisman, J. Liang and A. Krebber, Curr. Opin. Chem. Biol., 2010, 14, 122–129 CrossRef CAS PubMed
- S. K. Ma, J. Gruber, C. Davis, L. Newman, D. Gray, A. Wang, J. Grate, G. W. Huisman and R. A. Sheldon, Green Chem., 2010, 12, 81–86 RSC
- J. Liang, J. Lalonde, B. Borup, V. Mitchell, E. Mundorff, N. Trinh, D. A. Kochrekar, R. N. Cherat and G. G. Pai, Org. Process Res. Dev., 2010, 14, 193–198 CrossRef CAS
- J. E. Steves, Y. Preger, J. R. Martinelli, C. J. Welch, T. W. Root, J. M. Hawkins and S. S. Stahl, Org. Process Res. Dev., 2015, 19, 1548–1553 CrossRef CAS
- J. Liu, S. Wu and Z. Li, Curr. Opin. Chem. Biol., 2018, 43, 77–86 CrossRef CAS PubMed
- For a recent review see: R. S. Heath and N. J. Turner, Curr. Opin. Green Sustainable Chem., 2022, 38, 100693 CrossRef CAS
- R. S. Heath, W. Birmingham, M. P. Thompson, A. Taglieber, L. Daviet and N. J. Turner, ChemBioChem, 2019, 20, 276–281 CAS
- J. Vilim, T. Kraus and F. Mutti, Angew. Chem., Int. Ed., 2018, 57, 14240–14244 CrossRef CAS PubMed
- W. Zawodny and S. L. Montgmery, Catalysts, 2022, 12, 595 CrossRef CAS
- G. Grogan, Curr. Opin. Chem. Biol., 2018, 43, 15–22 CrossRef CAS PubMed
- A. K. Gilio, T. W. Thorpe, N. Turner and G. Grogan, Chem. Sci., 2022, 13, 4697–4713 RSC
-
B. Mijts, S. Muley, J. Liang, L. M. Newman, X. Zhang, J. Lalonde, M. D. Clay, J. Zhu, J. Gruber, J. Colbeck, J. D. Munger Jr., J. Mavinahalli and R. A. Sheldon, US Pat8574876B2, Codexis, 2013 Search PubMed
- M. J. Abrahamson, J. W. Wong and A. S. Bommarius, Adv. Synth. Catal., 2013, 355, 1780–1786 CrossRef CAS
- S. K. Au, B. R. Bommarius and A. S. Bommarius, ACS Catal., 2014, 4, 4021–4026 CrossRef CAS
- L. J. Ye, H. H. Toh, Y. Yang, J. P. Adams, R. Snajdrova and Z. Li, ACS Catal., 2015, 5, 1119–1122 CrossRef CAS
- T. Knaus, W. Böhmer and F. G. Mutti, Green Chem., 2017, 19, 453–463 RSC
- For a recent review see: J. Liu, W. Kong, J. Bai, Y. Li, L. Dong, L. Zou, Y. Liu, J. Gao, R. T. Bradshaw, N. J. Turner and Y. Jiang, Chem. Catal., 2022, 2, 1–27 CrossRef
- F. Zhou, Y. Xu, Y. Nie and X. Mu, Catalysts, 2022, 12, 380 CrossRef CAS
- O. Mayol, K. Bastard, L. Beloti, A. Frese, J. P. Turkenburg and J.-L. Petit,
et al.
, Nat. Catal., 2019, 2, 324–333 CrossRef CAS
- M. Höhne, Nat. Catal., 2019, 2, 841–842 CrossRef
- A. Bornadel, S. Bisagni, A. Pushpanath, S. L. Montgomery, N. J. Turner and B. A. Dominguez, Org. Process Res. Dev., 2019, 23, 1262–1268 CrossRef CAS
- J. H. Schrittwieser, S. Velikogne and W. Kroutil, Adv. Synth. Catal., 2015, 357, 1655–1685 CrossRef CAS
- J. Mangas-Sanchez, S. P. France, S. L. Montgomery, G. A. Aleku, H. Man, M. Sharma, J. L. Ramsden, G. Grogan and N. J. Turner, Curr. Opin. Chem. Biol., 2017, 37, 19–25 CrossRef CAS PubMed
- G. Grogan and N. J. Turner, Chem. – Eur. J., 2016, 22, 1900–1907 CrossRef CAS PubMed
- S. P. France, M. Howard, J. Steflik, N. J. Weise, J. Mangas-Sanchez, S. L. Montgomery, R. Crook, R. Kumar and N. J. Turner, ChemCatChem, 2018, 10, 510–514 CrossRef CAS
- J. R. Marshall, P. Yao, S. L. Montgomery, J. D. Finnigan, T. W. Thorpe and R. B. Palmer,
et al.
, Nat. Chem., 2021, 13, 140–148 CrossRef CAS PubMed
- M. Lenz, N. Borlinghaus, L. Weinmann and B. L. Nestl, World J. Microbiol. Biotechnol., 2017, 33(11), 1–10 CrossRef CAS PubMed
- M. Lenz, J. Meisner, L. Quertinmont, S. Lutz, J. Kastner and B. M. Nestl, ChemBioChem, 2017, 18, 253–256 CrossRef CAS PubMed
- P. Matzel, M. Gand and M. Hohne, Green Chem., 2017, 19, 385–389 RSC
- G. A. Aleku, S. P. France, H. Man, J. Mangas-Sanchez, S. L. Montgomery and M. Sharma,
et al.
, Nat. Chem., 2017, 9, 961–969 CrossRef CAS PubMed
- C. G. Connor, J. C. Deforest, P. Dietrich, N. M. Do and K. M. Doyle, Org. Process Res. Dev., 2021, 25, 608–615 CrossRef CAS
- F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525–1529 CrossRef CAS PubMed
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig and J. D. Hayler,
et al.
, Green Chem., 2018, 20, 5082–5103 RSC
- M. P. Thompson and N. J. Turner, ChemCatChem, 2017, 23, 3833–3836 CrossRef
- W. Böhmer, T. Kraus and F. Mutti, ChemCatChem, 2018, 10, 731–735 CrossRef PubMed
- R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC
- K. Chen and F. H. Arnold, Nat. Catal., 2020, 3, 203–213 CrossRef CAS
- D. C. Miller, S. V. Athavale and F. H. Arnold, Nat. Synth., 2022, 1, 8–23 CrossRef
- P. S. Coelho, E. M. Brustad, A. Kannan and F. H. Arnold, Science, 2013, 339, 307–310 CrossRef CAS PubMed
- Y. Yang and F. H. Arnold, Acc. Chem. Res., 2021, 54, 1209–1225 CrossRef CAS PubMed
- J. Zhang, A. O. Maggiolo, E. Alfonzo, R. Mao, N. J. Porter, N. Abney and F. H. Arnold, ChemRxiv, preprint, 2022.
- E. Alfonzo, A. Das and F. H. Arnold, Curr. Opin. Green Sustainable Chem., 2022, 38, 100701 CrossRef CAS PubMed
- F. Paradisi, Chimia, 2022, 76, 669–672 CrossRef CAS
- S. W. Snajdrova, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef PubMed
- X. Pei, Z. Luo, L. Qiao, Q. Xiao, P. Zhang, A. Wang and R. A. Sheldon, Chem. Soc. Rev., 2022, 51, 7281–7304 RSC
- K. Engelmark Cassimjee, M. Kadow, Y. Wikmark, M. Svedendahl Humble, M. L. Rothstein, D. M. Rothstein and J.-E. Backvall, Chem. Commun., 2014, 50, 9134 RSC
- For a recent review see: H. Gröger, F. Gallou and B. H. Lipshutz, Chem. Rev. DOI:10.1021/acs.chemrev.2c00416
-
R. A. Sheldon, in Multi-Step Enzyme Catalysis: Biotransformations and Chemoenzymatic Synthesis, ed. E. Garcia-Junceda, Wiley-VCH, Weinheim, 2008, pp. 109–135 Search PubMed
- K. Rosenthal, U. T. Bornscheuer and S. Lütz, Angew. Chem., Int. Ed., 2022, 61, e202208358 CrossRef CAS PubMed
- A. I. Benítez-Mateos, D. Roura Padrosa and F. Paradisi, Nat. Chem., 2022, 14, 489–499 CrossRef PubMed
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed
- D. T. Monterrey, I. Ayuso-Fernández, I. Oroz-Guinea and E. García-Junceda, Biotechnol. Adv., 2022, 60, 108016 CrossRef CAS PubMed
- N. Gaggero and D. C. M. Albanese, Curr. Org. Chem., 2021, 25, 2666–2675 CrossRef CAS
- S. Slagman and W. D. Fessner, Chem. Soc. Rev., 2021, 50, 1968–2009 RSC
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos and K. A. Canada,
et al.
, Science, 2019, 366, 1255–1259 CrossRef CAS PubMed
- J. A. McIntosh, T. Benkovics, S. M. Silverman, M. A. Huffman, J. Kong and P. E. Maligres,
et al.
, ACS Cent. Sci., 2021, 7, 1980–1985 CrossRef CAS PubMed
- L. Leemans Martin, T. Peschke, F. Venturoni and S. Mostarda, Curr. Opin. Green Sustainable Chem., 2020, 25, 100350 CrossRef
- M. P. Thompson, I. Peñafiel, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2019, 23, 9–18 CrossRef CAS
- M. Santi, L. Sancineto, V. Nascimento, J. Braun Azeredo, E. V. M. Orozco, L. H. Andrade, H. Gröger and C. Santi, Int. J. Mol. Sci., 2021, 22, 990 CrossRef CAS PubMed
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed
- B. Gutmann, D. Cantillo and O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed
- F. Paradisi, Chimia, 2022, 76, 669–672 CrossRef CAS
- V. De Vitis, F. Dall'Oglio, F. Tentori, M. L. Contente, D. Romano, E. Brenna, L. Tamborini and F. Molinari, Catalysts, 2019, 9, 208 CrossRef
- P. De Santis, L.-E. Meyer and S. Kara, React. Chem. Eng., 2020, 5, 2155–2184 RSC
- J. Nazor, J. Liu and G. Huisman, Curr. Opin. Biotechnol., 2021, 69, 182–190 CrossRef CAS PubMed
- P. Fernandes and C. C. C. R. de Carvalho, Processes, 2021, 9, 225 CrossRef CAS
- A. P. Mattey, G. J. Ford, J. Citoler, C. Baldwin and J. R. Marshall,
et al.
, Angew. Chem., Int. Ed., 2021, 60, 18660–18665 CrossRef CAS PubMed
- R. Wohlgemuth, New Biotechnol., 2021, 60, 113–123 CrossRef CAS PubMed
- W. Bohmer, T. Knaus, A. Volkov, T. K. Slota, N. R. Shijua, K. E. Cassimjee and F. G. Mutti, J. Biotechnol., 2019, 291, 52–60 CrossRef PubMed
- W. Böhmer, A. Volkov, K. Engelmark Cassimjee and F. G. Mutti, Adv. Synth. Catal., 2020, 362, 1858–1867 CrossRef PubMed
- R. Semproli, G. Vaccaro, E. E. Ferrandi, M. Vanoni, T. Bavaro and G. Marrubini,
et al.
, ChemCatChem, 2020, 12, 1359–1367 CrossRef CAS
- F. Croci, J. Vilím, T. Adamopoulou, V. Tseliou, P. J. Schoenmakers, T. Knaus and F. Mutti, ChemBioChem, 2022, 23, e202200549 CrossRef CAS PubMed
- J. P. Lange, Biofuels, Bioprod. Biorefin., 2007, 1, 39–48 CrossRef CAS
- X. Sun, H. K. Atiyeh, R. L. Huhnke and R. S. Tanner, Bioresour. Technol. Rep., 2019, 7, 100279 CrossRef
- U. Bornscheuer, K. Buchholz and J. Seibel, Angew. Chem., Int. Ed., 2014, 53, 10876–10893 CrossRef CAS PubMed
- L. T. Mika, E. Csefalvay and A. Nemeth, Chem. Rev., 2018, 118, 505–613 CrossRef CAS PubMed
- H. Guo, Y. Chang and D.-J. Lee, Bioresour. Technol., 2018, 252, 198–215 CrossRef CAS PubMed
- R. A. Sheldon, Appl. Microbiol. Biotechnol., 2011, 92, 467–477 CrossRef CAS PubMed
-
R. A. Sheldon, M. J. Sorgedrager and B. Kondor, Non-leachable magnetic cross-linked enzyme aggregate, PCT Int. Appl, CLEA Technologies, WO2012/023847A2, 2012 Search PubMed
- W.-C. Tu and J. P. Hallett, Curr. Opin. Green Sustainable Chem., 2019, 20, 11–17 CrossRef
- E. C. Achinivu, M. Cabrera, A. Umar, M. Yang, N. Raj Baral, C. D. Scown, B. A. Simmons and J. M. Gladden, ACS Sustainable Chem. Eng., 2022, 10, 12090–12098 CrossRef CAS
- P. Y. S. Nakasu, P. V. Barbara, A. E. J. Firth and J. P. Hallett, Trends Chem., 2022, 4, 175–178 CrossRef CAS
- R. A. Sheldon, J. Mol. Catal. A: Chem., 2016, 422, 3–12 CrossRef CAS
- J. A. Posada, A. D. Patel, A. Roes, K. Blok, A. P. C. Faaij and M. K. Patel, Bioresour. Technol., 2013, 135, 490–499 CrossRef CAS PubMed
- Y. Wang, J. Lu, M. Xu, C. Qian, F. Zhao, Y. Luo and H. Wu, ACS Sustainable Chem. Eng., 2022, 10, 13857–13864 CrossRef CAS
- N. Ladygina, E. G. Dedyukhina and M. B. Vainshtein, Proc. Biochem., 2006, 41, 1001–1014 CrossRef CAS
- J. N. Chheda and J. A. Dumesic, Catal. Today, 2007, 123, 59–70 CrossRef CAS
- R. Beerhuis, G. Rothenburg and N. R. Shiju, Green Chem., 2015, 17, 1341–1361 RSC
- N. Guajardo and P. Dominguez de Maria, Molecules, 2021, 26, 736 CrossRef CAS PubMed
- J.-P. Lange, E. van der Heide, J. van Buijtenen and R. Price, ChemSusChem, 2012, 5, 150–166 CrossRef CAS PubMed
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed
- Z. Zhang and K. Deng, ACS Catal., 2015, 5, 6529–6544 CrossRef CAS
- A. D. K. Deshan, L. Atanda, L. Moghaddam, D. W. Rackemann, J. Beltramini and W. O. S. Doherty, Front. Chem., 2020, 8, 659 CrossRef CAS PubMed
- W. P. Dijkman, C. Binda, M. W. Fraaije and A. Mattevi, ACS Catal., 2015, 5, 1833–1839 CrossRef CAS
- S. M. McKenna, P. Mines, P. Law, K. Kovacs-Schreiner, W. R. Birmingham, N. J. Turner, S. Leimkühler and A. J. Carnell, Green Chem., 2017, 19, 4660–4665 RSC
- A. Serrano, E. Calviño, J. Carro, M. I. Sanchez-Ruiz, F. Javier Cañada and A. T. Martínez, Biotechnol. Biofuels, 2019, 12, 217 CrossRef PubMed
- M. M. Cajnko, U. Novak, M. Grilc and B. Likozar, Biotechnol. Biofuels, 2020, 13, 66 CrossRef CAS PubMed
- A. Karich, S. B. Kleeberg, R. Ullrich and M. Hofrichter, Microorganisms, 2018, 6, 5 CrossRef CAS PubMed
- H. Cong, H. Yuan, Z. Tao, H. Bao, Z. Zhang and Y. Jiang,
et al.
, Catalysts, 2021, 11, 1113 CrossRef CAS
- S. P. Simeonov, J. A. S. Coelho and C. A. M. Afonso, ChemSusChem, 2013, 6, 997–1000 CrossRef CAS PubMed
- M. Mascal, ACS Sustainable Chem. Eng., 2019, 7, 5588–5601 CrossRef CAS
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed
- H. Sun, C. Rosenthal and L. D. Schmidt, ChemSusChem, 2012, 5, 1883–1887 CrossRef CAS PubMed
- N. M. Wang, G. Strong, V. DaSilva, L. Gao, R. Huacuja and I. A. Konstantinov,
et al.
, J. Am. Chem. Soc., 2022, 144, 18526–18531 CrossRef CAS PubMed
- See also: R. J. Conk, S. Hanna, J. X. Shi, J. Yang, N. R. Ciccia and L. Qi,
et al.
, Science, 2022, 377, 1561–1566 CrossRef CAS PubMed
- E. F. Lutz, J. Chem. Educ., 1986, 63, 202–203 CrossRef CAS
- R. Wei, T. Tiso, J. Bertling, K. O'Connor, L. M. Blank and U. T. Bornscheuer, Nat. Catal., 2020, 3, 867–871 CrossRef CAS
- See also: C. Jönsson, R. Wei, A. Biundo, J. Landberg, L. Scwarz Bour and F. Pezzotti,
et al.
, ChemSusChem, 2021, 14, 4028–4040 CrossRef PubMed
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, Science, 2016, 351, 1196–1198 CrossRef CAS PubMed
- See also: U. Bornscheuer, Science, 2016, 351, 1154–1155 CrossRef CAS PubMed
- S. Joo, I. J. Cho, H. Seo, H. F. Son, H.-Y. Sagong, T. J. Shin, S. Y. Choi, S. Y. Lee and K.-J. Kim, Nat. Commun., 2018, 9, 382 CrossRef PubMed
- F. Kawai, T. Kawabata and M. Oda, ACS Sustainable Chem. Eng., 2020, 8, 8894–8908 CrossRef CAS
- W. Zimmerman, Philos. Trans. R. Soc., A, 2020, 378, 1–7 CrossRef PubMed
- H. P. Austin, M. D. Allen, B. S. Donohoe, N. A. Rorrer and F. L. Kearns,
et al.
, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4350–E4357 CrossRef CAS PubMed
- R. Wei and W. Zimmerman, Microb. Biotechnol., 2017, 10, 1302–1307 CrossRef CAS PubMed
- R. Wei, D. Breite, C. Song, D. Gräsing and T. Plos,
et al.
, Adv. Sci., 2019, 6, 1900491 CrossRef PubMed
- S. Sulaiman, S. Yamoto, E. Kanaya, J.-J. Kim, Y. Koga, K. Takano and S. Kanaya, Appl. Environ. Microbiol., 2012, 78, 1556–1562 CrossRef CAS PubMed
- V. Tournier, C. M. Topham, A. Gilles, B. David and C. Folgoas,
et al.
, Nature, 2020, 580, 216–219 CrossRef CAS PubMed
- F. Yan, R. Wei, Q. Cui, U. T. Bornscheuer and Y.-J. Liu, Microb. Technol., 2021, 14, 374–385 CAS
- F. Kawai, Catalysts, 2021, 11, 206 CrossRef CAS
- L. DeFrancesco, Nat. Biotechnol., 2020, 38, 665–668 CrossRef CAS PubMed
- H. Lu, D. J. Diaz, N. J. Czarnecki, C. Zhu, W. Kim and R. Shroff,
et al.
, Nature, 2022, 604, 662–667 CrossRef CAS PubMed
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed
- A. Gandini, T. M. Lacerda, A. J. F. Carvalho and E. Trovatti, Chem. Rev., 2016, 116, 1637–1669 CrossRef CAS PubMed
- M. Carus, Ind. Biotechnol., 2017, 13(2), 41–51 CrossRef
- L. Shen, E. Nieuwlaar, E. Worrell and M. K. Patel, Int. J. Life Cycle Assess., 2011, 6, 522–536 CrossRef
- See also: L. Chen, R. E. O. Pelton and T. M. Smith, J. Cleaner Prod., 2016, 317, 667–676 CrossRef
- V. Siracusa and I. Blanco, Polymer, 2020, 12, 1641 CAS
- D. A. Fereirr-Filipe, A. Paco, A. C. Duarte, T. Rocha-Santos and A. L. Patricio-Silva, Int. J. Environ. Res. Public Health, 2021, 18, 7729 CrossRef PubMed
- S. RameshKumar, P. Shaiju, K. E. O'Connor and R. Babu, Curr. Opin. Green Sustainable Chem., 2020, 21, 75–81 CrossRef
- G. Coppolla, M. T. Gaudio, C. G. Lopresto, V. Calabro, S. Curcio and S. Chakraborty, Earth Syst. Environ., 2021, 5, 231–251 CrossRef
- S. V. Mohan, J. A. Modestra, K. Amulya, S. K. Butti and G. Velvizhi, Trends Biotechnol., 2016, 34, 506–519 CrossRef PubMed
- R. Hatti-Kaul, L. J. Nilsson, B. Zhang, N. Rehnberg and S. Lundmark, Trends Biotechnol., 2020, 38, 50–67 CrossRef CAS PubMed
- P. Scarfato, L. DiMaio and L. Incarnato, J. Appl. Polym. Sci., 2015, 132, 42597 CrossRef
- J. Esteban and M. Ladero, Food Sci. Technol., 2018, 53, 1095–1108 CAS
- C. Zhang, P.-L. Show and S.-H. Ho, Bioresour. Technol., 2019, 289, 121700 CrossRef CAS PubMed
- G. Q. Chen and M. K. Patel, Chem. Rev., 2011, 112, 2082–2099 CrossRef PubMed
- X. Wang, S. Gao, J. Wang, S. Xu, H. Li, K. Chen and P. Ouyang, Chin. J. Chem. Eng., 2021, 30, 4–13 CrossRef CAS
- For a recent review see: H. Wang, H. Li, C. K. Lee, N. S. M. Nanyan and G. S. Tay, Polymer, 2022, 14, 5059 CAS
- H. Nakajima, P. Dijkstra and K. Loos, Polymer, 2017, 9, 523 Search PubMed
- M. Volanti, D. Cespi, F. Passarini, E. Neri and F. Cavani,
et al.
, Green Chem., 2019, 21, 885–896 RSC
- D. I. Collias, A. M. Harris, V. Nagpal, I. W. Cottrel and M. W. Schultheis, Ind. Biotechnol., 2014, 10, 91–105 CrossRef CAS
- https://www.avantium.com .
- A. J. J. E. Eerhart, A. P. C. Faaij and M. K. Patel, Energy Environ. Sci., 2012, 5, 6407–6422 RSC
- X. Fei, J. Wang, J. Zhu, X. Wang and X. Liu, ACS Sustainable Chem. Eng., 2020, 8, 8471–8485 CrossRef CAS
- G. Z. Papageorgiou, D. G. Papageorgiou, V. Tsanaktsis and D. N. Bikiaris, Polymer, 2015, 62, 28–38 CrossRef CAS
- E. Korkakaki, M. Mulders, A. Veeken, R. Rozendal, M. C. van Loosdrecht and R. Kleerebezem, Water Res., 2016, 96, 74–83 CrossRef CAS PubMed
- T. Pittman and H. Steinmetz, Bioengineering, 2017, 4, 54 CrossRef PubMed
- J. Tamis, M. Mulders, H. Dijkman and R. Rozendal, J. Environ. Eng., 2018, 144(10), 04018107 CrossRef
- B. Johnston, I. Radecker, D. Hill, E. Chiellini and V. Ivanova Illieva,
et al.
, Polymer, 2018, 10, 957–979 Search PubMed
- https://www.plasticstoday.com/materials/newlight-licenses-aircarbon-ikea/57710583724253 , last accessed 12.12.2022.
- M. K. M. Smith, D. M. Paleri, M. Abdelwahab, D. F. Mielewski, M. Misrah and M. Lumar Mohanti, Green Chem., 2020, 22, 3906–3916 RSC
- L. G. Hong, N. Y. Yuhana and E. Z. E. Zawawi, AIMS Mater. Sci., 2021, 8, 166–184 CAS
- C. Kourmentza, J. Plácido, N. Venetsaneas, A. Burniol-Figols, C. Varrone, H. Gavala and M. Reis, Bioengineering, 2017, 4, 55 CrossRef PubMed
- S. Spierling, E. Knüpffer, H. Behnsen, M. Mudersbach and H. Krieg,
et al.
, J. Cleaner Prod., 2018, 185, 476–491 CrossRef
- J. Wu, K. Zhang, N. Girouard and J. C. Meredith, Biomacromolecules, 2014, 15, 4614–4620 CrossRef CAS PubMed
- C. C. Satam, C. W. Irvin, A. W. Lang, J. C. R. Jallorina and M. L. Shofner,
et al.
, ACS Sustainable Chem. Eng., 2018, 6, 10637–10644 CrossRef CAS
- M. A. R. Maier, Macromol. Rapid. Commun., 2019, 40, 1800524 CrossRef PubMed
- R. K. Donato and A. Mija, Polymer, 2020, 12, 32 CAS
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS PubMed
- J. R. Phillips, R. L. Huhnke and H. K. Atiyeh, Fermentation, 2017, 3, 28 CrossRef
- R. A. J. Verlinden, D. J. Hill, M. A. Kenward, C. D. Williams and I. Radecka, J. Appl. Microbiol., 2007, 102, 1437–1449 CrossRef CAS PubMed
- B. W. Brook, A. Alonso, D. A. Menely, J. Misak, T. Blees and J. B. van Erp, Sustainable Mater. Technol., 2014, 1–2, 8–16 CrossRef
- V. Knapp, M. Matijevic, D. Pevec, B. Cmobrnja and D. Lale, J. Energy Power Eng., 2016, 10, 651–659 CAS
- S. Mallapaty, Nature, 2021, 597, 311–312 CrossRef CAS PubMed
- C. Wulf, P. Zapp and A. Schreiber, Front, Energy Res, 2020, 8, 191 CrossRef
- R. Schlögl, Green Chem., 2021, 23, 1584–1593 RSC
- F. J. De Sisternes, J. D. Jenkins and A. Botterud, Appl. Energy, 2016, 175, 368–379 CrossRef
- Z. Chehade, C. Mansilla, P. Lucchese, S. Hilliard and J. Proost, Int. J. Hydrogen Energy, 2019, 44, 27637–27655 CrossRef CAS
- R. Daiyan, I. MacGill and R. Amal, ACS Energy Lett., 2020, 5, 3843–3847 CrossRef CAS
- A. Bhaskar, M. Assadi and H. Nikpey Somehsaraei, Energies, 2020, 13, 758 CrossRef CAS
- T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Nat. Catal., 2018, 32, 32–39 CrossRef
- J. D. Medrano-Garcia, M. A. Charalambous and G. Guillen-Gosalbez, ACS Sustainable Chem. Eng., 2022, 10, 11751–11759 CrossRef CAS PubMed
- M. E. H. Tijani, H. Zondag and Y. van Delft, ACS Sustainable Chem. Eng., 2022, 10, 16070–16089 CrossRef CAS
- G. A. Olah, A. Goeppert and G. K. S. Prakash, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed
-
G. A. Olah, A. Goeppert and G. K. Surya Prakash, Beyond Oil and Gas: The Methanol Econom, Wiley, 2nd edn, 2009 Search PubMed
- D. R. MacFarlane, J. Choi, B. H. R. Suryanto, R. Jalili, M. Chatti, L. M. Azofra and A. N. Simonov, Adv. Mater., 2020, 32, 1904804 CrossRef CAS PubMed
- H.-L. Du, M. Chatti, R. Y. Hodgetts, P. V. Cherepanov, C. K. Nguyen, K. Matuszek, D. R. MacFarlane and A. N. Simonov, Nature, 2022, 609, 722–727 CrossRef CAS PubMed
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358–3375 RSC
- M. C. Leech and K. Lam, Nat. Rev. Chem., 2022, 6, 275–286 CrossRef
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC
- M. C. Leech, A. D. Garcia, A. Petti, A. P. Dobbs and K. Lam, React. Chem. Eng., 2020, 5, 977–990 RSC
- S. B. Beil, D. Pollok and S. R. Waldvogel, Angew. Chem., Int. Ed., 2021, 60, 14750–14759 CrossRef CAS PubMed
- D. Pletcher, Electrochem. Commun., 2018, 88, 1–4 CrossRef CAS
- A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406–410 CrossRef CAS PubMed
- F. Harnisch and C. Urban, Angew. Chem., Int. Ed., 2018, 57, 10016–10023 CrossRef CAS PubMed
- D. Massazza, A. J. Robledo, C. N. R. Simon, S. J. Busalmen and S. Bonanni, Bioresour. Technol., 2021, 342, 125893 CrossRef CAS PubMed
- H. Chen, F. Dong and S. D. Minteer, Nat. Catal., 2020, 3, 225–244 CrossRef
- V. M. Dembitsky, Tetrahedron, 2003, 26, 4701–4720 CrossRef
- C. Kohlmann and S. Lütz, Eng. Life Sci., 2006, 6, 170–174 CrossRef CAS
- B. Cheng, L. Wan and F. A. Armstrong, ChemElectroChem, 2020, 7, 4672–4678 CrossRef CAS PubMed
- N. Carrillo and E. A. Ceccarelli, Eur. J. Biochem., 2003, 270, 1900–1915 CrossRef CAS PubMed
- C. F. Megarity, B. Siritanaratkul, R. S. Heath, L. Wan, G. Morello and S. R. Fitzpatrick,
et al.
, Angew. Chem., Int. Ed., 2019, 58, 4948–4952 CrossRef CAS PubMed
- G. Morello, C. F. Megarity and F. A. Armstrong, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed
- L. Wan, R. S. Heath, C. F. Megarity, A. J. Sills, R. A. Herold, N. J. Turner and F. A. Armstrong, ACS Catal., 2021, 11, 6526–6533 CrossRef CAS
- I. Penafiel, R. A. W. Dryfe, N. J. Turner and M. F. Greany, ChemCatChem, 2021, 13, 864–867 CrossRef CAS
- R. A. Sheldon, ChemSusChem, 2022, 15(9), e202102628 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2023 |