 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dielectric-free electrowetting on graphene
Athanasios A.
Papaderakis
 *ab,
Ji Soo
Roh
cd,
Kacper
Polus
ae,
Jing
Yang
ab,
Mark A.
Bissett
cd,
Alex
Walton
ae,
Anne
Juel
f and
Robert A. W.
Dryfe
*ab
*ab,
Ji Soo
Roh
cd,
Kacper
Polus
ae,
Jing
Yang
ab,
Mark A.
Bissett
cd,
Alex
Walton
ae,
Anne
Juel
f and
Robert A. W.
Dryfe
*ab
aDepartment of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: athanasios.papaderakis@manchester.ac.uk; robert.dryfe@manchester.ac.uk
bHenry Royce Institute, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cNational Graphene Institute, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
dDepartment of Materials, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
ePhoton Science Institute, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
fDepartment of Physics and Astronomy, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 27th March 2023
Abstract
Electrowetting is a simple way to induce the spreading and retraction of electrolyte droplets. This method is widely used in “device” applications, where a dielectric layer is applied between the electrolyte and the conducting substrate. Recent work, including contributions from our own laboratory, have shown that reversible electrowetting can be achieved directly on conductors. We have shown that graphite surfaces, in particular when combined with highly concentrated electrolyte solutions, show a strong wetting effect. The process is driven by the interactions between the electrolyte ions and the surface, hence models of double-layer capacitance are able to explain changes in the equilibrium contact angles. Herein, we extend the approach to the investigation of electrowetting on graphene samples of varying thickness, prepared by chemical vapor deposition. We show that the use of highly concentrated aqueous electrolytes induces a clear yet subtle electrowetting response due to the adsorption of ions and the suppression of the negative effect introduced by the surface impurities accumulating during the transfer process. The latter have been previously reported to fully hinder electrowetting at lower electrolyte concentrations. An amplified wetting response is recorded in the presence of strongly adsorbed/intercalated anions in both aqueous and non-aqueous electrolytes. The phenomenon is interpreted based on the anion–graphene interactions and their influence on the energetics of the interface. By monitoring the dynamics of wetting, an irreversible behaviour is identified in all cases as a consequence of the irreversibility of anion adsorption and/or intercalation. Finally, the effect of the underlying reactions on the timescales of wetting is also examined.
Introduction
The wettability of solids provides macroscopic insights into the molecular forces at the interface between adjoining bulk phases.1,2 These interfacial forces (van der Waals forces, electrostatic interactions and structural forces, e.g., hydrogen bonding) dictate the types of interactions between the solid|liquid phases, which strongly influence the overall wetting behaviour of the system.3 Consequently, the understanding of wetting phenomena on a molecular level can progressively tackle fundamental challenges in modern chemical physics and hence pave the way towards the elaborate control of various physicochemical phenomena underpinning the operation of electrofluidic systems with diverse functional surfaces and a broad range of applications.Two-dimensional (2D) materials, exhibiting unique physical properties have attracted interest in a vast range of research fields.4–6 In many of these areas, the surface interactions between the material of interest and a liquid, e.g. an electrolyte, is pivotal for the elucidation of fundamental phenomena underlying various physicochemical processes. The most basic interaction of a solid surface with a liquid is wetting. The extent of the latter is governed by the properties of the individual phases as well as the interfacial forces arising between them upon contact. In the 2D-materials family, graphene as the prototypical 2D-material and the building block of sp2-carbon allotropes, has been investigated more extensively than any other member of the group. Its unique surface and chemical properties arising from the high charge carrier mobility7 render it an ideal model for the mechanistic studies of its physicochemical interactions with liquids. The wetting properties of graphene are a matter of ongoing debate with significant advances being reported and continuously revisited throughout the last decade, from both theoretical and experimental perspectives.3,8–10 Despite a series of studies on the influence of various factors, such as substrate nature and doping,11,12 on the wetting behaviour of graphene, very few works on the effect of external potential bias are reported.13,14
In this work, we revisit electrowetting directly on graphene of varied thickness by investigating the effect of electrolyte concentration and identity on the overall process. Changes in contact angle with the applied potential bias are monitored in the presence and absence of specifically adsorbed ions using both aqueous and non-aqueous electrolytes. The results are interpreted in terms of the effect of the underlying potential-dependent physicochemical reactions on the surface energetics of the system. The dynamics of the wetting process are also probed and further insights into the influence of the underlying reactions on the mechanism of the phenomenon are given. We anticipate that the reported data will contribute towards better understanding of graphene–electrolyte interactions under potential bias and will complement the scarce literature in the area.
Experimental
Electrolytes
Anhydrous KF (BioUltra ≥99.5% (F)) from Sigma-Aldrich, lithium bis(trifluoromethanesulfonyl) imide (99%) from Fluorochem, lithium perchlorate (98%) from Alfa Aesar and propylene carbonate (anhydrous 99%) from Alfa Aesar were used for the preparation of the electrolytes. Ultra-pure water (18.2 MΩ cm resistivity at 25 °C, Milli-Q Direct 8) was used in all studies involving aqueous electrolytes.Synthesis of monolayer graphene
Monolayer graphene was synthesized on 25 μm-thick Cu foil (99.999% purity, Alfa Aesar) using a chemical vapor deposition (CVD) furnace system with a quartz tube of 1-inch diameter. Cu foils were pre-cleaned with nitric acid, acetone, and propan-2-ol before use, as described in a previous report.15 The Cu foils were heated to 1020 °C and annealed for 2 h under 40 sccm of hydrogen flow (99.999%). After the annealing step, 1.5 sccm of methane (99.999%) was introduced into the tube to grow graphene. The growth step lasted for 20 min, and then the furnace was allowed to cool to room temperature.Preparation of multilayer graphene
Synthesized monolayer graphene on Cu was transferred to a SiO2/Si wafer using the general wet transfer method, assisted by a poly(methyl methacrylate) (PMMA) layer. 2 wt% of PMMA (MW 350k, Sigma Aldrich) dissolved in chlorobenzene (anhydrous, 99.8%, Sigma Aldrich) was spin-coated on synthesized graphene on Cu foil. The Cu foil was removed using 0.5 M FeCl3 aqueous solution, and the PMMA/graphene layer was rinsed with deionized water 5 times before it was scooped up with another graphene/Cu layer. To stack graphene to the desired layer number, the same transfer process was repeated, but the PMMA/graphene layer was replaced as the substrate with a SiO2/Si wafer. After each “scoop” step, the samples were dried at 80 °C for 12 h and baked at 120 °C for 1 h to improve the contact between individual layers. The PMMA layer was removed using acetone at room temperature.Raman spectroscopy
Raman spectra were collected with 633 nm excitation using the 500× objective of a Renishaw 2000 spectrometer to confirm the layer number of the prepared graphene samples and their defect density.Electrowetting setup configuration and electrochemical procedures
The setup used for the electrowetting experiments has been previously described in detail.16,17 CVD graphene of varied layer thickness on Si/SiO2 substrates served as working electrodes (WE). Electrical connection was achieved by stripping an enamelled Cu wire (RS components, UK) for about 1 cm at each end and adhering one side to the edge of the graphene samples with silver conductive epoxy (RS components, UK). Following a 24 h curing step, the silver epoxy was covered by an insulating resin to establish a robust mechanical connection. Prior to the experiments the samples were thoroughly rinsed with water and propan-2-ol to remove any impurities adsorbed from the atmosphere. The electrolyte droplet (with a diameter in the range of ca. 100–300 μm) was deposited on the graphene by the controlled flow of the electrolyte through a micropipette using a microinjector (PV820 Pneumatic PicoPump, from World Precision Instruments, FL, US). A Sutter P-97 Flamming/Brown micropipette puller was employed to fabricate the micropipette from a borosilicate capillary (inner diameter 0.84 mm, outer diameter 1.5 mm, length 10.16 cm, from World Precision Instruments, UK). A platinum wire (99.99% purity, 0.05 mm diameter, from Advent, UK), placed on the upper, inner part of the micropipette was used as a counter and pseudo-reference electrode. The inner diameter of the tip in the resultant micropipettes was ca. 5–6 μm. The position of both the graphene samples and the micropipette was controlled using manual micro-positioners (Thor Labs).Contact angle variations during the electrowetting experiments were monitored using a Photron FASTCAM SA3 high speed camera controlled via Photron FASTCAM Viewer and a Storz Xenon Nova 300 light source. Image of the droplet were recorded under both static and dynamic conditions. In the former case, an equilibrium contact angle was considered following the attainment of steady-state conditions based on preliminary dynamic measurements (an equilibrium time of 3 s proved to be sufficient; the timescale of the advancing/receding motion was less than 500 ms in all systems under study). For the dynamic measurements, the frame rate was adjusted to 50 fps for the cycling experiments and 5000 fps for the high-resolution data used to investigate the timescale of the underlying phenomena during electrowetting. The characteristic times of the droplet’s motion used to determine the timescales were taken to be 90% of the corresponding equilibrium contact angles. Contact angle values were extracted from the recorded images using a custom-made edge detection algorithm in MATLAB® (MathWorks Inc., Natick, MA, USA) (for details see ref. 16).
All electrochemical experiments were performed on an Autolab PGSTAT302N potentiostat from Metrohm, operated using Nova 1.11.2 software. The experimental protocol followed for the equilibrium contact angle measurements (static conditions) was composed of consecutive potential pulses within the required potential region using a potential step of 100 mV. A similar approach was adopted for the dynamic measurements, in which the potential was stepped between the values of interest multiple times. Each repetition represents one cycle. Unless specified otherwise, the duration of the pulses for both static and dynamic conditions was adjusted to 3 s. Electrolyte concentrations throughout the manuscript are reported as molality, m, i.e., mol per kg of solvent.
Results and discussion
Physicochemical characterization of the prepared graphene layers
The quality and layer number of the prepared graphene samples were confirmed by Raman spectroscopy. Fig. 1 shows the recorded Raman spectra for the 1-, 3- and 5-layer samples. 2D bands near 2650 cm−1 were shifted to higher wavenumber with the increase in the number of graphene layers due to the reduced 2D1A component.18 The G/2D band intensity ratio was increased in 3- and 5-layers graphene samples compared to monolayer graphene, but the increment is inconsistent, which could be due to the random stacking order between graphene layers.19 The monolayer graphene was high quality with low defect density, having a low D/G ratio of 0.05. A higher D band intensity was observed in thicker graphene samples because of accumulated defects per unit area.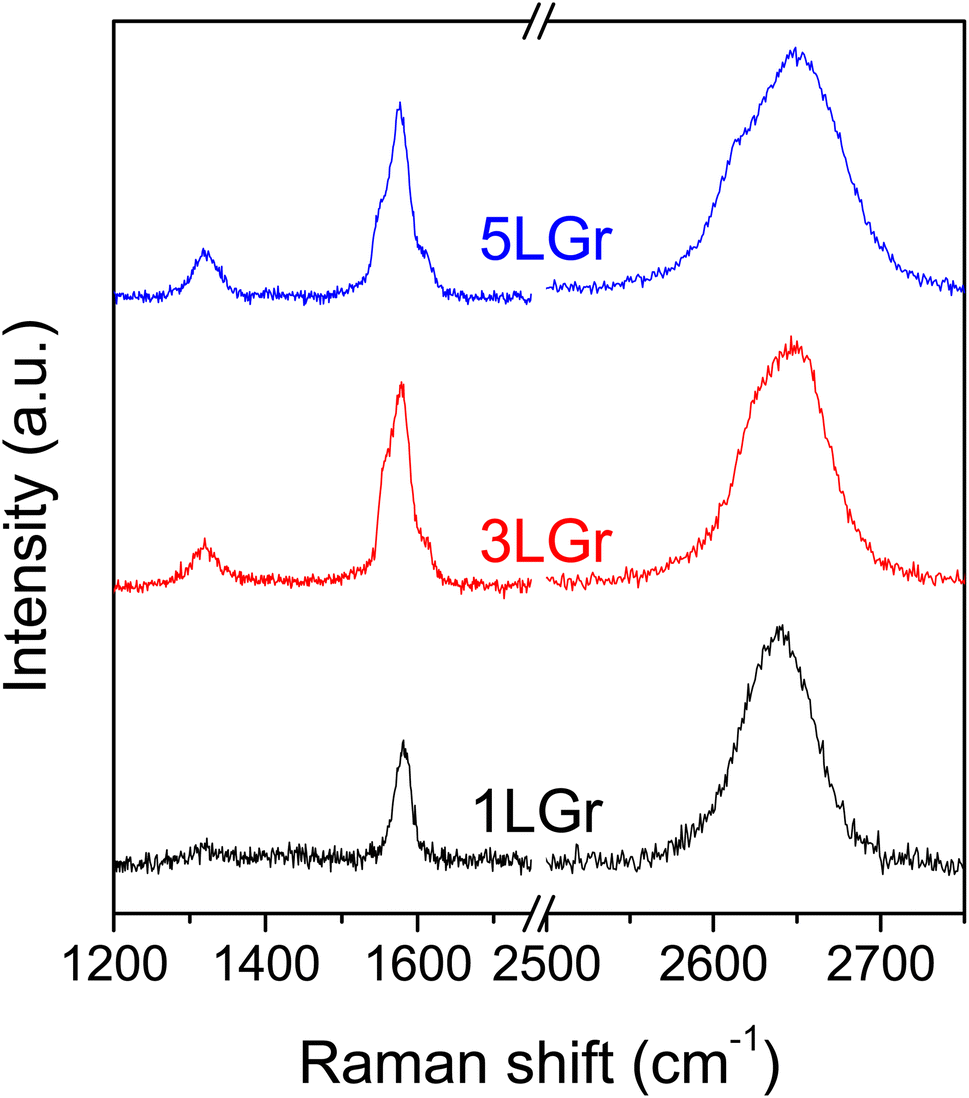 | ||
| Fig. 1 Raman spectra of the 1-, 3- and 5-layers graphene samples on Si/SiO2 wafer, denoted 1LGr, 3LGr and 5LGr, respectively. | ||
Electrowetting using aqueous electrolytes
The contact angle of aqueous KF electrolyte (denoted hereafter as KF(aq)) droplets deposited on CVD graphene samples of varied thickness was monitored as a function of the potential, E, applied vs. the Pt wire pseudo-reference using the pipette contact shown in Fig. 2 and following the experimental protocol described in the Experimental section. Fig. 2a shows snapshots of 1 m KF(aq) droplets on 1- and 5-layers graphene samples at selected applied biases. It is evident that in both cases no significant electrowetting response is recorded. In a first approach and in line with what has been previously reported for aqueous halide electrolytes on CVD graphene, the apparent complete suppression of electrowetting might be related to the presence of intrinsic defects and/or the adsorption of surface impurities, e.g., polymer particles, both introduced during the transfer process.14 In the first case, the decreased substrate coverage of the graphene layer(s) promotes wetting transparency, while on the other hand the adsorption of polymer residues is expected to reduce the effect of the substrate and hence result in a higher degree of wetting opacity.3,20,21 Furthermore, perturbation of the graphene crystal structure induced by the inherent polycrystallinity of the CVD graphene samples and the increase in surface roughness due to adsorbed impurities can lead to droplet pinning and surface charge heterogeneity that may impede the motion of the droplet during the electrowetting experiments.17,22,23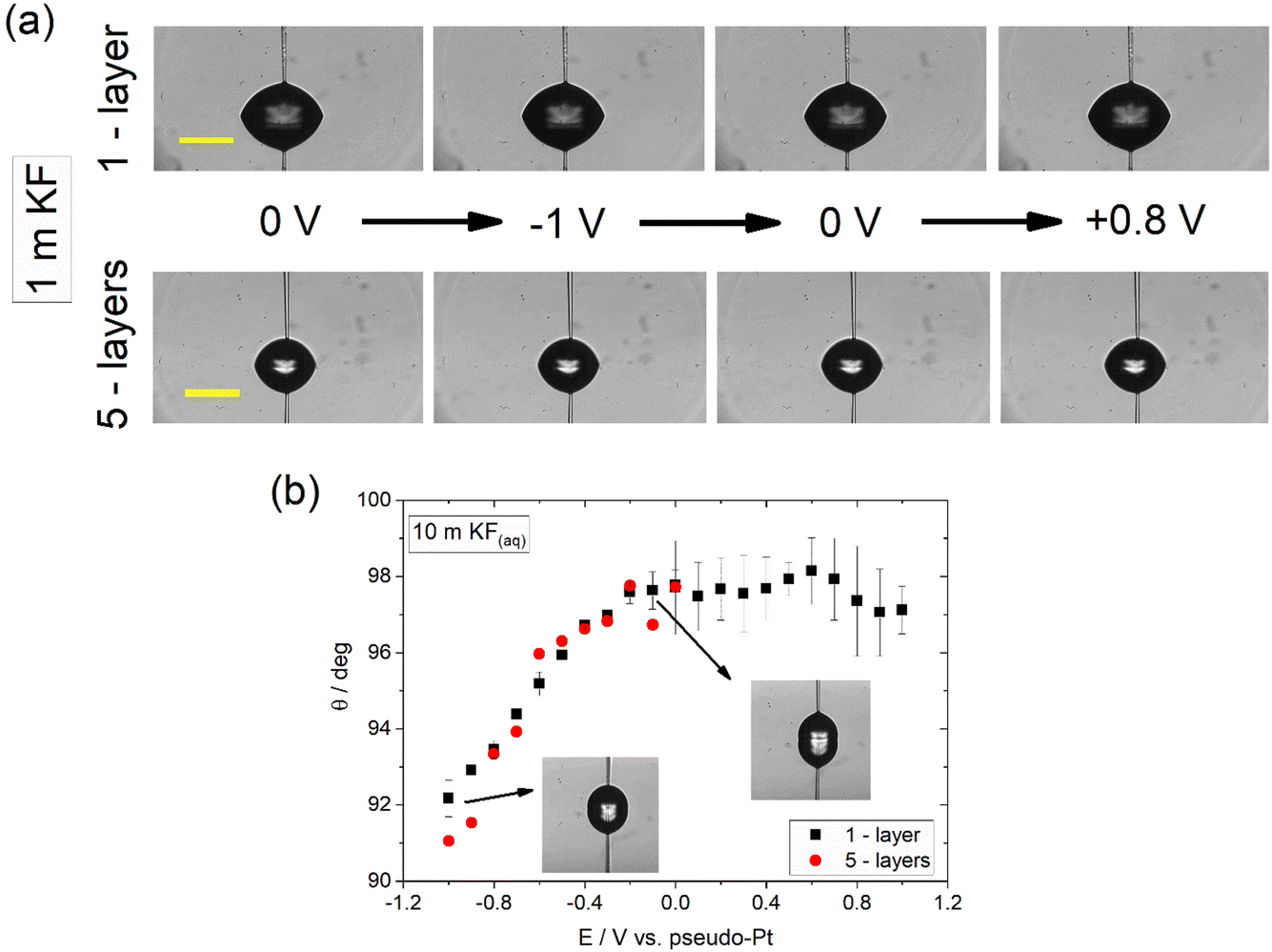 | ||
| Fig. 2 (a) Images of 1 m KF(aq) droplets on CVD graphene of 1- (top row) and 5- (bottom row) layers at varied applied potentials. The scale bars correspond to 200 μm. (b) Change in apparent equilibrium electrowetting contact angle, θ, with the applied bias (values are reported vs. Pt wire pseudo-reference electrode) at the CVD graphene|10 m KF(aq) interface for 1- and 5-layers thickness. Measurements were conducted under static conditions based on the protocol described in the Experimental section. | ||
Recently, we have shown that electrowetting on graphite using alkali metal halides exhibits a strong dependence on electrolyte concentration due to the occurrence of underlying potential induced electrochemical processes.16 By increasing the electrolyte concentration towards the water-in-salt regime, parasitic processes occurring in series with electrowetting are suppressed and the interface approaches ideally polarizable behaviour. Furthermore, the effect of impurities, such as neutral organic molecules, which often persist in solution, is significantly diminished due to the high concentration of the electrolyte which lowers their solubility compared to lower electrolyte concentrations. In this respect, we explore the possibility of similar phenomena on graphene by monitoring the contact angle variations using 10 m KF(aq). Fig. 2b shows the dependence of the contact angle on E for 1- and 5-layers CVD graphene. Interestingly, it can be seen that a subtle (ca. 6–7° change in contact angle) yet clear electrowetting effect is recorded in both cases within the negative potential region. Note that, in line with what is reported in our previous work with graphite, the data was recorded within the capacitive window of the electrolyte. Based on the derived data, it appears that the use of highly concentrated electrolytes suppresses the effect of impurities as well as that of the underlying electrochemical reactions on electrowetting using graphene, in a similar way to what is reported for graphite.16 Additionally, the high electrolyte concentration leads to increased electrostatic interactions between the electrolyte ions and graphene that influences the potential-dependent solid|liquid interfacial surface tension and therefore affects electrowetting.
In more detail, it is well established in the literature that graphene exhibits specific cation effects in contact with alkali metal electrolytes.24–26 The effect is more pronounced as the size of the cation increases, i.e., going from Li+ to Cs+. On this basis, we attribute the observed electrowetting effect at E < 0 to the adsorption of K+ ions on graphene that decreases the solid|liquid interfacial surface tension and consequently induces droplet spreading.27 The dependence of the solid|liquid interfacial tension on the applied potential bias results from the Gibbs adsorption isotherm.28,29 The latter is used to derive the electrocapillary equation of a system (eqn (1)):
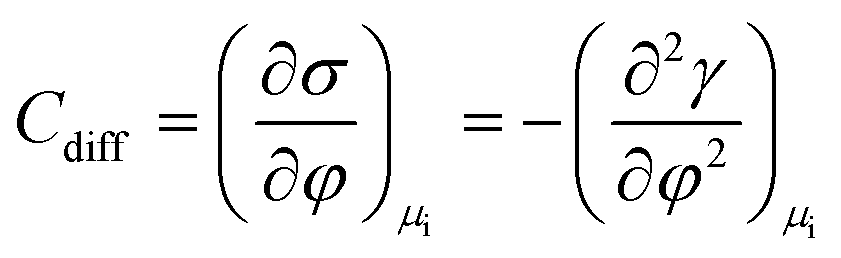 | (1) |
To provide insights into the effect of interfacial processes, such as ion adsorption, we proceed with investigating the electrowetting response of graphene in an electrolyte containing strongly adsorbed ions. Towards that direction we use the concentrated aqueous electrolyte 5 m lithium bis(trifluoromethanesulfonyl) imide (LiTFSI). The recorded electrowetting response is presented in Fig. 3a for 1-, 3- and 5-layers CVD graphene. The main feature of merit is that electrowetting appears to occur within the whole applied potential window for the 1- and 3-layers samples and for E > 0 for the 5-layers sample. The maximum changes in contact angle are observed for all samples at the most positive applied bias region and are of ca. 10° compared to the maximum value in the electrowetting plots. In order to reveal the underlying reasons for the observable potential-driven wetting, we investigate the electrochemical processes occurring on the surface of graphene using cyclic voltammetry (CV). From the derived data presented in Fig. 3a and b, a capacitive window in the ca. −0.6 V to +0.7 V range is identified, with a reduction and an oxidation process at more negative and positive potentials, respectively. We attribute the anodic process to the adsorption and/or intercalation of TFSI− on graphene in line with what is reported in the literature for highly concentrated aqueous electrolytes.36 The process is expected to be initiated through small vacancy defects on the surface of graphene.37 The cathodic reaction observed for E < −0.6 V can be assigned to hydrogen evolution reaction (HER) with a minor (if any) contribution from the cathodic decomposition of TFSI−.
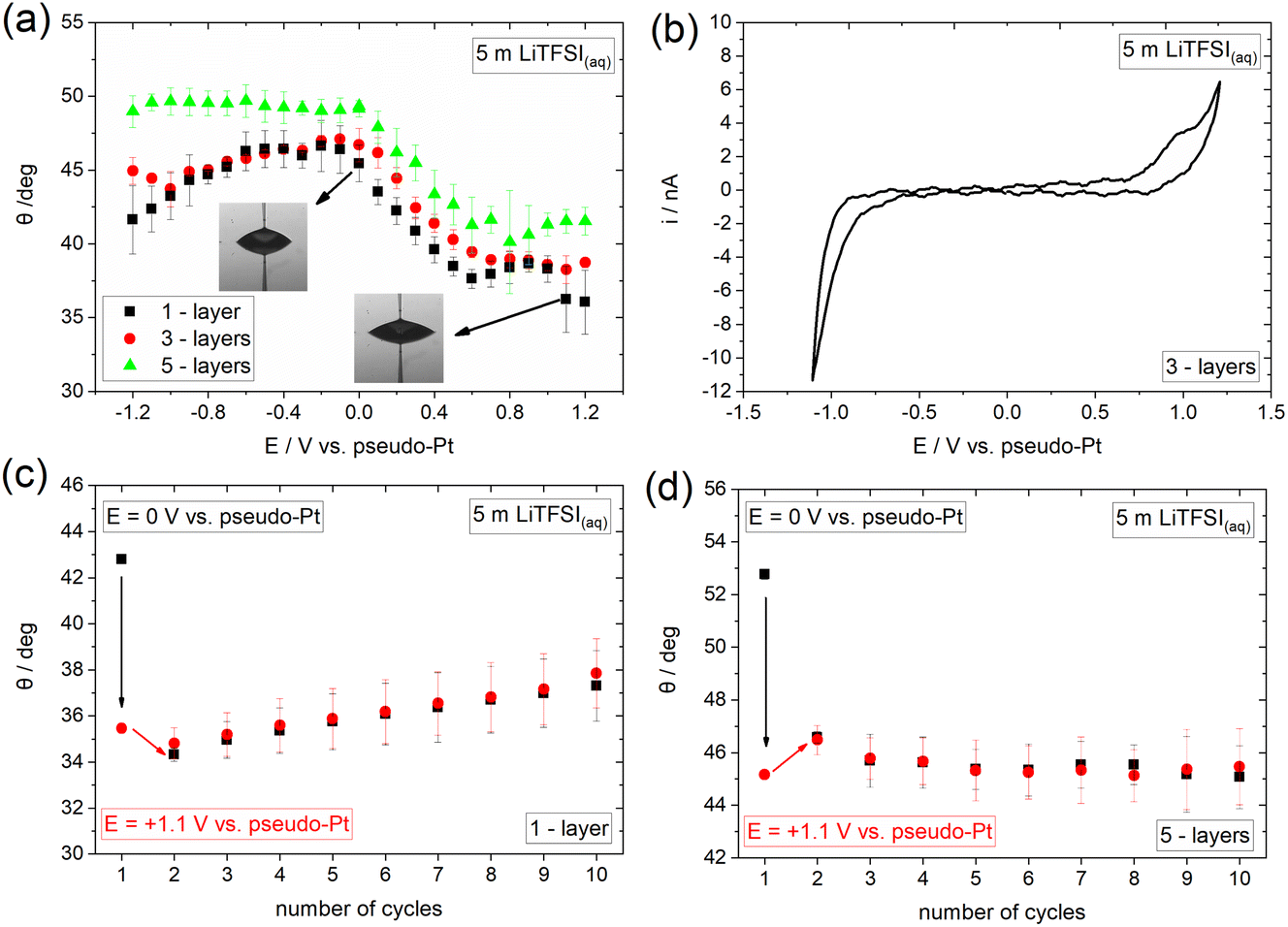 | ||
| Fig. 3 (a) Change in apparent equilibrium electrowetting contact angle, θ, with the applied bias (values are reported vs. Pt wire pseudo-reference electrode) at the CVD graphene|5 m LiTFSI(aq) interface for 1-, 3- and 5-layers thickness. Measurements were conducted under static conditions based on the protocol described in the Experimental section. (b) Cyclic voltammogram recorded at a 5 m LiTFSI(aq) droplet on 3-layers graphene using a scan rate of 100 mV s−1. Contact angle changes over ten consecutive cycles (see Experimental section) in 5 m LiTFSI(aq) droplets between 0 V and +1.1 V vs. pseudo-Pt on (c) 1- and (d) 5-layers CVD graphene. | ||
Having characterized the underlying electrochemical processes occurring at the graphene|5 m LiTFSI(aq) interface, we attempt to provide a possible mechanism for the overall electrowetting process. We propose that within the purely capacitive potential window, the strong interaction (i.e., adsorption) of TFSI− with graphene results in the decrease of the solid|liquid interfacial surface tension and hence induces the electrowetting response similar to what is observed for the concentrated KF electrolyte at E < 0 V (see Fig. 2). A similar effect has been also reported for the adsorption of Br− on Au.38,39 A notable observation when comparing the response of the 10 m KF(aq) (Fig. 2b) and 5 m LiTFSI(aq) (Fig. 3a) electrolytes in the potential range where ion adsorption occurs is that the changes in contact angle (relative to the maximum contact angle of each system) are larger in the LiTFSI solution. This finding might indicate that TFSI− anions are more strongly adsorbed on graphene compared to K+ cations and hence more markedly decrease the interfacial graphite|electrolyte tension. This phenomenon is depicted in the amplified electrowetting response for the LiTFSI(aq) electrolyte compared to KF(aq). On this basis, and analogously to the electrocapillary studies, the electrowetting curves could be used to provide quantitative insights into the interaction of various electrolytes with several solid substrates.27 For E > +0.7 V, TFSI− may start to intercalate between the graphene layers disrupting their crystal structure,37 a phenomenon that is depicted in the “irregular” variations of the contact angle with subsequent contact angle saturation within this potential region for all samples. Recently, we have shown that anion (i.e., TFSI− and ClO4−) intercalation from aqueous, non-aqueous and ionic liquid electrolytes into graphite drastically alters its wetting properties.40 Interestingly, from the data presented in Fig. 3a (see also Fig. 5a right below and the discussion therein), such an effect appears to also occur on graphene, however the electrowetting response is significantly weaker compared to graphite. At the negative extreme, i.e., E < −0.6 V the weaker electrowetting response compared to that for E > 0 V may be attributed to the more strongly bound aqueous network in excess of Li+ in the double layer with significant contributions from HER. The latter is expected to increase the ohmic drop at the interface, while simultaneously inducing surface defects that may lead to a transition to the Wenzel wetting state17,22,41 and impede droplet spreading.16 The reversibility of the phenomenon was also examined by monitoring the changes in contact angle over ten consecutive cycles from 0 V to +1.1 V (see Experimental section). From the data displayed in Fig. 3c and d it can be seen that, in line with KF(aq), the process seems to be fully irreversible. A possible explanation for this particularly interesting finding is that the adsorption and/or intercalation of TFSI− on/into graphene occurs in an irreversible way (i.e., TFSI− anions are not fully desorbed or deintercalated upon reversing the potential bias). Irreversible intercalation of other anions, such as PF6−, into defective graphene flakes prepared by electrochemical exfoliation has been previously identified via in situ Raman spectroscopy by our group.42 Considering the intrinsically defective nature of CVD graphene and the increased G/2D band intensity ratio on 3- and 5-layers graphene samples (see Fig. 1), such an assumption is plausible. In fact, as can be seen from the voltammogram presented in Fig. 3b, no cathodic counterpart is recorded for the anodic process identified at E > +0.7 V (assigned to TFSI− adsorption/intercalation), indicating the irreversibility of the interaction. In contrast, we have shown that intercalation of TFSI− into graphite using a highly concentrated aqueous electrolyte, i.e., 20 m LiTFSI, exhibits reversible behaviour, which results in a fully reversible and reproducible electrowetting response over consecutive cycles.40 The direct connection between the reversibility of the intercalation and electrowetting processes on graphite further supports the interpretation above regarding the electrowetting response of graphene within the potential region where TFSI− intercalation occurs. Furthermore, the disruption of graphene structure due to the adsorption and/or intercalation process of the large TFSI− anion can also inhibit the reversibility of electrowetting.
As previously mentioned, electrowetting can serve as a powerful tool to provide macroscopic insights into the electrochemically-induced surface processes occurring on various conducting substrates. Towards that direction, we monitor the dynamics of wetting by recording high resolution optical measurements (i.e., using 5000 fps; see Experimental section) and investigate the effect of the underlying potential-dependent reactions on the timescales of the overall process.16Fig. 4 shows the indicative timescales (highlighted regions) of the electrowetting process in the presence of TFSI− adsorption/intercalation (see Fig. 4b) on graphene samples of 1-, 3- and 5-layers thickness. The derived timescales are found to be 211 (±27.6) ms, 284 (±31.2) ms and 215 (±22.63) ms for the 1-, 3- and 5-layers samples, respectively. This finding suggests a very similar effect of the adsorption/intercalation process on the electrowetting response, irrespective of the graphene layer thickness. The determined timescales are slower by a factor of ca. 3.5 (as an average) compared to those recorded on graphite.40 This can be potentially attributed to the more defective nature of CVD graphene compared to the basal plane of highly oriented pyrolytic graphite (HOPG) used in our prior work,40 since the presence of surface defects has been proven to significantly increase the timescale of electrowetting during anion intercalation (see supporting information in ref. 40). At this point, we note that the timescales reported recently for graphite have been determined in a biphasic electrolyte|oil system,40 and therefore the comparison with the electrolyte|air configuration used in the present work is approximate (since any effect of the oil on the timescales of electrowetting needs to be considered; the latter could slow down the process due to the viscosity effects on hydrodynamics or accelerate the advancing motion of the droplet due to micro-, nano-lubrication). Furthermore, it is worth mentioning that the obtained timescales are significantly larger (by a factor of approximately 30) than those recorded on graphite during the formation of F− graphite intercalation compounds.16 This might be related to the stronger effect of the surface reconstruction occurring during the adsorption/intercalation process on the spreading of the electrolyte droplet at the much thinner graphene layers compared to bulk graphite. Another possible factor could be the larger size of the TFSI− anions compared to F−, which is expected to induce irreversible defects on the structure of graphene upon adsorption and/or intercalation in accordance with what is reported in the literature for graphite.36,43
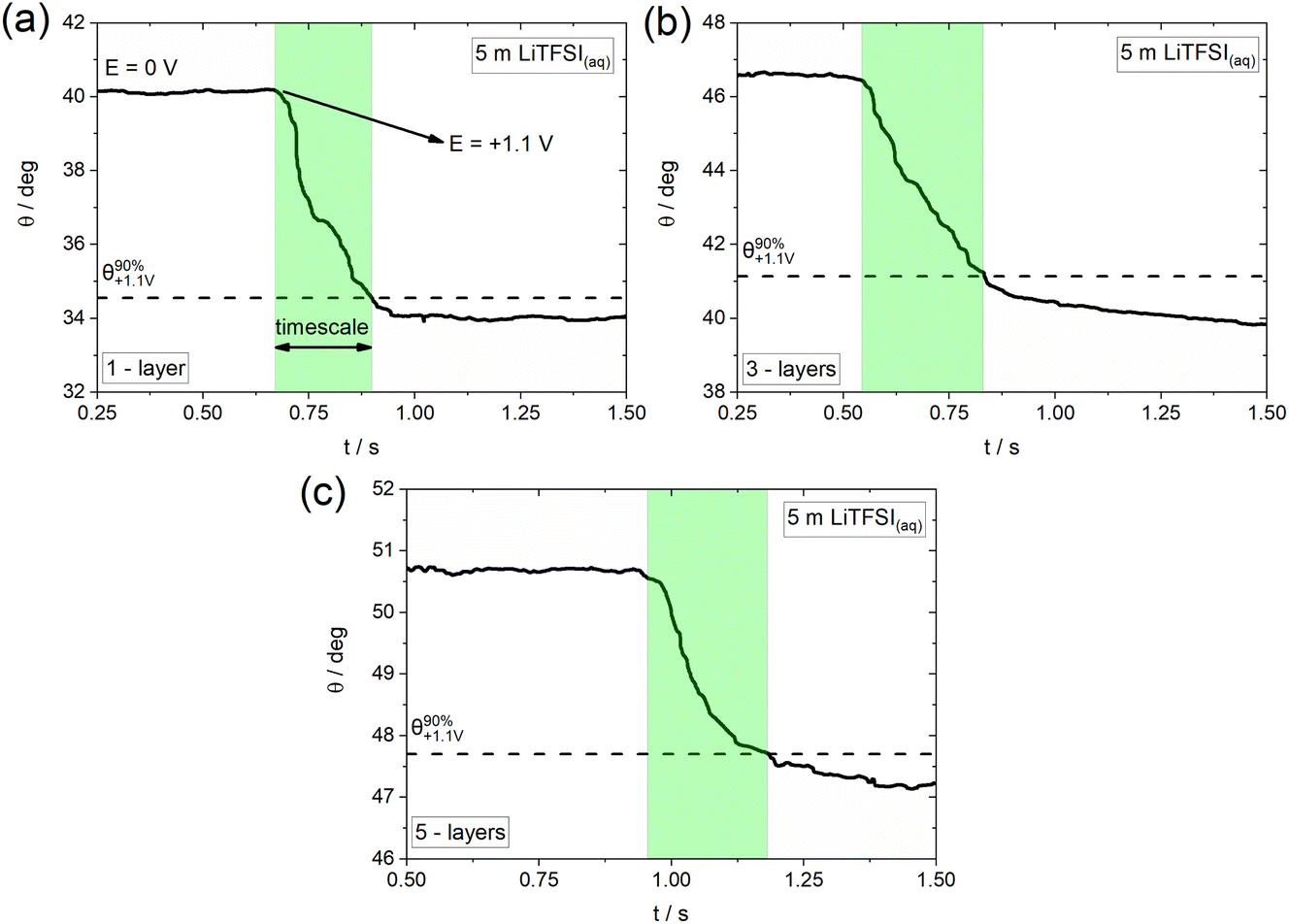 | ||
| Fig. 4 Change in apparent contact angle, θ, and timescales (highlighted regions) during a wetting cycle in 5 m LiTFSI(aq) following the protocol described in the Experimental section for (a) 1-, (b) 3- and (c) 5-layers CVD graphene. The depicted cycle corresponds to a potential pulse from 0 V to +1.1 V vs. pseudo-Pt, i.e., in the potential region where TFSI− anion adsorption/intercalation occurs. | ||
Electrowetting using non-aqueous electrolytes
Following the investigation of electrowetting on graphene of varied thickness in the presence and absence of specifically adsorbed ions using aqueous electrolytes, we aim to investigate the effect of the solvent on the overall process. Fig. 5a shows the dependence of the apparent equilibrium contact angle on E (recorded followed the same approach as that in Fig. 3a) for 1 m LiClO4 in propylene carbonate (denoted hereafter as LiClO4(PC)) using 1- and 5-layers CVD graphene. Similar to the observations reported in the previous section with concentrated aqueous solutions of LiTFSI, a clear electrowetting response is recorded in the whole applied potential range. In more detail, for E < 0, the changes in contact angle with applied bias are relatively low (less than 4°), while larger contact angle changes (up to 10° for the graphene monolayer) are seen for E > 0.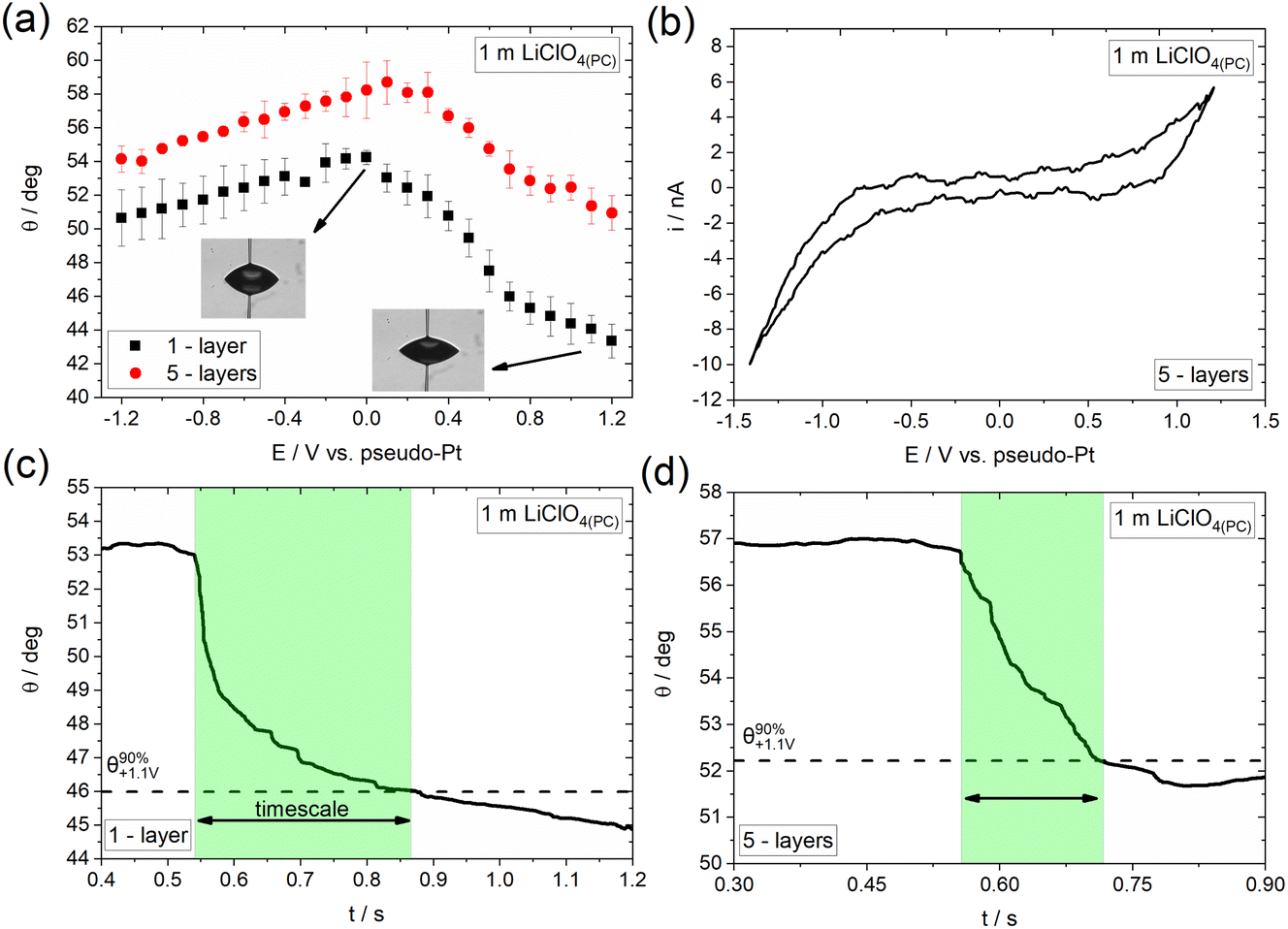 | ||
| Fig. 5 (a) Change in apparent equilibrium electrowetting contact angle, θ, with the applied bias (values are reported vs. Pt wire pseudo-reference electrode) at the CVD graphene|1 m LiClO4(PC) interface for 1- and 5-layers thickness. Measurements were conducted under static conditions based on the protocol described in the Experimental section. (b) Cyclic voltammogram recorded at a 1 m LiClO4(PC) droplet on 5-layers graphene using a scan rate of 100 mV s−1. Timescales (highlighted regions) during a wetting cycle in 1 m LiClO4(PC) following the protocol described in the Experimental section for (c) 1- and (d) 5-layers CVD graphene. The depicted cycle corresponds to a potential pulse from 0 V to +1.1 V vs. pseudo-Pt, i.e., in the potential region where ClO4− anion adsorption/intercalation occurs. | ||
Adopting the strategy described above, we proceed by studying the underlying processes occurring on the surface of graphene and their influence on the overall electrowetting response. Fig. 5b shows the CV recorded at a 1 m LiClO4(PC) droplet on 5-layers graphene. For E > +0.4 V the qualitative features of the voltammogram are reminiscent of the ClO4− adsorption/desorption process reported from both aqueous44–47 and non-aqueous electrolytes.48–50 The process is again expected to be driven through the small vacancies and defects on the surface of graphene layers37 and is considered to be the main factor for the relatively enhanced electrowetting response seen in this potential range. Zhang et al.51 have also reported a promoting effect of ClO4− anion adsorption/intercalation in aqueous electrolytes on the electrowetting response of graphite. The strong similarities between the electrowetting responses in the presence of anion adsorption/intercalation in aqueous (Fig. 3a) and non-aqueous (Fig. 5a) electrolytes suggests that the effect of solvent is less significant compared to the adsorption/intercalation reaction itself on the mechanism of the overall electrowetting process. For E < 0, electrowetting is less pronounced, again this may be due to the negative effect of electrolyte decomposition52,53 on the potential drop at the interface and the increase in solid|liquid surface tension resulting from the formation of PC decomposition by-products (often insoluble, e.g., carbonates).54 In line with the LiTFSI(aq) electrolyte, the electrowetting response appears to be irreversible (data not shown), most probably due to the irreversibility of the underlying processes, i.e., anion adsorption and intercalation, the disruption of graphene structure as well as the introduction of surface defects (see section above).
Fig. 5c and d present the dynamics of the electrowetting response on 1- and 5-layers graphene in the presence of ClO4− anion adsorption/intercalation. The determined timescales are 369 (±56.57) ms and 173 (±14.14) ms for the monolayer and 5-layers graphene, respectively, which shows that the advancing motion of the droplet is slower in the case of the monolayer compared to the thicker sample. We ascribe this finding to the weaker effect of the structural changes induced by the ClO4− anion adsorption/intercalation process on the electrowetting response of the thicker sample. Finally, as in the case of LiTFSI(aq) the derived timescales are slower compared to graphite by a factor of ca. 8.5 and 4 for the monolayer and 5-layers graphene, respectively (see supporting information in ref. 40).
Conclusions
The electrowetting behaviour of CVD graphene samples of varying thickness on Si/SiO2 was investigated using aqueous and non-aqueous electrolytes. No response was recorded at relatively low aqueous electrolyte concentrations for 1- to 5-layers graphene samples, which was attributed to the intrinsic defects and the impurities introduced during the transfer process. Increasing the electrolyte concentration to the water-in-salt regime induces a subtle electrowetting response restricted to the negative potential range, due to the adsorption of cations on graphene and the suppression of the impurity effect. In the presence of adsorbed/intercalated anions, electrowetting occurs even at lower concentrations in both aqueous and non-aqueous electrolytes within the whole applied potential region, with an amplified response observed for positive applied biases. This can be explained by the decrease in the solid|liquid interfacial surface tension as a consequence of the adsorption/intercalation processes. Under these conditions electrowetting is shown to be irreversible, most probably due to the irreversibility of the adsorption/intercalation reactions and the structural changes introduced during the overall process. By monitoring wetting dynamics, we demonstrated that the timescales of the adsorption/intercalation processes are independent of the graphene layers thickness when using aqueous electrolytes. In the case of the organic electrolyte, a decrease in the timescale is seen with thicker samples, most probably due to the weaker effect of the smaller anion used in these systems on the disruption of graphene’s structure compared to the aqueous solutions. Further studies aiming to decipher the mechanisms of these phenomena are currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. A. W. D. and A. A. P. would like to thank the Leverhulme Trust (RPG-2019-250) for financial support. Further support from the EPSRC (EP/T01816X/1) is also gratefully acknowledged.References
- P.-G. de Gennes, F. Brochard-Wyart and D. Quéré, in Capillarity and Wetting Phenomena: Drops, Bubbles, Pearls, Waves, ed. P.-G. de Gennes, F. Brochard-Wyart and D. Quéré, Springer, New York, NY, 2004, pp. 87–105 Search PubMed.
- D. Bonn, J. Eggers, J. Indekeu, J. Meunier and E. Rolley, Rev. Mod. Phys., 2009, 81, 739–805 CrossRef CAS.
- P. Snapp, J. M. Kim, C. Cho, J. Leem, M. F. Haque and S. Nam, NPG Asia Mater., 2020, 12, 1–16 CrossRef.
- S. Z. Butler, S. M. Hollen, L. Cao, Y. Cui, J. A. Gupta, H. R. Gutiérrez, T. F. Heinz, S. S. Hong, J. Huang, A. F. Ismach, E. Johnston-Halperin, M. Kuno, V. V. Plashnitsa, R. D. Robinson, R. S. Ruoff, S. Salahuddin, J. Shan, L. Shi, M. G. Spencer, M. Terrones, W. Windl and J. E. Goldberger, ACS Nano, 2013, 7, 2898–2926 CrossRef CAS PubMed.
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G.-H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed.
- S. Manzeli, D. Ovchinnikov, D. Pasquier, O. V. Yazyev and A. Kis, Nat. Rev. Mater., 2017, 2, 1–15 Search PubMed.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- J. Feng and Z. Guo, Nanoscale Horiz., 2019, 4, 339–364 RSC.
- L. A. Belyaeva and G. F. Schneider, Surf. Sci. Rep., 2020, 75, 100482 CrossRef CAS.
- E. Kim, D. Kim, K. Kwak, Y. Nagata, M. Bonn and M. Cho, Chem, 2022, 8, 1187–1200 CAS.
- R. Raj, S. C. Maroo and E. N. Wang, Nano Lett., 2013, 13, 1509–1515 CrossRef CAS PubMed.
- G. Hong, Y. Han, T. M. Schutzius, Y. Wang, Y. Pan, M. Hu, J. Jie, C. S. Sharma, U. Müller and D. Poulikakos, Nano Lett., 2016, 16, 4447–4453 CrossRef CAS PubMed.
- J. H. J. Ostrowski and J. D. Eaves, J. Phys. Chem. B, 2014, 118, 530–536 CrossRef CAS PubMed.
- K. Ounnunkad, H. V. Patten, M. Velický, A. K. Farquhar, P. A. Brooksby, A. J. Downard and R. A. W. Dryfe, Faraday Discuss., 2017, 199, 49–61 RSC.
- H. Park, C. Lim, C.-J. Lee, J. Kang, J. Kim, M. Choi and H. Park, Nanotechnology, 2018, 29, 415303 CrossRef PubMed.
- A. A. Papaderakis, K. Polus, P. Kant, F. Box, B. Etcheverry, C. Byrne, M. Quinn, A. Walton, A. Juel and R. A. W. Dryfe, J. Phys. Chem. C, 2022, 126, 21071–21083 CrossRef CAS PubMed.
- D. J. Lomax, P. Kant, A. T. Williams, H. V. Patten, Y. Zou, A. Juel and R. A. W. Dryfe, Soft Matter, 2016, 12, 8798–8804 RSC.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- Y.-S. No, H. K. Choi, J.-S. Kim, H. Kim, Y.-J. Yu, C.-G. Choi and J. S. Choi, Sci. Rep., 2018, 8, 571 CrossRef PubMed.
- C.-J. Shih, M. S. Strano and D. Blankschtein, Nat. Mater., 2013, 12, 866–869 CrossRef CAS PubMed.
- J. Rafiee, X. Mi, H. Gullapalli, A. V. Thomas, F. Yavari, Y. Shi, P. M. Ajayan and N. A. Koratkar, Nat. Mater., 2012, 11, 217–222 CrossRef CAS PubMed.
- A. A. Kornyshev, A. R. Kucernak, M. Marinescu, C. W. Monroe, A. E. S. Sleightholme and M. Urbakh, J. Phys. Chem. C, 2010, 114, 14885–14890 CrossRef CAS.
- N. E. A. Cousens and A. R. J. Kucernak, Faraday Discuss., 2017, 199, 63–73 RSC.
- C. Zhan, M. R. Cerón, S. A. Hawks, M. Otani, B. C. Wood, T. A. Pham, M. Stadermann and P. G. Campbell, Nat. Commun., 2019, 10, 4858 CrossRef PubMed.
- S. Yasuda, K. Tamura, M. Kato, H. Asaoka and I. Yagi, J. Phys. Chem. C, 2021, 125, 22154–22162 CrossRef CAS.
- C. D. Williams, J. Dix, A. Troisi and P. Carbone, J. Phys. Chem. Lett., 2017, 8, 703–708 CrossRef CAS PubMed.
- A. A. Papaderakis and R. A. W. Dryfe, Curr. Opin. Electrochem., 2023, 38, 101245 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, in Electrochemical Methods - Fundamentals and Applications, John Wiley & Sons, Inc., United States of America, 2nd edn, 2001, pp. 534–557 Search PubMed.
- D. C. Grahame, Chem. Rev., 1947, 41, 441–501 CrossRef CAS PubMed.
- A. Serva, N. Dubouis, A. Grimaud and M. Salanne, Acc. Chem. Res., 2021, 54, 1034–1042 CrossRef CAS PubMed.
- M. Turgeman, V. Wineman-Fisher, F. Malchik, A. Saha, G. Bergman, B. Gavriel, T. R. Penki, A. Nimkar, V. Baranauskaite, H. Aviv, M. D. Levi, M. Noked, D. T. Major, N. Shpigel and D. Aurbach, Cell Rep. Phys. Sci., 2022, 3, 100688 CrossRef CAS.
- R. Zhang, M. Han, K. Ta, K. E. Madsen, X. Chen, X. Zhang, R. M. Espinosa-Marzal and A. A. Gewirth, ACS Appl. Energy Mater., 2020, 3, 8086–8094 CrossRef CAS.
- C. H. Lui, L. Liu, K. F. Mak, G. W. Flynn and T. F. Heinz, Nature, 2009, 462, 339–341 CrossRef CAS PubMed.
- P. Iamprasertkun, A. Ejigu and R. A. W. Dryfe, Chem. Sci., 2020, 11, 6978–6989 RSC.
- E. Singh, A. V. Thomas, R. Mukherjee, X. Mi, F. Houshmand, Y. Peles, Y. Shi and N. Koratkar, ACS Nano, 2013, 7, 3512–3521 CrossRef CAS PubMed.
- Z. Huang, Y. Hou, T. Wang, Y. Zhao, G. Liang, X. Li, Y. Guo, Q. Yang, Z. Chen, Q. Li, L. Ma, J. Fan and C. Zhi, Nat. Commun., 2021, 12, 3106 CrossRef CAS PubMed.
- Y. Liu, X. Liu, C.-Z. Wang, Y. Han, J. W. Evans, A. Lii-Rosales, M. C. Tringides and P. A. Thiel, J. Phys. Chem. C, 2021, 125, 6954–6962 CrossRef CAS.
- T. Morooka, H. Tahara and T. Sagara, Electrochim. Acta, 2017, 251, 355–362 CrossRef CAS.
- T. Morooka and T. Sagara, Langmuir, 2020, 36, 9685–9692 CrossRef CAS PubMed.
- A. A. Papaderakis, A. Ejigu, J. Yang, A. Elgendy, B. Radha, A. Keerthi, A. Juel and R. A. W. Dryfe, J. Am. Chem. Soc., 2023, 145, 8007–8020 CrossRef CAS PubMed.
- C. Ishino and K. Okumura, Eur. Phys. J. E, 2008, 25, 415–424 CrossRef CAS PubMed.
- A. Ejigu, L. W. Le Fevre, K. Fujisawa, M. Terrones, A. J. Forsyth and R. A. W. Dryfe, ACS Appl. Mater. Interfaces, 2019, 11, 23261–23270 CrossRef CAS PubMed.
- G. Wang, M. Yu and X. Feng, Chem. Soc. Rev., 2021, 50, 2388–2443 RSC.
- B. Schnyder, D. Alliata, R. Kötz and H. Siegenthaler, Appl. Surf. Sci., 2001, 173, 221–232 CrossRef CAS.
- M. S. Jagadeesh, G. Bussetti, A. Calloni, R. Yivlialin, L. Brambilla, A. Accogli, E. Gibertini, D. Alliata, C. Goletti, F. Ciccacci, L. Magagnin, C. Castiglioni and L. Duò, J. Phys. Chem. C, 2019, 123, 1790–1797 CrossRef CAS.
- D. Alliata, R. Kötz, O. Haas and H. Siegenthaler, Langmuir, 1999, 15, 8483–8489 CrossRef CAS.
- R. Yivlialin, G. Bussetti, L. Brambilla, C. Castiglioni, M. Tommasini, L. Duò, M. Passoni, M. Ghidelli, C. S. Casari and A. Li Bassi, J. Phys. Chem. C, 2017, 121, 14246–14253 CrossRef CAS.
- R. Santhanam and M. Noel, J. Power Sources, 1997, 66, 47–54 CrossRef CAS.
- R. Santhanam and M. Noel, J. Power Sources, 1998, 76, 147–152 CrossRef CAS.
- J. Gao, S. Tian, L. Qi and H. Wang, Electrochim. Acta, 2015, 176, 22–27 CrossRef CAS.
- G. Zhang, M. Walker and P. R. Unwin, Langmuir, 2016, 32, 7476–7484 CrossRef CAS PubMed.
- M. Hahn, A. Würsig, R. Gallay, P. Novák and R. Kötz, Electrochem. Commun., 2005, 7, 925–930 CrossRef CAS.
- T. Melin, R. Lundström and E. J. Berg, Adv. Mater. Interfaces, 2022, 9, 2101258 CrossRef CAS.
- Y. Mukouyama and T. Shiono, J. Electrochem. Soc., 2016, 163, H36 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |