 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidative dehydrogenation of cyclohexene on atomically precise subnanometer Cu4−nPdn (0 ≤ n ≤ 4) tetramer clusters: the effect of cluster composition and support on performance†
Juraj
Jašík‡
 a,
Stanislav
Valtera‡
a,
Mykhailo
Vaidulych
a,
Muntaseer
Bunian
b,
Yu
Lei
b,
Avik
Halder
c,
Hana
Tarábková
d,
Martin
Jindra
de,
Ladislav
Kavan
d,
Otakar
Frank
d,
Stephan
Bartling
f and
Štefan
Vajda
*a
a,
Stanislav
Valtera‡
a,
Mykhailo
Vaidulych
a,
Muntaseer
Bunian
b,
Yu
Lei
b,
Avik
Halder
c,
Hana
Tarábková
d,
Martin
Jindra
de,
Ladislav
Kavan
d,
Otakar
Frank
d,
Stephan
Bartling
f and
Štefan
Vajda
*a
aDepartment of Nanocatalysis, J. Heyrovský Institute of Physical Chemistry v.v.i., Czech Academy of Sciences, Dolejškova 2155/3, 182 23 Prague 8, Czech Republic. E-mail: stefan.vajda@jh-inst.cas.cz
bDepartment of Chemical and Materials Engineering, The University of Alabama in Huntsville, Huntsville, Alabama 35899, USA
cMaterials Science Division, Argonne National Laboratory, 9600 South Cass Avenue, Lemont, Illinois 60439, USA
dDepartment of Electrochemical Materials, J. Heyrovský Institute of Physical Chemistry v.v.i., Czech Academy of Sciences, Dolejškova 2155/3, 182 23 Prague 8, Czech Republic
eDepartment of Physical Chemistry, University of Chemistry and Technology in Prague, Technická 5, 166 28 Prague, Czech Republic
fLeibniz Institute for Catalysis (LIKAT), Albert-Einstein-Strasse 29a, D-18059 Rostock, Germany
First published on 28th July 2022
Abstract
The pronounced effects of the composition of four-atom monometallic Cu and Pd and bimetallic CuPd clusters and the support on the catalytic activity and selectivity in the oxidative dehydrogenation of cyclohexene are reported. The ultra-nanocrystalline diamond supported clusters are highly active and dominantly produce benzene; some of the mixed clusters also produce cyclohexadiene, which are all clusters with a much suppressed combustion channel. The also highly active TiO2-supported tetramers solely produce benzene, without any combustion to CO2. The selectivity of the zirconia-supported mixed CuPd clusters and the monometallic Cu cluster is entirely different; though they are less active in comparison to clusters with other supports, these clusters produce significant fractions of cyclohexadiene, with their selectivity towards cyclohexadiene gradually increasing with the increasing number of copper atoms in the cluster, reaching about 50% for Cu3Pd1. The zirconia-supported copper tetramer stands out from among all the other tetramers in this reaction, with a selectivity towards cyclohexadiene of 70%, which far exceeds those of all the other cluster–support combinations. The findings from this study indicate a positive effect of copper on the stability of the mixed tetramers and potential new ways of fine-tuning catalyst performance by controlling the composition of the active site and via cluster–support interactions in complex oxidative reactions under the suppression of the undesired combustion of the feed.
Introduction
The catalytic oxidative dehydrogenation (ODH) of hydrocarbons is an energy-efficient analogy to nonoxidative dehydrogenation (DH).1,2 ODH is an exothermic reaction thanks to hydrogen being removed via water formation, in contrast to the endothermic DH route.2 ODH can be performed at lower temperatures, which not only leads to cost savings, but also prevents thermal cracking of the feedstock into lighter alkanes and coking.3,4 Catalytic dehydrogenation processes belong to the most important ones in the chemical industry for the production of olefins and aromatic intermediates for downstream processes, such as the production of fine chemicals or polymers, and there is an associated need for the identification and development of new highly active catalysts which perform at lower temperatures and with high selectivity.This study is focused on the catalytic dehydrogenation of cyclohexene, an intermediate in the dehydrogenation of cyclohexane, with cyclohexane also gaining increasing attention as a potential high density hydrogen storage medium.5 The catalytic dehydrogenation of cyclic hydrocarbons, like cyclohexane and cyclohexene, is a structure-sensitive reaction, as reported for extended crystalline surfaces,6–10 nanoparticulate catalysts and nanostructures.4,11–17 Recently, atomic size catalytic particles have also been studied for this reaction, with particles possessing a high fraction of undercoordinated atoms, extended interface in contact with the support and which are highly susceptible to support effects.18–24 In cyclohexane dehydrogenation, a cycloallylic (c-C6H9) intermediate is formed and the rate limiting step involved in benzene production is either the dehydrogenation of this intermediate to chemisorbed cyclohexadiene or the desorption of the final benzene product.9 Reactivity and adsorption studies that were carried out using all species involved in the multi-step dehydrogenation of cyclohexane, i.e. cyclohexane, cyclohexene, cyclohexadiene, and benzene, on a Pt(111) surface suggest that the other steps involved in the dehydrogenation to benzene, that is, cyclohexane conversion to cyclohexene and the dehydrogenation of cyclohexadiene to benzene, all proceed rapidly.8,9 This prompted the present study, in addition to its primary focus on the identification of highly active and selective dehydrogenation catalysts, to also explore the tunability of their selectivity towards the production of cyclohexadiene, thus the partial dehydrogenation to cyclohexadiene, a valuable industry precursor for polymer production.25 In addition to the dehydrogenation of saturated hydrocarbons, ODH is also used for the removal of unsaturated cyclic hydrocarbons which occur as by-products in the production of benzene via hydrocarbon reforming. The removal of these products is typically energy-intensive and challenging, so their conversion to benzene by efficient ODH catalysts can offer a viable alternative.26
Supported single atoms and subnanometer clusters of transition metals have proven to be promising catalysts in various reactions, including CO2 hydrogenation, the Fischer–Tropsch reaction and the hydrogenation/dehydrogenation of hydrocarbons.27–34 Thanks to the small size of the cluster, usually less than 50 atoms, quantum effects occur.35 By comparison with the bulk material, smaller clusters tend to have discrete electron levels and, as a result, they lose their metallic character and resemble isolated molecules, with their electronic (as well as geometric) structure altered by adding a single atom of the same or a different metal.36 In subnanometer clusters, most of the atoms are on the surface of the cluster. These atoms are often undercoordinated and therefore very reactive, which gives the possibility of creating new classes of catalysts with higher reaction rates and/or higher selectivity for a given product.37,38 A well-known example is gold, which is non-active in bulk, but in the form of molecular complexes, clusters or nanoparticles acts as an efficient and selective catalyst.39–44
The exact size of the cluster plays an important role. It is known that the addition or removal of even one atom can drastically change the cluster reactivity or selectivity.31 For example, Pt7 supported by Al2O3 is found to be significantly more active than the Pt8 cluster in ethylene dehydrogenation.45 Tang et al. describe a very small “window of opportunity” for CO oxidation-active gold clusters on TiO2, i.e., only clusters of six and seven gold atoms could be active in CO oxidation.46 Theoretical calculations of unsupported Cu clusters predict odd–even size dependent oscillations in the magnetic moment, detachment energy, ionization potential, and stability function due to the electron-pairing effect in terms of the HOMO–LUMO gap.47 Odd–even oscillations in the properties with the atomicity of the clusters were also theoretically predicted by Wang et al. for copper clusters supported by TiO2 (ref. 48) and experimentally demonstrated, for example in the ODH of cyclohexene,22 showing that clusters with an even number of atoms have 100% selectivity for benzene formation (except dimer) and clusters with odd numbers of copper atoms have lower selectivity.
Oxides, as well as carbon-based materials, are widely used materials for supporting metal clusters.28 The nature of the support plays an important role in determining the properties of clusters via cluster–support interactions,49 influencing the electronic structure and shape of the clusters,45 charge distribution inside the cluster and the charge transfer between the cluster and the support,50 all of which can strongly affect the catalytic properties.20,40,51–53 The choice of substrate provides one of the control knobs for tuning the performance of the catalyst.
