 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
H-Bonding leading to latent initiators for olefin metathesis polymerization†‡
Artur
Brotons-Rufes
a,
Naeimeh
Bahri-Laleh
 *b and
Albert
Poater
*a
*b and
Albert
Poater
*a
aInstitut de Química Computacional i Catàlisi, Departament de Química, Universitat de Girona, c/Maria Aurèlia Capmany 69, 17003 Girona, Catalonia, Spain. E-mail: albert.poater@udg.edu
bPolymerization Engineering Department, Iran Polymer and Petrochemical Institute (IPPI), P.O. Box 14965/115, Tehran, Iran. E-mail: n.bahri@ippi.ac.ir
First published on 16th January 2023
Abstract
Ruthenium–NHC based catalysts, with a chelated iminium ligand trans to the N-heterocyclic carbene (NHC) ligand, that polymerize dicyclopentadiene (DCPD) at different temperatures are monitored using Density Functional Theory calculations to unveil the reaction mechanism, and subsequently how important are the geometrical and electronic features vs. the non-covalent interactions in between. The balance is very fragile and H-bonds are fundamental to explain the different behaviour of latent catalysts. This computational study aims to facilitate future studies of new generations of latent initiators for olefin metathesis polymerization, with the 3D and mainly the 2D Non-Covalent Interaction plots the characterization tool for H-bonds.
Introduction
Among the wide variety of polymers, one property that can make them more special is that they have the ability to withstand aggressive temperatures.1 Thermoset polymers are a group of highly versatile materials due to their capability to harden in an irreversible curing process,2 which changes working materials from soft solids or resins to rigid networks.3 Phase transformation can be triggered by heat and pressure, radiation or when mixed with a catalyst,4 in particular by means of olefin metathesis reactions.5–12 A typical example of a thermoset polymer is poly(dicyclopentadiene) (PDCPD) which is synthesized through ring-opening metathesis (ROMP) of dicyclopentadiene (DCPD).13,14 ROMP changes the soft structure of monomeric DCPD or DCPD-derived linear oligomeric chains to a hard cross-linked polymer array. Derived materials are of great interest because of their thermal and corrosion resistance. Because of their mechanical properties, they also find applications in the materials, petrochemical and energy generating industries.15 Effective ROMP reactions of DCPD are successfully achieved via the reaction injection moulding (RIM) technique,16,17 in which the curing reaction is accomplished by mixing together of the DCPD monomer and a catalyst in the moulding process.15,18 Apart from preventing decomposition due to the presence of leaving groups such as phosphines trans to the N-heterocyclic carbene (NHC) ligand,19–22 or even getting rid of them,23–25 to prevent undesired premature polymerization, i.e. room temperature (rt) polymerization, the latent catalysts have been proposed as a potential solution,26 and therefore the interest in them has grown in recent years,27 knowing that a drawback of catalysts that initiate ROMP polymerization at rt is the requirement for a pretreatment, which is not suitable for RIM.28,29 Anyway, efficient latent catalysts have been described30–33 that work through thermal34–41 and (photo)chemical activation mechanisms.42–49Ruthenium catalysts bearing NHC and alkylidene ligands are found to be active at room temperature for ROMP of DCPD.31 Hejl et al. reported tuneable latency activity through alteration of the stereoelectronics of the substituted ligands. In these studies, alkylidene ROMP catalysts were tested, showing latent behaviour when an endocyclic imine moiety was present (Scheme 1A). Other studies reported that the nature of the imine substituents affected the activity of the catalyst, showing potential tuneability.30 All these results also coincide with the current mechanistic descriptions of ruthenium catalysed metathesis, where the removal of a donor ligand provides entrance to the catalytic cycle.50,51
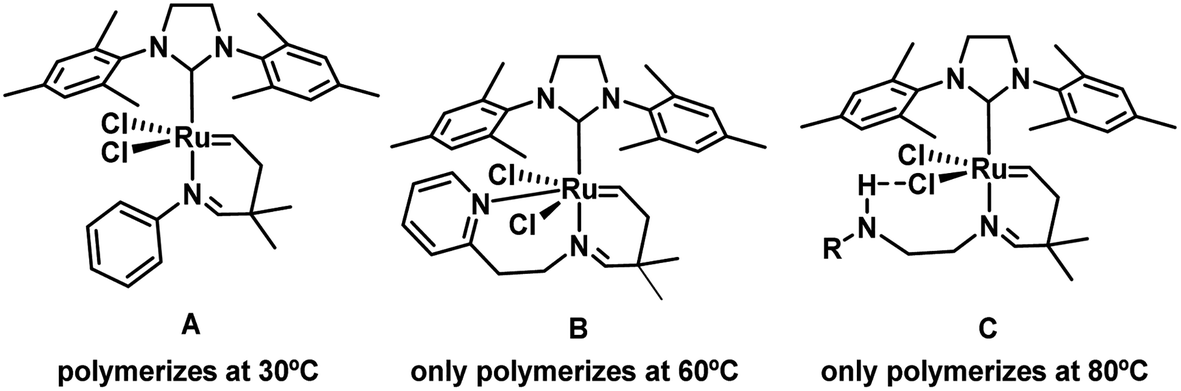 | ||
| Scheme 1 Different ruthenium–NHC catalysts for DCPD polymerization with their temperature of initiation. | ||
With the idea to favour the latency of the initiators it was not enough with the addition of donor groups such as methoxy or thioether substituents,30 but Kim and co-workers found it was mandatory to introduce a deeper skeletal modification, including pyridine based ligands on the ruthenium centre, facilitating the polymerization at 60 °C.39 Next, the same research group showed that the presence of a donor ligand with an additional potential binding capacity could enhance the process. The results clearly showed that for alkylidene-containing NHC–Ru complexes,52 the additional donor atom increases the initiation temperature, avoiding polymerization under 30 °C, but achieving normal reactivity when the temperature was increased up to 60 °C (Scheme 1B).34 Similar results were reported when a hydrogen donor such as carbamate or acetamide was introduced in the imine moiety, in detail preventing the dissociation of the imine nitrogen in the alkylidene moiety. Acting as a latent initiator, the hydrogen donor provides a weak hydrogen-bond interaction with the metal-bound chloride anion of the catalyst (Scheme 1C), which would stabilize sufficiently the precatalyst,53 and consequently the initiator remains unactive below 80 °C.35 In parallel, Czarnocki and co-workers claimed the enhancement of the latency with the insertion of a carbonyl ester group in a modified Hoveyda–Grubbs catalyst, replacing the oxygen by a nitrogen bonded to the ruthenium.54 Nevertheless, the conversion of carbonyl groups to carboxylates, with air and a strong base such as triethanolamine, cannot be extrapolated to any of the systems included in Scheme 1 for their lack of acidic protons to facilitate the intramolecular cyclization, apart from the low chances of a carbonyl group to generate 7-membered rings together with the metal centre and the chelated imine.
Inspired by this novel H-donor based latent strategy of Kim and co-workers,39 computational studies using Density Functional Theory (DFT) calculations and Non-Covalent Interactions (NCI) plots were conducted to delve into the origin of their increased thermal latency, trying to better define how this intramolecular interaction is able to increase the latent behaviour of these systems.
Results and discussion
The series of imine ylidene ligands 1P–5P in Scheme 2 was compared in terms of the activation pathway in olefin metathesis, following the mechanism in Scheme 2. Five different substituents were considered as imine derivatives in the calculations. Both methyl carbamate and amide, substituents 1P and 2P respectively, include a hydrogen donor moiety that allows a hydrogen bond with one of the chlorides of the metal centre. Substituent 3P also has a long chain as in the previously mentioned ones, but the lack of any additional functionality makes it unable to undertake additional electronic intramolecular interactions with the metal centre. Finally, substituents 4P and 5P were introduced in order to evaluate possible differences due to the length of the imine substituent. Additionally, system 4 contains the methoxy group that could electronically not only modify the electron donating capacity of the imine moiety, but also generate a potential hydrogen bond with its oxygen atom.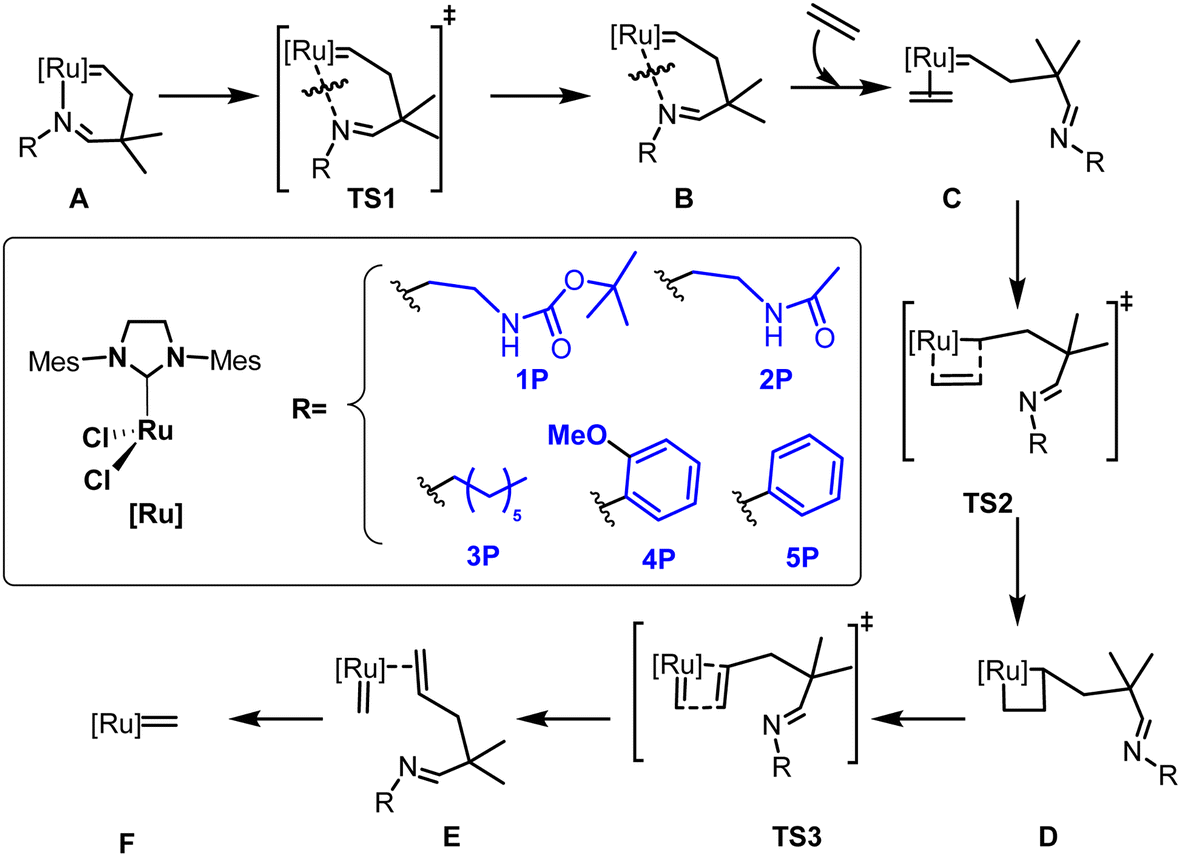 | ||
| Scheme 2 The studied activation pathway of the Ru based catalysts 1–5. | ||
From Scheme 2, starting with the imine based chelate precatalyst A, and finishing with the ylidene 14e active species F, the mechanism is somewhat analogous with respect to the Hoveyda–Grubbs catalysts,54 with the replacement of the oxygen atom bonded to ruthenium by nitrogen.55 Before a detailed structural and electronic analysis of the set of catalysts under analysis, the energetic values for the reaction pathway of the activation are included in Table 1.
| A | TS1 | B | C | TS2 | D | TS3 | E | F | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.0 | 21.5 | 14.9 | 27.4 | 25.2 | 23.2 | 29.5 | 26.9 | 23.9 |
| 2 | 0.0 | 22.9 | 21.8 | 27.8 | 29.6 | 25.7 | 31.9 | 30.0 | 25.0 |
| 3 | 0.0 | 19.4 | 19.3 | 25.3 | 25.8 | 22.3 | 30.1 | 26.5 | 27.7 |
| 4 | 0.0 | 17.9 | 16.0 | 22.2 | 27.0 | 20.3 | 26.7 | 27.9 | 23.6 |
| 5 | 0.0 | 15.8 | 15.7 | 21.6 | 23.4 | 19.8 | 22.2 | 23.8 | 21.6 |
The breaking of the trans Ru–N bond marks the beginning of preactivation, with a kinetic cost that involves a range of values that goes from 15.8 for complex 5 to 22.9 kcal mol−1 for complex 2, therefore with values similar to Hoveyda complexes,56,57 and even those that simply have a trans phosphine in the NHC.58 This first dissociative step seems trivial, but it is also framed by a relatively poor thermodynamic stabilization, since the later intermediate B shows that by Hammond's principle the transition state is a late transition state, with a destabilization of at least 14.9 kcal mol−1 in all cases. And even more is evident by incorporating ethylene into the equation, being trans to the NHC, in the coordination intermediate C, extending this value to at least 21.6 kcal mol−1 and worryingly to 27.8 kcal mol−1 for complex 2, although the correction of the entropic overestimation has been omitted here,59 in order to not treat the concentration at 1 M of solutes,60 and neither of solvents,61 which would cause a reduction of 2–6 kcal mol−1 depending on the nature of them. From C, TS2 leads to the metallacycle D overcoming the energy barriers of little importance, a maximum of 4.8 kcal mol−1, and likewise, the break in the second coordination intermediate E, homologous to C, is given by energy barriers that in no case exceed 7.8 kcal mol−1. However, this metallacycle opening TS3 defines the rate determining step (rds) of the precatalyst activation mechanism, which then ends up with the catalytic active species F, except for systems 4 and 5, where the rds switches to the formation of the metallacycle, TS2, however only by 0.3 and 1.2 kcal mol−1, respectively. On the other hand, it is also worth noting the relatively low stability of the intermediate E, which even when the solvent effect is added in the point energy calculation turns out to be unexpectedly more unstable than the previous TS3.
Once the cost of preactivation has been described, to determine the reason for the different behaviour of the five systems under study, it is necessary to put them under the microscope of structural and electronic factors, preferably emphasizing the imine group, and how it interacts with the rest of the system itself. Thus, the optimized geometries of precatalyst A in systems 1 and 2 demonstrate that the N–H bond is already oriented toward one of the chlorides of the ruthenium catalyst (Fig. 1). The distances between the polarized hydrogen and the nearest chloride are 2.210 and 2.193 Å, respectively. Thus, with the values obtained, and taking into account that an average hydrogen bond in water has a longer length, the additional intramolecular interaction was considered to be present for the precatalytic intermediates with substituents P1 and P2, and enjoying a considerable magnitude. Although the most important weight of H-bonds is installed in the first Ru–N bond cleavage step, they were studied for the rest of the intermediates in the reaction pathway. While for system 1, the (N)H⋯Cl interaction disappears for the rest of the mechanism, this is not the case for catalyst 2, where we still keep a rather weak H-bond in intermediate B (2.563 Å).
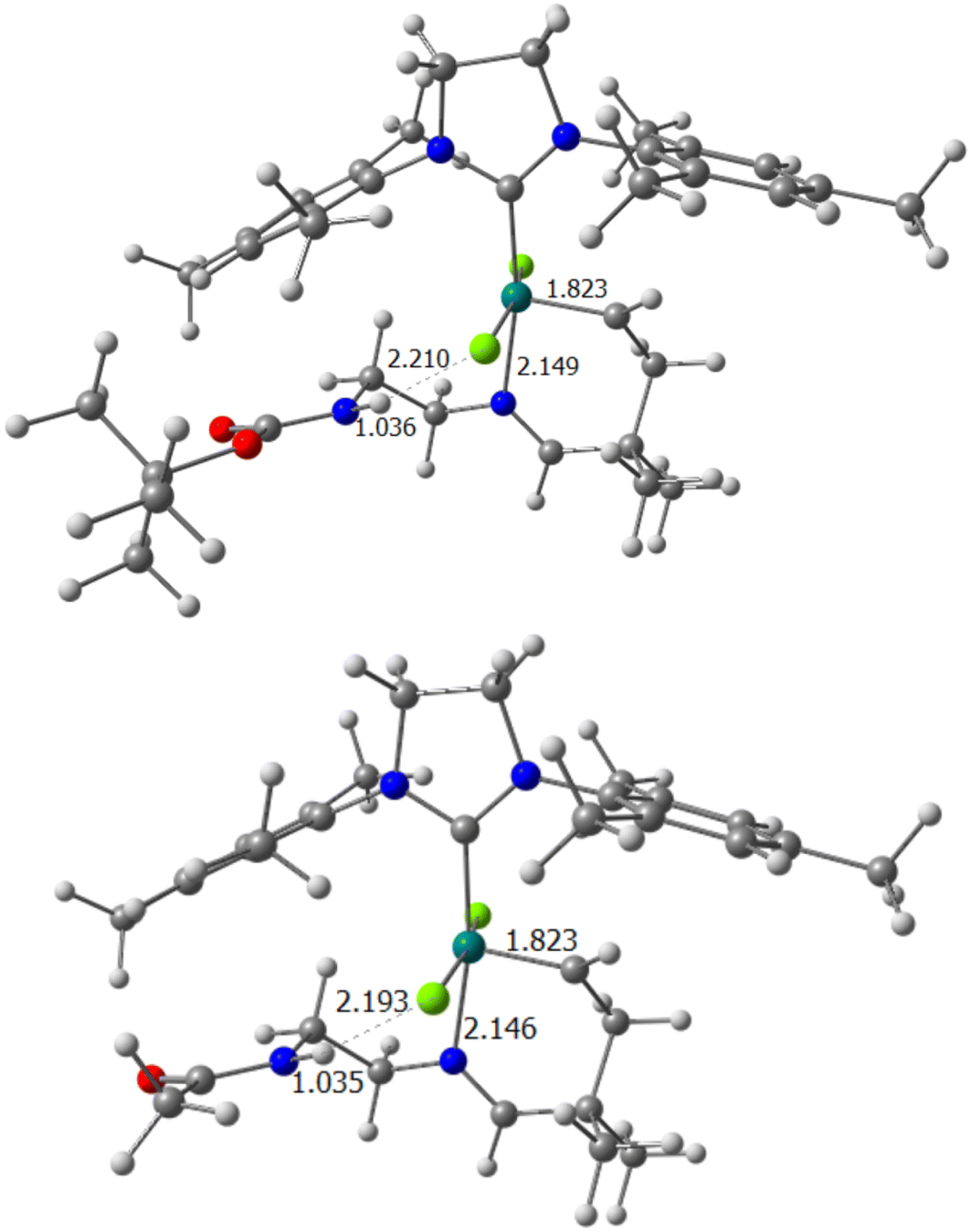 | ||
| Fig. 1 Precatalyst A for systems 1 (above) and 2 (below), main distances are shown in Å. | ||
Surprisingly, after unveiling the activation in Table 1, the above-mentioned hydrogen bond would not affect the overall thermodynamic pathway of the reaction. Although a relatively higher barrier is observed in the first transition state TS1 of systems 1 and 2, which belongs to the opening of the chelating ring from the initial precatalytic geometry, it would not affect it dramatically since it does not correspond to the rds. However, a lower kinetic cost is obtained for the systems with no internal hydrogen bond, not only for this TS1, but also for the next transition states, including the rds.
A more comparative analysis between the five systems, carried out with a tool capable of analysing the strength of bonds of different nature, such as the Mayer Bond Order (MBO) index,62 distils in Table 2 that the Ru–N bond in breaking follows the order of 1 ≅ 2 > 3 > 5 > 4, that is, we would expect it to be more difficult to break the Ru–N bond especially for the first two systems. It is also interesting in Table 2 to confirm the quite strong H-bond between the N–H moiety and the closest Cl ligand, with a MBO over 0.1 for systems 1 and 2. On the other hand, the MBOs for the metallacycle unveil that once the Ru–N bond has broken the next formation of the metallacycle D shows a ruthenacycle with a core nearly identical for the five systems.
| A | D | |||||
|---|---|---|---|---|---|---|
| Ru–N | (N)H⋯Cl | Ru–C1 | C1–C2 | C2–C3 | C3–Ru | |
| 1 | 0.489 | 0.117 | 1.091 | 0.879 | 0.955 | 1.185 |
| 2 | 0.490 | 0.119 | 1.093 | 0.877 | 0.955 | 1.183 |
| 3 | 0.480 | — | 1.092 | 0.877 | 0.955 | 1.184 |
| 4 | 0.465 | — | 1.093 | 0.878 | 0.955 | 1.184 |
| 5 | 0.474 | — | 1.093 | 0.879 | 0.955 | 1.185 |
To assess the lower sterical pressure on the metal centre, as well as to differentiate among the precatalysts A for systems 1–5, we performed steric maps following the scheme developed by Cavallo and co-workers.63 The results in Fig. 2 confirm the larger sterical hindrance on the metal centre due to the imine–ylidene ligands of systems 1 and 2.64 Actually, not only in the precatalyst A, but also throughout all the activation pathway (see ESI for further details‡).
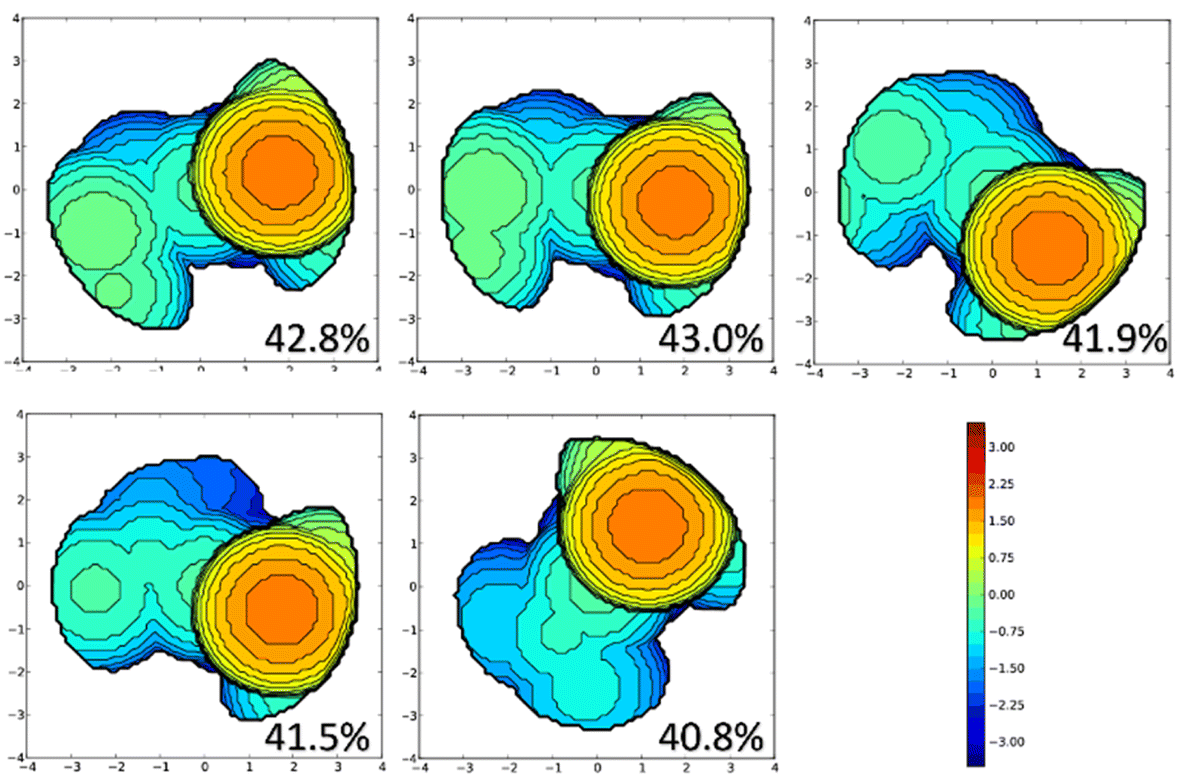 | ||
| Fig. 2 Topographic steric maps (xy plane) and %VBur of the imine–ylidene ligands of species A for systems 1–5. The Ru atom is at the origin and the Nimine atom is on the z axis, and the N atoms of the NHC ligand on the xz plane. The isocontour curves of the steric maps are given in Å. The radius of the sphere around the metal centre was set to 3.5 Å, while for the atoms we adopted the Bondi radii scaled by 1.17, and a mesh of 0.1 Å was used to scan the sphere for buried voxels (with a radius of 3.5 Å, the isocontour curves of the steric maps are given in Å). | ||
Electronically the Natural Bond Orbital analysis of the charges in Table 3 shows that there are no differences in the metallacycle, and no significant differences in the precatalyst A for systems 1 and 2 compared to the other systems, except for the interesting positive charge on the hydrogens of the N–H moieties in systems 1 and 2.
| A | D | |||||||
|---|---|---|---|---|---|---|---|---|
| Ru | N | N(H) | (N)H | Ru | C1 | C2 | C3 | |
| 1 | −0.290 | −0.399 | −0.691 | 0.431 | −0.300 | −0.125 | −0.486 | −0.322 |
| 2 | −0.323 | −0.403 | −0.675 | 0.416 | −0.298 | −0.125 | −0.487 | −0.322 |
| 3 | −0.303 | −0.396 | — | — | −0.300 | −0.124 | −0.486 | −0.323 |
| 4 | −0.276 | −0.396 | — | — | −0.300 | −0.125 | −0.487 | −0.322 |
| 5 | −0.275 | −0.392 | — | — | −0.282 | −0.129 | −0.489 | −0.322 |
A deeper analysis with the NBO approach of the NH⋯Cl interaction for intermediate A of system 1 unveiled the importance of the lone pair (LP) corresponding to both chlorides together with the antibonding character owed by the bonding orbital (BD*) for the NH moiety with a total energy of 11.6 kcal mol−1.
Before concluding that the electronic part does not affect the reactivity, taking into account the NBO charges, in Table 4 and Fig. 3, we proceeded to do a simple analysis of the frontier molecular orbitals together with conceptual DFT,65 with the surprise that even though it is about similar stability, corroborated by the almost identical chemical hardness for species A for all systems, the same does not happen with the chemical potential, nor with electrophilicity, but contrarily to what was predicted recently by Martínez and Trzaskowski with Grubbs–Hoveyda type catalysts, where the higher the electrophilicity the higher the reactivity was.66 Thus, a simple NH group modulates the reactivity, making systems 1 and 2 more electrophilic, explained by similar stabilization of both the HOMO and the LUMO.
| HOMO | LUMO | μ | η | ε | |
|---|---|---|---|---|---|
| 1 | −0.155 | −0.086 | −0.120 | 0.069 | 0.105 |
| 2 | −0.157 | −0.088 | −0.122 | 0.069 | 0.109 |
| 3 | −0.149 | −0.081 | −0.115 | 0.068 | 0.097 |
| 4 | −0.146 | −0.079 | −0.113 | 0.067 | 0.095 |
| 5 | −0.148 | −0.081 | −0.115 | 0.067 | 0.098 |
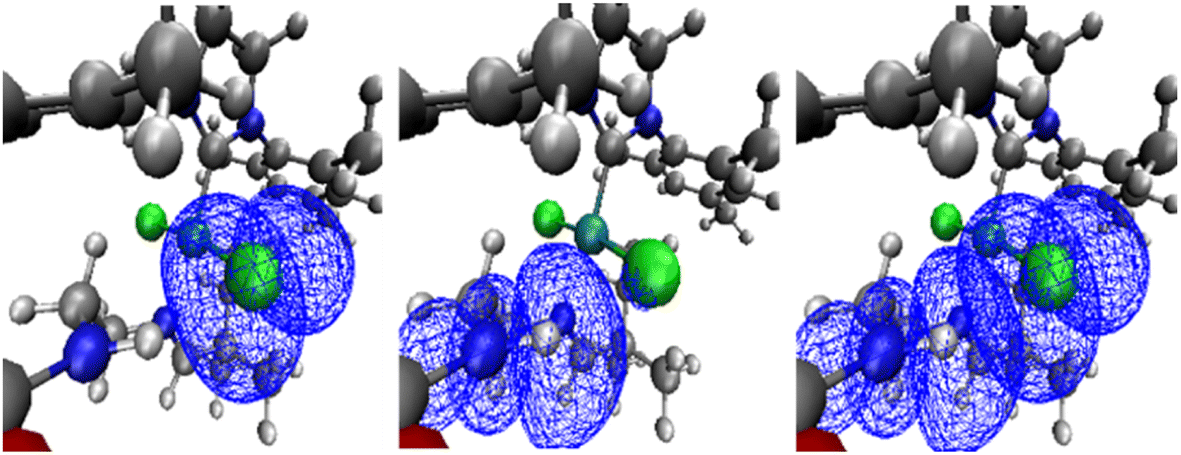 | ||
| Fig. 3 NBO orbitals for precatalyst A for system 1 involved in the NH⋯Cl interaction: LP (left), BD* (middle) and overlap of the latter orbitals (right), see ESI‡ for further details. | ||
To unveil how the H-bonds of the halides and the organic moiety of the imine ligand trans to the NHC affect the system, 3D and 2D NCI plots in Fig. 4 and 5 were calculated using the program developed by Contreras-García et al.67,68
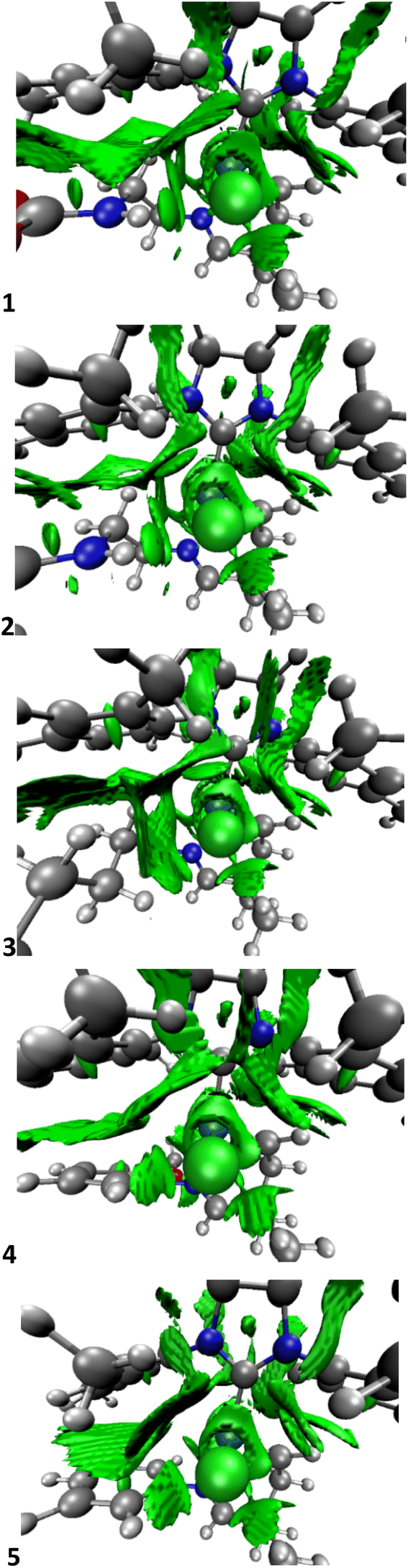 | ||
| Fig. 4 NCI plots of the intermediate A for systems 1–5 (orientated like Fig. 1, and the whole picture is truncated for the sake of clarity of the metal centre, see ESI‡ for the NCI plots of the whole molecule). | ||
 | ||
| Fig. 5 2D-NCI plots of reduced density gradient (s) vs. sign (λ2)ρ, in a.u., for the initial intermediate A for systems 2 (above) and 5 (below). | ||
The NCI plots help to understand here the different behaviours between catalysts, however only partially since energetics are still more important since interactions are less important than expected,69–71 especially compared to other olefin metathesis catalysts.72 Here, NCI interactions might not help to avoid decomposition, but help to influence the activation of the precatalyst. The NCI plots reveal how systems 1 and 2 have the H-bond between the H of the NH group and the nearest chloro ligand, which despite not being present in most of the following intermediates and transition states, does persist in some of them, although not so clearly. In addition, the bidimensional NCI plots in Fig. 5 clarify the difference in the first two systems compared to the other ones,73i.e. comparing systems 2 and 5, with a remarkable sign (λ2)ρ contribution for 2 in the region between −0.02 and −0.03 a. u. that for 5 is completely nonexistent.
Computational details
All the DFT static calculations were performed with the Gaussian16 set of programs.74 All geometry optimizations were performed in gas phase using the BP86 functional of Becke and Perdew,75–77 corresponding to the GGA level, including corrections due to dispersion through Grimme's method (GD3 keyword in Gaussian16).78,79 The electronic configuration of the molecular systems was described with the double-ζ basis set with the polarization of Ahlrichs for main group atoms from Karlsruhe (Def2SVP keyword in Gaussian),80,81 whereas for ruthenium, the small-core quasirelativistic Stuttgart–Dresden effective core potential was introduced, altogether with an associated valence basis set (standard SDD keyword in Gaussian).82–84 The geometry optimizations were performed without symmetry constraints, with analytical frequency calculations for the characterization of the located stationary points. These frequencies were used to calculate unscaled zero-point energies (ZPEs) as well as thermal corrections and entropy effects at 353.15 K. Energies were obtained through single-point calculations on the optimized geometries from the BP86/Def2SVP∼sdd employing the B3LYP functional,85,86 and the cc-pVTZ basis set for main atoms,87 while for ruthenium the same basis set as before. Also, dispersion effects were introduced with the Grimme D3 correction term to the electronic energy. Furthermore, the solvent effects were estimated with the PCM model,88–90 and Gibbs free energies have been computed at 353.15 K and atmospheric pressure reaction conditions.Conclusions
In a set of latent Ru based catalysts, the Ru–N bond cleavage is more favoured kinetically for systems 1 and 2, but this does not correspond to the rds, but the opening of the metallacycle. These results might seem somewhat in disagreement with experiments because kinetically systems 1 and 2 have larger energy barriers, and not only in the Ru–N cleavage, but also in the next transition states, including the rds. Actually, this is what really favours the latent activity in polymerization at over 353.15 K for both systems, whereas it starts at 303.15 K for the other systems of the set of catalysts tested experimentally. In the phase of characterisation, the non-covalent interactions play a key role between the NH moiety and the closest chloride ligand. And probably the potential H-bonds of the NH fragment with any other atom during the reaction pathway may also slow down the olefin metathesis process in the following steps. However, this was not confirmed for the described intermediates. Further studies are ongoing to confirm the validity of the results extending the analysis towards the catalytic cycle, and with the aim to check if the solvent energy calculations at the B3LYP/cc-pVTZ∼sdd level might either underestimate the kinetic Ru–N bond cleavage cost or overestimate the opening of the metallacycle.91Author contributions
Artur Brotons-Rufes performed all the calculations, the visualization and wrote the original draft together with Albert Poater. Naeimeh Bahri-Laleh and Albert Poater performed the analysis, supervision, review and editing of the text.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Spanish Ministerio de Ciencia e Innovación for project PID2021-127423NB-I00. A. P. is a Serra Húnter Fellow and ICREA Academia Prize 2019. N. B.-L. appreciates the support of the Iran Polymer and Petrochemical Institute.References
- I. D. Robertson, M. Yourdkhani, P. J. Centellas, J. E. Aw, D. G. Ivanoff, E. Goli, E. M. Lloyd, L. M. Dean, N. R. Sottos, P. H. Geubelle, J. S. Moore and S. R. White, Rapid energy-efficient manufacturing of polymers and composites via frontal polymerization, Nature, 2018, 557, 223–227 CrossRef CAS PubMed.
- D. Sivanesan, B. Seo, C.-S. Lim, S. Kim and H.-G. Kim, Trifunctional cycloaliphatic epoxy-based thermoset polymers: Synthesis, polymerization, and characterization, Polymer, 2021, 220, 123568 CrossRef CAS.
- P. Brøndsted, H. Lilholt and A. Lystrup, Composite materials for wind power turbine blades, Annu. Rev. Mater. Res., 2005, 35, 505–538 CrossRef.
- R. H. Grubbs and S. H. Pine, in Comprehensive Organic Synthesis, ed. B. M. Trost, I. Fleming and L. A. Paquette, Pergamon, New York, 1991, vol. 5 Search PubMed.
- K. J. Ivin and J. C. Mol, Olefin Metathesis and Metathesis Polymerization, Academic Press, San Diego, CA, 1997 Search PubMed.
- N. B. Nechmad, R. Phatake, E. Ivry, A. Poater and N. G. Lemcoff, Unprecedented Selectivity of Ruthenium Iodide Benzylidenes in Olefin Metathesis Reactions, Angew. Chem., Int. Ed., 2020, 59, 3539–3543 CrossRef CAS PubMed.
- C. Samojłowicz, M. Bieniek and K. Grela, Ruthenium-Based Olefin Metathesis Catalysts Bearing N-Heterocyclic Carbene Ligands, Chem. Rev., 2009, 109, 3708–3742 CrossRef PubMed.
- C. S. Higman, J. A. M. Lummiss and D. E. Fogg, Olefin Metathesis at the Dawn of Implementation in Pharmaceutical and Specialty-Chemicals Manufacturing, Angew. Chem., Int. Ed., 2016, 55, 3552–3565 CrossRef CAS PubMed.
- R. H. Grubbs, S. J. Miller and G. C. Fu, Ring-Closing Metathesis and Related Processes in Organic Synthesis, Acc. Chem. Res., 1995, 28, 446–552 CrossRef CAS.
- R. R. Schrock, J. S. Murdzek, G. C. Bazan, J. Robbins, M. DiMare and M. O'Regan, Synthesis of molybdenum imido alkylidene complexes and some reactions involving acyclic olefins, J. Am. Chem. Soc., 1990, 112, 3875–3886 CrossRef CAS.
- G. C. Bazan, E. Khosravi, R. R. Schrock, W. J. Feast, V. C. Gibson, M. B. O'Regan, T. K. Thomas and W. M. Davis, Living ring opening metathesis polymerization of 2,3-difunctionalized norbornadienes by Mo(:CHBu-tert)(:NC6H3Pr-iso2-2,6)(OBu-tert)2, J. Am. Chem. Soc., 1990, 112, 8378–8387 CrossRef CAS.
- G. C. Vougioukalakis and R. H. Grubbs, Ruthenium-Based Heterocyclic Carbene-Coordinated Olefin Metathesis Catalysts, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS PubMed.
- B. M. Novak, W. Risse and R. H. Grubbs, The development of well-defined catalysts for ring-opening olefin metathesis polymerizations (ROMP), Adv. Polym. Sci., 1992, 102, 47–72 CrossRef CAS.
- S. Kovačič and C. Slugovc, Ring-opening Metathesis Polymerisation derived poly-(dicyclopentadiene) based materials, Mater. Chem. Front., 2020, 4, 2235–2255 RSC.
- S. T. Nguyen, R. H. Grubbs and J. W. Ziller, Syntheses and activities of new single-component, ruthenium-based olefin metathesis catalysts, J. Am. Chem. Soc., 1993, 115, 9858–9859 CrossRef CAS.
- S. Naumann and M. R. Buchmeiser, Latent and delayed action polymerization systems, Macromol. Rapid Commun., 2014, 35, 682–701 CrossRef CAS PubMed.
- K. Denk, J. Fridgen and W. A. Herrmann, N-Heterocyclic Carbenes, Part 33.[1] Combining Stable NHC and Chelating Pyridinyl-Alcoholato Ligands: A Ruthenium Catalyst for Applications at Elevated Temperatures, Adv. Synth. Catal., 2002, 344, 666–670 CrossRef CAS.
- Ł. Gułajski, A. Michrowska, R. Bujok and K. Grela, New tunable catalysts for olefin metathesis: Controlling the initiation through electronic factors, J. Mol. Catal. A: Chem., 2006, 254, 118–123 CrossRef.
- G. A. Bailey, M. Foscato, C.S. Higman, C. S. Day, V. R. Jensen and D. E. Fogg, Bimolecular coupling as a vector for decomposition of fast-initiating olefin metathesis catalysts, J. Am. Chem. Soc., 2018, 140, 6931–6944 CrossRef CAS PubMed.
- S. Manzini, A. Poater, D. J. Nelson, L. Cavallo, A. M. Z. Slawin and S. P. Nolan, Insights into the decomposition of olefin metathesis pre-catalysts, Angew. Chem., Int. Ed., 2014, 53, 8995–8999 CrossRef CAS PubMed.
- S. Manzini, C. A. Urbina-Blanco, A. Poater, A. M. Z. Slawin, L. Cavallo and S. P. Nolan, From olefin metathesis catalyst to alcohol racemization catalyst in one step, Angew. Chem., Int. Ed., 2012, 51, 1042–1045 CrossRef CAS PubMed.
- A. Poater, N. Bahri-Lalehah and L. Cavallo, Rationalizing current strategies to protect N-heterocyclic carbene-based ruthenium catalysts active in olefin metathesis from C-H (de)activation, Chem. Commun., 2011, 47, 6674–6676 RSC.
- A. Poater, R. Credendino, C. Slugovc and L. Cavallo, Exploring new generations of ruthenium olefin metathesis catalysts: the reactivity of a bis-ylidene ruthenium complex by DFT, Dalton Trans., 2013, 42, 7271–7275 RSC.
- A. Poater, S. V. C. Vummaleti, E. Pump and L. Cavallo, Comparing Ru and Fe-catalyzed olefin metathesis, Dalton Trans., 2014, 43, 11216–11220 RSC.
- A. Poater, E. Pump, S. V. C. Vummaleti and L. Cavallo, The activation mechanism of Fe-based olefin metathesis catalysts, Chem. Phys. Lett., 2014, 610–611, 29–32 CrossRef CAS.
- A. Leitgeb, M. Abbas, R. C. Fischer, A. Poater, L. Cavallo and C. Slugovc, A latent ruthenium based olefin metathesis catalyst with a sterically demanding NHC ligand (pre)catalysts, Catal. Sci. Technol., 2012, 2, 1640–1643 RSC.
- I. Elser, B. R. Kordes, W. Frey, K. Herz, R. Schowner, L. Stöhr, H. J. Altmann and M. R. Buchmeiser, Latent and Air-Stable Pre-Catalysts for the Polymerization of Dicyclopentadiene: From Penta- to Hexacoordination in Molybdenum Imido Alkylidene N-Heterocyclic Carbene Complexes, Chem.–Eur. J., 2018, 24, 12652–12659 CrossRef CAS PubMed.
- I. Rozenberg, O. Eivgi, A. Frenklah, D. Butilkov, S. Kozuch, I. Goldberg and N. G. Lemcoff, Synthesis and Catalytic Properties of Sulfur-Chelated Ruthenium Benzylidenes Bearing a Cyclic (Alkyl)-(amino)carbene Ligand, ACS Catal., 2018, 8, 8182–8191 CrossRef CAS.
- A. Leitgeb, J. Wappel, C. A. Urbina-Blanco, S. Strasser, C. Wappl, C. S. J. Cazin and C. Slugovc, Two commercially available initiators for the retarded ring-opening metathesis polymerization of dicyclopentadiene, Monatsh. Chem., 2014, 145, 1513–1517 CrossRef CAS.
- A. Hejl, M. W. Day and R. H. Grubbs, Latent olefin metathesis catalysts featuring chelating alkylidenes, Organometallics, 2006, 25, 6149–6154 CrossRef CAS.
- S. Monsaert, A. Lozano Vila, R. Drozdzak, P. Van Der Voort and F. Verpoort, Latent olefin metathesis catalysts, Chem. Soc. Rev., 2009, 38, 3360–3372 RSC.
- P. A. van der Schaaf, A. Hafner and A. Mühlebach, Photoinduced Ring-Opening Metathesis Polymerization (PROMP) with Photochemically Generated Schrock-Type Catalysts, Angew. Chem., Int. Ed. Engl., 1996, 35, 1845–1847 CrossRef CAS.
- T. Ung, A. Hejl, R. H. Grubbs and Y. Schrodi, Latent Ruthenium Olefin Metathesis Catalysts that Contain an N-Heterocyclic Carbene Ligand, Organometallics, 2004, 23, 5399–5401 CrossRef CAS.
- D. Sivanesan, B. Seo, C.-S. Lim, D. Choi, T. Kim and H.-G. Kim, An Additional Potential Donor-bearing Alkylidene-containing Latent NHC-Ruthenium-based Catalyst for Olefin Metathesis Polymerization, Polym. Chem., 2020, 11, 2511–2518 RSC.
- B. Allaert, N. Dieltiens, N. Ledoux, C. Vercaemst, P. Van der Voort, C. V. Stevens, A. Linden and F. Verpoort, Synthesis and activity for ROMP of bidentate Schiff base substituted second generation Grubbs catalysts, J. Mol. Catal. A: Chem., 2006, 260, 221–226 CrossRef CAS.
- R. Drozdzak, N. Nishioka, G. Recher and F. Verpoort, Latent Olefin Metathesis Catalysts for Polymerization of DCPD, Macromol. Symp., 2010, 293, 1–4 CrossRef CAS.
- A. S. Chang, L. Jones, C. Wang, L. M. Henling and R. H. Grubbs, Synthesis and Characterization of New Ruthenium-Based Olefin Metathesis Catalysts Coordinated with Bidentate Schiff-Base Ligands, Organometallics, 1998, 17, 3460–3465 CrossRef.
- C. D. M. Hudson, E. J. Valente, J. Schachner, M. Limbach, K. Muller and H. J. Schanz, A Ru-vinylvinylidene Complex: Straightforward Synthesis of a Latent Olefin Metathesis Catalyst, ChemCatChem, 2011, 3, 297–301 CrossRef.
- D. Sivanesan, B. Seo, C.-S. Lim, H. Kim and H.-G. Kim, Intramolecular Hydrogen-Bond-Based Latent Initiator for Olefin Metathesis Polymerization, Organometallics, 2021, 40, 314–323 CrossRef CAS.
- Y. Vidavsky, A. Anaby and N. G. Lemcoff, Chelating alkylidene ligands as pacifiers for ruthenium catalysed olefin metathesis, Dalton Trans., 2012, 41, 32–43 RSC.
- A. Ben-Asuly, E. Tzur, C. E. Diesendruck, M. Sigalov, I. Goldberg and N. G. Lemcoff, A Thermally Switchable Latent Ruthenium Olefin Metathesis Catalyst, Organometallics, 2008, 27, 811–813 CrossRef CAS.
- E. Alessio, G. Mestroni, G. Nardin, W. M. Attia, M. Calligaris, G. Sava and S. Zorzet, Cis- and trans-dihalotetrakis(dimethyl sulfoxide)ruthenium(II) complexes (RuX2(DMSO)4; X = Cl, Br): synthesis, structure, and antitumor activity, Inorg. Chem., 1988, 27, 4099–4106 CrossRef CAS.
- A. Ben-Asuly, A. Aharoni, C. E. Diesendruck, Y. Vidavsky, I. Goldberg, B. F. Straub and N. G. Lemcoff, Photoactivation of Ruthenium Olefin Metathesis Initiators, Organometallics, 2009, 28, 4652–4655 CrossRef CAS.
- S. Monsaert, N. Ledoux, R. Drozdzak and F. Verpoort, A highly controllable latent ruthenium Schiff base olefin metathesis catalyst: Catalyst activation and mechanistic studies, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 302–310 CrossRef CAS.
- A. Hafner, A. Muhlebach and P. A. van der Schaaf, One-Component Catalysts for Thermal and Photoinduced Ring Opening Metathesis Polymerization, Angew. Chem., Int. Ed. Engl., 1997, 36, 2121–2124 CrossRef CAS.
- D. Wang, J. Unold, M. Bubrin, I. Elser, W. Fray, W. Kaim, G. Xu and M. R. Buchmeiser, Ruthenium–Triazene Complexes as Latent Catalysts for UV-Induced ROMP, Eur. J. Inorg. Chem., 2013, 2013, 5462–5468 CrossRef CAS.
- D. Wang, J. Unold, M. Bubrin, W. Fray, W. Kaim and M. R. Buchmeiser, Ruthenium(IV)–Bis(methallyl) Complexes as UV-Latent Initiators for Ring-Opening Metathesis Polymerization, ChemCatChem, 2012, 4, 1808–1812 CrossRef CAS.
- D. Wang, K. Wurst and M. R. Buchmeiser, Cationic versus Neutral RuII-N-Heterocyclic Carbene Complexes as Latent Precatalysts for the UV-Induced Ring-Opening Metathesis Polymerization, Chem.–Eur. J., 2010, 16, 12928–12934 CrossRef CAS PubMed.
- O. Eivgi, A. Vaisman, N. B. Nechmad, M. Baranov and N. G. Lemcoff, Latent Ruthenium Benzylidene Phosphite Complexes for Visible-Light-Induced Olefin Metathesis, ACS Catal., 2020, 10, 2033–2038 CrossRef CAS.
- M. S. Sanford, M. Ulman and R. H. Grubbs, New Insights into the Mechanism of Ruthenium-Catalyzed Olefin Metathesis Reactions, J. Am. Chem. Soc., 2001, 123, 749–750 CrossRef CAS PubMed.
- M. S. Sanford, J. A. Love and R. H. Grubbs, Mechanism and Activity of Ruthenium Olefin Metathesis Catalysts, J. Am. Chem. Soc., 2001, 123, 6543–6554 CrossRef CAS PubMed.
- A. Poater, F. Ragone, A. Correa and L. Cavallo, Comparison of different ruthenium–alkylidene bonds in the activation step with N-heterocyclic carbene Ru-catalysts for olefins metathesis, Dalton Trans., 2011, 40, 11066–11069 RSC.
- A. Poater and L. Cavallo, A comprehensive study of olefin metathesis catalyzed by Ru-based catalysts, Beilstein J. Org. Chem., 2015, 11, 1767–1780 CrossRef CAS PubMed.
- S. J. Czarnocki, I. Czelusniak, T. K. Olszewski, M. Malinska, K. Wozniak and K. Grela, Rational and Then Serendipitous Formation of Aza Analogues of Hoveyda-Type Catalysts Containing a Chelating Ester Group Leading to a Polymerization Catalyst Family, ACS Catal., 2017, 7, 4115–4121 CrossRef CAS.
- J. P. Martínez and B. Trzaskowski, Olefin Metathesis Catalyzed by a Hoveyda–Grubbs-like Complex Chelated to Bis(2-mercaptoimidazolyl) Methane: A Predictive DFT Study, J. Phys. Chem. A, 2022, 126, 720–732 CrossRef PubMed.
- F. Nunez-Zarur, X. Solans-Monfort, L. Rodríguez-Santiago and M. Sodupe, Differences in the Activation Processes of Phosphine-Containing and Grubbs–Hoveyda-Type Alkene Metathesis Catalysts, Organometallics, 2012, 31, 4203–4215 CrossRef CAS.
- X. Solans-Monfort, R. Pleixats and M. Sodupe, DFT Mechanistic Study on Diene Metathesis Catalyzed by Ru-Based Grubbs–Hoveyda-Type Carbenes: The Key Role of π-Electron Density Delocalization in the Hoveyda Ligand, Chem.–Eur. J., 2010, 16, 7331–7343 CrossRef CAS PubMed.
- C. A. Urbina-Blanco, A. Poater, T. Lebl, S. Manzini, A. M. Z. Slawin, L. Cavallo and S. P. Nolan, The Activation Mechanism of Ru–Indenylidene Complexes in Olefin Metathesis, J. Am. Chem. Soc., 2013, 135, 7073–7079 CrossRef CAS PubMed.
- J. A. Luque-Urrutia and A. Poater, The Fundamental Noninnocent Role of Water for the Hydrogenation of Nitrous Oxide by PNP pincer Ru-based catalysts, Inorg. Chem., 2017, 56, 14383–14387 CrossRef CAS PubMed.
- L. Falivene, V. Barone and G. Talarico, Unraveling the role of entropy in tuning unimolecular vs. bimolecular reaction rates: the case of olefin polymerization catalyzed by transition metals, Mol. Catal., 2018, 452, 138–144 CrossRef CAS.
- J. Serra, C. J. Whiteoak, F. Acuña-Parés, M. Font, J. M. Luis, J. Lloret-Fillol and X. Ribas, J. Am. Chem. Soc., 2015, 137, 13389–13397 CrossRef CAS PubMed.
- I. Mayer, Bond order and valence in the ab initio SCF theory, Chem. Phys. Lett., 1983, 97, 270–274 CrossRef CAS.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Towards the online computer-aided design of catalytic pockets, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- S. Kaur, V. Kumar, M. Chawla, L. Cavallo, A. Poater and N. Upadhyay, Pesticides Curbing Soil Fertility: Effect of Complexation of Free Metal Ions, Front. Chem., 2017, 5, 43 CrossRef PubMed.
- A. Poater, A. Gallegos Saliner, M. Solà, L. Cavallo and A. P. Worth, Computational Methods to Predict the Reactivity of Nanoparticles Through Structure-Property Relationships, Expert Opin. Drug Delivery, 2010, 7, 295–305 CrossRef CAS PubMed.
- J. P. Martínez and B. Trzaskowski, Electrophilicity of Hoveyda–Grubbs Olefin Metathesis Catalysts as the Driving Force that Controls Initiation Rates, ChemPhysChem, 2022, 23, 202200580 CrossRef PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. T. Yang, Revealing Noncovalent Interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. T. Yang, NCIPLOT: a program for plotting non-covalent interaction regions, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef CAS PubMed.
- J. Poater, M. Gimferrer and A. Poater, Covalent and Ionic Capacity of MOFs To Sorb Small Gas Molecules, Inorg. Chem., 2018, 57, 6981–6990 CrossRef CAS PubMed.
- C. J. Richmond, S. Escayola and A. Poater, Axial Ligand effects of Ru-BDA Complexes in the O-O Bond Formation via the I2M Bimolecular Mechanism in Water Oxidation Catalysis, Eur. J. Inorg. Chem., 2019, 2019, 2101–2108 CrossRef CAS.
- A. Vidal-López, S. Posada-Pérez, M. Solà, V. D’Elia and A. Poater, The Importance of the Bite Angle of Metal(III) Salen Catalysts in the Sequestration of CO2 with Epoxides in Mild Conditions, Green Chem. Eng., 2022, 3, 180–187 CrossRef.
- M. Gimferrer, P. Salvador and A. Poater, Computational Monitoring of Oxidation States in Olefin Metathesis, Organometallics, 2019, 38, 4585–4592 CrossRef CAS.
- R. A. Boto, F. Peccati, R. Laplaza, C. Quan, A. Carbone, J.-P. Piquemal, Y. Maday and J. Contreras-García, NCIPLOT4: Fast, Robust, and Quantitative Analysis of Noncovalent Interactions, J. Chem. Theory Comput., 2020, 16, 4150–4158 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. Becke, Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behaviour, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- J. P. Perdew, Erratum: Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 7406 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, A Thorough Benchmark of Density Functional Methods for General Main Group Thermochemistry, Kinetics, andNoncovalent Interactions, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- U. Häusermann, M. Dolg, H. Stoll and H. Preuss, Accuracy of Energy-Adjusted Quasirelativistic ab initio Pseudopotentials, Mol. Phys., 1993, 78, 1211–1224 CrossRef.
- W. Küchle, M. Dolg, H. Stoll and H. Preuss, Energy-Adjusted Pseudopotentials for the Actinides. Parameter Sets and Test Calculations for Thorium and Thorium Monoxide, J. Chem. Phys., 1994, 100, 7535–7542 CrossRef.
- T. Leininger, A. Nicklass, H. Stoll, M. Dolg and P. Schwerdtfeger, The accuracy of the pseudopotential approximation. II. A Comparison of Various Core Sizes for Indium Pseudopotentials in Calculations for Spectroscopic Constants of InH, InF, and InCl, J. Chem. Phys., 1996, 105, 1052–1059 CrossRef CAS.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects, J. Chem. Phys., 1981, 55, 117–129 Search PubMed.
- S. Miertus and J. Tomasi, Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes, J. Chem. Phys., 1982, 65, 239–245 CAS.
- J. L. Pascual-Ahuir, E. Silla and I. Tuñon, GEPOL: An improved description of molecular surfaces. III. A new algorithm for the computation of a solvent-excluding surface, J. Comput. Chem., 1994, 15, 1127–1138 CrossRef CAS.
- A. Poater, E. Pump, S. V. C. Vummaleti and L. Cavallo, The right computational recipe for olefin metathesis with Ru-based catalysts: the whole mechanism of ring-closing olefin metathesis, J. Chem. Theory Comput., 2014, 10, 4442–4448 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to the memory of our colleague and inspiration Prof. Dr Robert H. Grubbs. |
| ‡ Electronic supplementary information (ESI) available: XYZ coordinates and absolute energies of all species. See DOI: https://doi.org/10.1039/d2fd00163b |
| This journal is © The Royal Society of Chemistry 2023 |