
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mild and selective etherification of wheat straw lignin and lignin model alcohols by moisture-tolerant zirconium catalysis†
Cristiana
Margarita
 ,
Davide
Di Francesco
,
Hernando
Tuñon
,
Ivan
Kumaniaev
,
Carlos Jansson
Rada
and
Helena
Lundberg
*
,
Davide
Di Francesco
,
Hernando
Tuñon
,
Ivan
Kumaniaev
,
Carlos Jansson
Rada
and
Helena
Lundberg
*
Department of Chemistry, KTH Royal Institute of Technology, Teknikringen 30, S-10044 Stockholm, Sweden. E-mail: hellundb@kth.se
First published on 2nd March 2023
Abstract
The direct etherification of wheat straw lignin and lignin model compounds using alcohols as reagents and zirconocene triflate as moisture-tolerant Lewis acidic catalyst is herein described. Visual kinetic analysis was used to assess the average orders in the reaction components to map similarities between the model substrates and guide the method development. Full selectivity for the formation of benzylic ethers was obtained for models bearing free phenols and/or aliphatic alcohols, demonstrating that the present strategy enables a complementary reactivity to traditional phenolic lignin functionalisation. The reaction proceeds under mild conditions in absence of water-scavenging techniques and furnished a variety of ethers derived from lignin models with side chains bearing synthetic handles of relevance for thermoset applications. Finally, application of the Zr-mediated protocol on wheat straw-derived lignin resulted in the first example of metal-catalysed direct benzylic allylation of lignin using allyl alcohol as reagent, generating water as by-product.
Introduction
Efficient methods for the valorisation of lignocellulosic biomass to functional compounds and materials are required to enable the transition from a fossil to a renewables economy. While methods for upgrading the carbohydrate fraction to pulp and paper or to derive bioethanol have been known for long, efficient methods for conversion of lignin into valorised products are still scarce, yet crucial for the development of a sustainable society.1 Lignin is an underexploited source of bio-derived aromatics and a good candidate as a precursor for bioplastic production, including thermosets.2–7 Due to their availability, technical lignins, e.g. kraft lignin, soda lignin, and lignosulfonate, are typically used as substrates for such cross-linked materials. These technical lignins contain a higher amount of free phenolic moieties and a lower amount of β-O-4′ inter-unit linkages compared to the corresponding native lignins.8 Over the years, these characteristics have promoted the development of functionalisation strategies based on phenol/phenolate chemistry since the free phenol constitutes the most reactive functional group on those substrates. In the last decades, the development of fractionation strategies, e.g. organosolv, has enabled the production of technical lignins with a significant amount of intact phenyl ethers and benzylic alcohols that pave the way for alternative functionalisation strategies. An ideal functionalisation of the renewable feedstock should be carried out with sustainable methods: where catalytic transformations, bio-based solvents, high atom efficiency, and mild reaction conditions are to be prioritised in their design.9,10 Surprisingly, annual plants remain an underutilised source of lignocellulose despite their fast turnover and high abundance in e.g. agricultural non-food side streams.11 For this reason, crop residues such as wheat straw are expected to gain increased importance as a renewable feedstock for functional compounds and materials.In the context of lignin-based thermosets, the O-allylation of lignin phenols followed by cross-linking using thiol–ene click chemistry is an established approach. Among the allylation strategies, allyl halides have been reported as electrophiles under basic conditions.12–14 This methodology exploits the nucleophilic nature of alkoxides to react with the electrophilic allylating agent, typically allyl bromide or chloride (Fig. 1a). The major drawback of these reagents is their toxicity for both humans and the environment. Therefore, diallyl carbonate has been put forth as a greener alternative allylating agent (Fig. 1b).15 Nevertheless, the need for stoichiometric base to generate an alkoxide in situ results in a large amount of waste that decreases the atom economy of these processes unless efficiently recovered. These procedures, and other similar functionalisations,16 specifically target the formation of phenolic allyl ethers or demonstrate low selectivity towards different hydroxyl functions. In contrast, a Lewis acid-catalysed etherification procedure involving allyl alcohol as the allylating agent would overcome the issues associated with toxicity and atom economy (Fig. 1c). Additionally, these conditions selectively target the benzylic hydroxyl functionalities, in a complementary manner to precedent base-promoted strategies. While methods for the dehydrative etherification of benzylic alcohols have been reported with the use of several different catalytic systems,17–29 only a few examples are known of their direct condensation using allyl alcohol as the nucleophile.30–35
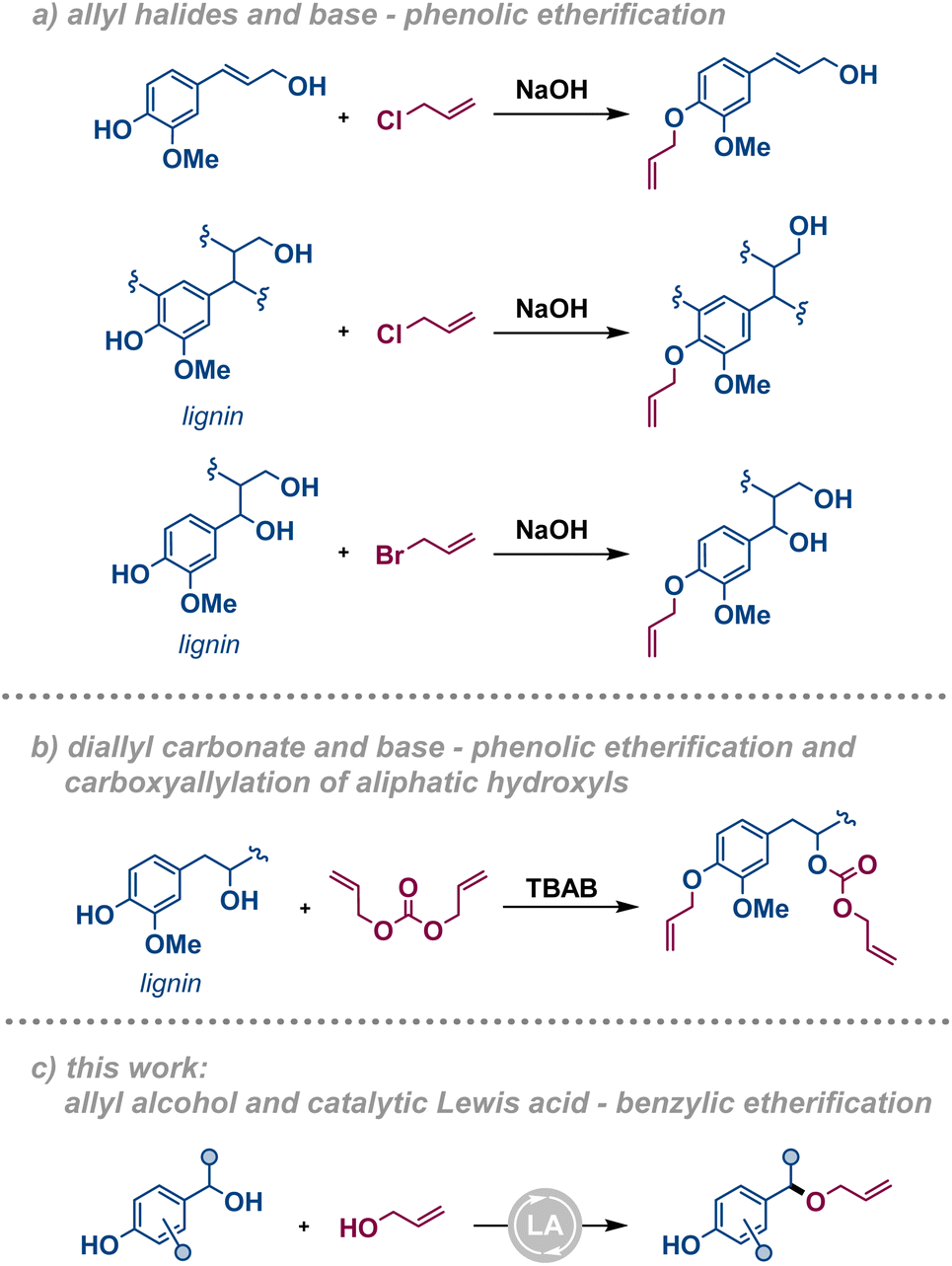 | ||
| Fig. 1 Approaches for allylation of lignin and model compounds. | ||
The use of Lewis acids as catalysts for direct etherification can be limited due to their sensitivity to the water produced during the transformation. However, some trivalent and tetravalent metal triflates constitute a moisture-tolerant class of Lewis acids that allow for procedures without water removal techniques.36–42 As part of our interest in this robust catalyst class,42,43 our group has recently studied the dehydrative formation of benzylic ethers from alcohols using the commercially available THF adduct of the moisture-tolerant Lewis acidic complex zirconocene triflate (Zr(Cp)2(CF3SO3)2·THF).44 In the present work, we demonstrate how time-resolved data enabled the extension of the Zr-catalysed methodology to the allylation of milled wheat straw lignin through a series of lignin model compounds of increasing complexity.
Results and discussion
In order to mimic the reactivity of our target lignin, the G-unit-like monomeric compound vanillyl alcohol (1a) and the more advanced model compound G–G′ β-O-4′ ether (1b) were chosen to study the direct etherification (Fig. 2).44 Initially, the catalytic etherification of the simplest compound 1a was conducted with 2-phenylethanol (2a) as model substrates and zirconocene triflate (Zr(Cp)2(CF3SO3)2·THF) as catalyst (Fig. 3). Kinetic analysis was carried out during the early optimisation phase to rationally aid the tuning of the reaction conditions. The monitoring of reaction mixtures was performed via periodical sampling and off-line HPLC analysis, using 4,4′-di-tert-butylbiphenyl (DTBB) as an internal standard. A solvent screening was performed, suggesting that ethereal solvents were optimal for the transformation. This solvent class grants high solubility of both catalyst and alcohol substrates, including lignin, while remaining relatively inert in the presence of Lewis acids, in contrast to EtOAc and sulfolane which can undergo undesired side-reactions, e.g. hydrolysis. While comparable yields of 3a were obtained in 1,4-dioxane (76%), THF (79%), and 2-methyltetrahydrofuran (Me-THF; 75%) after 5 h (ESI, Fig. S1†), the latter renewable solvent was chosen as the greener alternative.45,46 The yield obtained in Me-THF could be further improved by changing the reaction temperature from 30 °C to 40 °C (ESI, Fig. S2†).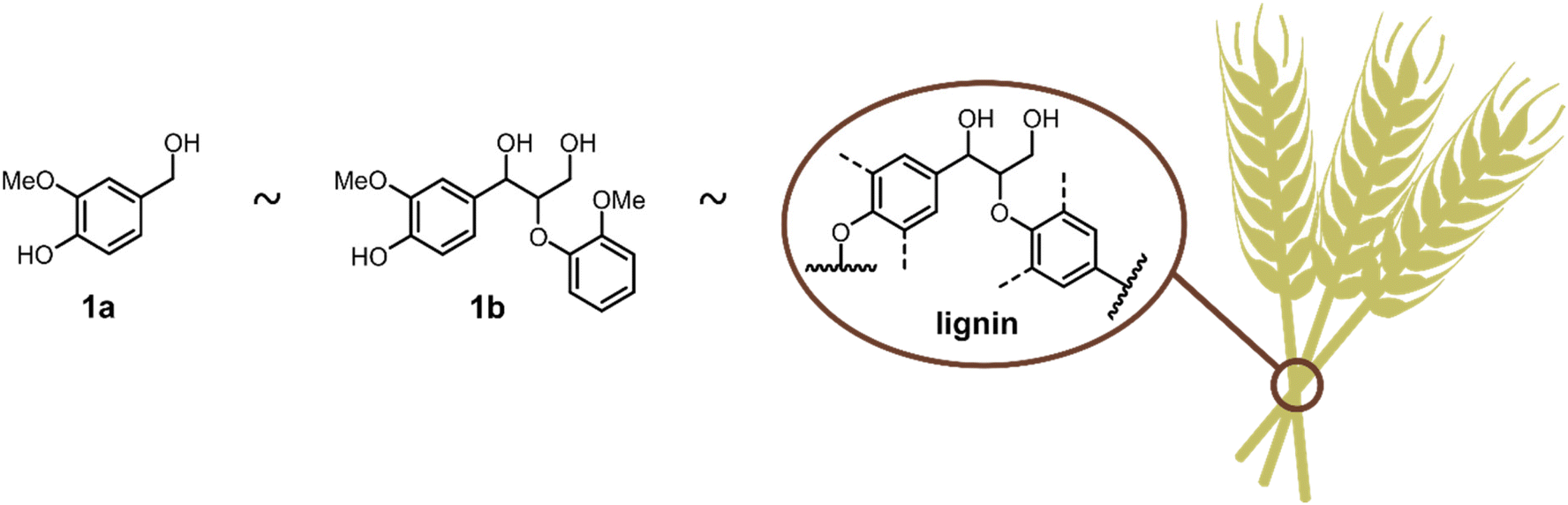 | ||
| Fig. 2 Lignin model compounds. | ||
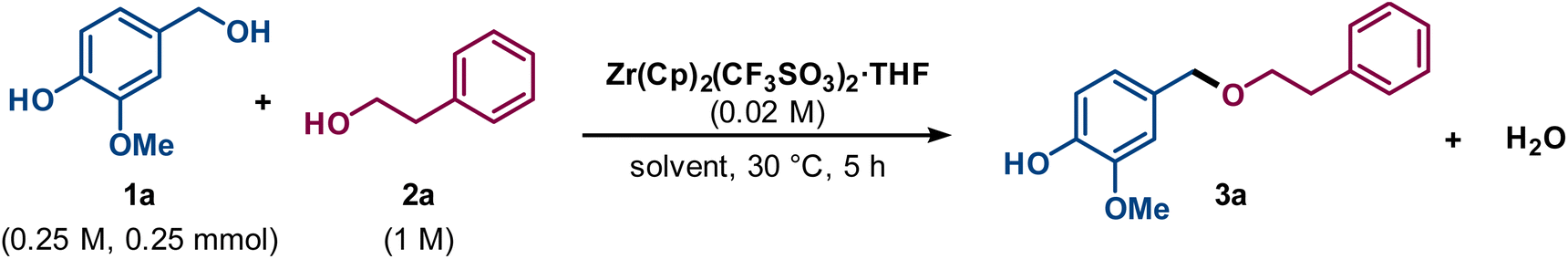 | ||
| Fig. 3 Initial conditions for evaluation of reaction parameters for direct etherification of lignin model compounds. | ||
Next, a group of Lewis acidic catalysts were screened to evaluate their activity in the formation of vanillyl ether (3a) (Fig. 4). While the use of titanocene triflate (Ti(Cp)2(OTf)2) or hafnium(IV) triflate (Hf(OTf)4) resulted in high reaction rates, decomposition of the product was also observed. In contrast, zirconocene triflate catalysed the etherification with a slightly lower rate but resulted in higher yield and slower decomposition of 3a over time. The observed reactivity differences between the catalysts are in accordance with their respective relative Lewis acidity. While Ti(Cp)2(OTf)2 is expected to possess a higher Lewis acidity compared to the zirconium analogue due to the intrinsic nature of the metal center,47 the Lewis acidity of Hf(OTf)4 is expected to exceed that of zirconocene triflate as a function of its greater number of electron withdrawing triflate ligands.48 Triflic acid (TfOH) and iron(III) triflate (Fe(OTf)3) resulted in lower product yields compared to the zirconocene triflate catalyst. While the former catalyst may enable a metal-free protocol, the difficult handling of TfOH makes it unsuitable from a scale-up perspective. In addition, strong Brønsted acids are known to promote lignin degradation and condensation.49,50 Thus, catalytic use of TfOH was not further investigated. Finally, the higher yields obtained using Zr(Cp)2(CF3SO3)2·THF compared to Fe(OTf)3 made us prioritise the former. No reaction was observed using zirconocene dichloride (Zr(Cp)2Cl2) in catalytic amounts or in the absence of any Lewis acid.
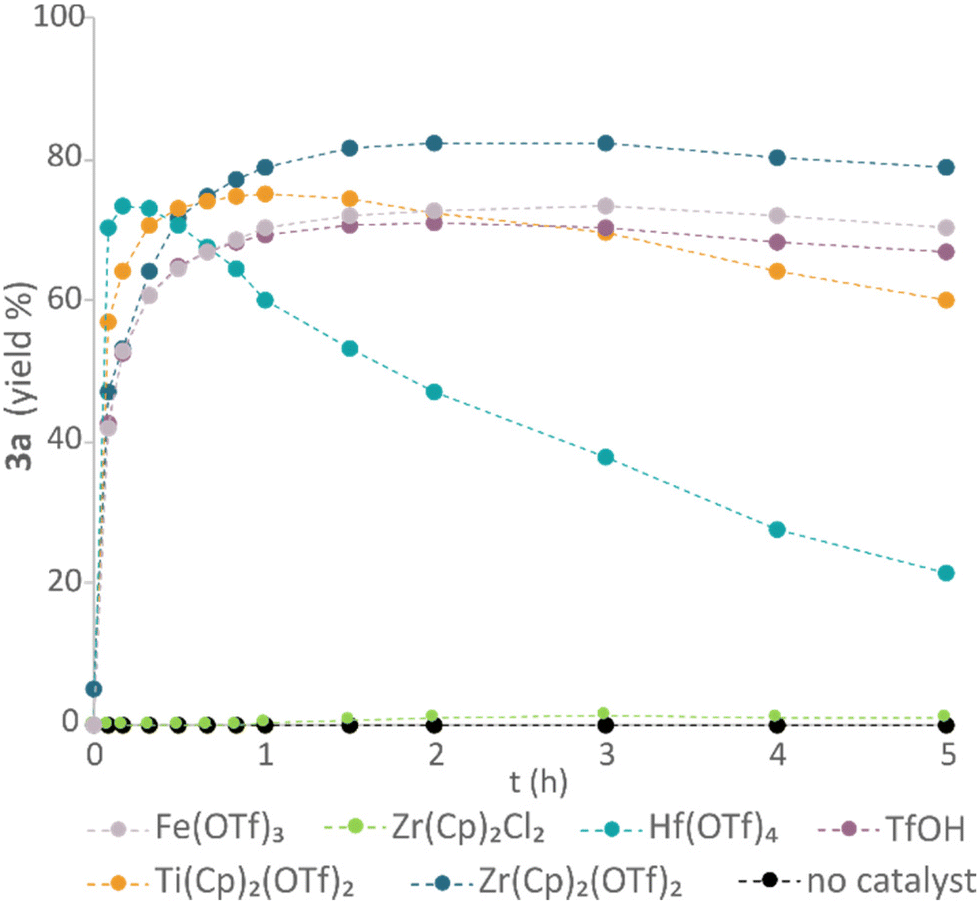 | ||
| Fig. 4 Catalyst screening. Conditions: vanillyl alcohol 1a (0.25 M, 0.25 mmol scale), 2-phenylethanol 2a (1 M), catalyst (0.02 M), DTBB as internal standard (0.01 M), Me-THF, 40 °C. | ||
A set of “same excess” experiments51,52 was performed on the model etherification reaction of vanillyl alcohol 1a with 2a, to evaluate possible catalyst deactivation and product inhibition. Two sets of reactions were carried out mimicking 25% conversion, both in the absence (Fig. 5a) and presence of the amount of H2O that the direct etherification would generate at this conversion (Fig. 5b). A faster rate was observed in the former case, while the addition of water resulted in the same reaction profile as that of standard conditions (visualized by operating a time shift), thus indicating minor catalyst inhibition by H2O.
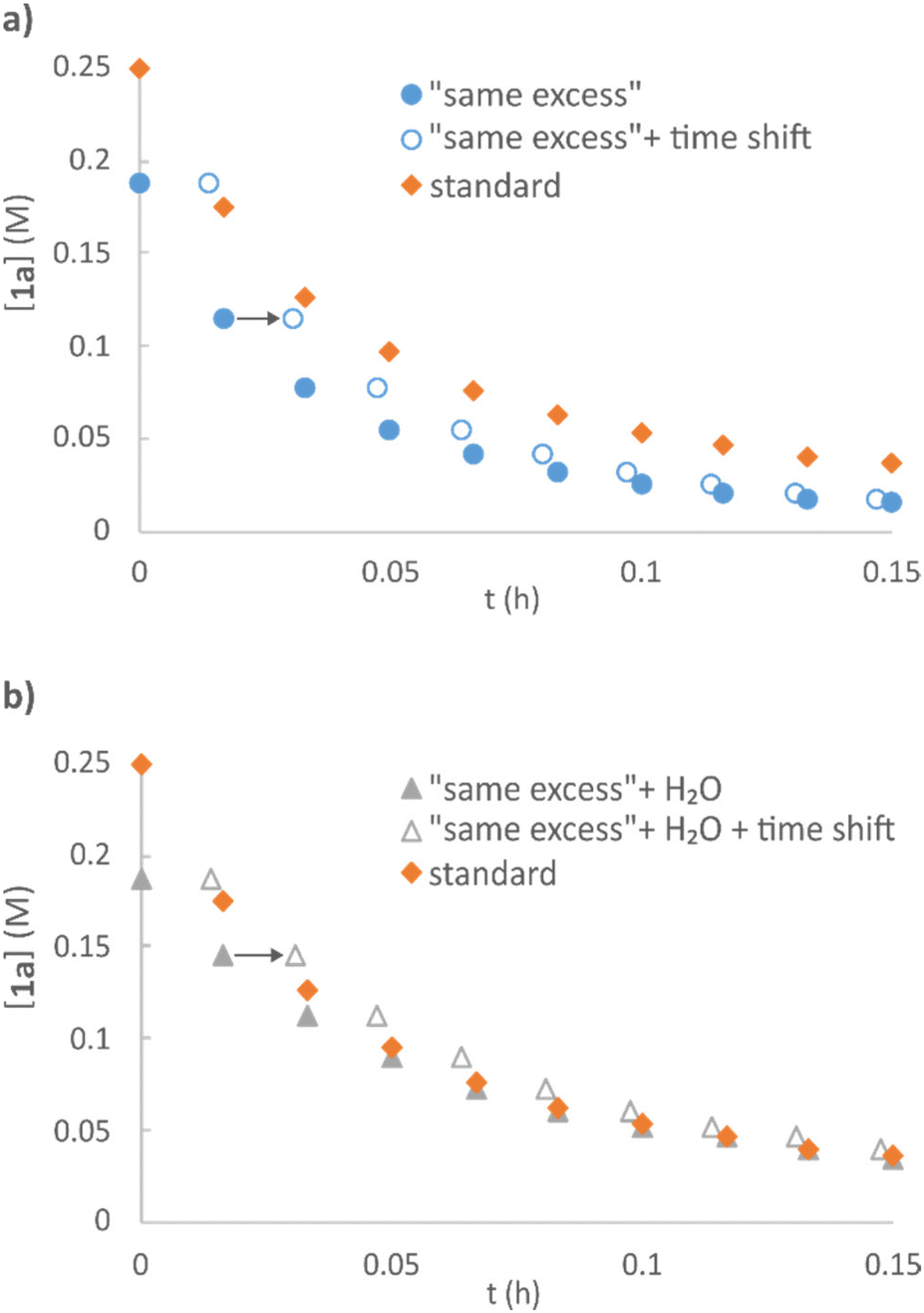 | ||
| Fig. 5 “Same excess” experiments with conditions mimicking 25% conversion of vanillyl alcohol in absence (a) and presence (b) of additional water. Standard conditions: 1a (0.25 M), 2a (1 M), Zr(Cp)2(CF3SO3)2·THF (0.02 M), DTBB as internal standard (0.01 M), Me-THF, 40 °C. | ||
The water tolerance of the catalytic system was assessed in a series of experiments in which the H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zr ratio was varied in the range of 25:1 to 250:1 (ESI, Fig. S8 and S9†). As expected from the inhibition observed in the “same excess” experiments, the reaction rate gradually decreased with a larger amount of H2O present. Nevertheless, good yields could be reached in presence of up to a 50:1 H2O:Zr ratio, albeit with a longer reaction time. The addition of molecular sieves in the reaction mixture strongly inhibited the etherification reaction, providing yields close to 25% after 5 hours (ESI, Fig. S10†). This finding stands in sharp contrast to the requirements of the related zirconium tetrachloride that does not catalyse dehydrative reactions in the absence of water scavenging.53 In accordance with the previously reported etherification of benzylic alcohols,44 keeping the concentration of electrophilic alcohol 1a low throughout the reaction by “slow” addition (5 portions over 40 minutes) improved the yield of ether 3a compared to the corresponding “batch” conditions, where all reactants were present from the outset of the reaction (ESI, Fig. S11†). Moreover, this allowed for the use of a lower catalyst loading (0.01 M, 4 mol%), which was found beneficial for product stability over time (Fig. 6). Thus, the optimised conditions were determined to be 3 hours reaction time, with the use of 0.01 M of Zr catalyst and a 0.25:1 M ratio of 1a:2a. Under these conditions, 3a was obtained in 85% isolated yield, while the “batch” reaction provided 68% isolated 3a. It is worth noting that a similar yield of 72% can be expected, according to HPLC quantification, when operating under “slow” addition with a ratio of 0.5:1 M 1a:2a and 5 hours reaction time, effectively improving the reactants’ global stoichiometry (ESI, Fig. S11†).
:Zr ratio was varied in the range of 25:1 to 250:1 (ESI, Fig. S8 and S9†). As expected from the inhibition observed in the “same excess” experiments, the reaction rate gradually decreased with a larger amount of H2O present. Nevertheless, good yields could be reached in presence of up to a 50:1 H2O:Zr ratio, albeit with a longer reaction time. The addition of molecular sieves in the reaction mixture strongly inhibited the etherification reaction, providing yields close to 25% after 5 hours (ESI, Fig. S10†). This finding stands in sharp contrast to the requirements of the related zirconium tetrachloride that does not catalyse dehydrative reactions in the absence of water scavenging.53 In accordance with the previously reported etherification of benzylic alcohols,44 keeping the concentration of electrophilic alcohol 1a low throughout the reaction by “slow” addition (5 portions over 40 minutes) improved the yield of ether 3a compared to the corresponding “batch” conditions, where all reactants were present from the outset of the reaction (ESI, Fig. S11†). Moreover, this allowed for the use of a lower catalyst loading (0.01 M, 4 mol%), which was found beneficial for product stability over time (Fig. 6). Thus, the optimised conditions were determined to be 3 hours reaction time, with the use of 0.01 M of Zr catalyst and a 0.25:1 M ratio of 1a:2a. Under these conditions, 3a was obtained in 85% isolated yield, while the “batch” reaction provided 68% isolated 3a. It is worth noting that a similar yield of 72% can be expected, according to HPLC quantification, when operating under “slow” addition with a ratio of 0.5:1 M 1a:2a and 5 hours reaction time, effectively improving the reactants’ global stoichiometry (ESI, Fig. S11†).
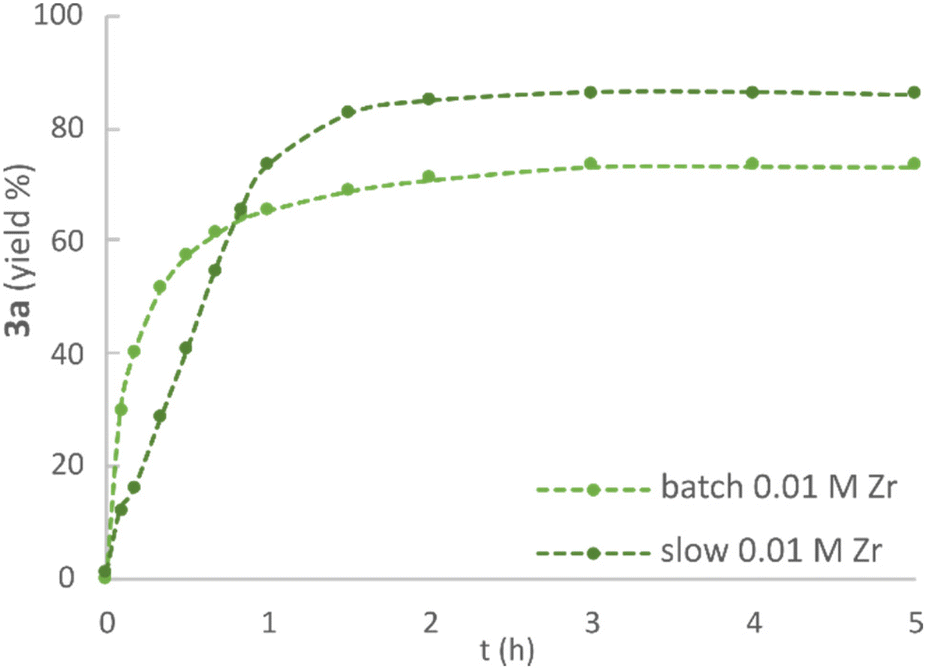 | ||
| Fig. 6 Effect of mode of addition on yield of 3a. Conditions: 1a (0.25 M), 2a (1 M), Zr(Cp)2(CF3SO3)2·THF (0.01 M), DTBB as internal standard (0.01 M), Me-THF, 40 °C. Slow addition: 1a added in 5 portions over 40 min. | ||
Variable Time Normalisation Analysis (VTNA)54,55 of kinetic data obtained in sets of “different excess” experiments51,52 helped us to evaluate the rate dependence on the concentration of the Zr catalyst and the average reaction orders in electrophile, i.e.1a and 1b, and nucleophile for the formation of ethers 3a and 3b (for details and VTNA plots see ESI, Fig. S4–S6 and Fig. S12–S14,† respectively). The summarised results in Fig. 7 demonstrate a close to first-order rate dependence on [Zr] and [electrophile] in both cases, while the average order in [nucleophile] was close to zero in both cases under synthetically relevant conditions, i.e. low [electrophile] and high [nucleophile]. In contrast, the average order in [nucleophile] increased at low concentrations (ESI, Fig. S6†), indicating that nucleophilic attack becomes more competitive as a rate-limiting step under such conditions. The lack of nucleophilic inhibition of the catalytic activity, which was previously observed for the same Zr complex in direct etherification in benzotrifluoride as reaction medium,44 was encouraging since it allowed for the concentration of the nucleophilic alcohol to be kept high with maintained reaction efficiency. Mechanistically, the kinetic results are consistent with a carbocationic route, in which the C–O bond activation is the turnover-limiting step of the catalytic cycle under standard conditions.44 The vicinal aromatic system is expected to result in enhanced thermodynamic stability of benzylic carbocations compared to carbocations that would result from primary aliphatic alcohols and phenols. This stability difference is reflected in the associated C–OH bond dissociation energies of the parent alcohols56 and is demonstrated by the selective etherification of 1b at the benzylic position while the phenolic and γ-OH groups remain unreacted.
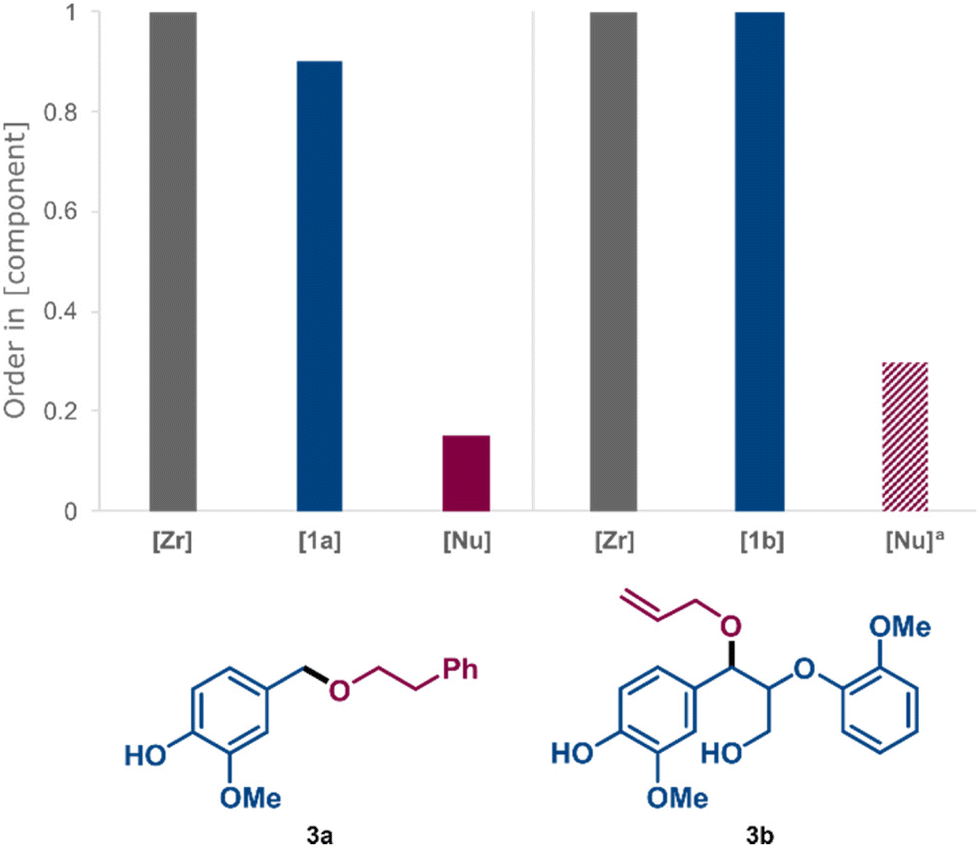 | ||
| Fig. 7 Order in [Zr], [1a], [Nu] (2a) for the formation of product 3a. Standard conditions: 1a (0.25 M), 2a (1 M), Zr(Cp)2(CF3SO3)2·THF (0.02 M), DTBB as internal standard Me-THF, 40 °C. Order in [Zr], [1b], [Nu] (allyl alcohol) for the formation of product 3b. Standard conditions: 1b (0.03 M), allyl alcohol (0.33 M), Zr(Cp)2(CF3SO3)2·THF (3 mM), DTBB as internal standard, Me-THF, 40 °C). aThe concentration of allyl alcohol over time was calculated based on its initial concentration and product formation, all other concentrations were monitored by off-line HPLC analysis. | ||
Using the optimised reaction conditions, a series of ethers were synthesized with different functionalities on the ether side chain (Scheme 1). Vanillyl alcohol 1a, 4-hydroxybenzyl alcohol 1c, and syringyl alcohol 1d were assessed to respectively mimic the G-, H-, and S-units in lignin, bearing both free phenols and benzylic alcohols. The allylation and propargylation of vanillyl alcohol proceeded in good yield to form ethers 3c and 3d (72% and 69%, respectively). Similar yields were obtained for the etherification products of 1a with longer chain alcohols substituted with terminal or internal unsaturated moieties as sites for possible further modification (3e–3g), as well as for the formation of ether 3h deriving from cyclopropylmethanol. The introduction of a terminal azide function was also tested, however, product 3i could only be isolated in low yield. Etherification of alcohol 1c provided the resulting allylated and propargylated products 3j and 3k in good yields, higher or comparable to the analogous transformation with vanillyl alcohol 1a. In contrast, the etherification outcome of the more electron-rich syringyl alcohol 1d depended on the nucleophilic partner. The use of 2a and allyl alcohol resulted in fair yields of around 50% for ethers 3l and 3m, whereas the propargylation turned out to be much less effective than the corresponding allylation, resulting in a low yield of 3n (25%). A similar outcome was observed when vanillyl alcohol was replaced for the dimethoxy-substituted analogue 1e in the reaction with 2a. Finally, an assessment of the catalytic conditions was carried out using the advanced lignin model compound 1b (Scheme 1), which bears a free primary γ-alcohol together with a free phenol and a benzylic alcohol. Gratifyingly, this model compound gave the corresponding benzylic ethers 3b and 3p in 75% and 50% yield while respectively reacting with allyl and propargyl alcohol.
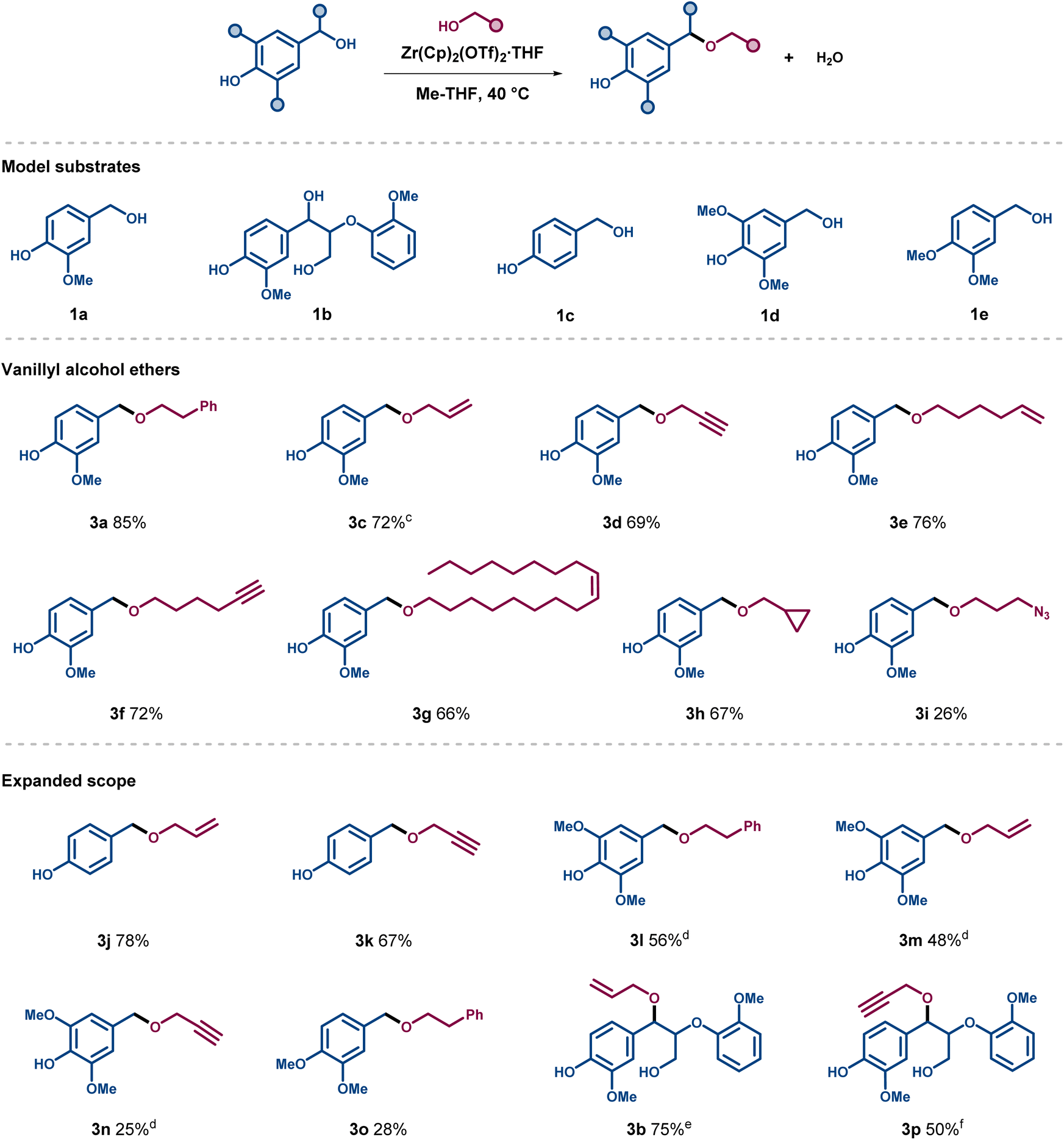 | ||
| Scheme 1 Scope of the etherification of lignin-derived alcohols. Conditions: 1a, 1c–e (0.25 M, added in 5 portions of 0.05 mmol over 40 min), 2a–h (1 M), Zr(Cp)2(CF3SO3)2·THF (0.01 M), Me-THF, 40 °C, 3 h. Isolated yields. a4 h. b1.5 h. c1b (0.22 mmol), 2b (1.46 mmol), 9 mol% Zr(Cp)2(CF3SO3)2·THF, Me-THF, 40 °C, 4 h. d1b (0.1 mmol), 2c (1 mmol), 1 mol% Zr(Cp)2(CF3SO3)2·THF, Me-THF, 40 °C, 3 h. | ||
With the promising results for model compound 1b at hand, we set out to assess our catalytic method in a biorefinery setting on wheat straw lignin (Fig. 8). The straw was determined to contain 15 wt% of Klason lignin with an estimated content of β-O-4′ inter-unit motif in the range of 75–79%.57 To preserve as many benzylic alcohol units as possible in our material, non-purified Milled Straw Lignin (MSL) was selected as the substrate of our methodology. The fractionation was performed according to the Björkman method (ESI, section 6†),58 which involved a preliminary dewaxing of the biomass followed by two subsequential milling processes and a final solvent extraction to yield 8 wt% of MSL versus the initial biomass. The reaction of MSL with allyl alcohol was performed with slightly modified standard catalytic conditions (ESI, section 6†). The solid product was freeze-dried and analysed by chromatographic and spectroscopic methods. Size exclusion chromatography (SEC) showed comparable molecular weight (MW) distributions between the unreacted MSL substrate and MSL from a control reaction performed without the catalyst, while the MSL subjected to the catalytic allylation protocol (AMSL) displayed a wider distribution including a higher molecular weight fraction (ESI, section 6†). Assessment with NMR (1H, 13C, and HSQC) indicated a volume decrease of the signal corresponding to the α-proton of the β-O-4′ moieties in lignin (MSL-α, δC/δH 71.8–71.1/4.8–4.7 ppm) while, in parallel, a signal associated to the C3 of the allyl ether was observed (AMSL-allyl, δC/δH 114.7–118.4/5.3–4.9 ppm) (Fig. 9a and b). The control reaction with allyl alcohol in the absence of catalyst, under otherwise identical conditions, showed no such decrease of α-proton signals and no formation of allyl ethers (Fig. 9c). The signal corresponding to the methoxy groups on the aromatic lignin units (OMe, δC/δH 57.8–54.1/4.0–3.1 ppm) was used as an internal standard to normalise against other integrals, as these methoxy groups were shown to be stable under comparable reaction conditions.44 These combined results suggest a similar reactivity of the wheat straw-derived lignin substrate in the desired transformation compared to the G–G′ β-O-4′ model 1b that, in turn, behaved similarly to the even simpler model compound 1a. This transferability of the catalytic protocol demonstrates how understanding the reactivity of the model compounds provides a good basis for synthetic method development for complex targets.
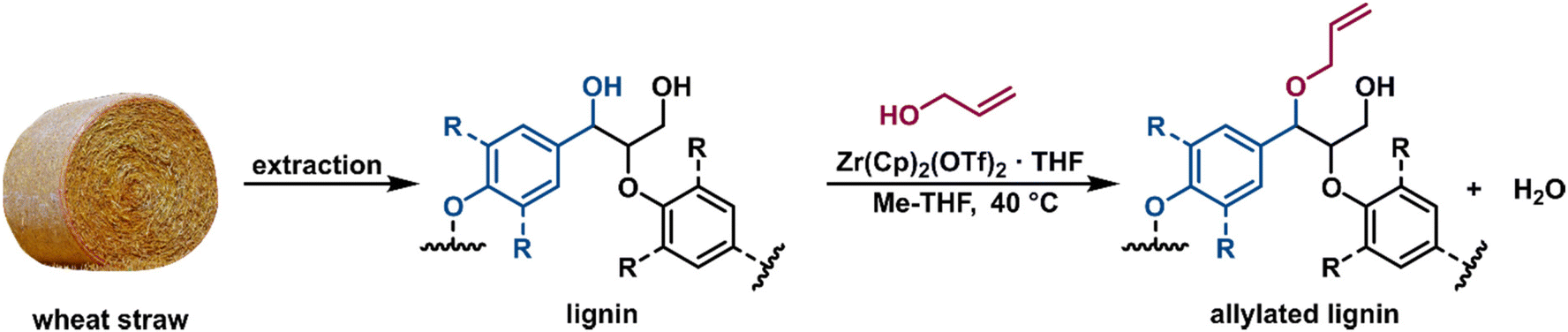 | ||
| Fig. 8 Wheat straw lignin extraction and functionalisation. | ||
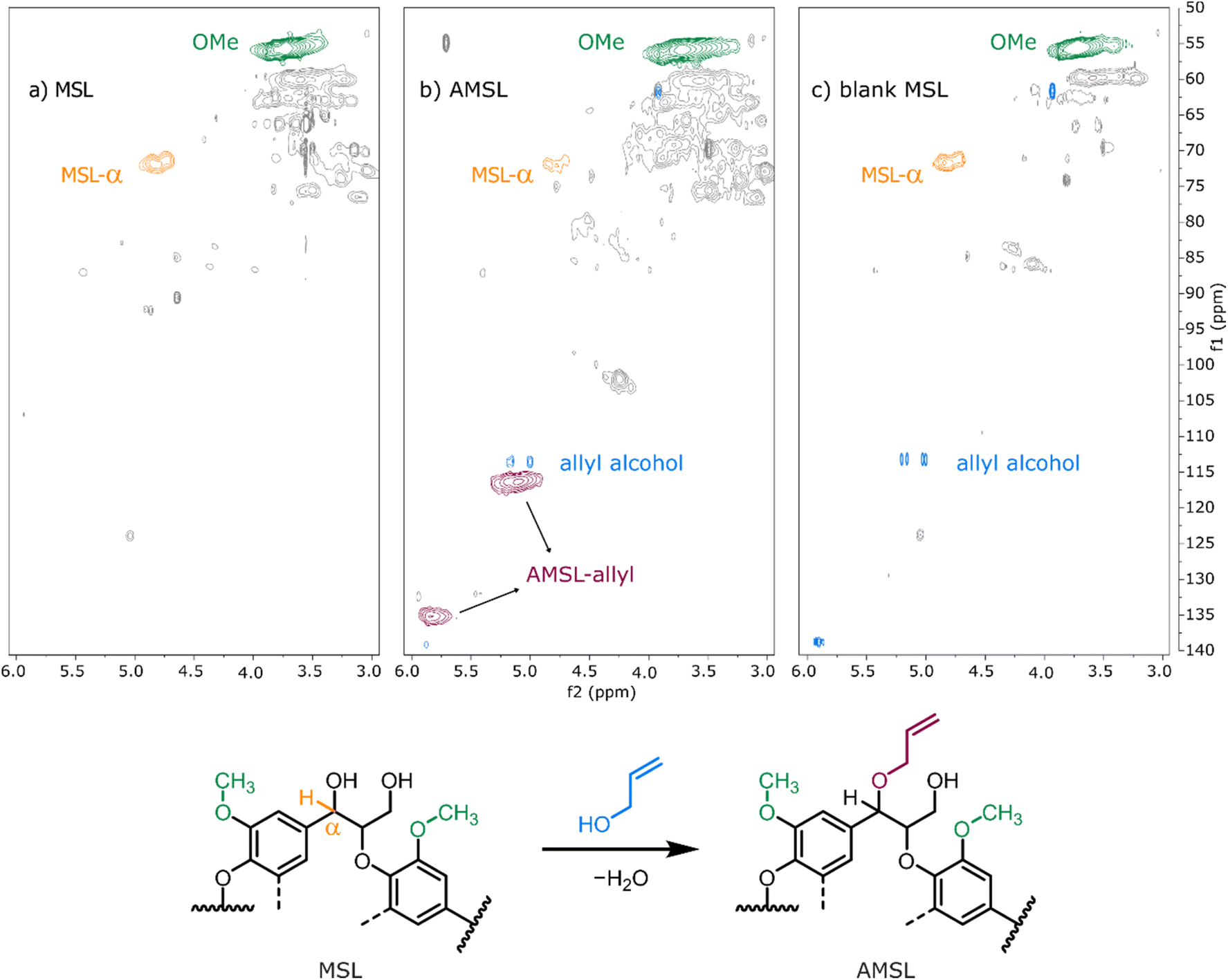 | ||
| Fig. 9 Magnified HSQC spectra of MSL, AMSL, and MSL reacted in absence of the catalyst. | ||
Conclusions
A zirconium-based homogeneous catalytic system able to promote the direct and selective etherification of benzylic alcohols is reported. The reaction occurs under particularly mild operating conditions in presence of free phenols and aliphatic alcohols, using a variety of alcohols as nucleophiles. VTNA was employed to map the kinetic driving forces of the transformation. The protocol was efficiently applied to different lignin model compounds and furnished a variety of ethers with different functionalities for future modifications. Finally, the proposed catalytic system was successfully utilised on wheat straw-derived milled lignin to produce a promising allylated precursor for lignin-based thermosets with allyl alcohol as allylating agent. To the best of our knowledge, this result represents the first example of a direct allylation of lignin with a benign reagent such as allyl alcohol producing water as side product. This work serves as a proof of concept for a lignin valorisation strategy with orthogonal reactivity compared to common phenolic functionalisations using disadvantageous chemicals. Further evaluation of lignins obtained by different fractionation methods is underway, along with assessments of the resulting material properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Lantmännens forskningsstiftelse, Formas (grant no. 2021-00678), the Swedish Foundation for Strategic Research (grant no. FFL21-0005), Stiftelsen Olle Engkvist Byggmästare, Frans Georg och Gull Liljenroths stiftelse, Magnus Bergvalls stiftelse, Stiftelsen Lars Hiertas Minne, and KTH Royal Institute of Technology is gratefully acknowledged.References
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- D. Di Francesco, D. Rigo, K. Reddy Baddigam, A. P. Mathew, N. Hedin, M. Selva and J. S. M. Samec, ChemSusChem, 2022, 15, e202200326 CrossRef CAS PubMed.
- M. Jawerth, M. Johansson, S. Lundmark, C. Gioia and M. Lawoko, ACS Sustainable Chem. Eng., 2017, 5, 10918–10925 CrossRef CAS.
- Y. Cao, Z. Liu, B. Zheng, R. Ou, Q. Fan, L. Li, C. Guo, T. Liu and Q. Wang, Composites, Part B, 2020, 200, 108295 CrossRef CAS.
- C. Gioia, G. Lo Re, M. Lawoko and L. Berglund, J. Am. Chem. Soc., 2018, 140, 4054–4061 CrossRef CAS PubMed.
- M. E. Jawerth, C. J. Brett, C. Terrier, P. T. Larsson, M. Lawoko, S. V. Roth, S. Lundmark and M. Johansson, ACS Appl. Polym. Mater., 2020, 2, 668–676 CrossRef CAS.
- Y. Xu, K. Odelius and M. Hakkarainen, ACS Sustainable Chem. Eng., 2019, 7, 13456–13463 CrossRef CAS.
- C. Crestini, H. Lange, M. Sette and D. S. Argyropoulos, Green Chem., 2017, 19, 4104–4121 RSC.
- P. Anastas and J. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, New York, 2000 Search PubMed.
- J. Sternberg, O. Sequerth and S. Pilla, Prog. Polym. Sci., 2021, 113, 101344 CrossRef CAS.
- L. Wang, J. Littlewood and R. J. Murphy, Renewable Sustainable Energy Rev., 2013, 28, 715–725 CrossRef CAS.
- M. Jawerth, M. Lawoko, S. Lundmark, C. Perez-Berumen and M. Johansson, RSC Adv., 2016, 6, 96281–96288 RSC.
- I. Ribca, M. E. Jawerth, C. J. Brett, M. Lawoko, M. Schwartzkopf, A. Chumakov, S. V. Roth and M. Johansson, ACS Sustainable Chem. Eng., 2021, 9, 1692–1702 CrossRef CAS.
- L. Zoia, A. Salanti, P. Frigerio and M. Orlandi, BioResources, 2014, 9, 6540–6561 CrossRef.
- L. C. Over and M. A. R. Meier, Green Chem., 2015, 18, 197–207 RSC.
- C. Dumont, R. M. Gauvin, F. Belva and M. Sauthier, ChemSusChem, 2018, 11, 1649–1655 CrossRef CAS PubMed.
- G. V. M. Sharma and A. K. Mahalingam, J. Org. Chem., 1999, 64, 8943–8944 CrossRef CAS PubMed.
- A. Corma and M. Renz, Angew. Chem., Int. Ed., 2007, 46, 298–300 CrossRef CAS PubMed.
- A. B. Cuenca, G. Mancha, G. Asensio and M. Medio-Simón, Chem. – Eur. J., 2008, 14, 1518–1523 CrossRef CAS PubMed.
- J. Kim, D.-H. Lee, N. Kalutharage and C. S. Yi, ACS Catal., 2014, 4, 3881–3885 CrossRef CAS.
- Y. Liu, X. Wang, Y. Wang, C. Du, H. Shi, S. Jin, C. Jiang, J. Xiao and M. Cheng, Adv. Synth. Catal., 2015, 357, 1029–1036 CrossRef CAS.
- R. M. P. Veenboer and S. P. Nolan, Green Chem., 2015, 17, 3819–3825 RSC.
- Q. Xu, H. Xie, P. Chen, L. Yu, J. Chen and X. Hu, Green Chem., 2015, 17, 2774–2779 RSC.
- Z. Lai, Z. Wang and J. Sun, Org. Lett., 2015, 17, 6058–6061 CrossRef CAS PubMed.
- L. Zhang, A. Gonzalez-de-Castro, C. Chen, F. Li, S. Xi, L. Xu and J. Xiao, Mol. Catal., 2017, 433, 62–67 CrossRef CAS.
- S.-S. Meng, Q. Wang, G.-B. Huang, L.-R. Lin, J.-L. Zhao and A. S. C. Chan, RSC Adv., 2018, 8, 30946–30949 RSC.
- P. K. Sahoo, S. S. Gawali and C. Gunanathan, ACS Omega, 2018, 3, 124–136 CrossRef CAS PubMed.
- S. Estopiñá-Durán, L. J. Donnelly, E. B. Mclean, B. M. Hockin, A. M. Z. Slawin and J. E. Taylor, Chem. – Eur. J., 2019, 25, 3950–3956 CrossRef PubMed.
- R. R. Singh, A. Whittington and R. S. Srivastava, Mol. Catal., 2020, 492, 110954 CrossRef CAS.
- A. Prades, R. Corberán, M. Poyatos and E. Peris, Chem. – Eur. J., 2008, 14, 11474–11479 CrossRef CAS PubMed.
- Y. Kayaki, T. Koda and T. Ikariya, J. Org. Chem., 2004, 69, 2595–2597 CrossRef CAS PubMed.
- Y. Kon, T. Fujitani, T. Nakashima, T. Murayama and W. Ueda, Catal. Sci. Technol., 2018, 8, 4618–4625 RSC.
- T. Watanabe, L. E. Conlon and J. C. H. Hwa, J. Org. Chem., 1958, 23, 1666–1668 CrossRef.
- K. V. Katkar, S. D. Veer and K. G. Akamanchi, Synth. Commun., 2016, 46, 1893–1901 CrossRef CAS.
- B. Das, B. Venkataiah and P. Madhusudhan, J. Chem. Res., 2000, 2000, 266–268 CrossRef.
- S. Kobayashi, S. Nagayama and T. Busujima, J. Am. Chem. Soc., 1998, 120, 8287–8288 CrossRef CAS.
- S. Kobayashi, M. Sugiura, H. Kitagawa and W. W.-L. Lam, Chem. Rev., 2002, 102, 2227–2302 CrossRef CAS PubMed.
- L. Yang, Y. Li and P. E. Savage, Ind. Eng. Chem. Res., 2014, 53, 2633–2639 CrossRef CAS.
- M. de Léséleuc and S. K. Collins, ACS Catal., 2015, 5, 1462–1467 CrossRef.
- X. Zhang, R. Qiu, C. Zhou, J. Yu, N. Li, S. Yin and X. Xu, Tetrahedron, 2015, 71, 1011–1017 CrossRef CAS.
- N. Li, L. Wang, L. Zhang, W. Zhao, J. Qiao, X. Xu and Z. Liang, ChemCatChem, 2018, 10, 3532–3538 CrossRef CAS.
- H. Lundberg, F. Tinnis and H. Adolfsson, Appl. Organomet. Chem., 2019, 33, e5062 CrossRef.
- P. Villo, O. Dalla-Santa, Z. Szabó and H. Lundberg, J. Org. Chem., 2020, 85, 6959–6969 CrossRef CAS PubMed.
- C. Margarita, P. Villo, H. Tuñon, O. Dalla-Santa, D. Camaj, R. Carlsson, M. Lill, A. Ramström and H. Lundberg, Catal. Sci. Technol., 2021, 11, 7420–7430 RSC.
- A. Pellis, F. P. Byrne, J. Sherwood, M. Vastano, J. W. Comerford and T. J. Farmer, Green Chem., 2019, 21, 1686–1694 RSC.
- V. Pace, P. Hoyos, L. Castoldi, P. Domínguez de María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369–1379 CrossRef CAS PubMed.
- R. Qiu, X. Xu, L. Peng, Y. Zhao, N. Li and S. Yin, Chem. – Eur. J., 2012, 18, 6172–6182 CrossRef CAS PubMed.
- H. Ishitani, H. Suzuki, Y. Saito, Y. Yamashita and S. Kobayashi, Eur. J. Org. Chem., 2015, 5485–5499 CrossRef CAS.
- P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, J. Am. Chem. Soc., 2015, 137, 7456–7467 CrossRef CAS PubMed.
- C. W. Lahive, P. J. Deuss, C. S. Lancefield, Z. Sun, D. B. Cordes, C. M. Young, F. Tran, A. M. Z. Slawin, J. G. de Vries, P. C. J. Kamer, N. J. Westwood and K. Barta, J. Am. Chem. Soc., 2016, 138, 8900–8911 CrossRef CAS PubMed.
- D. G. Blackmond, Angew. Chem., Int. Ed., 2005, 44, 4302–4320 CrossRef CAS PubMed.
- D. G. Blackmond, J. Am. Chem. Soc., 2015, 137, 10852–10866 CrossRef CAS PubMed.
- H. Lundberg, F. Tinnis and H. Adolfsson, Chem. – Eur. J., 2012, 18, 3822–3826 CrossRef CAS PubMed.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 2028–2031 CrossRef PubMed.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 16084–16087 CrossRef PubMed.
- P. Villo, A. Shatskiy, M. D. Kärkäs and H. Lundberg, Angew. Chem., Int. Ed., 2023, 62, e202211952 CrossRef CAS PubMed.
- L. Zhang, A. Larsson, A. Moldin and U. Edlund, Ind. Crops Prod., 2022, 187, 115432 CrossRef CAS.
- A. Björkman, Nature, 1954, 174, 1057–1058 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc04650d |
| This journal is © The Royal Society of Chemistry 2023 |