A possibility of preparing a combination of two or more elements with atomic precision in the form of mixed clusters further expands the available materials space in the quest for new catalyst formulas, including compositions that do not form as bulk alloys.54 The aim is to fabricate a catalyst which symbiotically combines the desired properties of its individual metal components while suppressing their shortcomings in a fashion not achievable with monometallic nanoparticles alone.17,55 For example, in case of ODH, the deactivation of the platinum catalyst by the strong binding of unsaturated hydrocarbons can be prevented by the addition of gallium to the catalyst.17 An example is the mixed clusters Ag9Pt2 and Ag9Pt3, which are not poisoned by CO, in contrast to monometallic Pt10 clusters,56 or the stabilization effect of tin on platinum clusters.57
A combination of copper and palladium offers an interesting mixture recently studied for the selective oxidation reactions of cyclopentene, cyclohexane and benzene.58 Supported Pd–Cu bimetallic nanocatalysts were used for the oxygenation of cyclopentene to cyclopentanone with a selectivity over 95%.59 Palladium–copper salts are known for the Wacker oxidation of cyclohexene. In the presence of Pd(NO3)2/CuSO4, the cyclohexene conversion was 13% with 50% selectivity to cyclohexanone.60 Tierney et al. showed that separated Pd atoms on a Cu support are surrounded by Cu and are electronically identical to Cu.61 In our recent study of the same reaction of cyclohexene ODH on subnanometer copper, it was demonstrated that the rate of benzene production can be boosted by an order of magnitude by changing the support.22 Palladium atoms and dimers, as well as clusters and nanoparticles, in addition to being very potent combustion catalysts, showed high activity in dehydrogenation reactions as well, such as nanodiamond–graphene supported Pd3 exceeding the performance of Pd nanoparticles and palladium single-atoms, Pd1, in ethylbenzene dehydrogenation to styrene62 or Pd1 being six times more active than 1–2 nm Pd particles in the dehydrogenation of propane.63 Subnanometer Cu, Pd and mixed CuPd clusters were found to also be active in cyclohexane21 and propane ODH,64 respectively. The subnanometer bimetallic copper–palladium size-selected clusters supported by Al2O3 showed propylene production rates in the order Pd4 ≪ Cu3Pd < Cu4Pd, and Cu+ species and structures with high Cu–Cu and low Cu–Pd coordination numbers were identified as the likely roots of the improved catalytic performance.64
In this study, the effect of the atomic composition of pure and mixed Cu, Pd and CuPd tetramer catalysts on their performance in the ODH of cyclohexene is addressed, along with the effect of the support. To investigate the effect of the support, three types of support films were chosen: ultra-nanocrystalline diamond (UNCD),19,21 non-reducible zirconia (ZrO2)65–67 and reducible titania (TiO2); the last two are known for their strong interactions exerted on the deposited metal.49,68 It is to be noted that even the prototypical irreducible zirconia may undergo superficial reduction as well when hydrogen spill-over takes place or metal atoms are present on its surface.49,69
Experimental
Atomic layer deposition – ALD – and ellipsometry
Naturally oxidized N-type phosphorus doped silicon chips (Si[100]) of 525 μm thickness and with a ∼2 nm thick native oxide top layer produced by an ON semiconductor were cleaned by sonicating them in acetone using a Branson 2800 sonicator for 20 minutes, and the thickness of the native oxide layer was measured on a J.A. Woollam α-SE variable angle spectroscopic ellipsometer (VASE) at an incident angle of 70° over the 380–900 nm wavelength range. ALD was performed in a viscous flow, benchtop atomic layer deposition reactor (Gemstar-6, Arradiance) under low pressure (∼1 Torr). Ultra high purity nitrogen (Nexair, 99.999%) was used as the carrier gas and purified further before entering the reactor using a Supelco (Sigma-Aldrich) gas purifier. For comparison, the thickness of the oxide layers was measured once more before and after reaction on an Accurion ep4 imaging ellipsometer at an incidence angle of 50° over 364–1000 nm wavelength range. The 3.5 Å ZrO2 film was prepared using alternating exposures of tetrakis(dimethylamido)zirconium(IV) (TDMAZ) as a Zr precursor and H2O, applying 7 cycles. Each cycle comprised a pulse–purge sequence: 0.05 s pulse of TDMAZ – 12 s N2 purge – 0.1 s pulse of H2O – 12 s N2 purge. The reaction chamber temperature was set to 140 °C. Ultrananocrystaline diamond (UNCD) supports (∼250 nm thick, on a Si(100) chip) were purchased from Advanced Diamond Technologies (UNCD, 25 Aqua DoSi). The 5.4 Å TiO2 film was prepared using alternating exposures of titanium tetraisopropoxide (TTIP) as a Ti precursor and H2O, applying 18 cycles. Each cycle comprised a pulse–purge sequence: 2 s pulse of TTIP – 12 s N2 purge – 15 s pulse of H2O – 12 s N2 purge. The reaction chamber temperature was set to 140 °C.Atomic force microscopy – AFM
The ZrO2, UNCD and TiO2 films were characterized ex situ under ambient conditions by atomic force microscopy (AFM, Dimension Icon, Bruker, USA) in tapping mode. Silicon cantilever VTESPA-300 with a resonance frequency of ≈ 300 kHz, spring constant k = 42 N m−1 and nominal tip radius 5 nm (Bruker, USA) was used. Gwyddion software (v. 2.60) was utilized for AFM image data processing. The roughness parameter Ra was calculated from the AFM height images (1 × 1 μm2). Selected samples were imaged after the catalytic tests as well.Deposition of size and composition selected clusters
The synthesis of atomically precise Cu4−nPdn (0 ≤ n ≤ 4) tetramer clusters was performed using molecular beams in the gas phase, utilizing two vacuum apparatuses. Both instruments consist of three interconnected vacuum chambers differentially pumped with turbomolecular pumps backed with fore-vacuum rotary pumps. The charged clusters were produced in the first chamber by means of a magnetron-based sputter source with a liquid nitrogen cooled gas aggregation chamber of the Haberland type70 powered by an Advanced Energy model MDX 500 DC Power Supply. Argon and helium were used as the sputtering and carrier gases, respectively, with a typical total flow in the range of 350 to 500 sccm. Two cluster instruments of analogue design were used to prepare the catalysts: a 2 inch diameter copper sputter target was used in cluster source 1 to prepare the pure copper tetramer, while in cluster source 2, 1 inch diameter pure palladium and copper–palladium alloy sputtering targets were used to fabricate the pure palladium and the mixed clusters, respectively. The beam of positively charged clusters produced in the source chamber was directed onto the entrance orifice of the ion guide chamber and was collected and guided downstream into two differentially pumped chambers by a series of conical and linear octupoles. Then, the molecular beam containing a distribution of cluster sizes entered the mass spectrometer and the output of the apparatus was optimized for the desired cluster size by on-line monitoring of the mass spectrum of the clusters produced (for typical mass spectra of the produced Cu4−nPdn (0 ≤ n ≤ 4) clusters used for the deposition of clusters, see Fig. S1–S3†). Next, the desired single size monometallic or single-size and single composition bimetallic clusters were filtered out by the mass spectrometer and directed towards the support in the deposition chamber. The flux of charged clusters landing on the support was measured in real-time using a picoammeter (Keithley 6487), which was also used to bias the substrate to control the impact energy of the clusters during their landing on the support, keeping their energy below 1 eV per metal atom. Using custom homemade software, written in Python, the deposition current was converted on-line into the total charge, providing the number of clusters (and atoms) deposited, as well as the atomic monolayer equivalent surface coverage. Typical deposition currents were in the range of several nA, corresponding to a flux of clusters landing on the surface at a rate in the order of 1010 clusters per second on an area 8 mm in diameter. In the magnetron based apparatus with the 1 inch targets,70 Pdn clusters of a single size and CumPdn clusters of a single size and composition were deposited on UNCD, ZrO2 and TiO2 substrates of 19.8 mm × 21.8 mm dimensions. The mixed tetramer CumPdn was deposited on two spots, each with a diameter of 0.8 cm (corresponding to an area of 0.5 cm2 per spot), at a surface coverage of 10% of the atomic monolayer equivalent of copper, corresponding to 1.77 × 1014 metal atoms, corresponding to 28.2, 25.02 and 21.9 ng metal for CuPd3, Cu2Pd2 and Cu3Pd1, respectively. Pd4 was deposited on 1 spot with 5% of the atomic monolayer equivalent surface coverage. For Pd1, 4 spots with 1.25% of the atomic monolayer equivalent surface coverage were chosen to have the same loading of atoms as for Pd4 and to avoid agglomeration of the clusters upon deposition. The total atom loading for the Pd1 and Pd4 samples was 3.83 × 1013 atoms, which is equal to 6.8 ng of Pd metal. The palladium dimer Pd2 was deposited on 4 spots at 2.5% of the atomic monolayer equivalent surface coverage to have the same number of particles as in the monoatomic Pd1 sample containing 7.65 × 1013 Pd atoms, equal to 13.5 ng of Pd. In the second apparatus, using the magnetron source with a 2 inch target, copper tetramers on ZrO2 and UNCD substrates were deposited on two spots with a diameter of 1.2 cm (corresponding to an area of 1.13 cm2 per spot) and amount corresponding to 10% of the atomic monolayer equivalent, which is equal to 42.2 ng of Cu or 4.0 × 1014 atoms of Cu. A detailed description of the cluster synthesis process can be found elsewhere.70 After deposition, the samples were transferred under air into the test setup. For the fabrication of the titania-supported CuPd3, Cu2Pd2 and Cu3Pd1 clusters, the same loading was used as applied for the clusters on titania and UNCD.Catalyst testing
The tests were performed in a custom reaction cell made of aluminium alloy (EN AW 6061) with an internal volume of 33 cm3 under continuous flow (17.5 sccm) of a reactant mixture containing 0.29% cyclohexene and 0.29% oxygen seeded in helium at a constant pressure of 1.07 bar, with the sample situated on top of a heater plate. The mixing of reactant gases seeded in helium was realized in a custom mixer setup equipped with mass-flow controllers (Brooks SLA5850). The pressure in the reaction cell was monitored using a pressure transducer (Omega PX209) and kept at a constant pre-set pressure by a downstream mass-flow controller (Brooks SLA5850) integrated in a regulation loop pumped by a diaphragm pump (Divac 1.4HV3) and controlled by a custom homemade software written in Python. The investigated sample was heated on a boron nitride heater (Momentive Performance Materials Inc.), using a Kepco power supply, with the temperature ramp set and regulated using a LakeShore 340 controller in combination with a K-type thermocouple attached to the body of the heater plate. The walls of the reaction cell were cooled by water circulating through it at a temperature of 20 °C maintained by an external chiller (Thermo AC200). Prior the experiment, the temperature on the sample surface was calibrated against the temperature of the body of the heater using a second K-type thermocouple in contact with the surface of a blank silicon chip under the same flow of He as during catalyst testing. A mass spectrometer with an electron impact ionization source (Pfeiffer Vacuum PrismaPlus QMS 220) differentially pumped with a turbostation (Pfeiffer HiCube) was used for online analysis of the composition of gas leaving the reaction cell. The gas in between the outlet of the reaction cell and the downstream mass-flow controller was sampled into the differentially pumped mass spectrometer chamber using an electronic needle control valve (Pfeiffer EVR 116), with the flowrate controlled by a regulator (Pfeiffer RVC 300) combined with a pressure gauge (Pfeiffer PKR 261) to keep a constant pressure set to 5.0 × 10−6 mbar in the mass spectrometer chamber. The mass spectrometer chamber was pumped by a turbo-molecular pump (Pfeiffer HiCube 80 Eco), which typically reached a background pressure of about 2 × 10−8 mbar. All upstream and downstream gas lines in path from mixer MFCs to the reaction cell and downstream from the cell were heated to 70 °C.The mass spectrometer was operated in the continuous mass scanning mode (2 scans per minute) in the range from 5 to 100 m/z controlled by PV MassSpec software (Pfeiffer). Electron impact energy for the ionization was set to 70 eV. The sensitivity of the mass spectrometer for the desired molecules (cyclohexene, cyclohexadiene, benzene, CO2, CO and O2) was determined using calibrated gas mixtures (certified analytical grade mixed gas – Siad, Messer, Air Products or Linde). The uncertainty of the measured concentrations was estimated to be ∼2%.
Before the catalytic test, and with the sample in the reactor, the reaction cell and all tubings were first evacuated and flushed several times with pure helium. Then, for 0.5 hour, a constant flow of helium (5 sccm) was maintained through the reaction cell at 800 Torr and 25 °C. After 0.5 hour of flushing with pure helium, the gas was switched to the reactant mixture (maintaining the 800 Torr pressure and 17.5 sccm flowrate) and the sampling of the gas to the mass spectrometer started. The flow of the reactant mixture was kept for 6 hours at 25 °C for the sample to stabilize the background in the mass spectrometer before the start of the temperature ramp. The reactant mixture, consisting of 0.29% cyclohexene and 0.29% oxygen in helium (i.e. a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cyclohexene to oxygen molar ratio), was obtained by mixing 12.50 sccm of 4000 ppm cyclohexene in helium (Air Products) with 5.00 sccm of 1.0% oxygen diluted in helium (Messer).
:1 cyclohexene to oxygen molar ratio), was obtained by mixing 12.50 sccm of 4000 ppm cyclohexene in helium (Air Products) with 5.00 sccm of 1.0% oxygen diluted in helium (Messer).
The performance of the catalysts was tested in a temperature range from 25 °C to 400 °C. The temperature program comprised 2 ramps, first with eight increasing temperature steps from 50 °C to 400 °C in increments of 50 °C, followed by eight decreasing steps by 50 °C from 400 °C down to 50 °C and the last step to 25 °C. Each temperature set point was approached with a ramp rate of 5 °C min−1 followed by a dwell time of 20 minutes at each temperature. The steps in the second ramp were identical to those in the first ramp, only the dwell time at the highest temperature was extended from 20 minutes to 120 minutes (see Fig. S4† for the applied temperature ramps). The second ramp started 120 min after the first ramp was finished. The continuous flow of reactants was kept in between the ramps.
Evaluation of the reactivity data
The relative concentrations of the reactants and products in the reaction mixture were determined from the measured mass spectra. The peak at m/z 44 was used to quantify CO2. The quantification of the products benzene and cyclohexadiene is complicated by fact that their dominant mass peaks (m/z 78 for benzene and m/z 79 and 80 for cyclohexadiene) overlap with the peaks of the fragments of cyclohexene (Fig. S5†). To resolve the contributions of individual compounds, the experimental mass spectra were fitted by a model based on a linear combination of cyclohexene, benzene and cyclohexadiene mass peaks patterns, used as the basis sets. The patterns were obtained from the mass spectra of calibrated gas mixtures measured during the determination of the mass spectrometer sensitivity (Fig. S5c and d†). Mass peaks with m/z 67, 77, 78, 79 and 80 were considered in the model. The peak at m/z 69 is the dominant peak of cyclohexene and the peaks at m/z from 77 to 80 correspond to benzene and cyclohexadiene overlapped by cyclohexene fragments. The fitting procedure was implemented in Python using the lmfit package.71 The fitted concentrations of the products were corrected to the linear background interpolated from the data collected at 25 °C prior to the start and after the end of the temperature ramp where no reaction products were formed (see Fig. S6†). The absolute concentrations were determined using relative concentrations normalized to cyclohexene values linearly interpolated from measurements at 25 °C, i.e. prior to the start and after the end of the temperature ramp, where the absolute concentration of cyclohexene was given by the composition of the reactant mixture. The concentrations obtained for the “blank”, which contain contributions from the blank support and the exposed internal parts of the reactor, were subtracted from the data obtained from the cluster-containing sample, thus yielding the sole contribution of the clusters. The carbon-based rates of the formation of the individual products were calculated by normalizing to the total number of metal atoms in the sample and multiplying by the number of carbon atoms in the given product. The sum of the relative concentrations of the carbonaceous reactants and products obtained from the fitting of the mass spectra is shown in Fig. S6f.† The sum equals to one over the entire temperature range, indicating that no major product remained unidentified.X-ray photoelectron spectroscopy – XPS
The samples with Pd1 and Pd2 on ZrO2 before and after catalyst testing were characterized by X-ray photoelectron spectroscopy (XPS). Measurements were performed on an ESCALAB 220iXL (Thermo Fisher Scientific) with monochromated Al Kα radiation (E = 1486.6 eV). Samples were prepared on a stainless steel holder with conductive double-sided adhesive carbon tape. The electron binding energies were obtained with charge compensation using a flood electron source and referenced to the C 1s core level of carbon at 284.8 eV (C–C and C–H bonds). For analysis, the peaks were deconvoluted with Gaussian–Lorentzian curves using the software Unifit 2021.Results and discussion
The ALD prepared layers were characterized by ellipsometry. For the ZrO2 layer, the thickness was found to be 3.5 Å and for ALD TiO2, the thickness was 5.4 Å. The characterization of the support films by ellipsometry before and after the catalytic testing did not reveal changes in the thickness of the films. AFM images of the UNCD, ALD ZrO2 and ALD TiO2 films on Si substrates collected before the catalytic testing are shown in Fig. S7a–c, S8b and S9a,† respectively. The UNCD surface composed of diamond nanograins is relatively rough, with a mean roughness of Ra = 7.5 nm. The phase image of UNCD shown in Fig. S7c† reveals additional morphological details, including nanograin boundaries, which could not be well resolved by topographic imaging.72 The ZrO2 film (Fig. S8b†) is rather smooth, (Ra = 0.08 nm) and copies the SiO2/Si support morphology (Fig. S8a†). The morphology of the ZrO2 film is similar to the one published by Jewel and Mamun for an ALD ZrO2 film on a silicon substrate.73,74 The TiO2 film is smooth as well, with a mean roughness of Ra = 0.12 nm without any indication of crystallinity. Neither a change in the supports’ nanomorphology after the catalyst testing, nor the formation of cluster aggregates with dimensions over a nanometer, is evident (Fig. S7d–f, S8d, e and S9c, d†).Catalytic performance of CuPd tetramers on UNCD and ZrO2 supports
Fig. 1 summarizes, for the first (i.e. “short”) temperature ramp of the applied double-ramp, the results from the catalytic testing of the mono- and bimetallic tetramers Cu4−nPdn (0 ≤ n ≤ 4), namely, the per metal atom calculated carbon-based rates of the formation of benzene, cyclohexadiene and carbon dioxide, as well as the total rate of product formation and selectivity at 400 °C for both UNCD- and ZrO2-supported four-atom clusters. At 400 °C, all samples achieve the highest reactivity, which, however, differs by the support. The reactivity of tetramers on the UNCD support is much higher, in the order of Pd4 ≫ Cu2Pd2 > Cu1Pd3 > Cu3Pd1, with rates of 347, 35, 14 and 4 per atom per s, respectively. As far as the production of cyclohexadiene is considered, the highest rate on UNCD-supported tetramers was observed for Pd4 at the rate of 5.6 per atom per s at 400 °C, see Fig. 1c and d. The mixed clusters show rates under 1 per atom per s at 400 °C. On the zirconia support, the rate of benzene formation follows the number of Pd atoms in the tetramer in the order of Pd4 ≫ Cu1Pd3 ≈ Cu2Pd2 > Cu3Pd1 > Cu4, with values of 109, 4.0, 3.8, 2.2 and 0.8 per atom per s, respectively. In comparison with the UNCD-supported clusters, the zirconia supported clusters produce significantly more cyclohexadiene. Pd4 produces cyclohexadiene at 350 °C already at a rate of 5.3 per atom per s and reaches 7 per atom per s at 400 °C. The mixed CuPd clusters and Cu4 produce cyclohexadiene at rates of around 2 per atom per s at 400 °C. All the investigated samples exhibit very low or no production of CO2 (see Fig. 1e and f) in comparison with the C6 products. For the UNCD supported samples, CO2 production was detected only in the case of Pd4 clusters, with a rate of 0.2 per atom per s at 400 °C and a selectivity below 1% (see Fig. 1e and i). In the case of the zirconia-supported clusters, the rate of CO2 production is around 0.2 per atom per s except for the most reactive Pd4, with CO2 formation at a rate of 0.8 per atom per s. From the selectivity point of view, in the case of ZrO2, the selectivity towards CO2 is below 1% for Pd4 and does not exceed 3% for the other tetramers (see Fig. 1f and j). An interesting trend of selectivity was observed in the case of the zirconia support: with an increasing number of Cu atoms in the tetramer, the selectivity gradually shifts in favour of cyclohexadiene, reaching a benzene:cyclohexadiene ratio of 1:1 for Cu3Pd1, and for pure Cu4, the selectivity towards cyclohexadiene rises ultimately to over 2:1. A desired but unexpected result in this case is the suppressed combustion by the investigated tetramers because Pd and Pd-containing catalysts are known for their potential for the full oxidation of hydrocarbons.75
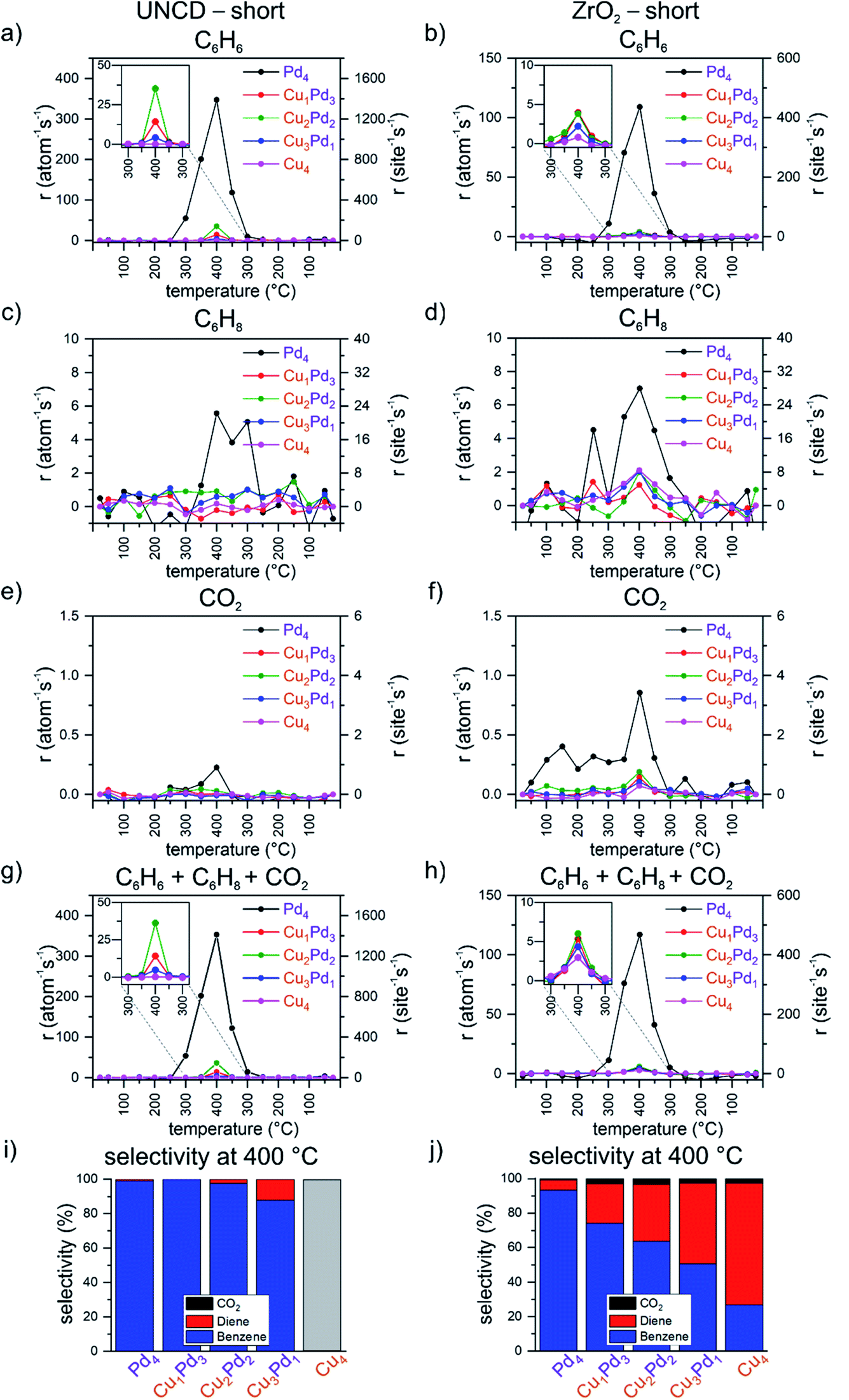 | ||
| Fig. 1 Per total atom and per site carbon-based rate of formation during the first (“short”) temperature ramp for (a) benzene on UNCD, (b) benzene on ZrO2, (c) cyclohexadiene on UNCD, (d) cyclohexadiene on ZrO2, (e) CO2 on UNCD, (f) CO2 on ZrO2; summed over products (g) on UNCD and (h) on ZrO2; carbon-based selectivity for benzene, cyclohexadiene and CO2 at 400 °C for all tetramers (i) on UNCD and (j) on ZrO2 (“short” refers to the first temperature ramp of the applied double ramp, with a dwell time of 20 minutes at 400 °C). | ||
Summing up the results shown in Fig. 1, on both supports, Pd4 is found to be the most active, with Cu4 the least active or even inactive (see Fig. 1g and h). On the UNCD support, by the sequential, atom-by-atom substitution of the Pd atoms in the palladium tetramer by Cu atoms, the change in activity does not linearly scale with the number of exchanged atoms, but shows an oscillatory behaviour instead (see the inset in Fig. 1g). No measurable catalytic activity is observed for the pure copper tetramer on the UNCD support. UNCD-supported tetramers containing two and three copper atoms also exhibit the formation of cyclohexadiene, with a selectivity of up to 10% on Cu3Pd1 (see Fig. 1i). In contrast to the UNCD support, Cu4 becomes active on zirconia. The switch to a zirconia support leads to a drop in the overall activity of the Pd4 and the mixed tetramers, accompanied by the increase of the activity of Cu4. While the differences in the overall activity of the mixed tetramers and Cu4 are much smaller on zirconia (see the inset in Fig. 1h), the composition-dependent oscillatory trend observed for UNCD-supported clusters is also preserved on the zirconia support. The most striking difference in the performance of the zirconia-supported clusters is the continuous shift of their selectivity towards cyclohexadiene with an increasing number of copper atoms in the cluster, reaching over 70% for Cu4 (see the inset in Fig. 1j).
