
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocatalytic hydroboration of olefins in pyrrolidinium ionic liquids†
Paweł
Huninik
 ab,
Jakub
Szyling
a,
Agnieszka
Czapik
b and
Jędrzej
Walkowiak
*a
ab,
Jakub
Szyling
a,
Agnieszka
Czapik
b and
Jędrzej
Walkowiak
*a
aCenter for Advanced Technology, Adam Mickiewicz University, Uniwersytetu Poznańskiego 10, 61-614 Poznań, Poland. E-mail: jedrzejw@amu.edu.pl
bFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland
First published on 24th April 2023
Abstract
An efficient, simple, metal and ligand free, regioselective hydroboration of alkenes has been developed using pinacolborane (HBpin) as a reducing reagent and 1-ethyl-methylpyrrolidinium triflate [EMPyrr][OTf] as a non-corrosive and recyclable organocatalyst. The described protocol is scalable and compatible with a variety of alkenes offering the desired organoboron compounds in moderate-to-excellent yields. The system was reused in 30 cycles with comparable effectivity and stable selectivity of the process, increasing its sustainability. The utility of the obtained alkylboronates was checked in various subsequent transformations such as Suzuki–Miyaura coupling, Zweifel olefination and oxidation. Moreover, a reaction mechanism based on previously reported literature and NMR studies was proposed.
Introduction
Metal-free catalysis has received a lot of attention in chemical transformations as it is a promising method for the synthesis of compounds uncontaminated by metals. Ionic liquids (ILs) constitute an attractive alternative for this explicit purpose. Thanks to their unique properties, such as negligible vapor pressure, incombustibility, high conductivity, tremendous structural variability and good solvent capabilities, ILs are considered to be one of the most significant media among the ecologically friendly solvents for designing green and sustainable processes.1 This group of substances, which consists of an organic or inorganic anion and a large organic cation, have found widespread application in industrial and academic laboratories. They can be deliberately constructed with specialized functions by selecting component ions that are task-specific.2 ILs have demonstrated interesting applications in chemical reactions as solvents,3 solutions for metal extraction,4 battery electrolytes,5–7 components of solar cells,8 catalysts in e.g., hydration of propargylic alcohols,9 oxidation,10 CO2 conversion,11–13 biomass processing14,15 and many others.In our research we are focusing on the development of effective catalytic methods for the synthesis of organoboron and organosilicon compounds.16–21 These processes are mainly catalyzed by expensive, moisture and air sensitive TM-complexes, which generate problems with process sustainability, catalyst stability and recyclability as well as the need to create inert conditions. Therefore, because of their high reactivity, low toxicity and ease of storage, organoboron compounds are valuable intermediates in C–C, C–N, C–O and C–X bond-forming reactions and other demetallation protocols, e.g., the Suzuki–Miyaura reaction,22–24 Zweifel olefination,25 halodemetallation,26,27 Chan–Lam coupling,28,29etc. Consequently, there is a need for the development of green techniques to synthesize these compounds.
Due to its 100% atom efficiency and high selectivity, hydroboration is one of the most important methods used to introduce boryl functionalities into the structure of organic compounds. Addition of B–H to alkenes depending on well-defined transition metal (TM) catalysts has advanced greatly in recent years.30 Several TM-complexes involving Ti,31,32 Zr,31,33 Mn,34,35 Fe,36,37 Ru,38 Co,39,40 Rh,41,42 Ir,43,44 Ni,45,46 Cu47,48 and Ag49 have shown remarkable catalytic activity, resulting in the formation of a wide range of valuable organoboranes (Scheme 1a). However, in addition to TM-precatalysts, additional ligands, bases and other additives are frequently required. Although compounds based on the main group elements are emerging as effective alkene hydroboration catalysts, the need for an excess of boronating agent, use of organic solvents, frequently low stability of catalyst, tedious workup and purification procedures and constrained scope of substrates limits the applications of these catalysts.50–55 To address these challenges, the development of green protocols for recyclable and sustained use, particularly in large-scale applications, is highly important. The goal of making hydroboration entirely metal-free was achieved by Nguyen et al. in their work on tropylium salts (Scheme 1b).56 The authors demonstrated the organocatalytic utility of the tropylium cation, which serves as a Lewis acid. Although this approach has its merits, this organocatalytic system was studied on narrow alkene scope and no effort was made to recycle the catalyst. Recently, our group reported that efficient immobilization of ruthenium catalyst in ILs provides the environmentally benign hydroboration of alkynes with multiple reuses of the IL and the catalyst, permitting the omission of waste-generating and cost-consuming purification steps.57 Herein, as a continuation of our work on the hydroboration of unsaturated C–C bonds,58,59 we report for the first time an ionic liquid organocatalyzed anti-Markovnikov selective hydroboration of mono- and 1,1-disubstituted terminal alkenes (Scheme 1c). Our approach aims to create a more environmentally friendly method by the elimination of TM-complexes, various additives and a huge amount of toxic organic solvents.
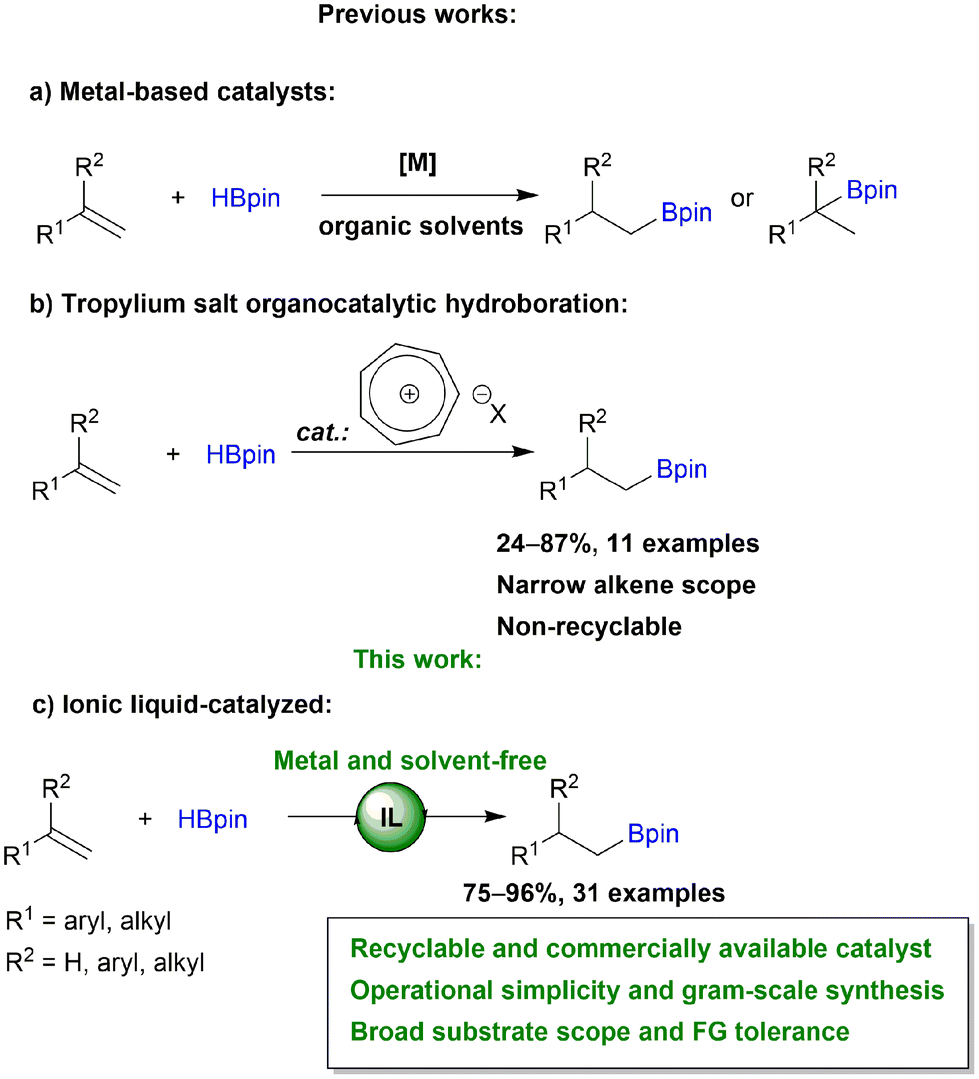 | ||
| Scheme 1 Catalytic hydroboration of alkenes. | ||
Results and discussion
We started our investigations by examining the hydroboration of styrene (1a) as a model substrate with 1.0 equiv. of pinacolborane (HBpin) to form alkylboronic ester (2a) – anti-Markovnikov product. Reactions were performed under completely organic solvent-free conditions using various commercially available ionic liquids (IL1–10) based on quaternary ammonium cations and inorganic or organic anions. At the outset, the control reaction between 1a and HBpin was conducted at 60 °C in the presence of IL1 (0.5 equiv.). After 24 h, product 2a was obtained with high selectivity, but low conversion of olefin was observed (Table 1, entry 1). Increasing the reaction temperature to 110 °C, conversion of 1a was improved to 95% (Table 1, entry 2). The process occurred with excellent regioselectivity, giving linear alkylboronic ester 2a as a single isomer. Slightly lower conversion of 1a was observed when other pyrrolidinium-based ionic liquids IL2 and IL3 were used (Table 1, entries 3 and 4). A significant drop in reaction yield was noted for the imidazolium-based and choline-based ionic liquids (IL3–9) for which the conversion did not exceed 60% (Table 1, entries 5–10). On the basis of IL screening, 1-ethyl-1-methylpyrrolidinium triflate [EMPyrr][OTf] (IL10) was chosen as the best catalyst for the B–H addition to styrene (1a), leading to its full conversion with excellent regioselectivity (Table 1, entry 11). When the reaction temperature was lowered to 80 °C, the efficiency of the process decreased, converting 1a at 91% (Table 1, entry 12). However, proceeding the reaction at 100 °C was sufficient for an excellent reaction yield (Table 1, entry 13). During the optimisation studies we established the lowest catalyst loading (40 mol%) by which the loss of product yield was not observed (Table 1, entries 15 and 16).|
|
|||||
|---|---|---|---|---|---|
| Entry | IL | Conv. of 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a [%] a [%] |
Temp. [°C] | Yield of 2aa [%] |
Selectivity (2a/3a)b |
| Reaction conditions: [IL]:[1a]:[HBpin] = 0.5:1:1, inert atmosphere, 24 h.a Determined by GC-MS analysis.b Determined by GC-MS and 1H NMR analyses.c [IL]:[1a]:[HBpin] = 0.3:1:1.d [IL]:[1a]:[HBpin] = 0.4:1:1.e 20 h reaction time.f 18 h reaction time. |
|||||
| 1 | IL1 | 26 | 60 | 25 | 98/2 |
| 2 | IL1 | 95 | 110 | 95 | >99/0 |
| 3 | IL2 | 80 | 110 | 78 | 98/2 |
| 4 | IL3 | 82 | 110 | 81 | 99/1 |
| 5 | IL4 | 11 | 110 | 10 | 94/6 |
| 6 | IL5 | 48 | 110 | 45 | 94/6 |
| 7 | IL6 | 56 | 110 | 53 | 96/4 |
| 8 | IL7 | 59 | 110 | 56 | 95/5 |
| 9 | IL8 | 10 | 110 | 10 | 99/1 |
| 10 | IL9 | 15 | 110 | 15 | 98/2 |
| 11 | IL10 | >99 | 110 | >99 | >99/0 |
| 12 | IL10 | 91 | 80 | 89 | 98/2 |
| 13 | IL10 | >99 | 100 | >99 | >99/0 |
| 14c | IL10 | 90 | 100 | 90 | >99/0 |
| 15d | IL10 | >99 | 100 | >99 | >99/0 |
| 16d,e | IL10 | >99 | 100 | >99 | >99/0 |
| 17f | IL10 | 96 | 100 | 96 | >99/0 |
| 18 | — | 8 | 110 | 7 | 93/7 |

The results obtained in Table 1 highlighted the importance of counterions in the structure of ionic liquids. Imidazolium-based ionic liquid containing triflate anion showed worse catalytic activity, which emphasizes the role of pyrrolidinium ion. One of the reasons for this occurrence could be that [EMPyrr]+ ion effectively coordinates with the oxygen atoms of HBpin (for details see mechanism discussion). Furthermore, simple triflate salt can also catalyze the reaction but with lower efficiency than [EMPyrr]+ ion (see Table S1 in the ESI† for more details on catalysts scope). To exclude the non-catalytic hydroboration of 1a, the control test was performed without IL10. As expected, the absence of IL10 resulted in only 8% conversion of 1a.
With the selected [EMPyrr][OTf] IL10 and optimized reaction conditions in hand, the substrate scope of the anti-Markovnikov hydroboration was investigated using a variety of olefins, including substituted styrenes (1a–1n), allylic derivatives (1p–1v) and 1,1-disubstituted alkenes (1x–1ae), (Scheme 2). Styrene derivatives bearing sterically hindered groups such as t-butyl (1b), 2,4,6-trimethyl (1c) or phenyl (1d) substituted at the para position showed very good reactivity and anti-Markovnikov selectivity. The reaction also worked efficiently for 2-vinylnaphthalene (1e), giving an excellent yield and regioselectivity. Smooth hydroboration of different aromatic alkenes with both electron-withdrawing and electron-donating functionalities (1f–1n) was observed. Lower efficiency was noted for styrene with ortho-substituted methoxy group (1l). The catalyst tolerates functional groups such as halogens (1f–j, 1r–1s, 1ab–ad), methoxy (1k–1l, 1t–1v, 1aa), tosyl (1w), methyl benzoate (1m) and amino (1n) presented in substrates, providing products capable of further transformations. The hydroboration of dimethylphenylvinylsilane (1o) allowed product 2o to possess silyl and boryl groups simultaneously at 91% yield. It should be highlighted that the reaction was not limited to styrenes, but allylic (1p–1v) and aliphatic (1w) olefins were also compliant to this procedure. Reaction with eugenol (1v) gave alkylboronic ester 2v with 81% yield, indicating that the catalyst is also tolerant to hydroxy groups. Product 2v was fully characterized for the first time. A crystal structure was also proved by X-ray analysis, confirming anti-Markovnikov addition of boryl moiety (Fig. 1). Moreover, this reaction demonstrated tolerance for bulky groups. It was proven by the hydroboration reactions of sterically demanding 1,1-disubstituted olefins (1x–1ae), which still proceeded smoothly, affording the corresponding products (2x–2ae) in high yields and selectivity.
 | ||
| Scheme 2 The scope of products obtained in the [EMPyrr][OTf] catalyzed hydroboration of alkenes. Reaction conditions: [EMPyrr][OTf]:[HBpin]:[1a–1ae] = 0.4:1:1, 100 °C, 20 h, Ar. Isolated yields are presented. The ratio of 2a–2ae/3a–3ae was determined by GC-MS and 1H NMR analyses of the crude product. a48 h. | ||
 | ||
| Fig. 1 Structural formula of product 2v. Crystal structure was confirmed by X-Ray analysis (ESI, Table S4†). | ||
In the next stage of our study, the recyclability of the catalytic system was verified in repetitive batch experiments with multiple reuses of IL10. The IL10 could easily be recycled several times, and when the reaction was completed, the products were extracted with n-heptane in almost quantitative yields and analysed by GC-MS and NMR, which showed excellent selectivity towards the anti-Markovnikov product in each run (≥97%). During the extraction process, the biphasic n-heptane/IL system was observed until most of the IL had become solidified due to the heat exchange with the solvent used for the extraction. After that, the products were easily separated from IL via decantation. The results of the hydroboration of 1a shown in Fig. 2 demonstrate the high stability and effectivity of the catalytic system used, IL10, which can be recycled 30 times before observation of a slight drop in its activity. Successful completion of 25 runs was also achieved when 0.4 equivalents of IL10 were used. However, a decrease in the conversion of reagents was noticed afterward (ESI, Plot S1†). Lowering the activity of the catalyst may be related to the presence of water in the system, which is accumulated during the extraction process. The controlled addition of a small amount of water significantly inhibited the system (ESI, Table S3,† entry 9).
 | ||
| Fig. 2 Hydroboration of styrene (1a) with HBpin under repetitive batch mode using [EMPyrr][OTf] (0.5 equiv.) at 100 °C for 20 h.aDetermined by GC-MS analysis. Selectivity of anti-Markovnikov/Markovnikov products was determined by GC-MS and 1H NMR analyses. | ||
A set of control experiments was conducted to obtain a better understanding of the possible mechanism for the [EMPyrr][OTf]-catalyzed hydroboration of alkenes using HBpin. Initially, we selected chloroform-d1 and benzene-d6 for NMR studies, as it was discovered that the hydroboration reaction was optimal in the presence of CHCl3 and C6H6 (ESI, Table S3†). The 1H and 11B NMR analyses revealed that the addition of styrene (1a) (0.2 mmol) to a mixture of IL10 (0.5 equiv.) and HBpin (1.0 equiv.) in chloroform-d1 at 100 °C led to the formation of the hydroboration product 2a (ESI, Fig. S80 and S81†). In order to gain a deeper understanding of the probable mechanism, it was crucial to carry out a comprehensive investigation into the activation mode of our ionic liquid catalyst. Thomas and co-workers made significant discoveries regarding previously unknown pathways in hydroboration chemistry, which involved the catalytic activity of BH3 generated in situ from nucleophilic catalysts or ligands and HBpin.60 To determine if the BH3 species formed in situ acts as a hidden catalyst driving the reaction, the [EMPyrr][OTf]-catalyzed hydroboration reaction of styrene (1a) was performed with the addition of N,N,N′,N′-tetramethylethylenediamine (TMEDA) (Table 2, entries 1–3). The addition of TMEDA, was expected to inhibit the reaction, as it can form mono- and bisadducts with BH3 that are much less catalytically active. Additionally, TMEDA adducts can be detected from characteristic signals in NMR spectra. The results of this mechanistic control study showed that it did not completely suppress the reaction, as hydroboration products 2a and 3a were still formed. Moreover, it was also possible to observe changes in the selectivity of the boronic esters that were produced. However, the NMR spectra revealed that BH3 had not been formed and coordinated with TMEDA, which ruled out the possibility of hidden borane catalysis (ESI, Fig. S101 and 102†). Rather, new and unidentified signals were detected, which could suggest the coordination of TMEDA to the [EMPyrr]+ ion or other intermediates formed during the process. Diazabicyclo[2.2.2]octane (DABCO) was also tested to see whether it inhibited the process (Table 2, entries 4 and 5). Presumably, the strong binding of TMEDA and DABCO effectively reduces the Lewis acidity of the [EMPyrr]+ ion or intermediates formed during the process, causing our catalytic system to become inactive.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Tertiary amine | Tertiary amine equiv. | Conv. of 1aa [%] |
Yield of 2aa [%] |
Selectivity (2a/3a)b |
| Reaction conditions: [IL]:[1a]:[HBpin] = 0.4:1:1, inert atmosphere, 100 °C, 20 h.a Determined by GC-MS analysis.b Determined by GC-MS and 1H NMR analyses. |
|||||
| 1 | TMEDA | 0.1 | 48 | 27 | 57/43 |
| 2 | TMEDA | 0.4 | 36 | 25 | 70/30 |
| 3 | TMEDA | 1.0 | 24 | 15 | 62/38 |
| 4 | DABCO | 0.4 | 26 | 24 | 93/7 |
| 5 | DABCO | 1.0 | 11 | 11 | 96/4 |

These results led us to the hypothesis that the [EMPyrr]+ ion could interact with the oxygen atoms of HBpin, enhancing the polarization of the B–H bond and promoting the addition to the double bond of alkene (Scheme 3a). Due to the delocalization of the positive charge of tetraalkylammonium ion on the α-hydrogen atoms, these atoms can interact with an oxygen atoms through hydrogen bonding.61–63 The 1H NMR spectrum of a 1:2 mixture of [EMPyrr][OTf] with HBpin at room temperature or after heating to 100 °C (to mimic reaction conditions) provided some indication to support this hypothesis. The same outcome was observed when an excess of ionic liquid was used or the solvent was switched to benzene-d6 (ESI, Fig. S82, S86, S89 and S92†). However, the spectroscopic data did not conclusively confirm our assumptions, as the presence of hydrogen bonds could not be ascertained through the 1H–1H NOESY NMR spectra (ESI, Fig. S100†). Additionally, we discovered that another significant transformation occurs between these two species. After the 1:2 mixture had been heated to 100 °C and this temperature maintained for 15 hours, the 11B NMR spectrum showed at least two new boron species had formed (ESI, Fig. S87†). The resonance at ∼21.1 ppm stems from a pinBOTf species, whereas the ∼34.2 ppm signal is more difficult to interpret. We believe it originates from newly formed intermediates or their decomposition under these conditions, as it was not visible during the experiments without heating or when an alkene was present. In the case of a mixture, where excess of [EMPyrr][OTf] was used, both peaks were significantly more pronounced (ESI, Fig. S90†). 19F NMR spectroscopic analyses of the reaction between IL10 and HBpin confirmed the formation of pinBOTf (ESI, Fig. S84, S88, S91 and S95†), and surprisingly, its formation was inhibited when TMEDA was present in the reaction mixture (ESI, Fig. S102†). The emergence of pinBOTf indicated that [EMPyrr][OTf] could form hydridic boronate species from nucleophilic addition to HBpin (Scheme 3b), as in the trialkoxyborohydride-catalyzed hydroboration of ketones reported by the Clark group.64 However, our 11B NMR analyses did not fully confirm the presence of this type of boronate species. This is likely due to the ion immediately reacting with an alkene in the reaction mixture to form a more stable alkyl boronic ester, or the species may simply have been undetectable in our experimental conditions. Based on our findings and prior published literature, we propose a possible catalytic cycle represented in Scheme 3c. The catalytic cycle is initiated by the interaction between [EMPyrr][OTf] (IL10) and HBpin to generate ionic intermediate A, which makes the B–H bond insertion to alkene more effective. The subsequent hydroboration of alkene with A creates alkylboronic ester Cvia intermediate B and regenerates the initial catalyst.
 | ||
| Scheme 3 Comprehensive mechanistic studies on catalytic transformations for hydroboration of alkenes with HBpin catalyzed by [EMPyrr][OTf]. | ||
To prove the utility of the IL catalyzed hydroboration of alkenes, we performed a gram-scale reaction of styrene (1a) with HBpin (Scheme 4). Product 2a was extracted with n-heptane at a high yield (2.25 g, 98%) with excellent anti-Markovnikov regioselectivity (2a/3a = >99/1). Due to its high extraction efficiency and excellent selectivity, product 2a was applied as an attractive building block for further processing in a crude form. Various transformations of the boryl group in 2a were conducted (Scheme 4). Two of them, providing products 4 and 5, were performed in a one pot strategy utilizing the in situ generated alkylboronic ester 2a without undesirable interferences from the ionic liquid, demonstrating an advantage of the presented methodology in practical applications. Pd-catalyzed Suzuki–Miyaura coupling of the in situ formed 2a with 4-iodotoluene proceeded successfully to give the cross-coupling product 4 at 76% yield. Treatment of the hydroboration reaction mixture of 2a with NaBO3 × 4H2O provided oxidation product 5 at 94% yield. The isolated 2a was treated with KHF2, delivering potassium trifluoroborate salt 6 at 97% yield. Finally, the reaction of 2a with vinylmagnesium bromide, I2 and MeONa produced olefination product 7 at 85% yield.
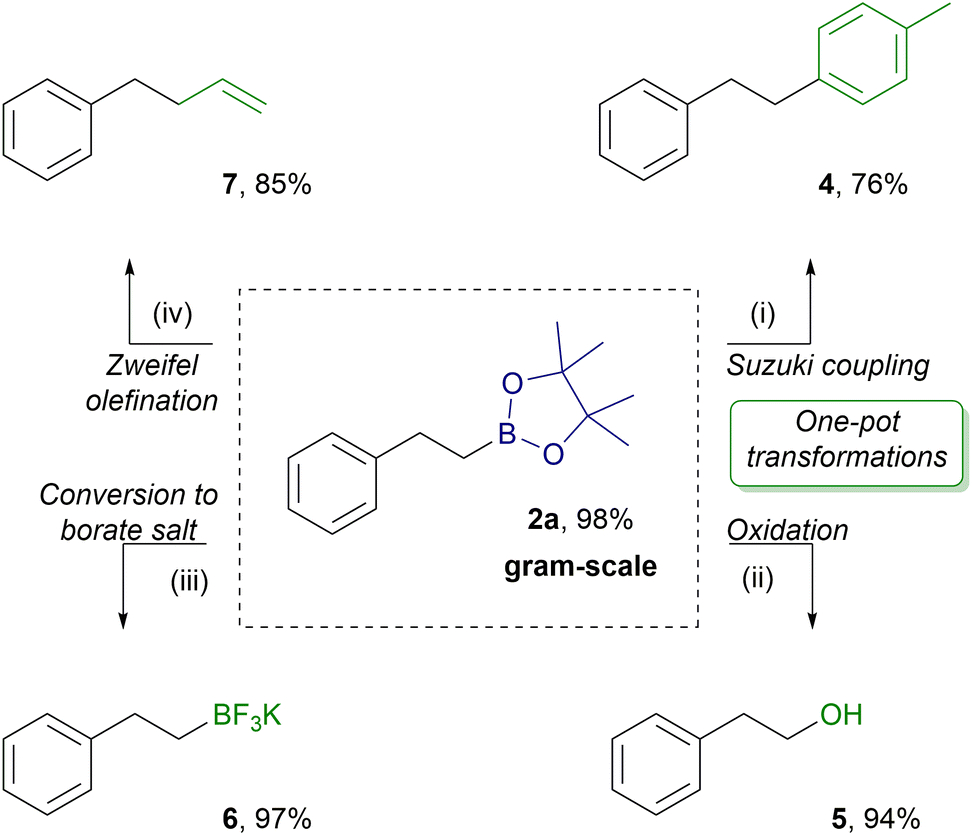 | ||
| Scheme 4 The application of 2a as a building block in various transformations. Reaction conditions: (i) Pd(PPh3)4 (5 mol%), 4-iodotoluene 1.2 equiv., THF (1 M), 3 M Cs2CO3, 70 °C, 24 h, argon. (ii) 3.0 equiv. NaBO3 × 4H2O, THF (1 M), H2O (1 M), rt., 5 h. (iii): 5.0 equiv. KHF2, H2O (2 M), rt., 24 h. (iv): Vinylmagnesium bromide (1.0 M in THF, 4.0 equiv.), I2 (4.0 equiv.) in MeOH (8 M), MeONa (8.0 equiv.) in MeOH (8 M), THF (10 M), −78 °C, 3 h. | ||
Conclusions
In conclusion, we have developed a sustainable, efficient and selective method for the synthesis of alkylboronic esters through ionic liquid organocatalyzed hydroboration of olefins. The protocol is effective for either monosubstituted or 1,1-disubstituted alkenes with various functional groups such as aryl, alkyl, alkoxy, amino, hydroxy, halogen and tosyl. The best results were obtained when [EMPyrr][OTf] was used at 100 °C for 20 h. The application of specially designed TM-complexes, structurally advanced ligands and sensitive activators was not required. Moreover, the equimolar ratio of reagents was used, and the process occurred with high anti-Markovnikov regioselectivity. A simple separation method from ionic liquid was applied using product extraction in nonpolar n-heptane. All these benefits make this catalytic system attractive for application in every synthetic laboratory providing borylated products at very good yields. The presented approach permitted the reuse of the IL, making this catalytic system uncommonly productive for the recyclable hydroboration of alkenes. The increase in the catalyst concentration to 0.5 equiv. allowed for the completion of 30 runs, which, according to our knowledge, is the best ever recyclable system for the hydroboration of alkenes.65–67 A combination of experimental studies and literature reviews provides intriguing insights into the mechanism of [EMPyrr][OTf]-catalyzed hydroboration reactions, where the various possible activation pathways of the ionic liquid and boronating agent result in the formation of alkylboronic esters. Additionally, the method was effectively scaled up to gram scale, followed by the further modifications of the boryl group in various transformations such as Suzuki–Miyaura coupling, oxidation, transformation to potassium organotrifluoroborate salt and Zweifel olefination, providing useful organic compounds. The elimination of metal complexes, high process effectivity and selectivity meet the green chemistry requirements for the hydroboration of olefins.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the National Science Centre in Poland, Grants No. UMO-2019/34/E/ST4/00068 and UMO-2019/32/C/ST4/00235.References
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772–3789 CrossRef CAS PubMed.
- Y. Zhao, B. Han and Z. Liu, Acc. Chem. Res., 2021, 54, 3172–3190 CrossRef CAS PubMed.
- T. Welton, Coord. Chem. Rev., 2004, 248, 2459–2477 CrossRef CAS.
- T. Vander Hoogerstraete, S. Wellens, K. Verachtert and K. Binnemans, Green Chem., 2013, 15, 919–927 RSC.
- Q. Yang, Z. Zhang, X.-G. Sun, Y.-S. Hu, H. Xing and S. Dai, Chem. Soc. Rev., 2018, 47, 2020–2064 RSC.
- G. A. O. Tiago, I. A. S. Matias, A. P. C. Ribeiro and L. M. D. R. S. Martins, Molecules, 2020, 25, 5812–5839 CrossRef CAS PubMed.
- W. Zhang, W. Huang and Q. Zhang, Chem. – Eur. J., 2021, 27, 6131–6144 CrossRef PubMed.
- Y. Zhao and T. Bostrom, Curr. Org. Chem., 2015, 19, 556–566 CrossRef CAS.
- Y. Zhao, Z. Yang, B. Yu, H. Zhang, H. Xu, L. Hao, B. Han and Z. Liu, Chem. Sci., 2015, 6, 2297–2301 RSC.
- C. Dai, J. Zhang, C. Huang and Z. Lei, Chem. Rev., 2017, 117, 6929–6983 CrossRef CAS PubMed.
- G. Shi, R. Zhai, H. Li and C. Wang, Green Chem., 2021, 23, 592–596 RSC.
- Y. Chen and T. Mu, Green Chem., 2019, 21, 2544–2574 RSC.
- T. Ding, J. Zha, D. Zhang, J. Zhang, H. Yuan, F. Xia and G. Gao, Green Chem., 2021, 23, 3386–3391 RSC.
- H. Tadesse and R. Luque, Energy Environ. Sci., 2011, 4, 3913–3929 RSC.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- J. Walkowiak, J. Szyling, A. Franczyk and R. L. Melen, Chem. Soc. Rev., 2022, 51, 869–994 RSC.
- J. Szyling, A. Szymańska, A. Franczyk and J. Walkowiak, J. Org. Chem., 2022, 87, 10651–10663 CrossRef CAS PubMed.
- T. Sokolnicki, J. Szyling, A. Franczyk and J. Walkowiak, Adv. Synth. Catal., 2020, 362, 177–183 CrossRef CAS.
- K. Stefanowska, A. Franczyk, J. Szyling, K. Salamon, B. Marciniec and J. Walkowiak, J. Catal., 2017, 356, 206–213 CrossRef CAS.
- K. Stefanowska, A. Franczyk, J. Szyling, M. Pyziak, P. Pawluć and J. Walkowiak, Chem. – Asian J., 2018, 13, 2101–2108 CrossRef CAS PubMed.
- K. Stefanowska, T. Sokolnicki, J. Walkowiak, A. Czapik and A. Franczyk, Chem. Commun., 2022, 58, 12046–12049 RSC.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- S. E. Hooshmand, B. Heidari, R. Sedghi and R. S. Varma, Green Chem., 2019, 21, 381–405 RSC.
- R. J. Armstrong and V. K. Aggarwal, Synthesis, 2017, 3323–3336 CAS.
- J. Szyling, A. Franczyk, P. Pawluć, B. Marciniec and J. Walkowiak, Org. Biomol. Chem., 2017, 15, 3207–3215 RSC.
- P. Pawluć, A. Franczyk, J. Walkowiak, G. Hreczycho, M. Kubicki and B. Marciniec, Org. Lett., 2011, 13, 1976–1979 CrossRef PubMed.
- J.-Q. Chen, J.-H. Li and Z.-B. Dong, Adv. Synth. Catal., 2020, 362, 3311–3331 CrossRef CAS.
- J. D. Grayson, F. M. Dennis, C. C. Robertson and B. M. Partridge, J. Org. Chem., 2021, 86, 9883–9897 CrossRef CAS PubMed.
- S. J. Geier, C. M. Vogels, J. A. Melanson and S. A. Westcott, Chem. Soc. Rev., 2022, 51, 8877–8922 RSC.
- E. A. Bijpost, R. Duchateau and J. H. Teuben, J. Mol. Catal. A: Chem., 1995, 95, 121–128 CrossRef CAS.
- J. F. Hartwig and C. N. Muhoro, Organometallics, 2000, 19, 30–38 CrossRef CAS.
- S. Pereira and M. Srebnik, J. Am. Chem. Soc., 1996, 118, 909–910 CrossRef CAS.
- S. Weber, D. Zobernig, B. Stöger, L. F. Veiros and K. Kirchner, Angew. Chem., Int. Ed., 2021, 60, 24488–24492 CrossRef CAS PubMed.
- J. R. Carney, B. R. Dillon, L. Campbell and S. P. Thomas, Angew. Chem., Int. Ed., 2018, 57, 10620–10624 CrossRef CAS PubMed.
- M. Espinal-Viguri, C. R. Woof and R. L. Webster, Chem. – Eur. J., 2016, 22, 11605–11608 CrossRef CAS PubMed.
- T. F. C. Cruz, L. C. J. Pereira, J. C. Waerenborgh, L. F. Veiros and P. T. Gomes, Catal. Sci. Technol., 2019, 9, 3347–3360 RSC.
- S. Kisan, V. Krishnakumar and C. Gunanathan, ACS Catal., 2017, 7, 5950–5954 CrossRef CAS.
- L. Zhang, Z. Zuo, X. Wan and Z. Huang, J. Am. Chem. Soc., 2014, 136, 15501–15504 CrossRef CAS PubMed.
- E. Bergamaschi, F. Beltran and C. J. Teskey, Chem. – Eur. J., 2020, 26, 5180–5184 CrossRef CAS PubMed.
- W. Zhao, K.-Z. Chen, A.-Z. Li and B.-J. Li, J. Am. Chem. Soc., 2022, 144, 13071–13078 CrossRef CAS PubMed.
- A. J. Bochat, V. M. Shoba and J. M. Takacs, Angew. Chem., Int. Ed., 2019, 58, 9434–9438 CrossRef CAS PubMed.
- M. Magre, M. Biosca, O. Pàmies and M. Diéguez, ChemCatChem, 2015, 7, 114–120 CrossRef CAS.
- C. Mazet and D. Gérard, Chem. Commun., 2011, 47, 298–300 RSC.
- G. Vijaykumar, M. Bhunia and S. K. Mandal, Dalton Trans., 2019, 48, 5779–5784 RSC.
- F. Ulm, Y. Cornaton, J.-P. Djukic, M. J. Chetcuti and V. Ritleng, Chem. – Eur. J., 2020, 26, 8916–8925 CrossRef CAS PubMed.
- J. Won, D. Noh, J. Yun and J. Y. Lee, J. Phys. Chem. A, 2010, 114, 12112–12115 CrossRef CAS PubMed.
- D. Noh, S. K. Yoon, J. Won, J. Y. Lee and J. Yun, Chem. – Asian J., 2011, 6, 1967–1969 CrossRef CAS PubMed.
- Y. Wang, R. Guan, P. Sivaguru, X. Cong and X. Bi, Org. Lett., 2019, 21, 4035–4038 CrossRef CAS PubMed.
- Z.-C. Wang, M. Wang, J. Gao, S.-L. Shi and Y. Xu, Org. Chem. Front., 2019, 6, 2949–2953 RSC.
- Y. Wu, C. Shan, J. Ying, J. Su, J. Zhu, L. L. Liu and Y. Zhao, Green Chem., 2017, 19, 4169–4175 RSC.
- D. H. Ma, A. K. Jaladi, J. H. Lee, T. S. Kim, W. K. Shin, H. Hwang and D. K. An, ACS Omega, 2019, 4, 15893–15903 CrossRef CAS PubMed.
- A. Bismuto, M. J. Cowley and S. P. Thomas, ACS Catal., 2018, 8, 2001–2005 CrossRef CAS.
- Q. Yin, S. Kemper, H. F. T. Klare and M. Oestreich, Chem. – Eur. J., 2016, 22, 13840–13844 CrossRef CAS PubMed.
- J. Liu, C. Wu, T. Hu, W. Yang, Y. Xie, Y. Shi, Q. Liu, Y. Shao and F. Zhang, J. Org. Chem., 2022, 87, 3442–3452 CrossRef CAS PubMed.
- N. N. H. Ton, B. K. Mai and T. V. Nguyen, J. Org. Chem., 2021, 86, 9117–9133 CrossRef CAS PubMed.
- J. Szyling, A. Franczyk, K. Stefanowska, H. Maciejewski and J. Walkowiak, ACS Sustainable Chem. Eng., 2018, 6, 10980–10988 CrossRef CAS.
- J. Szyling, A. Franczyk, K. Stefanowska and J. Walkowiak, Adv. Synth. Catal., 2018, 360, 2966–2974 CrossRef CAS.
- J. Szyling, A. Franczyk, K. Stefanowska, M. Klarek, H. Maciejewski and J. Walkowiak, ChemCatChem, 2018, 10, 531–539 CrossRef CAS.
- A. D. Bage, T. A. Hunt and S. P. Thomas, Org. Lett., 2020, 22, 4107–4112 CrossRef CAS PubMed.
- M. T. Reetz, S. Huette and R. Goddard, J. Am. Chem. Soc., 1993, 115, 9339–9340 CrossRef CAS.
- T. Nakamura, K. Okuno, R. Nishiyori and S. Shirakawa, Chem. – Asian J., 2020, 15, 463–472 CrossRef CAS PubMed.
- C. E. Cannizzaro and K. N. Houk, J. Am. Chem. Soc., 2002, 124, 7163–7169 CrossRef CAS PubMed.
- I. P. Query, P. A. Squier, E. M. Larson, N. A. Isley and T. B. Clark, J. Org. Chem., 2011, 76, 6452–6456 CrossRef CAS PubMed.
- M. L. Shegavi, S. Saini, R. Bhawar, M. D. Vishwantha and S. K. Bose, Adv. Synth. Catal., 2021, 363, 2408–2416 CrossRef CAS.
- X.-F. Zhou, Y.-Y. Sun, Y.-D. Wu, J.-J. Dai, J. Xu, Y. Huang and H.-J. Xu, Tetrahedron, 2016, 72, 5691–5698 CrossRef CAS.
- Q. Yao, X. Lu, K. Liu, C. Ma, J. Su, C. Lin, D. Li, J. Dou, J. Sun and W. Duan, Dalton Trans., 2019, 48, 5144–5148 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2216859. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2gc04163d |
| This journal is © The Royal Society of Chemistry 2023 |