
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimisation of the electrochemical conversion of CO2 into formate in a flow cell configuration using a bismuth-based electrocatalyst†
Matteo
Miola
a,
Donatella
Chillé
b,
Georgia
Papanikolaou
b,
Paola
Lanzafame
b and
Paolo P.
Pescarmona
 *a
*a
aChemical Engineering Group, Engineering and Technology Institute Groningen, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: p.p.pescarmona@rug.nl
bDepartment ChiBioFarAm, University of Messina, ERIC aisbl, INSTM/CASPE, V. le F. Stagno d'Alcontres 31, 98166 Messina, Italy
First published on 31st January 2023
Abstract
Highly selective and active electrocatalysts are essential for industrial application of the electrochemical conversion of CO2 into valuable products. The electrochemical cell configuration and the optimisation of its parameters are also of paramount importance to promote the conversion of CO2 and enhance the selectivity and stability of the electrocatalyst. Furthermore, the cell needs to be designed to enable the continuous conversion of CO2 in a flow cell configuration with optimal use of energy resources, to obtain a sustainable CO2-to-formate conversion process. In this work, we present an in-depth, systematic investigation of the crucial parameters for the optimum operation of a flow cell for the conversion of CO2 with a highly scalable Bi-based electrocatalyst at industrially-relevant current densities (j = 100 mA cm−2), achieving high Faradaic efficiency (FEformate > 70%) for a prolonged application time (t = 65 h). By studying and tuning the electrolyte and CO2 feed flow, the membrane configuration, the pH of the electrolyte and the effect of possible contaminants in the CO2 source, we were able to gain important insights into the large-scale application of the Bi-based electrocatalyst for the electrochemical reduction of CO2 into formate.
Introduction
The electrochemical reduction of CO2 is an enticing technology to address anthropogenic-driven climate change as it allows the utilisation of renewable energy sources to enable the conversion of this greenhouse gas through reactions that would otherwise be limited by unfavourable thermodynamics (ΔG° > 0).1–3 Among the CO2 electrochemical reduction products (CO, C2H4, HCOO−, etc.), formic acid is a valuable building block due to its existing or potential industrial applications in textile manufacturing, as an antibacterial agent, in fuel cells and as an additive in cement production.2,4 In basic aqueous media, the electrochemical reduction of CO2 to formate at the cathode of an electrochemical cell is described by eqn (1), and is coupled with the oxygen evolution reaction (OER) at the anode (eqn (2)).| CO2 + 2e− + H2O → HCO2− + OH− | (1) |
| 2OH− → 2e− + H2O + ½O2 | (2) |
Among the electrocatalysts that have been studied for promoting the electrochemical CO2-to-formate conversion (e.g. based on Bi, Sn, and In as active species),4 Bi-based catalysts are the most promising option with respect to Faradaic efficiency (FE) and current density (j), particularly in comparison with Sn-based ones, which show lower stability and activity (Table S1†).1 In order to achieve industrially-relevant production rates and selectivity (j ≥ 100 mA cm−2, FEformate ≥ 90%),1 it is not only important to develop new electrocatalytic materials seeking lower overpotentials (η) but also to implement them in electrochemical cells with similar features to large-scale, industrial electrolysers. More specifically, the industrialisation of the sustainable electrochemical conversion of CO2 necessitates green advances in terms of the optimisation of cell parameters related to the energy requirements, recycling of the electrolyte and the stability of the electrocatalyst over long operation times. In this context, the use of a flow cell based on gas diffusion electrodes (GDEs) is of paramount importance to overcome CO2 mass transport limitations.5,6 Moreover, a variety of parameters can influence electrocatalyst performance and greatly affect the ultimate performance of the system. These include electrocatalyst loading, electrode preparation methods, electrode and membrane configuration, the presence of contaminants in the CO2 feed, and the nature of the electrolyte solutions and their pH.2 The choice of a suitable membrane separator between cathodic and anodic compartments is important to avoid diffusion of the produced formate to the anode and its consequent re-oxidation. For example, the diffusion of formate can be mitigated by the use of a Nafion membrane separating the cell compartments, but it cannot be entirely prevented,7 and the extent of this issue depends on the thickness of the Nafion membrane.8 Moreover, the pH of the electrolyte affects the CO2-to-formate onset potential, shifting it to more positive potentials with alkaline electrolytes (e.g. KOH), and this has been reported to limit the competitive HER.5,9 In addition, the separation of dilute formate solutions from the electrolyte and their concentration and acidification are challenges to be faced when developing this process,10 in order to produce pure and concentrated formic acid solutions. Specific flow cell designs aimed at tackling these challenges have recently been proposed.8,11–15 Another crucial parameter to enable the large-scale application of Bi electrocatalysts is their stability under operating conditions. Although multi-hour stability tests (t > 10 h) are currently included in most studies, only a few reports investigate the stability of Bi electrocatalysts in prolonged tests (t > 50 h, Table S1†) at industrially-relevant current densities. Additionally, insufficient attention is dedicated to the nevertheless crucial optimisation of the cell parameters, and to the understanding of how the cell configuration affects the electrocatalyst efficiency, and most importantly, the stability. In this work, we present a systematic study of the cell parameters for prolonged application (t = 65 h) of a highly scalable Bi-based electrocatalyst4 at industrially-relevant current densities (j = 100 mA cm−2) on a GDE in a two-compartment flow cell (Fig. 1a). While the investigation and improvement of the cell parameters are often neglected in flow cell system studies, we demonstrate here that these are critical factors to achieve the optimal utilisation of an electrocatalyst at high current densities.
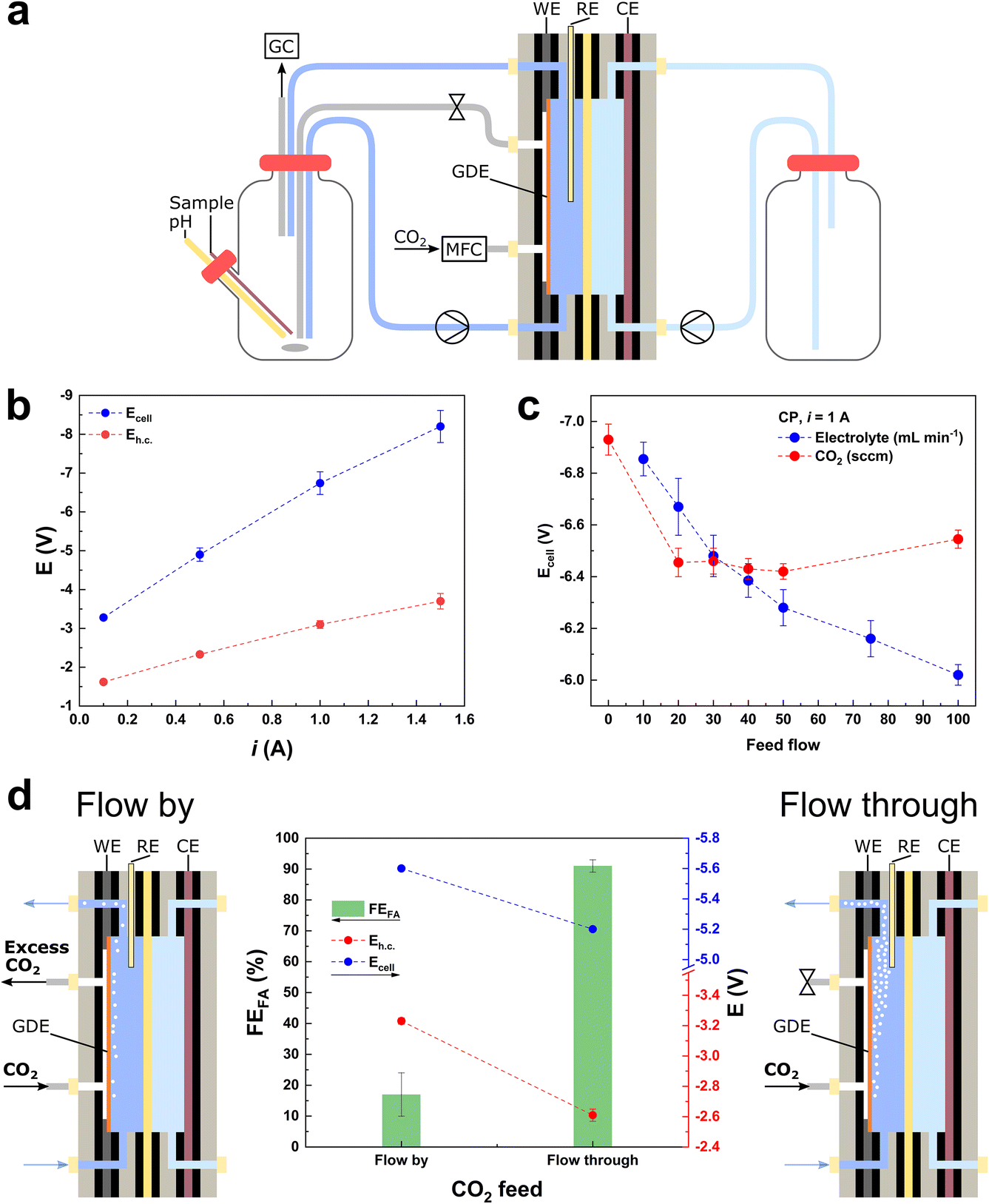 | ||
| Fig. 1 (a) Electrochemical flow cell system, equipped with a 10 cm2 GDE coated with the BiSub@AC-400 electrocatalyst, 0.5 M KHCO3 as the catholyte and 0.5 M H2SO4 as the anolyte. (b) i vs. Ecell dependency for the electrochemical reduction of CO2 in the flow cell setup (20 sccm CO2). (c) Ecell dependency on: (i) the electrolyte flow (blue) at constant 50 sccm CO2 feed flow and (ii) the CO2 feed flow (red) at constant 50 mL min−1 electrolyte flow in chronopotentiometric mode at i = 1 A (j = 100 mA cm−2). (d) Chronopotentiometry at i = 1 A (j = 100 mA cm−2), Faradaic efficiency (FE) towards formate (green bars), Ecell (blue dots) and Eh.c. (red dots). The “flow-by” configuration was held for 3 h and the “flow-through” configuration was held for 5 h, opening or closing the gas outlet of the flow cell, respectively (Fig. S1b and c†). | ||
Results and discussion
We started our investigation and optimisation of the electrocatalytic reduction of CO2 to formate in a flow cell set-up (Fig. 1a) by focusing on an evaluation of the general feasibility of the system at industrially-relevant current densities. The flow cell (FC) used in our study was equipped with an electrocatalyst recently developed by our group and consisting of highly dispersed, small Bi nanoparticles (average diameter: 5.5 nm) supported on activated carbon.4 The electrocatalyst was deposited on the GDE via spray coating, with a loading of 0.5 mg cm−2, in line with the majority of the literature reports for the electrochemical reduction of CO2 to formate (0.5–1 mg cm−2, Table S1†).2 The flow cell assembly was tested using 0.5 M KHCO3 as catholyte, 0.5 M H2SO4 as anolyte, and a Nafion membrane as separator between the two half-cells. The choice of such a flow-through set-up (Fig. 1a and S1b†) is based on the ease of the double compartment stacking, and the consideration that this is a mature technology for water electrolysers, which would facilitate the transfer of know-how to CO2 electrochemical conversion. In order to assess whether this system can sustain high cell current densities and thus be industrially applicable, it was tested in chronopotentiometric (CP) mode at increasing cell currents (i), from 0.1 to 1.5 A. The total cell potential (Ecell) increased at a higher rate with respect to the cathodic half-cell potential (Eh.c.), indicating that the major contribution to Ecell is due to the anodic half-cell potential and, therefore, to the OER (eqn (2) and Fig. 1b). This means that it is possible to use the proposed system at high currents, provided that the anodic electrocatalyst is optimised in order to decrease the Eanode and, therefore, the Ecell. After this proof of principle, we carried out a systematic and critical assessment of the parameters expected to exert the most relevant influence on the electrocatalyst performance (activity, selectivity and stability): (i) the CO2 feed (flow intensity, configuration and contaminants), (ii) the electrolyte feed (flow intensity, configuration, volume, pH and contaminants), and (iii) the membrane performance (catholyte/anolyte diffusion). In this context, it is important to take into account that the cell parameters and their effects on the electrocatalyst performance are often interlinked.Optimisation of the flows of CO2 and electrolyte
The behaviour of catalysts for the CO2 electrochemical reduction (CO2ER) is known to be greatly affected by the diffusion of the reactants and products (i.e., CO2, OH− and HCO2−) to and from the electrode.16 Therefore, we started our study by screening the flows of CO2 and electrolyte, with the purpose of ensuring that the measured electrocatalytic activity in the later experiments is not limited by the feed flows. Furthermore, the combined effect of these two feed flows plays a critical role in the wettability of the electrocatalyst surface and its exposure to the multiphasic reaction medium. Therefore, the optimisation of the flows determines the exposure of the surface of the electrocatalyst deposited on the GDE mesh to CO2 and to the electrolyte, which in turn affects the relative rates of the competing CO2ER and HER, and thus the overall cell efficiency.17 The effect of different CO2 and electrolyte feed flows was screened in CP (i = 1 A, Eh.c. = −3.0 ± 0.1 V) and chronoamperometric (CA) (Ecell = −6.5 V, Eh.c. = −2.9 ± 0.1 V) modes (Fig. 1c and S2,† respectively), revealing a monotonic increase in i and decrease in Ecell with the increment of the electrolyte flow up to 100 mL min−1. On the other hand, when screening the CO2 feed flow of up to 100 sccm, the highest i and lowest values of Ecell were obtained in the range of 20–50 sccm (Fig. 1c). Based on this result, we employed a value of CO2 flow within this range (50 sccm) in the rest of this work, to prevent limitations in the CO2 feed and to ensure the lowest Ecell (in CP mode). To give a measure of the importance of optimising these flows, it is worth noting that without changing the nature of the electrocatalyst and by tuning the feed flows, the Ecell can be decreased by 7–12% in CP mode (or current i can be increased by 18–24% in CA mode) in the tested flow ranges (electrolyte: 10–100 mL min−1 range and CO2: 0–100 sccm range). In addition, the CO2 flow can be fed to the flow cell in two configurations: flow-by and flow-through (Fig. 1d). The flow-through configuration showed the best performance (Fig. 1d), in terms of both Faradaic efficiency (FEformate = 90%, compared to FEformate = 15% for the flow-by configuration) and lowest cell potentials (−10% in Ecell and −23% in Eh.c.). The higher efficiency of the flow-through configuration compared to that of the flow-by one is in agreement with previous reports and has been ascribed to the combined optimisation of electrode wetting and local concentration of CO2 at the electrocatalyst surface.15,18 Given its better performance, the CO2 flow-through configuration was selected for the rest of the work. The fact that the flow-by and flow-through configurations lead to different performances is also important as an accessible probe of the mechanical integrity of the GDE: if a drastic increase in Eh.c. is observed during a flow-through measurement, a mechanical failure of the GDE (i.e., cracks) is likely to have occurred.Cell configuration and long-term stability
As the selected parameters of CO2 and electrolyte flows ensure that the electrocatalyst performance is not affected by diffusion limitations, we proceeded with the investigation of the role played by the electrolyte in combination with the cell membrane on the selectivity of the electrocatalyst. The electrolyte/membrane interplay controls the species that diffuse between the two cell compartments and their rate of transfer. If the transfer of the relevant species (OH− for reactions in alkaline medium) does not proceed optimally, the performance of the electrocatalyst can be negatively affected. For this set of experiments, we used KHCO3 as both the catholyte and the anolyte, in combination with a Nafion cationic exchange membrane (CEM), as this is the most commonly used combination in literature studies of the CO2ER. Under these conditions, our Bi-electrocatalyst displayed an excellent initial Faradaic efficiency towards formate (FEformate > 99%) at i = 0.5 A in CP mode (Fig. 2a).4 However, when prolonging the test at i = 0.5 A over 5 h (Fig. 2a), the values of FEformate and Eh.c. decreased, most likely due to the migration of K+ ions from the anolyte to the catholyte, which would increase the conductivity of the catholyte and decrease that of the anolyte. As a result, the anodic half-cell potential and, therefore, the Ecell drastically increased, with a major detrimental effect on the full cell efficiency. By restocking the electrolyte in both the catholyte and anolyte compartments every 5 h, the initial, optimum values of FE, Ecell and Eh.c. were restored (Fig. 2a, see values at 6 h and 11 h, i.e., just after restocking). This clearly shows that the observed drop in cell performance during the first 5 h of operation was not related to deactivation of the electrocatalyst but rather to the change in the composition of the electrolytes in the two compartments. It can be concluded that, although the use of the Nafion membrane prevents the diffusion of the formate product from the catholyte to the anolyte, it poses severe limitations on the species that migrate, allowing only the transfer of cations (K+ in this case) to ensure the charge balance within each compartment. This phenomenon was also recently reported by Kirner et al., but no solution to this issue was proposed.14 With the aim of solving the K+-diffusion issue, the Nafion membrane was substituted with an anion-exchange membrane (AEM) from Fumatech. The use of an AEM allows the diffusion of OH− species but also of the formate anion. As a consequence, the FEformate observed after 1 h of chronopotentiometry (70%, see Fig. 2b) was lower than the one previously reported with a Nafion membrane (Fig. 2a). Furthermore, formate was detected also in the anolyte, with the amount that diffused from the cathodic to the anodic compartment after 1 h of CP at i = 1 A corresponding to FEformate = 5%. The ratio between the Faradaic efficiency calculated from the formate quantified in the catholyte and anolyte (Fig. S3†) decreased from an initial value of 9.5 to 1.5 after 8 h and then remained stable for the next 14 h. These results prove that formate can diffuse through the AEM and then get oxidised at the anode. Furthermore, the fact that formate was observed in both compartments after prolonged testing indicates that the rate of production of formate at the cathode is higher than that of its diffusion through the membrane, which in turn is higher than the rate of oxidation at the anode. Despite the drawback of the undesired diffusion and oxidation of formate, the cathodic potential (Eh.c.) was more stable when using the AEM in comparison with the previous set-up with the Nafion membrane (Fig. 2a). In this test, the pH tended to stabilise at 8.2 ± 0.1 (Fig. S3†). It is worth noting that in the literature reports of long-time stability tests of the electrochemical reduction of CO2 to formate with KOH electrolyte and AEMs (e.g. Sustainion, Fumasep or Selemion, see Table S1†), the issues of formate diffusion and oxidation were never addressed.5,8,9,12,19–21 Only in one case was it specifically noted that the electrolyte was changed every 48 h.9 Discussion of the electrolyte volumes, electrolyte flows and recirculation is important to assess whether an electrochemical flow cell set-up can be scaled up to the industrial level. For example, a large electrolyte volume, combined with a low electrolyte flow and low CO2 consumption, can in principle allow the use of a CEM and pH > 7 without experiencing the above-mentioned issue related to cation-diffusion, or an AEM without the issue of major formate loss due to diffusion and oxidation. However, such a combination of parameters is not meaningful for large-scale application and should be discouraged. The cell configuration was further investigated using a Nafion membrane and 0.5 M H2SO4 as the anolyte. The choice of the acidic anolyte (pH = 0.3) leads to the local production of H+ during the OER at the anode and sustains their diffusion through the membrane. The FEformate remained > 90% for the first 18 h of chronopotentiometry at i = 1 A, and then gradually decreased to FEformate = 21% at 65 h of operation (Fig. 2c). The Ecell and Eh.c. remained constant during all the experiments at −6.4 ± 0.2 V and −3.0 ± 0.1 V, respectively (Fig. S4†). The decrease in Faradaic efficiency is attributed to the strong change recorded in the pH of the catholyte (2H+ form at the anode for each OH− formed at the cathode), which decreased from pH = 6.7 after 5 h to pH < 3.3 after 18 h. If on the one hand, the diffusion of H+ through the Nafion membrane towards the catholyte can sustain the cell operation for a much longer time with respect to the KHCO3 anolyte system (Fig. 2a), it acted detrimentally by facilitating the HER side-reaction and possibly dissolving the catalyst (Bi2O2CO3 or Bi2O3 surface layer4). To overcome the drawbacks brought about by the decrease in the pH of the catholyte, we decided to control the pHcatholyte (to pH = 7.5), via the manual injection of 5 M KOH every hour (Fig. 2d). This approach allowed the preservation of high Faradaic efficiency throughout the whole test, with only a relatively minor drop in the 65 h test at j = 100 mA cm−2 and with a final value of FEformate > 70%. This excellent performance allowed us to achieve the production of a 0.35 M HCOO− solution in 65 h. We envision that automatic pH control would be highly beneficial for the set-up, limiting or possibly even completely preventing the FEformate decrease. It is worth underlining that the visual inspection of the electrolyte throughout the 65 h test allowed excluding any detachment of the electrocatalyst from the GDE. | ||
| Fig. 2 Chronopotentiometry with 0.5 M KHCO3 catholyte at: (a) i = 0.5 A (j = 50 mA cm−2) with CEM and with 0.5 M KHCO3 as the anolyte; (b) at i = 1 A (j = 100 mA cm−2) with AEM (Fumatech) and with 0.5 M KHCO3 as the anolyte; (c) at i = 1 A (j = 100 mA cm−2) with CEM and with 0.5 M H2SO4 as the anolyte; and (d) at i = 1 A (j = 100 mA cm−2) with CEM and 0.5 M H2SO4 as the anolyte and pH = 7.5, controlled with injections of 5 M KOH every hour, increasing from 0.5 mL to 5 mL. Electrolyte flow at 35 mL min−1. FEformate based on formate detected in the catholyte (green bars), FEformate based on formate detected in the anolyte (red bars, only in b), pH (black dots), Ecell (blue dots) and Eh.c. (red dots). | ||
CO2 contaminants
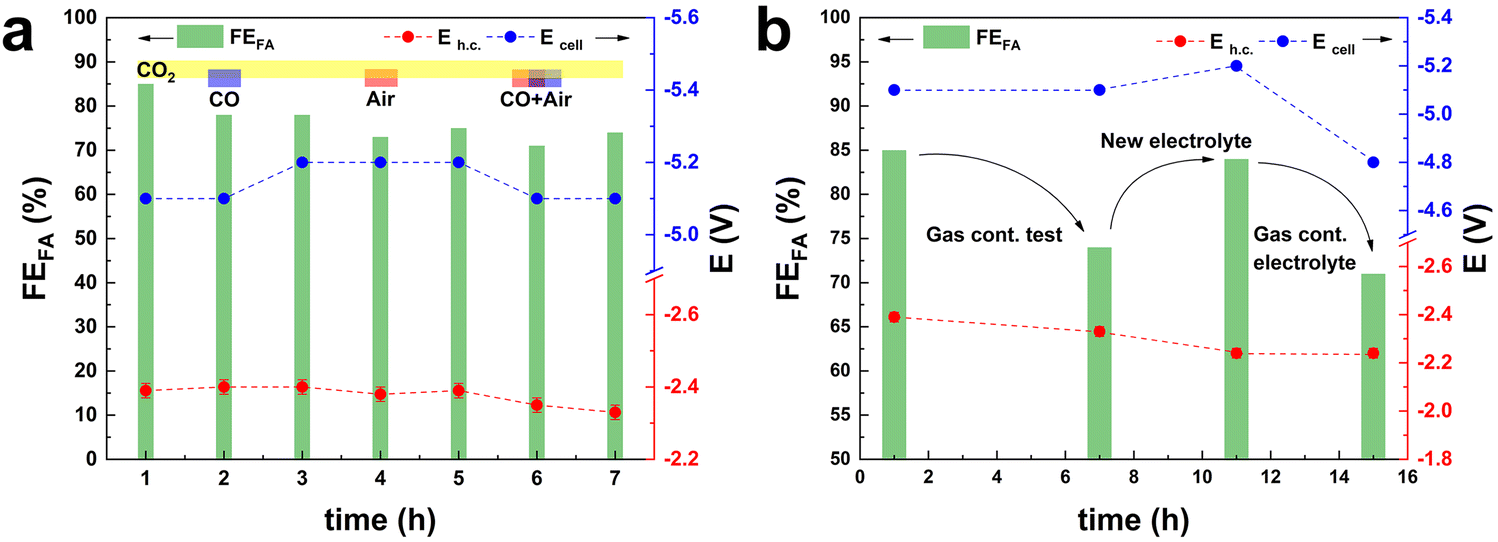
Another usually overlooked factor in the investigation of the CO2ER is the purity of the CO2 feed. The utilisation of CO2 from industrial flue gas as reactant source entails the presence of contaminants, such as CO and air (O2 and N2), that could act as poisoning agents and be detrimental to the electrocatalyst performance. This work was part of a H2020 project (Recode, https://recodeh2020.eu/), that aimed at capturing and utilising the CO2 emitted by a cement production factory (Titan Cement group, Kamari, Greece). In the composition of this industrial flue gas source (Table S2†), O2, N2 and CO account for the highest concentrations among the contaminants in the emitted CO2. Therefore, we chose to investigate the effect of CO and air (O2 and N2) on the electrocatalyst activity. CO poisoning was assessed by performing preliminary tests at −1.6 V vs. Ag/AgCl (j = 10 mA cm−2) over 5 h (Fig. S5†), and testing different CO concentrations in the range from 300 ppm up to 1200 ppm. No significant variation of FEformate was observed up to 750 ppm of CO, while a 10% decrease was recorded when the CO concentration reached 1200 ppm. O2 contamination tests were performed at −1.6 V vs. Ag/AgCl, for a total charge of 2 kC. The presence of O2 in the CO2 stream had a drastic impact on the Faradaic efficiency towards formate. In the presence of a 2.6% v/v concentration of O2, FEformate decreased to 37%. An increase in O2 concentration to 5.3% v/v led to a further, though less dramatic, decrease to FEformate = 12% and a further increase to 10.5% v/v led to no substantial further drop in terms of FEformate (Fig. S6†). The drastic decay of FEformate was accompanied by a net increase in current density (from 13 to 50 mA cm−2), indicating that the bismuth-based electrocatalyst displays high activity in the oxygen reduction reaction (ORR). This study of the effect of the contaminants present in CO2 streams is of great importance for the scale-up applicability of such an electrocatalyst. The obtained results prove that the presence of O2 in the catholyte due to possible cross-over of O2 produced at the anode or due to air leaks in the CO2 feed line would drastically decrease the CO2-to-formate conversion efficiency. In order to study the effect of the contaminants under our industrially-relevant conditions (j = 100 mA cm−2 at 50 mL min−1 of CO2), contamination tests were performed by adding 1200 ppm CO, 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm O2 and their combination to the pure CO2 inlet (Fig. 3a). The drop in FEformate (−8.2% in the presence of CO, −6.4% in the presence of O2 and −5.3% in the presence of CO and O2) cannot be solely ascribed to the decrease in CO2 concentration in the feed due to the dilution effect caused by the contaminant gas(es). The FEformate was only partially recovered after a pure CO2 stream was restored. When the gas feed was changed to Ar, no gas products or formate were observed (Fig. S7†), indicating that formate formation is exclusively ascribed to CO2 reduction. Remarkably, the replacement of the electrolyte after the test with the contaminants had a strong impact on the measured electrocatalyst selectivity: the original Faradaic efficiency towards formate was completely recovered when fresh electrolyte was employed in both the cathodic and anodic compartments (Fig. 3b). This suggests that contamination of the electrolyte takes place during these tests. This hypothesis is supported by the fact that removing the fresh electrolyte and reusing the electrolyte employed during the gas-contamination test led to a noticeable decrease in FEformate (−15%, Fig. 3b), similar to the one recorded during the gas-contamination test (−13%). This phenomenon prompted us to study more in-depth the effect of possible species present in the electrolyte. In particular, we investigated whether formic acid, which might be present in solution as a consequence of protonation of the produced formate, could affect the electrocatalyst. For this purpose, the GDEs coated with the Bi electrocatalyst (BiSub@AC-400) were treated with a 0.5 M solution of either HCOOK or HCOOH for 24 h under stirring prior to use in the flow cell. 0.5 M HCOO− was chosen as a representative final product concentration obtained with BiSub@AC-400/GDE (Fig. 2d). The untreated (Fig. 4a) and the treated GDEs (Fig. 4b and c) required similar Ecell and Eh.c. to sustain a current density of 100 mA cm−2. The HCOOK-treated GDE also showed a similar FEformate to the untreated one (with an average variability of ±4%). On the other hand, the HCOOH-treated GDE displayed consistently lower FEformate (−20% after 2 h and −50% after 10 h, Fig. 4d). ICP-OES elemental analysis of the electrolytes and treatment solutions (Table S3†) showed that Bi leaching was observed only upon treatment with HCOOH, whereas no Bi species were detected in the electrolyte solutions after the tests. XRD characterisation of the GDEs after the CO2ER tests (Fig. S8†) showed that the electrocatalyst is not affected by the treatment with HCOOK, whereas the treatment with HCOOH led to dissolution of the [Bi2O2CO3] phase present at the electrocatalyst surface.4 Therefore, we hypothesise that this is an active phase for the CO2ER and its dissolution is the cause of the partial loss of activity towards the reduction of CO2 to formate. This phenomenon was not observed during the stability tests (Fig. 2d), probably as a consequence of the pH control of the catholyte solution, which led to neutralisation of (most) formic acid, and thus to a much lower concentration of formic acid in the catholyte than in the HCOOH-contamination test.
000 ppm O2 and their combination to the pure CO2 inlet (Fig. 3a). The drop in FEformate (−8.2% in the presence of CO, −6.4% in the presence of O2 and −5.3% in the presence of CO and O2) cannot be solely ascribed to the decrease in CO2 concentration in the feed due to the dilution effect caused by the contaminant gas(es). The FEformate was only partially recovered after a pure CO2 stream was restored. When the gas feed was changed to Ar, no gas products or formate were observed (Fig. S7†), indicating that formate formation is exclusively ascribed to CO2 reduction. Remarkably, the replacement of the electrolyte after the test with the contaminants had a strong impact on the measured electrocatalyst selectivity: the original Faradaic efficiency towards formate was completely recovered when fresh electrolyte was employed in both the cathodic and anodic compartments (Fig. 3b). This suggests that contamination of the electrolyte takes place during these tests. This hypothesis is supported by the fact that removing the fresh electrolyte and reusing the electrolyte employed during the gas-contamination test led to a noticeable decrease in FEformate (−15%, Fig. 3b), similar to the one recorded during the gas-contamination test (−13%). This phenomenon prompted us to study more in-depth the effect of possible species present in the electrolyte. In particular, we investigated whether formic acid, which might be present in solution as a consequence of protonation of the produced formate, could affect the electrocatalyst. For this purpose, the GDEs coated with the Bi electrocatalyst (BiSub@AC-400) were treated with a 0.5 M solution of either HCOOK or HCOOH for 24 h under stirring prior to use in the flow cell. 0.5 M HCOO− was chosen as a representative final product concentration obtained with BiSub@AC-400/GDE (Fig. 2d). The untreated (Fig. 4a) and the treated GDEs (Fig. 4b and c) required similar Ecell and Eh.c. to sustain a current density of 100 mA cm−2. The HCOOK-treated GDE also showed a similar FEformate to the untreated one (with an average variability of ±4%). On the other hand, the HCOOH-treated GDE displayed consistently lower FEformate (−20% after 2 h and −50% after 10 h, Fig. 4d). ICP-OES elemental analysis of the electrolytes and treatment solutions (Table S3†) showed that Bi leaching was observed only upon treatment with HCOOH, whereas no Bi species were detected in the electrolyte solutions after the tests. XRD characterisation of the GDEs after the CO2ER tests (Fig. S8†) showed that the electrocatalyst is not affected by the treatment with HCOOK, whereas the treatment with HCOOH led to dissolution of the [Bi2O2CO3] phase present at the electrocatalyst surface.4 Therefore, we hypothesise that this is an active phase for the CO2ER and its dissolution is the cause of the partial loss of activity towards the reduction of CO2 to formate. This phenomenon was not observed during the stability tests (Fig. 2d), probably as a consequence of the pH control of the catholyte solution, which led to neutralisation of (most) formic acid, and thus to a much lower concentration of formic acid in the catholyte than in the HCOOH-contamination test.
 | ||
| Fig. 3 (a) Gas-contamination test (CO2 in yellow, CO in blue, and air in orange). (b) FEformate before and after the gas-contamination test, after restocking of the electrolyte (3 h) and after the reuse of the electrolyte utilised during the gas-contamination test (3 h). Conditions: chronopotentiometry at i = 1 A (j = 100 mA cm−2) for 7 h, 0.5 M KHCO3 as the catholyte and 0.5 M H2SO4 as the anolyte. Faradaic efficiency towards formate (green bars), Ecell (blue dots) and Eh.c. (red dots). In the presence of CO2 at pH = 7.5, controlled with injections of 5 M KOH every 2 h, increasing from 0.5 mL to 5 mL. | ||
 | ||
| Fig. 4 Chronopotentiometry at i = 1 A (j = 100 mA cm−2), 0.5 M KHCO3 as the catholyte and 0.5 M H2SO4 as the anolyte. FEformate (green bars), Ecell (blue dots) and Eh.c. (red dots). pH = 7.5, controlled with injections of 5 M KOH every hour, increasing from 0.5 mL to 5 mL for: (a) BiSub@AC-400 GDE; (b) BiSub@AC-400 GDE treated with a 0.5 M HCOOK bath for 24 h prior to FC use; and (c) BiSub@AC-400 GDE treated with a 0.5 M HCOOH bath for 24 h prior to FC use. (d) FEformate comparison between GDEs based on BiSub@AC-400 (black), BiSub@AC-400/HCOOK-treated (blue) and BiSub@AC-400/HCOOH-treated (red). | ||
Conclusions
In this work, we demonstrated that the implementation of the electrochemical reduction of CO2 in a flow cell can greatly benefit from the careful tuning of (i) electrolytes and CO2 feed flows and (ii) the cell configuration and the combined optimisation of electrolytes and membranes, including the control of pH. This allowed us to maximise the activity and selectivity of a Bi-based electrocatalyst for the reduction of CO2 to formate in a long (65 h) test at an industrially-relevant current density of j = 100 mA cm−2. Under the optimum conditions identified in this work, it was possible to convert CO2 into formate with high Faradaic efficiency throughout the whole test (FEformate > 70%) and in this way to produce a 0.35 M HCOO− solution in 65 h. Besides this notable green advance, our study also proved the limitations posed by the use of a cationic (Nafion) membrane in combination with 0.5 M KHCO3 electrolytes, as under these conditions the diffusion of K+ through the membrane hampers the long-term performance of the cell and, therefore, strongly limits the large-scale application of the process. We investigated the use of an anionic membrane (Fumasep) as an alternative, but this leads to the diffusion of formate towards the anodic compartment and its consequent oxidation, leading to a marked drop in Faradaic efficiency. With the current design of the flow cell, the best option identified in this work consists of combining the use of a cationic membrane (Nafion) and 0.5 M H2SO4 as the anolyte, with a system to control the pH of the catholyte. Further improvements can be expected by investigating the effects of highly recirculated electrolytes vs. semi-batch experiments. Future studies should also focus on the exploration of novel flow cell designs such as 3-chamber electrolysers (Table S1†)8 and membrane-less electrolysers.22 Another factor that should be increasingly investigated is the influence of the contaminants that are often present in industrial CO2 streams. Here, we studied the performance of our Bi-based electrocatalyst (BiSub@AC-400) with CO2 containing the contaminants typically present in CO2 emissions of the cement industry (CO and O2). It was concluded that the electrocatalyst is also active in the oxygen reduction reaction, which means that this reaction strongly competes with the reduction of CO2 in the presence of an O2 concentration as low as 2.6% v/v. This observation is of great importance for scaling up the system, as it indicates that special care should be taken to prevent cross-diffusion of O2 towards the catholyte or gas (air) leaks in the CO2 feeding line. Altogether, this study allowed us to achieve important enhancements in the flow cell application of the electrocatalytic reduction of CO2 to formate using an affordable, up-scalable and selective Bi-based electrocatalyst, and at the same time it led to the identification of the main limitations in the current flow cell design for this reaction. Therefore, we believe that this work can inspire future developments towards the large-scale application of the electrochemical reduction of CO2 to valuable products, which is a very attractive target reaction in the context of green chemistry.Experimental section
Materials
Bismuth(III) subsalicylate 99.9% (BiSub), ethanol 96%, dimethyl sulphoxide 99.9% (DMSO), sodium bicarbonate >99.7% (NaHCO3), potassium bicarbonate >99.7% (KHCO3), formic acid >95%, potassium formate >99%, KOH pellets, conc. H2SO4 solution, and Nafion perfluorinated ion-exchange resin, 10 wt% dispersion in water, were purchased from Sigma Aldrich. Norit SX1G activated carbon (AC) was purchased from Cabot. The Nafion 115 and the Fumasep (FAA-3-pk-75, reinforced) membranes were purchased from the Fuel Cell Store.Electrocatalyst synthesis
The electrocatalyst was prepared by scaling up a synthesis method reported previously by our group.4 In a 200 mL round-bottom flask, 1.50 g of activated carbon was mixed with 1.76 g of bismuth(III) subsalicylate and 75 mL of ethanol. After stirring overnight (8 h), the slurry was dried via rotaevaporation and dried overnight (8 h) at 90 °C in an oven. The dried powder was divided into two quartz boats (1.5 g each) and pyrolysed under N2 atmosphere (flow: 100 mL min−1) at 400 °C for 1 h (ramp: 25 °C min−1) in a tubular oven, with a final mass yield of 84%. The obtained material was labelled BiSub@AC-400. The detailed physicochemical characterisation of this material before and after electrocatalytic testing has been reported previously.4GDE preparation
The gas diffusion electrodes (GDEs) were prepared as follows: first, 117.6 mg of the electrocatalyst powder was dispersed in 13.8 mL of acetone (or ethanol) with 177 μL (159 mg) of Nafion (5 wt%) suspension and sonicated for 30 min in an ultrasonic bath. The suspension was then loaded onto an Iwata Eclipse HP BCS airbrush and the catalyst was spray-coated on a carbon paper (39.3 mm × 43 mm; GDL 29 BC Sigracet), up to a deposition of 9 mg (0.5 mg cm−2) of the electrocatalyst (based on the dried GDE).Nafion membrane activation
The Nafion 115 membrane was purchased from the Fuel Cell Store and, before use, it was treated with 3 wt% of aqueous H2O2 at 80 °C for 1 h in order to remove organic impurities. Then, it was activated using a 0.5 M aqueous solution of H2SO4 at the same temperature for 1 h. Finally, the Nafion membrane was washed with deionised H2O until a neutral pH was reached.Flow cell set-up
The experiments were performed with a Microflow cell from Electrocell. The flow cell is made from PTFE (body and spacers) and is equipped with a 10 cm2 (34 mm × 30 mm) active area GDE as working electrode (WE), a Mixed Metal Oxide Dimensionally Stable Anode (MMO DSA) as counter electrode (CE), leak-free Ag/AgCl (3.5 M KCl, Alvatek: LF-1-100) as reference electrode (RE) and a Nafion or a Fumatech (Fumasep FAA-3-pk-75, reinforced) membrane. The gas and electrolyte lines were connected to the cell as depicted in Fig. 1a and S1.† The CO2 line tubing was provided with a transparent plastic section to monitor the possible presence of any trace of liquid or salt crystals during the experiments. Additionally, the gas chamber (back of the GDE) was visually inspected after every experiment. In this way, we determined that no flooding occurred during our experiments.If not otherwise specified, 0.5 M KHCO3 (205 g) and 0.5 M H2SO4 (210 g) were respectively used as the catholyte and anolyte. Unless otherwise specified, the electrolyte flow was set to 50 mL min−1 and the CO2 feed was set to 50 sccm. The catholyte and anolyte were continuously recirculated. In the experiments in which the pH was controlled, the catholyte pH was maintained constant by the addition of small aliquots of 5 M concentrated KOH (0.5–5 mL). The flow cell was connected to an Autolab potentiostat (10 A booster) with Nova software and the total cell potential was measured with a multimeter (WE to CE).
Product characterisation
The analysis of the liquid products was carried out via HPLC on an Agilent Technologies 1200 series, equipped with a Bio-Rad Aminex HPX-87H 300 × 7.8 mm column at T = 60 °C with 0.5 mM aqueous H2SO4 as eluent (0.55 mL min−1) and a refractive index detector. The HPLC samples were prepared by mixing 1.0 g of sample and 0.5 g of a standard aqueous solution containing DMSO (0.03 M) and NaHCO3 (0.5 M). The liquid products derived from the contamination tests were analysed using HPLC (Shimadzu Nexera-I LC-2040C), equipped with an Aminex HPX-87H column at T = 40 °C with 8 mM H2SO4 as eluent (0.8 ml min−1) and a photodiode array (PDA) detector. The detection of the gas products was carried out with an online compact GC (CGC) from Interscience, equipped with Paramond (CO2, C2+) and Molsieve (H2, O2, N2, CO, CH4) columns, while for the contamination tests a micro gas chromatograph (Micro GC Agilent) with Molsieve 5 Å and PoraPLOT Q columns was used.Based on the concentration of formate determined using HPLC, the Faradaic efficiency (FEformate) was calculated using the following equation:
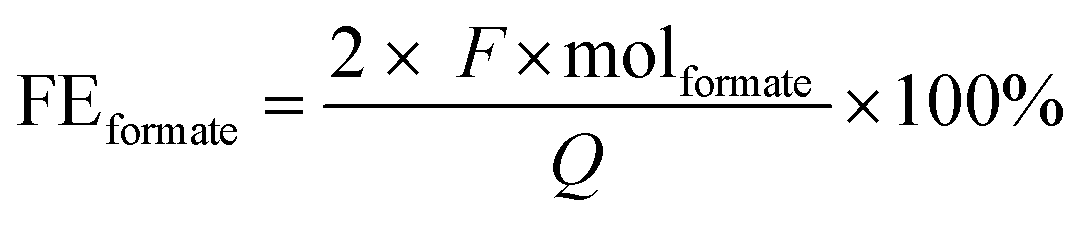 485.3 C mol−1), Q is the charge measured during the test (C) and molformate are the produced moles of formate (mol). The Faradaic efficiency is thus a measure of the selectivity of the electrochemical process towards the synthesis of formate. Besides formate, H2 was the only other product that was observed in this study (no other gaseous or liquid side products were detected). Since formate was the only carbon-containing product detected during the electrocatalytic tests, the selectivity relative to CO2 was >99.9% towards formate.
485.3 C mol−1), Q is the charge measured during the test (C) and molformate are the produced moles of formate (mol). The Faradaic efficiency is thus a measure of the selectivity of the electrochemical process towards the synthesis of formate. Besides formate, H2 was the only other product that was observed in this study (no other gaseous or liquid side products were detected). Since formate was the only carbon-containing product detected during the electrocatalytic tests, the selectivity relative to CO2 was >99.9% towards formate.
Based on the Bi loading, the turnover frequency (TOF) of the GDE containing BiSub@AC-400 at j = 100 mA cm−2 (i = 1 A) was calculated to be TOFformate = 3.49 s−1 × FEformate (see Fig. S9 and the related text in the ESI† for further information).23,24
ICP-OES analysis
Analysis by inductively coupled plasma–optical emission spectrometry (ICP-OES) was carried out using an Optima 7000 from PerkinElmer. For the analysis, 1.0 g of the sample was diluted with 1% HNO3 until a volume of 11 mL was obtained. The ICP-OES measurements were carried out in duplicate, from which the average and standard deviation were calculated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the EU H2020 project RECODE (grant number 768583, https://www.recodeh2020.eu) for the funding of this project, Titan Cement for the collaboration and for providing the flue gas samples, and Avantium for useful discussion. We acknowledge Marcel de Vries and Erwin Wilbers for technical support and Hans van der Velde for the ICP-OES analysis.References
- R. I. Masel, Z. Liu, H. Yang, J. J. Kaczur, D. Carrillo, S. Ren, D. Salvatore and C. P. Berlinguette, Nat. Nanotechnol., 2021, 16, 118–128 CrossRef CAS PubMed.
- M. F. Philips, G.-J. M. Gruter, M. T. M. Koper and K. J. P. Schouten, ACS Sustainable Chem. Eng., 2020, 8, 15430–15444 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- M. Miola, B. C. A. de Jong and P. P. Pescarmona, Chem. Commun., 2020, 56, 14992–14995 RSC.
- T. Fan, W. Ma, M. Xie, H. Liu, J. Zhang, S. Yang, P. Huang, Y. Dong, Z. Chen and X. Yi, Cell Rep., 2021, 2, 100353 CAS.
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC.
- D. Kopljar, A. Inan, P. Vindayer, N. Wagner and E. Klemm, J. Appl. Electrochem., 2014, 44, 1107–1116 CrossRef CAS.
- H. Yang, J. J. Kaczur, S. D. Sajjad and R. I. Masel, J. CO2 Util., 2017, 20, 208–217 CrossRef CAS.
- L. Li, A. Ozden, S. Guo, A. d. A. F. P. Garci, C. Wang, M. Zhang, J. Zhang, H. Jiang, W. Wang, H. Dong, D. Sinton, E. H. Sargent and M. Zhong, Nat. Commun., 2021, 12, 5223 CrossRef CAS PubMed.
- M. Ramdin, A. R. T. Morrison, M. de Groen, R. van Haperen, R. de Kler, E. Irtem, A. T. Laitinen, L. J. P. van den Broeke, T. Breugelmans, J. P. M. Trusler, W. d. Jong and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2019, 58, 22718–22740 CrossRef CAS.
- C. Xia, P. Zhu, Q. Jiang, Y. Pan, W. Liang, E. Stavitski, H. N. Alshareef and H. Wang, Nat. Energy, 2019, 4, 776–785 CrossRef CAS.
- L. Fan, C. Xia, P. Zhu, Y. Lu and H. Wang, Nat. Commun., 2020, 11, 3633 CrossRef CAS PubMed.
- W. Lee, Y. E. Kim, M. H. Youn, S. K. Jeong and K. T. Park, Angew. Chem., Int. Ed., 2018, 57, 6883–6887 CrossRef CAS PubMed.
- J. Kirner, Y. Chen, H. Liu, J. Song, J. Liao, W. Li and F. Zhao, J. Electrochem. Soc., 2022, 169, 054511 CrossRef.
- D. Ma, T. Jin, K. Xie and H. Huang, J. Mater. Chem. A, 2021, 9, 20897–20918 RSC.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- M. E. Leonard, L. E. Clarke, A. Forner-Cuenca, S. M. Brown and F. R. Brushett, ChemSusChem, 2020, 13, 400–411 CrossRef CAS PubMed.
- M. Duarte, B. De Mot, J. Hereijgers and T. Breugelmans, ChemElectroChem, 2019, 6, 5596–5602 CrossRef CAS.
- X. Wang, W.-J. Yin, Y. Si, X. Wang, X. Guo, W. Guo and Y. Fu, J. Mater. Chem. A, 2020, 8, 19938–19945 RSC.
- Q. Gong, P. Ding, M. Xu, X. Zhu, M. Wang, J. Deng, Q. Ma, N. Han, Y. Zhu, J. Lu, Z. Feng, Y. Li, W. Zhou and Y. Li, Nat. Commun., 2019, 10, 2807 CrossRef PubMed.
- J. Yang, X. Wang, Y. Qu, X. Wang, H. Huo, Q. Fan, J. Wang, L. M. Yang and Y. Wu, Adv. Energy Mater., 2020, 10, 2001709 CrossRef CAS.
- M. M. Monroe, P. Lobaccaro, Y. Lum and J. W. Ager, J. Phys. D: Appl. Phys., 2017, 50, 154006 CrossRef.
- P. Lamagni, M. Miola, J. Catalano, M. S. Hvid, M. A. H. Mamakhel, M. Christensen, M. R. Madsen, H. S. Jeppesen, X. M. Hu, K. Daasbjerg, T. Skrydstrup and N. Lock, Adv. Funct. Mater., 2020, 30, 1910408 CrossRef CAS.
- S. Anantharaj, P. E. Karthik and S. Noda, Angew. Chem., Int. Ed., 2021, 60, 23051–23067 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc04482j |
| This journal is © The Royal Society of Chemistry 2023 |