Catalytic performance of CuPd tetramers over time on UNCD and ZrO2 supports
To investigate the evolution of the performance of the tetramers with time, a second temperature ramp (i.e. “long”, Fig. S4†) was applied without exposing the samples to air between the first and the second ramp, keeping the samples under the flow of the reactant mixture at 25 °C. On the UNCD support, the main difference found is the increase in the total rates at 400 °C of the mixed clusters in comparison with the first ramp by a factor of 2 to 4. The increase is more pronounced with an increasing number of palladium atoms in the cluster, with Pd2Cu2 remaining the most active among the mixed clusters (Fig. 2g), with an increase in benzene production driving up these rates (Fig. 2a). There is a slight increase by about 1 per atom per s observed in the rate of cyclohexadiene formation (Fig. 2c), while combustion to CO2 remains suppressed (Fig. 2e). Fig. S10† demonstrates the absence of CO2 production by Pd4 clusters on UNCD as compared to the CO2 formation on the reference blank UNCD support. In the second temperature ramp, the selectivity of UNCD-supported clusters remains dominated by benzene formation. On the zirconia support, there is an approximately 25% drop observed in the total activity of Pd4 in comparison with that of the first “short” ramp and no change for Cu4. However, there is an increase of approximately 50% and 100% for Cu3Pd1 and Cu2Pd2, respectively (Fig. 2h). The opposite trend is observed for pure Pd4, which has a lower benzene rate during the second ramp of all the tested catalysts, lower by about a quarter. For the mixed clusters, the rate of cyclohexadiene formation increases by about 50% to approximately 3 per atom per s. The production of CO2 is comparable to that of the first ramp, with the highest combustion rate for Pd4 (Fig. 2f), as also shown in Fig. S10† compared to CO2 formation on the reference blank ZrO2 support. While the overall trends in the order of total activities in the second (“long”) ramp are the same as observed in the first (“short”) ramp, there are some subtle differences evident for the individual clusters; in the second ramp, there is about a 10% increase observed for Pd4 on both supports during the second ramp, no activity of Cu4 on UNCD and comparable activity of Cu4 on zirconia, contrasted by an up to approximately 120–400% and 75–100% increase for the mixed clusters on UNCD and zirconia, respectively. Based on the available data, it is not possible to unambiguously identify the roots of changes in the performance of the catalyst with elapsed time. AFM images (Fig. S7 and S8†) of the most active mixed clusters did not reveal the formation of nanosized aggregates and previous in situ synchrotron small-angle X-ray scattering studies of similar clusters on various supports and under a variety of reaction conditions, including oxidative dehydrogenation, did not indicate sintering of the clusters either.21,64,76–79 A possible cause could be the temperature-dependent alteration in the oxidation state of Cu and Pd or changes in the chemical composition of the clusters revealed in other studies by X-ray absorption spectroscopy, reported to be cluster size, composition and support dependent.21,34,64,65,76,77,80,81 These studies indicated the evolution of the oxidation state of the metal components during the course of the increasing and decreasing phase of the temperature ramp, as well as change in the chemical composition of the catalyst, like copper in the form of a hydroxide in the as made catalyst, an evolution of the nature of the catalytic metal with temperature/time through oxidized and reduced forms. These phenomena can mirror, for example, the apparent hysteresis visible in some of the data presented in this study. The partial reduction of zirconia49,69 or an encapsulation of the clusters by it cannot be fully excluded either.82–84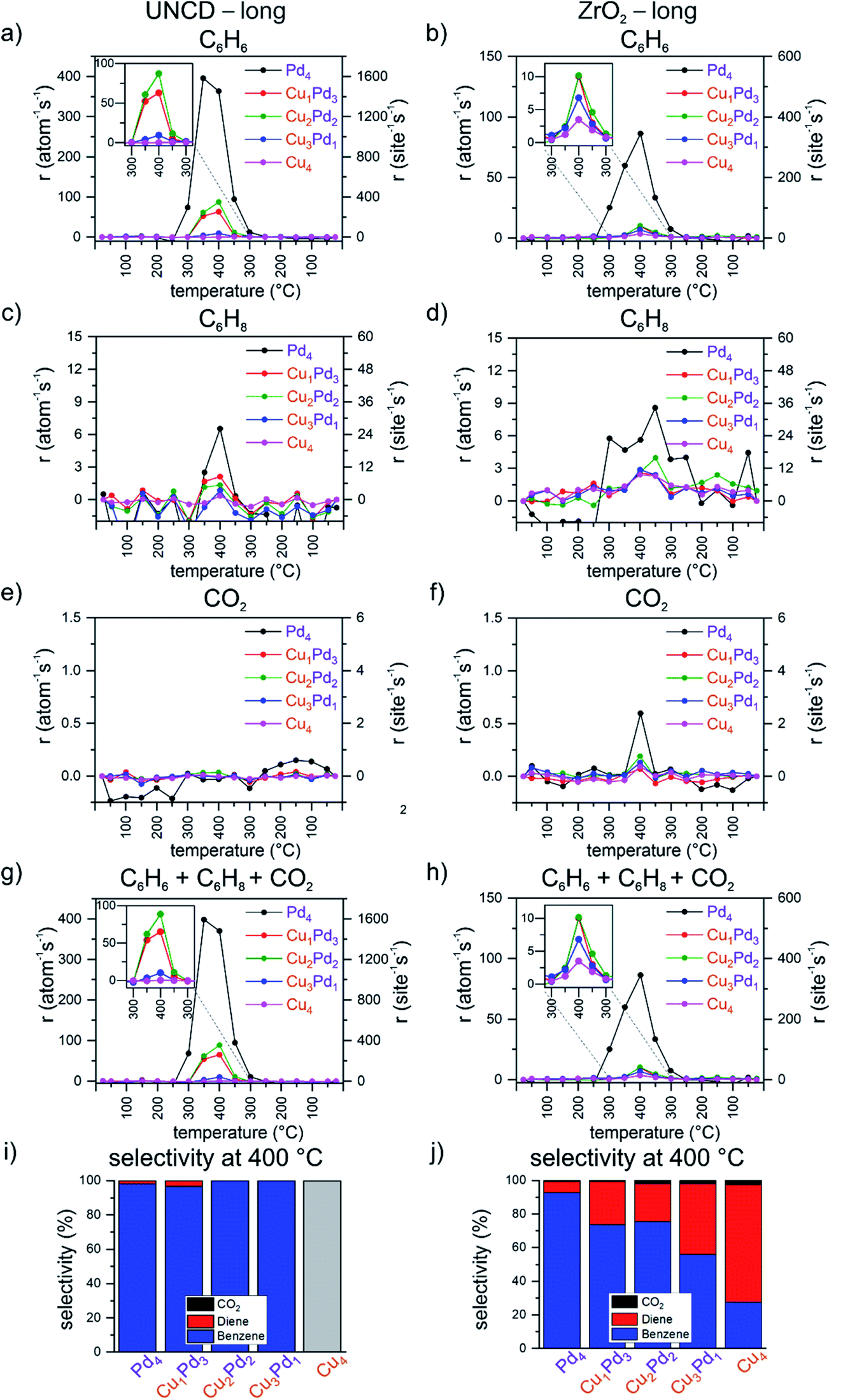 | ||
| Fig. 2 Per total atom and per site carbon-based rate of formation during the second (“long”) temperature ramp for (a) benzene on UNCD, (b) benzene on ZrO2, (c) cyclohexadiene on UNCD, (d) cyclohexadiene on ZrO2, (e) CO2 on UNCD, (f) CO2 on ZrO2; summed over products (g) on UNCD and (h) on ZrO2; carbon-based selectivity for benzene, cyclohexadiene and CO2 at 400 °C for all tetramers (i) on UNCD and (j) on ZrO2 (“long” refers to the second temperature ramp with 2 hours extended dwell time at 400 °C). | ||
In summary, a strong effect of the support on the performance of the clusters was observed when switching from the carbon-based UNCD support to the non-reducible zirconia one. The experimentally obtained selectivities of the individual samples in the second ramp are an important indicator that they are stable catalysts in this reaction.
Catalytic performance of the Pd1 and Pd2 subunits on UNCD and ZrO2 supports
With the Pd4 cluster exhibiting the highest activity, questions arise about the effect of Cu on mixed tetramer clusters, causing a significant drop in the catalytic activity in comparison with Pd4 on both UNCD and zirconia supports, together with a strong effect on the selectivity as well. Along the same lines, it was unclear how the monoatomic Pd (Pd1) and Pd dimer (Pd2) would perform alone, i.e. without their copper neighbours, in comparison to the mixed Cu3Pd1 and Cu2Pd2 clusters, which were the most selective against cyclohexadiene. To evaluate such an effect, copper-free monoatomic Pd (Pd1) and Pd dimer (Pd2) were compared with the catalytically most active tetramer (Pd4) as well as with mixed Cu3Pd1 and Cu2Pd2 clusters. which were the most selective for cyclohexadiene. The results for Pd1 and Pd2, compared with the performance of Pd4, are summarized in Fig. S11 and S12† for the UNCD and zirconia-supported species, respectively. In contrast to Pd4, a much smaller support effect is observed for Pd1 during the first ramp, while the Pd tetramer at 400 °C is about 3 times more active on UNCD than on the ZrO2 support (∼350 vs. 110 per atom per s). In the case of the monoatomic Pd, this difference amounts to only about 15% (∼175 vs. 150 per atom per s). This indicates that the performance of the monomer is not much affected by the support; an expected outcome considering the different nature of carbon-based and oxidic supports. The formation rates for cyclohexadiene show no support effect, with the same rates for UNCD and zirconia supports of around 6 and 3 per atom per s for the tetramer and the monomer, respectively. The monomer on UNCD produces CO2 at a rate of 0.5 per atom per s and negligible amount on zirconia. The selectivities shown in Fig. S11g and S12g† indicate the necessity of the presence of copper atoms in the catalytic site to achieve improved selectivity towards cyclohexadiene because a single Pd atom or a Pd dimer does not yield this product. The activity of Pd2 (80 per atom per s) is approximately half of that of Pd1 (150 per atom per s), which provides almost identical rates considering the active sites: 160 per site per s (Pd2) and 150 per atom per s (Pd1) (here, active sites correspond to the number of deposited Pd particles). While Pd1 on UNCD retains its activity in the second temperature ramp, its activity drops by a factor of 6 during the second extended ramp.For this reason, a Pd1/ZrO2 sample was characterized with XPS before and after exposure to the reactants and heating to 400 °C (Fig. S13†) in an attempt to determine whether palladium may get immersed in the zirconia support, eventually becoming overgrown by the support oxide. However, no clear conclusion on the destiny of Pd atoms could be made from this analysis. XPS spectra of the Pd 3d region for Pd1 on ZrO2 before and after the catalytic testing are shown in Fig. S13.† The Pd 3d region strongly overlaps with Zr 3p, which complicates the deconvolution of the spectra and the precise quantitative analysis of Pd oxidation states. Nevertheless, the spectra provide qualitative evolution of the chemical composition. Hence, Pd 3d5/2 and Pd 3d3/2 peaks with a lower intensity can be detected between the Zr 3d peaks. For the fresh sample, Pd 3d5/2 reveals a binding energy of 338.0 eV (with 1.7 eV shift compared to the reference XPS spectra85). Whereas after the reaction, two components of the Pd 3d5/2 peak were detected with binding energies of 337.1 and 338.5 eV. We assigned these peaks to Pd0 and Pd2+, respectively, considering a shift in binding energies of about 2.1 eV. Typical binding energies for bulk Pd0, Pd2+ and Pd4+ are recorded as 335.1, 336.3, and 337.9 eV, respectively.86 In the present case, such a significant shift in binding energies in comparison with those of bulk Pd can be explained by the size effect of subnanometer clusters that has already been reported in the literature. For example, Wu et al. showed that monoatomic Pd1 on alumina/NiAl(110) can be shifted up to 1.8 eV to higher binding energies compared to those of the bulk structures.87 This leads to the conclusion that, in this case, Pd can be in the oxidation state +2 and in the metallic form but the difference in the support used must be noted. Also, 1–1.4 eV shifts in the binding energies were reported for subnanometer Pd4 clusters on Al2O3 compared to Pd foil.81 Interestingly, the shift of the Pd 3d5/2 peak for the fresh sample and the one after the testing procedure diverge, with a difference of around 0.5 eV. This can be explained by the influence of the O2/CO environment during the reaction and, thus, a greater shift in the binding energy of Pd for the tested sample.81 The above-mentioned XPS related studies can confirm our assumption that Pd particles after the reaction are composed of the oxidation state Pd2+ and metallic Pd0. Such an assumption can be also supported by a XANES study, showing the evolution of the average oxidation state of Pd in Pd4 clusters supported on UNCD during the ODH of cyclohexane.21 The reported data reveal that, at 200 °C, Pd has an average oxidation state of +2. As the temperature increases, the oxidation number decreases to ∼+1.2 and reactivity is observed. After cooling to room temperature, the oxidation state does not return to +2, but stays at +1.6, which is in a reasonable agreement with the results from XPS studies after the reaction. Thus, we can hypothesise that a reduction of the catalyst takes place during the dehydrogenation reaction and is confirmed with the presence of metallic Pd0. It is also important to point out that Pd reduces under the X-ray beam, which was also observed to a certain extent with our Pd1 samples after about 1 hour of measurement. The position of the Pd peaks can also depend on the substrate, as shown by a comparison with the Pdx on TiO2 support.50 In the latter case, it turns out that the activity in CO oxidation correlates with the binding energy shift, which varies with the change in the shielding effect of the electrons. The switch to a zirconia support affects the activity of the monomer and dimer differently in the second (“long”) temperature ramp. While Pd1 on UNCD maintains the same activity, Pd1 and Pd2 on ZrO2 suffer a drop to the same value of 25 per atom per s. Compared to the first ramp, where the activity was the same per site (double for the monomer on the atom basis), in the second ramp, the observed reactivity is the same on the atom basis, which could be caused either by the formation of similar structures in both cases, or as a consequence of the disintegration of Pd2 owing to its lowest stability from among Pd2–7 clusters,88 and previous studies reported the redispersion of palladium under O2 on SiO2.89–92 Considering the abovementioned results, it follows that Pd1 and Pd2 are not stable on the ZrO2 support, reflected in the significant decrease in their catalytic activity during the long ramp. It is possible that the initially active sites can be passivated through the spillover effect and/or due to strong metal–support interactions.82–84,95,96 Moreover, aggregation can be an additional factor; even though we did not observe Pd agglomerating in the AFM images, one cannot fully exclude agglomeration to some extent. It has also been shown that Pd1 is stable on nanodiamonds under propane dehydrogenation conditions.63
Catalytic performance of the Cu1Pd3, Cu2Pd2 and Cu3Pd1 mixed tetramers on the TiO2 support
From the UNCD and ZrO2-supported mixed four-atom clusters, only the zirconia-supported Cu1Pd3, Cu2Pd2 and Cu3Pd1 tetramers produced cyclohexadiene; moreover, they exhibited a strongly composition-dependent selectivity for the C6 products cyclohexadiene and benzene. In order to further explore the effect of the support, a reducible oxide support TiO2 was chosen to complement the UNCD and the non-reducible zirconia supports. The results of the catalytic tests from the double temperature ramp are shown in Fig. 3. In the first ramp, the most active catalysts were Cu1Pd3 and Cu2Pd2, with comparable rates of 17 and 15 per atom per s, followed by Cu3Pd1, with a rate of 5 per atom per s. During the second ramp, a similar odd–even oscillatory pattern in the activity was observed, as in the case of these clusters on the UNCD support, namely Cu2Pd2 > Cu1Pd3 > Cu3Pd1, accompanied by a substantial increase in the activity of these clusters in the second ramp: a 2-, 10- and 15-fold increase for Cu3Pd1, Cu2Pd2 and Cu1Pd3, respectively. In addition, with an increasing number of Pd atoms in the mixed tetramers, an increasingly slower convergence in activity within the two hour dwell-time at 400 °C was observed during the second ramp, where Cu1Pd3 presents an extreme case with a steep increase during the entire time spent at this temperature (Fig. 3). In order to address the evolution of the activity of this sample with time, another double ramp was applied to the catalyst without exposing it to air and keeping it in the cell under a flowing stream of the reactant mixture for 6 hours. The evolution of the benzene formation rate with time in the course of the two double ramps is plotted in Fig. S14.† The rate of benzene formation during the first “short” temperature ramp shows measurable activity at 350 °C (Fig. S14c†), followed by a skyrocketing increase in activity at 400 °C during the two hours of the first “long” ramp without any indication of levelling off by the end of the two hours (Fig. S14d†). A convergence of activity is seen at the end of the second “long” ramp (Fig. S14f†). From the activity data alone, it is not possible to identify the driving force which is causing such an abrupt boost in the activity. Based on in situ studies reported on Cu, Pd and CuPd clusters on various supports and the reactions mentioned above, an agglomeration of clusters is unlikely for these coverages, as well as not finding measurable nanometer size aggregates by AFM. One cause could be the change in the oxidation state of Cu and Pd with temperature, reported to be size, composition and support dependent for similar clusters in the above-mentioned studies, as well as by XPS.50 Another cause could be the change in the oxidation state of titania. The titania support was used as prepared by ALD, without any annealing or pretreatment after exposure to air, and it can undergo reduction and/or reoxidation under the mixture of reactants with changing temperature. Palladium, present in this sample at the highest loading, corresponding to 7.5% atomic monolayer equivalent coverage, can improve the reduction of titania with some of the hydrogen created during the multistep dehydrogenation process, as also reported, for example, for subnanometer and nanometer size copper clusters on an iron oxide support in CO2 or in the case of hydrogen spillover from nanoparticles.93 Lastly, titania can overgrow palladium or intermix with it.94–97 The 50% drop in the activity from 170 to 120 per atom per s between the first “long” and the second “short” ramp most likely reflects the change in the oxidation state of palladium or titania or both during the 6 h long exposure of the sample to the reactant mixture at 25 °C. The overlapping rising edge of the benzene formation rate in the initial 20 minutes of the “short” and “long” ramp of the second run at 400 °C indicates the reversibility of the properties of the system in this time window. By the end of the following 100 minutes at 400 °C of the “long” ramp, the benzene formation rate starts levelling off at around 240 per atom per s, most likely an indication of the system converging to its final state. A similar trend was reported for alumina-supported size-selected AgPt clusters in CO oxidation.98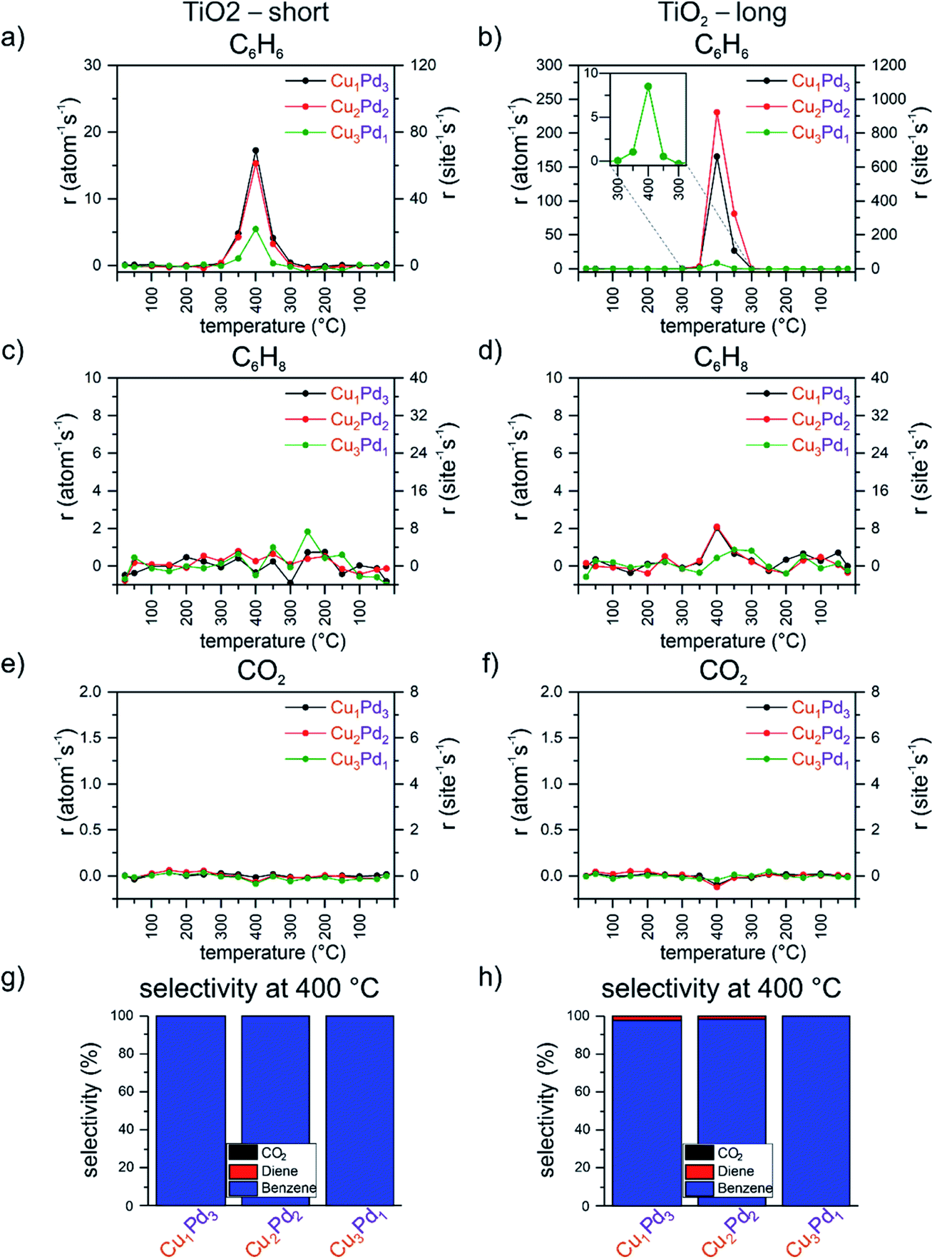 | ||
| Fig. 3 Performance of Cu1Pd3, Cu2Pd2 and Cu3Pd1 clusters on TiO2. Carbon-based per total atom and per site rates of CumPdn+ on TiO2 for the “short” and “long” ramps: (a) benzene – short, (b) benzene – long, (c) cyclohexadiene – short, (d) cyclohexadiene – long, (e) CO2 – short, (f) CO2 – long; carbon-based selectivity for benzene, cyclohexadiene and CO2 at 400 °C during (g) the short ramp and (h) the long ramp (“short” refers to the first temperature ramp of the applied double ramp, with a dwell time of 20 minutes at 400 °C; “long” refers to the second temperature ramp with a 2 hour dwell time at 400 °C). | ||
The zirconia and titania substrates are made of a naturally oxidized n-type Si wafer coated with an ultrathin oxide film by ALD. While AFM shows a homogenous zirconia layer, the base Si support could also interact with particles through the subnanometer ALD layers, with the possibility of charge transfer to the cluster through the thin oxide film,44,99 ultimately affecting the catalyst’s performance. In the same reaction, Lee et al. reported a benzene production rate of 25 per atom per s reached at 300 °C for a catalyst based on MgO supported Co27±4 clusters.18 On the contrary, the selectivity towards benzene was only around 25% and the rest was CO2. The authors also reported a solid solution of oxidized cobalt clusters and a magnesia support yielding dynamically formed transient ∼2.5 nm size aggregates at 300 °C. Rioux et al. reported a benzene production rate of 0.12 per atom per s (a per atom estimate calculated from the published weight of the used catalyst) for 8–10 nm Pd particles on a TiO2 support.4 The example of two orders of magnitude more active subnanometer size cobalt clusters indicated the advantage of using subnanometer clusters when all or almost all atoms of the cluster are exposed to the reactants, in contrary to a precious metal-based large nanoparticle with a small fraction of surface atoms. The rates of benzene production by the Cu4/ZrO2 tetramer reported in the current study are in the same order of magnitude as those found in our current previous study of the ODH of cyclohexene on Cu4 clusters supported on thin Al2O3, SiO2, SnO2, and TiO2 layers under similar reaction conditions with a slightly higher ratio of cyclohexene to oxygen1:1.1, in comparison with the currently applied 1:1 ratio but otherwise identical reaction conditions.22 The observed reaction rates are orders of magnitude higher for all other CuPd tetramers along with their size, composition and support-dependent selectivity, underlining the dramatic impact of mixing, or alloying, at the subnanometer scale.
As discussed above, the search for the main driving force or forces which ultimately define the performance of subnanometer clusters poses a major challenge. However, there have been reports dealing with the properties of subnanometer clusters, some of those identical to the clusters in this study, which reveal some specific features of these materials. In general, the inclusion of a second metal to form a bimetallic particle leads to the redistribution of the electron density within the particle and on its individual atoms, changes in interatomic bond lengths and restructuring and, as a consequence, the reactivity of the clusters may change. These changes can be expected, and have already been shown on numerous examples, to be much more pronounced at the subnanometer scale than in larger particles or in bulkier materials. For example, according to density functional theory (DFT) calculations on isolated copper–palladium tetramers, Pd4, Cu1Pd3 and Cu2Pd2 possess a three-dimensional structure with HOMO orbitals located in the atoms, particularly with a larger lobe for the palladium atoms; thus, there is a higher electronic probability over palladium atoms and, consequently, the interaction of the reactant over this atom could be preferred to that on other sites/atoms in the cluster.100 The largest Cu–Cu distance and the highest charge polarization was calculated for the Cu2Pd2 cluster, which could in part explain its highest reactivity observed among those of the mixed clusters, and at the same time mixed Cu2Pdx cluster compositions come with the highest binding energy and maximal stabilization effect.101 On the other hand, Cu3Pd1 and Cu4 clusters with low reactivity in the present study show up in the calculations as planar structures where the HOMO orbital is distributed in the lateral zones, including the bridge sites. This indicates that the reactivity of monometallic and bimetallic clusters is not always located in atoms, and the bridge sites should be considered as well. Electronic and structural effects are expected to be further modulated by the support used to hold such clusters. Interesting trends in selectivity are observed in the case of mixed copper–palladium tetramers, dominated by benzene production on UNCD, benzene production with totally suppressed combustion on TiO2, and towards cyclohexadiene shifted selectivity on ZrO2 (Fig. 1–3). The differences in activity and selectivity may be related, for example, to findings which emerged from DFT calculations on isolated CuPd tetramers100,101 showing increasing electron density on the Pd atoms and the equivalent positive charge on Cu atoms as a linear function of the Cu:Pd ratio, which could affect the adsorption energy of the cyclohexadiene intermediate and its desorption prior to the ultimate dehydrogenation step to benzene. The effect of charge can be strongly affected by the properties of the support. On an oxide support, there could be a higher charge transfer in comparison to that on UNCD, reducing the electron density on Pd atoms and, thus, lowering the HOMO orbital energy, which can lead to decreased activity.99 Thus, the combination of charge distribution in the cluster, the effect of composition and support on the bond length between atoms, and the cluster structure itself, including transitions from 3D to 2D structures, can all affect the performance of subnanometer clusters in the structure-sensitive dehydrogenation of cyclohexene, which ultimately are reflected in differences in the selectivity as well as the activity, the latter summarized in Tables S1 and S2 of the ESI† for the catalysts studied in this paper.
Conclusions
Pronounced effects of the composition of the four-atom Cu, CuPd and Pd clusters and the support on the catalytic activity and selectivity in the oxidative dehydrogenation of cyclohexene were observed. The UNCD-supported clusters are highly active and dominantly produce benzene, some of the mixed clusters also produce cyclohexadiene, and all clusters have a much suppressed combustion channel, while the also highly active TiO2-supported tetramers solely produce benzene, without any combustion to CO2. The selectivity of the ZrO2-supported mixed CuPd and monometallic Cu clusters is entirely different. Though they have lower activity in comparison with those on other supports, these clusters produce significant fractions of cyclohexadiene, with their selectivity towards cyclohexadiene gradually increasing with an increasing number of copper atoms in the cluster, reaching about 50%. The zirconia-supported copper tetramer stands out from all the other tetramers in this reaction, with a selectivity towards cyclohexadiene of 70%, far exceeding those of all other cluster–support combinations. The findings from this study indicate a positive effect of copper on the stability of the mixed tetramers and potential new ways of fine-tuning catalyst performance by controlling the composition of the active site and via cluster–support interactions in complex oxidative reactions under the suppression of the undesired combustion of the feed.Author contributions
MB and YL prepared the ALD supports and characterized the as prepared oxide films by ellipsometry. HT performed the AFM characterization of the supports and cluster samples. JJ and MV synthesized the samples of the deposited clusters. JJ and StV performed catalyst testing, AH performed initial experiments. MJ performed the characterization of the film thickness by ellipsometry of the samples after tests. SB and StV performed XPS characterization, JJ developed software for data processing and analysis. JJ, StV, MV, SV, YL, AH, LK, OF and SB discussed the results. StV wrote the first draft of the manuscript. StV, JJ and SV finalized the manuscript with inputs from all other authors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to thank the Argonne National Laboratory for facilitating the use of the cluster synthesis and testing equipment for this study. JJ, StV, MV and SV acknowledge support from the European Union’s Horizon 2020 Research and Innovation Program under grant agreement no. 810310, which corresponds to the J. Heyrovský Chair project (“ERA Chair at J. Heyrovský Institute of Physical Chemistry AS CR – the institutional approach towards ERA”). The funders had no role in the preparation of the article. StV also acknowledges DAAD for financial support through a short-term research grant for his stay at the University of Rostock and the Leibniz-Institut für Katalyse e.V. AH acknowledges the support of the U.S. Department of Energy, BES Materials Sciences, under Contract DEAC02-06CH11357 with UChicago Argonne, LLC, operator of Argonne National Laboratory during the initial exploratory experiments. MJ and OF acknowledge the support of the European Regional Development Fund; OP RDE; Project: “Carbon allotropes with rationalized nanointerfaces and nanolinks for environmental and biomedical applications” (No. CZ.02.1.01/0.0/0.0/16_026/0008382). The authors thank ON Semiconductor Czech Republic for providing Si wafers free of charge in support of fundamental research. YL would like to thank the support from NSF CBET-1606117. MB is grateful for the support from the Alabama EPSCoR Graduate Research Scholars Program.References
- A. Guerrero-Ruiz and I. Rodríguez-Ramos, Carbon, 1994, 32, 23–29 CrossRef CAS.
- F. Cavani and F. Trifirò, Appl. Catal., A, 1995, 133, 219–239 CrossRef CAS.
- M. Lezanska, G. S. Szymanski, P. Pietrzyk, Z. Sojka and J. A. Lercher, J. Phys. Chem. C, 2007, 111, 1830–1839 CrossRef CAS.
- N. F. Dummer, S. Bawaked, J. Hayward, R. Jenkins and G. J. Hutchings, Catal. Today, 2011, 160, 50–54 CrossRef CAS.
- P. M. Modisha, C. N. M. Ouma, R. Garidzirai, P. Wasserscheid and D. Bessarabov, Energy Fuels, 2019, 33, 2778–2796 CrossRef CAS.
- R. K. Herz, W. D. Gillespie, E. E. Petersen and G. A. Somorjai, J. Catal., 1981, 67, 371–386 CrossRef CAS.
- K. M. Bratlie, L. D. Flores and G. A. Somorjai, Surf. Sci., 2005, 599, 93–106 CrossRef CAS.
- J. L. Gland, K. Baron and G. A. Somorjai, J. Catal., 1975, 36, 305–312 CrossRef CAS.
- B. E. Koel, D. A. Blank and E. A. Carter, J. Mol. Catal. A: Chem., 1998, 131, 39–53 CrossRef CAS.
- C. E. Smith, J. P. Biberian and G. A. Somorjai, J. Catal., 1979, 57, 426–443 CrossRef CAS.
- R. M. Rioux, B. B. Hsu, M. E. Grass, H. Song and G. A. Somorjai, Catal. Lett., 2008, 126, 10–19 CrossRef CAS.
- Y. Yao, Z. Yan, L. Chen, Z. Zhou, L. Liu and D. W. Goodman, Catal. Lett., 2012, 142, 1437–1444 CrossRef CAS.
- S. Goergen, C. Yin, M. Yang, B. Lee, S. Lee, C. Wang, P. Wu, M. B. Boucher, G. Kwon, S. Seifert, R. E. Winans, S. Vajda and M. Flytzani-Stephanopoulos, ACS Catal., 2013, 3, 529–539 CrossRef CAS.
- E. C. Tyo, C. Yin, M. Di Vece, Q. Qian, G. Kwon, S. Lee, B. Lee, J. E. DeBartolo, S. Seifert, R. E. Winans, R. Si, B. Ricks, S. Goergen, M. Rutter, B. Zugic, M. Flytzani-Stephanopoulos, Z. W. Wang, R. E. Palmer, M. Neurock and S. Vajda, ACS Catal., 2012, 2, 2409–2423 CrossRef CAS.
- H. Feng, J. W. Elam, J. A. Libera, M. J. Pellin and P. C. Stair, J. Catal., 2010, 269, 421–431 CrossRef CAS.
- P. A. Sermon, G. Georgiades, M. S. W. Vong, M. A. Martin-Luengo, P. N. Reyes and J. M. Thomas, Proc. R. Soc. London, A, 1987, 410, 353–372 CAS.
- W. Yu, M. D. Porosoff and J. G. Chen, Chem. Rev., 2012, 112, 5780–5817 CrossRef CAS.
- S. Lee, M. D. Vece, B. Lee, S. Seifert, R. E. Winans and S. Vajda, Phys. Chem. Chem. Phys., 2012, 14, 9336 RSC.
- S. Lee, D. M. Vece, B. Lee, S. Seifert, R. E. Winans and S. Vajda, ChemCatChem, 2012, 4, 1632–1637 CrossRef CAS.
- A. S. Crampton, M. D. Rötzer, U. Landman and U. Heiz, ACS Catal., 2017, 7, 6738–6744 CrossRef CAS.
- A. Halder, M.-A. Ha, H. Zhai, B. Yang, M. J. Pellin, S. Seifert, A. N. Alexandrova and S. Vajda, ChemCatChem, 2020, 12, 1307–1315 CrossRef CAS.
- S. Valtera, J. Jasik, M. Vaidulych, J. E. Olszówka, M. Zlámalová, H. Tarábková, L. Kavan and S. Vajda, J. Chem. Phys., 2022, 156, 114302 CrossRef CAS PubMed.
- S. L. Nauert, F. Schax, C. Limberg and J. M. Notestein, J. Catal., 2016, 341, 180–190 CrossRef CAS.
- S. Lee, A. Halder, G. A. Ferguson, S. Seifert, R. E. Winans, D. Teschner, R. Schlögl, V. Papaefthimiou, J. Greeley, L. A. Curtiss and S. Vajda, Nat. Commun., 2019, 10, 954 CrossRef.
- D. T. Williamson, J. F. Elman, P. H. Madison, A. J. Pasquale and T. E. Long, Macromolecules, 2001, 34, 2108–2114 CrossRef CAS.
- J. C. Gentry, Asia-Pac. J. Chem. Eng., 2007, 2, 272–277 CrossRef CAS.
- K. An and G. A. Somorjai, Catal. Lett., 2015, 145, 233–248 CrossRef CAS.
- M. A. Gawish, Q. A. Drmosh and S. A. Onaizi, Chem. Rec., 2022, 22, e202100328 CrossRef CAS.
- J. Jašik, A. Fortunelli and S. Vajda, Phys. Chem. Chem. Phys., 2022, 24, 12083–12155, 10.1039/d1cp05677h.
- S. Vajda, M. J. Pellin, J. P. Greeley, C. L. Marshall, L. A. Curtiss, G. A. Ballentine, J. W. Elam, S. Catillon-Mucherie, P. C. Redfern, F. Mehmood and P. Zapol, Nat. Mater., 2009, 8, 213–216 CrossRef CAS PubMed.
- E. C. Tyo and S. Vajda, Nat. Nanotechnol., 2015, 10, 577–588 CrossRef CAS PubMed.
- S. Vajda and M. G. White, ACS Catal., 2015, 5, 7152–7176 CrossRef CAS.
- S. Lee, B. Lee, S. Seifert, R. E. Winans and S. Vajda, J. Phys. Chem. C, 2015, 119, 11210–11216 CrossRef CAS.
- C. Liu, B. Yang, E. Tyo, S. Seifert, J. Debartolo, B. Von Issendorff, P. Zapol, S. Vajda and L. A. Curtiss, J. Am. Chem. Soc., 2015, 137, 8676–8679 CrossRef CAS.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- E. Fernandez and M. Boronat, J. Phys.: Condens. Matter, 2019, 31, 013002 CrossRef.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- M. Peng, C. Dong, R. Gao, D. Xiao, H. Liu and D. Ma, ACS Cent. Sci., 2021, 7, 262–273 CrossRef CAS PubMed.
- B. C. Gates, Chem. Commun., 2013, 49, 7876 RSC.
- A. Sanchez, S. Abbet, U. Heiz, W. D. Schneider, H. Häkkinen, R. N. Barnett and U. Landman, J. Phys. Chem. A, 1999, 103, 9573–9578 CrossRef CAS.
- M. D. Hughes, Y.-J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132–1135 CrossRef CAS.
- S. Lee, C. Fan, T. Wu and S. L. Anderson, J. Chem. Phys., 2005, 123, 124710 CrossRef PubMed.
- L. M. Molina, S. Lee, K. Sell, G. Barcaro, A. Fortunelli, B. Lee, S. Seifert, R. E. Winans, J. W. Elam and M. J. Pellin, Catal. Today, 2011, 160, 116–130 CrossRef CAS.
- G. Pacchioni and H.-J. Freund, Chem. Soc. Rev., 2018, 47, 8474–8502 RSC.
- E. T. Baxter, M.-A. Ha, A. C. Cass, A. N. Alexandrova and S. L. Anderson, ACS Catal., 2017, 7, 3322–3335 CrossRef CAS.
- X. Tang, J. Schneider, A. Dollinger, Y. Luo, A. S. Wörz, K. Judai, S. Abbet, Y. D. Kim, G. F. Ganteför, D. H. Fairbrother, U. Heiz, K. H. Bowen and S. Proch, Phys. Chem. Chem. Phys., 2014, 16, 6735–6742 RSC.
- A. S. Chaves, G. G. Rondina, M. J. Piotrowski, P. Tereshchuk and J. L. F. Da Silva, J. Phys. Chem. A, 2014, 118, 10813–10821 CrossRef CAS.
- R.-Y. Wang, J.-X. Wang, J. Jia and H.-S. Wu, Appl. Surf. Sci., 2021, 536, 147793 CrossRef CAS.
- S. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef CAS.
- W. E. Kaden, T. Wu, W. A. Kunkel and S. L. Anderson, Science, 2009, 326, 826–829 CrossRef CAS PubMed.
- S. Nigam and C. Majumder, J. Phys. Chem. C, 2012, 116, 2863–2871 CrossRef CAS.
- H. Tao, Y. Li, X. Cai, H. Zhou, Y. Li, W. Lin, S. Huang, K. Ding, W. Chen and Y. Zhang, J. Phys. Chem. C, 2019, 123, 24118–24132 CrossRef CAS.
- J. Hulva, M. Meier, R. Bliem, Z. Jakub, F. Kraushofer, M. Schmid, U. Diebold, C. Franchini and S. Parkinson Gareth, Science, 2021, 371, 375–379 CrossRef CAS.
- J. Rodriguez, Surf. Sci. Rep., 1996, 24, 223–287 CrossRef CAS.
- D. Bazin, C. Mottet and G. Tréglia, Appl. Catal., A, 2000, 200, 47–54 CrossRef CAS.
- C. Yin, F. R. Negreiros, G. Barcaro, A. Beniya, L. Sementa, E. C. Tyo, S. Bartling, K.-H. Meiwes-Broer, S. Seifert, H. Hirata, N. Isomura, S. Nigam, C. Majumder, Y. Watanabe, A. Fortunelli and S. Vajda, J. Mater. Chem. A, 2017, 5, 4923–4931 RSC.
- T. J. Gorey, B. Zandkarimi, G. Li, E. T. Baxter, A. N. Alexandrova and S. L. Anderson, J. Phys. Chem. C, 2019, 123, 16194–16209 CrossRef CAS.
- M. Gholinejad, F. Khosravi, M. Afrasi, J. M. Sansano and C. Nájera, Catal. Sci. Technol., 2021, 11, 2652–2702 RSC.
- W.-W. Liu, Y.-S. Feng, G.-Y. Wang, W.-W. Jiang and H.-J. Xu, Chin. Chem. Lett., 2016, 27, 905–909 CrossRef CAS.
- Y. Kim, H. Kim, J. Lee, K. Sim, Y. Han and H. Paik, Appl. Catal., A, 1997, 155, 15–26 CrossRef CAS.
- H. L. Tierney, A. E. Baber and E. C. H. Sykes, J. Phys. Chem. C, 2009, 113, 7246–7250 CrossRef.
- L. Wang, J. Diao, M. Peng, Y. Chen, X. Cai, Y. Deng, F. Huang, X. Qin, D. Xiao, Z. Jiang, N. Wang, T. Sun, X. Wen, H. Liu and D. Ma, ACS Catal., 2021, 11, 11469–11477 CrossRef CAS.
- M. Peng, Z. Jia, Z. Gao, M. Xu, D. Cheng, M. Wang, C. Li, L. Wang, X. Cai, Z. Jiang, H. Jiang, N. Wang, D. Xiao, H. Liu and D. Ma, ACS Catal., 2022, 12, 2244–2252 CrossRef CAS.
- Y. Liu, A. Halder, S. Seifert, N. Marcella, S. Vajda and A. I. Frenkel, ACS Appl. Mater. Interfaces, 2021, 13, 53363–53374 CrossRef CAS PubMed.
- A. Halder, C. Lenardi, J. Timoshenko, A. Mravak, B. Yang, L. K. Kolipaka, C. Piazzoni, S. Seifert, V. Bonačić-Koutecký, A. I. Frenkel, P. Milani and S. Vajda, ACS Catal., 2021, 11, 6210–6224 CrossRef CAS.
- Y. Wang and H. Gao, J. Phys. Chem. B, 2017, 121, 2132–2141 CrossRef CAS PubMed.
- S. Letichevsky, P. C. Zonetti, P. P. P. Reis, J. Celnik, C. R. K. Rabello, A. B. Gaspar and L. G. Appel, J. Mol. Catal. A: Chem., 2015, 410, 177–183 CrossRef CAS.
- M. Bowker, P. Stone, P. Morrall, R. Smith, R. Bennett, N. Perkins, R. Kvon, C. Pang, E. Fourre and M. Hall, J. Catal., 2005, 234, 172–181 CrossRef CAS.
- V. Korpelin, M. M. Melander and K. Honkala, J. Phys. Chem. C, 2022, 126, 933–945 CrossRef CAS.
- C. Yin, E. Tyo, K. Kuchta, B. von Issendorff and S. Vajda, J. Chem. Phys., 2014, 140, 174201 CrossRef CAS PubMed.
- M. Newville, R. Otten, A. Nelson, A. Ingargiola, T. Stensitzki, D. Allan, A. Fox, F. Carter, Michał, R. Osborn, D. Pustakhod, lneuhaus, S. Weigand, Glenn, C. Deil, Mark, A. L. R. Hansen, G. Pasquevich, L. Foks, N. Zobrist, O. Frost, A. Beelen, Stuermer, Azelcer, A. Hannum, A. Polloreno, J. H. Nielsen, S. Caldwell, A. Almarza and A. Persaud, Zenodo, 2021, DOI:10.5281/ZENODO.5570790.
- G. K. H. Pang, K. Z. Baba-Kishi and A. Patel, Ultramicroscopy, 2000, 81, 35–40 CrossRef CAS PubMed.
- M. U. Jewel, M. S. Mahmud, M. A. Monne, A. Zakhidov and M. Y. Chen, RSC Adv., 2019, 9, 1841–1848 RSC.
- M. A. Mamun, H. Baumgart and A. A. Elmustafa, ECS J. Solid State Sci. Technol., 2015, 4, Q35–Q37 CrossRef CAS.
- J. A. Enterkin, W. Setthapun, J. W. Elam, S. T. Christensen, F. A. Rabuffetti, L. D. Marks, P. C. Stair, K. R. Poeppelmeier and C. L. Marshall, ACS Catal., 2011, 1, 629–635 CrossRef CAS.
- B. Yang, X. Yu, A. Halder, X. Zhang, X. Zhou, G. J. A. Mannie, E. Tyo, M. J. Pellin, S. Seifert, D. Su and S. Vajda, ACS Sustainable Chem. Eng., 2019, 7, 14435–14442 CrossRef CAS.
- B. Yang, C. Liu, A. Halder, E. C. Tyo, A. B. F. Martinson, S. Seifert, P. Zapol, L. A. Curtiss and S. Vajda, J. Phys. Chem. C, 2017, 121, 10406–10412 CrossRef CAS.
- S. Lee, B. Lee, F. Mehmood, S. Seifert, J. A. Libera, J. W. Elam, J. Greeley, P. Zapol, L. A. Curtiss, M. J. Pellin, P. C. Stair, R. E. Winans and S. Vajda, J. Phys. Chem. C, 2010, 114, 10342–10348 CrossRef CAS.
- V. Habibpour, C. Yin, G. Kwon, S. Vajda and R. E. Palmer, J. Exp. Nanosci., 2013, 8, 993–1003 CrossRef CAS.
- B. Zandkarimi, G. Sun, A. Halder, S. Seifert, S. Vajda, P. Sautet and A. N. Alexandrova, J. Phys. Chem. C, 2020, 124, 10057–10066 CrossRef CAS.
- B.-H. Mao, R. Chang, S. Lee, S. Axnanda, E. Crumlin, M. E. Grass, S.-D. Wang, S. Vajda and Z. Liu, J. Chem. Phys., 2013, 138, 214304 CrossRef PubMed.
- Y. Cao, R. Ran, X. Wu, Z. Si, F. Kang and D. Weng, J. Environ. Sci., 2023, 125, 401–426 CrossRef PubMed.
- G. W. Graham, A. E. O’Neill and A. E. Chen, Appl. Catal., A, 2003, 252, 437–445 CrossRef CAS.
- H. P. Sun, X. P. Pan, G. W. Graham, H. W. Jen, R. W. McCabe, S. Thevuthasan and C. H. F. Peden, Appl. Phys. Lett., 2005, 87, 201915 CrossRef.
- J. F. Moulder, J. Chastain and R. C. King, Handbook of x-ray photoelectron spectroscopy: a reference book of standard spectra for identification and interpretation of XPS data, Physical Electronics, Eden Prairie Minnesota, 1995 Search PubMed.
- NIST X-ray Photoelectron Spectroscopy Database, NIST Standard Reference Database Number 20, National Institute of Standards and Technology, Gaithersburg MD, 20899, 2000, DOI:10.18434/T4T88K.
- T. Wu, W. E. Kaden, W. A. Kunkel and S. L. Anderson, Surf. Sci., 2009, 603, 2764–2770 CrossRef CAS.
- V. Bertani, C. Cavallotti, M. Masi and S. Carrà, J. Phys. Chem. A, 2000, 104, 11390–11397 CrossRef CAS.
- C. Y. Seo, X. Chen, K. Sun, L. F. Allard, G. B. Fisher and J. W. Schwank, Catal. Commun., 2018, 108, 73–76 CrossRef CAS.
- M. A. Newton, C. Belver-Coldeira, A. Martínez-Arias and M. Fernández-García, Angew. Chem., 2007, 119, 8783–8785 CrossRef.
- S. T. Homeyer and W. M. H. Sachtler, Appl. Catal., 1989, 54, 189–202 CrossRef CAS.
- H. Lieske and J. Voelter, J. Phys. Chem., 1985, 89, 1841–1842 CrossRef CAS.
- W. Karim, C. Spreafico, A. Kleibert, J. Gobrecht, J. VandeVondele, Y. Ekinci and J. A. van Bokhoven, Nature, 2017, 541, 68–71 CrossRef CAS PubMed.
- W. E. Kaden, W. A. Kunkel, F. S. Roberts, M. Kane and S. L. Anderson, Surf. Sci., 2014, 621, 40–50 CrossRef CAS.
- M. Bowker, Phys. Chem. Chem. Phys., 2007, 9, 3514–3521 RSC.
- M. Bowker and E. Fourré, Appl. Surf. Sci., 2008, 254, 4225–4229 CrossRef CAS.
- R. A. Bennett, C. L. Pang, N. Perkins, R. D. Smith, P. Morrall, R. I. Kvon and M. Bowker, J. Phys. Chem. B, 2002, 106, 4688–4696 CrossRef CAS.
- F. R. Negreiros, A. Halder, C. Yin, A. Singh, G. Barcaro, L. Sementa, E. C. Tyo, M. J. Pellin, S. Bartling, K. H. Meiwes-Broer, S. Seifert, P. Sen, S. Nigam, C. Majumder, N. Fukui, H. Yasumatsu, S. Vajda and A. Fortunelli, Angew. Chem., Int. Ed., 2018, 57, 1209–1213 CrossRef CAS PubMed.
- L. Giordano, G. Pacchioni, J. Goniakowski, N. Nilius, E. D. L. Rienks and H.-J. Freund, Phys. Rev. Lett., 2008, 101, 026102 CrossRef PubMed.
- A. Alvarez-Garcia, E. Flórez, A. Moreno and C. Jimenez-Orozco, Mol. Catal., 2020, 484, 110733 CrossRef CAS.
- I. Efremenko and M. Sheintuch, Chem. Phys. Lett., 2005, 401, 232–240 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The mass spectra of the molecular beam optimized for the production and deposition of Cum, CumPdn and Pdn clusters, temperature ramp sequence, mass spectra of the ODH of the cyclohexene reaction with Pd4/ZrO2, normalized concentrations of the ODH of the cyclohexene reaction with Pd4/ZrO2, AFM height and phase images of supports, comparison of CO2 production on different supports, temperature-averaged data and selectivity for Pdn, XPS of Pd1/ZrO2 and performance of CumPdn+ clusters on TiO2, tables summarizing benzene production. See https://doi.org/10.1039/d2fd00108j |
| ‡ Equally contributing (first) authors. |
| This journal is © The Royal Society of Chemistry 2023 |