 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the degree of CO2 activation by carbon doping Cun− (n = 3–10) clusters: an IR spectroscopic study†
Olga V.
Lushchikova
 ab,
Máté
Szalay
d,
Tibor
Höltzl
cd and
Joost M.
Bakker
*a
ab,
Máté
Szalay
d,
Tibor
Höltzl
cd and
Joost M.
Bakker
*a
aRadboud University, Institute for Molecules and Materials, FELIX Laboratory, Toernooiveld 7, 6525 ED Nijmegen, The Netherlands. E-mail: joost.bakker@ru.nl
bInstitut für Ionenphysik und Angewandte Physik, Universität Innsbruck, Technikerstraße 25, 6020 Innsbruck, Austria
cMTA-BME Computation Driven Chemistry Research Group, Department of Inorganic and Analytical Chemistry, Budapest University ofTechnology and Economics, Muegyetem rkp. 3, Budapest 1111, Hungary
dFurukawa Electric Institute of Technology, Késmárk Utca 28/A, 1158 Budapest, Hungary
First published on 3rd November 2022
Abstract
Copper clusters on carbide surfaces have shown a high catalytic activity towards methanol formation. To understand the interaction between CO2 and the catalytically active sites during this process and the role that carbon atoms could play in this, they are modeled by copper clusters, with carbon atoms incorporated. The formed clusters CunCm− (n = 3–10, m = 1–2) are reacted with CO2 and investigated by IR multiple-photon dissociation (IR-MPD) spectroscopy to probe the degree of CO2 activation. IR spectra for the reaction products [CunC·CO2]−, (n = 6–10), and [CunC2·CO2]−, (n = 3–8) are compared to reference spectra recorded for products formed when reacting the same cluster sizes with CO, and with density functional theory (DFT) calculated spectra. The results reveal a size- and carbon load-dependent activation and dissociation of CO2. The complexes [CunC·CO2]− with n = 6 and 10 show predominantly molecular activation of CO2, while those with n = 7–9 show only dissociative adsorption. The addition of the second carbon to the cluster leads to the exclusive molecular activation of the CO2 on all measured cluster sizes, except for Cu5C2− where CO2 dissociates. Combining these findings with DFT calculations leads us to speculate that at lower carbon-to-metal ratios (CMRs), the C can act as an oxygen anchor facilitating the OC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond rupture, whereas at higher CMRs the carbon atoms increasingly attract negative charge, reducing the Cu cluster’s ability to donate electron density to CO2, and consequently its ability to activate CO2.
O bond rupture, whereas at higher CMRs the carbon atoms increasingly attract negative charge, reducing the Cu cluster’s ability to donate electron density to CO2, and consequently its ability to activate CO2.
Introduction
The continuous growth of carbon emissions has led, and continues to lead, to an accumulation of CO2 in the atmosphere, and the concomitant global warming. To mitigate this, an urgent solution is needed to reduce atmospheric CO2 concentrations. At the same time, CO2 is the largest carbon source on Earth, and it could be used as a feedstock for many valuable chemicals, such as alcohols and other higher hydrocarbons. The bottleneck lies in the high kinetic and thermal stability of the CO2 molecule, expressed in the CO bond energy of more than 7 eV.1
One promising way for CO2 utilization is its hydrogenation to simple alcohols like methanol. Industrially this process takes place over a Cu/ZnO/Al2O3 catalyst at temperatures of 200–300 °C and pressures of 50–100 bar.2 The energy required to drive this reaction leads to additional CO2 emissions and elevated costs, while the one-pass selectivity towards methanol formation is rather low, with larger concentrations of CO and H2O formed. Thus, for direct CO2 conversion, a more selective catalyst is required. Many different catalyst materials have been tested to reduce reaction temperatures and increase the methanol yield.
One issue that seems not to affect relatively well-functioning CO2 hydrogenation catalysts is poisoning due to coke formation, a process that hampers the reverse reaction during methanol decomposition.3 Rather, metal carbide catalyst materials have shown enhanced activity in CO2 activation. This enhancement has been rationalized by increased catalyst stability upon adding carbon to the metal, and by the modification of the metal’s electronic and structural properties, changing its catalytic activity.4,5 Transition metal carbides (TMCs) have even demonstrated chemical activity similar, and sometimes even better, than that of the platinum group metals in the transformation of hydrocarbons and oxygen-containing molecules.6–8 TMCs are also considered to be promising catalysts for CO2 conversion by H2.8 However, experimentally, it was shown that, for example, Mo2C is quite aggressive and leads to CO2 dissociation, forming CO and H2O, via the reverse water gas shift (rWGS) reaction.9,10 Based on density functional theory (DFT) calculations, it is suggested that Mo8C12 nanoparticles are better catalysts, considering their high stability and moderate chemical activity in comparison to that of bulk Mo (too low) and Mo2C (too high).11 Similar conclusions were drawn for Ti carbides.11 Some transition metal catalysts with a carbon-to-metal ratio (CMR) below 1 easily break the C–O bond leading to CO formation.6,7 An increase in carbon content may enhance catalyst stability, it also leads to a decrease of the chemical activity, for instance observed for TiC and MoC, due to the ligand effect expressed in a downshift of the metal d-band center affecting the charge transfer from the metal to CO2 and, thus, also adsorption energy.7,11–13 Accordingly, the activity of a metal carbide catalyst towards CO2 dissociation can be tuned by varying the CMR of the catalyst.14
TMC catalytic activity can also be altered by the deposition of other metals on the TMC surface.5,7,10,14 Cu-promoted carbides exhibit enhanced selectivity towards methanol formation in comparison to both bare copper and bare TMC surfaces, like TaC, SiC, TiC, Mo2C, and MoC.5,7,14 It is also pointed out that the Cu–TMCs interface plays a crucial role. The hydrogenation of the reaction intermediates, such as HCO and H2CO, is energetically more favorable on the Cu sites, making small Cu clusters more suitable catalysts for methanol formation.5,14 Cu4/TiC has an even higher CO2 adsorption energy, and therefore methanol formation rate, than the commercial Cu/ZnO catalyst.5,15,16 However, most Cu/TMC catalyzed reactions proceed via CO formation and its subsequent hydrogenation, rather than via direct CO2 hydrogenation and a formate intermediate, as is the widely accepted reaction pathway over Cu catalysts.2 The high selectivity towards CO results in an elevated CO yield at the expense of methanol production. Therefore, it is important to understand what the direct influence of carbon is on the deposited clusters and how it influences CO2 activation.
The fine details of a reaction proceeding on the active sites can be studied at the molecular level using isolated clusters. Mass-selective gas-phase spectroscopy allows studying the interaction of CO2 with clusters of well-defined elemental composition. For example, photoelectron and IR spectroscopy have been used to study the interaction between CO2 and metal anions.17–27 This interaction can be characterized by four binding motifs: bidentate (η2), metal formate, oxalate, and dissociative adsorption. The variety in CO2 adsorption motifs is related to the difference in the degree of electron transfer from the metal center into the antibonding 2πu LUMO of CO2.28 For Cu− and other coinage metal ions in particular, it was found that CO2 binds in a formate-like fashion, where the metal ion replaces the hydrogen.17,19 The reaction of CO2 with isolated anionic metal clusters was also studied with flow tube reactor mass spectrometry,29,30 and IR spectroscopy.31,32 The nature of CO2 adsorption on these clusters appears to be size-dependent, for example on Ptn− clusters, where Pt4− shows CO2 activation, whereas Pt5−, Pt6− and Pt7− show dissociation.31 Another example is Con−, where the Co− ion binds two CO2 molecules in a bidentate configuration,27 and clusters with n > 7 dissociate CO2.32
In this work, we investigate how the carbon doping of anionic Cu clusters will influence the degree of CO2 activation, where the anionic charge state was chosen to facilitate electron transfer to the CO2 molecule. Anionic copper clusters containing 5–10 Cu atoms were doped with one or two C atoms and reacted with CO2. The formed [CunCm·CO2]− species, with n = 3–10 and m = 1–2, were studied by mass-selective IR spectroscopy. The obtained spectra are compared to reference IR spectra of the clusters reacted with CO and with DFT calculated spectra.
Methods
The experiment is shown schematically in Fig. 1. Carbon-doped anionic copper clusters CunCm− are produced in a Smalley-type laser ablation source.33 An isotopically enriched Cu-65 (99.9%) target is ablated by the focused second harmonic of a Nd:YAG laser (532 nm, 14 mJ) in the presence of a He carrier gas that is injected by a pulsed valve (General Valve series 9, stagnation pressure 6 bar). Carbon-doped copper clusters were formed due to the presence of trace carbon in the carrier gas or residual pump oil vapour. The formed clusters are reacted with CO2 or CO by injecting the pure gases through a second pulsed valve with a 1 bar stagnation pressure into an extension of the clustering channel. The CunCm− clusters react with CO2 or CO at room temperature in a reaction channel, forming [CunCm·CO2]− or [CunCm·CO]− complexes with n = 3–10, and m = 1–2. The bracket notation implies that no prior knowledge of the adsorption form of CO or CO2 is assumed. The gas mixture of helium and clusters is expanded into vacuum, forming a molecular beam that is collimated by a 2 mm diameter skimmer and an 8 × 2 mm slit aperture. The ions are then irradiated by IR laser light, and subsequently pulse-extracted by high voltage plates into an orthogonal time-of-flight mass spectrometer (TOF-MS), where they are mass separated and detected by a multichannel plate (MCP) detector. For the spectroscopic experiments, the molecular beam overlaps between the extraction plates at a 35° angle with the IR laser beam of the free-electron laser for intracavity experiments (FELICE).34 IR light is produced in the form of macropulses, consisting of an approximately 10 μs pulse train of transform-limited ps duration micropulses, and is characterized by a spectral bandwidth of ∼0.7% of the central frequency. The frequency range covered in this work is 320–2120 cm−1, with typical macropulse energies of 0.06–0.8 J, and fluences of 0.7–6.1 J cm−2. The experiment is operated at twice the IR laser frequency, and mass spectra with and without irradiation are recorded to correct for the fluctuations during cluster production. IRMPD spectra are obtained by calculating the depletion yield Y(ν) at frequency ν, defined as:where Ion(ν) is the ion intensity of the irradiated complex, Ioff the ion intensity without irradiation, and P the macropulse energy. Spectra displaying the depletion Ion(ν)/Ioff can be found in Fig. S1† of the ESI.
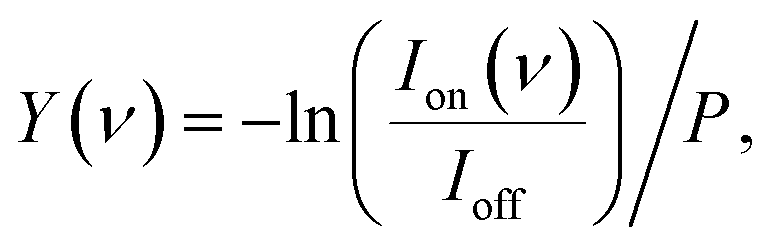
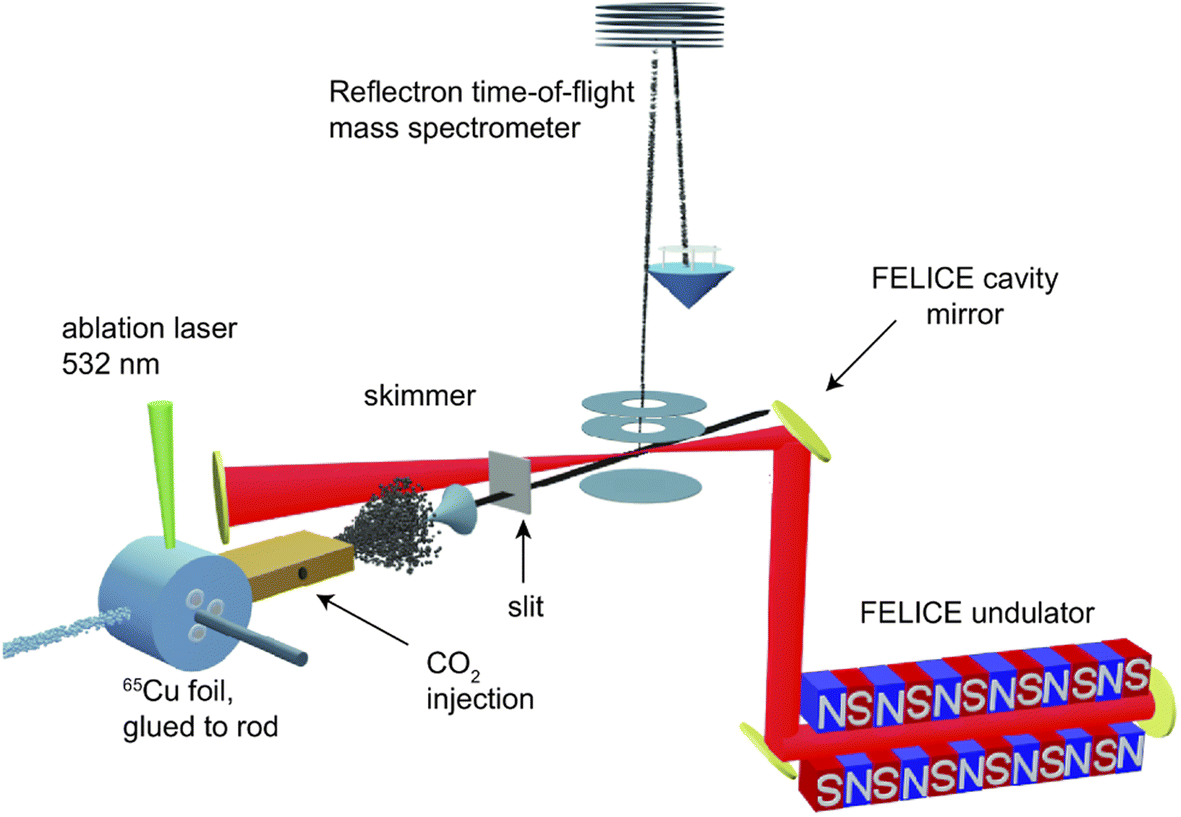 | ||
| Fig. 1 Schematic of experimental setup. | ||
For the structural assignment of selected spectra (for Cu6C− and Cu7C−), DFT calculations of different isomers were done using the Q-Chem 5.3 program package.35 Stable structures were identified using the TPSSh/def2-TZVP + D3 level of theory,36 as described in the ESI.† A detailed description of the search procedure is given in the ESI.† This combination of functional, basis set and dispersion correction was chosen to be able to directly compare the results with our previous work on the adsorption of CO2 on cationic Cu clusters,37 where adsorption was limited to physisorption. The accuracy of this method was carefully evaluated compared to a CCSD(T)/def2-QZVPPD benchmark.37 In other studies it has been found that the precise mode of physisorption found in these calculations can markedly be influenced by the choice of dispersion correction.38 However, since the interaction for the species under study here is significantly stronger, as seen by the activated and dissociated products discussed below, we believe that these influences are not decisive here. This is confirmed by further DFT based computations with D3 or D4 and without dispersion correction (Table S1 in the ESI†).
Only the lowest spin multiplicities were considered in the calculations, so doublet for Cu6C−, and singlet for Cu7C−. Harmonic vibrational frequencies of these structures are convoluted with 20 cm−1 Gaussian line shape function and compared to the experimental data for final assignment. No frequency scaling has been applied for the comparison.
Results and discussion
To understand how CO2 binds to anionic carbon-doped copper clusters, IRMPD spectra of products formed upon reacting CunCm− (n = 3–10) with different carbon loading m = 1–2 are recorded. A mass spectrum of all species formed is shown in Fig. 2. From this figure, one can see that for each size n, a distribution of CunCm− clusters is formed, that for larger clusters (n ≥ 9) is essentially limited to m = 0–3. Pure Cun− clusters are lower in intensity than the carbon doped clusters, which we attribute to a more facile formation of a CuC− seed as a nucleation core than Cu2−.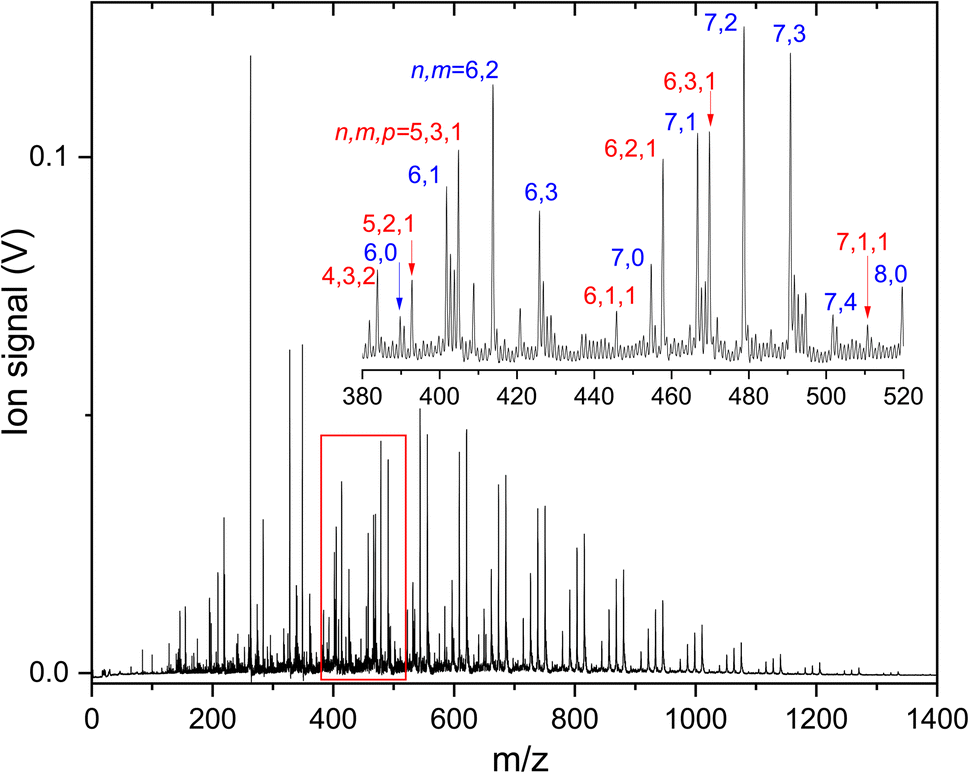 | ||
| Fig. 2 Mass spectrum of the products formed when reacting anionic CunCm− clusters with CO2. Mass peaks in the inset are labeled by (n and m) or (n, m, and p) for CunCm(CO2)p−. | ||
In the inset, a close-up of the mass spectrum in the region close to Cu6− and Cu7− is shown, with individual mass peaks corresponding to CunCm(CO2)p− labeled by (n and m) or (n, m and p). It can be seen that the pure Cu clusters are produced in this mass spectral region too, albeit at lower intensity than the CunC−, CunC2−, and CunC3− signals. Unfortunately, CO2 adsorption on the pure Cun− clusters is negligible, precluding spectroscopic characterization.
CO2 activation by CunC−
The IRMPD spectra of low-carbon loading products [CunC·CO2]− (n = 6–10) are shown in Fig. 3 (panels a–e). The complexes with n = 7–9 show only one main band around 2000 cm−1, which is fairly close to the frequency of the free C–O stretch vibration at 2143 cm−1.39 We can thus speculate that these cluster sizes adsorb CO2 dissociatively, leading to the formation of CO. In contrast, the spectra of complexes with n = 6 and 10 are dominated by two bands around 720 and 1630 cm−1, with a weaker band visible around 1100 cm−1. Earlier, experimental IR studies of the CO2− radical embedded in alkali halide matrices have revealed a characteristic band at 1671 cm−1.40 The presence of a band in this region in the IRMPD spectra suggests that CunC− clusters with n = 6 and 10 induce a distortion of CO2, activating it. Bands at similar frequencies were observed when the Pt4− ion was reacted with CO2, which was also concluded to lead to activation.31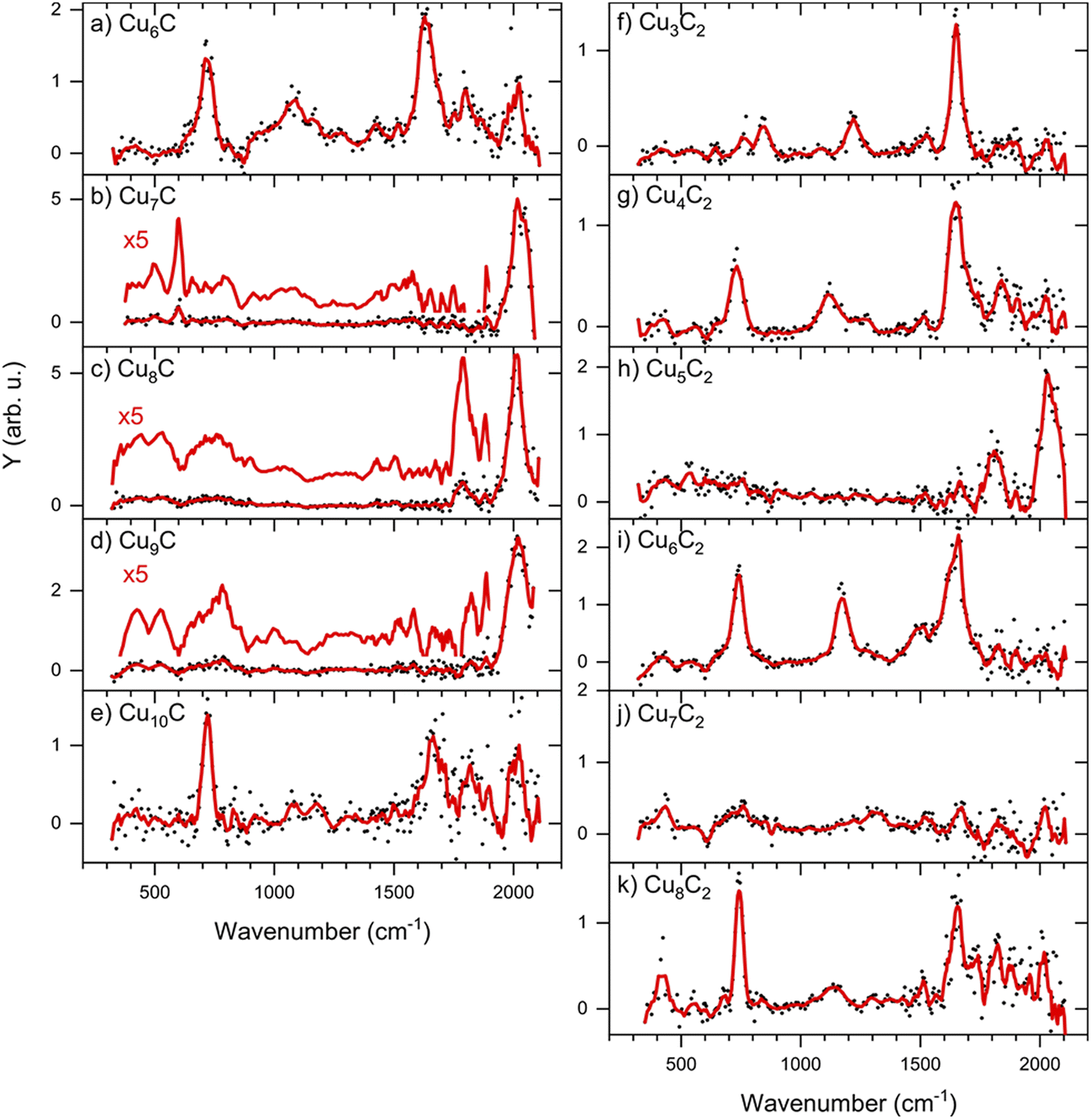 | ||
| Fig. 3 IRMPD spectra of [CunC·CO2]− (left) and [CunC2·CO2]− (right). The red lines are five-point adjacent-averages of the solid dots. | ||
From here, we focus on the species [CunC·CO2]− with n = 6 and 7 since their spectra are representative of two modes of adsorption, activated and dissociated. The suspicion that CO2 adsorbs dissociatively on CCu7− can be verified by reference spectra of carbon monoxide (CO) adsorbed to Cu7C−, which is compared to the IRMPD spectrum of [Cu7C·CO2]− in Fig. 4b (second panel from the top). For this, we reacted the clusters with CO and recorded IRMPD spectra in the 650–2100 cm−1 range for the [CunC·CO]− (n = 4–10) species formed. The resulting spectra for all measured cluster sizes are dominated by a strong band around 2020 cm−1, characteristic for the C–O stretch. This band shows an excellent agreement with the bands observed when Cu7C− is reacted with CO2 (Fig. 4b, top panel), strengthening the hypothesis that CO2 adsorbs dissociatively on CunC− (n = 7–9). In contrast, the mismatch between spectra for [Cu6C·CO2]− and [Cu6C·CO]− (Fig. 4a) suggests that CO2 adsorbs predominantly molecularly on Cu6C− and, by extension, also on Cu10C−.
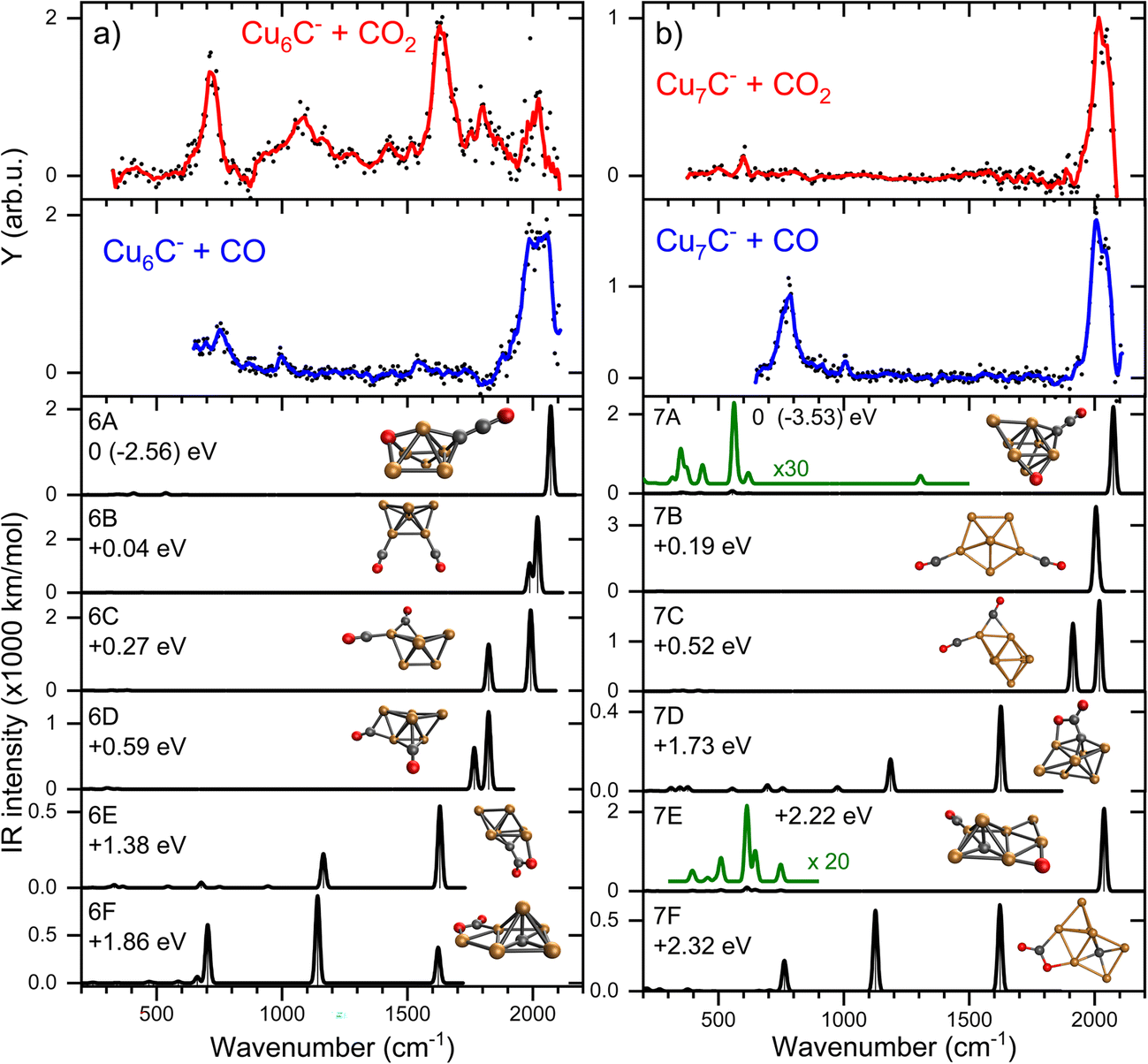 | ||
| Fig. 4 IRMPD spectra of: (a) Cu6C−, and (b) Cu7C−, reacted with CO2 (red) and CO (blue), and calculated spectra (black) of the lowest energy structure. All calculated spectra are convoluted with a 20 cm−1 FWHM Gaussian line shape function, and are accompanied by the geometrical structure (Cu, C and O atoms represented by orange, black and red spheres) and relative energy. The green curves are vertical zooms with the multiplication factors indicated. The CO2 binding energy is given for the lowest energy structure, relative energies for other isomers. | ||
DFT calculations were performed to further interpret these observations. For this, we first searched for bare CunCm− clusters. In our global optimization routine, we have identified the lowest energy structures shown in Fig. 5. The lowest energy structures found are not unlike those predicted for the pure clusters. Cu6C− is similar to the predicted Cu6− octahedron,41 with the C substituting one of the Cu atoms in the octahedron, and the sixth Cu atom bound to one of the Cu–Cu edges. Cu7C− looks like the boat structure proposed for Cu6−,42 with a C bound on a hollow site, and the last Cu atom bound both to the Cu–Cu edge and to the C atom. Interestingly, the structures for Cu5C2− and Cu6C2− are planar and are similar to the structure predicted for pure Cu5−, a 2D trapezoid.41,42 In both clusters, the C atoms are bound together in a C2 unit that forms one corner of a planar hexagon for Cu6C2−, with one Cu atom missing for Cu5C2−.
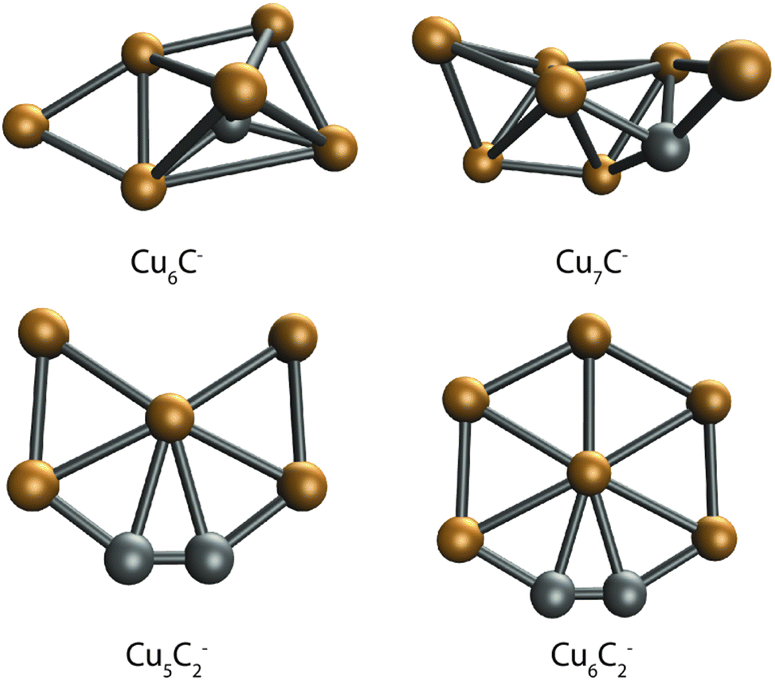 | ||
| Fig. 5 Lowest energy structures of the single C-doped clusters Cu6C− and Cu7C−, and double C-doped clusters Cu5C2− and Cu6C2−. | ||
Using the structures found for the bare clusters, a search was done for structures of the reaction product of Cu6C− and Cu7C−. An extensive search has identified over sixty stable structures for each species. Many of these structures have a similar binding motif of C and O atoms to the cluster leading to only minor differences in IR spectra, which are indistinguishable in the currently applied experimental method. Therefore, the structures were grouped, first using their spectral properties only. Visual inspection of all structures per group allowed to conclude that they all shared similar binding motifs. For the CO2 reaction products with Cu6C−, sixteen unique spectral patterns were identified, for Cu7C− only nine. The spectra of the lowest energy representative from each group are compared to the experimental data in Fig. S2.† A selection of the most promising candidates is shown in Fig. 4 (black traces).
In the search, structures are found both with dissociated and molecular CO2, where structures with CO2 dissociated are generally lowest in energy. The spectra of structures with dissociated CO2 are dominated by high-frequency bands originating from the C–O stretch vibration(s), with frequencies ranging from 1800–2100 cm−1, depending on the binding site of the CO. Spectra of clusters with molecularly bound CO2 have multiple bands typically at the lower frequencies associated with the CO2 bending mode, and the C–Obound and C–Ofree stretch.
For both n = 6 and 7, the largest number of structures found is with CO2 dissociatively adsorbed, where CO is bound in an on-top configuration (μ1) to a Cu atom, and the O eliminated from CO2 bound to another Cu; an example is structure 7E. However, this binding motif is not the energetically most favorable lying at least 1.78 (2.19) eV higher in energy for n = 6 (7) than the lowest energy structure. We attribute this simply to a computational sampling effect, where the probability to find a Cu atom in a Cu6C− cluster is six times higher than finding a C atom.
For n = 6, the spectra of structures from this most populated group are very similar to that of structure 6A, the lowest energy structure, whereas this has a different structural motif. In structure 6A, the cluster structure contains a Cu5 square pyramid, with the sixth Cu bridge-bound (μ2) to one of the edges, and the C bound to a hollow (μ3) Cu3 site. Of the dissociated CO2, the CO is μ1-bound to the cluster’s C atom, forming a linear C–C–O moiety, and the O to a hollow Cu site. The spectrum has its main band, the CO stretch vibration, close to 2100 cm−1, or blue-shifted from the observed band frequency by about 60 cm−1. The same motif of CO binding to the lone C is found for the lowest energy group for n = 7 (e.g. structure 7A). 7A is a Cu6 octahedron capped by the seventh Cu and the C, both in μ3 configuration. The CO2 is dissociated with the single O capping a third octahedron plane, and the CO bound to the C dopant.
For both n = 6 and 7 the second group (e.g. 6B and 7B) is formed by structures where CO2 dissociation leads to an O atom migrating to the C dopant atom, forming two CO molecules that are μ1-bound to a Cu. For 6B this is in the form of a Cu6 boat structure with the CO molecules attached to the bow and the stern, while for 7B it is a Cu7 pentagonal bipyramid. The frequencies of both CO stretch vibrations are close to each other resulting in one merged vibrational band, which is found at frequencies of about 2000 cm−1, slightly lower than that for the C–C–O group in 6A and 7A. Group 3 (e.g. 6C/7C) is characterized by two CO molecules, with one μ1- and the other μ2-bound. In 7C, the same capped octahedron is recognized, with the bridge-bound CO close to the on-top CO. In this case, two distinct C–O stretch bands are seen, one around 2000 cm−1 for the μ1 bound CO, and one around 1900 cm−1 for the μ2 bound CO. For n = 6, group 4 has CO molecules bound in μ2 and μ3 configurations, with again clearly two distinct bands, now at 1800 and 1900 cm−1, consistent with a weakening of the C–O bond when it coordinates to multiple Cu atoms. An interesting motif found is the linking of two CO molecules resulting in an O–C–C–O chain. However, for both cluster sizes, this last group is lowly populated, relatively high in energy, and presents no match for the experimental spectrum.
Structures with molecularly bound CO2 are typically much higher in energy (>1.4 eV) for both n = 6 and 7. The structures found are always di-σ-bound via the C and the O. Formation of a C–C bond is favorable resulting in the preferential binding of the CO2 carbon atom to the cluster’s carbon atom, with one of the O atoms binding to a Cu atom. The lowest energy structures displaying this binding motif are 6E and 7D.
Structures are assigned based on the comparison between calculated and experimental spectra. The IRMPD spectrum of [Cu6C·CO2]− shows two major bands at 718 and 1630 cm−1 with weaker bands at 1094, 1426, 1802, and 2016 cm−1. This number of bands cannot be explained by a single structure. The major band at 718 cm−1 agrees best with the CO2 bending mode predicted at 705 cm−1 for structure 6F, where CO2 is intact, but bent as a result of charge transfer. The CO2 molecule is bound with a C and the O to two neighboring Cu atoms in a bidentate bridging configuration. Two more intense bands are predicted at 1141 and 1620 cm−1 offering a good match for the experimental bands at 1094 and 1630 cm−1. These correspond to the C–O stretching vibrations involving the bound and free O atoms, respectively. Although the band frequencies of 6F match the experiment very well, the relative intensities are less convincing. The experimental band at 718 cm−1 has the highest intensity, even though it does not dominate any of the calculated spectra. We do not have a good explanation for this observation; we can speculate that it is related to the excitation mechanism in IRMPD spectroscopy, which may cause discrepancies with calculated intensities.
The search for low energy structures for Cu6C− yielded a Cu5 square pyramid structure shown in Fig. 5, with the C atom fourfold coordinated to the base, and an additional Cu atom μ2-bound on one of the edges. In structure 6F, this structure is retained, making this a likely entrance complex. Despite the higher energy of structure 6F (+1.86 eV relative to the lowest energy structure) it is the easiest formed. The formation of other more stable isomers necessarily proceeds via either C–O bond cleavage or structural rearrangement of the cluster. Both processes require additional energy to overcome a barrier associated with the transition state.
The depletion spectrum of [Cu6C·CO2]− (Fig. S1†) does not rule out a mixture of different isomers present in the molecular beam, since none of the bands reach 100% depletion. The enhanced intensity of the 1630 cm−1 experimental band could then potentially be explained by the presence of an isomer like 6E with predicted bands at 1163 and 1629 cm−1. Structure 6E appears also relatively easily formed as an entrance complex, where CO2 also binds in a bidentate bridging configuration, now with the C to the cluster’s carbon atom. However, the bare cluster’s bridging Cu is now bound on a hollow site.
The dominant experimental bands can thus be explained by molecularly adsorbed CO2, but minor bands above 1750 cm−1 more likely originate from the carbonyl C–O stretch, resulting from CO2 dissociation. The band at 1802 cm−1 might be due to structure 6C’s 1825 cm−1 predicted mode but could also be assigned to the doublet from structure 6D at 1764 and 1825 cm−1, which could have merged into one band due to band broadening. The 2016 cm−1 band could be the second band of structure 6C at 1990 cm−1 or 6A and 6B’s bands at 2069 and 2018 cm−1, respectively. The offset for 6A makes 6B, 6C and 6D the more likely candidates to explain the high-frequency bands.
All in all, it can be concluded that CO2 binds mostly molecularly to Cu6C−, but in an activated form via the C atom either to C or to Cu atoms. Dissociation may occur, but it is certainly not the dominant motif, which is underlined by the significantly higher predicted IR intensities of the carbonyl C–O stretch modes.
This dominance of molecular binding is in sharp contrast to what happens after adsorption of CO2 on Cu7C−. The IRMPD spectrum of [Cu7C·CO2]− has one dominant band at 2033 cm−1 and a low-intensity band at 598 cm−1. From a comparison with low-energy isomers, it is quite clear that CO2 is dissociated since no bands associated with activated complexes (7D and 7F) are present in the experimental spectrum. Structure 7C has two CO binding motifs, and its spectrum has separate bands at 1915 (μ2, Cu–Cu) and 2017 cm−1 (μ1, Cu). Because this does not match the experimental spectrum, we must look for structures with only μ1 binding. Spectra calculated for such structures (7A, 7B, and 7E) are dominated by one band, depending on the adsorption site of CO, at 2078 (μ1 to C), 2017 (twice μ1 to Cu) and 2038 cm−1 (μ1 to Cu), respectively. Each of them could explain the main experimental band at 2033 cm−1. The low-intensity band at 598 cm−1 could be due to the weak bands around 558 and 600 cm−1 for structures 7A and 7E, respectively. These bands are more than twenty times lower in intensity than the C–O stretch. We therefore assign the spectrum to either 7A or 7E, corresponding to structures where the CO formed from dissociating CO2 is μ1-bound either to the cluster’s C or Cu atom, and O bound separately to the cluster. Both structures appear not easily formed, because the lowest energy structure for Cu7C− (Fig. 5) requires substantial re-arrangement.
CO2 activation by CunC2−
If CO2 is reacted with Cu clusters containing a second carbon, the picture changes. The spectra for the [CunC2·CO2]− (n = 3–8) species are presented in Fig. 3 (right panels). Due to a different production efficiency for clusters with two C atoms, a slightly different range of n is presented. In contrast to the CunC− species, where only [Cu6C·CO2]− and [Cu10C·CO2]− show molecular adsorption, all spectra except that for [Cu5C2·CO2]− show bands around 740, 1150, and 1650 cm−1 indicating activated, but molecular adsorption of CO2. For all these species, bands above 1800 cm−1, attributed to dissociative CO2 adsorption, have disappeared, suggesting that only molecularly bound CO2 complexes are left. Only the spectrum of [Cu5C2·CO2]− shows an intense band at 2034 cm−1, suggesting dissociative adsorption. From this, it can be concluded that, overall, the addition of the second C atom reduces the cluster reactivity.Again, we selected two cluster sizes for DFT calculations, [Cu5C2·CO2]− and [Cu6C2·CO2]−. From the glancing overview in Fig. 3 they are representative for dissociative and molecular binding of CO2. The comparison of the spectra for these structures with spectra of the clusters reacted with CO confirms this conclusion, because the 2034 cm−1 band for [Cu5C2·CO2]− perfectly matches that observed for [Cu5C2·CO]−. In contrast, the spectrum for [Cu6C2·CO]− does not show any counterpart for the bands dominating the spectrum of [Cu6C2·CO2]−.
The calculated structures were again categorized following their spectral and adsorption motifs, as illustrated in Fig. 6. The full list of the lowest energy structures of the identified groups can be found in Fig. S3.† Interestingly, for higher carbon loading, the most common motif found is still the dissociation of CO2 to CO and O (5C and 6.2C), both bound to Cu atoms. The lowest energy structures 5A and 6.2A are formed by the adsorption of CO to one of the cluster’s C atoms and O to the other, forming a C–C–O and a CO moiety. These structures are comparable to the CO motif of Cu6C− and Cu7C−, suggesting it is overall more favorable for CO2 to dissociate and bind to the C atoms of the cluster, rather than to Cu.
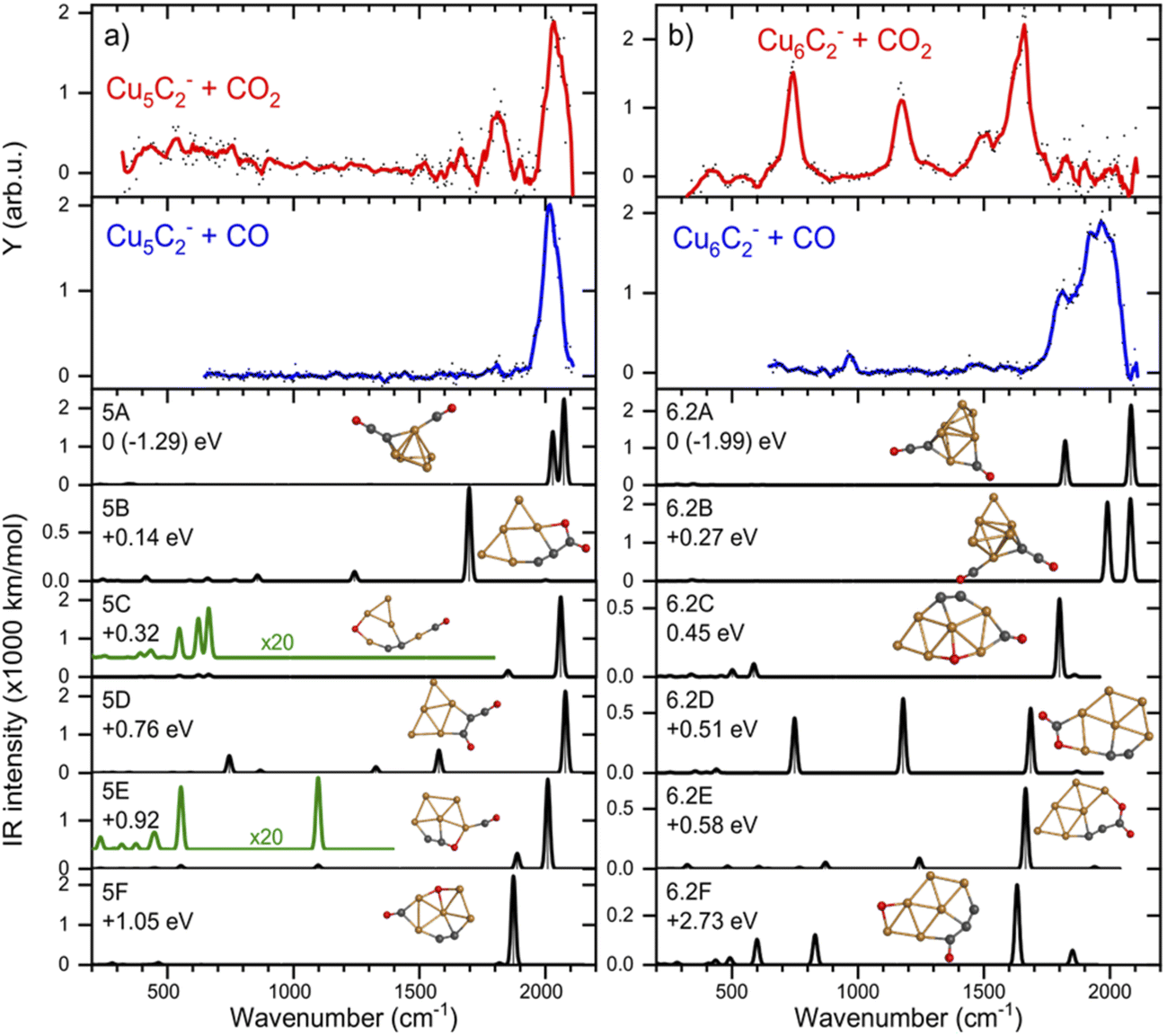 | ||
| Fig. 6 IRMPD spectra of (a) Cu5C2− and (b) Cu6C2− reacted with CO2 (red) and CO (blue), and calculated spectra (black) of the lowest energy structure. For details, see caption Fig. 4. | ||
The experimental spectrum of [Cu5C2·CO2]− is dominated by the strong band at 2035 cm−1 and has two more bands at 1667 and 1810 cm−1. The maximum depletion observed (∼45%) allows room for assignment to multiple isomers. All structures shown could potentially be present in the molecular beam since they all have bands that might overlap with the experimentally detected. The experimental band at 2035 cm−1 could be explained either by structure 5A with two close by bands predicted at 2077 and 2027 cm−1, or by structures 5C, 5D, and 5E with bands at 2063, 2078, and 2012 cm−1, respectively. The second band, at 1810 cm−1, could be explained by the main band of structure 5F at 1865 cm−1, or by the minor bands predicted for structures 5C and 5E. The only serious contender for the weakest band observed at 1667 cm−1 is structure 5B with the band at 1698 cm−1. All except for 5B, have CO2 adsorbed dissociatively on the cluster. 5B has molecularly adsorbed CO2 bound with the one of the C atoms attached to one of the cluster’s C atoms, and the O to a Cu.
If we compare the structures shown in Fig. 6a to the lowest energy structure for Cu5C2−, predicted to have a 2D, wheel-like structure, it appears that all structures apart from 5A could be formed without all too large structural re-arrangements of the original cluster: all retain a planar structure with the two C atoms of the bare clusters close together. Only the formation of 5A upon CO2 adsorption requires a major structural re-arrangement.
In contrast to the spectrum of [Cu5C2·CO2]−, the spectrum of [Cu6C2·CO2]− does not exhibit any bands above 1780 cm−1, which is indicative that CO2 is adsorbed molecularly. Therefore, structures 6.2A, 6.2B and 6.2C can be ruled out since they all have high-intensity bands at 1800 cm−1 or above. Three of the four experimental bands (742, 1173, and 1648 cm−1) could quite well be explained by isomer 6.2D with bands at 748, 1179, and 1685 cm−1. The band at 1648 cm−1 could also be explained by the 1648 cm−1 band of structure 6.2E or by structure 6.2F’s 1634 cm−1 band, but these structures lack intense bands at lower frequencies. Only the experimental band at 1484 cm−1, a shoulder of the 1648 cm−1 band, cannot be explained by isomer 6.2D. Interestingly, it appears quite similar to the weaker band at 1426 cm−1 in the [Cu6C·CO2]− spectrum, which may have a similar origin. Another parallel drawn from the spectrum of [Cu6C·CO2]− is that the relative intensities of the middle band are not reflecting the calculated ratios.
Reactive potential energy surface
It is now of interest to understand what factors could determine dissociation or lack thereof. For this, we attempted to calculate reaction pathways for two representative clusters. The complexity associated with reconstructing the reactive pathway of CO2 adsorption and subsequent activation over carbon-doped Cu clusters is enormous. First, as experimental evidence for the structures of the bare clusters is unknown, the starting point is ill-defined. Nevertheless, using the minima found in Fig. 5, we calculated a reaction pathway for CO2 dissociation. It is clear that the structural phase-space for CO2 initial adsorption is large, and the pathways to dissociation are plenty. We therefore limit ourselves to potential pathways that could illuminate whether there could exist a difference in barrier for even or odd numbers of Cu atoms.In the top panel of Fig. 7, the adsorption and subsequent dissociation of CO2 over Cu6C− is shown. Adsorption leads to the formation of structure 6F, already thought to be an entrance complex, with an adsorption energy of 0.73 eV. The CO2 is adsorbed via the C to one of the Cu atoms forming the base of the octahedron, and the O atom to the capping extra Cu atom. From here, CO is abstracted towards the base Cu atom, after which it is bound to the next Cu–Cu bridge, overall gaining little energy with respect to the entrance complex. The transition state associated with abstracting the CO is relatively high at 1.18 eV above the reactants. Such a barrier is insurmountable, certainly under the room-temperature near-thermal conditions of the reaction channel used here.
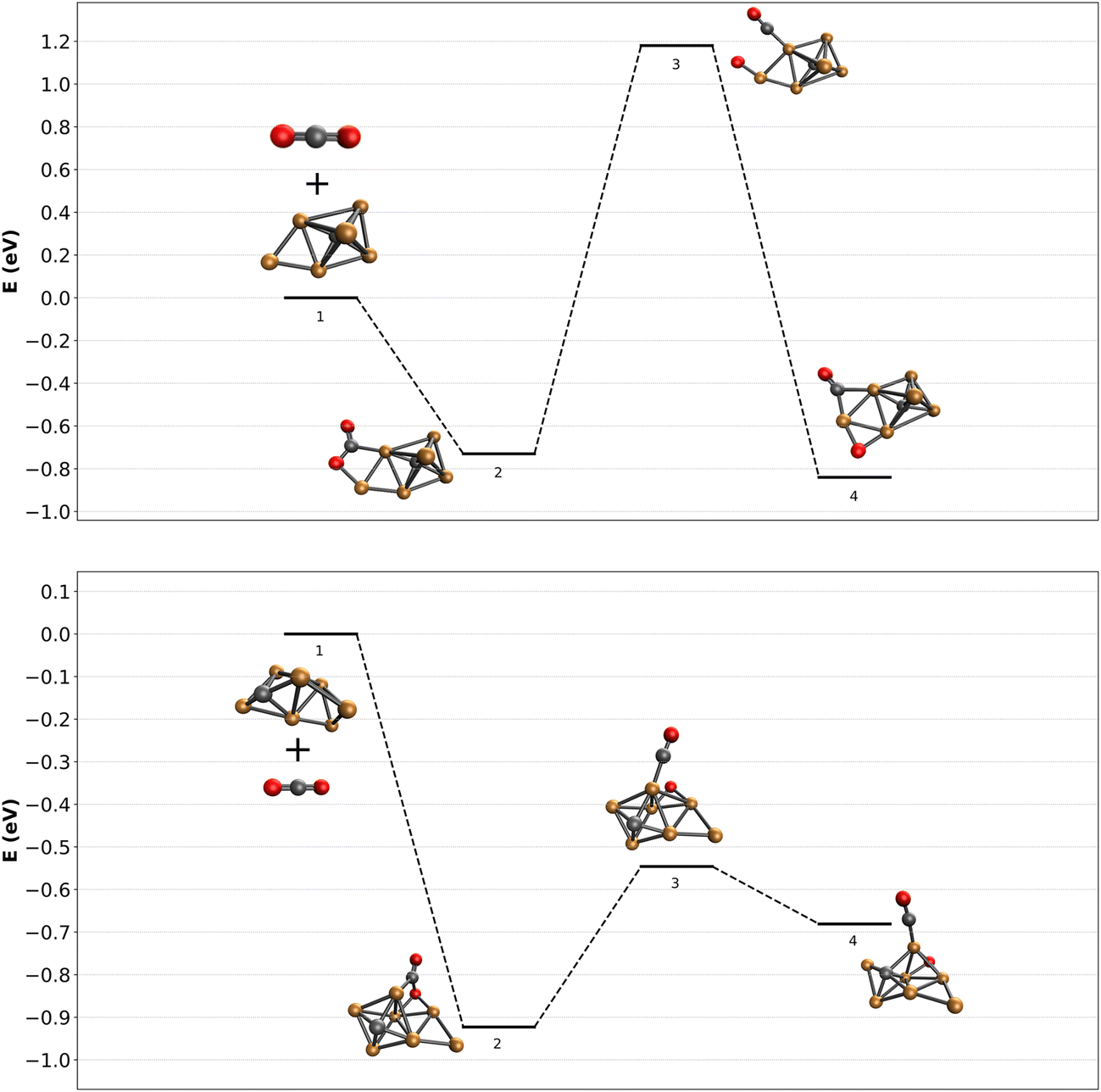 | ||
| Fig. 7 Reactive potential energy surface for CO2 dissociation over the lowest energy structures found for Cu6C− and Cu7C−, calculated at the TPSSh/def2-TZVPD level of theory. Energies (in eV) are given with respect to the bare clusters and free CO2. | ||
The bottom panel of Fig. 7 shows the same reaction path, now calculated for CO2 adsorbing onto Cu7C−. The entrance complex found is much higher in energy than anything shown in Fig. 4b, at about 2.5 eV higher than structure 7A, the lowest energy structure. The cluster is an octahedral Cu5C, with the sixth Cu (denoted Cu(6)) bridge-bound to a Cu–Cu edge, and the seventh to the newly formed Cu–Cu(6) edge. CO2 is adsorbed via its C atom to an octahedron Cu atom, and via an O atom to the other Cu–Cu(6) edge. CO abstraction then leads to an energetically still not very favorable structure with the CO bound on top of a Cu atom, and the O atom on a hollow Cu–Cu–Cu site. The initial adsorption energy of CO2 onto Cu7C− is with 0.93 eV not much different from that of Cu6C−. In contrast, the dissociation reaction over Cu7C− is much more facile than over Cu6C−, and with an energy barrier 0.55 eV lower than the energy of the reactants, this dissociation reaction is well possible.
These two reaction paths are by no means a complete description of the reactions taking place. However, they do allow to draw initial conclusions. (1) The observation of dominant molecular adsorption in the experimental spectrum of [Cu6C·CO2]− is consistent with the high barrier calculated from structure 6F in Fig. 7; (2) finding a dissociation barrier below the energy of the reactants for Cu7C− demonstrates that dissociation should occur, also consistent with the observed spectrum. It may not be this pathway, as lower barrier pathways may exist, but if an exothermic reaction with a barrier below the reactants exists, the system should find it.
So what is now the influence of the carbon atom on the dissociation propensity? We recall that for CunC− the dominant adsorption form is dissociative and for CunC2− molecular. If extra carbon atoms have a decisive influence, one would compare the outcome of the CO2 adsorption reaction for clusters with the same number of atoms, where Cu atoms are exchanged for C atoms. For this comparison, we evaluate the spectra in Fig. 3, comparing Cu6C− with Cu5C2−, Cu7C− with Cu6C2−, and so on. If anything, one sees that this comparison gives opposite outcomes: Cu6C− molecular, Cu5C2− dissociative; Cu7C− dissociative, Cu6C2− molecular; Cu8C− dissociative, Cu7C2− inconclusive; Cu9C− dissociative, Cu8C2− molecular. Then, if we compare clusters with the same number of Cu atoms: Cu6C− and Cu6C2− molecular, Cu7C− dissociative, Cu7C2− inconclusive; Cu8C− dissociative, Cu8C2− molecular. The only trend, if one may call it this: the addition of the second carbon to the cluster inhibits the dissociative adsorption for all cluster sizes except for n = 5. We can only speculate that the addition of an extra C atom, which has the higher electron affinity of C (2.55) compared to Cu (1.90),43 leads to a decrease of the charge located on the Cu atoms thereby reducing the electron donation from the cluster into the CO2 antibonding orbital. Of course, such a hypothesis requires a more detailed investigation, including spectroscopic characterization of CO2 activation by the pure Cun− clusters that was elusive in this work.
Conclusion
IRMPD spectroscopy of the CO2 adsorbed on the anionic copper carbides reveals a size-dependent bonding nature. Single-carbon doped Cu clusters CunC− with n = 6 and 10 show predominantly activated adsorption, but dissociative adsorption of CO2 for n = 7–9. These results were verified with control experiments of CO adsorbed on the same cluster sizes and with DFT calculations. The exact position of the C and O atoms on the clusters could not be determined precisely, since the bands for specific structures are very similar in frequency. Therefore, only the main binding motifs have been analyzed. Doubly-carbon doped Cu clusters CunC2− with n = 3–8 show predominantly molecular adsorption, excepting Cu5C2− which shows dissociative adsorption of CO2. DFT calculations of the dissociation reaction of CO2 over Cu6C− and Cu7C− are consistent with the observed IR spectra, showing only activation for the former and dissociation for the latter. To understand why the addition of a second C atom apparently throttles CO2 dissociation requires further extensive computational work, evaluating reactive pathways over other cluster compositions. We speculate that the higher electron affinity of C relative to that of Cu will reduce the capacity of Cu atoms in higher C/Cu ratio clusters to donate electron density to CO2, reducing the activation capabilities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Netherlands Organization for Scientific Research (NWO) under grant no. 739.017.008 (Mat4Sus), and the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 955650. We further thank NWO for the support of the FELIX Laboratory and for CPU time at the Dutch National Supercomputers Cartesius and Snellius (2021.055). TH is grateful for the János Bolyai Research Scholarship (BO/00642/21/7) of the Hungarian Academy of Sciences. OVL acknowledges the Austrian Science Fund (FWF) for a Lise Meitner Scholarship (M3229-N). We thank Prof. László Nyulászi for fruitful discussions.Notes and references
- S. Xie, Q. Zhang, G. Liu and Y. Wang, Chem. Commun., 2016, 52, 35–59 RSC.
- M. Behrens, F. Studt, I. Kasatkin, S. Kuhl, M. Havecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS.
- M. V. Twigg and M. S. Spencer, Top. Catal., 2003, 22, 191–203 CrossRef CAS.
- H. H. Hwu and J. G. Chen, Chem. Rev., 2005, 105, 185–212 CrossRef CAS.
- S. Posada-Pérez, F. Viñes, J. A. Rodriguez and F. Illas, Top. Catal., 2015, 58, 159–173 CrossRef.
- Q. Zhang, L. Pastor-Pérez, S. Gu and T. R. Reina, Catalysts, 2020, 10, 1–23 CrossRef.
- A. L. Stottlemyer, T. G. Kelly, Q. Meng and J. G. Chen, Surf. Sci. Rep., 2012, 67, 201–232 CrossRef CAS.
- M. D. Porosoff, S. Kattel, W. Li, P. Liu and J. G. Chen, Chem. Commun., 2015, 51, 6988–6991 RSC.
- X. Liu, C. Kunkel, P. Ramírez De La Piscina, N. Homs, F. Viñ and F. Illas, ACS Catal., 2017, 7, 4323–4335 Search PubMed.
- J. A. Rodriguez, J. Evans, L. Feria, A. B. Vidal, P. Liu, K. Nakamura and F. Illas, J. Catal., 2013, 307, 162–169 CrossRef CAS.
- P. Liu and J. A. Rodriguez, J. Chem. Phys., 2004, 120, 5414–5423 CrossRef CAS.
- L. K. Ono, D. Sudfeld and B. Roldan Cuenya, Surf. Sci., 2006, 600, 5041–5050 CrossRef CAS.
- J. A. Rodriguez and F. Illas, Phys. Chem. Chem. Phys., 2012, 14, 427–438 RSC.
- S. Posada-Pérez, P. J. Ramírez, J. Evans, F. Viñes, P. Liu, F. Illas and J. A. Rodriguez, J. Am. Chem. Soc., 2016, 138, 8269–8278 CrossRef.
- A. B. Vidal, L. Feria, J. Evans, Y. Takahashi, P. Liu, K. Nakamura, F. Illas and J. A. Rodriguez, J. Phys. Chem. Lett., 2012, 3, 2275–2280 CrossRef CAS PubMed.
- J. A. Rodriguez, D. Stacchiola, S. Senanayake, P. Liu and J. Evans, ACS Catal., 2015, 5, 6696–6706 CrossRef CAS.
- X. Zhang, E. Lim, S. K. Kim and K. H. Bowen, J. Chem. Phys., 2015, 143, 174305 CrossRef.
- D. W. Arnold, S. E. Bradforth, E. H. Kim and D. M. Neumark, J. Chem. Phys., 1995, 102, 3510–3518 CrossRef CAS.
- B. J. Knurr and J. M. Weber, J. Phys. Chem. A, 2014, 118, 10246–10251 CrossRef CAS.
- B. J. Knurr and J. M. Weber, J. Phys. Chem. A, 2014, 118, 8753–8757 CrossRef CAS PubMed.
- B. J. Knurr and J. M. Weber, J. Am. Chem. Soc., 2012, 134, 18804–18808 CrossRef CAS.
- A. D. Boese, H. Schneider, A. N. Glöβ and J. M. Weber, J. Chem. Phys., 2005, 122, 154301 CrossRef.
- B. J. Knurr and J. M. Weber, J. Phys. Chem. A, 2013, 117, 10764–10771 CrossRef CAS.
- M. C. Thompson, L. G. Dodson and J. M. Weber, J. Phys. Chem. A, 2017, 121, 4132–4138 CrossRef CAS PubMed.
- M. C. Thompson, J. Ramsay and J. M. Weber, J. Phys. Chem. A, 2017, 121, 7534–7542 CrossRef CAS PubMed.
- M. C. Thompson and J. M. Weber, J. Phys. Chem. A, 2018, 122, 3772–3779 CrossRef CAS PubMed.
- B. J. Knurr and J. M. Weber, J. Phys. Chem. A, 2014, 118, 4056–4062 CrossRef CAS.
- J. M. Weber, Int. Rev. Phys. Chem., 2014, 33, 489–519 Search PubMed.
- P. A. Hintz and K. M. Ervin, J. Chem. Phys., 1995, 103, 7897–7906 CrossRef CAS.
- J. Ho, K. M. Ervin and W. C. Lineberger, J. Chem. Phys., 1990, 93, 6987–7002 CrossRef CAS.
- A. E. Green, J. Justen, W. Schöllkopf, A. S. Gentleman, A. Fielicke and S. R. Mackenzie, Angew. Chem., Int. Ed., 2018, 57, 14822–14826 CrossRef CAS.
- A. Yanagimachi, K. Koyasu, D. Y. Valdivielso, S. Gewinner, W. Schöllkopf, A. Fielicke and T. Tsukuda, J. Phys. Chem. C, 2016, 120, 14209–14215 CrossRef CAS.
- T. G. Dietz, M. A. Duncan, D. E. Powers and R. E. Smalley, J. Chem. Phys., 1981, 74, 6511–6512 CrossRef CAS.
- J. M. Bakker, V. J. F. Lapoutre, B. Redlich, J. Oomens, B. G. Sartakov, A. Fielicke, G. von Helden, G. Meijer and A. F. G. van der Meer, J. Chem. Phys., 2010, 132, 074305 CrossRef PubMed.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kuś, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C.-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T.-C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S.-P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O’Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y.-C. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z.-Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J.-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C.-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- F. Liu, E. Proynov, J.-G. Yu, T. R. Furlani and J. Kong, J. Chem. Phys., 2012, 137, 114104 CrossRef.
- O. V. Lushchikova, M. Szalay, H. Tahmasbi, L. B. F. Juurlink, J. Meyer, T. Höltzl and J. M. Bakker, Phys. Chem. Chem. Phys., 2021, 23, 26661–26673 RSC.
- J. B. A. Davis, F. Baletto and R. L. Johnston, J. Phys. Chem. A, 2015, 119, 9703–9709 CrossRef CAS PubMed.
- N. Mina-Camilde, C. I. Manzanares and J. F. Caballero, J. Chem. Educ., 1996, 73, 804 CrossRef CAS.
- K. O. Hartman and I. C. Hisatsune, J. Chem. Phys., 1966, 44, 1913–1918 CrossRef CAS.
- S. Li, M. M. G. Alemany and J. R. Chelikowsky, J. Chem. Phys., 2006, 125, 034311 CrossRef.
- K. Jug, B. Zimmermann, P. Calaminici and A. M. Köster, J. Chem. Phys., 2002, 116, 4497 CrossRef CAS.
- A. Louis Allred, J. Inorg. Nucl. Chem., 1961, 17, 215–221 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2fd00128d |
| This journal is © The Royal Society of Chemistry 2023 |