
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst-free amino-alcoholysis depolymerization strategy: a facile and powerful tool for chemical recycling of poly(bisphenol A carbonate)†
Xianyue
Zhou‡
a,
Maoqing
Chai‡
a,
Guangqiang
Xu
 *bc,
Rulin
Yang
bc,
Hongguang
Sun
*a and
Qinggang
Wang
*bc
*bc,
Rulin
Yang
bc,
Hongguang
Sun
*a and
Qinggang
Wang
*bc
aCollege of Polymer Science and Engineering, Qingdao University of Science and Technology, Qingdao 266042, P. R. China. E-mail: hgsun816@qust.edu.cn
bKey Laboratory of Biobased Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao, 266101, P. R. China. E-mail: xu_gq@qibebt.ac.cn; wangqg@qibebt.ac.cn
cCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
First published on 12th January 2023
Abstract
A catalyst-free amino-alcoholysis depolymerization protocol was explored for the chemical recycling of poly(bisphenol A carbonate) (BPA-PC). Under mild conditions, the initial monomer bisphenol A (BPA) and value-added oxazolidinone chemicals were recovered with high efficiency and yield. The chemical cycle was closed by producing new BPA-PC from recovered BPA.
Plastic has been deeply integrated into the basic necessities of daily life. Originally, plastics based on fossil resources were designed for performance and durability, rather than degradability and recyclability.1–3 As a result, plastic waste is now one of the major crises threatening sustainable development.4,5 Plastic sustainability is an inevitable requirement to solve the problem of the plastic crisis. Therefore, transformation from the traditional “linear plastic economy mode” into the promising “circular plastic economy mode” should be paid more and more attention.6–9
When considering the circular plastic economy mode, mechanical recycling of plastics is first taken into consideration.10,11 However, thermomechanical degradation caused by the harsh remelting conditions eventually results in material downcycling.12 In contrast, through chemical recycling, plastics can be converted into virgin monomer or useful synthetic chemicals/feedstocks to realize polymer recycling and upcycling.13–15 Focusing on chemical recycling, one approach is to design and synthesize chemically recyclable polymers through a concept of “chemical recycling to monomers (CRM)”.16–18 These new polymers exhibit simultaneously high degradability and excellent thermal–mechanical properties, thus providing an opportunity to achieve the closed-loop mode for polymer material usage. There has been a flurry of examples in this area in recent years.19–37 However, most of the research design is still in the laboratory stage, and there is a long path to achieve industrialization due to the complicated monomer preparation steps and high catalyst cost, and so on. In addition to designing new polymers, attention should also be paid to the chemical recycling of plastics that are being used on large scale, which is of great significance to solve the current plastic crisis caused by the accumulation of polymer wastes. A prominent representative is poly(bisphenol A carbonate) (BPA-PC). As an engineering plastic with many desirable properties, the production of BPA-PC has gradually increased up to 5 million tons per year.38 However, the potential environmental hazards due to the release of BPA make the recovery of BPA-PC particularly important.39,40
Tremendous progress has been made to achieve chemical recycling of BPA-PC. It mainly includes pyrolysis, hydrolysis, alcoholysis and aminolysis.41–43 The pyrolysis and hydrolysis methods are energy-intensive and poorly selective due to the harsh reaction conditions, such as extremely high temperature and pressure.44,45 However, alcoholysis and aminolysis can not only recover BPA but also capture the carbonyl in BPA-PC and yield the corresponding carbonyl products, such as dimethyl carbonate, ethylene carbonate, urea, etc.46,47 For example, Kim reported that upon employing 1,5,7-triazabicyclo[4.4.0]-dec-5-ene (TBD) as an organocatalyst, a complete degradation of BPA-PC occurred, resulting in BPA monomers and carbonates.48 Besides, Dove and Sardon demonstrated that functionalized cyclic carbonates and BPA can be obtained when using a combination of TBD with MSA (methane sulfonic acid) as the catalytic system.49 Although great progress has been made, the participation of a catalyst or elevated temperature was required. Liu reported the methanolysis process in ionic liquids.50 Although no additional catalyst was involved, actually ionic liquids acted as both the solvent and the catalyst. The high cost of ionic liquids and the complex preparation processes limited their applications. In contrast, catalyst-free depolymerization processes assisted by the use of low-cost conventional solvents or even solvent-free depolymerization have never been reported.
This work aims to develop an economical, practical and green route for the chemical recycling of BPA-PC. For a depolymerization process in which a catalyst is involved, the disadvantages are manifold: (1) the preparation of the catalyst increases the cost; (2) the reaction is not easy to scale up; and (3) the residual catalyst reduces the purity of the product and post-treatment purification is a necessity. While, catalyst-free has always been one of the criteria for green chemistry,51 in this study, we propose that by choosing a stronger nucleophile and promoting the solvation effect, it is possible to depolymerize BPA-PC without a catalyst. Herein, a catalyst-free amino-alcoholysis depolymerization protocol was explored. Under mild conditions, the initial monomer BPA and high value-added oxazolidinone chemicals were recovered from BPA-PC with high efficiency and yield (Scheme 1). The recycled BPA could be polymerized to prepare new BPA-PC materials, thus realizing real chemical recycling. Besides, oxazolidinones with different structures were obtained, showing the universality of this strategy. As far as we know, oxazolidinones have been extensively used as pharmaceutical intermediates in medical chemistry.52,53 Therefore, a chemical upcycling progress was achieved.
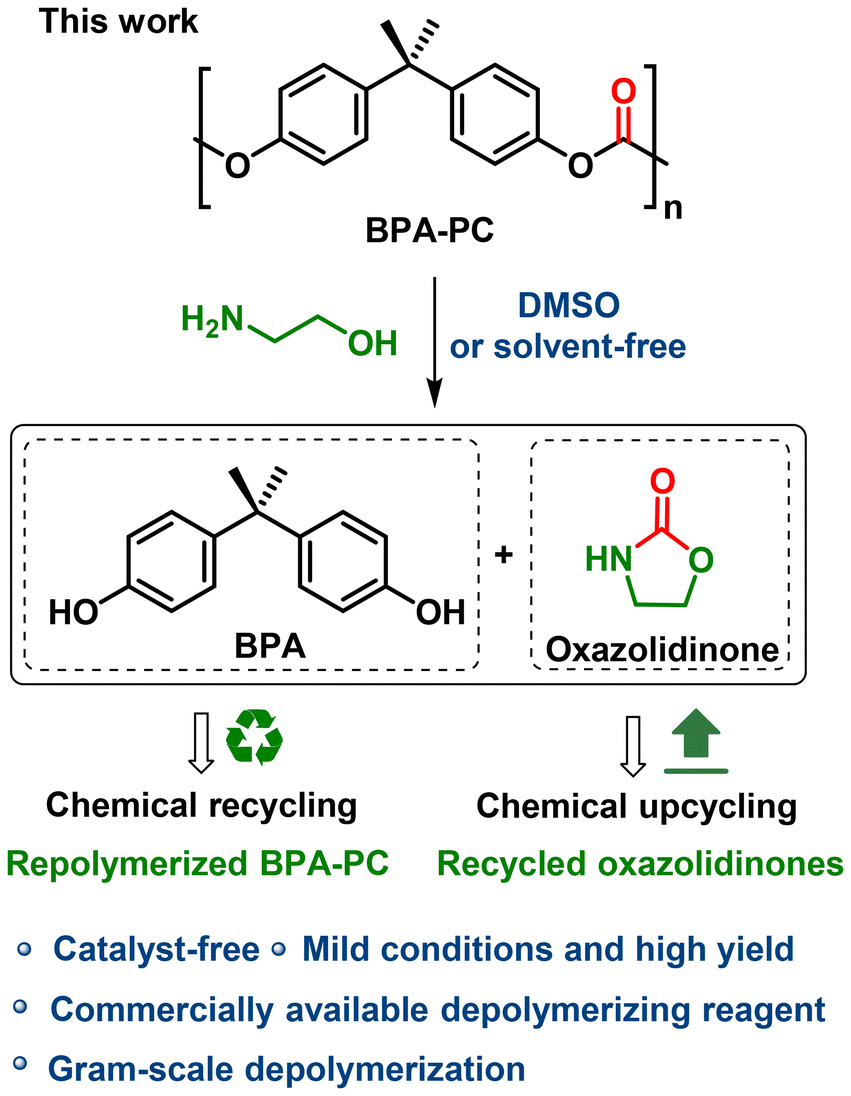 | ||
| Scheme 1 The catalyst-free amino-alcoholysis depolymerization of BPA-PC. | ||
Primarily, the catalyst-free degradation process of BPA-PC was systematically examined using ethanolamine as the depolymerizing reagent. No moisture-sensitive reagents were involved; therefore, the reactions did not require the protection of inert gases. Firstly, under bulk conditions, 5 equiv. ethanolamine was employed as both the depolymerization reagent and solvent, without the participation of auxiliary solvents. As shown in Fig. 1, the complete conversion of BPA-PC was achieved after 57 h. The relatively low reaction activity may be due to the poor solubility of BPA-PC in ethanolamine, thus the heterogeneous reaction hindered the contact between reagents and polymers. Therefore, the influence of adding the assisted solvent was investigated. Ethyl acetate (EtOAc) was the first to be considered. Two equivalents of ethanolamine relative to the BPA-PC repeating unit were applied. However, under these conditions, the yield of BPA and 2-oxazolidone could only reach 60%, and the reaction could not proceed to the end even with a prolonged reaction time. Similar results were also found when dichloromethane (DCM) was used as the solvent, in which BPA-PC showed good solubility. In these processes, products without ring closure were recognized (Fig. S1†). These products were observed to precipitate from EtOAc and DCM during the reaction, thus making the reaction difficult to proceed. More common solvents were then brought into account. The reaction in toluene required 20 h to complete the depolymerization process. While for the oxygenated solvent tetrahydrofuran (THF) and 2-methyl tetrahydrofuran (2-Me-THF), the reaction time was further extended to 61 h and 74 h. Throughout the above results, ethanolamine as a strong nucleophile enabled a catalyst-free depolymerization process that was realized but was less efficient. Therefore, efforts to further improve efficiency were implemented.
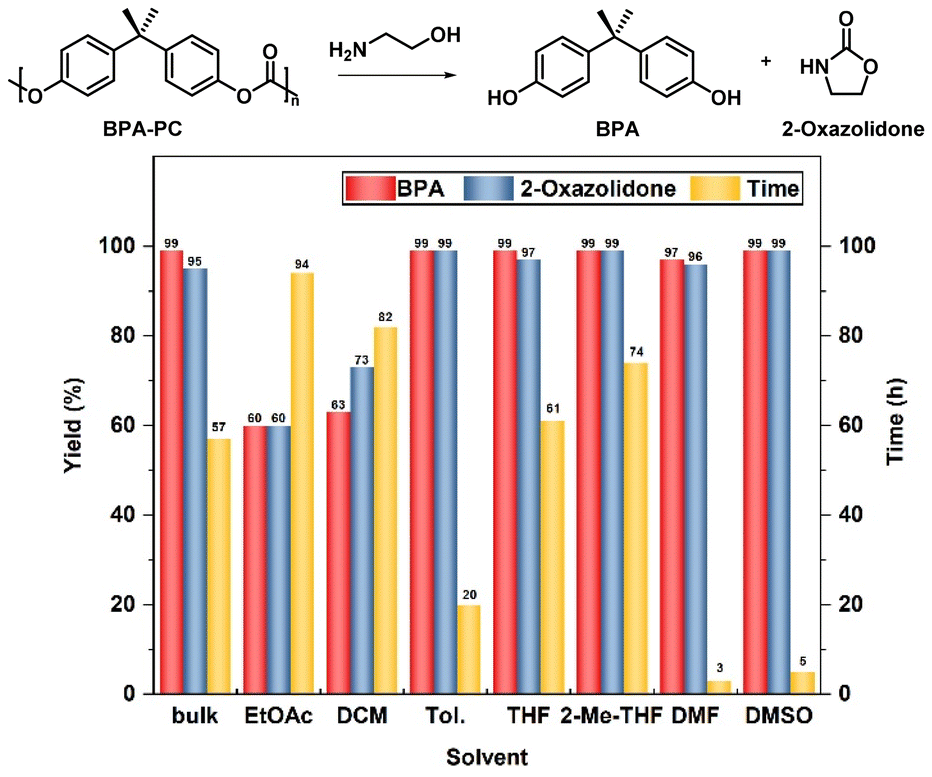 | ||
| Fig. 1 The degradation of BPA-PC in different solvents. Depolymerization conditions: BPA-PC pellets (sizes: 3–4 mm, Mn = 25.8 kg mol−1, Đ = 2.27, 254 mg, 1 mmol based on BPA unit), ethanolamine (122 μL, 2 mmol), hexamethylbenzene (32 mg, 0.2 mmol) as an internal standard, acetic acid as the quencher, Vsolvent = 1 mL, 25 °C. Bulk condition: 5 equiv. ethanolamine. | ||
In previous reports, the participation of highly polar solvents facilitated the reaction.54,55 Hence, the dipolar solvents N,N-dimethylformamide (DMF) and methyl sulfoxide (DMSO) were further tried. To our surprise, in DMF, after only 5 h full conversions were achieved, and near-quantitative yields of BPA (>99%) and 2-oxazolidone (>99%) were obtained without any by-products (entry 1). Almost identical results were obtained in DMSO (entry 2). In addition, to improve the economy of the reaction process, the amount of ethanolamine added was reduced. When the equivalent of ethanolamine was decreased from 2.0 to 1.1, the reaction activity decreased only slightly (entries 3 and 4). Furthermore, upon reducing the depolymerizing reagent to 1.0 equivalent amount, an obvious increase in degradation time was observed (6 h vs. 14 h, 7 h vs. 48 h, entries 5 and 6). Intriguingly, increasing the temperature from room temperature to 50 °C, resulted in a significant increase in depolymerization activity (14 h vs. 3 h, entries 5, 7). These data show that the high depolymerization performance was still obtained even if the amount of ethanolamine was decreased to 1 equivalent (entries 7 and 8). Considering that the degradation efficiency was higher in DMSO than in DMF, DMSO was determined to be the best solvent. Besides, to get closer to the principles of green chemistry, the solvent-free depolymerization reaction was further explored under different conditions. When 1 equiv. of ethanolamine was introduced, the reaction was difficult to proceed at 50 °C (entry 9). After 36 h, a large number of BPA-PC particles remained undissolved. This may be because it was difficult for the small amount of ethanolamine to make adequate contact with BPA-PC. Considering this reason, the temperature was further increased to 120 °C at which point the depolymerization process was smoothly achieved even with as few as 1 equiv. of ethanolamine (entry 10) (Table 1).
| Entrya | Temp. (°C) | Solvent | Ethanolamine (equiv.) | Time (h) | BPAb (%) | 2-Oxazolidoneb (%) |
|---|---|---|---|---|---|---|
| a Depolymerization conditions: BPA-PC pellets (sizes: 3–4 mm, Mn = 25.8 kg mol−1, Đ = 2.27, 254 mg, 1 mmol based on BPA unit), dimethylsulfoxide and N,N-dimethylformamide as the solvent, Vsolvent = 1 mL. b Determined by 1H NMR spectroscopy. | ||||||
| 1 | R.T. | DMSO | 2 | 5 | >99 | >99 |
| 2 | R.T. | DMF | 2 | 5 | >99 | >99 |
| 3 | R.T. | DMSO | 1.1 | 6 | >99 | >99 |
| 4 | R.T. | DMF | 1.1 | 7 | 90 | 87 |
| 5 | R.T. | DMSO | 1 | 14 | >99 | 98 |
| 6 | R.T. | DMF | 1 | 48 | 96 | 96 |
| 7 | 50 | DMSO | 1 | 3 | >99 | >99 |
| 8 | 50 | DMF | 1 | 3 | 82 | 83 |
| 9 | 50 | Bulk | 1 | 36 | — | — |
| 10 | 120 | Bulk | 1 | 44 | 99 | 87 |
To clarify the degradation mechanism, diphenyl carbonate was used as a simplified model to simulate the reaction process (Fig. 3). The reaction results with 1 equiv. ethanolamine in DMSO versus chloroform were explored separately. As shown in Fig. 3a and b, diphenyl carbonate was rapidly converted into the ring-opened product and the oxazolidinone in DMSO, and then the ring-opened product was further converted into the oxazolidinone. Whereas in chloroform, a gradual conversion of ethanolamine into the ring-opened product was observed; however, conversion to the oxazolidinone was difficult. Moreover, the stability of the ring-opened product in different deuterated reagents was also verified. A gradual conversion of the ring-opened product into phenol and oxazolidinone was observed in deuterated DMSO (Fig. S2†). However, in deuterated chloroform, the ring-opened product was stable (Fig. S3†). These results amply illustrated the promotion effect of DMSO for the aminolysis of carbonate and ring-closure to generate oxazolidinones. When it comes to solvent-free depolymerization, the ring-opened product was heated to 120 °C, and after 2 h, the production of phenol and 2-oxazolidone was similarly observed, which proved that temperature also played a key role in this process (Fig. 3c). Based on the above experimental phenomenon, a possible mechanism was proposed. At the beginning of the reaction, the amino group in ethanolamine acted as a strong nucleophile that attacks the carbonyl group in BPA-PC. Then, the hydroxyl group further attacked the ester group to generate oxazolidinone through the ring-closing reaction. In the second step, the ring-closing reaction was the rate determining step. In dipolar solvents, such as DMSO, the presence of hydrogen bonds and dipolar interactions between DMSO and the hydroxyl groups promoted the ring-closing reaction.56 While under solvent-free conditions, heating was necessary to promote the ring-closing reaction to generate oxazolidinone.
Encouraged by these results, eight common BPA-PC commodities were also explored (Fig. 2). It is noteworthy that these BPA-PC commodities, varying in molecular weight (Mn = 16.1 kg mol−1–33.0 kg mol−1) and dispersity (Đ = 1.99–2.86), are suitable for this strategy. All samples were depolymerized completely, affording the corresponding BPA with 94–99% yields and 2-oxazolidone with 93–99% yields, respectively. When the reaction was carried out at room temperature, the conversion could be completed within 8 h. As anticipated, a more efficient process was obtained when raising the temperature to 50 °C and the polymers could be completely depolymerized in 3 h. Under solvent-free conditions at 120 °C, the BPA-PC commodities achieved the degradation process as well. These experimental results showed that even if there are different additives in the commercial BPA-PC products, they barely have an obvious influence on this depolymerization approach.
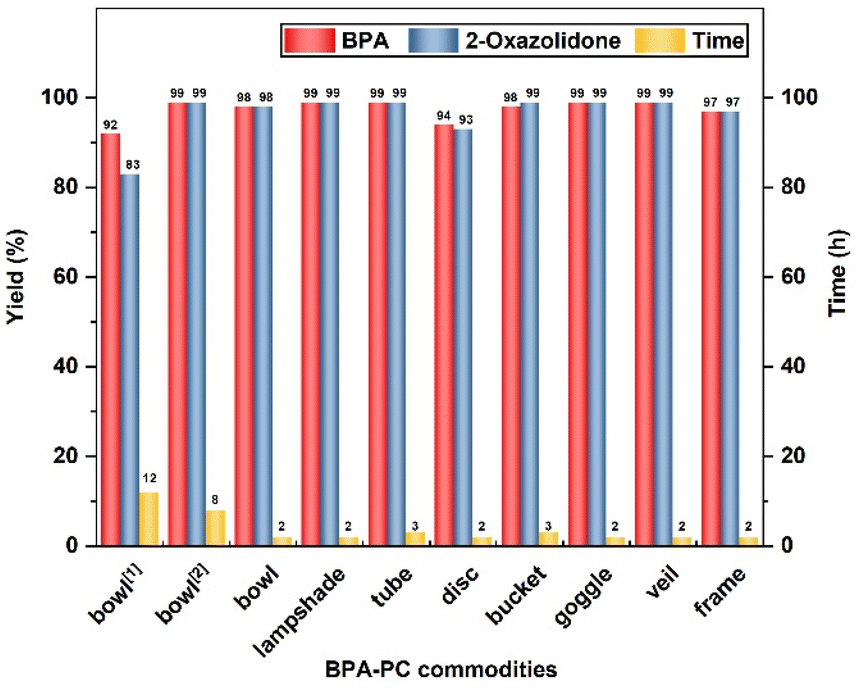 | ||
| Fig. 2 The degradation of BPA-PC commodities. Depolymerization conditions: BPA-PC commodities (254 mg, 1 mmol based on BPA unit), ethanolamine (61 μL, 1 mmol) in DMSO (1 mL) at 50 °C, dibromomethane (145 μL, 2 mmol) as an internal standard, acetic acid as the quencher.1 The reaction was conducted at 120 °C, solvent-free, 1 equiv. ethanolamine.2 The reaction was conducted at room temperature. | ||
Moreover, to further explore the practical application of this strategy, the BPA-PC tube was subjected to gram-scale depolymerization under standard conditions (Fig. S4 and 4†). Firstly, 100 g of BPA-PC tube was subjected to DMSO and 1 equiv. of ethanolamine was added simultaneously. As represented in Fig. S4,† the depolymerization process was completed within 2 h at 80 °C. Then, DMSO was recovered by vacuum distillation with a recovery rate of 93%. The NMR spectrum showed that the recovered DMSO was pure without impurities (Fig. S17†). After that, a certain amount of water was introduced to the remaining solids under uniform stirring. The suspension was filtered to collect the precipitate and filtrate. Quite interestingly, BPA is insoluble in water, while 2-oxazolidone is soluble, which means that the resulting products can be easily separated under convenient-to-perform conditions. The precipitate was vacuum dried, and then 88 g of BPA with a yield of 98% was obtained. After evaporative concentration and ether washing of the filtrate, 28 g of 2-oxazolidone was afforded with a yield of 83%. The chemical structures and high purity of the recovered BPA and 2-oxazolidone were characterized by IR, 1H NMR spectra, 13C NMR spectra and HPLC (Fig. S18–24†). A gram-scale depolymerization experiment under solvent-free conditions was also performed (Fig. 4). 30 g of PC tubes was fully degraded by 1 equiv. of ethanolamine under heating to 120 °C. Since there was no excess solvent, catalyst as well as other reagents present, the post-treatment process was greatly simplified. The separation process of BPA and 2-oxazolidone could be realized by directly adding water after degradation. The final yields of these products were 98% and 71%, respectively. The prices of ethanolamine and 2-oxazolidone on the market were checked (Table S5†), which illustrated that the depolymerization products were relatively expensive compared with ethanolamine (ethanolamine vs. 2-oxazolidone; TCI, $ 23.00 vs. $ 284.00), which further proved the economic benefit of the depolymerization process. This gram-scale depolymerization process unequivocally confirmed that this strategy is an economical, practical and green route to convert BPA-PC into its monomers and high value-added products.
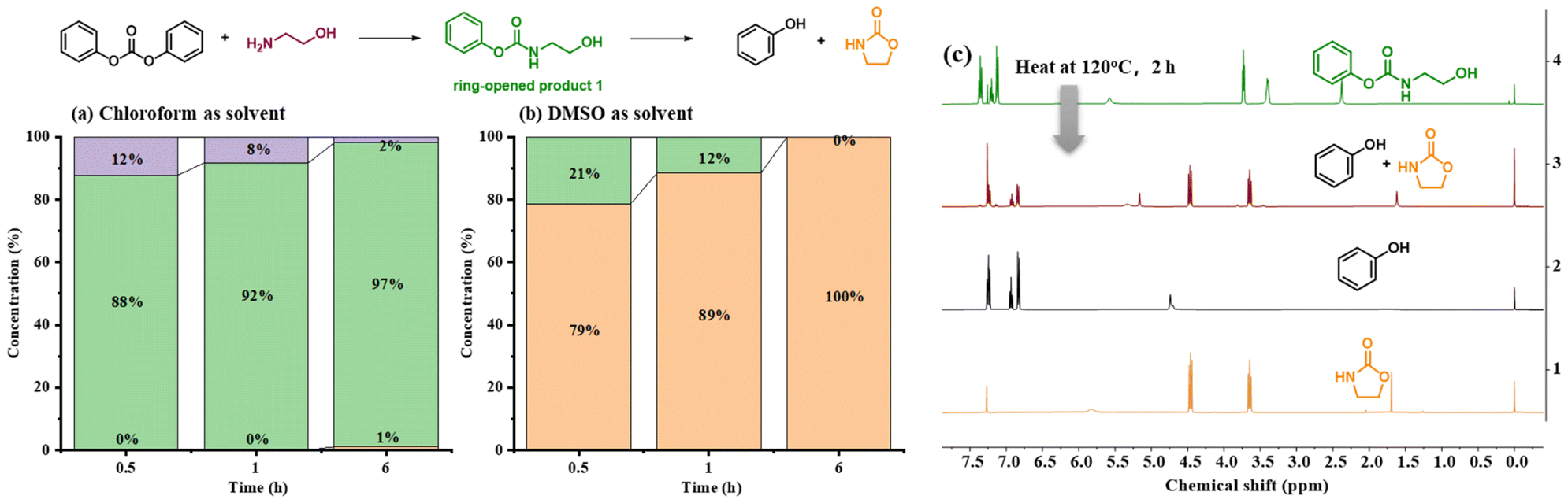 | ||
| Fig. 3 (a) Results of the reaction of diphenyl carbonate with 1 equiv. of ethanolamine in chloroform (purple column is the concentration of ethanolamine, green column is the concentration of the ring-opened product, orange column is the concentration of 2-oxazolidone). (b) Results of the reaction of diphenyl carbonate with 1 equiv. of ethanolamine in DMSO. (c) Heating promoted the ring-closing process to generate oxazolidinone under solvent-free conditions. | ||
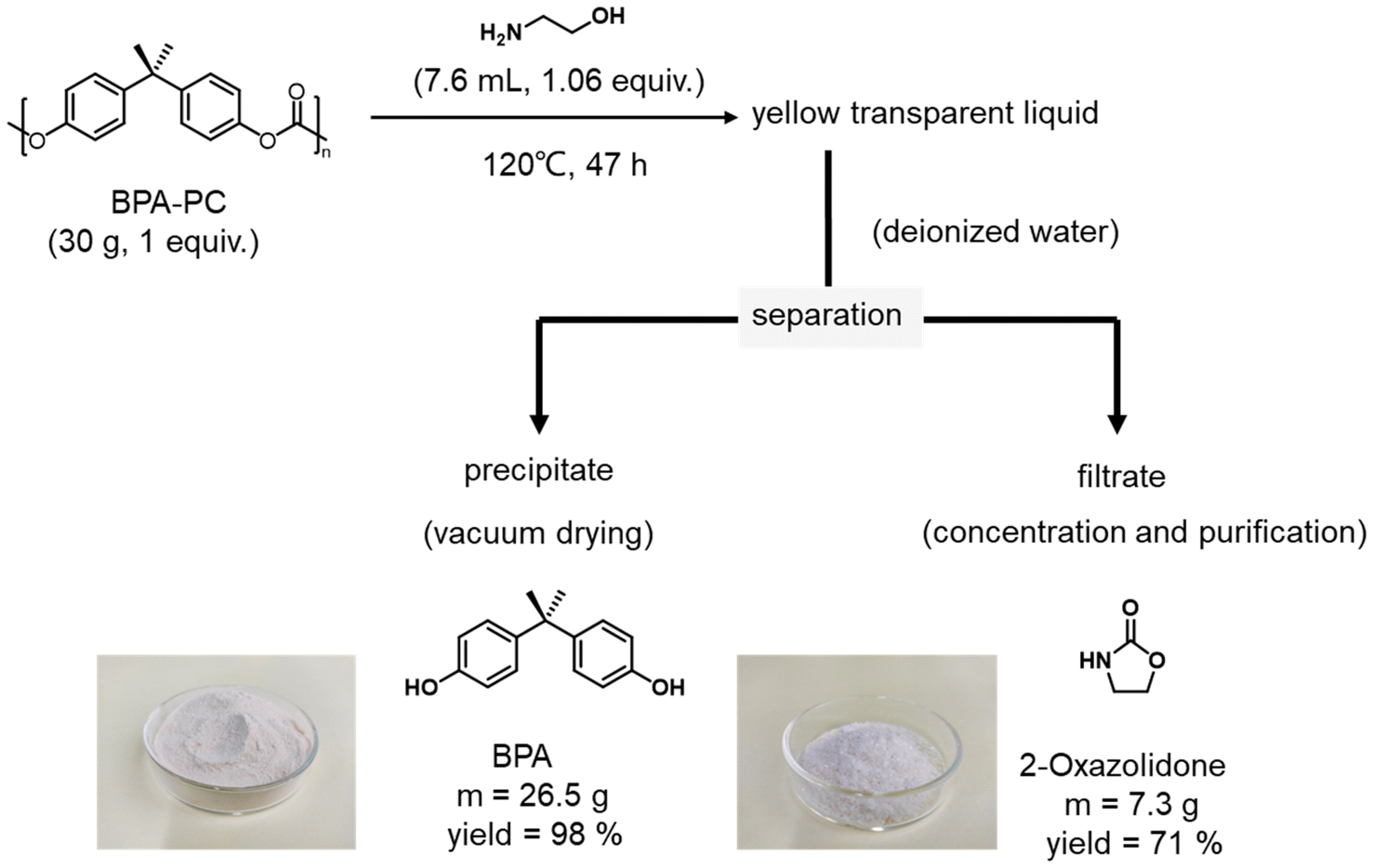 | ||
| Fig. 4 The gram-scale depolymerization of BPA-PC tube by using ethanolamine. | ||
To verify the purity of recovered BPA, the preparation of new BPA-PC by repolymerization was explored. A melt transesterification method was used by employing KOH as the catalyst (BPA/DPC = 1/1.1, 0.01 mol% KOH, melt transesterification at 170 °C, polycondensation at 260 °C and 0.001 mbar).57,58 Under these conditions, the 1H NMR spectra of the obtained polymers proved that BPA-PC with high purity was successfully synthesized (Fig. S26†). As demonstrated in Fig. 5, the properties of BPA-PC from recovered BPA were comparable to that prepared using commercially available BPA. The number-average molecular weights of 14.3 kg mol−1 and 13.5 kg mol−1 were obtained by GPC tests, respectively. In addition, a similar glass transition was observed (137 °C vs. 138 °C), indicating that the polymers had consistent thermal properties. Moreover, the transmittance of regenerated BPA-PC was tested via spectrophotometer (Table S6†). The BPA-PC prepared from recovered BPA and commercial BPA have high transmittance values of 98.17% and 98.19%, respectively. Thus, the comparable high transparency of the regenerated BPA-PC was proved. The slightly lower values than the original BPA-PC tube (99.10%) may be due to the differences in the process of BPA-PC synthesis in our laboratory compared to industrialized conditions. These results fully demonstrated the high purity of the recovered BPA while also achieving the closed-loop chemical recycling process.
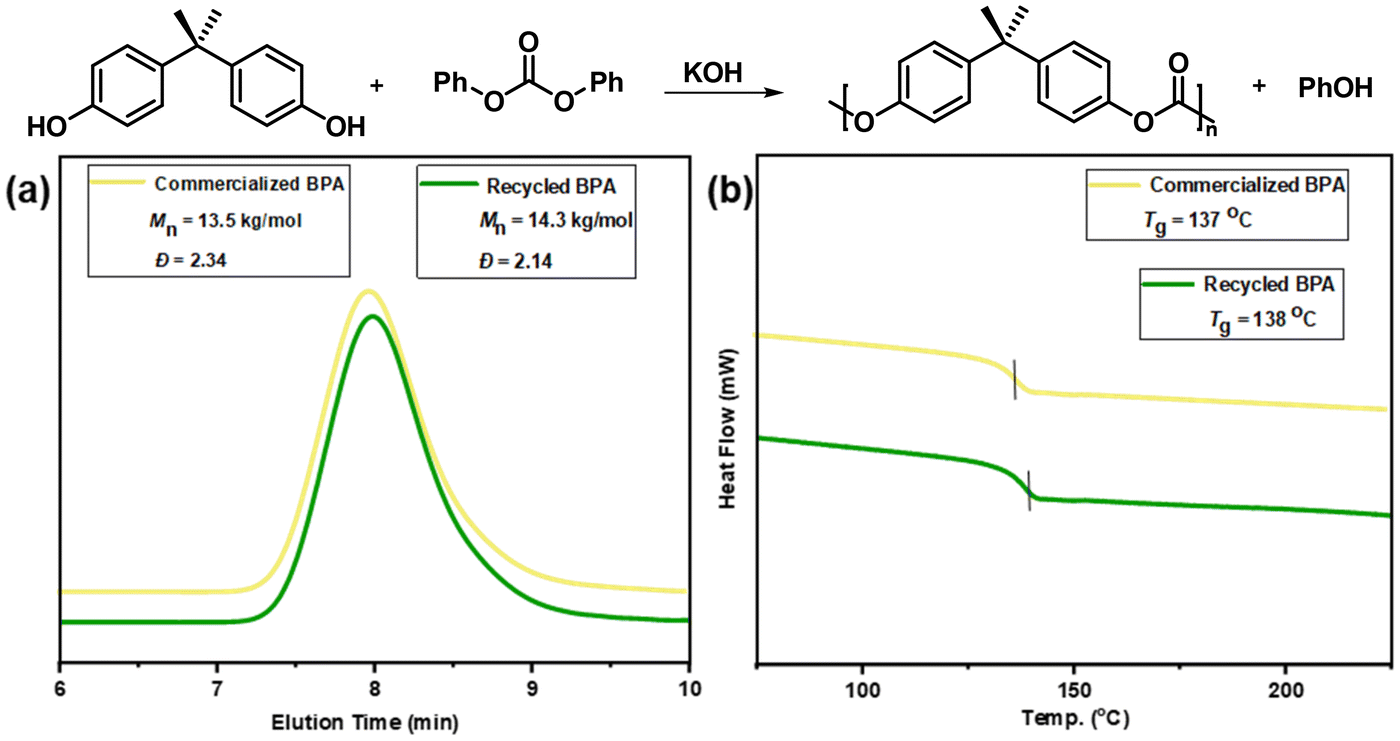 | ||
| Fig. 5 (a) GPC traces and (b) DSC analysis of BPA-PC from commercialized BPA (yellow line) and recycled BPA (green line). | ||
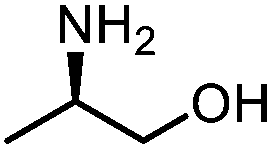
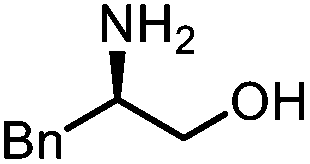
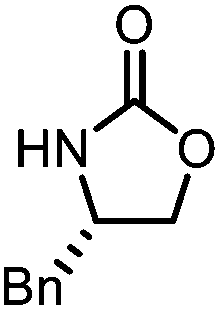
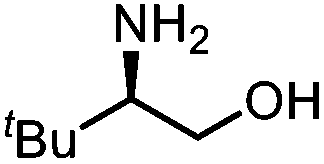
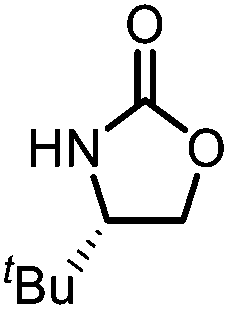
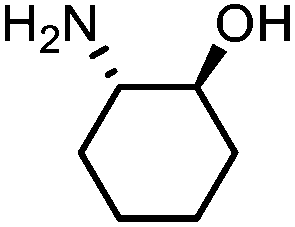
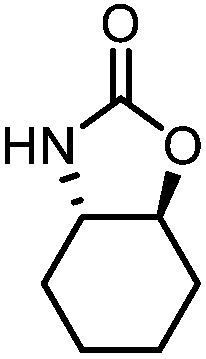
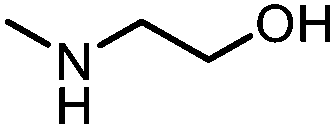
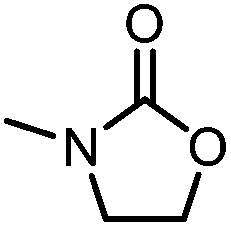
Oxazolidinones were important intermediates in organic synthesis.59,60 However, their synthesis usually required the participation of toxic reagents such as phosgene.61,62 Through this protocol, a range of organic oxazolidinones from polycarbonate waste were attempted to be synthesized by varying the amino alcohols. As shown in Table 2, when (R)-2-amino-1-propanol and (R)-phenylalaninol were employed as depolymerizing reagents, the depolymerization reactions were completed efficiently within 5 h and the yields of the products were more than 98% (entries 1 and 2). When an amino-alcohol-bearing sterically demanding tert-butyl substituent was used, quantitative yield was still achieved, albeit with a slight decrease in activity (entry 3). In addition, with the participation of (S,S)-2-aminocyclohexanol, cyclohexyl oxazolidinone was also obtained with a high yield (99%) (entry 4). Besides, secondary amino alcohol was considered as well. The depolymerization efficiency declined significantly, indicating that the steric effect of amine moieties had a great influence on the depolymerization. To our delight, high yields were still obtained (95% and 91%, entry 5). Furthermore, depolymerization attempts utilizing different amino-alcohols under solvent-free conditions were also made (Table 2, numbers in parentheses). Within 12 h, the polymers were all completely degraded even with different depolymerization reagents. Notably, the depolymerization efficiencies of different substituents under temperature-increasing conditions are approximate, which indicates that temperature played a decisive role in this process. Overall, all these successful cases highlighted the broad adaptability of this strategy.
| Entrya | Reagent | Products | Time (h) | BPAb (%) | Productsb (%) |
|---|---|---|---|---|---|
| a Depolymerization conditions: BPA-PC pellets (sizes: 3–4 mm, Mn = 25.8 kg mol−1, Đ = 2.27, 254 mg, 1 mmol based on BPA unit), amino alcohol (1 mmol), DMSO as the solvent at 50 °C, Vsolvent = 1 mL. b The yields were determined by 1H NMR spectroscopy in DMSO-d6 using dibromomethane as an internal standard. c Reaction was carried out at 100 °C. d Numbers in parentheses are reaction results under solvent-free conditions: 120 °C, 1 equiv. ethanolamine, 12 h. | |||||
| 1 |
|
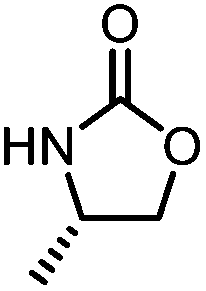
|
4 | 99(93)d | 99(99) |
| 2 |
|
|
5 | 99(95) | 98(99) |
| 3 |
|
|
25 | 97(96) | 99(93) |
| 4 |
|
|
4 | 99(99) | 99(58) |
| 5c |
|
|
84 | 95(95) | 91(98) |
Compared with other BPA-PC depolymerization processes, this work mainly has the following environmental benefits: (1) The reaction could be carried out under mild conditions without harsh conditions such as high temperature and high pressure. (2) The reaction did not require the participation of the catalyst, which avoided the environmental damage caused by the metal residue of the catalyst and reduced the energy consumption of the catalyst preparation process. (3) Oxazolidinones were recovered by capturing carbonyl groups in BPA-PC through amino alcohol. By this method, oxazolidinone could be prepared under more environmentally friendly conditions without the participation of toxic reagents such as phosgene. (4) The reaction could proceed smoothly with the participation of 1 equivalent of ethanolamine rather than the involvement of excess reagents; hence the atom utilization of the whole process was 100%, conforming to the principles of green chemistry. (5) The reaction could also be completed under solvent-free conditions, which was more consistent with the principles of green chemistry.
In addition, other commercial polyester materials, such as PLA cups, PBS straws and PET bottles, were also investigated to verify the broad polymer adaptability of this strategy. Different aminolysis products were obtained under solvent-free conditions with high yields (Fig. 6). As depicted in Fig. 6a, PLA cups exhibited a high degradation reactivity and were transformed into 2-hydroxy-N-(2-hydroxyethyl)propanamide in less than 1 h with a yield of 97%, indicating that PLA degrades faster than BPA-PC under the same conditions. For the aminolysis depolymerization of PBS straw, it took 5 h to achieve high conversion under the same conditions, producing the resultant degradation product with high yield (98%) (Fig. 6b). In addition, PET bottles were consumed completely within 24 h under bulk depolymerization conditions to produce bis(2-hydroxyethyl) terephthalamide (BHETA) with a yield of 96% (Fig. 6c). Although the degradation products were not the initial monomers, the introduction of NH made it possible to synthesize polymers with exceptional properties compared to the initial monomers.63,64 Further developmental applications of these monomers are under investigation in our laboratory.
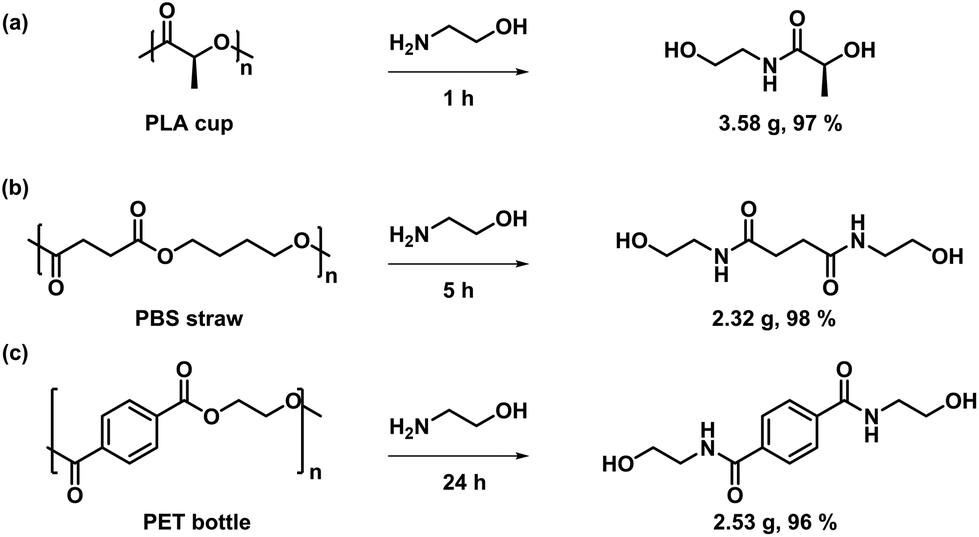 | ||
| Fig. 6 Degradation of (a) PLA, (b) PBS and (c) PET by using ethanolamine in bulk. Conditions: plastics (2 g), ethanolamine (5 mL), 80 °C. | ||
Conclusions
In summary, an economical, practical and green route for the chemical recycling of BPA-PC has been developed through a catalyst-free amino-alcoholysis depolymerization strategy. Using the strong nucleophilic ethanolamine as the depolymerization reagent and promotion by a highly polar solvent, the reaction could be carried out efficiently under mild conditions. Moreover, BPA-PC could also be degraded with high yields under solvent-free conditions with only 1 equivalent of depolymerization reagent. These strategies proved to be applicable to different types of commercial BPA-PCs. The feasibility of the practical applications of this method was further verified by the scale-up reaction. Besides, the recovered high-purity BPA could be repolymerized to prepare new polymer materials, realizing a closed-loop recycling process. In addition, oxazolidinones with various structures were recovered from this process. These strategies are of great significance for plastic recycling and economic sustainability. Further investigations of this new strategy using different plastics are in progress in our laboratory.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21901249), the Taishan Scholars Program of Shandong Province (tsqn201812112), the Scientific Research and Innovation Fund Project of the Shandong Energy Research Institute (SEI I202004), and the Zhoushan Science and Technology Project (2020C02001).References
- M. Hong and E. Y.-X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- J. M. Millican and S. Agarwal, Macromolecules, 2021, 54, 4455–4469 CrossRef CAS.
- X. Tang and E. Y.-X. Chen, Chem, 2019, 5, 284–312 CAS.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- A. J. Martín, C. Mondelli, S. D. Jaydev and J. Pérez-Ramírez, Chem, 2021, 7, 1487–1533 Search PubMed.
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310–6322 RSC.
- J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041–4070 CrossRef CAS PubMed.
- M. Hong and E. Y.-X. Chen, Trends Chem., 2019, 1, 148–151 CrossRef CAS.
- B. M. Stadler, C. Wulf, T. Werner, S. Tin and J. G. de Vries, ACS Catal., 2019, 9, 8012–8067 CrossRef CAS.
- Z. Ma, M. W. Ryberg, P. Wang, L. Tang and W.-Q. Chen, ACS Sustainable Chem. Eng., 2020, 8, 16861–16868 CrossRef CAS.
- T. Chilton, S. Burnley and S. Nesaratnam, Resour., Conserv. Recycl., 2010, 54, 1241–1249 CrossRef.
- J. D. Badia and A. Ribes-Greus, Eur. Polym. J., 2016, 84, 22–39 CrossRef CAS.
- R. Petrus, D. Bykowski and P. Sobota, ACS Catal., 2016, 6, 5222–5235 CrossRef CAS.
- C. Jehanno, I. Flores, A. P. Dove, A. J. Müller, F. Ruipérez and H. Sardon, Green Chem., 2018, 20, 1205–1212 RSC.
- P. McKeown, M. Kamran, M. G. Davidson, M. D. Jones, L. A. Román-Ramírez and J. Wood, Green Chem., 2020, 22, 3721–3726 RSC.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- X.-B. Lu, Y. Liu and H. Zhou, Chem. – Eur. J., 2018, 24, 11255–11266 CrossRef CAS PubMed.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- G. Xu and Q. Wang, Green Chem., 2022, 24, 2321–2346 RSC.
- Y. Liu, H. Zhou, J.-Z. Guo, W.-M. Ren and X.-B. Lu, Angew. Chem., Int. Ed., 2017, 56, 4862–4866 CrossRef CAS PubMed.
- Y.-M. Tu, X.-M. Wang, X. Yang, H.-Z. Fan, F.-L. Gong, Z. Cai and J.-B. Zhu, J. Am. Chem. Soc., 2021, 143, 20591–20597 CrossRef CAS PubMed.
- H. Chen, Z. Shi, T.-G. Hsu and J. Wang, Angew. Chem., Int. Ed., 2021, 60, 25493–25498 CrossRef CAS PubMed.
- Y. Yu, L.-M. Fang, Y. Liu and X.-B. Lu, ACS Catal., 2021, 11, 8349–8357 CrossRef CAS.
- C. Li, R. J. Sablong, R. A. T. M. van Benthem and C. E. Koning, ACS Macro Lett., 2017, 6, 684–688 CrossRef CAS PubMed.
- W. Xiong, W. Chang, D. Shi, L. Yang, Z. Tian, H. Wang, Z. Zhang, X. Zhou, E.-Q. Chen and H. Lu, Chem, 2020, 6, 1831–1843 CAS.
- M. Hong and E. Y.-X. Chen, Nat. Chem., 2016, 8, 42–49 CrossRef CAS.
- M. Hong and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2016, 55, 4188–4193 CrossRef CAS PubMed.
- X. Tang, M. Hong, L. Falivene, L. Caporaso, L. Cavallo and E. Y.-X. Chen, J. Am. Chem. Soc., 2016, 138, 14326–14337 CrossRef CAS PubMed.
- Y. Shen, W. Xiong, Y. Li, Z. Zhao, H. Lu and Z. Li, CCS Chem., 2020, 2, 620–630 Search PubMed.
- J.-B. Zhu and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2019, 58, 1178–1182 CrossRef CAS PubMed.
- J. Yuan, W. Xiong, X. Zhou, Y. Zhang, D. Shi, Z. Li and H. Lu, J. Am. Chem. Soc., 2019, 141, 4928–4935 CrossRef CAS PubMed.
- C. Shi, Z.-C. Li, L. Caporaso, L. Cavallo, L. Falivene and E. Y.-X. Chen, Chem, 2021, 7, 670–685 CAS.
- B. A. Abel, R. L. Snyder and G. W. Coates, Science, 2021, 373, 783–789 CrossRef CAS PubMed.
- Y. Wang, M. Li, J. Chen, Y. Tao and X. Wang, Angew. Chem., Int. Ed., 2021, 60, 22547–22553 CrossRef CAS PubMed.
- L.-G. Li, Q.-Y. Wang, Q.-Y. Zheng, F.-S. Du and Z.-C. Li, Macromolecules, 2021, 54, 6745–6752 CrossRef CAS.
- D. Sathe, J. Zhou, H. Chen, H.-W. Su, W. Xie, T.-G. Hsu, B. R. Schrage, T. Smith, C. J. Ziegler and J. Wang, Nat. Chem., 2021, 13, 743–750 CrossRef CAS PubMed.
- M. Häußler, M. Eck, D. Rothauer and S. Mecking, Nature, 2021, 590, 423–427 CrossRef PubMed.
- J. G. Kim, Polym. Chem., 2020, 11, 4830–4849 RSC.
- D. Li and S. Suh, Environ. Int., 2019, 123, 580–587 CrossRef CAS PubMed.
- C. Brede, P. Fjeldal, I. Skjevrak and H. Herikstad, Food Addit. Contam., 2003, 20, 684–689 CrossRef CAS PubMed.
- Z. Wang, R. Yang, G. Xu, T. Liu and Q. Wang, ACS Sustainable Chem. Eng., 2022, 10, 4529–4537 CrossRef CAS.
- R. Singh, S. Shahi and Geetanjali, ChemistrySelect, 2018, 3, 11957–11962 CrossRef CAS.
- K. Saito, C. Jehanno, L. Meabe, J. L. Olmedo-Martínez, D. Mecerreyes, K. Fukushima and H. Sardon, J. Mater. Chem. A, 2020, 8, 13921–13926 RSC.
- G. Grause, R. Kärrbrant, T. Kameda and T. Yoshioka, Ind. Eng. Chem. Res., 2014, 53, 4215–4223 CrossRef CAS.
- E. Quaranta, E. Mesto, M. Lacalamita, C. Malitesta, E. Mazzotta, E. Scelsi and E. Schingaro, Waste Manage., 2021, 120, 642–649 CrossRef CAS PubMed.
- L.-C. Hu, A. Oku and E. Yamada, Polymer, 1998, 39, 3841–3845 CrossRef CAS.
- F. Iannone, M. Casiello, A. Monopoli, P. Cotugno, M. C. Sportelli, R. A. Picca, N. Cioffi, M. M. Dell'Anna and A. Nacci, J. Mol. Catal. A: Chem., 2017, 426, 107–116 CrossRef CAS.
- T. Do, E. R. Baral and J. G. Kim, Polymer, 2018, 143, 106–114 CrossRef CAS.
- C. Jehanno, J. Demarteau, D. Mantione, M. C. Arno, F. Ruipérez, J. L. Hedrick, A. P. Dove and H. Sardon, ACS Macro Lett., 2020, 9, 443–447 CrossRef CAS PubMed.
- F. Liu, Z. Li, S. Yu, X. Cui and X. Ge, J. Hazard. Mater., 2010, 174, 872–875 CrossRef CAS PubMed.
- A. Sarkar, S. Santra, S. K. Kundu, A. Hajra, G. V. Zyryanov, O. N. Chupakhin, V. N. Charushin and A. Majee, Green Chem., 2016, 18, 4475–4525 RSC.
- J. Lee, J. Lee, H. Jung, D. Kim, J. Park and S. Chang, J. Am. Chem. Soc., 2020, 142, 12324–12332 CrossRef CAS PubMed.
- D. J. Kerr, J. M. White and B. L. Flynn, J. Org. Chem., 2010, 75, 7073–7084 CrossRef CAS PubMed.
- Y.-J. Guo, S.-J. Li, Y.-L. Sun, L. Wang, W.-M. Zhang, P. Zhang, Y. Lan and Y. Li, Green Chem., 2021, 23, 7041–7052 RSC.
- H. Liao and Q. Zhu, Org. Biomol. Chem., 2020, 18, 215–219 RSC.
- C. Jehanno, J. Demarteau, D. Mantione, M. C. Arno, F. Ruiperez, J. L. Hedrick, A. P. Dove and H. Sardon, Angew. Chem., Int. Ed., 2021, 60, 6710–6717 CrossRef CAS PubMed.
- S. Fukuoka, M. Kawamura, K. Komiya, M. Tojo, H. Hachiya, K. Hasegawa, M. Aminaka, H. Okamoto, I. Fukawa and S. Konno, Green Chem., 2003, 5, 497–507 RSC.
- S. Fukuoka, M. Tojo, H. Hachiya, M. Aminaka and K. Hasegawa, Polym. J., 2007, 39, 91–114 CrossRef CAS.
- B.-L. Deng, T. L. Hartman, R. W. Buckheit, C. Pannecouque, E. De Clercq, P. E. Fanwick and M. Cushman, J. Med. Chem., 2005, 48, 6140–6155 CrossRef CAS PubMed.
- G.-C. Chu, M. Pan, J. Li, S. Liu, C. Zuo, Z.-B. Tong, J.-S. Bai, Q. Gong, H. Ai, J. Fan, X. Meng, Y.-C. Huang, J. Shi, H. Deng, C. Tian, Y.-M. Li and L. Liu, J. Am. Chem. Soc., 2019, 141, 3654–3663 CrossRef CAS PubMed.
- Y. Liu, Z. Yi, X. Yang, H. Wang, C. Yin, M. Wang, X.-Q. Dong and X. Zhang, ACS Catal., 2020, 10, 11153–11161 CrossRef CAS.
- H. Sakaguchi, H. Tokuyama and T. Fukuyama, Org. Lett., 2007, 9, 1635–1638 CrossRef CAS PubMed.
- J. Natarajan, G. Madras and K. Chatterjee, ACS Appl. Mater. Interfaces, 2017, 9, 28281–28297 CrossRef CAS.
- T. H. N. Nguyen, F. Balligand, A. Bormann, V. Bennevault and P. Guégan, Eur. Polym. J., 2019, 121, 10314 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for the products. See DOI: https://doi.org/10.1039/d2gc03568e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |