
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous flow synthesis of 1,4-disubstituted 1,2,3-triazoles via consecutive β-azidation of α,β-unsaturated carbonyl compounds and CuAAC reactions†
Giulia
Brufani‡
a,
Federica
Valentini‡
a,
Gabriele
Rossini
a,
Luigi
Carpisassi
a,
Daniela
Lanari
*b and
Luigi
Vaccaro
 *a
*a
aLaboratory of Green S.O.C. – Dipartimento di Chimica, biologia e Biotecnologie, Università degli Studi di Perugia, Via Elce di Sotto 8, 06123 Perugia, Italy. E-mail: luigi.vaccaro@unipg.it; Web: https://greensoc.chm.unipg.it
bDipartimento di Scienze Farmaceutiche, Università degli Studi di Perugia, Via del Liceo 1, 06123 Perugia, Italy
First published on 27th February 2023
Abstract
We herein report a multi-step flow protocol for the synthesis of 1,4-disubstituted 1,2,3-triazoles starting from α,β-unsaturated carbonyls. Best results, both in terms of chemical productivity and sustainability, were obtained using a sequential combination of two flow reactors: one packed with a tailor-made POLITAG-F organocatalytic system and a copper tube apparatus. Moreover, the use of aqueous acetonitrile azeotrope led to excellent catalytic performances in both the developed processes while affording a waste-minimized multi-step protocol. Indeed, the azide intermediates subsequently reacted without the need for an additional purification step, and the aqueous acetonitrile azeotrope could be largely recovered/reused at the end of the process. 1,4-disubstituted β-keto 1,2,3- triazoles were obtained with high yields, which can be associated with low E-factor values. Finally, the green metrics evaluation of the process is also given to quantify the advantages associated with the use of our flow strategy.
Introduction
Modern synthetic chemistry aims to meet the urgent need for a shift toward more sustainable industrial production by designing efficient and benign chemical processes.1 In this context, different principles of green chemistry are fulfilled by click chemistry. This approach was introduced in 2001 by Sharpless as a novel organic synthesis strategy to obtain complex molecules by employing simple reaction conditions that produce innocuous by-products.2 Among click chemistry reactions, Copper-Catalyzed Azide–Alkyne Cycloaddition (CuAAC), which was developed independently by the groups of Sharpless3 and Meldal4 in 2002, is the simplest method for generating architecturally complex functional organic molecules.The widespread utilization of the CuAAC strategy in different fields for the synthesis of valuable molecules has been acknowledged by the Nobel prize in Chemistry 2022.5
The Cu(I)-catalyzed cycloaddition of organic azides and alkynes leads to the formation of 1,2,3-triazoles with exclusive selectivity to the 1,4-disubstituted isomer. This class of compounds has gained increasing interest as they show anti-HIV, antibiotic, antiviral and antibacterial activities.6
Although the CuAAC reaction is a benign synthetic approach, different strategies have been developed to improve the sustainability of the protocol by further reducing the waste production.7
Particular attention has been paid to the recovery and reuse of both the catalytic system and reaction media,7a,b as well as the implementation of continuous flow techniques to improve large-scale production.8
In the context of heterogeneous copper catalysis under continuous flow conditions, using copper tubes (Cu-tubes), which are generally referred to as copper tube flow reactors (CTFRs), is a solid option. They are commercially available and can easily be incorporated into a flow line while allowing rapid heating and cooling owing to their highly increased thermal conductivity.9 This kind of reactor has been widely used in different transformations.9,10 Thanks to the above-mentioned advantages, different groups have exploited Cu-Tubes in the CuAAC reaction for the assembly of high-value products, such as macrocycles, peptoids and API rufinamide, with high yields.11
Despite the ready availability of alkynes, most organic azides are not commercially available and difficult to manipulate due to their explosive character and instability. Moreover, organic azides are usually obtained using a combination of sodium azide and strong acid as hydrazoic sources, which render unsuitable conditions for the safety of the process.12 For this purpose, different approaches have been developed for the synthesis of 1,2,3-triazoles, such as the utilization of multicomponent reactions13 and multi-step flow protocols.11c,14
Flow chemistry has become a key tool to enhance the selectivity, safety, and efficiency of synthetic chemical processes.15 Indeed, the shift from batch to flow ensures easy scale-up16 and safer handling of hazardous reagents and intermediates.17 Moreover, the flow technology allows better control of reaction parameters, even for gas phase reagents,18 by providing a high area-to-volume ratio, which increases thermal contact and mass transfer.19 The automation of the flow process is also responsible for enhanced reproducibility.20 Other advantages are the facilitated work-up procedure along with a decrease in environmental footprint.21
Herein, we report a waste-minimized multi-step flow protocol for the synthesis of 1,4-disubstituted β-keto triazoles from enones. The two-step protocol consists of the azido-Michael reaction of α,β-unsaturated carbonyl compounds to yield the corresponding β-azidocarbonyl products, which are readily converted to β-keto triazoles via the CuAAC reaction.12b,c
Trimethylsilyl azide (TMSN3) has been selected as a safer N3− source in comparison with hydrazoic acid and sodium azide.9,22 TMSN3 is widely applied in the synthesis of organic azides in combination with metal-based catalysts23 and under metal-free conditions,24 and in a catalyst-free protocol for the azidation of perfluoroalkyl α,β-unsaturated ketones.25
However, some procedures report the use of TMSN3 under acidic conditions to generate HN3in situ.24d–f Due to the high toxicity and explosive character of hydrazoic acid, it is necessary to minimize its formation. In this context, it has been demonstrated that the combination of a fluoride-based catalyst with TMSN3 shows a safer profile compared with both these procedures and the use of other azido sources, such as sodium azide.22a Although a holistic evaluation has revealed that the preparation of TMSN3 from trimethylsilyl chloride and sodium azide12a is disadvantageous, and the presence of the trimethylsilyl group certainly affects the Atom Economy (AE) of the process and the mass of byproduct formed (trimethylsilanol), in comparison with NaN3, the use of additives can be avoided when TMSN3 is used as the azido source. Moreover, considering the risks associated with the use of TMSN3 and the lack of data regarding its hazardous attributes, such as explosive power and occupational exposure limits, the global environmental impact and safety profile are lower when TMSN3 is used.22a
POLITAG-F is an organocatalytic system,26 wherein F refers to the fluoride counter anion of the ionic resin, has been chosen as the catalyst for the first step as fluoride is known to effectively interact with silicon in TMSN3, activating the Si–N bond and thereby the nucleophilic ability of the azido ion.27
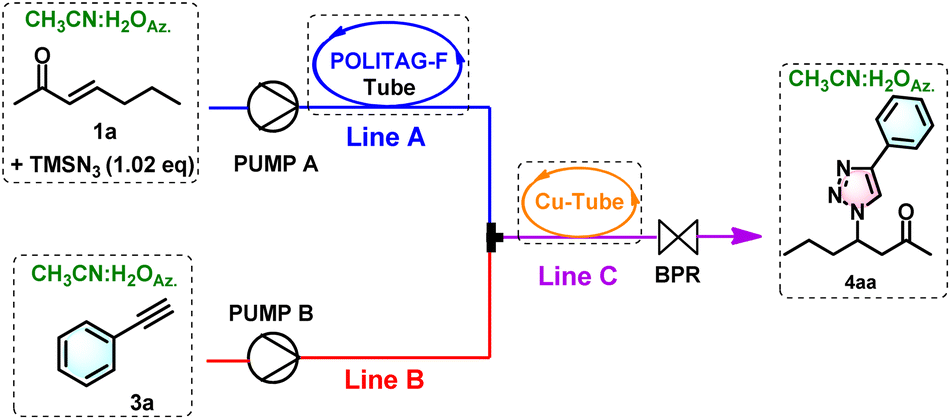
Therefore, in our strategy, α,β-unsaturated carbonyl compounds and TMSN3 are flowed through the first POLITAG-F-packed reactor to yield the corresponding azides, which without purification undergo the CuAAC reaction in the flow when mixed with an alkyne substrate in the Cu-tube. While this strategy can effectively pave the way to a waste-minimized green route for the synthesis of the target 1,4-disubstituted β-keto 1,2,3-triazoles, it requires the delicate tuning of the two separate reactors and flows to achieve the highest chemical efficiency.
For the minimization of waste, in this process, we have focused on the use of an aqueous acetonitrile azeotropic mixture as the medium as it brings the benefits of both aqueous and organic media, while it can also be efficiently recovered and reused.28
A final comparison of the different green metrics (Atom Economy (AE), Mass Recovery Parameter (MRP), Reaction Mass Efficiency (RME), Stochiometric Factor (1/SF) and E-factor) highlights the advantages of the flow protocol developed herein, compared to batch, as it features a low environmental footprint for the telescoped synthesis of 1,4-disubstituted β-keto 1,2,3-triazoles with minimal metal contamination.
Results and discussion
POLITAG-F has previously shown superior results in the cyanosilylation of aldehydes compared with commercially available F-based catalysts of both homogeneous and heterogeneous nature.26 In addition, the “SPACER”-type polymeric support has shown enhanced stability and better recyclability in comparison with commercially available chloromethylated resins.26,28b For these reasons, we began our investigation with the polymer-supported bis-imidazolium pincer ligand, POLITAG, which contained fluoride as the counterion26 and SPACER as the cross-linker of the polymeric support29 (Fig. 1).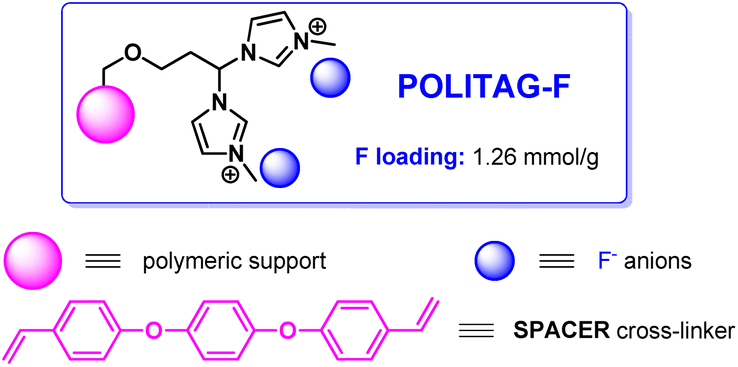 | ||
| Fig. 1 Representation of POLITAG-F organocatalyst. | ||
The loading of the fluoride organocatalyst (1.26 mmol g−1), as per the previously reported procedure,26 was calculated by elemental analysis based on the number of bis-imidazolium units (see ESI for further details†).
The consecutive batch protocol
A preliminary optimization for the definition of our synthetic strategy, including the study of the consecutive protocol, was performed under batch conditions.Initially, the POLITAG-F catalyst was tested in the β-azidation reaction of (E)-3-hepten-2-one (1a) in different reaction media (Table 1).
|
|
|||
|---|---|---|---|
| Entry | Reaction medium | T (h) | Conv.b (%) |
a Reaction conditions: 1a (1 mmol), TMSN3 (1.05 eq.), POLITAG-F (5 mol%), reaction medium (0.5 mL), 60 °C.
b Conversion determined by GC analysis, the remaining material was unreacted 1a.
c CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O 83.7: 16.3% w/w.
d Reaction medium (0.2 mL, 5M).
e Reaction medium (0.1 mL, 10 M). :H2O 83.7: 16.3% w/w.
d Reaction medium (0.2 mL, 5M).
e Reaction medium (0.1 mL, 10 M).
|
|||
| 1 | SolFC | 8 | 35 |
| 2 | CH2Cl2 | 2.5 | 48 |
| 3 | CH3CN | 2.5 | 53 |
| 4 | CH3CN:H2OAz.c |
2.5 | 86 |
| 5d | CH3CN:H2OAz.c |
3 | >99 |
| 6e | CH3CN:H2OAz.c |
3 | >99 |
| 7 | CPME | 2.5 | 32 |
| 8 | TAME | 7 | 23 |
| 9 | 2-MeTHF | 2.5 | 56 |
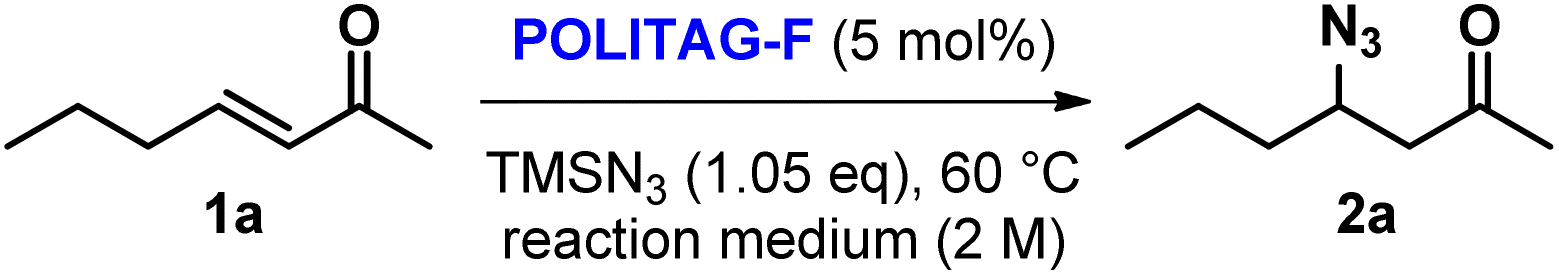
With the intention to develop a waste-minimized process, we started by performing the reaction under solvent-free conditions (SolFC). However, in these conditions, the conversion to product 2a was poor even after 8 h reaction time (entry 1, Table 1).
By employing cyclopentylmethylether (CPME) and tert-amyl methyl ether (TAME) as reaction media, only small amounts of the desired product could be detected (entries 7 and 8, Table 1). On the contrary, when dichloromethane, acetonitrile and 2-methyl tetrahydrofuran (2-MeTHF) were used, more efficient conversion was observed in a short time (2.5 h).
The use of an aqueous acetonitrile azeotropic mixture gave the best results in terms of conversion and yield (entry 4, Table 1). Indeed, in comparison with the reaction performed in acetonitrile, the process in aqueous acetonitrile azeotrope presented enhanced catalytic performance.
When the reaction medium amount was reduced to increase the mixture concentration from 2 M to 5 M, a complete conversion of 1a was detected in 3 h. Comparable results were also obtained at 10 M concentration. However, under batch conditions, for better mixing of the reaction mixture in the presence of the heterogeneous catalytic system, we selected 5 M concentration as the best to balance efficiency and waste minimization.
With these optimized conditions, we proceeded with the study of the consecutive CuAAC reaction by reacting intermediate 2a with phenylacetylene 3a in aqueous acetonitrile azeotrope for testing the possibility of avoiding the intermediate purification step.
After the completion of the first β-azidation step, POLITAG-F was recovered by filtration and washed with aqueous acetonitrile azeotrope. To the resulting mixture, copper rods (10 mol%) and 3a (1 eq.) were added, and the reaction mixture was heated at 60 °C for 24 h.
With the use of a metallic copper catalyst, we could avoid the utilization of additional additives that are generally needed in homogeneous catalytic conditions. Indeed, the CuAAC reaction performed using homogenous Cu(II) salts required the use of reductive agents to generate the catalytically active species Cu(I).
By adding specific amounts of the azeotropic mixture when washing the first mixture after the first azidation step, different concentrations were screened to select the best condition for the consecutive process (Table 2).
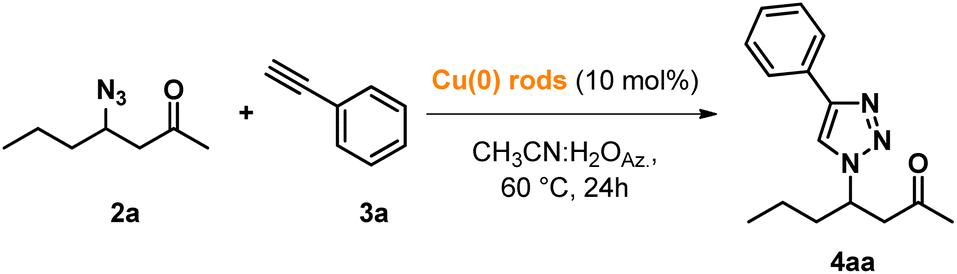
The reaction showed quantitative conversion in the concentration range from 4M to 1M, while a drop in the formation of 1,4-disubstituted β-keto 1,2,3-triazole 4aa was observed at 0.5M concentration.
At the end of the process, the metallic copper rods were filtered and washed with 2 mL of the aqueous azeotrope. The overall reaction medium was then recovered by distillation (89%), and the obtained crude product was washed with additional cold azeotrope (0.5 mL) to afford pure product 4aa. Even at this stage, it could be noticed that the protocol is promising for overall waste minimization.
Depending on the concentration used, we observed important differences in the isolated yields. Indeed, partial precipitation of the product in the reaction mixture at high concentrations (entries 1 and 2, Table 2) led to lower yields in comparison with the reaction performed using 1 mL mmol−1 (1M) azeotropic mixture (entry 3, Table 2), which led to the isolation of product 4aa with an excellent overall yield of 94%.
The multi-step protocol in flow
At this stage, we focused our attention on using these preliminary data obtained in the batch protocol for the definition of a multi-step continuous-flow protocol for the synthesis of β-keto 1,2,3-triazoles starting from enones, without the isolation and manipulation of the azide intermediates.The first step was optimized by packing a PTFE coil reactor with the heterogenous organocatalyst POLITAG-F blended with quartz powder to guarantee a homogenous flow and pressure in the reactor. Several parameters were varied to achieve the complete conversion of 1a to the desired azide 2a before proceeding to the optimization of the consecutive CuAAC process.
The adoption of a 10-atm back pressure regulator (BPR) was crucial. Indeed, in the process performed with a BPR at 6 atm, only a trace amount of product 2a was detected (entry 1, Table 3).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Length (m) | POLITAG-F (g) | Conc. (M) | Flow rate (mL min−1) | T (°C) | Conv.b (%) |
| a Reaction conditions: 1a (5 mmol), TMSN3 (1.05 eq.), reactor diameter (4.5 mm), BPR (10 atm). b Conversion determined by GC analysis, the remaining material was unreacted 1a. c BPR 6 atm. d Reactor inner diameter: 1/8′′. e Reaction performed with TMSN3 (1.02 eq.). | ||||||
| 1c | 0.25 | 0.4 | 10 | 0.25 | 60 | Traces |
| 2 | 0.25 | 0.4 | 10 | 0.25 | 60 | 12 |
| 3 | 0.25 | 0.4 | 10 | 0.05 | 60 | 20 |
| 4 | 0.25 | 0.8 | 5 | 0.05 | 60 | 22 |
| 5 | 0.25 | 0.8 | 5 | 0.10 | 60 | 28 |
| 6 | 0.50 | 1.6 | 5 | 0.10 | 60 | 38 |
| 7 | 0.50 | 1.6 | 10 | 0.10 | 60 | 30 |
| 8 | 0.50 | 1.6 | 10 | 0.10 | 75 | 26 |
| 9 | 0.50 | 1.6 | 10 | 0.20 | 60 | 10 |
| 10d | 1.60 | 1.0 | 1 | 0.10 | 60 | 35 |
| 11d | 1.60 | 1.0 | 5 | 0.10 | 60 | 63 |
| 12d | 3.20 | 2.0 | 5 | 0.10 | 60 | >99 |
| 13d,e | 3.20 | 2.0 | 5 | 0.10 | 60 | >99 |
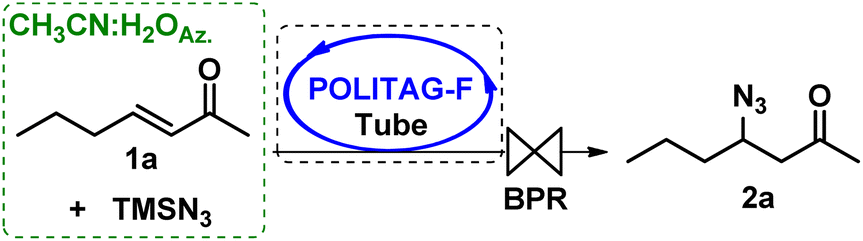
A positive effect was observed by setting the flow rate at 0.1 mL min−1 and the aqueous acetonitrile azeotrope concentration at 5M. Conversion to product 2a gradually increased depending on the reactor length (entries 5 and 6, Table 3) rather than on the catalyst amount (entries 6 and 11, Table 3). Increasing the temperature to 75 °C showed a negligible effect on the process (entry 8, Table 3).
Complete conversion of 1a was obtained by using a PTFE tube reactor with a length of 3.2 m and an internal diameter of 1/8′′ packed with 2.0 g of the POLITAG-F organocatalyst.
After this preliminary set-up of the first part of the flow apparatus, we connected the second reactor for the optimization of the consecutive CuAAC process. Notably, the employment of a consecutive flow protocol required further optimization to combine the two single processes (see Table ESI-1 in ESI†). We selected 1a ketone and phenylacetylene 3a as model substrates for the optimization of the continuous-flow synthesis of 1,4-disubstituted 1,2,3-triazoles.
The metallic copper heterogeneous catalyst used under batch condition was replaced with a Cu tube, which is commercially available and largely used for setting up gas lines and other general purposes, with an internal diameter of 1/32′′ and a length of 10 m, providing a volume of 2 mL.
The two different lines equipped with two HPLC pumps were connected using a T joint, and the resultant mixture flowed through the Cu tube. The reactors were thermostated in two different isolated boxes: the PTFE-packed reactor with POLITAG-F at 60 °C and the Cu tube at 100 °C. In this modified configuration, the BPR was placed at the end of the Cu-tube.
Due to the pressure generated in the second copper tube reactor, we started the optimization using a single BPR at 6 atm (Table ESI-1†). To allow the efficacious combination and mixing of the two reaction mixtures, the flow rate of line A was slowed down to 0.05 mL min−1, and the line B flow was set at 0.1 mL min−1 (entry 1, Table ESI-1†). However, these flow rates were insufficient for the mixture to properly flow through the reactors. Even after increasing these values to 0.25 mL min−1, unsuccessful results were obtained (entries 2 and 3, Table ESI-1†). Optimal conditions were obtained by setting both lines at 0.1 mL min−1 (entry 4, Table ESI-1†). However, due to the poor yield of 4aa even after varying the concentration and an excess of 3a (entries 5 and 6, Table ESI-1†), different conditions were evidently needed.
Particularly, unreacted TMSN3 and excess 3a led to the rapid deactivation of the copper tube. This phenomenon was clearly visible as a greenish solution was recovered at the end of these optimization experiments. Anyway, we confirmed that the Cu-tube could be regenerated8f by using 5 mL of H2O2. To minimize this deactivation effect, TMSN3 was reduced to 1.02 eq., which in combination with the optimal flow rates used, achieved very high efficiency in the first azidation process (entry 13, Table 3).
However, for the consecutive process, the 6 atm BPR was inadequate to achieve a satisfactory result (entry 1, Table 4). Indeed, only 34% of product 4aa was detected at the flow outlet; the remaining material was a 2:1 mixture of unreacted ketone 1a and the azide intermediate 2a.
|
|
|||||
|---|---|---|---|---|---|
| Entry | BPR | Conc. Ab (M) | Conc. Bc (M) | Conc. Cb (M) |
4aad (%) |
|
a Line A: 1a (5 mmol), TMSN3 (1.02 eq.), CH3CN:H2OAz.; line B: 3a (1.2 eq.) and CH3CN:H2OAz.; flow rate 0.1 mL min−1; 1st reactor (POLITAG-F, loading: 1.26 mmol g−1) thermostated at 60 °C; Cu-tube thermostated at 100 °C.
b Referred to 1a.
c Referred to 3a.
d Determined by GC analysis.
e The remaining material was a 2:1 mixture of 1a and 2a.
f The remaining material was a 1:1 mixture of 1a and 2a.
g The remaining material was unreacted 2a.
|
|||||
| 1 | 6 atm | 5 | 1.2 | 0.83 | 34e |
| 2 | 10 atm | 1 | 1.2 | 0.5 | 42f |
| 3 | 10 atm | 5 | 1.2 | 0.83 | >99 |
| 4 | 10 atm | 5 | 6 | 2.5 | 57g |
At this stage, a change of the first reactor was needed, and different polymer-supported fluoride-based catalysts were used to achieve the best performance (entries 7–9, Table ESI-1†). Although high catalyst loading showed beneficial effects in the single process,24c in the consecutive azidation-CuAAC protocol, poor results were obtained. For this reason, we proceeded with further optimization of the POLITAG-F catalyst (Table 4).
Since under batch conditions, both processes were sensitive to the mixture concentration in aqueous acetonitrile azeotrope, we investigated this effect in the combined consecutive flow protocol using a BPR at 10 atm (entries 2–4, Table 4).
Performing the reaction with both reactants at low concentrations led to only 42% product 4aa in the final mixture (entry 2, Table 4). Further, when the concentration was increased in both lines to achieve a final concentration of 2.5M in line C, the high-density mixture also displayed a lower conversion of azide 2a to the desired product, affording a low yield of 4aa (entry 4, Table 4).
The use of 5M concentration in the aqueous acetonitrile azeotrope in line A and 1.2M in line B boosted the quantitative conversion of (E)-3-hepten-2-one to the desired triazole 4aa (entry 3, Table 4). Furthermore, we also attempted to reduce the Cu-tube length from 10 m to 5 m (entries 10 and 11, Table ESI-1†), which also led to poor results. Indeed, even when the BPR was increased to 17 atm, the product yield did not exceed 70% (entry 11, Table ESI-1†).
The optimized consecutive protocol (entry 3, Table 4) afforded 4aa at 1.5 mmol h−1. The reaction time in flow was also drastically reduced compared with the small-scale batch protocol. Remarkably, under continuous flow conditions, it is possible to keep the heterogeneous catalyst POLITAG-F safely in the reactor and reuse it efficiently, as proven by our test. On the contrary, its recovery under batch conditions proved to be more complicated due to the crushing of the material to a finely dispersed powder under the classic stirring conditions.
Once the pumping of the reagents was stopped, and the reaction mixture was completely eluted, the lines and reactors were washed with 15 mL of the CH3CN:H2O azeotropic mixture, which could mostly be recovered (94%) by distillation and reused.
After the removal and recovery of the azeotropic mixture, the desired product 4aa was collected as a pure solid product (97% yield) without further purification. Furthermore, copper leaching reduced from 87.7 ppm in the batch process to 2.4 ppm under flow conditions (after 20 mmol conversion), thus avoiding the additional purification of the product. The product isolated from the flow protocol featured minimal copper contamination (41.1 μg g−1), which is below the limits permitted in drug production.30 To give more insights into the long-term stability of the consecutive flow protocol, we further stressed the system by pumping the reagent mixtures for a longer time (see Figure ESI-1 in ESI†). After the steady state was reached, 60 mmol of α,β-unsaturated ketones (1) were efficiently converted to the desired triazoles. After 40 h, copper leaching increased to 19 ppm accompanied by a drop in the conversion of the azides 2 to 45%, and then, a further increase in leaching up to 45 ppm was measured. At this point, the Cu-tube was treated with H2O2, and the initial efficiency along with the low Cu leaching (3.0 ppm) could be regenerated. This approach was preventively employed after every 60 mmol reactant was processed to keep the efficiency of the system constant (Figure ESI-1†).
These optimized conditions were then extended to the consecutive β-azidation of α,β-unsaturated carbonyls (1a–b) followed by the CuAAC reaction with different terminal alkynes (3b–j). The substrates tested showed good isolated yields in the range of 83% to 99% (Scheme 1).
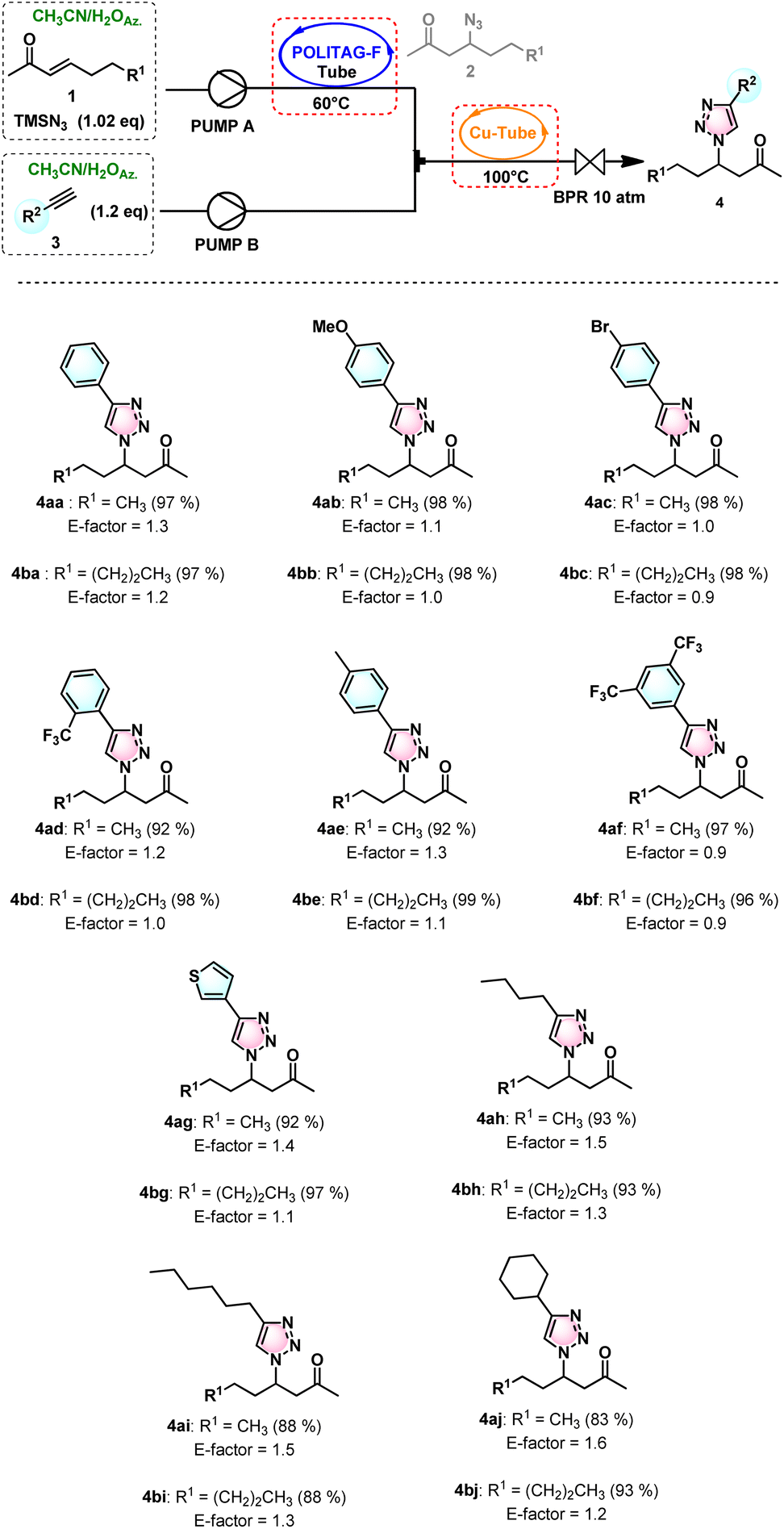 | ||
| Scheme 1 The 1,4-disubstituted β-keto-1,2,3-triazoles synthesized from β-azido-ketones and functionalized alkynes in continuous flow. The isolated yields are given in parentheses. | ||
Up to 185 mmol α,β-unsaturated carbonyl compounds were efficiently converted to various functionalized 1,4-disubstituted 1,2,3-triazoles with the optimized flow apparatus.
The high efficiency of this consecutive flow protocol and the recovery of the reaction medium led to the fast and waste-minimized synthesis of triazoles associated with very low E-factor values (0.9–1.6) (see ESI for further details†). These values are extremely low compared with those of other previously developed waste-minimized CuAAC protocols.7b,c
To better comprehend the main factors that led to the reduced generation of waste, we performed a green metrics assessment by evaluating AE, RME, MRP, 1/SF and the E-factor distribution profile (see Fig. 1 and ESI for further details†).31
The advantages of the flow protocol not only include the reduction of reaction time but also the easy manipulation of the reaction mixture, which allows the avoidance of any additional steps and materials necessary to separate the heterogeneous catalyst from the reaction mixture. Moreover, the flow protocol led to an enhancement of the sustainability of the process, as visualized and quantified by the comparison of the radial polygons in Fig. 2. When the reaction is scaled up from batch to flow, the red line (right panel) fits the ideal situation better (green line). MRP and RME are doubled in the flow protocol (see ESI for further details†) mainly due to the high recovery of the selected azeotropic mixture.
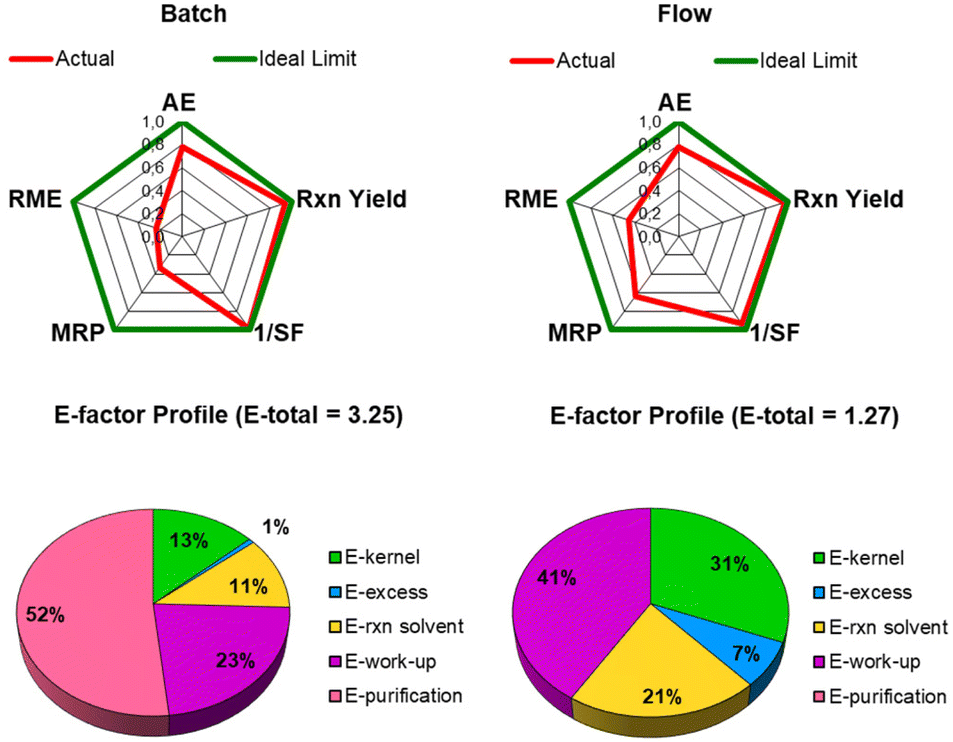 | ||
| Fig. 2 A comparison of the green metrics (AE: Atom economy; MRP: Mass recovery parameter, RME: Reaction mass efficiency, SF: Stochiometric factor; E-factor: Environmental factor) between the batch and consecutive-flow β-azidation/CuAAC reaction protocols. | ||
This improvement is also reflected in the E-factor value, which was reduced by 60% when the protocol was switched from batch to flow (Fig. 2).
The main contribution that influenced the E-factor of the batch protocol was the final purification step, which contributed 52% to the total waste generation. Thanks to the high efficiency of the flow protocol and low Cu contamination, this step is avoided.
Moreover, the kernel contribution to the total E-factor values increased up to 31% in the flow protocol compared with 13% in the batch process (Fig. 2). Indeed, with all the auxiliary substances in the kernel E-factor excluded from the calculation,31 the enhanced E-factor profile further confirms the waste minimization of the optimized consecutive protocol.
Conclusions
With the consecutive use of POLITAG-F as a heterogeneous organocatalyst for the β-azidation of α,β-unsaturated carbonyls and the use of Cu-tube as a heterogenous metallic copper catalyst for the CuAAC reaction, we have defined a very efficient protocol for the preparation of 1,4-disubstituted β-keto 1,2,3-triazoles. After a preliminary optimization of the batch protocol, we developed a consecutive flow protocol performed in a CH3CN/H2O azeotropic mixture, featuring significant waste reduction.The developed two-step consecutive protocol in flow allows for a drastic reduction in reaction time compared with the batch mode, while also allowing safe and complete recovery and reuse of the catalytic system, and affording a productivity of 1.5 mmol h−1.
The high efficiency of the telescoped flow synthesis method avoids purification steps while maintaining the copper contamination level below the permitted limit for pharmaceutical compounds. This is reflected in the minimization of the environmental footprint in comparison with the batch process, as demonstrated by the different green metrics (AE, RME, MRP, 1/SF, and E-factor).
Starting from different α,β-unsaturated carbonyls, high isolated yields of various 1,4-disubstituted 1,2,3-triazoles were obtained, resulting in very low E-factor values (0.9–1.6).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Università degli Studi di Perugia and MIUR are acknowledged for financial support to the project AMIS, through the program “Dipartimenti di Eccellenza – 2018–2022”. G.B. and L.V. wish also to thank INPS and Sterling SpA for the PhD grant and training offered to G.B. This work has been funded by the European Union – NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 – VITALITY. We acknowledge Università degli Studi di Perugia and MUR for support within the project Vitality.References
- (a) G. Guillén-Gosálbez, A. González-Garay, P. Limleamthong, Á. Galán-Martín and C. Pozo, Systematic Multi Objective Life Cycle Optimization Tools Applied to the Design of Sustainable Chemical Processes, in Sustainable Nanoscale Engineering, Elsevier, 2020, pp. 435–449 Search PubMed; (b) L. Vaccaro, Eur. J. Org. Chem., 2020, 4273–4283 CrossRef CAS; (c) F. Ferlin, D. Lanari and L. Vaccaro, Green Chem., 2020, 22, 5937–5955 RSC; (d) P. Lanzafame, S. Perathoner, G. Centi, S. Gross and E. J. M. Hensen, Catal. Sci. Technol., 2017, 7, 5182–5194 RSC; (e) J. F. Jenck, F. Agterberg and M. J. Droescher, Green Chem., 2004, 6, 544–556 RSC; (f) P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed; (g) L. Vaccaro, M. Curini, F. Ferlin, D. Lanari, A. Marrocchi, O. Piermatti and V. Trombettoni, Pure Appl. Chem., 2017, 90, 21–23 CrossRef.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., 2001, 113, 2056–2075 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- https://www.nobelprize.org/prizes/chemistry/2022/summary/ .
- (a) A. Rani, G. Singh, A. Singh, U. Maqbool, G. Kaur and J. Singh, RSC Adv., 2020, 10, 5610–5635 RSC; (b) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed.
- (a) K. S. Rodygin, D. E. Samoylenko, M. M. Seitkalieva, K. A. Lotsman, S. A. Metlyaeva and V. P. Ananikov, Green Chem., 2022, 24, 1132–1140 RSC; (b) L. Luciani, E. Goff, D. Lanari, S. Santoro and L. Vaccaro, Green Chem., 2018, 20, 183–187 RSC; (c) D. Rasina, A. Lombi, S. Santoro, F. Ferlin and L. Vaccaro, Green Chem., 2016, 18, 6380–6386 RSC.
- (a) D. S. de Sá, R. de Andrade Bustamante, C. E. Rodrigues Rocha, V. D. da Silva, E. J. da Rocha Rodrigues, C. Djenne Buarque Müller, K. Ghavami, A. Massi and O. Ginoble Pandoli, ACS Sustainable Chem. Eng., 2019, 7, 3267–3273 CrossRef; (b) S. B. Ötvösa and F. Ferenc, Catal. Sci. Technol., 2015, 5, 4926–4941 RSC; (c) A. C. Varas, T. Noël, Q. Wang and V. Hessel, ChemSusChem, 2012, 5, 1703–1707 CrossRef CAS PubMed; (d) L. Kupracz, J. Hartwig, J. Wegner, S. Ceylan and A. Kirschning, Beilstein J. Org. Chem., 2011, 7, 1441–1448 CrossRef CAS PubMed; (e) S. Ceylan, T. Klande, C. Vogt, C. Friese and A. Kirschning, Synlett, 2010, 2009–2013 CAS; (f) M. Fuchs, W. Goessler, C. Pilger and C. O. Kappe, Adv. Synth. Catal., 2010, 352, 323–328 CrossRef CAS.
- J. Bao and G. K. Tranmer, Chem. Commun., 2015, 51, 3037–3044 RSC.
- A. R. Bogdan and K. James, Chem. – Eur. J., 2010, 16, 14506–14512 CrossRef CAS PubMed.
- (a) G. Brufani, F. Valentini, G. Rossini, L. Rosignoli, Y. Gu, P. Liu and L. Vaccaro, Green Synth. Catal., 2023 DOI:10.1016/j.gresc.2023.01.004; (b) M. Z. Hatit, L. F. Reichenbach, J. M. Tobin, F. Vilela, G. A. Burley and A. J. Watson, Nature commun., 2018, 9, 4021 CrossRef PubMed; (c) C. E. M. Salvador, B. Pieber, P. M. Neu, A. Torvisco, C. Kleber, Z. Andrade and C. O. Kappe, J. Org. Chem., 2015, 80, 4590–4602 CrossRef CAS PubMed; (d) P. Zhang, M. G. Russell and T. F. Jamison, Org. Process Res. Dev., 2014, 18, 1567–1570 CrossRef CAS; (e) A. R. Bogdan and K. James, Org. Lett., 2011, 13, 4060–4063 CrossRef CAS PubMed; (f) A. R. Bogdan and K. James, Chem. – Eur. J., 2010, 16, 14506–14512 CrossRef CAS PubMed.
- (a) S. Brase, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef CAS PubMed; (b) M. S. Taylor, D. N. Zalatan, A. M. Lerchne and E. N. Jacobsen, J. Am. Chem. Soc., 2005, 127, 1313–1317 CrossRef CAS PubMed; (c) L. W. Xu, C. G. Xia, J. W. Li and S. L. Zhou, Tetrahedron Lett., 2004, 45, 1219–1221 CrossRef CAS.
- (a) G. Vilé, G. Di Liberto, S. Tosoni, A. Sivo, V. Ruta, M. Nachtegaal, A. H. Clark, S. Agnoli, Y. Zou, A. Savateev, M. Antonietti and G. Pacchioni, ACS Catal., 2022, 12, 2947–2958 CrossRef; (b) G. Vilé, J. Liu and Z. Zhang, React. Chem. Eng., 2021, 6, 1878–1883 RSC; (c) S. Elavarasan, A. Bhaumik and M. Sasidharan, ChemCatChem, 2019, 11, 4340 CrossRef CAS; (d) A. Mandoli, Molecules, 2016, 21, 1174 CrossRef PubMed; (e) M. K. Barman, A. K. Sinha and S. Nembenna, Green Chem., 2016, 18, 2534–2541 RSC; (f) J. García-Álvarez, J. Díez, J. Gimeno, F. J. Suárez and C. Vincent, Eur. J. Inorg. Chem., 2012, 5854–5863 CrossRef; (g) P. Appukkuttan, W. Dehaen, V. V. Fokin and E. Van der Eycken, Org. Lett., 2004, 6(23), 4223–4225 CrossRef CAS PubMed.
- (a) A. M. Pandey, S. Mondal and B. Gnanaprakasam, J. Org. Chem., 2022, 87, 9926–9939 CrossRef CAS PubMed; (b) M. Escribà-Gelonch, G. A. de Leon Izeppi, D. Kirschneck and V. Hessel, ACS Sustainable Chem. Eng., 2019, 7, 17237–17251 CrossRef PubMed; (c) P. Zhang, N. Weeranoppanant, D. A. Thomas, K. Tahara, T. Stelzer, M. G. Russell, M.O’ Mahony, A. S. Myerson, H. Lin, L. P. Kelly, K. F. Jensen, T. F. Jamison, C. Dai, Y. Cui, N. Briggs, R. L. Beingessner and A. Adamo, Chem. – Eur. J., 2018, 24, 2776–2784 CrossRef CAS PubMed; (d) S. Borukhova, T. Noël, B. Metten, E. de Vos and V. Hessel, Green Chem., 2016, 18, 4947–4953 RSC.
- (a) L. Wan, G. Kong, M. Liu, M. Jiang, D. Cheng and F. Chen, Green Synth. Catal., 2022, 3, 243–258 CrossRef; (b) F. Ferlin, D. Lanari and L. Vaccaro, Green Chem., 2020, 22, 5937–5955 RSC; (c) M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2020, 10(24), 1802–1813 CrossRef; (d) H. Ishitani, Y. Saito, B. Laroche, X. Rao and S. Kobayashi, RSC Green Chem. Ser., 2020, 62, 1–49 CAS; (e) A. R. Bogdan and A. W. Dombrowski, J. Med. Chem., 2019, 62(14), 6422–6468 CrossRef CAS PubMed; (f) U. K. Sharma and E. V. Van der Eycken, Top. Heterocycl. Chem., 2018, 56, 343–373 Search PubMed; (g) J. Britton and C. L. Raston, Chem. Soc. Rev., 2017, 46, 1250–1271 RSC.
- (a) L. Chen, L. M. Barton, J. C. Vantourout, Y. Xu, C. Chu, E. C. Johnson, J. J. Sabatini and P. S. Baran, Org. Process Res. Dev., 2021, 12(25), 2639–2645 CrossRef; (b) A. Steiner, P. M. C. Roth, F. J. Strauss, G. Gauron, G. Tekautz, M. Winter, J. D. Williams and C. O. Kappe, Org. Process Res. Dev., 2020, 24(10), 2208–2216 CrossRef CAS.
- (a) M. Tissot, J. Jacq and P. Pasau, Org. Process Res. Dev., 2020, 24, 802–806 CrossRef CAS; (b) M. T. Rahman and T. Wirth, Top. Heterocycl. Chem., 2018, 56, 343–373 CAS; (c) H. Lehmann, Green Chem., 2017, 19(6), 1449–1453 RSC.
- (a) F. Ferlin, I. Anastasiou, L. Carpisassi and L. Vaccaro, Green Chem., 2021, 23, 6576–6582 RSC; (b) V. Trombettoni, F. Ferlin, F. Valentini, F. Campana, M. Silvetti and L. Vaccaro, Mol. Catal., 2021, 509, 111613–111622 CrossRef CAS; (c) C. J. Mallia and I. R. Baxendale, Org. Process Res. Dev., 2016, 20(2), 327–360 CrossRef CAS; (d) M. Brzozowski, M. O'Brien, S. V. Ley and A. Polyzos, Acc. Chem. Res., 2015, 48(2), 349–362 CrossRef CAS PubMed.
- A. Adeyemi, J. Bergman, J. Brånalt, J. Sävmarker and M. Larhed, Org. Process Res. Dev., 2017, 21, 947–955 CrossRef CAS.
- A. C. Bedard, A. Adamo, K. C. Aroh, M. G. Russell, A. A. Bedermann, J. Torosian, B. Yue, K. F. Jensen and T. F. Jamison, Science, 2018, 361, 1220–1225 CrossRef CAS PubMed.
- (a) F. Ferlin, F. Valentini, D. Sciosci, M. Calamante, E. Petricci and L. Vaccaro, ACS Sustainable Chem. Eng., 2021, 9, 12196–12204 CrossRef CAS; (b) V. Hessel, M. Escribà-Gelonch, J. Bricout, N. N. Tran, A. Anastasopoulou, F. Ferlin, F. Valentini, D. Lanari and L. Vaccaro, ACS Sustainable Chem. Eng., 2021, 9, 9508–9540 CrossRef CAS.
- (a) J. Andraos, E. Ballerini and L. Vaccaro, Green Chem., 2015, 17, 913–925 RSC; (b) F. Gonzalez-Bobes, N. Kopp, L. Li, J. Deerberg, P. Sharma, S. Leung, M. Davies, J. Bush, J. Hamm and M. Hrytsak, Org. Process Res. Dev., 2012, 16, 2051–2057 CrossRef CAS; (c) M. Jafarzadeh, Synlett, 2007, 2144–2145 CrossRef CAS.
- (a) X.-H. Ouyang, R.-J. Song, Y. Liu, M. Hu and J.-H. Li, Org. Lett., 2015, 17, 6038–6041 CrossRef CAS PubMed; (b) L. Chen, H. Xing, H. Zhang, Z.-X. Jiang and Z. Yang, Org. Biomol. Chem., 2016, 14, 7463–7467 RSC; (c) J. K. Myers and E. N. Jacobsen, J. Am. Chem. Soc., 1999, 121, 8959–8960 CrossRef CAS; (d) P. Zhou, L. Lin, L. Chen, X. Zhong, X. Liu and X. Feng, J. Am. Chem. Soc., 2017, 139, 13414–13149 CrossRef CAS PubMed; (e) P. Zhou, X. Liu, W. Wu, C. Xu and X. Feng, Org. Lett., 2019, 21, 1170–1175 CrossRef CAS PubMed.
- (a) H. Han, B. Zhu, X. Du, Y. Zhu, C. Yu and X. Jiang, Green Chem., 2021, 23, 10132 RSC; (b) J. Humbrías-Martín, M. C. Pérez-Aguilar, R. Mas-Ballesté, A. Dentoni Litta, A. Lattanzi, G. Della Sala, J. A. Fernández-Salas and J. Alemán, Adv. Synth. Catal., 2019, 361, 4790–4796 CrossRef; (c) T. Angelini, D. Lanari, R. Maggi, F. Pizzo, G. Sartori and L. Vaccaro, Adv. Synth. Catal., 2012, 354, 908–916 CrossRef CAS; (d) T. E. Horstmann, D. J. Guerin and S. J. Miller, Angew. Chem., 2000, 112, 3781–3784 CrossRef; (e) D. J. Guerin and S. J. Miller, J. Am. Chem. Soc., 2002, 124, 2134–2136 CrossRef CAS PubMed; (f) D. J. Guerin, T. E. Horstmann and S. J. Miller, Org. Lett., 1999, 1, 1107–1109 CrossRef CAS PubMed.
- Y. Tu, H. Dong, H. Wang, Y. Ao and Y. Liu, Chem. Commun., 2021, 57, 4524–4527 RSC.
- F. Ferlin, F. Valentini, G. Brufani, D. Lanari and L. Vaccaro, ACS Sustainable Chem. Eng., 2021, 9, 5740–5749 CrossRef CAS.
- (a) S. Rendler and M. Oestreich, Synthesis, 2005, 11, 1727–1747 Search PubMed; (b) A. D. Dilman and S. L. Ioffe, Chem. Rev., 2003, 103, 733–772 CrossRef CAS PubMed; (c) C. Chuit, R. J. P. Corriu, C. Reye and J. C. Young, Chem. Rev., 1993, 93, 1371–1448 CrossRef CAS.
- (a) F. Ferlin, D. Sciosci, F. Valentini, G. Cravotto, J. Menzio and K. Martina, Green Chem., 2021, 23, 7210–7218 RSC; (b) F. Valentini, F. Ferlin, E. Tomarelli, H. Mahmoudi, M. Bagherzadeh, M. Calamante and L. Vaccaro, ChemSusChem, 2021, 14, 3359 CrossRef CAS PubMed; (c) F. Valentini and L. Vaccaro, Molecules, 2020, 25, 5264 CrossRef CAS PubMed.
- (a) M. Tassi, E. Bartollini, P. Adriaensens, L. Bianchi, B. Barkakaty, R. Carleer, J. Chenc, D. K. Hensley, A. Marrocchi and L. Vaccaro, RSC Adv., 2015, 5, 107200–107208 RSC; (b) A. Marrocchi, P. Adriaensens, E. Bartollini, B. Barkakaty, R. Carleer, J. Chen, D. K. Hensley, C. Petrucci, M. Tassi and L. Vaccaro, Eur. Polym. J., 2015, 73, 391–401 CrossRef CAS.
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Q3D on elemental impurities - Step 5 - Revision 2. https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-16.pdf.
- J. Andraos and A. Hent, J. Chem. Educ., 2015, 92, 1820–1830 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General procedures, green metrics calculation, full characterization of the compounds and copies of the 1H, 13C and 19F NMR spectra. See DOI: https://doi.org/10.1039/d2gc04672e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |