
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioavailability and systemic transport of oleanolic acid in humans, formulated as a functional olive oil†
Aída
García-González
a,
Juan Manuel
Espinosa-Cabello
 b,
Isabel
Cerrillo
a,
Emilio
Montero-Romero
c,
Juan José
Rivas-Melo
d,
Andrea
Romero-Báez
b,
María Dolores
Jiménez-Andreu
c,
Carmen Amelia
Ruíz-Trillo
c,
Ana
Rodríguez-Rodríguez
c,
Antonio Jesús
Martínez-Ortega
c,
María
Del Carmen Roque-Cuellar
c,
Silvia
García-Rey
c,
Andrés
Jiménez-Sánchez
c,
Miguel Ángel
Mangas-Cruz
c,
José Luis
Pereira-Cunill
c,
Javier S.
Perona
b,
Pedro Pablo
García-Luna
*c and
José María
Castellano
*b
b,
Isabel
Cerrillo
a,
Emilio
Montero-Romero
c,
Juan José
Rivas-Melo
d,
Andrea
Romero-Báez
b,
María Dolores
Jiménez-Andreu
c,
Carmen Amelia
Ruíz-Trillo
c,
Ana
Rodríguez-Rodríguez
c,
Antonio Jesús
Martínez-Ortega
c,
María
Del Carmen Roque-Cuellar
c,
Silvia
García-Rey
c,
Andrés
Jiménez-Sánchez
c,
Miguel Ángel
Mangas-Cruz
c,
José Luis
Pereira-Cunill
c,
Javier S.
Perona
b,
Pedro Pablo
García-Luna
*c and
José María
Castellano
*b
aUniversity Pablo de Olavide, Department of Molecular Biology and Biochemical Engineering, 41013-Seville, Spain
bSpanish Scientific Research Council, Instituto de la Grasa-CSIC, 41013-Seville, Spain. E-mail: jmcas@ig.csic.es
cRegional Andalusian Health Service, Service of Endocrinology and Nutrition, University Hospitals Virgen del Rocío, 41013-Seville, Spain. E-mail: garcialunapp@yahoo.es
dUniversity of Sassari, Faculty of Medicine, 07100 Sassari, Italy
First published on 2nd October 2023
Abstract
Evidence of the pharmacological activity of oleanolic acid (OA) suggests its potential therapeutic application. However, its use in functional foods, dietary supplements, or nutraceuticals is hindered by limited human bioavailability studies. The BIO-OLTRAD trial is a double-blind, randomized controlled study with 22 participants that received a single dose of 30 mg OA formulated as a functional olive oil. The study revealed that the maximum serum concentration of OA ranged from 500 to 600 ng mL−1, with an AUC0−∞ value of 2862.50 ± 174.50 ng h mL−1. Furthermore, we discovered a physiological association of OA with serum albumin and triglyceride-rich lipoproteins (TRL). UV absorption spectra showed conformational changes in serum albumin due to the formation of an adduct with OA. Additionally, we demonstrated that TRL incorporate OA, reaching a maximum concentration of 140 ng mL−1 after 2–4 hours. We conjecture that both are efficient carriers to reach target tissues and to yield high bioavailability.
1. Introduction
Oleanolic acid (OA; PubChem CID: 10494) (Fig. 1) is a hydroxy-pentacyclic triterpene widely found in the plant kingdom, where it occurs naturally in both free form and as the aglycone of triterpenoid saponins. The scientific community has focused its attention on OA due to its potential therapeutic applications against a variety of diseases. Evidence suggests that OA may be effective in treating dyslipidemia, insulin resistance, hypertension, diabetes, cardiovascular and neurodegenerative disorders, as well as different types of cancer. Additionally, OA has been shown to possess antiviral, antibacterial, antifungal, anti-cariogenic, anti-inflammatory, hepato-protective, gastro-protective, and anti-atherosclerotic properties.1 | ||
| Fig. 1 Molecular structure of oleanolic acid. | ||
OA is present in many medicinal herbs and foods. Among them, it is a natural constituent of olive oils, with concentrations ranging from 30 mg kg−1 in virgin olive oil to 190 mg kg−1 in pomace olive oil.2 Therefore, OA is an usual ingredient of the human diet.3
Current understanding of OA bioavailability in humans is derived from a very limited number of studies. As a strong hydrophobic compound, OA is almost insoluble in water. It is classified as a class IV drug in the biopharmaceutical classification system, signifying limited pharmacological effects attributable to its low solubility in aqueous solutions and theoretical difficulties in permeating biological membranes.4 These characteristics have led to the belief that OA has poor bioavailability. However, in 2015, we demonstrated that the oral bioavailability of this triterpene may be highly increased if it is dissolved in a lipid matrix,5 when compared with the intake of solid micronized formulations.6,7 These findings corroborated that the OA bioavailability is dependent on the formulation in which it is administered.
On the other hand, there is also very scarce knowledge regarding the mechanisms by which OA is transported in the bloodstream when orally ingested. Serum proteins play a key role in binding and transporting endogenous and exogenous molecules, including hormones, fatty acids, and xenobiotics. The interactions of these substances with serum proteins strongly influence their distribution, free concentration, and metabolism.8 In a previous study,5 we reported that OA had a high affinity to be linked (>90%) to human serum proteins (HSP), presumably albumin (HSA). HSA is responsible for binding mainly acidic drugs.9 Furthermore, considering the highly lipophilic nature of OA, we now propose that OA may also be transported within postprandial lipoproteins. In a recent investigation, we discovered that OA efficiently associates with laboratory-made triglyceride-rich lipoproteins (TRL),10 suggesting that its presence in human postprandial TRL after oral OA administration is highly plausible.
In view of this background, we have developed a randomized and controlled postprandial trial with adult individuals of both sexes, following the oral intake of a single dose of OA. This study has been designed and executed with two main objectives: (1) to contribute with more robust data to the understanding of the pharmacokinetics of OA in humans, extending the study also to women, and using in the triterpene formulation a commercial olive oil widely consumed; and (2) to deepen in the knowledge of the association of OA with serum proteins. We hypothesize that the bioavailability of OA can be very substantially improved by administering the triterpenoid dissolved in a functional olive oil. Furthermore, we propose that after ingestion, a portion of OA binds to HSA, while another fraction integrates into postprandial TRL. We suggest that both pathways are efficient routes for OA to reach its target tissues and achieve high bioavailability.
2. Materials and methods
2.1. Materials and reagents
We obtained OA from olive leaves with a purity of ≥95% in our laboratory based on the Spanish Patent ES2160553.11 Betulinic acid (BA) with a purity of 90% was purchased from Sigma-Aldrich (St Louis, MO, USA). A commercial-grade olive oil was kindly provided by ACESUR Group S.L. Methanol of chromatographic grade and other reagents of analytical grade were used. Milli Q water (Milli Q Direct; Millipore Merck, Darmstadt, Germany) was used throughout the study.2.2. Elaboration of functional olive oil enriched in OA
The commercial olive oil (control oil) was enriched up to 600 mg kg−1 by adding high purity OA, following the procedure described in the European Patent EP18382510.12 No additional substances were added as adjuvants. The quality indexes and chemical composition of both control and functional oils are provided in Appendix C Table S1 (ESI†). Both oils were bottled in 100 mL amber-colored flasks and labeled with alphanumeric codes for blinding. The codes were kept confidential from participants, physicians, and nurses.2.3. Trial design, settings, and participants
The BIOavailability of Oleanolic acid in the TReAtment of Diabetes (BIO-OLTRAD) trial (ClinicalTrials.gov CNT05529953) was designed as a randomized, crossover, double-blinded, and controlled trial in the postprandial phase. The trial was conducted from March to June 2021 at the Virgen del Rocío University Hospitals (HUVR) in Seville, Spain. The participants were healthy men and women, aged 18–30 years, residing in the community. They had a body mass index (BMI) in the range of 18.5–29.9 kg m−2 and fasting serum triglyceride, cholesterol, and glucose concentrations within the normal ranges. Participants with digestive or metabolic disorders, allergies or intolerances related to food consumption, and pregnant or breastfeeding women within 3 months postpartum were excluded. Individuals with alcohol or drug dependence and physical or intellectual limitations were also excluded. All participants provided informed consent after receiving detailed information about the trial's objectives and methodology.2.4. Determination of sample size
The primary quantitative variable of interest was the serum OA concentration. Due to the lack of consistent published data, we referred to the PREDIABOLE study13 to estimate the maximal concentration of serum OA in a cohort from the control group of this trial, which was 155 ± 72 ng OA per mL. Considering this, we postulated that the OA content in serum would be increased by at least 50% as a consequence of the functional olive oil intake. We tested this null hypothesis of unilateral contrast, comparing two paired means (repeated in a group), with a confidence level of 99% (α = 0.01) and a power of 95% (β = 0.05), assuming as tolerable participant losses of less than 15%. We used the “GRANMO Sample Size Calculator” software (https://www.imim.cat/ofertadeserveis/software-public/granmo/), determining a sample size of 18 volunteers.2.5. Recruitment and intervention
Twenty-two young and healthy individuals of both sexes were recruited from the university academic environment for the trial. They were instructed to abstain from consuming alcoholic beverages and tobacco the day before the experiments. On the test days, the participants arrived after a 12-hour overnight fasting period, and blood samples were collected from their cubital vein. These samples were analyzed for general, liver, and kidney biochemistry parameters at the Clinical Biochemistry and Analysis Laboratory of the Virgen del Rocío University Hospitals.Following the initial blood sampling, the participants were randomly divided into two groups of eleven individuals each. One group received a breakfast consisting of 50 g of OA-enriched olive oil (equivalent to a dose of 30 mg of OA), 208 g of skimmed milk, 71 g of whole meal bread, and 20 g of mashed tomato, providing a total energy content of 2806 kJ. The other group received the same quantity of the control olive oil as part of the experimental meal. After consuming their respective breakfasts, blood samples were collected from the participants every hour throughout a 6 hours postprandial period. The participants had unrestricted access to water for hydration during this time.
Following the completion of the first test session, a washout period of four weeks was implemented before the second session was conducted. During the second session, the study groups switched and ingested the type of oil they had not consumed in the first session. This crossover study design allowed each participant to serve as their own control, minimizing potential confounding factors related to inter-individual variations. The study protocol is detailed in Appendix B of ESI.†
2.6. Analysis of blood biochemical parameters of the participants
Blood samples were analyzed using standard automated assays at the Clinical Biochemistry and Analysis Laboratory of the Virgen del Rocío University Hospitals. Enzymatic methods were used to determine serum glucose, total cholesterol, HDL and triglycerides, whereas LDL was estimated by the Friedewald formula.14 Total lipoprotein Apo B (Apo B48 + Apo B100) was quantified by an immunoturbidimetric assay (Tinaquant; Roche Diagnostics, Mannheim, Germany), and serum insulin using an ELISA kit (Diaclone, Besançon, France).2.7. Serum sample preparation for OA analysis
Serum was obtained by centrifugation (1700g, 20 min, 15 °C) from blood aliquots drawn every hour throughout the postprandial period. Sodium azide, pheny-methyl-sulfonyl-fluoride and aprotinin, at final concentrations of 1 mmol L−1, 10 mol L−1, and 28 mg L−1, respectively, were added to the serum to prevent the proteolytic degradation of the apolipoproteins. Sera were stored at −80 °C until used.To 1 mL of serum, 15 μL of a betulinic acid (BA, internal standard, IS) solution (0.1 mg mL−1 in methanol) was added. The mixture was stirred for 60 s and dry nitrogen was passed to remove traces of methanol. Subsequently, it was filtered through a cellulose acetate membrane filter with a pore size 0.20 μm (AC02047BL), and extracted with 3.0 mL of 5% isopropyl alcohol in diethyl ether, with vigorous vortex mixing for 3 min followed by centrifugation at 3000 rpm for 10 min. These extracting steps were repeated three times. Supernatants were combined and brought to dryness under a dry nitrogen stream. The residue was resuspended and derivatized with BSTFA [N,O-bis-(trimethylsilyl) trifluoroacetamide +1% TMCS (trimethylchlorosilane)] in pyridine (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v), assisted by ultrasound (70 W; 40 °C, 15 min) (ULTRASONS H-D, P-Selecta, Spain). A 1 μL aliquot of the derivatized sample was analyzed by gas chromatography.
:1 v/v), assisted by ultrasound (70 W; 40 °C, 15 min) (ULTRASONS H-D, P-Selecta, Spain). A 1 μL aliquot of the derivatized sample was analyzed by gas chromatography.
2.8. Isolation of TRL and determination of OA
The TRL fraction was obtained from human serum by ultracentrifugation at 356000g (18 h at 15 °C), using a Beckman L8-70M preparative ultracentrifuge and a SW 41 Ti rotor (Beckman Instruments, Palo Alto, CA, USA), as previously described by ref. 15. The TRL layer was carefully transferred to a vial and 20 μL of the 0.1 mg mL−1 BA solution were added. The mixture was vortexed for 1 min and the solvent removed with dry nitrogen. The lipid content of TRL was extracted with 5 mL of chloroform:methanol 2:1 (v/v) and vortexed for 2 min. Subsequently, 2 mL of saline was introduced in the vial and vortexed for other 1 min. The mixture remained at rest for 5 min and then centrifuged at 1700g for 15 min at room temperature. The organic phase was recovered. This procedure was repeated with the resulting aqueous phase. The two organic phases were combined, reduced to dryness under nitrogen, and re-dissolved in 200 μL of the mixture BSTFA/pyridine for silylation.
2.9. Gas chromatography-mass spectrometry (GC-MS) determination of OA
The identification and quantification of OA in the serum and TRL samples were performed using a gas chromatography-mass spectrometry (GC-MS) system, following a modification of the method established and validated in our laboratory.5 A 1 μL aliquot of the silylated sample was injected into a coupled QP2010 Plus GC-MS (Shimadzu Europa GmbH), equipped with a Rtx-65TG column (30 m × 0.25 mm I.D.; 0.10 mm stationary phase thickness) (Restek, Co., Bellefonte, PA, USA) and fitted with an AOC-20i autosampler, an ion source of electron impact and a quadrupole detector. Injection was made in a split-mode (split ratio 5:1) and the injector temperature was maintained at 290 °C. Helium was used as carrier gas at a constant flow of 1 mL min−1. The oven temperature was programmed: 90 °C for 1 min; 90–250 °C at 40 °C min−1; 250–310 °C at 10 °C min−1; and finally held for 2 min. Total run time was 13.00 min. The MS operating conditions were: interface temperature, 290 °C; ion source temperature, 250 °C; electron impact, 70 eV; acquisition in scan mode (m/z 50–600). Identification of the chromatographic peaks was made by GC retention times and also by comparison of mass spectra with those provided by the standards substances. Specifically, the retention times of OA and BA (IS) trimethylsilyl derivatives were 8.97 and 9.07 min, respectively, whereas the identification/quantification of these triterpenes was accomplished based in the abundance of two major fragments ions (203 and 189 m/z). This method has yielded a limit of detection (LOD) of 10.7 ng mL−1. The response factor OA/BA was determined as 1.96. The blank serum chromatogram shows no peak at these retention times (data not shown), revealing that the method is free of interferences from other compounds present in the samples.
2.10. Pharmacokinetic analysis of serum OA concentrations
The one-compartment pharmacokinetic model was assumed, and the serum OA concentration-time curve plotted. The maximum serum OA concentration (Cmax) and the time taken to reach it (tmax) were directly determined from the curve. The apparent elimination rate constant (Ke) was calculated by fitting mean data at four terminal points of the serum concentration profile using a log-linear regression equation with the least-squares method. The half-life (t1/2) was determined as 0.693/Ke. The area under the serum concentration-time curve from zero to the last measurable sample (AUC0–6) was obtained using the linear-trapezoidal rule up to the final sampling point. The area under the serum concentration-time curve from zero to infinity (AUC0–∞) was calculated using the trapezoidal rule with extrapolation to infinity using Ke. The mean residence time (MRT) was estimated as the ratio of the area under the first moment curve (AUMC0–6) to AUC0–6. The apparent total body clearance (CL) was calculated as dose/AUC0–6, whereas the apparent volume of distribution (Vd) was assessed as dose/C0.2.11. Study of OA association with human serum albumin
To investigate the binding of OA to serum proteins, particularly HSA, a modification of the procedure established by16 for the in vitro interaction of OA with bovine serum albumin was employed. Serum was diluted 1:50 with 0.1 M Tris-HCl buffer (pH 7.4) containing 0.9% NaCl (w/w), to achieve an albumin concentration of approximately 20 μM (molar extinction coefficient, ε, of 35700 M−1 cm−1 at 280 nm). UV absorption spectra in the wavelength range of 250–310 nm (slit width 5 nm) were recorded at room temperature using a NanoDrop 2000c spectrophotometer (Thermo, Waltham, MA, USA) with quartz cells. Blank samples consisting of buffer alone were assayed and subtracted to correct for background absorbance.
2.12. Chemical composition of the control and OA-enriched olive oils
:1, v/v) at a flow rate of 1 ml min−1. Detection was performed by fluorescence with excitation and emission wavelengths set at 290 nm and 330 nm, respectively, using an RF-10AXL Shimadzu fluorescence detector (Shimadzu, Columbia, MD, USA). For quantitative determinations, solutions of tocopherol standards in hexane (1–6 μg ml−1) were analyzed. Data were processed using 32 Karat 5.0 system software.
:methylene chloride (90:10, v/v) and hexane:ethyl acetate (40:60, v/v). The corresponding eluates were discarded. Finally, the fraction containing the triterpenes was eluted with diethyl ether:acetic acid (98:2, v/v), and the eluate was evaporated under a nitrogen stream until dryness. The residue was re-dissolved and derivatized with 200 μL of the BSTFA/pyridine reagent, and a 1 μL aliquot of the silylated solution was analyzed by GC-FID using an Agilent 6890N GC (Agilent Technologies, CA) equipped with an FID and a Rtx-65TG column (30 m × 0.25 mm I.D.; 0.1 mm film thickness) (Restek, Co., Bellefonte, PA, USA). The injection was performed in split mode (1:5 split ratio), and hydrogen was used as the carrier gas. The oven temperature was initially set at 90 °C for 1 min and programmed to increase first to 250 °C at a rate of 40 °C min−1 and then to 310 °C at a rate of 10 °C min−1, maintaining this temperature for 4 min. The injector and detector temperatures were isothermally set at 300 °C.
2.13. Ethics
The protocols of the study were approved by the Institutional Bioethics Committee of the CSIC during its session on February 12th, 2021. Prior to participating in the trial, all participants were fully informed about the objectives and methodology of the study, and they provided their consent by signing a consent form. The study was conducted in adherence to the guidelines and principles outlined in the Helsinki Declaration, the Council of Europe Convention, and the UNESCO Universal Declaration.2.14. Statistical analysis
The trial was designed and conducted following the intention-to-treat principle. All the experiments included in the present work were performed in triplicate. Data were analyzed using SPSS Statistics version 26 (IBM Corp., Armonk, NY, USA) and GraphPad Prism version 9.0 (GraphPad Software, San Diego, CA, USA). Normality of data distribution was assessed using the Kolmogorov–Smirnov test. The variables were expressed as mean ± standard deviation (SD), unless otherwise stated. Differences between the study groups were evaluated by one- or two-way ANOVA, following by Bonferronís multiple-comparison test or Kruskal–Wallis test. A p-value of less than 0.05 was considered statistically significant.3. Results
3.1. Eligibility/recruitment/drops and baseline characteristics of participants
From December 2020 to February 2021, 47 men and women from the university academic environment were screened for eligibility to participate in this trial, and 22 volunteers who fulfilled the requirements to be included were recruited. The participants were highly homogeneous with respect to all the variables considered, including clinical characteristics (Table 1). The first sessions of the study were performed in March–April and the second ones during May–June 2021. No significant differences were observed in the baseline characteristics of the participants at the start of the two sessions. The volunteers did not report significant changes in physical activity during the entire study period, nor significant changes in their diets. One drop was recorded in the second session of the trial. It was not possible to identify any adverse effect attributable to the dietary intervention.| Parameter | Value | ||
|---|---|---|---|
| 1st session | 2nd session | ||
| Anthropometric measurements | Sex (n, %) | ||
| Women | 11 (50%) | 10 (48%) | |
| Men | 11 (50%) | 11 (52%) | |
| Age (years) | 22.0 ± 2.6 | 21.9 ± 2.6 | |
| Height (cm) | 173.1 ± 7.9 | 172.8 ± 7.9 | |
| Weight (kg) | 70.8 ± 11.7 | 70.1 ± 12.0 | |
| Muscle mass (kg) | 53.0 ± 11.2 | 52.4 ± 11.3 | |
| Bone mass | 2.8 ± 0.5 | 2.8 ± 0.6 | |
| Fat mass | 14.1 ± 7.9 | 14.0 ± 7.7 | |
| BMI (kg m−2) | 23.2 ± 2.8 | 23.1 ± 2.8 | |
| SBP (mm Hg) | 114 ± 15 | 113 ± 15 | |
| DBP (mm Hg) | 69 ± 10 | 68 ± 10 | |
| Pulse (bpm) | 78 ± 12 | 78 ± 12 | |
| Waist circumference (cm) | 76.6 ± 8.0 | 76.0 ± 8.2 | |
| Hip circumference (cm) | 101.1 ± 8.0 | 100.9 ± 7.8 | |
| Blood biochemistry | Glucose (mg dL−1) | 78.0 ± 8.2 | 76.3 ± 8.9 |
| Insulin (μIU mL−1) | 6.0 ± 2.6 | 6.0 ± 4.3 | |
| CT (mg dL−1) | 169.2 ± 33.2 | 169.6 ± 29.8 | |
| HDL (mg dL−1) | 64.3 ± 16.4 | 63.8 ± 15.4 | |
| LDL (mg dL−1) | 94.2 ± 23.5 | 92.1 ± 21.0 | |
| TG (mg dL−1) | 65.3 ± 22.3 | 68.6 ± 21.0 | |
| Protein (mg dL−1) | 7.4 ± 0.4 | 7.6 ± 0.5 | |
| Urea (mg dL−1) | 31.7 ± 7.7 | 30.4 ± 7.1 | |
| Creatinine (mg dL−1) | 0.90 ± 0.15 | 0.89 ± 0.14 | |
| Total bilirubin (mg dL−1) | 0.69 ± 0.38 | 0.85 ± 0.45 | |
| AST (IU L−1) | 21.2 ± 5.6 | 21.9 ± 2.6 | |
| ALT (IU L−1) | 15.9 ± 5.0 | 16.2 ± 8.9 | |
| GGT (IU L−1) | 12.9 ± 4.1 | 12.1 ± 4.8 | |
| Alcaline phosphatase (IU L−1) | 67.5 ± 20.8 | 69.2 ± 26.1 | |
| Na (mEq L−1) | 141.9 ± 2.2 | 140.5 ± 2.4 | |
| K (mEq L−1) | 4.1 ± 0.2 | 4.1 ± 0.6 | |
| Ca (mg dL−1) | 9.7 ± 0.3 | 9.8 ± 0.3 | |
| P (mg dL−1) | 3.7 ± 0.6 | 3.8 ± 0.6 | |
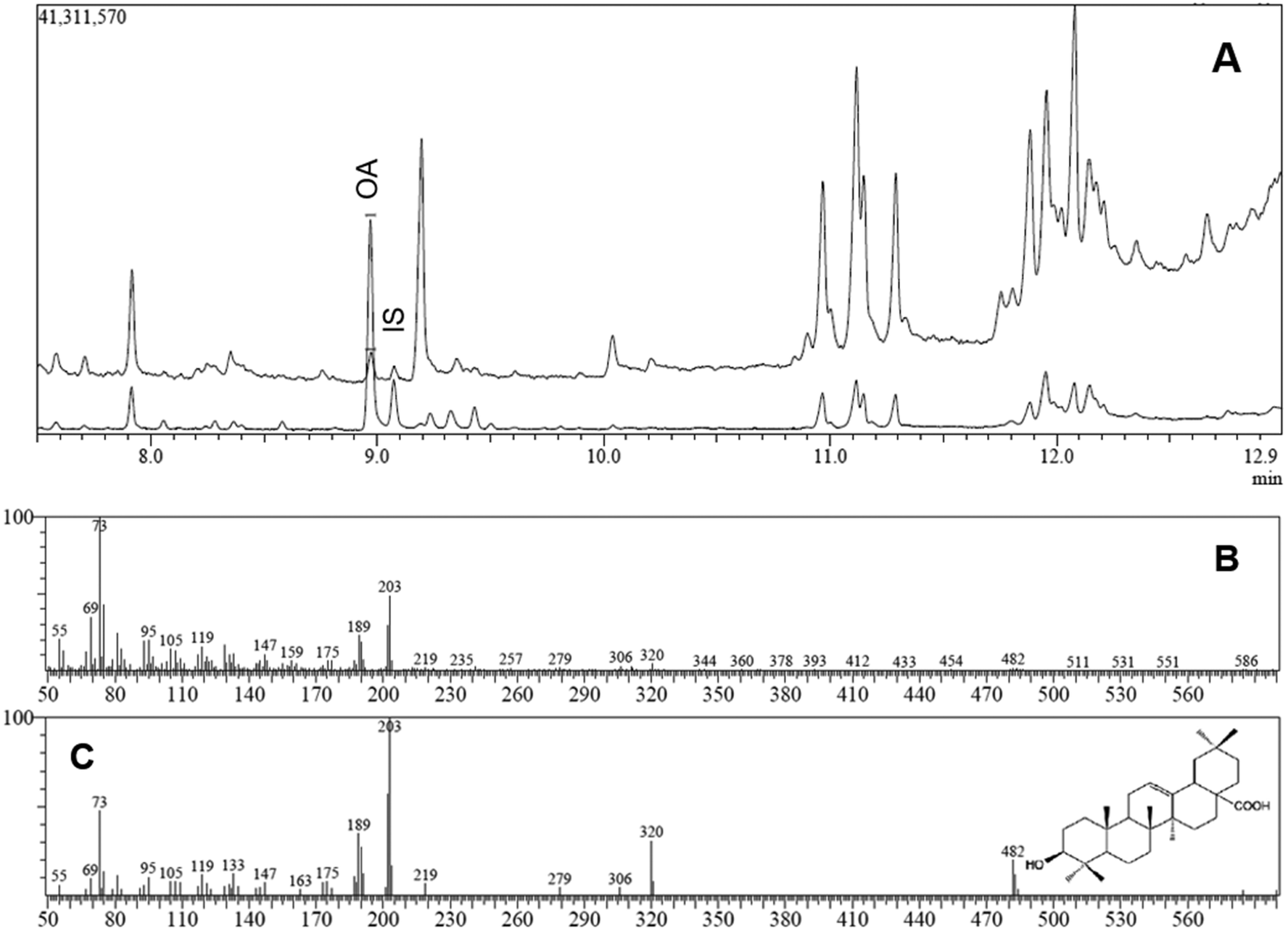 | ||
| Fig. 2 OA analysis by GC-MS. (A) Typical chromatogram of the triterpenoid fraction isolated from a serum aliquot obtained within the postprandial phase from an individual who consumed the OA- enriched functional olive oil. The peak eluted at 8.97 min corresponded to oleanolic acid (OA), whereas that at 9.07 min corresponded to Betulinic acid (BA), used as internal standard (IS). (B) Experimental mass spectrum for chromatographic peak eluted at 8.97 min. (C) Mass spectrum for standard OA in the Mass Spectra Library NIST 08. | ||
The serum OA concentration versus time profiles for both oil types (functional and control) throughout the 6 h postprandial phase are shown in Fig. 3A, and the main pharmacokinetic parameters are listed in Table 2. Serum OA concentration remained above the limit of detection (LOD) during the 6 h postprandial period analyzed. Baseline serum OA concentrations were around 25 ng mL−1, and the maximal concentration of the triterpene (524 ± 110 ng mL−1) was achieved at 3 h after the intake of the functional olive oil. The AUC0–6 value was determined as 2019.30 ± 164.20 ng h mL−1, and Vd in 47.99 ± 8.90 L. Fig. 3B displays the ln[OA] − t profile for this OA-enriched olive oil which was obtained to estimate parameters related to the elimination phase. The elimination rate constant (Ke) was obtained as the slope of the linear regression of this natural logarithm-transformed concentration values versus time data in the terminal phase, yielding a value of 0.28 ± 0.02 h−1. AUC0−∞ was calculated using the trapezoidal rule with extrapolation to infinity with Ke obtaining a value of 2862.50 ± 174.50 ng h mL−1. On the other hand, the calculated values of MRT and CL were 3.19 ± 0.39 h and 14.96 ± 1.22 L h−1, respectively.
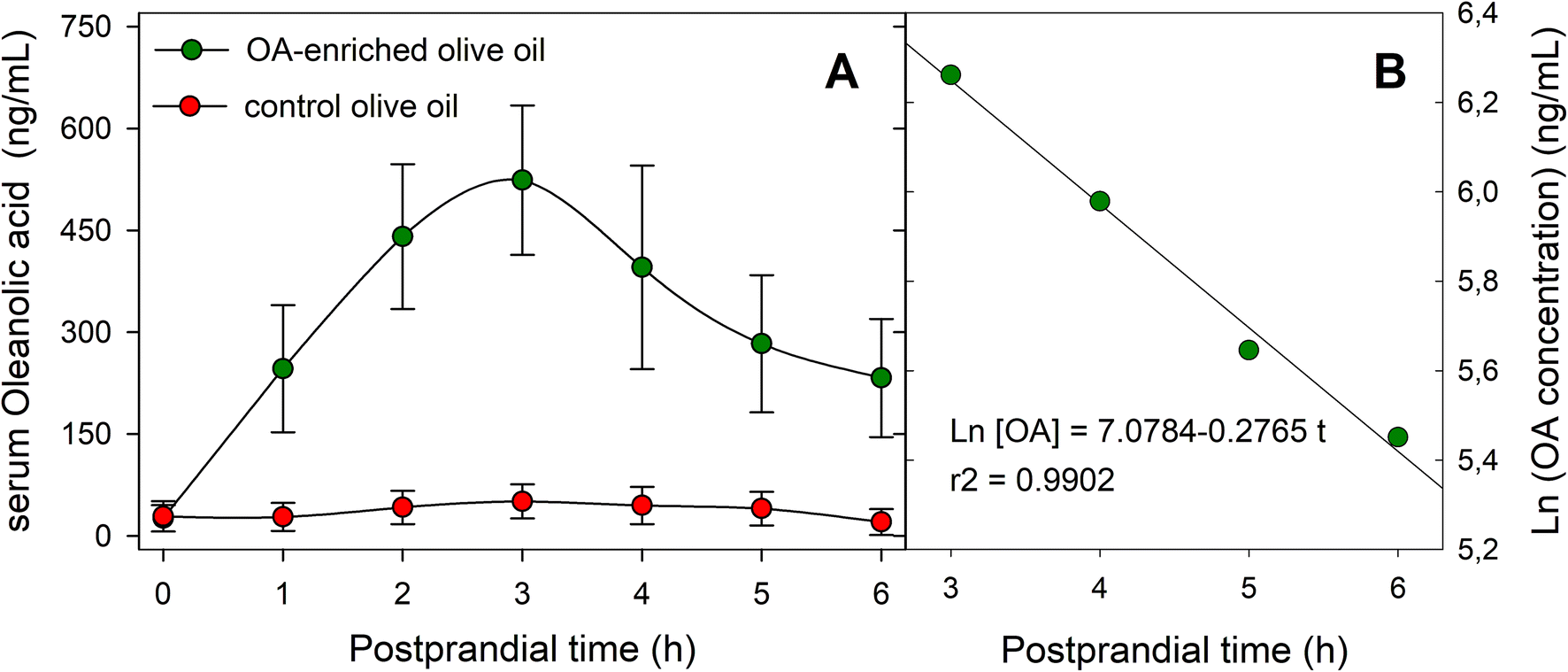 | ||
| Fig. 3 Serum levels of OA after the ingestion of 50 g of OA-enriched olive oil (equivalent dose 30 mg OA) and control oil. (A) Serum concentration-time profiles; (B) semi-logarithm concentration-time curse for the last four postprandial sampling in individuals consuming the OA-enriched olive oil. | ||
| Parameter | Value (mean ± SD) |
|---|---|
| C max (ng mL−1) | 524.00 ± 110.20 |
| t max (h) | 3.00 ± 0.00 |
| k e (h−1) | 0.28 ± 0.02 |
| t 1/2 (h) | 5.49 ± 0.02 |
| AUC0–6 (ng h mL−1) | 2019.30 ± 164.20 |
| AUC0–∞ (ng h mL−1) | 2862.50 ± 174.50 |
| AUMC0–6 (ng h2 mL−1) | 6395.00 ± 573.40 |
| MRT (h) | 3.19 ± 0.39 |
| CL/F (L h−1) | 14.96 ± 1.22 |
| V d (L) | 47.99 ± 8.90 |
3.2. Association of OA to HSA
In the present work we have analyzed the UV spectra of aliquots of human serum obtained throughout the postprandial phase. Fig. 4A shows the changes in the UV absorbance-wavelength profile of serum proteins after ingesting 30 mg OA included in the experimental meal as functional olive oil, and displays that maximal absorbance intensity, estimated as both area under the absorbance-wavelength curve and the absorbance at λmax (278–280 nm), were achieved around 2–4 hours postprandial, a lapse of time in which both parameters increased by 12–14%. By contrast, when the control olive oil was introduced in the meal no rise in the absorbance intensity was observed (Fig. 4B). In fact, both area under the absorbance-wavelength curve and the absorbance at λmax remained in the range 95–102% of the baseline values. To corroborate the suitability of the spectrophotometric method used, analogous preliminary tests were carried out using bovine serum albumin (BSA), as model albumin protein, and blank human serum, which were spiked with different concentrations of standard OA (Fig. 4C and D). In all cases, a hyperchromic effect was reproduced, which indicates conformational changes of serum proteins, mainly albumin, as a consequence of the formation of adducts with OA. Fig. 4C also shows that both OA, TRIS-HCl buffer and DMSO did not exhibit significant UV absorption in the range 250–310 nm.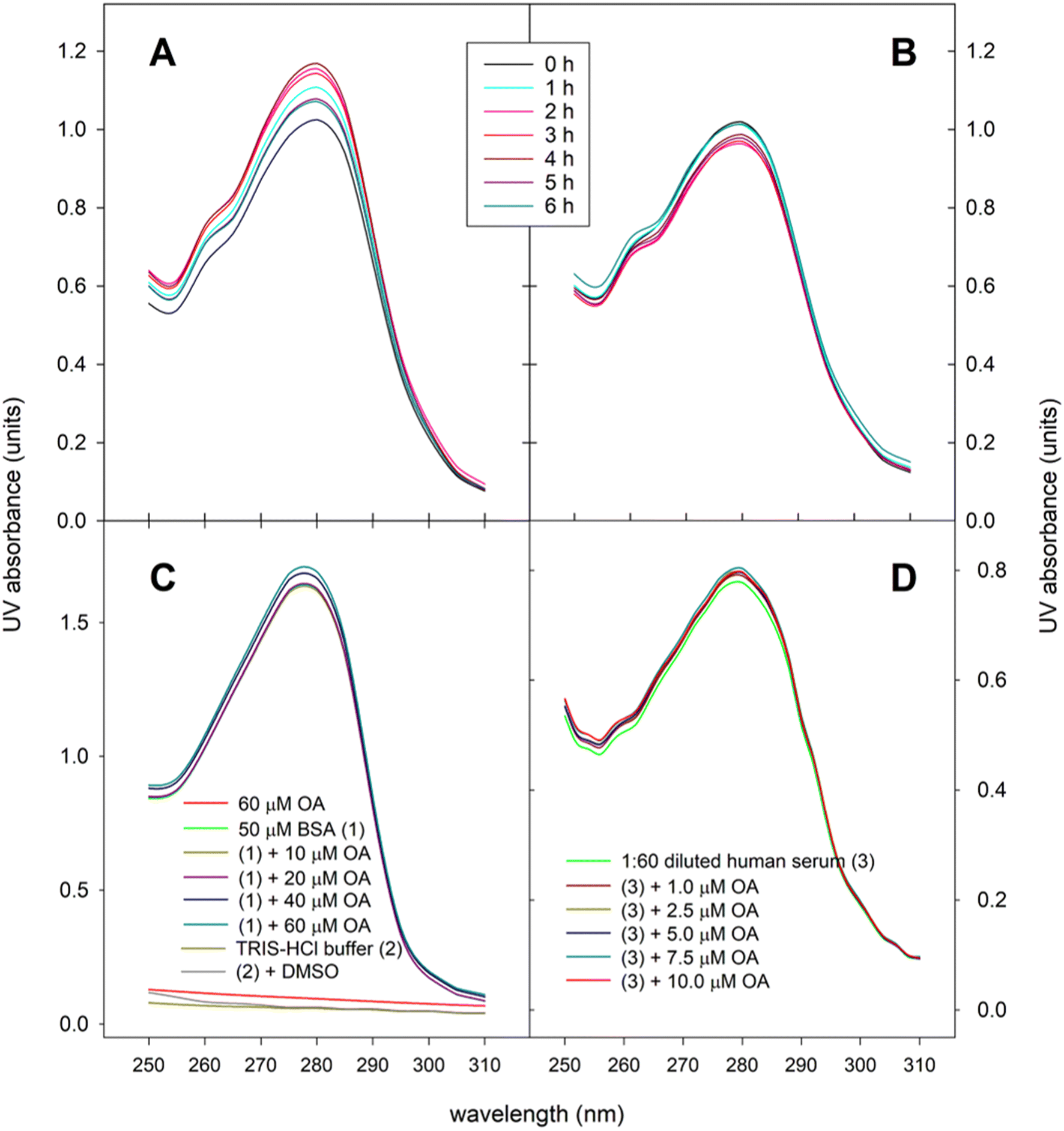 | ||
| Fig. 4 UV Absorption spectra at 25 °C of serum samples (diluted 1:50 in 0.1 M Tris-HCI buffer at pH 7.4) obtained before and after the ingestion of the experimental meal containing (A) the OA-enriched olive oil (equivalent dose 30 mg OA) or (B) the control olive oil. (C and D) shows the UV absorption spectra in experiments with standard BSA and blank human serum, respectively, spiked with different concentrations of standard OA. | ||
3.3. Association of OA to triglyceride-rich lipoproteins
The TRL fraction was isolated from the serum aliquots by ultracentrifugation, their lipid content extracted, and the presence of OA analyzed. Fig. 5 displays for the first time to the best of our knowledge the physiological inclusion of OA in the core of human TRL generated during the postprandial phase. In addition, the figure also shows that the highest OA contents in TRL were reached between 2 and 4 hours after the intake of the functional olive oil, mirroring the evolution of the serum OA concentration-time curse. In this period of time, the OA contents in the TRL obtained from one milliliter of serum were close to 140 ng in the volunteers who consumed the functional olive oil, while in the individuals who ingested the control olive oil these values reached a maximum of 45 ng.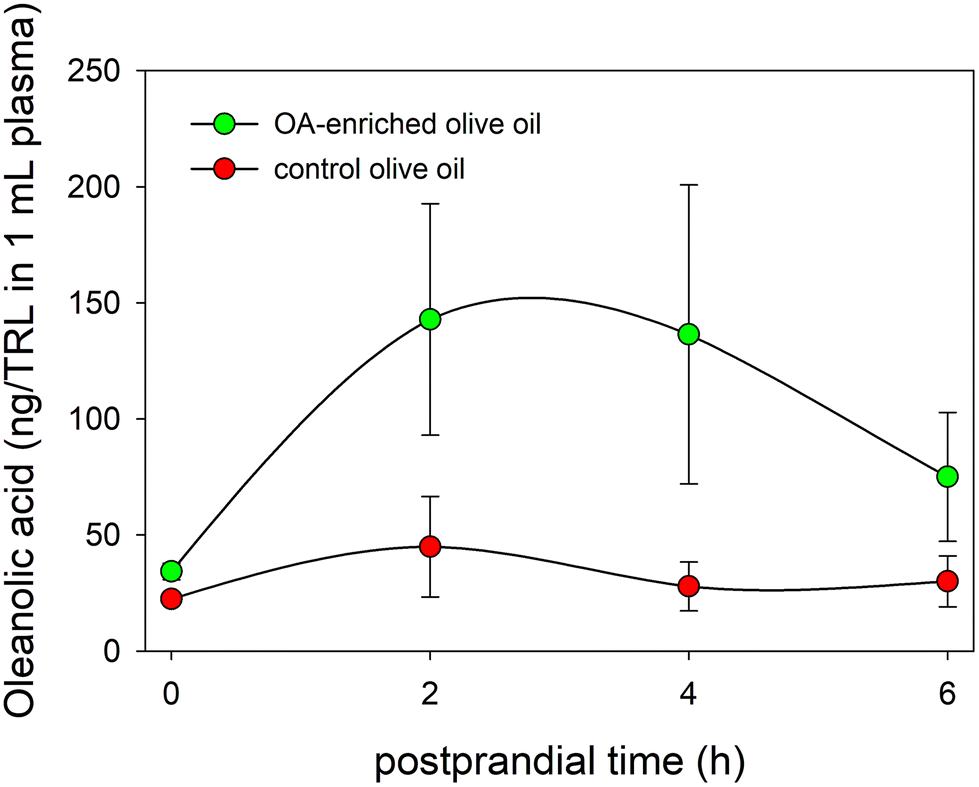 | ||
| Fig. 5 Evolution of OA concentrations in TRL throughout the postprandial period after the intake of the OA-enriched olive oil and the control one. | ||
3.4. Effects of the experimental meals on serum biochemistry of the participants during the postprandial phase
Fig. 6 shows the evolution of a series of serum parameters related to glycemic and lipid metabolisms. Glycemia remained practically stable throughout the postprandial period, although a relative maximum few prominent was observed one hour after the ingestion of the experimental meal. No significant differences were found in blood glucose values when comparing the intake of the two types of oil (Fig. 6A). The evolution of serum insulin levels is presented in Fig. 6B. It showed a marked maximum value at 60 minutes after the ingestion, as response to the increase in glucose. No significant differences were detected between both types of oils. Regarding serum concentrations of total cholesterol, HDL-cholesterol and LDL-cholesterol, no notable changes were found throughout the entire postprandial period (Fig. 6C–E), with no significant differences between the oils. On the other hand, serum triglycerides experienced a slight increasing trend throughout the postprandial period, but neither could significant differences be observed between the tested oils (Fig. 6F).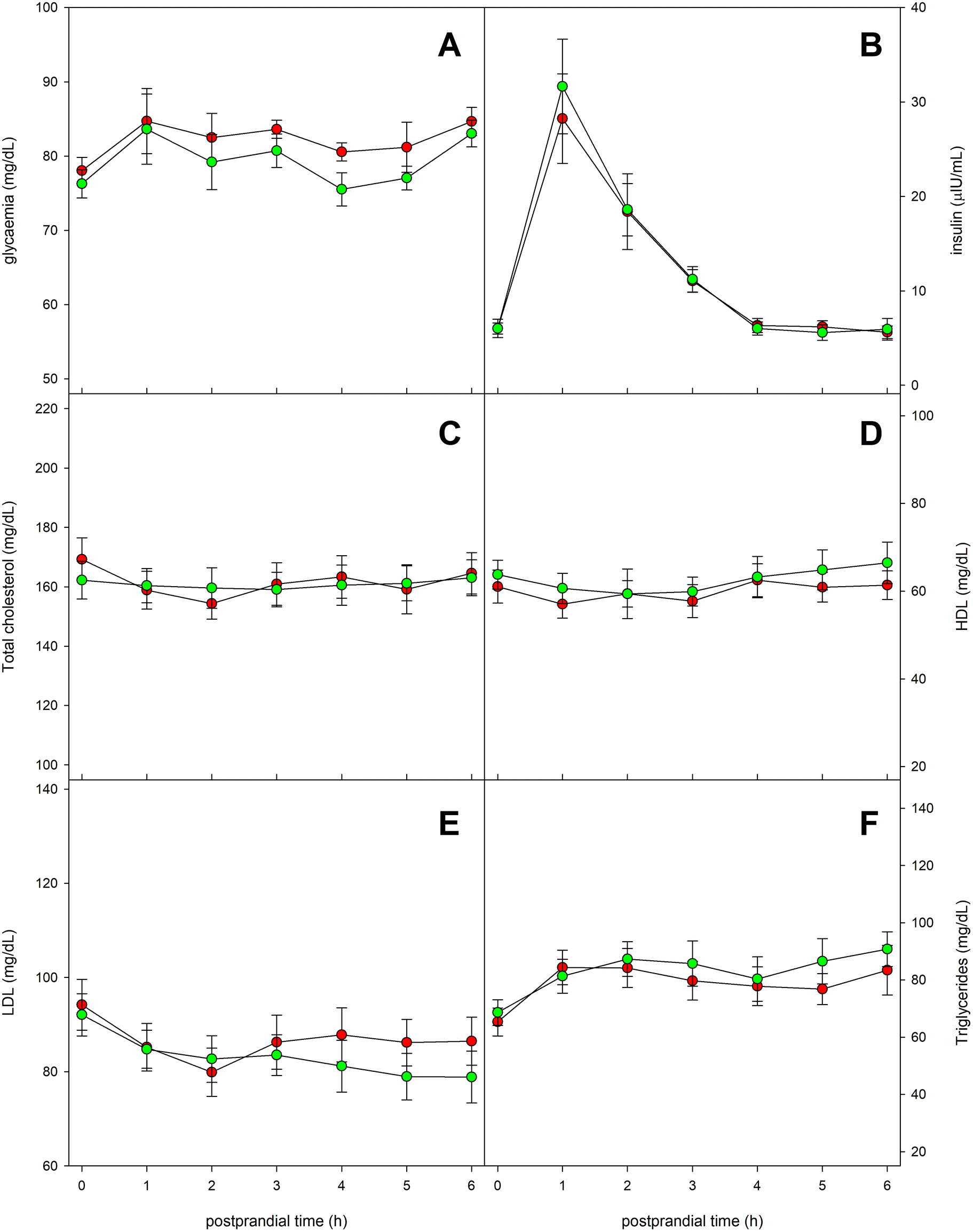 | ||
| Fig. 6 Time-course of blood biochemical parameters related to glycemic and lipid metabolisms. (A) glycaemia; (B) insulinemic; (C) total cholesterol; (D) HDL; (E) LDL; and (F) triglycerides. Red circles correspond to the OA-enriched olive oil and the green ones to the control olive oil. | ||
4. Discussion
To fully exploit the pharmacological potential of OA in therapies for the management of varied diseases is necessary to establish firmly a deeper knowledge of its bioavailability. Only with solid data from human studies will be possible its successful use in functional food, dietary supplements or nutraceutical.Previous human studies in vivo have shown that OA bioavailability differs when it is administered as pure compound or in a complex matrix, such as a food item. Indeed, Song et al.6 conducted a study with 18 healthy men in China who received a single 40 mg dose of pure solid micronized OA in a soft capsule. The analysis of the OA contents in postprandial venous blood samples revealed an area under the plasma OA concentration-time curve (AUC) of 124.3 ± 106.7 ng h mL−1, with a maximum OA concentration of 12.1 ± 6.8 ng mL−1. In a subsequent study, Chen et al.7 performed a randomized, crossover, self-controlled trial involving 18 healthy Chinese male volunteers who normal and dispersible tablet forms, reporting similar pharmacokinetic parameters, including AUC values of 112.2 ± 56.6 and 109.7 ± 41.6 ng h mL−1 for normal and dispersible tablets, respectively, and received 20 mg of solid micronized OA orally in both peak OA concentrations of 18.9 ± 8.0 and 17.8 ± 7.5 ng mL−1. These human trials, besides some physicochemical features of OA, led to the belief that this triterpene has poor bioavailability. However, we executed in 2015 a trial with nine Spanish adult men who were dosed with 30 mg of OA dissolved in 70 g of pomace olive oil, and determined an AUC value of 3181.9 ± 894.3 ng h mL−1, with a peak OA concentration of 598.2 ± 176.7 ng mL−1, reached at 3.0 hours postprandially.5 These findings demonstrated that dissolving the triterpene in an edible vegetable oil the bioavailability of OA in humans can be significantly increased. Moreover, in the PREDIABOLE Study (Current Controlled Trials number ISRCTN03372660), we proved that this same OA dose, formulated as OA-enriched functional olive oil, was safe and therapeutically effective in the prevention of Type 2 Diabetes Mellitus in prediabetic patients with impaired fasting glucose (IFG) and impaired glucose tolerance (IGT).13
Now, in the BIO-OLTRAD trial we have also used a single dose of 30 mg OA (equivalent to 50 g functional olive oil) to confirm those earlier results. In this trial, we have recruited individuals of both sexes (n = 22) and, in addition, the randomized crossover design has allowed us to compare the effects of the olive oil enriched with OA versus the control olive oil within the same individuals. Therefore, the trial has provided reliable data on the pharmacokinetics of OA and on the impact of the triterpene on various physiological parameters during the postprandial phase.
Our results corroborate that OA bioavailability may be markedly enhanced when it is dissolved in a lipid matrix, such as an olive oil, in comparison with reported data on the administration of OA capsules or tablets.
The presence of dietary fat seems of major importance, since the solubilization and micellization of OA together with other lipids are necessary steps prior to its absorption.21 The activities of gut microbiota and digestive enzymes may also influence the OA absorption in the gastrointestinal tract surface.22 Although absorption and metabolism processes are not well established, permeability studies in Caco-2 cells have revealed that both passive and P-glycoprotein-mediated active transport are involved in the intestine brush border absorption of OA.23 In addition, it has been proposed that this process takes place through the paracellular route, through the relaxation of the intercellular tight junctions.24
In this work, we have confirmed that the maximal serum OA concentration achieved levels in the range 500–600 ng mL−1, with an AUC0−∞ value of 2862.50 ± 174.50 ng h mL−1 when administrated within a lipid matrix (Table 2). This means that the total OA exposure was 112-fold higher than when the triterpene was administered as a soft capsule.6 Furthermore, we have calculated an apparent volume of distribution, Vd, of 47.99 ± 8.90 L. This parameter does not correspond to a “true volume”, but it relates the concentration of the compound in serum to the total amount of it in the body. The Vd is conditioned by binding to serum proteins, among other aspects. Thus, if a biomolecule/drug is highly bound to serum protein, its Vd will be small. On the other hand, depending on the value of Vd, the distribution of the drug in the body could be roughly deduced: a Vd around 40 L indicates that the drug is able to cross cell membranes and to access the intracellular space. Furthermore, a Vd value higher than 40 L suggest that the drug is concentrated in certain organs.25
The available evidence suggests that, once OA first passed through the liver, it circulates in the bloodstream mainly associated with serum proteins, although detectable levels of OA in its free acid form are also observed.26 The OA affinity toward serum proteins is an important question when determining its pharmacokinetic profile. HSA, the most abundant protein in human serum (≈600 μM), has large binding capacity that allows to enhance the effective solubility of bioactive substances in serum. HSA may enhance their distribution and bioaccessibility, modulate their delivery rate and therapeutic efficiency.27
The nature of OA–HSA interaction has not been fully explored, although it is known that the native conformation of the protein is affected by the binding with the triterpene.28 Thermodynamic analyses have demonstrated that the basic forces acting between triterpenes and BSA are hydrogen bonds, van der Waals forces, and hydrophobic interactions.29 These molecular interactions are often monitored by spectroscopic techniques because these methods are sensitive and easy to use.
Fluorescence quenching spectroscopy revealed a single binding site in HSA for OA, which caused a significant alteration in the native tertiary protein structure.27 This finding contrasts with studies of the OA interaction with BSA, in which OA was found to bind tryptophan residues, causing minimal conformational changes in the protein.30 Measurement of absorbance spectra in the UV region is also an applicable method to explore the structural change and to display the formation of complexes.31,32 Proteins present intrinsic UV absorbance at approximately 280 nm. On the basis of these considerations, a previous research of our group26 studied the in vitro formation of OA–HSP complexes by UV spectrophotometry, observing that the spiking of OA to HSP dose-dependently increased the absorption intensity at λmax (278–280 nm), indicating a change in the protein conformation as a consequence of the OA binding.
In the present work, we recorded the UV absorption spectra of serum aliquots and we verified that they indeed fluctuate during the postprandial period, when the participants in the trial ingested the OA-enriched olive oil (Fig. 4). Maximal absorbance intensity was determined at 2–4 hours postprandially, a lapse of time in which UV absorption (assessed as both the area under the absorbance-wavelength curve and the absorbance at λmax) increased by 12–14%. This hyperchromic effect reflects the conformational change of the protein by the formation of the OA–HSP adduct. The progression of that changes mimics the evolution of the serum OA concentration-time curve quantified by GC-MS (Fig. 3). To our knowledge, this is the first time that the physiological formation of OA–HSP complexes has been reported in real postprandial serum samples.
In addition to albumin, other serum proteins, such as glycoproteins, lipoproteins and, to a lesser extent, globulins, might have the ability to bind biomolecules and drugs. Indeed, it has been previously described that different phenolic compounds are transported associated to human serum proteins, such as albumin33,34 and lipoproteins,35,36 but also into exosomes.37,38 Regarding OA, and given that it is a strictly hydrophobic substance, we hypothesized that, after ingesting a functional olive oil enriched in this triterpene, a significant fraction of it could also be integrated into postprandial TRL, along with triglycerides, cholesterol, and cholesterol esters. In this regard, our laboratory previously developed a procedure to elaborate synthetic TRL with lipid composition on demand,15 and more recently we reported that these laboratory-made TRL can harbor OA in their lipophilic core.10 We also demonstrated that administering these artificial lipoproteins to THP-1 macrophages and BV-2 microglial cells, OA can exert its pharmacological potential (specifically antioxidant and anti-inflammatory activities).10,39
In the present investigation, we report, for the first time to the best of our knowledge, that physiological TRL assembled after ingestion of functional olive oil incorporate the administered OA. From Fig. 4 it can also be inferred that the maximum OA concentration reached in these lipoprotein particles would be of the order of 140 ng mL−1, at 2–4 hours postprandially.
In a previous work from our laboratory,5 blank human sera were spiked with different concentrations of standard OA (1–20 μg mL−1), and after equilibration at 37 °C, an Amicon (Millipore, Bedford, MA, USA) YM-30 system with a molecular cut-off of 30000 was used to separate the protein-bound from the free OA, by centrifuge ultrafiltration (3000g for 30 min, at 37 °C). Aliquots of each ultrafiltrate was subjected HPLC analysis to determine the concentration of free OA. In that study we estimated than more 90% of the OA provided was bound to HSP.
Alternatively, Zhang et al.40 established a procedure which combined equilibrium dialysis with LC-MS to quantify both the total concentration and the free form percentage of OA in human serum. The authors applied the method with three different OA concentrations, and determined that the degree of OA association with human serum proteins could be estimated at a 60–80%, while the specific association with HSA would account for 45–55% of the triterpene concentration.
The results of the present work agree with these antecedents. Quantification of the OA content in the isolated fraction of postprandial TRL would indicate that a portion around 20–25% of the circulating triterpene would be associated with these lipoproteins, whereas the remaining 75–80% would remain bound to other HSPs, mainly HSA.
5. Conclusions
In summary, with this work we provide new data on the bioavailability of OA in humans, and corroborate previous results showing that it depends on the nature of the formulation in which the triterpene is administered. We report that the administration of OA integrated in fatty foods, such as an olive oil, promotes high circulating levels of the compound during the postprandial phase. On the other hand, we provide new evidence of the physiological association of triterpenes with serum albumin and TRL (chylomicrons and VLDL) as systemic transport mechanisms.We conjecture that the involvement of HSA and postprandial TRL, as biological Trojan horse-like carriers of OA, affects the solubility and bioaccessibility of the triterpene, and that these mechanisms may be an efficient way of delivering OA to target tissues and produce high bioavailability.
Therefore, the findings reported here are of high biological importance, since the interaction of OA with serum proteins was only described in experimental models until now, but not in a human trial. In our opinion, this work is also relevant in nutritional research because it sheds light on the metabolic pathway of triterpenoids, a fundamental item to exploit the pharmacological potential of OA in the human health. Furthermore, the recent use of proteins as vehicles for bioactive molecules has revealed as a promising approach in this field.41–44
Informed consent statement
All the participants signed their consent to participate after being informed orally and documentarily of the objectives and methodology of the trial (see Appendix A. ESI†).Data availability statement
The datasets generated during the trial are available in DIGITAL CSIC repository (https://hdl.handle.net/10261/335582) upon reasoned request to the corresponding author.Author contributions
Conceptualization, I. C., J. S. P., P. P. G. L. and J. M. C.; methodology, I. C. G., E. M. R., A. R. R., J. S. P. and J. M. C.; investigation, A. G. G., J. M. E. C., I. C. G., J. J. R. M., A. R. B., M. D. J. A., C. A. R. T., A. R. R., J. S. P. and J. M. C.; resources, I. C. G., E. M. R., A. J. M. O., M. C. R. C., S. G. R., A. J. S., M. A. M. C., J. L. P. C.; formal análisis, A. G. G., J. M. E. C., I. C. G., J. J. R. M., A. R. B., A. R. R., J. S. P. and J. M. C.; validation, J. M. C.; data curating, J. M. C.; visualization, J. S. P. and J. M. C.; writing – original draft preparation, J. M. C.; writing – review and editing, I. C. G., J. S. P., P. P. G. L.; supervisión, E. M. R., P. P. G. L. and J. M. C.; funding adquisition, P. P. G. L. and J. M. C.; project administration, P. P. G. L. and J. M. C.; all the authors have read and agreed to the published version of the manuscript.Conflicts of interest
All the authors declare that they have no conflicts of interest.Acknowledgements
This research is part of the R+D+i project PID2019-107837RB-I00, funded by the Spanish Ministry of Science and Innovation/Spanish National Research Agency, grant number MCIN/AEI/10.13039/501100011033/. A. G.-G. is grateful for funding received from the “Next Generation EU” funds, the European Union through the Recovery, Transformation and Resilience Plan and by the Ministry of Universities, in the framework of the Margarita Salas, Maria Zambrano grants for the Requalification of the Spanish University System 2021–2023, organized by the Pablo de Olavide University, Seville. J. J. R.-M. obtained an Erasmus+ scholarship (No. 2021-1-IT02-KA131-HED-000008483) from the University of Sassari (ITALY), for a stay at the Department of Food and Health of the Instituto de la Grasa-CSIC. The authors especially thank the ACESUR Group (Dos Hermanas, Seville, Spain), which donated the commercial olive oil for the trial. This collaborator had no role in the design, collection, analysis or interpretation of the data or in the decision to submit the manuscript for publication.References
- J. M. Castellano, S. Ramos-Romero and J. S. Perona, Oleanolic Acid: Extraction, Characterization and Biological Activity, Nutrients, 2022, 14, 623 CrossRef CAS PubMed , 1–29.
- M. C. Pérez-Camino and A. Cert, Quantitative Determination of Hydroxy Pentacyclic Triterpene Acids in Vegetable Oils, J. Agric. Food Chem., 1999, 47, 1558–1562 CrossRef PubMed.
- S. Jäger, H. Trojan, T. Kopp, M. N. Laszczyk and A. Scheffler, Pentacyclic Triterpene Distribution in Various Plants—Rich Sources for a New Group of Multi-Potent Plant Extracts, Molecules, 2009, 14, 2016–2031 CrossRef PubMed.
- W. Jinhua, Ursolic acid: Pharmacokinetics process In Vitro and In Vivo, a mini review, Arch. Pharm., 2019, 352, e1800222 CrossRef PubMed , 1–5.
- M. Rada, J. M. Castellano, J. S. Perona and Á. Guinda, GC-FID determination and pharmacokinetic studies of oleanolic acid in human serum, Biomed. Chromatogr., 2015, 29, 1687–1692 CrossRef CAS PubMed.
- M. Song, T.-J. Hang, Y. Wang, L. Jiang, X.-L. Wu, Z. Zhang, J. Shen and Y. Zhang, Determination of oleanolic acid in human plasma and study of its pharmacokinetics in Chinese healthy male volunteers by HPLC tandem mass spectrometry, J. Pharm. Biomed. Anal., 2006, 40, 190–196 CrossRef CAS PubMed.
- R.-J. Chen, X. Liu, P.-M. Li, L. Zhang, L. Zhao and X.-L. Zhang, Pharmacokinetic profiles of oleanolic acid formulations in healthy Chinese male volunteers, Chin. Pharm. J., 2010, 45, 621–626 CAS.
- P. Lee and X. Wu, Review: Modifications of Human Serum Albumin and Their Binding Effect, Curr. Pharm. Des., 2015, 21(14), 1862–1865 CrossRef CAS PubMed.
- N. Husain, R. A. Agbaria and I. M. Warner, Spectroscopic analysis of the binding of doxorubicin to human R-1 acid glycoprotein, J. Phys. Chem., 1993, 97, 10857–10861 CrossRef CAS.
- J. M. Espinosa, J. M. Castellano, S. Garcia-Rodriguez, A. Quintero-Flórez, N. Carrasquilla and J. S. Perona, Lipophilic Bioactive Compounds Transported in Triglyceride-Rich Lipoproteins Modulate Microglial Inflammatory Response, Int. J. Mol. Sci., 2022, 23, 7706 CrossRef CAS PubMed , 1–16.
- Á. Guinda, T. Albi and A. Lanzón, Procedure for the obtaining of terpenic acids from the leaf of Olea europaea, Spanish Patent, ES2160553, 2001 Search PubMed.
- J. M. Castellano, M. Rada, Á. Guinda, J. M. Santos-Lozano and J. Lapetra, Olive oil enriched with oleanolic acid, process for its preparation and use thereof, European Patent, EP18382510, 2018 Search PubMed.
- J. M. Santos-Lozano, M. Rada, J. Lapetra, Á. Guinda, M. C. Jiménez-Rodríguez, J. A. Cayuela, A. Ángel-Lugo, A. Vilches-Arenas, A. M. Gómez-Martín, M. Ortega-Calvo and J. M. Castellano, , Prevention of type 2 diabetes in prediabetic patients by using functional olive oil enriched in oleanolic acid: The PREDIABOLE study, a randomized controlled trial, Diabetes, Obes. Metab., 2019, 21, 2526–2534 CrossRef CAS PubMed.
- W. T. Friedewald, R. J. Levy and D. S. Fredrickson, Estimation of the concentration of low-density lipoprotein cholesterol in plasma without use of the preparative ultracentrifuge, Clin. Chem., 1972, 18, 499–502 CrossRef CAS.
- J. S. Perona and V. Ruiz-Gutierrez, Quantification of major lipid classes in human triacylglycerol-rich lipoproteins by high-performance liquid chromatography with evaporative light-scattering detection, J. Sep. Sci., 2004, 27, 653–659 CrossRef CAS PubMed.
- Z. Cheng and Y. Zhang, Fluorometric investigation on the interaction of oleanolic acid with bovine serum albumin, J. Mol. Struct., 2008, 879, 81–87 CrossRef CAS.
- EU European Union, Commission Delegated Regulation (EU) 2022/2104 of 29 July 2022 supplementing Regulation (EU) No 1308/2013 of the European Parliament and of the Council as regards marketing standards for olive oil, and repealing Commission Regulation (EEC) No 2568/91 and Commission Implementing Regulation (EU) No 29/2012., Off. J. Eur. Union, 2022, 284/1(1234), L284/1–L284/22 Search PubMed.
- Commission THEE., Commission Implementing Regulation (EU) 2022/2105 of 29 July 2022, Off. J. Eur. Union, 2022, 284/23(1234), 23–48 Search PubMed.
- IUPAC Standard Method 2.432, Standard methods for the analysis of oils, fats and derivatives, Determination of tocopherol and tocotrienols in vegetable oils and fats by HPLC, Blackwell Scientific, Oxford, U.K., 1987 Search PubMed.
- International Olive Council, Determination of the composition and content of sterols, triterpenic dialcohols and aliphatic alcohols by capillary column gas chromatography, Int. Olive Counc., 2020, Doc. No. 26(Rev. 4), 1–31 Search PubMed.
- J. M. Castellano, J. M. Espinosa and J. S. Perona, Modulation of Lipid Transport and Adipose Tissue Deposition by Small Lipophilic Compounds, Front. Cell Dev. Biol., 2020, 8, Article 555359 CrossRef PubMed , 1–15.
- M. J. Rein, M. Renouf, C. Cruz-Hernandez, L. Actis-Goretta, S. K. Thakkar and M. da Silva Pinto, Bioavailability of bioactive food compounds: A challenging journey to bioefficacy, Br. J. Clin. Pharmacol., 2013, 75, 588–602 CrossRef CAS PubMed.
- M. Wang, T. Zhao, Y. Liu, Q. Wang, S. Xing, L. Li, L. Wang, L. Liu and D. Gao, Ursolic acid liposomes with chitosan modification: promising antitumor drug delivery and efficacy, Mater. Sci. Eng., C, 2017, 71, 1231–1240 CrossRef CAS PubMed.
- B. Srinivasan, A. R. Kolli, M. B. Esch, H. E. Abaci, M. L. Shuler and J. J. Hickman, TEER measurement techniques for in vitro barrier model systems, J. Lab. Autom., 2015, 20, 107–126 CrossRef CAS PubMed.
- A. Canut-Blasco, L. Aguilar-Alfaro, J. Cobo-Reinoso, M. J. Giménez-Mestre and A. Rodríguez-Gascón, Pharmacokinetic-pharmacodynamic analysis in Microbiology: tools to evaluate antimicrobial treatment, Enferm. Infecc. Microbiol. Clin., 2015, 33(1), 48–57 CrossRef PubMed.
- M. Rada, V. Ruiz-Gutiérrez and Á. Guinda, Determination of triterpenic acids in human serum by high-performance liquid chromatography: triterpenoid interaction with serum protein, J. Agric. Food Chem., 2011, 59, 2308–2313 CrossRef CAS PubMed.
- K. Dopierała, M. Krajewska and M. Weiss, Physicochemical Characterization of Oleanolic Acid−Human Serum Albumin Complexes for Pharmaceutical and Biosensing Applications, Langmuir, 2020, 36, 3611–3623 CrossRef PubMed.
- R. Abboud, C. Charcosset and H. Greige-Gerges, Interaction of Triterpenoids with Human Serum Albumin: A Review, Chem. Phys. Lipids, 2017, 207, 260–270 CrossRef CAS PubMed.
- W. Peng, F. Ding, Y. T. Jiang and Y. K. Peng, Bioavailability and Activity of Natural Food Additive Triterpenoids as Influenced by Protein, J. Agric. Food Chem., 2014, 62, 2271–2283 CrossRef CAS PubMed.
- H. Yang, Q. Liu, L. Zhao, Y. Yuan, D. Fan, J. Deng and R. Zhang, Fluorescence Spectroscopic Studies on the Interaction of Oleanolic Acid and Its Triterpenoid Saponins Derivatives with Two Serum Albumins, J. Solution Chem., 2014, 43, 774–786 CrossRef CAS.
- F. L. Cui, J. Fan, J. P. Li and Z. D. Hu, Interactions between 1-benzoyl-4-p-chlorophenyl thiosemicarbazide and serum albumin: investigation by fluorescence spectroscopy, Bioorg. Med. Chem., 2004, 12, 151–157 CrossRef CAS PubMed.
- Y.-J. Hu, Y. Liu, J.-B. Wang, X.-H. Xiao and S.-S. Qu, Study of the interaction between monoammonium glycyrrhizinate and bovine serum albumin, J. Pharm. Biomed. Anal., 2004, 36, 915–919 CrossRef CAS PubMed.
- H. Cao, X. Liu, N. P. Ulrih, P. K. Sengupta and J. Xiao, Plasma protein binding of dietary polyphenols to human serum albumin: A high performance affinity chromatography approach, Food Chem., 2019, 270, 257–263 CrossRef CAS PubMed.
- M.-J. Bae, T. Ishii, K. Minoda, Y. Kawada, T. Ichikawa, T. Mori, M. Kamihira and T. Nakayama, Albumin stabilizes (-)-epigallocatechin gallate in human serum: binding capacity and antioxidant property, Mol. Nutr. Food Res., 2009, 53, 709–715 CrossRef CAS PubMed.
- V. Ivanov, A. C. Carr and B. Frei, Red wine antioxidants bind to human lipoproteins and protect them from metal ion-dependent and -independent oxidation, J. Agric. Food Chem., 2001, 49, 4442–4449 CrossRef CAS PubMed.
- M. Urpí-Sardá, O. Jaúregui, R. M. Lamuela-Raventós, W. Jaeger, M. Miksits, M. I. Covas and C. Ancrés-Lacueva, Uptake of diet Resveratrol into the human low-density lipoprotein. Identification and quantification of Resveratrol metabolites by liquid chromatography with tandem mass spectrometry, Anal. Chem., 2005, 77, 3149–3155 CrossRef PubMed.
- A. Arola-Arnal, M. C. López de las Hazas, L. Iglesias-Carres, D. C. Mantilla-Escalante, M. Suárez, R. Busto, F. Visioli, C. Bladé and A. Dávalos, Exosomes transport trace amounts of (poly)phenols, Food Funct., 2020, 2020(11), 7784–7792 RSC.
- C. E. Iglesias-Aguirre, M. A. Ávila-Gálvez, M. C. López de las Hazas, A. Dávalos and J. C. Espín, Exosome-containing extracelular vesicles contribute to the transport of Resveratrol metabolites in the bloodstream: a human pharmacokinetic study, Nutrients, 2022, 14, Article 3632 CrossRef PubMed , 1–14.
- Á. Fernández-Aparicio, J. S. Perona, J. M. Castellano, M. Correa-Rodríguez, J. Schmidt-RioValle and E. González-Jiménez, Oleanolic Acid-Enriched Olive Oil Alleviates the Interleukin-6 Overproduction Induced by Postprandial Triglyceride-Rich Lipoproteins in THP-1 Macrophages, Nutrients, 2021, 13, Article 3471 CrossRef PubMed , 1–17.
- H. Zhang, H.-F. Zhang, H.-C. Chang, X. Han, K.-S. Bi and X.-H. Chen, Determination of protein binding rate of oleanolic acid in human plasma and serum albumin, Yaoxue Xuebao, 2011, 46, 243–246 CAS.
- Y. D. Livney, Milk Proteins as Vehicles for Bioactives, Curr. Opin. Colloid Interface Sci., 2010, 15, 73–83 CrossRef CAS.
- A. O. Elzoghby, W. M. Samy and N. A. Elgindy, Protein-Based Nanocarriers as Promising Drug and Gene Delivery Systems, J. Controlled Release, 2012, 161, 38–49 CrossRef CAS PubMed.
- G. M. Tavares, T. Croguennec, A. F. Carvalho and S. Bouhallab, Milk Proteins as Encapsulation Devices and Delivery Vehicles: Applications and Trends, Trends Food Sci. Technol., 2014, 37, 5–20 CrossRef CAS.
- A. Chakraborty and P. Dhar, A Review on Potential of Proteins as an Excipient for Developing a Nano-Carrier Delivery System, Crit. Rev. Ther. Drug Carrier Syst., 2017, 34, 453–488 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3fo02725b |
| This journal is © The Royal Society of Chemistry 2023 |