 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural characterisation of nanoalloys for (photo)catalytic applications with the Sapphire library
Robert M.
Jones
 a,
Kevin
Rossi
b,
Claudio
Zeni
c,
Mirko
Vanzan
d,
Igor
Vasiljevic
e,
Alejandro
Santana-Bonilla
a and
Francesca
Baletto
ae
a,
Kevin
Rossi
b,
Claudio
Zeni
c,
Mirko
Vanzan
d,
Igor
Vasiljevic
e,
Alejandro
Santana-Bonilla
a and
Francesca
Baletto
ae
aPhysics Department, King's College London, Strand WC2R 2LS, UK. E-mail: robert.m.jones@kcl.ac.uk
bEcole Polytechnique Federale de Lausanne, Laboratory of Nanochemistry for Energy, 1950, Sion, Switzerland. E-mail: kevin.rossi@epfl.ch
cInternational School for Advanced Studies, Via Bonomea, 265, 34136 Trieste, TS, Italy. E-mail: czeni@sissa.it
dDepartment of Chemical Sciences, University of Padovua, Via Marzolo1, 2, 35131,22, Padova, Italy
ePhysics Department, Universitá di Milano “La Statale”, Via Celoria 16, I-20133, Italy. E-mail: francesca.baletto@unimi.it; igor.Vasiljevic@studenti.unimi.it
First published on 28th June 2022
Abstract
A non-trivial interplay rules the relationship between the structure and the chemophysical properties of a nanoparticle. In this context, characterization experiments, molecular dynamics simulations and electronic structure calculations may allow the variables that determine a given property to be pinpointed. Conversely, a rigorous computational characterization of the geometry and chemical ordering of metallic nanoparticles and nanoalloys enables discrimination of which descriptors could be linked with their stability and performance. To this end, we introduce a modular and open-source library, Sapphire, which may classify the structural characteristics of a given nanoparticle through several structural analysis techniques and order parameters. A special focus is geared towards using geometrical descriptors to make predictions on a given nanoparticle's catalytic activity.
1 Introduction
Mono-, bi-, and poly-metallic nanoparticles (MNPs), or nanoalloys (NAs), find a wealth of potential uses across disciplines ranging from sensing1,2 to drug-delivery,3 memory-storage4,5 and optics.6,7 Among the most prominent applications, MNPs further play a significant role as thermal,8–10 electro-chemical,11,12 and photo-chemical13,14 catalysts.The need for a detailed characterization of the MNP's morphology stems from the delicate interplay among size, geometrical features, chemical composition and ordering, and the chemo-physical (e.g., optical, catalytic, etc.) properties of the MNP itself.1,15–17 Focusing on catalytic applications, the role of the MNP's and NA's surface is central. In this context, electronic structure calculations represent an established route to infer the structure–property relationships which rule the activity, selectivity, and stability of the catalyst.18–21 A knowledge of robust structure–property relationships, and of the finite temperature probability of observing MNPs and NAs with a given architecture, in turn, allows the drawing of design rules, prediction of the activity, and forecast of the ageing of a nanocatalyst.
One challenge in such a process is the lack of an automated and agnostic characterization of the MNP's and NA's morphology, with a special focus on its surface. Developing a comprehensive and robust set of taxonomy rules to classify and characterise MNPs and NAs could similarly be beneficial across their large number of multi-disciplinary applications. The objective of this communication is to offer the community an open-source and user-friendly package for the structural and chemical analysis of MNPs and NAs, released as Sapphire. Whilst there exists software and libraries to characterise structural motifs in molecular and periodic systems,22–24 we believe that Sapphire fills a gap in the computational chemical physics community by providing an open-source, modular, and documented library dedicated to the computational characterisation of metallic nanoparticles and nanoalloys. Sapphire is a Python library publicly available at https://github.com/kcl-tscm/Sapphire,25 which provides a unified framework to calculate various structural descriptors, from the pair-distance distribution function to the identification of surface atoms. Sapphire extends the common neighbour analysis with the introduction of atomic patterns, allowing a classification into the morphological families, e.g. icosahedra (Ih), decahedra (Dh) and FCC (both with and without stacking faults), as well as amorphous/molten-like motifs. In case of multiple independent simulations and/or long trajectories, Sapphire offers a robust statistical evaluation through the use of ensemble averages and well-known statistical quantities, such as the Kullback–Leibler divergence, and statistical tests, like the Kolmogorov–Smirnov one. We recommend the use of Sapphire only for metallic nanoparticles and nanoalloys, usually made of at least one chemical species adopting a FCC structure in the bulk. Although algorithms are independent of the chemistry of the system, the interpretation of results obtained from Sapphire analysis for periodic structures, and finite-size objects whose components may span the whole of the periodic table is far from trivial.
The next section of the manuscript is dedicated to discussing the philosophy and workflow of Sapphire, focusing primarily on the nature of its hierarchical design, and how we implement the library to be flexible and facile in its use. We provide a retrospective overview of Sapphire’s capabilities. Each section presents a didactic introduction to the theory and practical use of each of the characterisation tools. Among them, it is worth noting that Sapphire provides a Metadata section, consistent with the philosophy of the FAIR (Findable, Accessible, Interoperable and Reusable) principles.26 We thus hope Sapphire will help this community to more easily create FAIR databases in future contributions. As paradigmatic examples, we apply Sapphire to characterise individual snapshots as well as molecular dynamics trajectories of Au, Pt, AuPt, CuPt, and AuRh nanoalloys.
2 Workflow
The aim of this section is to briefly discuss Sapphire's architecture. The interested reader is referred to the Github main page, and the tutorial folder in particular, to obtain a complete overview of how Sapphire practically exploits each of its modules. Indeed, details on how to use the software are available within the tutorial at /main/Sapphire/Tutorials/. Furthermore, these web resources will allow the prospective user to reproduce the material in this manuscript with Sapphire 1.0.0 given as a free-standing version to accurately reproduce the presented figures.Fig. 1 provides the structure of Sapphire and shows the nature of the decentralised analysis tools. Sapphire requires as input only the atomic coordinates for the MNP. Given that Sapphire leverages the input/output (IO) stream of ASE,27 many common formats (e.g., .xyz, .exyxyz, etc.) are compatible. Note, the coordinate file can be obtained from experimental reconstructions – e.g., from a tomography experiment – as well as from numerical simulations. As for the nature of the analysis, one may provide a single snapshot, an entire trajectory, or a set of independent trajectories.
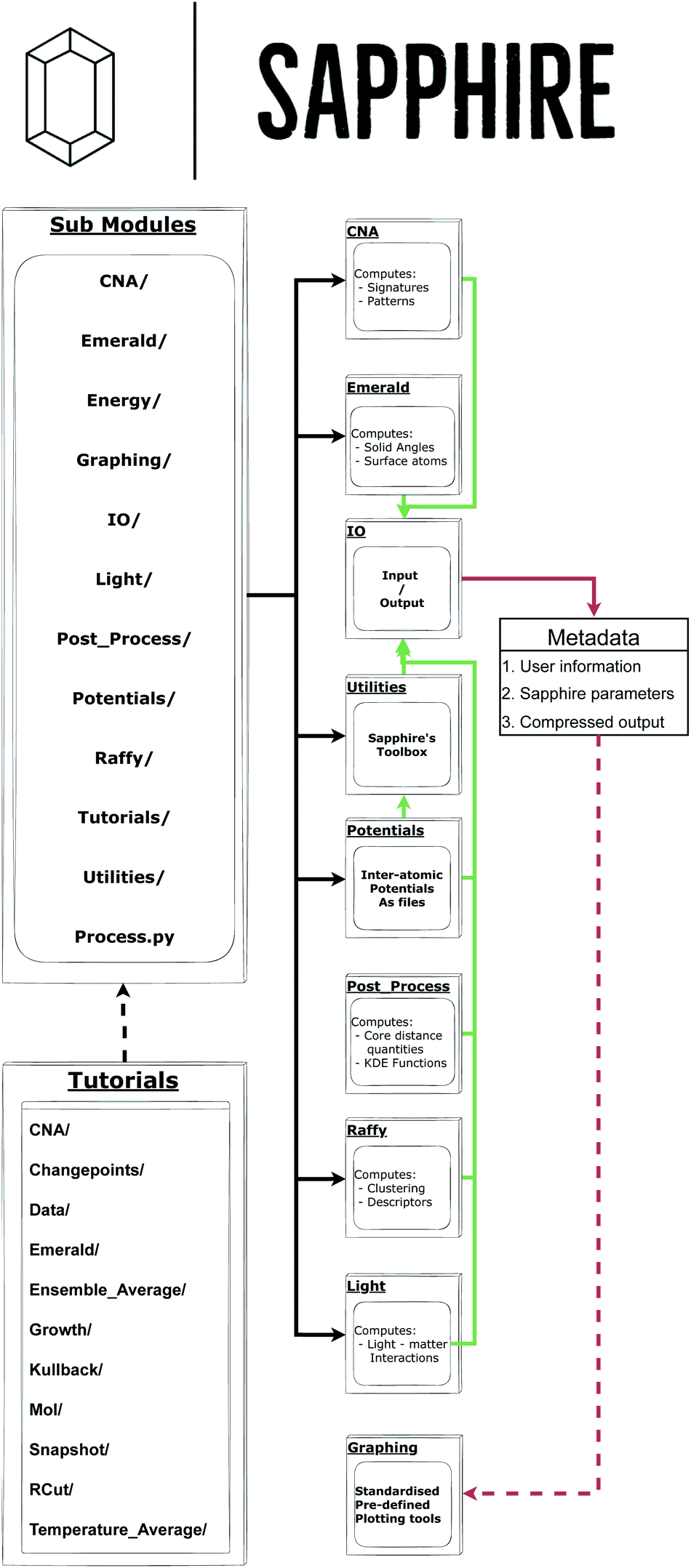 | ||
| Fig. 1 Flowchart detailing the scheme under which Sapphire is executed. Black lines indicate module import directionality. Green lines indicate IO streams, red lines indicate data flow, solid lines indicate hard-coded streams, and dashed lines highlight user choice. | ||
From here an interconnected nest of sub-modules, organised by common theme, may be accessed. It is worth noting that because the Process class does not directly interact with the IO stream, one may simply call a sub-module to begin the desired analysis.
After gathering the set of configurations to be analysed, Sapphire creates an extended xyz file, which encodes user-selected single-atom labels, e.g., its radial distance from the centre of mass, coordination number, and functions thereof. Global quantities are both stored in separate plain text files – for the users who like to plot immediately via Gnuplot or the user's plotting tool of choice – on a build an architecture for matplotlib, and in the dedicated Metadata object. In the following, we describe the quantities, aka descriptors, Sapphire calculates.
A crucial feature of Sapphire is its data storage philosophy. Each calculation using Sapphire's Process creates a Metadata in agreement with EMMO28 and EMMC29 rules, and the FAIR26 principles. Sapphire is amongst the first materials modelling post-processing tools to automatically write metadata in a standardised format, to our knowledge. Contained within the metadata are, among other pieces of information, the following:
• Compressed forms of the written output, to better facilitate a streamlined integration with potential subsequent interactive MNP and NA databases.
• User specific information, to enable the community to monitor the provenance of a given data set.
• Specifically chosen parameters for post-processing tools (each parameter is described in detail in the online manual).
The final stage of the flowchart in Fig. 1 lies in the use of Graphing tools. This is a library of pre-prepared matplotlib templates which may be called by the Plot_Funcs object defined within the library. From this, the Figures object is constructed from the collected metadata and the user requested input quantities. This parses the lists of input parameters for each requested plotting function and introduces Sapphire's defaults, should any arguments be omitted. We then call the Make_Plots function of the Figures object which iterates over all of the plotting functions, passing into each one the relevant list of input arguments.
3 Sapphire descriptors
In this section, we list and explain the analysis that Sapphire performs. We first discuss the distance related properties as they are fundamental to introduce a cutoff and hence the concept of local environment for any further analysis.3.1 Distance distribution functions
The distribution of pair-atomic distances (pair-distance distribution function, PDDF) is a crucial quantity to characterise the geometry and chemical ordering of an NA. Further, the PDDF is a directly measurable quantity via X-ray techniques.30,31To define the PDDF, let dij be the pair-distance between atoms i and j:
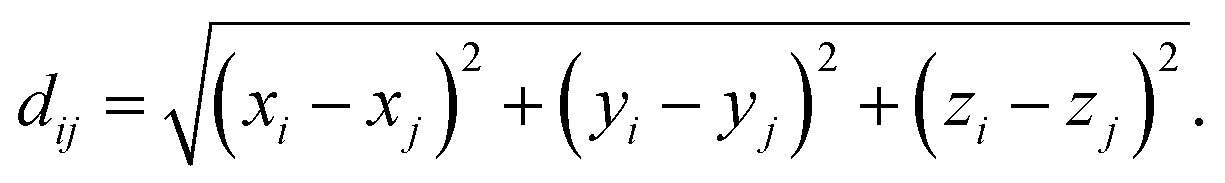 | (1) |
We then calculate the PDDF via a Kernel density estimate (KDE), constructed from n observations:
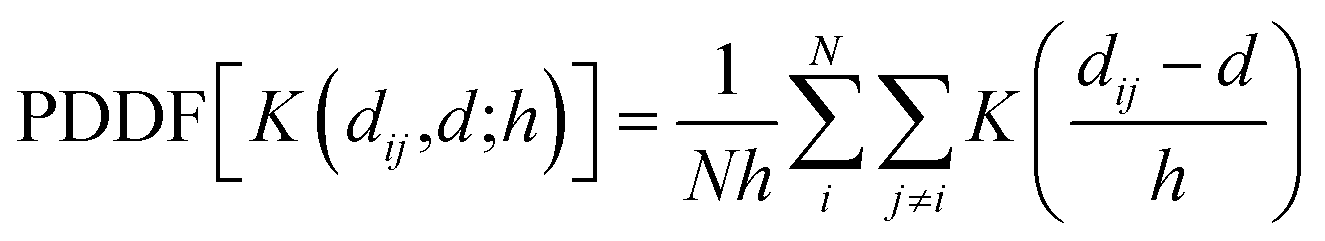 | (2) |
Fig. 2 shows the effect of the K and h choice in the PDDF calculation, as a function of the independent parameter d, written in terms of the lattice bulk a0. Setting the bandwidth, h, is a delicate step. The h-choice should balance a too fine resolution – where each distance might be present a single time, and below any reasonable resolution – and a too large one – where different neighbour shells are projected onto the same distance width. As a default, we have set the bandwidth to be 0.05 of the bulk lattice, which we have found to be sufficiently broad to smooth the sharp Dirac peaks from having a finite sample, and to sufficiently resolve key features, i.e., the first peak, minimum, and the second peak (orange line in Fig. 2). In principle, one may consider the h parameter to be comparable to the resolution in various microscopy techniques. As such, deviating from the default value must be done whilst considering the physical meaning of this parameter. That is to say that measuring interatomic distances to arbitrary precision is not possible at finite temperatures due to lattice vibrations.
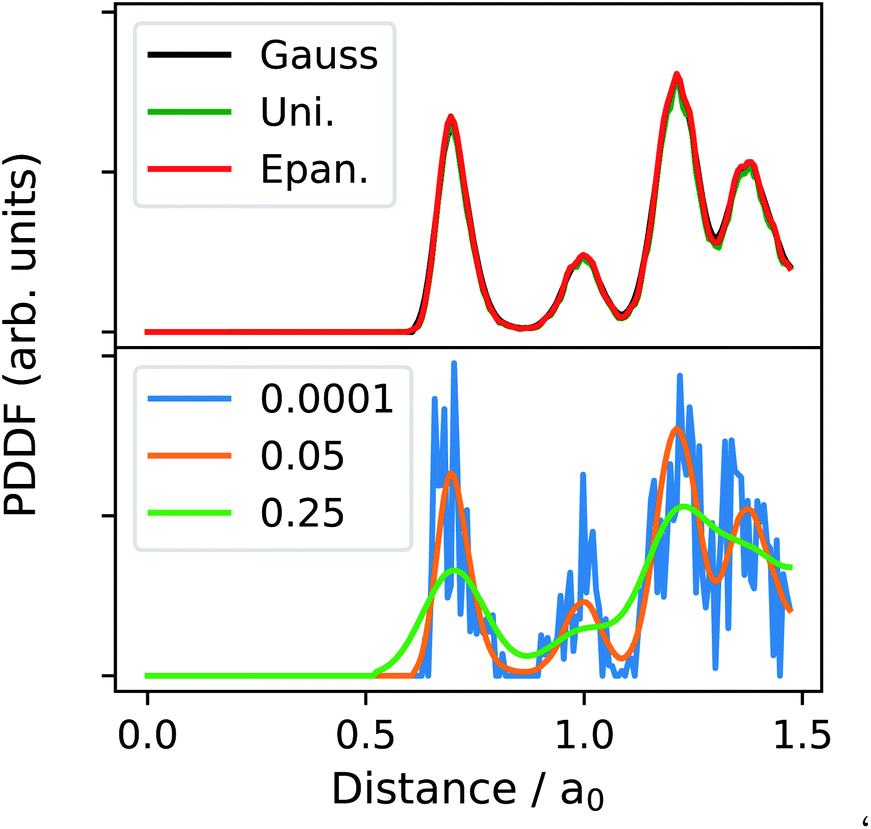 | ||
Fig. 2 The three possible parameterisations for calculating the PDDF using eqn (2). The top panel demonstrates the agreement between available kernels. The bottom panel illustrates the dependence on the bandwidth, h, orange (0.05a0 Å), blue (0.0001a0 Å) and (0.25a0 Å) green. We have selected a randomly alloyed AuPt NA of 1415 atoms arranged as an icosahedron with a ratio of Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt of 4:1. :Pt of 4:1. | ||
Given the analytical form of the KDE, derivatives can be easily calculated, and the approximate location of minima (and maxima) in the distribution can be identified. This is a key utility. The first PDDF minimum is instrumental to a robust determination of nearest neighbour distance. The position of the second maximum may also allow identification of whether the NA loses its symmetry or undergoes a structural or phase change. The latter should be associated to an overall radial breathing of the NA.
The evolution of the pair-distance distribution function (PDDF), independently of the chemical label, enables the:
• Identification of a cut-off distance (Rcut) set as where the first minimum in the PDDF falls. Such a definition of radial distance allows the subsequent definition of a local environment, and the construction of the adjacency matrix.
• Detection of solid, liquid, and amorphous, depending on the position of the second peak of the PDDF. A robust definition of “amorphous/molten-like” shape is where there is not a maximum in correspondence with the bulk lattice, a0, and the maximum radial distance is comparable with other well ordered shapes. The same feature and a radial breathing refers to a solid to liquid transition.33
To clarify further this point, Fig. 3 details the PDDFs of pure MNPs with 1415 atoms of Au, Pt, and AuPt with two different compositions. The reported simulation data are obtained from iterative molecular dynamics runs, as available in LoDiS,34–36 where the temperature is increased by 50 K every 1 ns, from 300 K to 1000 K.
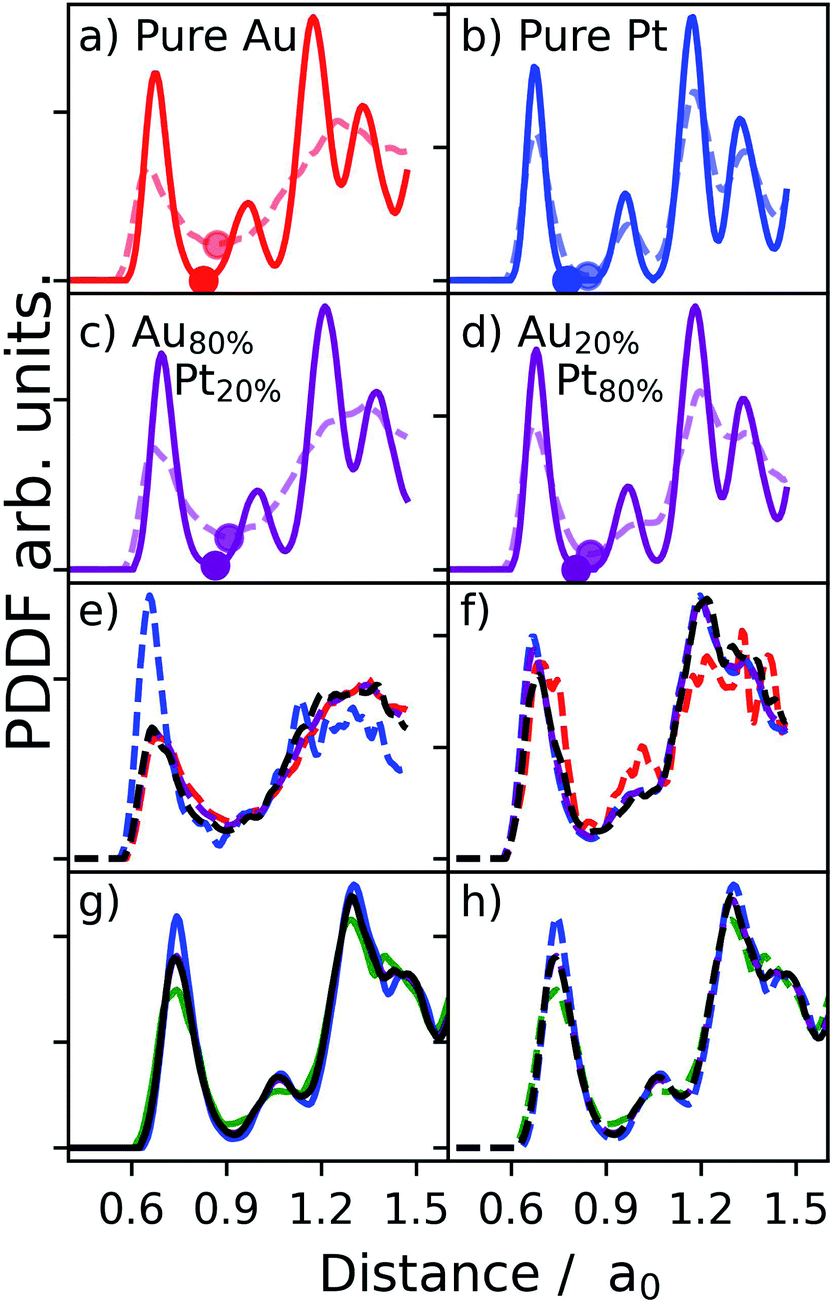 | ||
| Fig. 3 PDDF computed with Sapphire for a set of pure Au, Pt, and AuPt NAs containing 1415 atoms and adopting initially an icosahedral (Ih) shape. Distances are scaled with respect to the bulk lattice distance a0. For NAs, this measure is taken as the average of the two pure metals' bulk lattice distances. Panels (a) and (b) refer to pure Au and pure Pt MNPs, respectively. Panels (c) and (d) refer to randomly alloyed AuPt NAs at different chemical compositions, as indicated in the inset. Solid lines correspond to the PDDF sampled at 300 K while dashed lines are taken at 1000 K. Dots represent the position of the first PDDF minimum, corresponding to Rcut. Panels (e)–(h) present the PDDF for homo interactions (Au – red, Pt – blue, Cu – green), and hetero interactions (black). | ||
We show PDDFs sampled at both cold (solid lines in panels (a)–(d), Fig. 3) and high temperatures (dashed lines in the same panels). We note a broadening of the first peak at higher T, for both pure and alloyed nanosystems. At the same time, it is evident that the position of the first peak does not change significantly with the temperature. We stress that a radial cut-off can be defined independently of the temperature, as highlighted by the full-circle dots marking the position of the first minimum.33,37 The position of the second peak instead clearly changes between cold and hot temperatures. This is particularly evident in the case of pure Au and Au-based nanoalloys. Pt-NAs just show a minor effect because they are still approaching their melting point. Indeed, the pure Pt-NP PDDF still presents a clear peak in correspondence with its bulk lattice distance. For NAs with a large amount of Pt, the second peak also still presents a shoulder in correspondence with the bulk lattice, suggesting that the phase change starts mainly in the gold network (see the lines corresponding to Au–Au and Au–Pt bonding in panel (f) of Fig. 3).
Sapphire also supports the pair correlation function, g(r), which is used to describe long-range order in liquids38 and is related to the structure factor.
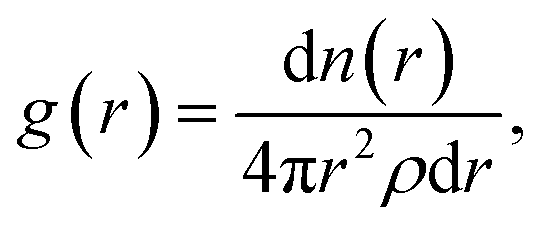 | (3) |
 | (4) |
![[x with combining circumflex]](https://www.rsc.org/images/entities/i_char_0078_0302.gif) α, ŷα, and ẑα of the atom-i and chemical species α = A, B are re-scaled with respect to the centre of mass chosen, namely the whole NA, or the A and B sub-regions. As was the case for computing the PDDF, we may also apply the KDE approach to compute the RDFs for each of the three types of radial distribution described above:
α, ŷα, and ẑα of the atom-i and chemical species α = A, B are re-scaled with respect to the centre of mass chosen, namely the whole NA, or the A and B sub-regions. As was the case for computing the PDDF, we may also apply the KDE approach to compute the RDFs for each of the three types of radial distribution described above: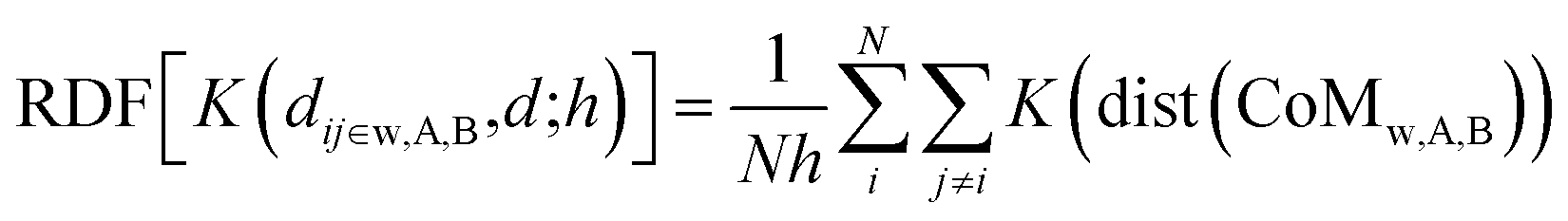 | (5) |
In principle, eqn (5) can be extended to more than two chemical species, where we consider all the independent sub-regions occupied by each element.
For binary nanoalloys, aside from how the atoms are radially distributed, a handy and easy quantity to characterise the chemical ordering is the relative distance between the centres of mass of the two chemical sub-regions, Δ(CoM). This quantity is a easy and robust descriptor to monitor the formation and the evolution to/from Janus and quasi-Janus chemical orderings.39
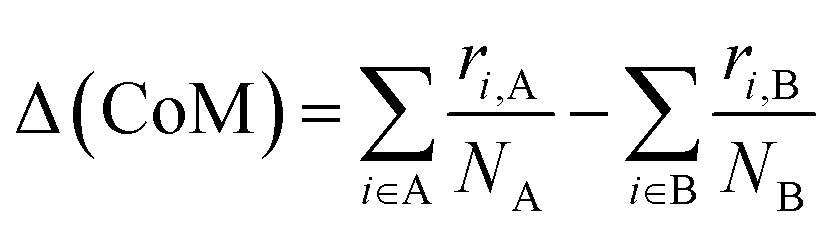 | (6) |
3.2 Adjacency matrix based descriptors
The definition and characterization of nearest neighbour networks has been largely adopted to classify MNPs' and NAs' morphology and draw structure–property relationships.40–42To evaluate nearest neighbour networks and quantities deriving from the latter, solely according to a distance criterion, the adjacency matrix A is defined as:
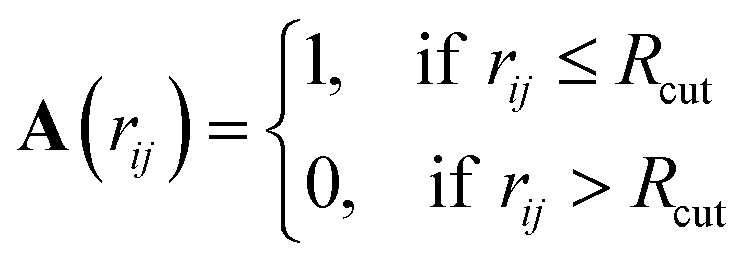 | (7) |
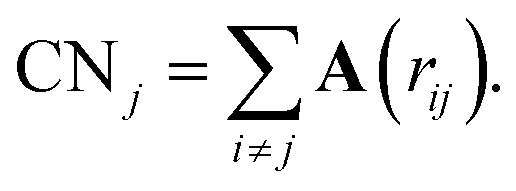 | (8) |
Nonetheless, this definition might suffer in finding a suitable cut-off that might not always be easy to select, especially for nanoalloys with a large mismatch.
As the identification of the local environment of each atom is of paramount importance, Sapphire also contains a solid-angle nearest-neighbour criterion, as in van Meel's paper43 (VMCN), to estimate, count, and list the nearest-neighbours of each atom i. The VMCN algorithm lists the nearest-neighbours of the atom i as the atoms j such that the solid-angle θi,j – defined by the cone with atom i at the apex and j at the center of the cone's base – equals 4π. The solid angle can be replaced by the ratio 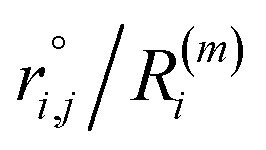 , where the inter-particle distances
, where the inter-particle distances  are ordered such that
are ordered such that 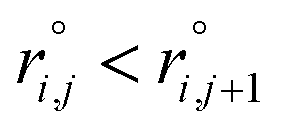 . R(m)i is the neighbour-shell radius,
. R(m)i is the neighbour-shell radius,
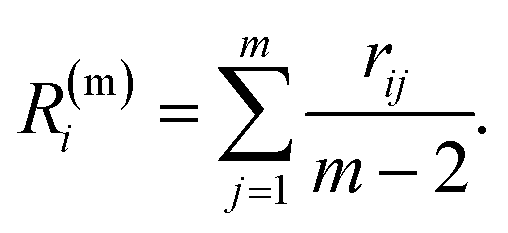 | (9) |
The minimum value of m, the number of neighbours, is 3. In a FCC bulk, the maximum is expected to be 12. The smallest m, ![[m with combining circumflex]](https://www.rsc.org/images/entities/i_char_006d_0302.gif) that satisfies
that satisfies 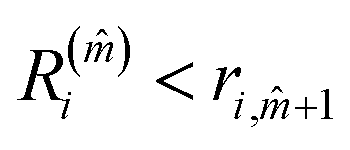 identifies the number of neighbours of the atom i, and hence its coordination. The corresponding
identifies the number of neighbours of the atom i, and hence its coordination. The corresponding 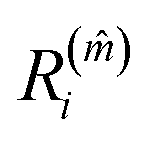 is the shell-radius that identifies the neighbourhood of atom i. The second step is to calculate the solid angle as
is the shell-radius that identifies the neighbourhood of atom i. The second step is to calculate the solid angle as
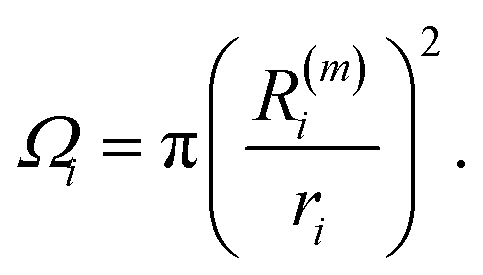 | (10) |
Both algorithms, the one with a fixed cut-off from the PDDF and the adjacency matrix, and the VMCN, can be used simultaneously to define the number of neighbours and hence to perform the following analysis.
The nominal CN definition and the one from van Meel's algorithm generally agree well. The latter tends to show higher values of the coordination than the former; see the lighter blue atoms in Fig. 4. In both cases, it is possible to have atoms that are more than 12-coordinated, even for metals that are FCC in the bulk. This is due to internal distortion and not to a faulty algorithm.
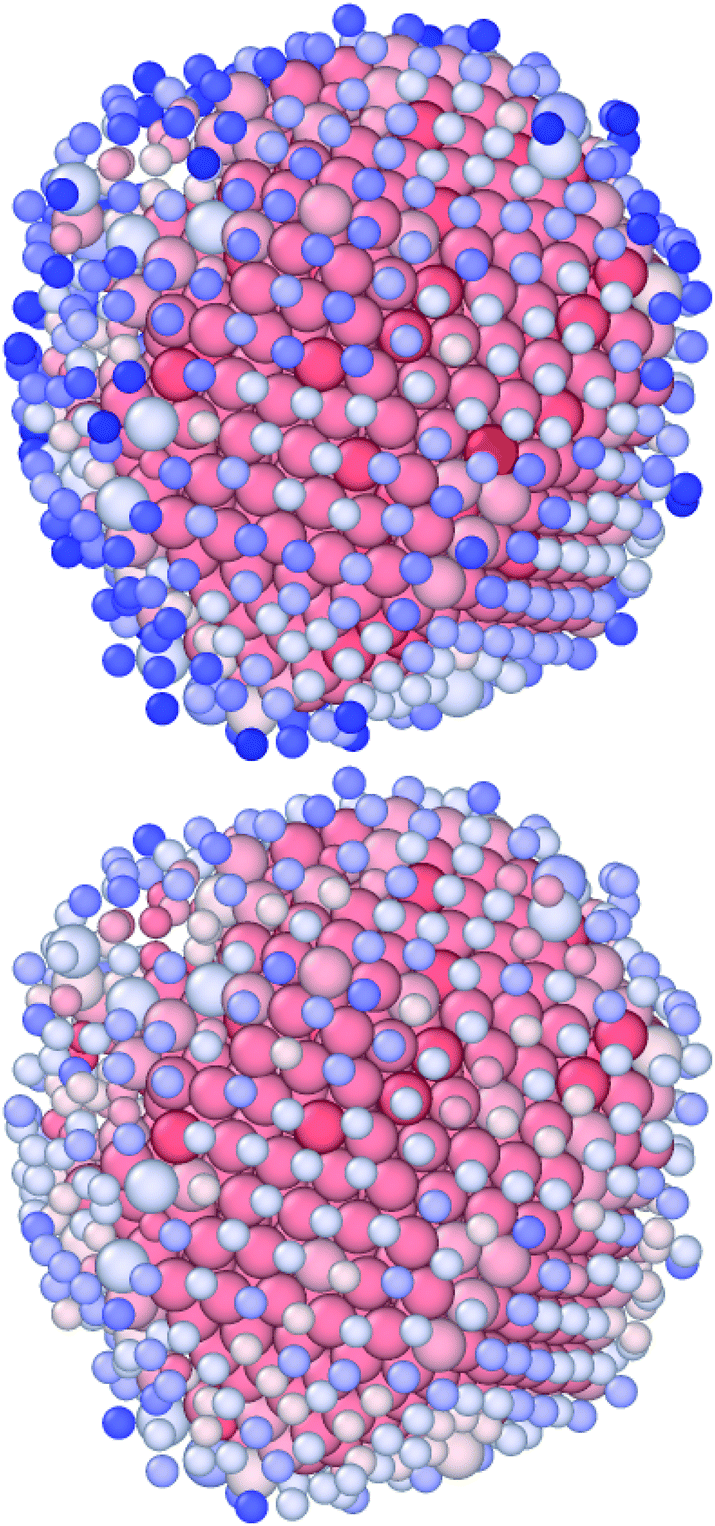 | ||
| Fig. 4 Au283Pt1132 NAs where atoms are coloured based on their nominal CN (top, with a cutoff of 3.55 Å) and the VMCN (bottom). Atoms are coloured with respect to their CN (or VMCN), ranging from 16 (full red) to 3 (dark blue). Au atoms are represented as smaller than Pt, to improve visualization. Minor discrepancies between the two algorithms occur. Using the solid angle condition, we count 561 atoms with a CN less than 10, 170 with CN = 11, and 684 with a CN above or equal to 12; using van Meel's algorithm, their occurrence is 477, 216, and 722, respectively. | ||
The atop generalised coordination number (aGCN)44 is defined as:
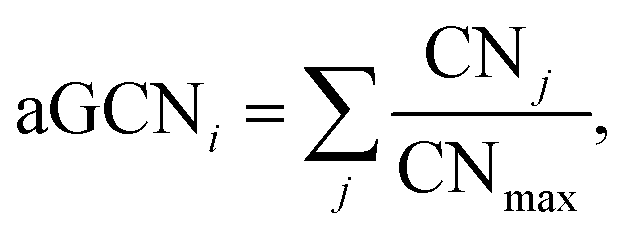 | (11) |
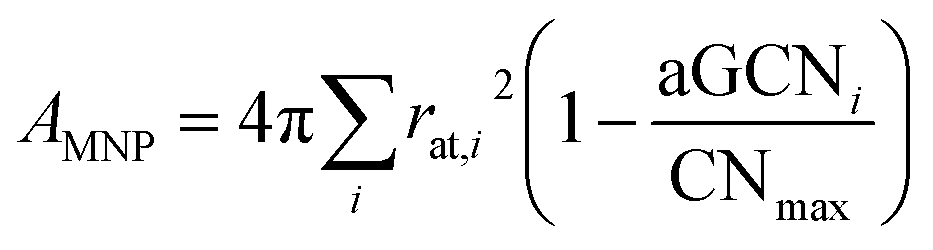 | (12) |
A full comparison of the surface area calculated for closed shell geometries using cluster spherical approximation, using geometrical consideration, and based on the aGCN has been reported in ref. 47.
In the case of nanoalloys, we can easily separate the local environment of each atom between homo-pairs and hetero-pairs. Counting the A–A, NBAA, B–B, NBBB, and A–B pairs, NBAB enables evaluation of the mixing parameter μ. The latter is a useful parameter for a fast characterization of the NA’s chemical ordering:
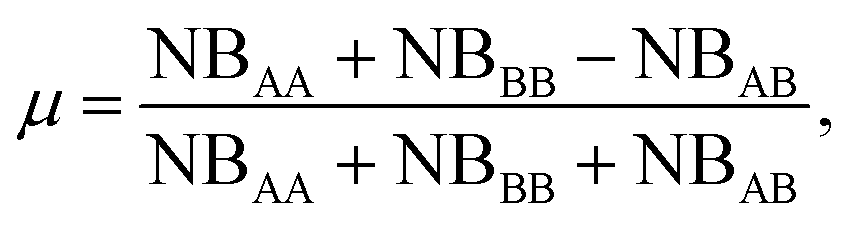 | (13) |
3.3 Concertedness and collectivity of a morphology rearrangement
Monitoring time-dependent changes in the adjacency matrix, over successive time-steps, allows characterization of whether structural rearrangements took place, and if these involve concerted and/or collective rearrangements. In this context, we adopt the definitions first put forward in ref. 35.Let AAij(Δt) be a matrix counting the absolute number of bonds formed or lost between each ij pair of atoms, within a characteristic time length Δt:
| AAij(Δt) = |Aij(t + Δt) − Aij(t)| | (14) |
From this quantity, the system mobility, R(t, t + Δt), is measured by summing over single atom mobility:
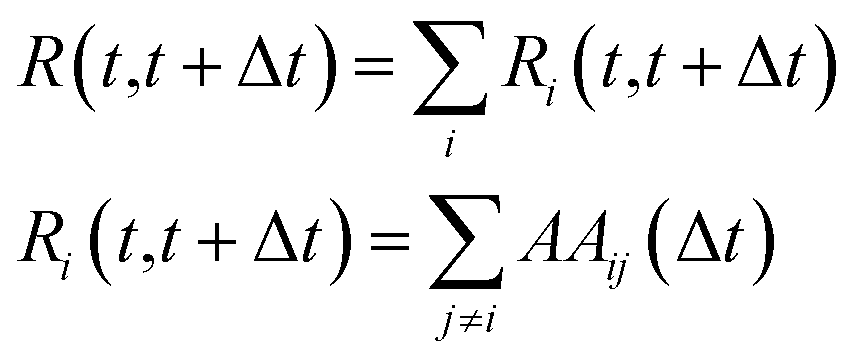 | (15) |
To measure the collectivity of a mechanism, H, one then calculates the ratio of atoms which change at least one neighbour within a Δt interval, and the total number of atoms in the MNP:
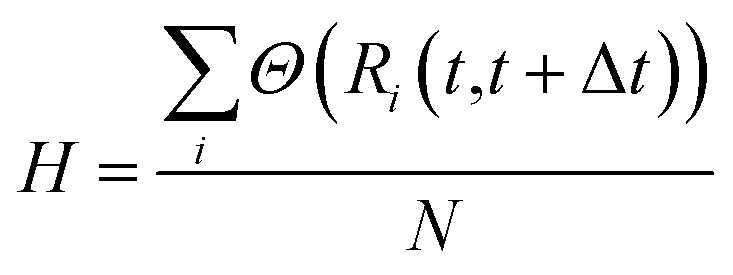 | (16) |
| C(t − Δt,t,t + Δt) = |H(t − Δt,t) − H(t,t + Δt)| | (17) |
When all the atoms in the MNP change their local connectivity at time t, but the connectivity remains stable at the successive time point t + Δt, C(t − Δt, t, t + Δt) reaches its maximum value, 1.
Note that all the descriptors of mobility, concertedness, and collectivity discussed in this section display a dependence on the magnitude of Δ(t). A too long Δt may affect the H and C estimate by coarsening many atomic rearrangements into a single one. A too short Δt may unfaithfully describe a single step rearrangement as a multi-step one. The suggested (and default) value for the choice of this quantity is Δ(t) = 10 ps, which is consistent with the time-scale of adatom diffusion on low Miller index surfaces. We consider the jumping of adatoms as one of the fastest mechanisms that can take place during MNPs’ structural rearrangements.
All the quantities discussed above can be readily modified to account for the presence of multiple chemical species, chem = AA, BB, AB, in the NA:
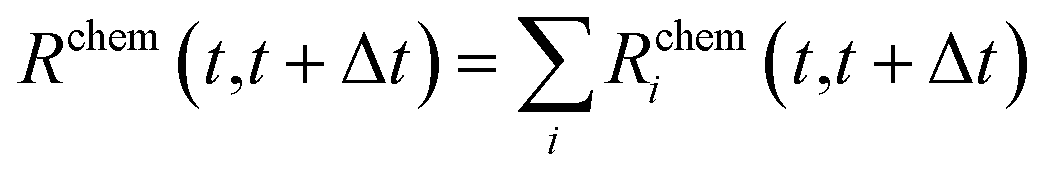 | (18) |
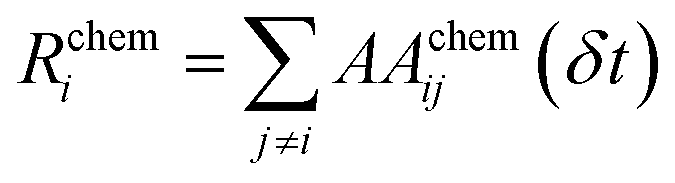 | (19) |
| Cchem(t − Δt,t,t + Δt) = |Hchem(t − Δt,t) − Hchem(t,t + Δt)|. | (20) |
Single-step mechanisms involving only a section of the cluster, or multi-step processes are characterized by lower values of C(t − Δt,t,t + Δt), while continuous atomic rearrangements display C ∼ 0. This is precisely exemplified in Fig. 6, which characterizes the concertedness and collectivity of rearrangements in the AuPd NA undergoing a solid–liquid phase change when heated above 700 K.
The C statistic is consistently near 0 whilst the H statistic monotonically increases as a function of temperature. This is indicative of a highly dynamical object whose constituent atoms are regularly rearranging with respect to one another. At each subsequent 10 ps window beyond 600 K, half of the atoms have a minimum of one change to their nearest-neighbour network. By the same token, we also draw attention to the sharp peak in the C statistic at the beginning of the dynamics. In this instance, this is representative of the collective motion of the Pt atoms within the cluster, which quickly rearrange into a more energetically favourable geometry shortly after thermal equilibration of the NA.
3.4 Common neighbour analysis: signatures and patterns
To classify the geometrical environments of core and surface atoms, we evaluate all of the common neighbour analysis (CNA) signatures attributed to each pair of nearest-neighbour atoms. CNA signatures are of the form (rst) such that r is the number of nearest-neighbours common to both atoms in the pair; s is the number of bonds between shared neighbours and t is the longest chain which can be made from bonding s atoms if they are nearest neighbours. While an individual CNA signature describes the local environment of a pair of neighbours, the estimate of the percentage of how many pairs contribute to selected CNA signatures provides information on the overall nanoparticle's structure, allowing a fast classification into the expected geometrical family, as icosahedral, decahedral, or FCC.48 The main CNA signatures are (555), (422), and (421) for chemical species with a FCC bulk lattice. Although varying with the NPs' size, a high concentration of (555) stands for Ih, while FCC morphologies are expected to have 0% of that signature. As seen for structural rearrangements, the nearest-neighbour pair can be labelled as homo (A–A and B–B type), and hetero (A–B) when needed. However, to describe the overall geometry of a NA, we weight A and B atoms in the same manner.While proven to be successful in many cases, the CNA is not a property of an atom i. Some commercial software, i.e.Ovito,49 offers a basic characterisation of each atom i looking at its relative contribution to a few known CNA signatures. Usually this criterion is able to classify only atoms in the core and when there is a clear character of each atomic environment.42Sapphire extends the idea behind the assignment of a local crystal structure to an atom using the common neighbour analysis.
First of all, we list all the possible CNA signatures and we limit them to only a few values important in the bulk. For each atom, we list all the CNA signatures it contributes to, and count how many times the same signature occurs (frequency chart). The local environment of each atom-i is, therefore, the collection of those signatures, and their frequency, into a CNA-pattern (CNAP). On top of the mapping following the CNAP, as highlighted in the snapshots reported in Table 1, we note that we can have insights on the NP-morphology on the basis of the number of different CNAPs. In principle, each atom has its own CNAP, leading to N independent CNA-patterns for a nanoparticle with N atoms. However, atoms in a similar environment will display the same CNAP. Therefore, the number of different CNA-patterns will decrease when the symmetry of the shape increases. Furthermore, with the CNAP being sensitive to the surface, and not only to the core, nanoparticles limited by different facets will have more CNAPs. With this is mind, an octahedron presents five different CNA patterns while a Marks decahedron has at least 11. Table 1 reports the CNAP distributions for common geometries classifying the most commonly observed CNAPs, and a short description of each CNAP. We do stress that no restriction to any specific signature occurs here. However, as the geometries shown are highly symmetric, overall we provide a description for a limited number of CNAPs. The CNAP is given as a string [(%i, (risiti))], where %i is the number of times the atom participates in the (rst) signature. The sum of %i is expected to be 12 if the atom i is in the core, and less for surface atoms as this sum is simply the coordination number of the atom with that given CNAP.
| Pattern | CNAP-label | Description | Surface |
|---|---|---|---|
| [(1,(100)), (2,(211)), (1,(322)), (1,(422))] | CNAP1 | Vertex from CNAP16 bordering two (111) facets and a (100) | Y |
| [(1,(200)), (2,(211)), (2,(311)), (1,(421))] | CNAP2 | Edge between a (100) and a slightly distorted (111) facet | Y |
| [(10,(422)), (2,(555))] | CNAP3 | Atoms lying on a (555) symmetry axis | N |
| [(12,(421))] | CNAP4 | FCC bulk | N |
| [(12,(555))] | CNAP5 | Interception of six five-fold axes | N |
| [(2,(100)), (2,(211)), (2,(422))] | CNAP6 | Edge between (100) facets | Y |
| [(2,(200)), (1,(300)), (2,(311)), (1,(322)), (1,(422))] | CNAP7 | Vertex lying on twinning planes shared by (111) facets | Y |
| [(2,(200)), (4,(311)), (1,(421))] | CNAP8 | Edge between (111) re-entrances and (111) facets | Y |
| [(2,(300)), (4,(311)), (2,(421)), (2,(422))] | CNAP9 | Re-entrance delimited by (111) facets | Both |
| [(3,(211)), (2,(311)), (2,(421))] | CNAP10 | Edge between (100) and (111) facets | Y |
| [(4,(211)), (1,(421))] | CNAP11 | Vertex shared by (100) and (111) facets | Y |
| [(4,(211)), (4,(421))] | CNAP12 | (100) facet | Y |
| [(4,(311)), (2,(322)), (2,(422))] | CNAP13 | Five-fold symmetry axes (no centre) | Y |
| [(5,(322)), (1,(555))] | CNAP14 | Five-fold vertex | Y |
| [(6,(311)), (3,(421))] | CNAP15 | (111) facet | Y |
| [(6,(421)), (6,(422))] | CNAP16 | Twinning planes | N |
Given the evident sensitivity to local geometry, the next question to ask is how sensitive CNA-patterns are to imperfections in the crystalline structure and if they could be used to classify a MNP or NA into a certain family. While we discussed already how we expect the number of independent CNAPs to be dependent on the geometrical symmetry, here we would like to stress that similar edges such as the one between (100) and (111) can be identified by two patterns, namely CNAP2 and CNAP10, depending on whether they are from a Dh structure or from a FCC structure. Generally speaking, an automatic classification of NAs into the distinguished three main morphology families, namely FCC-like, decahedra (Dh), and icosahedra (Ih), is desirable. The CNA-patterns [10,(422), 2,(555)]; [4,(311), 2,(322), 2,(422)] occur in both Dh and Ih. The latter, if sufficiently symmetric, has a [12,(555)] pattern too. Line displacement in a FCC arrangement arises as a mixture of [(l,(422)), (k,(421))] with different l, k frequency, such as l + k = 12.
4 Surface identification
As many physical and chemical processes occur at the surface,50–52 an automated classification of which atoms are at the surface, subsurface, and core for a given NA is welcome. The classification of core and subsurface atoms will be facile following a “peeling” of the outermost layer. Core atoms are designated as those atoms that do not belong to the surface or sub-surface. The reader should note that combining the identification of atoms in the surface, subsurface, and core shells will permit the calculation of other atomistic properties, i.e., those related to the energy when calculated as atomistic contributions.While on perfectly built geometries the identification of atoms at the surface follows their coordination number, i.e., imposing CN ≤ 10 to define atoms at the surface. For unknown, low-symmetry, or amorphous morphologies, this task is far from trivial. Any classification simply based on the coordination number fails, independently of how the coordination number is calculated, as both low and high coordinated atoms can be present at the surface; see the heatmap in Fig. 5.
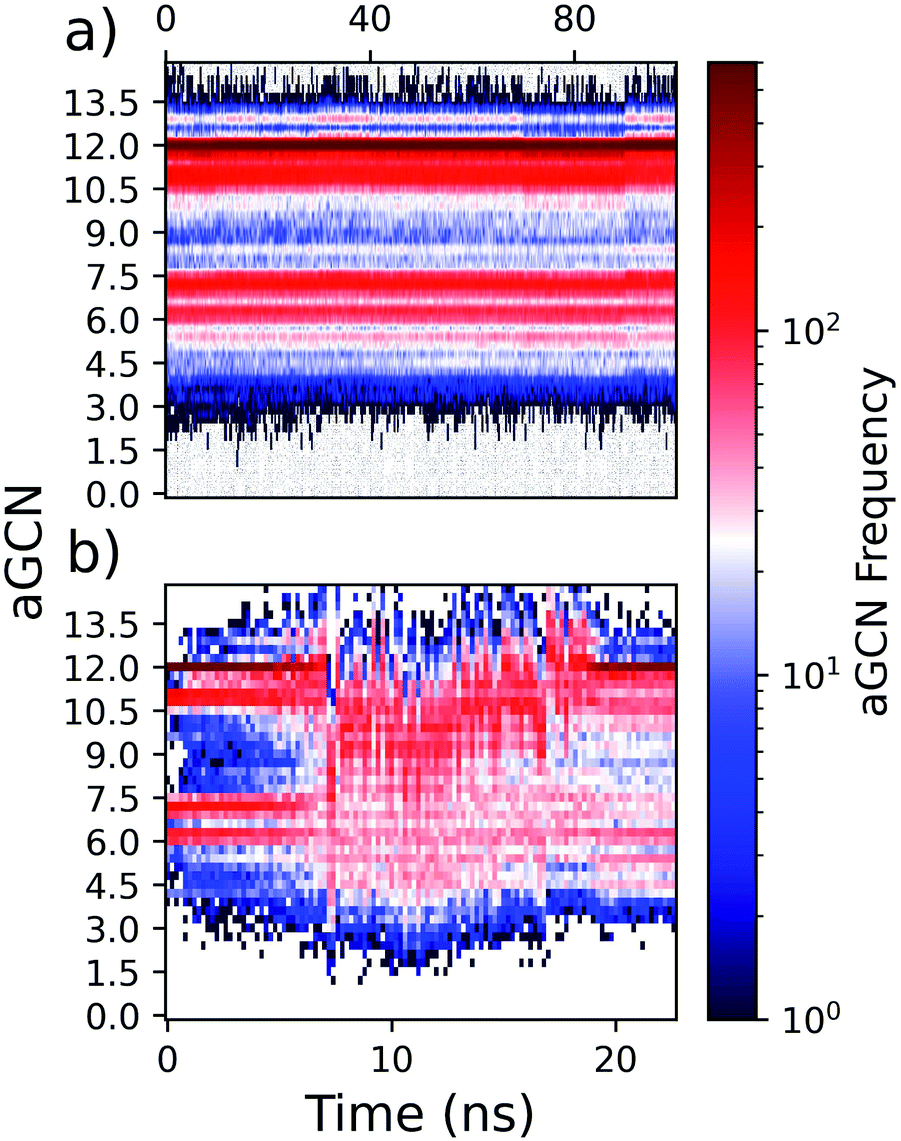 | ||
| Fig. 5 Heat-maps demonstrating the evolution of the aGCN distribution, as calculated via the adjacency matrix, with respect to time for two types of dynamical processes. Panel (a): two PtIh55 NAs deposited atop an AuIh2057 MNP maintained at 600 K for 100 ns. Panel (b): randomly alloyed Au1283Pt132 NAs with the initial morphology of an Ih heated from 300 K (t = 0) to 1000 K (t = 12 ns) and then cooled back to 300 K (t = 24 ns). | ||
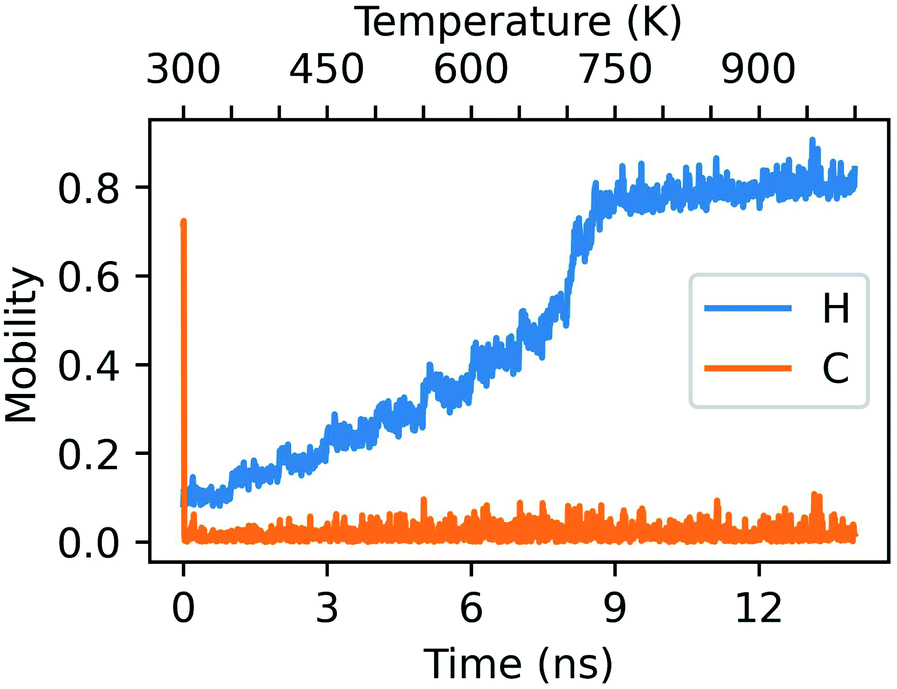 | ||
| Fig. 6 Mobility descriptors for a randomly alloyed Au1149Pt266 NA in the configuration of an Ih which has been rapidly heated from 300 K to 1000 K over the span of 14 ns. Δt has been set to 10 ps. | ||
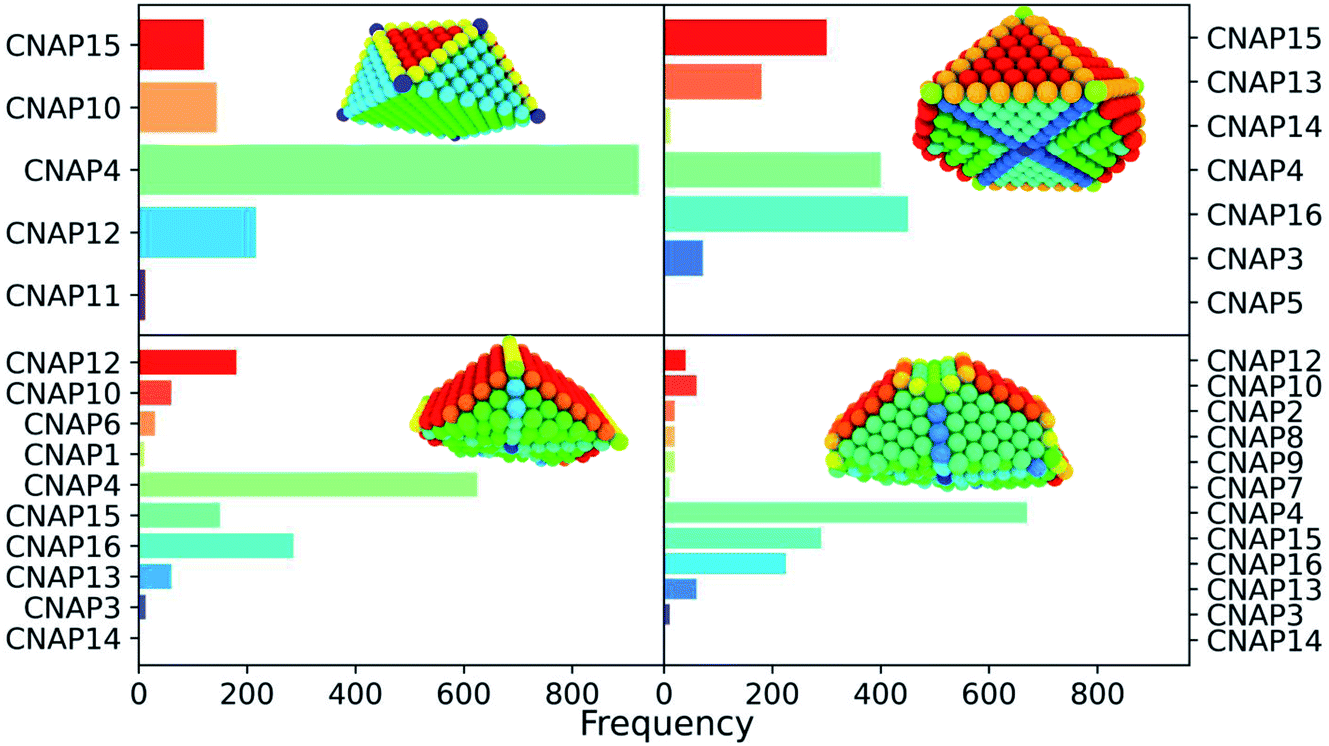 | ||
| Fig. 7 Frequency chart of regularly-occurring CNA patterns in closed-shell geometries, namely a cuboctahedron (Co), an icosahedron (Ih), an Ino-decahedron (InoDh) with 1415 atoms, and a Marks-decahedron (mDh) with 1428 atoms. Snapshots of cut-in-half MNPs are shown in the insets, and atoms are coloured in agreement with their CNA-pattern, as listed in Table 1. | ||
A combination of a condition on CN and radial distance will work only for spherical MNPs, but will fail for prolate/oblate systems. Hence, there is a need to find alternative approaches which require no a priori information on the target system. For these reasons, we provide three algorithms to identify, list, and classify atoms at the surface. (i) The first is based on imposing a threshold on the solid angle Ωi, as calculated viaeqn (10), combined with the radial distance of atoms i. This method reproduces data for closed shell geometries, and provides a good estimate for the surface sites of melted/low symmetry MNPs and NAs as illustrated in Fig. 4. However, it suffers from the introduction of a threshold value on the solid angle which depends inversely on the size of the MNP.
(ii) Sapphire offers the “Layers algorithm”, usually adopted to calculate the accessible area in macromolecules.53 This algorithm creates three cylinders for each atom along the x, y, z Cartesian components, and records the lowest and highest atoms in each cylinder. In this case, there is only a free parameter tuning the radius of the cylinder. As a rule of thumb, choosing 1.5 times the atomic radius (or times the average atomic radius for binary systems) provides a good definition. A comparison of the two algorithms is shown in Fig. 8 for Au and CuPt cases with 1415 atoms.
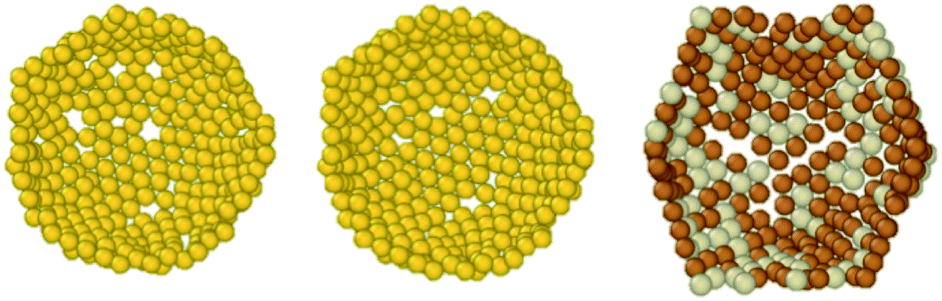 | ||
| Fig. 8 Comparison between the solid-angle (left) and the Layers (middle and right) algorithms to identify the surface layer. Here we display only half of the external shell of low symmetry Au1415. We use the condition on the solid angle as Ωi ≤ 0.05 combined with ri ≥ 15 Å (left). On the right, the Layers method is applied to low symmetry CuPt1415 with a chemical composition of 0.3 for Pt (silver-ish coloured and Cu is orange). | ||
(iii) The final algorithm is a clustering approach based on local atomic environment descriptors.37 The clustering approach was used to classify atoms in Au NPs, and implemented in the RAFFY Python package,54 and labels atoms in an unsupervised manner via hierarchical k-means clustering. The atoms from one or more MD snapshots are fingerprinted using local descriptors based on the atomic cluster expansion55 (ACE) framework. The latter encodes information on local atomic environments by approximating the local atomic density via spherical harmonics and radial basis expansions, and then constructing rotation-invariant representations from the coefficients of such expansions. A cut-off radius, alongside other parameters, must be specified for the ACE descriptors; typical cut-off values are the distances between the second and third shells of neighbours. This approach unbiasedly distinguishes between low-coordination surfaces, highly coordinated surfaces, and bulk atoms.37 Moreover, it enables the discernment of locally melted and locally solid atomic environments, therefore providing a robust measure of melting and surface rearrangement temperatures.
5 Averaging routes
There is support for creating plots for a single analysed trajectory or to consider the average over multiple, independent simulations. One can toggle between these alternatives by setting the single_file flag to be true and iter_dir to be false, or by setting the single_file flag to be false and setting the iter_dir flag to be a list of relative paths from the defined base directory to each of the iterations to be considered.It should be noted that Sapphire provides the arithmetic average for each of the requested quantities. Caution is advised to users who take advantage of this feature as it may not always be meaningful to compute or present such averages. Indeed, these meta-analyses are only meaningful if the ensemble being sampled across is consistent over time, and if the dynamics are at equilibrium.
When reporting and presenting these ensemble averages, Sapphire computes uncertainties across samples as the standard error. That is to say that across the ensemble, we assume that all deviations are independently and identically distributed (i.i.d.). Indeed, the former is trivial to guarantee if multiple independent realisations of a given dynamical process have been computed. For the extensive phase space of dynamical systems, a Boltzmann distribution is an adequate candidate for systems close to equilibrium. However, if one is intending to probe non-equilibrium dynamics for which a steady state distribution cannot be uniquely defined at a given point in the dynamics, then the choice of distribution for describing the observables as random variables becomes more nuanced and additional care is necessary to ensure that the phase space has been adequately sampled. Nonetheless, when the dynamics of the system are not far from equilibrium, this scheme provides a fast and intuitive method to visualise variance and spread in the descriptors. We have elected to present a AuPt system to demonstrate the utility of the Sapphire averaging scheme.
Fig. 9 reports four ensemble averaged quantities for coalesced AuIh923PtIh55 held at 450 K for 500 ns. Classical molecular dynamics simulations are done using the software package LoDiS.36 This temperature is too low to activate structural rearrangements within the timescale sampled by the molecular dynamics trajectory.
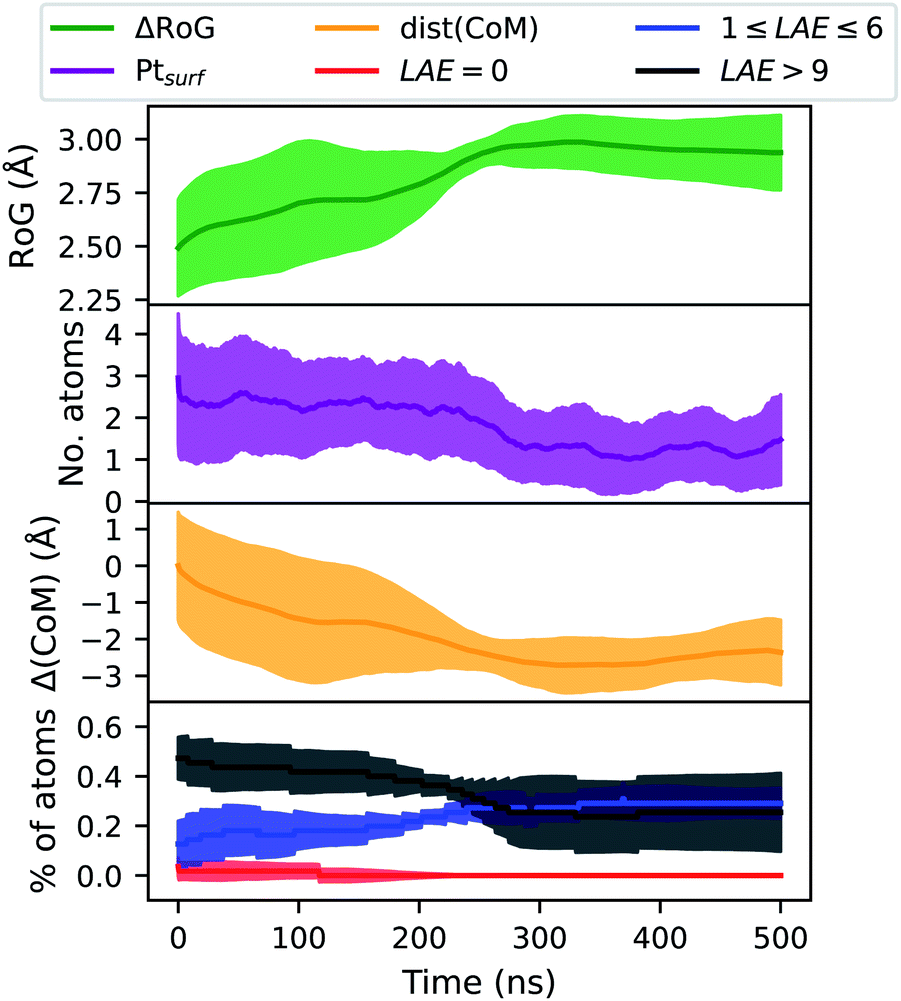 | ||
| Fig. 9 Ensemble averaged evolution of atomic descriptors for a coalesced system of AuIh923PtIh55 thermalised at 600 K for 500 ns. From top to bottom, the radius of gyration of the Pt sub-region only; the number of Pt atoms at the surface, based on the definition from the CNAP; the Δ(CoM) between the centres of mass of the two sub-regions; and the LAE of Pt atoms. | ||
We define the local heterogeneous atomic environment (LAE) as being the percentage of species B = Pt with LAE neighbours of species A = Au. LAE = 0 indicates that these atoms create no heterogeneous bonds. 1 ≤ LAE ≤ 6 describes a mixed environment; while LAE > 9 suggests that these atoms (Pt) are almost totally encapsulated by Au atoms. Initially, we note that all descriptors are found to have only small inter-sample variations at approximately 10% of the mean value. All the descriptors considered, namely the radius of gyration (RoG), the number of Pt atoms at the surface (Ptsurf), local atomic environment (LAE) of Pt, and the distance between the sub-region centres of mass Δ(CoM), versus time, show that a structural transformation occurs at the beginning (below 100 ns), with Pt incorporating inside the Au core and decreasing the number of Pt atoms at the surface. At the same time, we recognise two other steps. At the small plateau between 100 and 200 ns where Δ(CoM) is almost flat a small change in the LAE occurs. After 200 ns, we observe an increment in the Pt mobility and a change in the chemical distribution of Pt atoms in the NA.
We do not present these averaging techniques to serve as a replacement for an individual's choice of averaging routine, as these are unique and particular to the dynamics and quantities under consideration. Rather, we present this utility as a means to explore and present variations across i.i.d. samples.
6 Structure–property relationships
The Sapphire library focuses on the characterisation of MNP and NA morphology. Nonetheless, Sapphire can be used in synergy with available codes geared toward the prediction of chemophysical properties of MNPs and NAs. In the next sub-sections we briefly discuss three examples, where:• Sapphire leverages the ASE environment to move back to energy-related properties;
• Sapphire exploits a link with a microkinetic model56 to estimate the activity over certain reduction reactions;
• Sapphire is connected with the pyGDM57 library to qualitatively estimate the optical properties, and absorption and extinction spectra, of mono- and bimetallic nanoparticles.
6.1 From structure to energy
Often it is desirable to check the energy stability of a certain shape or chemical ordering. Currently, ASE supports effective medium (EMT), the embedded atom method (EAM), and the kimpy calculators. EAM benefits from the Interatomic Potentials Repository Project available at https://www.ctcms.nist.gov/potentials/, while OpenKIM is a recent NSF project devoted to providing access to classical interatomic potentials. Unfortunately, second moment approximations of the tight binding (SMATB) calculators are not currently available. They were part of the ASAP3 distribution,58 which has since been deprecated and discontinued. In the manual and tutorials we demonstrate how Sapphire may interface with ASE and its library of calculators to compute energetics. Given the broad support for potential calculators in the ASE distribution, one is at liberty to select their own theory of choice for their particular system.6.2 Electrocatalytic properties via a microkinetic model
Let α be the value of a descriptor which encodes an explicit scaling between the structural properties of an adsorption site and the reaction free energy associated to the rate-limiting step of an electrochemical reaction occurring at that site. A simple micro-kinetic model for the current density jnanoparticle(t,T,U) produced by a nanoparticle, for such an electro-chemical process, can be written as: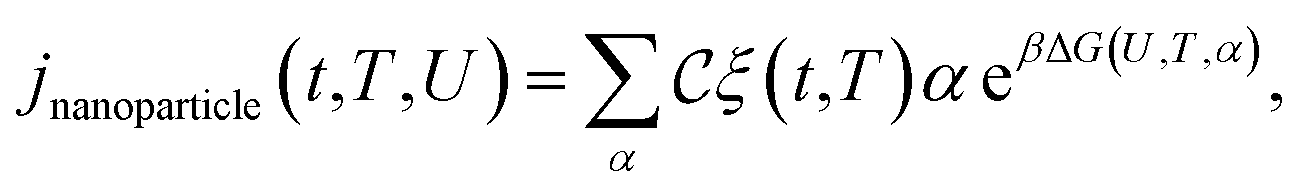 | (21) |
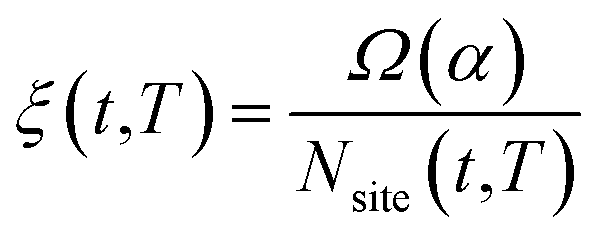 is the fraction of non-equivalent adsorption sites Fnanoparticle to the total number of sites available, Nsite, and the sum runs over the collection of the non-equivalent sites appearing in the MNP under consideration. The latter are also categorized by their α values.
is the fraction of non-equivalent adsorption sites Fnanoparticle to the total number of sites available, Nsite, and the sum runs over the collection of the non-equivalent sites appearing in the MNP under consideration. The latter are also categorized by their α values.
Note that in eqn (21) the effect of the potential is treated as an a posteriori correction, in line with the computational hydrogen electrode model.59 The aGCN, eqn (11), of a surface site is an accurate descriptor to predict the activity of a metallic adsorption site, as demonstrated for Au, Cu, Pt, and PtNi, catalysing the electrochemical reduction of oxygen or carbon dioxide.44,45,60 Assuming a volcano-plot relationship, the reaction free energy ΔG and the aGCN are related by the following general expression:
 | (22) |
Once the Gibbs free energy is defined in terms of the aGCN, a quantity that can be monitored during the lifetime of a MNP, we can establish relationships between the ageing of a MNP, referring to its structural stability, and its activity. Examples have been reported previously by our group for the ORR on Pt,47 and for the conversion of CO2 into methane on Cu NPs.56 Under the assumption of identifying robust structure–activity relationships, we do not see any limitation in using the scheme for NAs and for other chemical reactions.
6.3 Optical properties via semi-classical methods
For a fast evaluation and screening of the extinction spectra of MNPs and NAs, one can adopt a classical approach, via Green's Dyadic Method (GDM),57,62 as implemented in the pyGDM code.63,64 At its core, this library constructs a refractive environment via the construction of a 3D mesh of dipole oscillators whose physics is predicted within the quasi-static coupled dipole approximation. It then uses an efficient and generalised propagator to predict the extinction spectrum associated to the user-defined mesh. For example, this mesh may be defined from a series of coordinates consisting of multiple constitutive components.To evaluate the extinction spectrum, we need only to consider the interaction of the dipole moment and total field in each discretised volume cell:
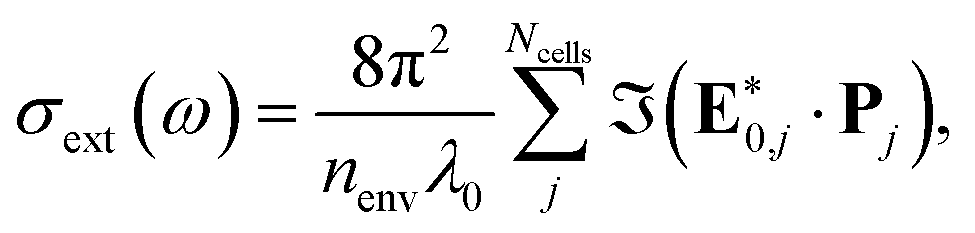 | (23) |
where GEEtot(ri,rj,ω) is the Green dyadic to be solved, χ is the metal's susceptibility, and Vcell is the volume of a unit cell. From this field, we then calculate the effective dipole moments, Pj = Vcellχj·Ej, from the internal field distribution.

From a knowledge of the functional form of the incident field, and a sufficiently converged internal field distribution, one is able to compute the extinction spectrum for a given finite system held within a dispersive medium, as described by eqn (23). All metallic dielectric parameters used in these calculations are provided by either experimental work,66–68 or ab initio investigations.69 The interested reader is referred to previous literature for further detail.57,63,64
Any behaviour that may arise from quantum many-body effects is neglected. Furthermore, when considering structures with size ∼ 1 nm illuminated in the ultraviolet-visible-near-infrared, the incident field will only weakly couple to the structure. This results in non-trivial internal field enhancement.
Fig. 10 reports the dipole mesh reconstructed from atomic coordinates and species in the left set of panels, and the near-field enhancement at 520 nm plane wave illumination on the right side. Intensity, as described by the colour bar, is reported as |E(r)|2/|E0|2, the strength of the field relative to the amplitude of the illuminating wave. In the bottom panel, we present the extinction spectra computed viaeqn (23) for each of the four different systems taken into consideration. We have elected to present these systems to demonstrate the utility in being able to directly map a set of atomic coordinates, as may be parsed from a structural file, to a mesh of coupled dipoles with pre-defined dielectric properties.
 | ||
| Fig. 10 An illustration of the interaction between Sapphire and pyGDM. Panel (a): AuIh1132PtIh283 consisting of Au (orange), and Pt (green) showing a Janus chemical ordering. Panel (c): AuIh1132PtIh283 consisting of Au (orange), and Pt (green) showing a shell–core chemical ordering. Panels (e) and (g): pure MNPs of Au1415 and Pt1415, respectively. Panels (b), (d), (f) and (h): near-field enhancement of the structure when illuminated by plane waves at 550 nm. Panel (i): photo-extinction spectra as computed viaeqn (23) for the examples showcased in panels (a) (black), (c) (blue), (e) (red), and (g) (green). | ||
7 Conclusions
The characterization and classification of the morphology and chemical ordering in mono-, bi-, and multi-metallic NAs is a key ingredient towards rationalizing their chemo-physical properties.The necessity of an open-source robust, reproducible, and FAIR compliant post-processing tool, which caters to the needs of the nanoalloy computational modelling community, is more and more evident. Sapphire, the software we presented here, is tailored specifically to address this need, by providing a library of standardised analysis tools for NA characterization. Sapphire is indeed an open-source, modular, user-friendly platform to characterise a nanoparticle's or NA’s architecture observed during, e.g., atom coordinate reconstructions from experiment, molecular dynamics, or Monte Carlo based sampling.
Within Sapphire, several order-parameters and descriptors can be calculated, namely: pair distance and radial distribution functions, inertia tensors and quantities derived from the latter, coordination distributions and other topological descriptors (generalized coordination and common neighbour analysis) derived from adjacency matrices. Sapphire also offers tools for detailed analysis of the surface of MNPs and NAs. In the long term, it is our vision that Sapphire may serve to create a standardised, meaningful characterisation and classification tool for MNPs and NAs. This will be the first critical stage in creating a comprehensive community database for such complex systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project is funded by the Engineering and Physical Sciences Research Council. Via our membership of the UK's HEC Materials Chemistry Consortium, which is funded by the EPSRC (EP/L000202), this work used the UK Materials and Molecular Modelling Hub for computational resources, MMM Hub, which is partially funded by the EPSRC (EP/P020194 and EP/T022213). This work used the ARCHER2 UK National Supercomputing Service (https://www.archer2.ac.uk). R. Jones acknowledges funding by the Engineering and Physical Sciences Research Council (EPSRC) through the Centre for Doctoral Training Cross-Disciplinary Approaches to Non-Equilibrium Systems (CANES, Grant No. EP/L015854/1).Notes and references
- B. A. Prabowo, A. Purwidyantri and K.-C. Liu, Surface plasmon resonance optical sensor: A review on light source technology, Biosensors, 2018, 8, 3 CrossRef PubMed.
- A. A. Yaqoob, H. Ahmad, T. Parveen, A. Ahmad, M. Oves, I. M. I. Ismail, H. A. Qari, K. Umar and M. N. Mohamad Ibrahim, Recent advances in metal decorated nanomaterials and their various biological applications: A review, Front. Chem., 2020, 8, 2296–2646 Search PubMed.
- M. Rahman, K. Alam, A. Hafeez, R. Ilyas and S. Beg, Chapter 7 – metallic nanoparticles in drug delivery and cancer treatment, in Nanoformulation Strategies for Cancer Treatment, ed. S. Beg, M. Rahman, H. Choudhry, E. B. Souto and F. J. Ahmad, Micro and Nano Technologies, Elsevier, 2021, pp. 107–119 Search PubMed.
- H. Chen, Y. Zhou and S.-T. Han, Recent advances in metal nanoparticle-based floating gate memory, Nano Sel., 2021, 2(7), 1245–1265 CrossRef CAS.
- H. Ditlbacher, J. R. Krenn, B. Lamprecht, A. Leitner and F. R. Aussenegg, Spectrally coded optical data storage by metal nanoparticles, Opt. Lett., 2000, 25, 563–565 CrossRef CAS.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, The optical properties of metal nanoparticles: The influence of size, shape, and dielectric environment, J. Phys. Chem. B, 2003, 107(3), 668–677 CrossRef CAS.
- E. A. Coronado, E. R. Encina and F. D. Stefani, Optical properties of metallic nanoparticles: manipulating light, heat and forces at the nanoscale, Nanoscale, 2011, 3, 4042–4059 RSC.
- J.-D. Xiao and H.-L. Jiang, Metal–organic frameworks for photocatalysis and photothermal catalysis, Acc. Chem. Res., 2019, 52(2), 356–366 CrossRef CAS.
- D. Mateo, J. L. Cerrillo, S. Durini and J. Gascon, Fundamentals and applications of photo-thermal catalysis, Chem. Soc. Rev., 2021, 50, 2173–2210 RSC.
- K. Lorber and P. Djinović, Accelerating photo-thermal CO2 reduction to CO, CH4 or methanol over metal/oxide semiconductor catalysts, iScience, 2022, 25(4), 104107 CrossRef CAS PubMed.
- Y. Tang and W. Cheng, Key parameters governing metallic nanoparticle electrocatalysis, Nanoscale, 2015, 7, 16151–16164 RSC.
- S. D. Lacey, Q. Dong, Z. Huang, J. Luo, H. Xie, Z. Lin, D. J. Kirsch, V. Vattipalli, C. Povinelli, W. Fan, R. Shahbazian-Yassar, D. Wang and L. Hu, Stable multimetallic nanoparticles for oxygen electrocatalysis, Nano Lett., 2019, 19(8), 5149–5158 CrossRef CAS PubMed , PMID: 31313586..
- J. Yu, Q. Liu, W. Qiao, D. Lv, Y. Li, C. Liu, Y. Yu, Y. Li, H. Niemantsverdriet, B. Zhang and R. Su, Catalytic role of metal nanoparticles in selectivity control over photodehydrogenative coupling of primary amines to imines and secondary amines, ACS Catal., 2021, 11(11), 6656–6661 CrossRef CAS.
- H. Song, Metal hybrid nanoparticles for catalytic organic and photochemical transformations, Acc. Chem. Res., 2015, 48(3), 491–499 CrossRef CAS , PMID: 25730414..
- T. P. Rossi, P. Erhart and M. Kuisma, Hot-carrier generation in plasmonic nanoparticles: The importance of atomic structure, ACS Nano, 2020, 14(8), 9963–9971 CrossRef CAS.
- V. Amendola, R. Pilot, M. Frasconi, O. M. Maragò and M. A. Iatì, Surface plasmon resonance in gold nanoparticles: a review, J. Phys.: Condens. Matter, 2017, 29(20), 203002 CrossRef PubMed.
- V. G. Rao, U. Aslam and S. Linic, Chemical requirement for extracting energetic charge carriers from plasmonic metal nanoparticles to perform electron-transfer reactions, J. Am. Chem. Soc., 2019, 141(1), 643–647 CrossRef CAS PubMed.
- W. Hong and C. W. Li, Microstructural evolution of AuPt core–shell nanoparticles under electrochemical polarization, ACS Appl. Mater. Interfaces, 2019, 11(34), 30977–30986 CrossRef CAS.
- L.-M. Guo, D.-F. Zhang and L. Guo, Structure design reveals the role of Au for ORR catalytic performance optimization in PtCo-based catalysts, Adv. Funct. Mater., 2020, 30, 2001575 CrossRef CAS.
- M. S. P. Jagannath, S. Divi and A. Chatterjee, Kinetic map for destabilization of Pt-skin Au nanoparticles via atomic scale rearrangements, J. Phys. Chem. C, 2018, 122(45), 26214–26225 CrossRef CAS.
- D. Chen, Z. Lai, J. Zhang, J. Chen, P. Hu and H. Wang, Gold segregation improves electrocatalytic activity of icosahedron Au@Pt nanocluster: Insights from machine learning, Chin. J. Chem., 2021, 39(11), 3029–3036 CrossRef CAS.
- V. Ramasubramani, B. D. Dice, E. S. Harper, M. P. Spellings, J. A. Anderson and S. C. Glotzer, freud: A software suite for high throughput analysis of particle simulation data, Comput. Phys. Commun., 2020, 254, 107275 CrossRef CAS.
- R. J. Gowers, M. Linke, J. Barnoud, T. J. E. Reddy, M. N. Melo, S. L. Seyler, D. Jan, D. L. Dotson, S. Buchoux, I. M. Kenney and B. Oliver, MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations, in Proceedings of the 15th Python in Science Conference, ed. S. Benthall and S. Rostrup, 2016, pp. 98–105 Search PubMed.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, Mdanalysis: A toolkit for the analysis of molecular dynamics simulations, J. Comput. Chem., 2011, 32(10), 2319–2327 CrossRef CAS PubMed.
- R. M. Jones, Sapphire, 2022, https://github.com/kcl-tscm/Sapphire.git Search PubMed.
- C. Draxl and M. Scheffler, “Nomad: The Fair Concept for Big-Data-Driven Materials Science”, 2018 Search PubMed.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, The atomic simulation environment—a python library for working with atoms, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef.
- B. Stiller, T. Bocek, F. Hecht, G. Machado, P. Racz and M. Waldburger, Mobile Systems IV, Tech. Rep., University of Zurich, Department of Informatics, 2010 Search PubMed.
- N. Adamovic, Documentation on Materials Modelling Ontology in UML, Tech. Rep., TU WIEN, 2016 Search PubMed.
- R. B. Neder and V. I. Korsunskiy, Structure of nanoparticles from powder diffraction data using the pair distribution function, J. Phys.: Condens. Matter, 2005, 17, S125–S134 CrossRef CAS.
- R. B. Neder and T. Proffen, Exact and fast calculation of the X-ray pair distribution function, J. Appl. Crystallogr., 2020, 53, 710–721 CrossRef CAS.
- V. A. Epanechnikov, Non-parametric estimation of a multivariate probability density, Theory Probab. Its Appl., 1969, 14(1), 153–158 CrossRef.
- L. Delgado-Callico, K. Rossi, R. Pinto-Miles, P. Salzbrenner and F. Baletto, A universal signature in the melting of metallic nanoparticles, Nanoscale, 2021, 13(2), 1172–1180 RSC.
- K. Rossi, L. Pavan, Y. Soon and F. Baletto, The effect of size and composition on structural transitions in monometallic nanoparticles, Eur. Phys. J. B, 2018, 91, 33 CrossRef.
- K. Rossi, L. B. Pártay, G. Csányi and F. Baletto, Thermodynamics of CuPt nanoalloys, Sci. Rep., 2018, 8, 9150 CrossRef CAS PubMed.
- F. Baletto, Lodis, 2021, https://github.com/kcl-tscm/LoDiS Search PubMed.
- C. Zeni, K. Rossi, T. Pavloudis, J. Kioseoglou, S. de Gironcoli, R. E. Palmer and F. Baletto, Data-driven simulation and characterisation of gold nanoparticle melting, Nat. Commun., 2021, 12(1) CAS.
- Powder Diffraction, ed. R. E. Dinnebier and S. J. L. Billinge, The Royal Society of Chemistry, 2008 Search PubMed.
- B. Farkaš and N. H. de Leeuw, AuCo nanoparticles: ordering, magnetisation, and morphology trends predicted by DFT, Phys. Chem. Chem. Phys., 2022, 24, 10451–10464 RSC.
- S. Mourdikoudis, R. M. Pallares and N. T. K. Thanh, Characterization techniques for nanoparticles: comparison and complementarity upon studying nanoparticle properties, Nanoscale, 2018, 10, 12871–12934 RSC.
- D. E. Perea, I. Arslan, J. Liu, Z. Ristanović, L. Kovarik, B. W. Arey, J. A. Lercher, S. R. Bare and B. M. Weckhuysen, Determining the location and nearest neighbours of aluminium in zeolites with atom probe tomography, Nat. Commun., 2015, 6, 7589 CrossRef PubMed.
- A. Stukowski, Structure identification methods for atomistic simulations of crystalline materials, Modell. Simul. Mater. Sci. Eng., 2012, 20(4), 045021 CrossRef.
- J. A. van Meel, L. Filion, C. Valeriani and D. Frenkel, A parameter-free, solid-angle based, nearest-neighbor algorithm, J. Chem. Phys., 2012, 136, 234107 CrossRef PubMed.
- F. Calle-Vallejo, J. I. Martínez, J. M. García-Lastra, P. Sautet and D. Loffreda, Fast prediction of adsorption properties for platinum nanocatalysts with generalized coordination numbers, Angew. Chem., Int. Ed., 2014, 53(32) Search PubMed.
- F. Calle-Vallejo, D. Loffreda, M. T. M. Koper and P. Sautet, Introducing structural sensitivity into adsorption–energy scaling relations by means of coordination numbers, Nat. Chem., 2015, 7, 403–410 CrossRef CAS PubMed.
- K. Rossi, G. G. Asara and F. Baletto, A genomic characterisation of monometallic nanoparticles, Phys. Chem. Chem. Phys., 2019, 21(9) RSC.
- K. Rossi, G. G. Asara and F. Baletto, Structural screening and design of platinum nanosamples for oxygen reduction, ACS Catal., 2020, 10(6), 3911–3920 CrossRef CAS.
- F. Baletto, Structural properties of sub-nanometer metallic clusters, J. Phys.: Condens. Matter, 2019, 31, 113001 CrossRef PubMed.
- A. Stukowski, Visualization and analysis of atomistic simulation data with OVITO-the Open Visualization Tool, Modell. Simul. Mater. Sci. Eng., 2010, 18, 015012 CrossRef.
- J. Greeley, I. Stephens, A. Bandarenka, T. Johansson, H. Hansen, I. Chorkendorff and J. Norskov, Alloys of platinum and early transition metals as oxygen reduction electrocatalysts, Nat. Chem., 2009, 1, 552 CrossRef CAS PubMed.
- T. P. Rossi, P. Erhart and M. Kuisma, Hot-carrier generation in plasmonic nanoparticles: The importance of atomic structure, ACS Nano, 2020, 14, 9963–9971 CrossRef CAS PubMed.
- C. D. Paola, R. D'Agosta and F. Baletto, Geometrical effects on the magnetic properties of nanoparticles, Nano Lett., 2016, 16, 2885 CrossRef PubMed.
- N. Karampudi and R. P. Bahadur, Layers: A molecular surface peeling algorithm and its applications to analyze protein structures, Sci. Rep., 2015, 5, 16141 CrossRef CAS PubMed.
- C. Zeni, K. Rossi, A. Glielmo and S. De Gironcoli, Compact atomic descriptors enable accurate predictions via linear models, J. Chem. Phys., 2021, 154(22), 224112 CrossRef CAS PubMed.
- R. Drautz, Atomic cluster expansion for accurate and transferable interatomic potentials, Phys. Rev. B, 2019, 99, 014104 CrossRef CAS.
- E. Gazzarrini, K. Rossi and F. Baletto, Born to be different: The formation process of Cu nanoparticles tunes the size trend of the activity for CO2 to CH4 conversion, Nanoscale, 2021, 13(11), 5857–5867 RSC.
- O. J. F. Martin, C. Girard and A. Dereux, Generalized field propagator for electromagnetic scattering and light confinement, Phys. Rev. Lett., 1995, 74, 526–529 CrossRef CAS PubMed.
- Asap3 software, https://gitlab.com/asap/asap Search PubMed.
- M. Mavrikakis, B. Hammer and J. K. Nørskov, Effect of strain on the reactivity of metal surfaces, Phys. Rev. Lett., 1998, 81, 2819–2822 CrossRef.
- Z. Zhao, Z. Chen, X. Zhang and G. Lu, Generalized Surface Coordination Number as an Activity Descriptor for CO2 Reduction on Cu Surfaces, J. Phys. Chem. C, 2016, 120(49) CrossRef.
- F. Calle-Vallejo and A. S. Bandarenka, Enabling generalized coordination numbers to describe strain effects, ChemSusChem, 2018, 11(11), 1824–1828 CrossRef CAS.
- C. Girard, Near fields in nanostructures, Rep. Prog. Phys., 2005, 68, 1883–1933 CrossRef.
- C. Girard, Pygdm, 2021, https://pypi.org/project/pyGDM2/ Search PubMed.
- P. R. Wiecha, pygdm—a python toolkit for full-field electro-dynamical simulations and evolutionary optimization of nanostructures, Comput. Phys. Commun., 2018, 233, 167–192 CrossRef CAS.
- B. T. Draine, The Discrete-Dipole Approximation and Its Application to Interstellar Graphite Grains, Am. Phys. J., 1988, 333, 848 CAS.
- P. B. Johnson and R. W. Christy, Optical constants of the noble metals, Phys. Rev. B: Solid State, 1972, 6, 4370–4379 CrossRef CAS.
- A. D. Rakić, A. B. Djurišić, J. M. Elazar and M. L. Majewski, Optical properties of metallic films for vertical-cavity optoelectronic devices, Appl. Opt., 1998, 37, 5271–5283 CrossRef PubMed.
- J. H. Weaver, C. G. Olson and D. W. Lynch, Optical investigation of the electronic structure of bulk Rh and Ir, Phys. Rev. B: Solid State, 1977, 15, 4115–4118 CrossRef CAS.
- W. S. M. Werner, K. Glantschnig and C. Ambrosch-Draxl, Optical constants and inelastic electron-scattering data for 17 elemental metals, J. Phys. Chem. Ref. Data, 2009, 38(4), 1013–1092 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |