 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Disproportional surface segregation in ligand-free gold–silver alloy solid solution nanoparticles, and its implication for catalysis and biomedicine†
Frederic
Stein
 a,
Sebastian
Kohsakowski
b,
Ricardo
Martinez-Hincapie
c,
Sven
Reichenberger
a,
Christoph
Rehbock
a,
Viktor
Colic
c,
Daniel
Guay
d and
Stephan
Barcikowski
*a
a,
Sebastian
Kohsakowski
b,
Ricardo
Martinez-Hincapie
c,
Sven
Reichenberger
a,
Christoph
Rehbock
a,
Viktor
Colic
c,
Daniel
Guay
d and
Stephan
Barcikowski
*a
aTechnical Chemistry I, Center for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, D-45141 Essen, Germany. E-mail: stephan.barcikowski@uni-due.de
bLaufenberg GmbH, D-47839 Krefeld, Germany
cMax Planck Institute for Chemical Energy Conversion, D-45470 Mülheim an der Ruhr, Germany
dInstitut National de la Recherche Scientifique, INRS-Énergie, Matériaux et Télécommunications, Varennes, Québec J3X 1P7, Canada
First published on 2nd August 2022
Abstract
Catalytic activity and toxicity of mixed-metal nanoparticles have been shown to correlate and are known to be dependent on surface composition. The surface chemistry of the fully inorganic, ligand-free silver–gold alloy nanoparticle molar fraction series, is highly interesting for applications in heterogeneous catalysis, which is determined by active surface sites which are also relevant for understanding their dissolution behavior in biomedically-relevant ion-release scenarios. However, such information has never been systematically obtained for colloidal nanoparticles without organic surface ligands and has to date, not been analyzed in a surface-normalized manner to exclude density effects. For this, we used detailed electrochemical measurements based on cyclic voltammetry to systematically analyze the redox chemistry of particle-surface-normalized gold–silver alloy nanoparticles with varying gold molar fractions. The study addressed a broad range of gold molar fractions (Ag90Au10, Ag80Au20, Ag70Au30, Ag50Au50, Ag40Au60, and Ag20Au80) as well as monometallic Ag and Au nanoparticle controls. Oxygen reduction reaction (ORR) measurements in O2 saturated 0.1 M KOH revealed a linear reduction of the overpotential with increasing gold content on the surface, probably attributed to the higher ORR activity of gold over silver, verified by monometallic Ag and Au controls. These findings were complemented by detailed XPS studies revealing an accumulation of the minor constituent of the alloy on the surface, e.g., silver surface enrichment in gold-rich particles. Furthermore, highly oxidized Ag surface site enrichment was detected after the ORR reaction, most pronounced in gold-rich alloys. Further, detailed CV studies at acidic pH, analyzing the position, onset potential, and peak integrals of silver oxidation and silver reduction peaks revealed particularly low reactivity and high chemical stability of the equimolar Au50Ag50 composition, a phenomenon attributed to the outstanding thermodynamic, entropically driven, stabilization arising at this composition.
Introduction
When discussing real-life applications of nanoparticles (NPs), catalysis and biomedicine are frequently considered highly relevant fields though they are mostly treated independently as the research communities from both areas usually have minor interactions. However, some phenomena in biomedicine, e.g., the formation of reactive oxygen species (ROS) in the presence of nanoparticles, is closely linked to catalysis. Here, libraries of metal oxide nanoparticles were synthesized, and cytotoxic effects on bacteria and mammalian cells were correlated to low hydration energies and bandgap energies in the same region as the redox potentials of biomolecules.1 In a more recent study, doped oxide nanoparticles were examined for catalytic features in in vivo skin lesion assays in zebrafish. ROS generation causing the observed cell damage was correlated with (I) charge transfer through different oxidation states in the doped nanoparticles, (II) charge transfer from the space charge layer to the electron sink in tightly coupled elements, and (III) ion release.2 Next to these unintended use scenarios, nanoparticle-induced ROS is further discussed as one of the most critical pathways in the anti-tumor efficiency of sensitizers in irradiation therapy.3 Here, a recent study suggested a correlation between therapy-enhancing effects of Pt and Au nanoparticles in proton therapy and catalytic surface reactions.4 Another important research field is single atom catalysts which are discussed as relevant materials for biomedical applications.5 An important common feature that bridges both fields would be surface chemistry and active surface sites relevant to heterogeneous catalysis and interactions at the nano–bio interface. In this context, bimetallic nanoparticles with heterogeneous surface sites are particularly interesting as model materials.Bimetallic nanoparticles composed of gold and silver have been studied in detail, as they possess several attractive properties that may be utilized in a broad scope of applications in optics, biosensing, biomedicine, and catalysis.6 In this context, the AgAu system is a particularly interesting reference system, as its constituents are fully miscible on the complete composition range. Merging them into an alloy is an easy way to combine advantageous features in one nanoparticle and simultaneously tune these properties precisely to their respective use.7 In optics, the AgAu-nano-system is attractive due to its composition-tuneable surface-plasmon resonance (SPR).8 Here, a higher gold fraction will shift the SPR to longer wavelengths, while a higher silver content will shift it to lower wavelengths between 410–520 nm, representing an exciting feature for multiplexing in sensing.9 The second important application is biomedicine, where an active and more soluble antibacterial/cytotoxic material (silver) is combined with an inert material (gold), which allows controlled release of silver ions from these AgAu alloy nanoparticles. Here, disproportional passivation of silver ion activity by alloying with gold was found.10 This was confirmed by other studies where antimicrobial activity and cytotoxicity of AgAu nanoparticles were examined11 and by related work on the functionality of spermatozoa and the maturation of oocytes.12,13 All these studies discovered a disproportional effect, revealing a non-linear dependency of bioactivity/toxicity on the gold molar fraction due to the passivation of silver ion release by gold. However, this effect was only observed and correlated with Ag![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au bulk composition, while a correlation with AgAu surface properties is underexplored. The kinetics of silver release from AgAu alloy nanoparticles has recently been elucidated by utilizing hyperspectral single-particle spectroscopy.14 In that study, the Ag ion release from the alloy kinetically showed two leaching stages, with the initial shrinking-particle stage having a rate constant that exponentially depends on the silver content, and the slower ion leaching stage was controlled by the electrochemical oxidation potential of the alloy being steadily increased by the change in relative gold content and diffusion of silver atoms through the lattice.
:Au bulk composition, while a correlation with AgAu surface properties is underexplored. The kinetics of silver release from AgAu alloy nanoparticles has recently been elucidated by utilizing hyperspectral single-particle spectroscopy.14 In that study, the Ag ion release from the alloy kinetically showed two leaching stages, with the initial shrinking-particle stage having a rate constant that exponentially depends on the silver content, and the slower ion leaching stage was controlled by the electrochemical oxidation potential of the alloy being steadily increased by the change in relative gold content and diffusion of silver atoms through the lattice.
As a final application area, a variation in the silver to gold ratio within and on the AgAu alloy NP surface, respectively, while maintaining the particle size and mass-specific surface area, allows further systematic studies of their catalytic activity in different reactions and can help to identify the related active sites. Exemplary reactions that were investigated in this regard are glucose oxidation,15 aerobic alcohol oxidation,16 CO oxidation,17 oxidative coupling of methanol,18 CO2-reduction,19 and, as published recently, the selective oxidation of 5-(hydroxymethyl)furfural (HMF).20 Nevertheless, all these previously mentioned catalytic investigations resemble thermally driven chemical model reactions. In comparison, the electrocatalytic behavior of AgAu nanoparticles, e.g., under oxygen reduction conditions, is much less investigated.
The surface chemistry of chemically synthesized bimetallic gold–silver alloy nanoparticles (AgAu NP) has been previously investigated, and it was found that NP generated in this way have a significant silver enrichment on the surface, and inherit a distinct deviation from Vegard’s rule, which results in a high defect density.21,22 However, chemically generated AgAu NP from co-reduction of precursor materials like HAuCl4 and AgNO3, possess two disadvantages that impair systematic studies: (I) the bulk composition is inhomogeneous due to different reduction rates of gold and silver precursors;23 (II) their synthesis necessitates the presence of organic ligands, e.g., the reducing agent sodium citrate, which strongly affects the surface chemistry of the NP.24
To overcome the previously mentioned knowledge gaps and related limitations of ‘traditional’ (wet-chemical) nanoparticle synthesis, the laser-based synthesis allows the creation of fully inorganic, solid-solution AgAu NPs without the explicit need to add stabilizing agents.25,26 In contrast to traditional wet chemical synthesis routes that run at thermodynamic equilibrium, LAL is a kinetically controlled synthesis method27,28 with nano-thermodynamic contributions.29
Previous problems that complicated the use of laser ablation in liquids (LAL) to synthesize nanoparticles, like reproducibility, production scale, and heterogeneity in size and composition, were mainly solved with the improved mechanistic understanding of the ablation and cavitation dynamics, nanoparticle formation, and growth quenching.27,30 Recent studies reveal significant progress in the scale-up of laser-based synthesis processes,25 yielding gold nanoparticle productivities up to 8 g h−1.31 Furthermore, size control has been greatly improved by utilizing particle growth quenching via the addition of ions, the variation of pH during synthesis, and centrifugal purification, allowing the generation of monodisperse and monomodal nanoparticle colloids.32–35 Nowadays, LAL can produce spherical AgAu nanoparticles of the same overall composition as the target,14,20,36 down to the single-particle level.8
Laser-fabricated, totally ligand-free AgAu nanoparticles have been widely utilized in biomedical research, e.g., when examining toxicological13 and antimicrobial effects,11 in fundamental studies elucidating AgAu NP dissolution on a single particle level,14 as well as catalytic applications.20 Preliminary studies using XPS already revealed a substantial deviation between bulk and surface composition in laser-generated AgAu NPs, with a surface enrichment of silver in gold-rich particles and gold in their silver-rich analogues.20 However, the surface chemistry of these fully ligand-free AgAu NPs, with features of high interdisciplinary relevance in catalysis and biomedicine, and particularly how they are affected by composition, is vastly underexplored.
In this work, we used cyclic voltammetry in O2 saturated alkaline electrolyte (ORR conditions), as well as in the presence of chloride ions at acidic pH (HCl), to characterize the surface chemistry and surface reactions on fully inorganic ligand-free AgAu alloy NPs in correlation with particle composition, using a broad range of gold molar fractions (GMF) (Ag90Au10, Ag80Au20, Ag70Au30, Ag50Au50, Ag40Au60, and Ag20Au80) as well as monometallic Ag and Au NP as controls. XPS measurements complemented these studies to obtain additional information on the atomic surface composition and surface oxidation. To assess the intrinsic properties of each type of NP, all electrochemical measurements were normalized to the calculated surface area of the applied particles. It could be shown that the activity of the particle surface is connected to the composition in a multifaceted way, especially for the equimolar composition.
Results and discussion
Characterization of the colloidal nanoparticles
The nanoparticles in this study were synthesized via LAL. They were obtained with a monomodal size distribution after centrifugation with an average hydrodynamic diameter of 6–11 nm based on an analytical disc centrifuge (ADC) analysis, as shown in Fig. 1a. The total nanoparticle surface area that was drop-cast onto the electrode, shown in Fig. 1b, was used to normalize the currents during consecutive electrochemical measurements. The raw mass- and number-weighted distributions can be seen in the ESI (Fig. S2†). To better compare the different compositions, we used the gold molar fraction (GMF) in the following to characterize sample composition. For instance, Ag80Au20 would have a GMF of 0.2.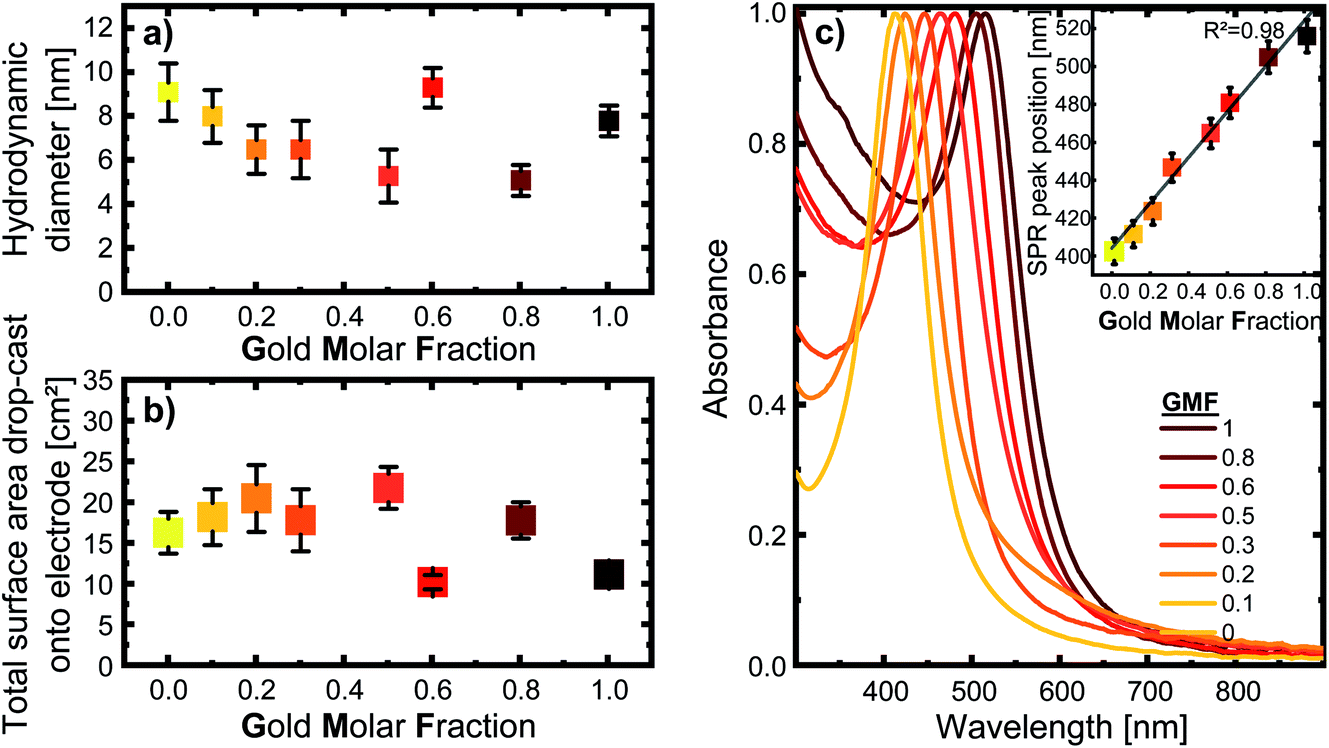 | ||
| Fig. 1 (a) Mean hydrodynamic particle diameter of the colloidal silver–gold alloy nanoparticles based on ADC, with the error bars indicating the distribution width; (b) total nanoparticle surface area drop-cast onto the respective electrode, calculated from the hydrodynamic diameter (determined by ADC) and mass concentration (determined by UV/vis); (c) UV/vis spectra of the respective colloids. The inset show how the SPR peak position correlates with the gold molar fraction. The error-bars show standard deviations from sample triplicates. The color code is used to better illustrate differentiation between samples, while colors are chosen based on the color of the corresponding AgAu colloids. | ||
The UV/vis spectra in Fig. 1c, show a single, distinct peak originating from each composition’s surface plasmon resonance, indicating alloy formation. However, a single SPR peak is not direct proof of homogenous elemental distribution, as bimetallic nanoparticles with elemental gradients would also show only a single SPR peak. However, the UV-vis spectrum allows us to exclude the presence of a variety of particle shapes that differ from the spherical form, such as nanorods or highly segregated particle structures, like Janus particles, which would show a broader SPR band, or even separate silver and gold nanoparticles in the colloid.8,22,37 Furthermore, the peaks are sharp and show negligible extinction in the long-wavelength region around 800–900 nm, indicating the absence of substantial concentrations of agglomerates and aggregates in the samples. Based on a previous publication by Neumeister et al., showing in-depth TEM/EDX characterization of LAL-synthesized gold–silver alloy nanoparticles generated under similar conditions, we can assume that an overall crystalline solid solution structure with homogeneous elemental distribution and compositions matching those of the target can be found in the AgAu alloy nanoparticles synthesized for this study.8 This has been revealed for laser-generated colloidal AgAu NPs by previous extensive single-particle EDX-line scans and EM images, verifying their spherical shape.8 Additionally, crystallographic analysis of these LAL-made NPs by Prymak et al.36 showed that they follow Vegard’s rule, proving uniform alloy crystal formation.
For all following experiments, it is vital to keep in mind that when nanoparticles are investigated, the relevant factor to understanding their functionality and catalytic activity is their effective surface area. This is because a nanoparticles’ surface-to-volume ratio increases as the particle size decreases, directly affecting the effective surface area. Since the direct electrochemical determination of the electrochemically active surface area (ECSA) of AgAu NPs is not trivial, an alternative approach was implemented. For this reason, the mass concentration and size distribution of the colloids were used to calculate the surface-area-concentrations of every sample. As explained in more detail within the methods section, this was done by taking the fraction of particles at every measured diameter and calculating the surface area of a corresponding sphere of each fraction. When these surface areas are summed up for each increment in diameter, we get the total surface area in the 0.1 ml sample (see Fig. 1b). With the mass concentration determined by UV/vis and the density of each composition, we can calculate the surface-area-concentration (see Fig. S3†).
Surface composition and surface oxidation of AgAu nanoparticles
To deepen our understanding of the processes occurring at the surface of AgAu NPs, we analyzed the surface composition and oxidation of the laser-fabricated AgAu alloy NP with XPS. These studies complement previous XPS examinations on LAL-generated gold–silver alloy nanoparticles published by Schade et al.20 Raw data taken from this study are re-plotted for illustrative purposes (Fig. 2a) together with our recent results (see Fig. S4† for the raw XPS data). While Schade et al. studied freeze-dried AgAu NP deposited on ZrO2 microparticles (MP) and used them to evaluate the selective oxidation of 5-(hydroxymethyl)furfural, in the present study, we directly drop-cast the AgAu NP on glassy carbon electrodes and evaluated them in ORR. In this context, it should be noted that the particles and reactions in the two studies substantially deviated. A common trend in the measured particles is observable despite the substantial differences. We can observe enrichment of the less abundant element in the surface-near-area (gold enriched on the surface of silver-rich particles and vice versa), a surprisingly consistent trend in both experimental series (Fig. 2a). This finding only partially agrees with the literature. According to Guisbiers et al., the surface segregation of two completely miscible elements, A and B, in one nanoparticle follows two specific rules:38(1) If the bulk melting temperature of element A is higher than that of element B, element A will deposit at the surface.
(2) If the bulk melting temperature of both elements is in the same order of magnitude, the surface energy of the respective elements will determine which element segregates to the surface.
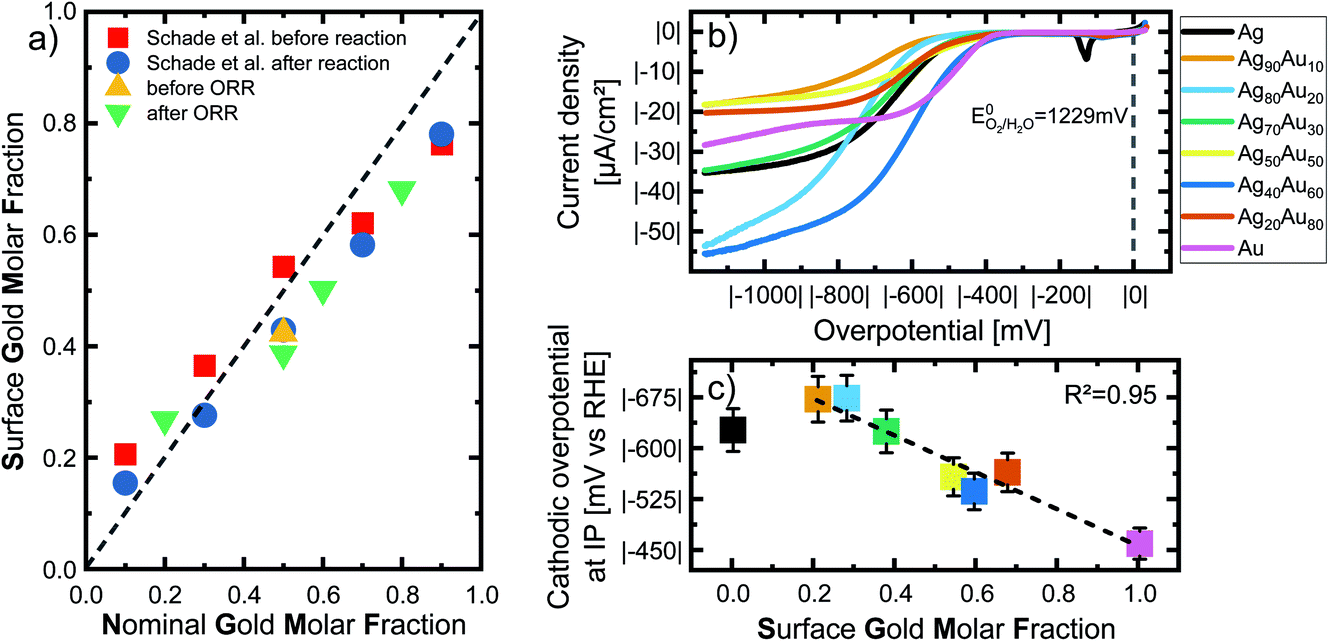 | ||
| Fig. 2 (a) Nominal gold molar fraction (x-axis) plotted against the measured surface gold molar fraction (SGMF) in the surface-near volume (y-axis) of silver–gold alloy nanoparticles, analyzed via XPS. The green and yellow symbols show the results of XPS measurements within the context of this study, while the data of Schade et al. of AgAu NP on ZrO2 before and after selective oxidation of 5-(hydroxymethyl)furfural (shown with red and blue symbols) has been adapted from ref. 13. Copyright 2014 CC BY 4.0 https://creativecommons.org/licenses/by/4.0/legalcode. Wiley-VCH GmbH. See Fig. S5† for details on nominal-surface GMF conversion. (b) Analysis of the oxygen reduction reaction (ORR) of the LAL-generated AgAu-NP in oxygen saturated 0.1 M KOH. The current density is normalized as mentioned above. (c) Overpotential determined by inflection point (IP) analysis. Overpotential in O2 saturated 0.1 M KOH is plotted as a function of the surface gold molar fraction (SGMF). The dotted line shows a linear trend with increasing gold content. The error bars show standard deviations from repeating the ORR three times. The color code in (b) is the same in (c). | ||
The authors state that the difference would have to be approximately less than 10% for the second rule to apply. Literature tells us that the bulk melting temperature of pure gold is 1337 K, while that of pure silver is 1235 K, which means the difference is ∼6.7%. Hence, the second rule applies.39 Both elements crystallize in fcc unit cells, simplifying the comparison of surface energies. Surface energy of Au is always higher than that of Ag,40 indicating that silver should always enrich on the surface. Deng et al. applied extensive Monte-Carlo simulations of AgAu NPs to investigate the surface segregation and always found silver enrichment on the surface.41 To explain this obvious deviation from our experimental results, we hypothesize that enrichment of the less abundant element on the surface is a phenomenon driven by mixing entropy. The stabilizing effects from mixing entropy are higher the closer the system is to a 50:50 Ag:Au ratio. When we assume a AgAu nanoparticle with a strong silver enrichment (e.g. GMF = 0.2) all silver atoms have a driving force to assume lattice positions neighboring gold atoms, though this is not possible due to gold deficiencies. However, the surface atoms of the nanoparticles obtained from a fast-cooling laser ablation in liquids synthesis under kinetic control are particularly rich in surface defects due to extremely high cooling rates,27,28 a thermodynamically unfavorable state. Hence, it is conceivable that particularly these strongly disfavoured surface sites have a higher driving force to enrich with the minority element (in this case Au) to reap the benefits of entropic stabilization. A way to verify the validity of this hypothesis would be atomistic simulations in which high energy states are assumed for surface atoms, though this exceeds the scope of this work.
To be able to correlate the results with the actual surface composition, we will refer to the samples by their surface gold molar fraction (SGMF). Please note that SGMF based on XPS studies does not refer to the composition of only the outer atom layer but is determined by the penetration depth of the XPS, which is commonly in the range of a few nm. By correlating surface composition with catalytic activity, Schade et al. found favorable synergetic effects on the catalytic activity for Ag10Au90 (SGMF 0.76 to 0.78) and Ag30Au70 (SGMF of 0.62 to 0.58).20
We performed the oxygen reduction reaction (ORR) in O2-saturated 0.1 M KOH at a scan rate of 50 mV s−1 and a rotating disc electrode at a speed of 1600 rpm to analyze the electrochemical properties and possible catalytic applications of the gold–silver alloy nanoparticles. These experimental conditions, particularly the high spinning rate of the electrode, ensure that measurements are independent of transport limitations and are a good representation of the chemical activity of the surface atoms. The current density is normalized to the nanoparticle surface area, as specified above, and explained in the Methods section in more detail. Fig. 2b shows linear sweep voltammograms of all tested compositions in an overpotential range of 0 to |−1200| mV recorded after 20 cycles. No distinctive linear trend is visible for either the gold or silver content. On the other hand, samples with a GMF of 0.1 and 0.5 specifically exhibit particularly low overall current densities. Pure silver and Ag70Au30 show very similar trends, even though the latter consists of 30 mol% gold. This could be a valuable insight for further studies on the catalytic properties of these alloys.
As the ORR polarization curves alone do not follow a clear trend showing a 1-to-1 correspondence between GMF and the catalytic activity, we applied inflection point (IP) analysis to extract more information from these measurements. IP analysis is an approach introduced by Schuhmann et al. to compare the composition-dependent electrocatalytic activities of complex nanoparticle materials (complex solid solution, particularly high-entropy alloys), primarily when multiple active centres are to be expected.42 To perform IP analysis, we fitted the ORR LSV curves with suitable polynomial functions at the interval around the inflection points of the curves. The inflection point is located where the second derivate of the fitted functions reaches zero. As the overpotential at the inflection point corresponds to the stage at which the diffusion of O2 does not yet limit the current to the electrode surface, it can directly correlate with the surface reaction at the most active surface sites. Generally, a low overpotential and high current density at the IP during ORR reactions are indicators of high catalytic activity and optimally binding surface sites. Only pure gold shows more than one IP, which would indicate a second active centre, while the other alloy materials show no obvious secondary IPs.
The extrapolated overpotentials at the IP, as a function of the SGMF, are shown in Fig. 2c. Therein we expressed the overpotential at the IP as a function of the SGMF deducted from XPS studies (Fig. 2a and S5†) since the potential at the IP is connected to the reduction activity for a specific surface reaction.43 Previous studies on the catalytic properties of laser-generated AgAu NPs involved extensive XPS studies and found up to 15% deviations between the surface composition and the nominal bulk composition of the nanoparticles. In this study, the silver enriched the surface in the case of nanoparticles with high molar gold content in the bulk and vice versa.20,42 We used this data and applied a polynomial fit (R2 = 1) to calculate the expected surface composition (SGMF) from the nominal composition (GMF) (Fig. S5†), with the SGMF used in Fig. 2c.
The overpotential decreases with increasing SGMF, starting at |−629 mV| for pure silver to |−462 mV| for pure gold. For the alloy nanoparticles, the overpotential shows a linear trend with increasing molar surface gold content above an SGMF of 20%. Yet, the SGMF between 20–30% appears even less active than for pure silver. We observed a similar synergetic trend with gold–silver surface composition in HMF oxidation with laser-generated AgAu alloy nanoparticles, although in this case, at an SGMF of ∼50–70%.20 Nonetheless, we can conclude from this data that the catalytic activity of gold in the ORR reaction is higher than that of silver indicating a more potent kinetic inhibition of the reaction on silver surface sites. This finding is in good accordance with the literature. Praveen et al. found out that a gold-rich composition of Ag25Au75 showed the highest activity among their tested compositions compared to the more silver-rich compositions.44 The linear decrease of the overpotential at the inflection point (increase in catalytic activity) with the SGMF indicates that catalytic activity is a direct function of the number of more active gold surface atoms.
Furthermore, we utilized XPS to characterize the oxidation state of the surface silver and gold atoms in relation to the SGMF after ORR for Ag80Au20, Ag50Au50, Ag40Au60, and Ag20Au80, while the equimolar 50:50 composition was also analyzed before it reacted (Fig. 3a). Please note that we used the actual SGMF instead of the nominal composition (GMF). Here, we observe that the noble gold is not prone to oxidation, even if alloyed with silver in every ratio. The percentage of gold oxidation varies only by 2%, while the most gold-rich composition has the highest level of oxidation with 9.5%. This finding is in good accordance with previous XPS studies on monometallic LAL-generated Au NPs, where surface oxidation values of 3–6%45 and 5–10%33 were reported. However, recent studies highlighted the difficulties of XPS in verifying the presence of surface oxidation for free-standing Au NPs.46
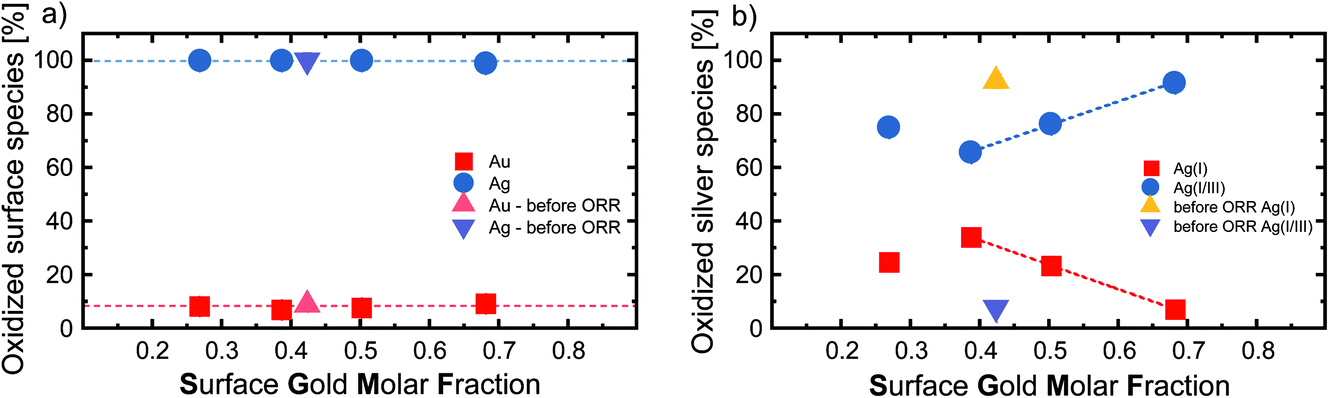 | ||
| Fig. 3 (a) Percentage of oxidized Ag and Au surface species (y-axis) and (b) percentage of oxidized Ag(I) and Ag(I/III) species on the NP surface after ORR (y-axis) plotted against the surface gold molar fraction (x-axis) of silver–gold alloy nanoparticles, analysed via XPS. For the equimolar Ag50Au50 (GMF 0.5) compositions we also plotted the results before ORR. | ||
On the other hand, the surface silver atoms are 100% oxidized, which is consistent with the fact that silver has a lower redox potential than gold and that the ORR reaction was carried out in the presence of the potent oxidant O2. This is in contrast to the previously shown XPS measurements of Schade et al., which showed that a GMF of 0.3 had the smallest proportion of oxidized silver on the particle surface.20 When the gold content is increased above 0.5, their study shows that the proportion of oxidized Ag atoms at the surface does not change significantly anymore, while the proportion of oxidized Au atoms continues to decrease. This clear dependence of gold and silver oxidation on the GMF, not found in our study, even for a reference sample taken before reaction, might be attributed to the AgAu NPs adsorption on ZrO2 supports in the study of Schade et al. To better evaluate the oxidation states of the surface silver atoms, we performed an in-depth analysis of the silver oxidation peaks by deconvolution to differentiate between Ag(I) oxides and mixed Ag(I/III) surface states.47 Exemplary XPS spectra illustrating the peak analysis can be found in Fig. S4.† The resulting Ag(I) and Ag(I/III) surface ratios in correlation with the GMF are found in Fig. 3b. The most striking trend observable here is the change in oxidation state before and after ORR, observed for the nominal Ag50Au50 composition (SGMF 0.42). While this sample remains fully oxidized after ORR (regardless of the reductive conditions therein) (compare with Fig. 3a), the Ag(I)/Ag(I/III) atomic ratio substantially deviates. Before ORR, the surface silver mainly consisted of Ag(I) (92.3%), with the mixed Ag(I/III) oxide (7.7%) being the minor constituent. After ORR the Ag(I/III) oxide makes up 65.8% of the surface atoms.
The corresponding current densities at the IP strongly fluctuate with increasing gold content (Fig. S6†), reaching in total three very similar minima at SGMFs of 0.21, 0.54, 0.68, with |−6.6| μA cm−2, |−6.2| μA cm−2, and |−6.3| μA cm−2 respectively. Ag40Au60 shows that the addition of 59.3 mol% gold to silver leads to the highest measured current density at an IP of |−14.5| μA cm−2, almost identical to the current density of pure silver at |−14.3| μA cm−2. Ag40Au60 has the highest current density and a low overpotential of |−538| mV, indicating this composition has the highest density of active sites at the IP.
Our study also reveals the high current densities for the gold-rich compositions Ag30Au70, Ag20Au80 and Ag50Au50 with SGMFs of 0.59, 0.68, and 0.54, respectively (see Fig. 2b). Even though the results on the GMF 0.5 samples slightly deviated, we observe an unexpected consistency in catalytic activity in fundamentally different reactions from thermal catalysis (Schade et al.) and electrocatalysis (this work) catalysed by laser-fabricated AgAu NPs.
Based on these findings compared with the XPS results, it can be inferred that the ORR reaction favors the formation of Ag(I/III) surface sites. If we compare the silver oxidation trend from Fig. 3b with the overpotential from Fig. 2c, we see that the Ag(I) silver species decreases similarly to the overpotential, indicating that an increased fraction of higher oxidized silver species coincides with higher surface activity. It has been previously shown that surface oxides play an active role in the mechanism of certain reactions like OER.48 It is conceivable that surface oxides may also play a vital role in the reaction observed here. However, this interpretation should be carefully evaluated considering the following points: (I) while the trends from XPS only depict the portion of higher oxidized silver species, the total number of silver atoms would still decrease in the particles due to an overall increasing SGMF; (II) as stated above, the catalytic activity (overpotential) also linearly scales with the SGMF, and Au atoms are more active than silver atoms (Fig. 2c). For specific reactions like OER, it has been shown that the surface oxygen can play an active role in the reaction mechanism, which could be a valuable insight into the mechanistic sequence behind these reactions. Even though we could not prove that this also plays a role in these experiments, it should be considered for future studies.48 It may be concluded that we cannot justify a correlation between the Ag(I)/Ag(III) surface sites and the catalytic activity in ORR based on this data. While a high amount of high valence silver species was created during this process, we can primarily observe increased catalytic activity with increasing gold content. This finding indicates that gold surface atoms are the catalytically most active species for this specific reaction.
Surface chemistry of AgAu nanoparticles in correlation with composition in an acidic anaerobic solvent
To potentially intensify and observe further correlations between surface chemistry and GMF in AgAu alloy nanoparticles, we conducted electrochemical measurements in an acidic medium and in the absence of oxygen. Furthermore, these measurements were conducted in the presence of chloride ions, prone to form strong bonds with silver sites,49 which more closely represent conditions observable in real-life applications, during ion release in a biological environment, or in actual water samples, where chloride is always present. Hence, we examined the electrochemistry of pure Au and Ag NPs and their intermediate alloys in 0.1 M HCl saturated with N2. The measured voltammograms are shown in Fig. 4. All CVs were performed in a potential range of 0–1.4 V, however, as the Ag50Au50 composition showed no activity in this range, the upper limit was increased to 1.7 V in order to pinpoint whether higher potentials might initiate a surface reaction. Please note that the voltammograms are not plotted with a unified y-axis scaling. This presentation was used to illustrate the corresponding peak more clearly. Fig. S7† shows the voltammograms but with the same scaling on both axes to better illustrate pronounced differences in overall current densities between the compositions. As described in more detail in the method section, all measured current values are normalized by the total particle surface area to compare surface-atom-controlled reactions better. | ||
| Fig. 4 CVs of pure Au and Ag NPs and their intermediate alloys, 1st cycle (in black) and 20th cycle (in the respective colors from Au in red to Ag in yellow) were measured under a N2 atmosphere with a scan rate of 100 mV s−1 in 0.1 M HCl. The current density is normalized to the applied nanoparticle surface area, while the potential is calculated against RHE @pH 1. | ||
Interestingly, the oxidation of gold could be detected in the first cycle for all gold-containing samples emerging at ∼1240 mV, except in the equimolar composition Ag50Au50 and the pure gold sample. After the 20th cycle, the gold oxidation peak vanished completely from all compositions. This is especially unexpected in a medium containing chloride, as Cl− promotes the dissolution of gold.50 One reason for the vanishing or flat-out missing gold peak could be the enrichment of the surface with silver. However, this contradicts the recent XAS and XPS results shown by Schade et al. for AgAu alloy nanoparticles from a laser ablation approach,20 and the XPS results in the alkaline electrolyte from the previous chapter. Here, changes in surface composition before and after the reaction (similar to 1 vs. 20 cycle data) were minimal. Hence, substantial enrichment with silver cannot be the reason for the missing oxidation of surface gold atoms. Another more probable explanation could be the dissolution of gold species on the surface, or the surface restructuring of more active structural gold surface defects during continuous cycling. In the case of pure gold, the Au oxidation peak barely begins to show in the observed potential window. The absence of gold oxidation in the GMF of 0.5 samples could be due to the mixing-entropy-driven thermodynamic stability of equiatomic AgAu alloys. According to Guisbiers et al., the equimolar 50:50 composition is, in regards to their thermodynamic properties, the most stable composition of this material system.38
An oxidation peak that can be attributed to silver can be observed between 364 mV to 677 mV for pure Ag and all alloys, but not pure Au. These peaks indicate the oxidation of Ag0 to Ag+ and the subsequent formation of water-insoluble AgCl.51 The first silver oxidation peak of the pure silver sample reaches its maximum at 422 mV.
| Ag + Cl− ↔ AgCl + e− | (1) |
This first peak can also be an overlap of this reaction with other processes. According to Chen et al., the first reaction that would take place would be the electrolytic dissolution of Ag to Ag(OH)2− by the absorption of OH− and the subsequent desorption and diffusion of the soluble species.52 This would only happen to a minor part of the silver atoms in an acidic environment, and this reaction is barely visible. Furthermore, the broad peak could be based on the formation of a monolayer of electrolytically formed Ag2O. According to Saw et al., alloying silver with gold will generally lead to a broadening of this oxidation peak with increasing gold content as the silver is rendered less thermodynamically active.53 A third silver oxidation process can be seen between 700 and 800 mV in all silver-containing samples, except pure silver and Ag50Au50. This third peak is characteristic of AgAu alloys. As the nanoparticle sizes were in a similar range, this peak cannot be attributed to a size effect but rather to the oxidation of silver, whose activity has been decreased due to the stabilization by the gold matrix,52,54 a phenomenon previously observed in leaching experiments with AgAu single particles analysed by hyperspectral spectroscopy.14 An explanation for the lack of this reaction with Ag50Au50, is the higher thermodynamic stability of the equimolar composition, as discussed above, which would inhibit complex oxidation processes. Besides that, it is interesting to observe that this reaction is not observable in pure Ag NPs. As Rurainsky et al. did not register this peak when they tested pure Ag and Ag80Au20 NPs, its absence could be attributed to the stabilizing ligand we also had to use to produce the pure Ag NPs.55 Pure Ag NPs are not stable in 0.1 mM NaCl, which forced us to add 0.1 mM trisodium citrate during particle storage, while Rurainsky et al. used a substantially higher citrate concentration of 1.17 mM in their study.55 Here, sodium citrate, as a known reducing agent for noble metal ions, could have inhibited this silver oxidation process.56 When inspecting the negative sweep from 1–4000 mV, we could only observe gold oxide reduction in samples with a GMF between 0.8 down to 0.5, with an increasing potential from 962 mV (GMF = 0.8) to 1173 mV (GMF = 0.5) after the first cycle. After 20 cycles, gold oxide reduction was only detectable in Ag20Au80. Silver oxide reduction starts at ∼300 mV for all silver-containing samples in the first cycle. Ag shows a vast and indistinct peak that seems to be composed of several overlapping reactions. Similar behaviour can be observed for Ag90Au10, while Ag80Au20 only shows one distinct peak. All other alloys produce a double peak in this area, which can be attributed to the reduction of the alloyed AgAu, followed by the reduction of Ag+ to Ag0.57 It is not clear why Ag80Au20 does not show the alloy reduction peak, as former studies have shown all compositions to be homogenous alloys over the whole particle, except for the surface-near volume, which showed the already mentioned enrichment of the nominally scarcer element.8,20 After 20 cycles, all reduction reactions occur at a lower potential, indicating stronger inhibition or higher electrochemical stability, while overall peaks are much smaller. For biomedical applications, particularly nano-toxicity, this could indicate that potential adverse effects would deteriorate with increasing contact time between nanoparticles and cells or tissue.
Additionally, we could find reduction peaks, which can be attributed to the alloyed silver in particles with a gold content between 0.2–0.8 (Fig. S8†). This peak position shifts from 567 mV at a gold content of 0.2 to a maximum of 794 mV at a GMF of 0.6, and decreases afterward. Unfortunately, the position of this peak is not easy to determine precisely, as the peak has a low intensity or is split into multiple peaks. Either way, their presence indicates a pronounced change in alloy structure after cycling and the formation of an alloy sub-population with deviating composition or structure. To aid in maintaining an overview of the various reactions, we have plugged an overview graph of the CVs of Ag80Au20 into the ESI,† in which the regions where the addressed reactions happen are highlighted by color and number (see Fig. S9†).
The results seem to indicate that the gold oxidation in all samples is irreversible and requires a higher potential to occur with an increase in gold content. In previous studies, Schade et al. saw a reduction of the noble properties of gold by the addition of silver, and our findings confirm these results.20 Grasmik et al. saw the gold reduction in all tested AgAu alloy NPs, but it is unclear how many cycles were conducted in their cyclic voltammetry measurements.22 In contrast, Saw et al. could not identify a gold reduction peak in their work after the second cycle.53 This opens the possibility that the oxidative dissolved gold might have separated from the electrode and thus could not be reduced any more, even during the first cycle.
For further analysis, we extracted the silver oxidation and reduction peak positions from the voltammograms for the first and 20th cycles (Fig. 5). Examination of the position of the silver oxidation peaks (solid black and red symbols) shows that the positions do not significantly change, either as a function of the gold molar fraction nor due to multiple cycling. Considering the error bars, the median peak potential is 416 mV. Based on these results it may be concluded that the potential required to oxidize silver does not depend on the GMF. This finding would have a pronounced impact on these particles’ utilization in biological systems. Based on this, we may conclude that Ag ion release would always be initiated independently of composition, though its rate over time would depend on GMF or SGMF. This finding is in good accordance with our XPS results from the ORR reaction, where surface silver oxidation was independent of the GMF and constantly at 100% (Fig. 3a).
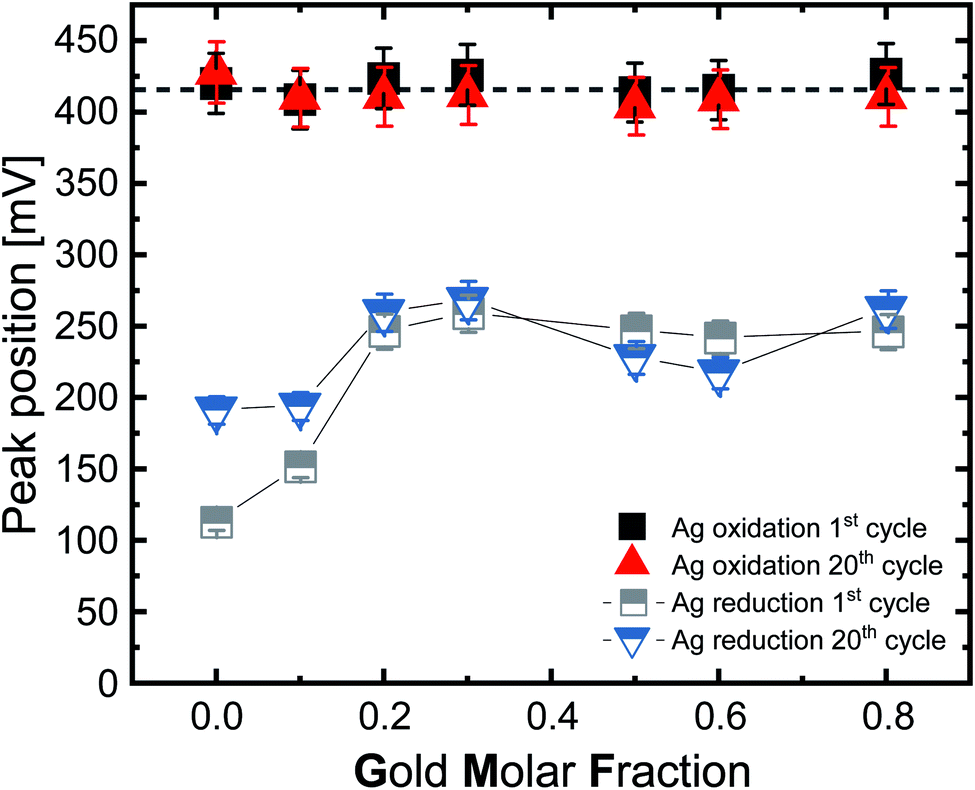 | ||
| Fig. 5 Position of the silver-oxidation (solid symbols) and reduction (semi-solid symbols) peak as a function of the gold molar fraction of the surface atoms in 0.1 M HCl under a N2 atmosphere with a scan rate of 100 mV s−1. The lines guide the eye. The error bars were calculated by repeating the experiments three times. | ||
Comparing the values of the first broad silver oxidation peak with the literature shows that much higher potentials are required to cause oxidation of the silver in the case of laser synthesized, ligand-free nanoparticles. According to Grasmik et al. and Snyder et al., wet-chemically synthesized silver nanoparticle oxidation occurs at 120 mV and 150 mV, respectively.22,58 Additionally, Grasmik et al. noted that the peaks of the alloy nanoparticles are shifted to higher potentials with a broadening of the peaks due to the reduction in silver dissolution if compared to pure silver. This indicates a constraint of Ag leaching due to the alloy character of these particles. Thus, the AgAu-NP fabricated by LAL behave very differently from the chemically synthesized AgAu-NP of Grasmik et al.22 However, it should be noted that Grasmik et al. used an ion concentration of 3 M KCl. The use of KCl instead of HCl could for once influence the cycle stability since, according to Zhu et al., KCl has the lowest long-term stability of the common electrolytes (KCl < NaCl < LiCl < HCl).59 The stability of the electrolyte depends more on the potential window than the electrode material, while the electrode material has to be explicitly chosen to show the desired reaction in the potential stability window of the electrolyte.60 Furthermore, KCl yields a neutral pH, while our examinations were conducted in the acidic regime, where different surface reactions may occur.
The reduction peaks of silver behave similarly to the oxidation peaks (Fig. 5, semi-solid grey and blue symbols) but differ by showing a higher deviation between the first and 20th cycle and by exhibiting a distinct impact of the composition on the peak potential, particularly in the silver-rich samples. Silver oxidation occurs on average at 416 mV, with minor variations of <20 mV, while the reduction occurs between 114–259 mV (1st cycle) and 192–269 mV (20th cycle). When we compare the ranges between the first and 20th cycle, we can see that the peaks get progressively closer as the number of cycles increases, which seems to indicate the formation of an electrochemically stable state, probably due to the disappearance of soluble silver species from the vicinity of the electrode or a surface restructuring. We want to emphasize that the silver reduction peaks seem to consist of multiple peaks. This is important to mention as all samples, except Ag80Au20, showed at least a double peak, and the two most silver-rich compositions, especially Ag, produced very irregular peaks. We used the largest of these irregular peaks to compare the potentials, which may explain the stronger deviations in silver-rich samples. According to the literature, the reduction of pure silver nanoparticles takes place at a wide range between 543–626 mV, but in our study and according to Fig. 5, the reduction for pure Ag occurs at 110 mV in the first cycle and at 192 mV in the 20th cycle, which is significantly lower than the literature (on wet-chemically prepared particles) indicates.22,55 Such a decisive shift of the reduction potential to lower values could be due to the formation of passivating layers of silver chloride on the electrode surface; as in the case of reverse scanning, a lower potential means a higher potential difference needs to be applied to start the reduction reaction. Furthermore, previously mentioned differences in particle surface structure could be contributing factors.
The next step is to look at the area under the curve, or rather the integral of the first silver oxidation peak. When this value is divided by the scan rate, we obtain the total charge transferred during the oxidation reaction. Fig. 6a shows the resulting transferred charge in μA s for the 1st and 20th cycles. We used only the potential range (364 mV to 677 mV) of silver oxidation of the pure silver nanoparticles for integration (including the tailing) since the peak width deviated strongly from this, especially for the gold-rich compositions. We used the same principle for all integrations in this study. When analyzing reactions that can be attributed to gold, we used the peak widths of the pure gold as a reference.
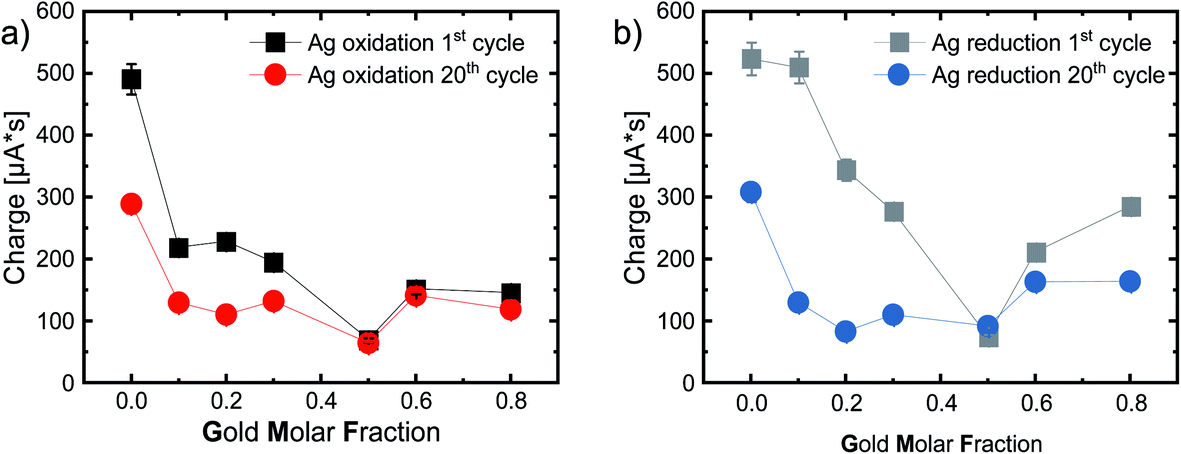 | ||
| Fig. 6 (a) Intercalated charge during the 1st (or rather main) silver oxidation and (b) during silver reduction. Calculated by integrating the Ag oxidation peak and afterwards divided by the scan rate, for the 1st (black) and 20th (colored) cycle, as a function of the gold molar fraction in 0.1 M HCl under a N2 atmosphere with a scan rate of 100 mV s−1. The error bars were calculated by repeating the experiments three times. The lines guide the eye. | ||
Fig. 6a shows the charge connected to silver oxidation, while Fig. 6b shows the charge connected to the reduction reaction, with very similar trends. Ag NPs have the highest charge transfer, most likely due to the high reactivity of pure silver NPs, while the charge decreases with increasing GMF. Furthermore, the equimolar Ag50Au50 composition stands out with a minimum of the transferred charge. After 20 cycles, we observe a substantial loss in activity for pure Ag (490 μA s to 289 μA s) and the following alloys with a GMF of 0.1, 0.2, and 0.3 (see Table S2†). This can be explained by the aforementioned irreversible reactions and the disappearance of active surfaces, e.g., defects (in the surface-near particle volume) restructured by multiple cycling. Losses are also substantially lower for the gold-rich samples than for the silver-rich alloys, indicating that primarily active silver and not gold atoms are restructured by cycling – a potentially highly relevant finding for catalyst durability.
Interestingly, the charge transfer processes in these experiments, related to surface site reactivity and stability, stand out for the equimolar 50:50 composition. Here we observe a particularly low surface reactivity and high structural stability. Peculiar properties of this intermediate composition are frequently reported in the literature and associated with thermodynamically driven processes, particularly the favorable mixing entropy at a 1:1 Ag:Au molar ratio. This trend was also observable when the crystal structure, particularly the lattice constants of AgAu alloy molar series, are analyzed with XRD.36,61 Here a pronounced contraction of the lattice constants and strong negative deviations from Vegard’s law with a minimum at the 50:50 composition was found in chemically-synthesized AgAu NPs, which the authors associated with microstrain, which could not be attributed to a size effect, as pure Ag and Au NPs did not show any variation in their lattice parameters compared to the bulk material.61 These findings could be due to the inhomogeneous internal phase structure of the chemically-synthesized AgAu NPs with strong elemental gradients. Hence, the intraparticle deviation from the entropically favored ideal mixing ratio of 50:50 caused contraction and strain on the crystal structure. In the AgAu molar series from kinetically-controlled LAL synthesis, the deviations from Vegard’s law were far less pronounced,36 attributed to the fact that these particles have an entirely homogeneous bulk elemental distribution. Other studies also saw that a laser-induced reduction method yielded fully homogeneous AgAu NPs that precisely followed Vegard’s law.62 On the other hand, deviations with a minimum lattice constant at the 50:50 composition were also reported for particles from chemical reduction, consecutively thermally annealed on a carbon black support.62
In another study, AgAu NPs with different GMF, synthesized by CVD and supported on PTFE, were analyzed for their dissolution and ion release properties. It was found that the Ag50Au50 nanoparticles exhibited a pronounced silver ion burst release in the first 40 min, higher than in the corresponding Ag70Au30 and Ag90Au10, while on longer timescales, the ion release from the equimolar composition slowed down. Both phenomena were explained by the favorable entropic stabilization of the 50:50 composition. During the initial phase, residual silver atoms causing a deviation from the favorable equimolar composition were most easily removed from the crystal due to the good entropic stabilization of this composition. For the same reason, further ion release was inhibited on longer time scales as its release from the 50:50 composition is disfavored.63 These findings are in good accordance with our electrochemical measurements, where particularly the equimolar composition (Ag50Au50) had the lowest transfer rate and the best chemical stability, as it is already in an optimal thermodynamic state.
To verify the high electrochemical stability of Ag50Au50, we performed additional CV measurements with 380 additional cycles for the equimolar composition as well as Ag80Au20 and Ag20Au80 for reference. The results are shown in Fig. S10.† Fig. S10a, b and c† show the respective CVs of Ag80Au20, Ag50Au50 and Ag20Au80 after 1, 20, 50, 100, 150, 200, 300 and 400 cycles (Fig. S10a–c†). The CVs were again normalized to the applied NP surface area. The repeated experiments show that the current per applied surface area for Ag50Au50 and Ag80Au20 is in a similar range, while the gold-rich Ag20Au80 samples’ highest measured current in the first cycle reaches only ∼1/10th of the current of both other samples. An extrapolation of the silver oxidation peak currents can be found in Fig. S10d.† While comparing the changes in current per nanoparticle area of all compositions with the previous experiment (see Fig. 6), it is obvious that the stability indicated by the current reduction for both, Ag50Au50 and Ag80Au20, is lower than anticipated from the previous measurements. Meanwhile, the current in Ag20Au80 seems to be constant, even though the values are comparably low. For the gold-rich sample, we attribute this difference to a measurement error. The normalized currents here are not only substantially lower than those of the two other samples, but we also see a pronounced increase in the current and integral area of the silver reduction peak, which cannot be explained.
However, what is obvious from these measurements is that the silver-rich composition Ag80Au20, as well as the gold-rich Ag20Au80 shows a comparably broad Ag oxidation peak in the beginning in comparison to the equimolar composition, which overlaps with a second peak that can be attributed to alloyed silver, according to Saw et al.53 With increasing cycle number, this double peak continues to broaden and shift to higher potentials. The silver reduction peak also consists of multiple peaks for Ag20Au80 and Ag80Au20, possibly due to dealloying or surface segregation of the elements involved, but additional characterizations like XRD and TEM would be necessary to conclude the actual reason for this change. Nonetheless, we can conclude that these processes seem to be less pronounced in the equimolar composition, which points to improved stability.
In their study, Saw et al. only saw a broad Ag oxidation peak for the first cycle, which vanished by the second cycle. According to the authors, this would exclude mass-transport limitations because the silver reduction transported almost the same charge in both cycles.53 However, the same cannot be said for the silver-rich sample measured in our work, as areas of the silver oxidation and reduction peak get much smaller with increasing cycle number, indicating irreversible losses. If we compare Ag50Au50 with Ag80Au20, we can observe that Ag80Au20 initially has a higher current than the equimolar composition, but both samples show the same proportional current decrease in the first 50 cycles (see Fig. S10d).† In this region, Ag80Au20 shows a change in current from 0.105 mA cm−2 to 0.066 mA cm−2, translating to a reduction of 37%. Ag50Au50, on the other hand, changes from 0.067 mA cm−2 to 0.043 mA cm−2 in the first 50 cycles, making up a reduction of 36%. When the samples are measured with a higher number of cycles, the current reduction of Ag50Au50 slows down remarkably, leading to a higher current than Ag80Au20 after 300 cycles. This trend continues with more cycles, leading to a total current reduction of 60% for Ag50Au50 compared to the current of the first cycle, while the Ag80Au20 current reduces by 71%. In addition, the charge transfer for the silver reduction of Ag80Au20 almost vanishes completely after 400 cycles, showing that this process is highly irreversible. Ag50Au50 has a similar charge transfer between silver oxidation and reduction from the first to the 400th cycle, proving that the reaction is reversible.
In the next step, we examined the onset potentials of our tested samples. The onset potential of the silver oxidation peaks shows us the threshold that needs to be overcome to start the oxidation reaction. To determine the onset potential, we draw tangents in the non-faradaic zone (the zone in which no charge transfer can be determined) and the faradaic zone (where a charge is transferred between the electrodes). The potential on which those two lines intersect gives the onset potential (see Fig. S11a).† As this determination is prone to inaccuracy, mainly due to shifting baselines, and because there are multiple processes like diffusion, which overshadow the reactions when using a static electrode at high scan rates, we decided to put this data into the ESI† and only use it as a reference point to support the more accurately determinable results. These onset potentials can be seen in Fig. S11b.† The onset potential increases with increasing gold content in the first cycle, starting at 369 mV up to a GMF of 0.3. The more gold-rich Ag70Au30 experiences an enhanced kinetic inhibition, which increases the onset potential.
Further increasing the GMF to 0.6 leads to a local minimum, followed by a slight increase with higher GMF. After 20 cycles, the observable onset potential follows a similar trend, while the differences between the compositions is reduced. The silver onset reduction potential in Fig. S12† shows even less difference between the compositions.
Conclusions
AgAu alloy nanoparticles are one of the most frequently studied binary alloy systems, applicable in optics, catalysis, and biomedicine. Here, it was found that the biological activity of AgAu alloy nanoparticles (toxicity, antimicrobial activity) disproportionally changes with the GMF, a finding which necessitates a more detailed study of the surface structure in particular to identify the most active surface atoms. Furthermore, the surface structure of AgAu nanoparticles has a similarly strong impact on the catalytic activity of these particles. Consequently, the surface properties at the nanoparticle-solvent-interface are particularly relevant for both application areas, biomedicine and catalysis. It is especially interesting to pinpoint how surface structure and hence the type and concentration of active surface site changes with time and upon multiple electrochemical cycles, which will heavily impact the activity and durability of an electrochemical catalyst and their dissolution behavior at the nano–bio interface.To explore this interdisciplinary topic, we used cyclic voltammetry in conjunction with in-depth XPS studies to analyze the surface chemistry and surface composition of a AgAu alloy nanoparticle molar series. We used a fully-inorganic model of AgAu nanoparticles with 8 different compositions generated by the laser ablation in liquids synthesis method, avoiding cross-effects from organic surface ligands, and we applied surface-normalized data interpretation. Based on the main findings obtained, we can draw the following major conclusions:
(I) The activity at the first inflection point on AgAu alloy particles during ORR is linearly dependent on the surface gold molar fraction. Hence Au surface sites are probably more active than Ag surface sites in the corresponding reaction. Ag40Au60 has the highest current density and a low overpotential of |−538| mV, indicating this composition has the highest density of active sites at the inflection point.
(II) XPS measurements of surface composition in correlation with the gold molar fraction during oxygen reduction reactions have yielded a trend that shows an enrichment of the minor component in the surface of the nanoparticle – gold-rich AgAu particles have a surface enriched with silver and vice versa. This trend seems to be universally applicable and is found in electrocatalytic reactions on electrodes and thermal catalysis with particles supported on ZrO2. It could also help explain the disproportional trends observed in the biomedical activity of these particles.
(III) During ORR, we observe total oxidation of silver surface atoms with a high abundance of highly oxidized (Ag(I)/Ag(III)) oxide–hydroxide surface atoms after the reaction, particularly at gold-rich nanoparticles. Even though a direct correlation between catalytic activity and the abundance of these surface sites could not be observed, their presence may be an important finding to elucidate the mechanism of ORR on AgAu nanoparticles. Additionally, high surface oxidation and the presence of more active surface species would also strongly influence ion release kinetics from these particles and affect the biological activity.
(IV) A detailed electrochemical analysis of AgAu nanoparticles in an acidic chloride-containing medium revealed a complex dependency of surface activity on the particle composition. We particularly document a pronounced change in surface activity with multiple cycling, leading to a restructuring of surface atoms. One main effect is a loss of electrochemical activity in the reduction sweep. This could strongly affect these particles’ toxicity and indicate that adverse effects to tissue and cells could wear off with time, which is in line with the fact that the position of the silver oxidation peak is independent of the GMF and SGMF, meaning that ion release and toxicity is probably strong initially and wears off on a longer timescale. These phenomena should go along with a deactivation of surface sites and a reduction of catalyst durability for catalytic applications.
(V) In particular, the Ag50Au50 composition reveals surprisingly low reactivity and high chemical stability, probably attributed to an entropic stabilization of this intermediate composition. This finding is in good accordance with previous studies where this intermediate composition had a initial burst of kinetics followed by high chemical stability on longer timescales. This could be relevant for AgAu nanoparticles for medical imaging, as they would feature pronounced structural and hence optical stability, while on the other hand, a burst release directly after injection could be beneficial to sterilize the insertion wound.
Experimental
Synthesis of silver–gold alloy nanoparticles
Colloidal gold, silver, and silver–gold alloy nanoparticles were produced via laser ablation in liquids (LAL) in 0.1 mM NaCl aqueous solution, using an Nd-YAG laser (Rofin Powerline E) with 10 ns pulse length, 10 kHz repetition rate, a wavelength of 1064 nm, and a fluence of 23.5 J cm−2. Targets of the respective pure metals and alloys (0.5 mm thickness, 99.99% purity, Institute for Noble Metal and Metal Chemistry, Schwäbisch-Gmünd, Germany) were fixed in a self-constructed PTFE batch chamber with a total volume of 30 ml.64 The ablation was performed using a highly dilute electrolyte in Milli-Q water (0.1 mM NaCl) for size quenching and electrostatic stabilization.32–34 As the samples were analyzed cross-continentally, they required high long-term stability. For this reason, we had to make an exception for the pure silver nanoparticles, as those show very low colloidal stability when ablated in 0.1 mM NaCl. Consequently, we used 0.1 mM trisodium citrate instead.The liquid was stirred continuously during the process to minimize shielding effects and re-irradiation of already produced NPs. An F-theta lens (F = 100 mm) was used to focus the laser pulses onto the target in the batch chamber, while a galvanometric scanner system (ScanLab, SCANcube10) guided the pulses in a spiral pattern with a diameter of 6 mm over the target surface. Each ablation was carried out for 10 min. This process yields spherical nanoparticles with a AgAu solid solution structure.8
Characterization of alloy nanoparticles
Mass concentrations of the colloids were determined gravimetrically by weighing the targets before and after the ablation process using a micro-balance (Precisa XT 220A) and UV/vis extinction photo spectroscopy (Thermo Scientific Evolution 201) before and after centrifugation. UV/vis spectra were acquired in the range 300–900 nm by diluting the colloids, where necessary, and transferring them into a quartz glass cuvette with 10 mm path length. To correlate the absorbance of the prominent surface plasmon resonance (SPR) peak with mass concentrations, calibration curves were collected beforehand with known concentrations (see Fig. S1†). The position of the SPR-peak depends on the composition of the nanoparticles (which allows an allocation to a composition), and its intensity corresponds directly to the mass concentration via Lambert–Beer’s-Law.Collective size characterizations were done with an analytical disc centrifuge (ADC, DC 24000, CPS Instruments) at 24000 rpm. This device measures the density-selective sedimentation of particles in the centrifugal field of a rotating disk. The main readout for the sedimenting particle fronts is the attenuation of an incident laser beam with a wavelength of 405 nm, while size-dependent optical properties of the nanoparticles based on Mie theory are used to convert intensities into mass fractions. Measurements were conducted against a D(+)-sucrose density gradient, and a sample volume of 0.1 ml was used. The mass- and number-weighted hydrodynamic particle diameter distributions represent spheres with equal sedimentation characteristics. Together with the mass concentrations, these distributions were used to calculate the overall surface concentration in cm2 ml−1 in every colloid.
The surface composition, surface oxidation, and possible alteration due to the electrochemical tests were investigated with a ULVAC-PHI VersaProbe II setup using a monochromatic Al anode with a Kα line at 1486.6 eV and a spot size of 100 μm. A hemispherical analyzer (with an angle of 45° between the sample surface and the analyzer) and dual-beam charge neutralization were used. All spectra are referenced to the 1s peak of graphitic carbon. Data processing was conducted using CasaXPS software. After Shirley background subtraction65 the Au4f and Ag3d spectra were fitted. For the deconvolution of the individual oxidization states of Au4f and Ag3d, constraints were chosen in accordance with the literature.66 The constraints used during the fit are summarized in Table S3.† Peak splitting was fixed to 3.67 eV (Au4f7/2 and Au4f5/2) and 6.0 eV (Ag3d5/2 and Ag3d3/2) according to the literature.66 The peak type was chosen as symmetric Gauss–Lorentzian with 50% Gaussian, 50% Lorentzian in the case of symmetric oxide peaks and asymmetric Lorentzian (1.9:1.8:1) for asymmetric elementary peaks Au0 and asymmetric Lorentzian (1.9:2:20) for Ag0.
Cyclic voltammetry measurements
The electrochemical properties of the bare nanoparticles were initially investigated by utilizing a standard three-electrode setup Potentiostat (Metrohm Autolab PGSTAT 302N), while further experiments were conducted using another three-electrode setup potentiostat (VersaSTAT 3F, AMETEK Scientific Instruments, and Bio-Logic VSP-3). A Pt wire was used as the counter electrode, Ag/AgCl or MMS as the reference electrode, and a glassy carbon electrode, drop cast with the respective nanoparticles as the working electrode. To coat the glassy carbon electrode with the nanoparticles, 30 μg of AgAu alloy NPs were drop-cast onto the electrode and allowed to dry for 20 min while the electrode was rotated at 1600 rpm. The electrolyte solutions with 0.1 M HCl (pH = 1) and 0.1 M KOH (pH = 13.5) were prepared using Millipore water (Merck-Millipore, Mill-Q IQ 7003, resistivity = 18.2 MΩ cm−1) and KOH pellets (Sigma-Aldrich, Germany) or concentrated HCl (37%, Sigma-Aldrich, Germany). The electrolyte was purged directly before the measurement with either oxygen (6.0, Air Liquide, Germany) or nitrogen for 15 min. This purging was reduced during the measurements to prevent disturbing the data acquisition. The ORR measurements were performed with a spinning electrode at 1600 rpm, forming a rotating disc electrode (RDE) (Pine Instruments, USA) and a sweep rate of 50 mV s−1. Cyclic sweep voltammograms without a rotating disc electrode were collected with a sweep rate of 100 mV s−1 and a potential ramp between 0.12 V and 1.6 V, while all potentials were corrected to obtain the potential against the reversible hydrogen electrode (RHE), using the Nernst equation:| E(RHE) = EAg/AgCl + 0.059 pH + E0Ag/AgCl | (2) |
Surface normalization
Even though the same mass was drop cast on the electrode for each experiment, following routine protocols in nanoparticle electrochemistry, we decided to deviate from the commonly applied mass-based normalization. Instead, we used nanoparticle surface-area normalization for all measured data, as the observed reactions are primarily surface-area-driven. This is vital for comparing nanoparticle samples with strongly deviating densities or particle size distributions. In this study, size deviations between samples were minimal, but density differences between the particles were substantial (10.49 g cm−3:19.3 g cm−3 for Ag:Au). For example, if we compare the Ag and Au nanoparticles at identical mass loading, the Ag sample will have a surface area more than 1.8 times higher than its Au counterpart, making a meaningful comparison between samples impractical. To compensate for this effect, we used the combination of two analysis techniques to calculate the total nanoparticle surface area in the colloids deposited on the electrodes. At first, we performed UV/vis measurements of the colloids and used these to determine the mass concentrations based on Lambert–Beer’s-Law, using calibrations based on gravimetry. We then determined the nanoparticle size distributions using analytical disk centrifugation (ADC) to convert the mass concentration into surface concentration. The known mass concentrations from UV/vis measurements allowed us to determine the exact mass injected into the ADC, while the ADC gave us information on the distribution of mass-weighted particle size. Based on this, we estimated the mass of nanoparticles found in the respective size fraction in a reliable range from 5–500 nm with varying bin widths. By dividing the measured mass of every size fraction by the mass of a single nanoparticle of the corresponding size, while assuming the ideal spherical shape of the particles and the bulk density of the corresponding composition, we can calculate the total number of particles in the corresponding size fraction and thus in the whole injected sample. A simple calculation of the surface area of a single nanoparticle of the corresponding size, assuming an ideal sphere, and multiplication of this value with the number of particles in each fraction, allows us to yield an absolute surface-weighted particle size distribution. The surface concentration of the entire sample was then calculated by integrating the surface distribution curve.
Author contributions
Frederic Stein: investigation, formal analysis writing – original draft, methodology, visualization. Sebastian Kohsakowski: formal analysis, investigation. Ricardo Martinez-Hincapie: investigation, writing – reviewing and editing. Sven Reichenberger: resources, investigation, formal analysis, writing – reviewing and editing. Christoph Rehbock: conceptualization, resources, writing – reviewing and editing, supervision. Viktor Colic: resources, writing – reviewing and editing. Daniel Guay: resources, writing – reviewing and editing. Stephan Barcikowski: resources, writing – reviewing and editing, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG) (Project BA 3580/16-1).Notes and references
- (a) A. Ivask, T. Titma, M. Visnapuu, H. Vija, A. Kakinen, M. Sihtmae, S. Pokhrel, L. Madler, M. Heinlaan, V. Kisand, R. Shimmo and A. Kahru, Curr. Top. Med. Chem., 2015, 15, 1914–1929 CrossRef CAS PubMed; (b) C. Kaweeteerawat, A. Ivask, R. Liu, H. Zhang, C. H. Chang, C. Low-Kam, H. Fischer, Z. Ji, S. Pokhrel, Y. Cohen, D. Telesca, J. Zink, L. Mädler, P. A. Holden, A. Nel and H. Godwin, Environ. Sci. Technol., 2015, 49, 1105–1112 CrossRef CAS PubMed.
- G. Peng, Y. He, X. Wang, Y. Cheng, H. Zhang, K. Savolainen, L. Mädler, S. Pokhrel and S. Lin, ACS Nano, 2020, 14, 4166–4177 CrossRef CAS PubMed.
- C. Sicard-Roselli, E. Brun, M. Gilles, G. Baldacchino, C. Kelsey, H. McQuaid, C. Polin, N. Wardlow and F. Currell, Small, 2014, 10, 3338–3346 CrossRef CAS PubMed.
- S. Zwiehoff, J. Johny, C. Behrends, A. Landmann, F. Mentzel, C. Bäumer, K. Kröninger, C. Rehbock, B. Timmermann and S. Barcikowski, Small, 2022, 18, e2106383 CrossRef.
- H. Xiang, W. Feng and Y. Chen, Adv. Mater., 2020, 32, e1905994 CrossRef PubMed.
- (a) M. B. Cortie and A. M. McDonagh, Chem. Rev., 2011, 111, 3713–3735 CrossRef CAS; (b) S. Kadkhodazadeh, F. A. A. Nugroho, C. Langhammer, M. Beleggia and J. B. Wagner, ACS Photonics, 2019, 6, 779–786 CrossRef CAS; (c) A. Loiseau, V. Asila, G. Boitel-Aullen, M. Lam, M. Salmain and S. Boujday, Biosensors, 2019, 9, 78 CrossRef CAS; (d) L. Wang, M. Hasanzadeh Kafshgari and M. Meunier, Adv. Funct. Mater., 2020, 30, 2005400 CrossRef CAS; (e) Z. Zhang and K. Deng, ACS Catal., 2015, 5, 6529–6544 CrossRef CAS.
- A. V. Girão, P. C. Pinheiro, M. Ferro and T. Trindade, RSC Adv., 2017, 7, 15944–15951 RSC.
- A. Neumeister, J. Jakobi, C. Rehbock, J. Moysig and S. Barcikowski, Phys. Chem. Chem. Phys., 2014, 16, 23671–23678 RSC.
- G. Doria, M. Larguinho, J. T. Dias, E. Pereira, R. Franco and P. V. Baptista, Nanotechnology, 2010, 21, 255101 CrossRef CAS PubMed.
- G. A. Sotiriou, G. D. Etterlin, A. Spyrogianni, F. Krumeich, J.-C. Leroux and S. E. Pratsinis, Chem. Commun., 2014, 50, 13559–13562 RSC.
- S. Grade, J. Eberhard, J. Jakobi, A. Winkel, M. Stiesch and S. Barcikowski, Gold Bull., 2014, 47, 83–93 CrossRef CAS.
- U. Taylor, D. Tiedemann, C. Rehbock, W. A. Kues, S. Barcikowski and D. Rath, Beilstein J. Nanotechnol., 2015, 6, 651–664 CrossRef CAS PubMed.
- D. Tiedemann, U. Taylor, C. Rehbock, J. Jakobi, S. Klein, W. A. Kues, S. Barcikowski and D. Rath, Analyst, 2014, 139, 931–942 RSC.
- A. Al-Zubeidi, F. Stein, C. Flatebo, C. Rehbock, S. A. Hosseini Jebeli, C. F. Landes, S. Barcikowski and S. Link, ACS Nano, 2021, 15, 8363–8375 CrossRef CAS PubMed.
- T. Benkó, A. Beck, K. Frey, D. F. Srankó, O. Geszti, G. Sáfrán, B. Maróti and Z. Schay, Appl. Catal., A, 2014, 479, 103–111 CrossRef.
- X. Huang, X. Wang, X. Wang, X. Wang, M. Tan, W. Ding and X. Lu, J. Catal., 2013, 301, 217–226 CrossRef CAS.
- C.-W. Yen, M.-L. Lin, A. Wang, S.-A. Chen, J.-M. Chen and C.-Y. Mou, J. Phys. Chem. C, 2009, 113, 17831–17839 CrossRef CAS.
- A. Wittstock, V. Zielasek, J. Biener, C. M. Friend and M. Bäumer, Science, 2010, 327, 319–322 CrossRef CAS.
- J. W. Park, W. Choi, J. Noh, W. Park, G. H. Gu, J. Park, Y. Jung and H. Song, ACS Appl. Mater. Interfaces, 2022, 14, 6604–6614 CrossRef CAS.
- O. R. Schade, F. Stein, S. Reichenberger, A. Gaur, E. Saraci, S. Barcikowski and J.-D. Grunwaldt, Adv. Synth. Catal., 2020, 362, 5681–5696 CrossRef CAS.
- O. Prymak, V. Grasmik, K. Loza, M. Heggen and M. Epple, Cryst. Growth Des., 2020, 20, 107–115 CrossRef CAS.
- V. Grasmik, C. Rurainsky, K. Loza, M. V. Evers, O. Prymak, M. Heggen, K. Tschulik and M. Epple, Chem. - Eur. J., 2018, 24, 9051–9060 CrossRef CAS.
- (a) D. Mahl, J. Diendorf, S. Ristig, C. Greulich, Z.-A. Li, M. Farle, M. Köller and M. Epple, J. Nanopart. Res., 2012, 14(10), 1153 CrossRef; (b) T. Li, B. Albee, M. Alemayehu, R. Diaz, L. Ingham, S. Kamal, M. Rodriguez and S. W. Bishnoi, Anal. Bioanal. Chem., 2010, 398, 689–700 CrossRef CAS; (c) N. Blommaerts, H. Vanrompay, S. Nuti, S. Lenaerts, S. Bals and S. W. Verbruggen, Small, 2019, 15, e1902791 CrossRef PubMed.
- (a) R. G. Saratale, G. D. Saratale, H. S. Shin, J. M. Jacob, A. Pugazhendhi, M. Bhaisare and G. Kumar, Environ. Sci. Pollut. Res. Int., 2018, 25, 10164–10183 CrossRef CAS; (b) D.-B. Grys, B. de Nijs, A. R. Salmon, J. Huang, W. Wang, W.-H. Chen, O. A. Scherman and J. J. Baumberg, ACS Nano, 2020, 14, 8689–8696 CrossRef CAS; (c) J.-W. Park and J. S. Shumaker-Parry, J. Am. Chem. Soc., 2014, 136, 1907–1921 CrossRef CAS.
- R. Streubel, S. Barcikowski and B. Gökce, Opt. Lett., 2016, 41, 1486–1489 CrossRef CAS.
- C. Rehbock, J. Jakobi, L. Gamrad, S. van der Meer, D. Tiedemann, U. Taylor, W. Kues, D. Rath and S. Barcikowski, Beilstein J. Nanotechnol., 2014, 5, 1523–1541 CrossRef.
- V. Amendola, D. Amans, Y. Ishikawa, N. Koshizaki, S. Scirè, G. Compagnini, S. Reichenberger and S. Barcikowski, Chem. - Eur. J., 2020, 26, 9206–9242 CrossRef CAS.
- C.-Y. Shih, M. V. Shugaev, C. Wu and L. V. Zhigilei, Phys. Chem. Chem. Phys., 2020, 22, 7077–7099 RSC.
- (a) J. Johny, M. Kamp, O. Prymak, A. Tymoczko, U. Wiedwald, C. Rehbock, U. Schürmann, R. Popescu, D. Gerthsen, L. Kienle, S. Shaji and S. Barcikowski, J. Phys. Chem. C, 2021, 125, 9534–9549 CrossRef CAS; (b) A. Tymoczko, M. Kamp, O. Prymak, C. Rehbock, J. Jakobi, U. Schürmann, L. Kienle and S. Barcikowski, Nanoscale, 2018, 10, 16434–16437 RSC; (c) A. Tymoczko, M. Kamp, C. Rehbock, L. Kienle, E. Cattaruzza, S. Barcikowski and V. Amendola, Nanoscale Horiz., 2019, 4, 1326–1332 RSC.
- (a) D. Zhang, B. Gökce and S. Barcikowski, Chem. Rev., 2017, 117, 3990–4103 CrossRef CAS PubMed; (b) J. Zhang, M. Chaker and D. Ma, J. Colloid Interface Sci., 2017, 489, 138–149 CrossRef CAS.
- F. Waag, R. Streubel, B. Gökce and S. Barcikowski, Appl. Nanosci., 2021, 11, 1303–1312 CrossRef CAS.
- C. Rehbock, V. Merk, L. Gamrad, R. Streubel and S. Barcikowski, Phys. Chem. Chem. Phys., 2013, 15, 3057–3067 RSC.
- V. Merk, C. Rehbock, F. Becker, U. Hagemann, H. Nienhaus and S. Barcikowski, Langmuir, 2014, 30, 4213–4222 CrossRef CAS PubMed.
- C. Pfeiffer, C. Rehbock, D. Hühn, C. Carrillo-Carrion, D. J. de Aberasturi, V. Merk, S. Barcikowski and W. J. Parak, J. R. Soc. Interface, 2014, 11, 20130931 CrossRef PubMed.
- F. Bonaccorso, M. Zerbetto, A. C. Ferrari and V. Amendola, J. Phys. Chem. C, 2013, 117, 13217–13229 CrossRef CAS.
- O. Prymak, J. Jakobi, C. Rehbock, M. Epple and S. Barcikowski, Mater. Chem. Phys., 2018, 207, 442–450 CrossRef CAS.
- D. S. Chauhan, R. Prasad, J. Devrukhkar, K. Selvaraj and R. Srivastava, Bioconjugate Chem., 2018, 29, 1510–1518 CrossRef CAS.
- G. Guisbiers, R. Mendoza-Cruz, L. Bazán-Díaz, J. J. Velázquez-Salazar, R. Mendoza-Perez, J. A. Robledo-Torres, J.-L. Rodriguez-Lopez, J. M. Montejano-Carrizales, R. L. Whetten and M. José-Yacamán, ACS Nano, 2016, 10, 188–198 CrossRef CAS PubMed.
- W. Martienssen and H. Warlimont, Springer Handbook of Condensed Matter and Materials Data, Springer Berlin Heidelberg, 2005 Search PubMed.
- L. Vitos, A. V. Ruban, H. L. Skriver and J. Kollár, Surf. Sci., 1998, 411, 186–202 CrossRef CAS.
- L. Deng, W. Hu, H. Deng, S. Xiao and J. Tang, J. Phys. Chem. C, 2011, 115, 11355–11363 CrossRef CAS.
- T. Löffler, F. Waag, B. Gökce, A. Ludwig, S. Barcikowski and W. Schuhmann, ACS Catal., 2021, 11, 1014–1023 CrossRef.
- M. Lau, S. Reichenberger, I. Haxhiaj, S. Barcikowski and A. M. Müller, ACS Appl. Energy Mater., 2018, 1(10), 5366–5385 CAS.
- R. Praveen and R. Ramaraj, Mater. Chem. Phys., 2019, 238, 121915 CrossRef CAS.
- H. Muto, K. Yamada, K. Miyajima and F. Mafuné, J. Phys. Chem. C, 2007, 111, 17221–17226 CrossRef CAS.
- M. de Anda Villa, J. Gaudin, D. Amans, F. Boudjada, J. Bozek, R. Evaristo Grisenti, E. Lamour, G. Laurens, S. Macé, C. Nicolas, I. Papagiannouli, M. Patanen, C. Prigent, E. Robert, S. Steydli, M. Trassinelli, D. Vernhet and A. Lévy, Langmuir, 2019, 35, 11859–11871 CrossRef CAS.
- (a) S. W. Gaarenstroom and N. Winograd, J. Chem. Phys., 1977, 67, 3500–3506 CrossRef CAS; (b) G. B. Hoflund, Z. F. Hazos and G. N. Salaita, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 11126–11133 CrossRef CAS.
- Y.-R. Zheng, J. Vernieres, Z. Wang, K. Zhang, D. Hochfilzer, K. Krempl, T.-W. Liao, F. Presel, T. Altantzis, J. Fatermans, S. B. Scott, N. M. Secher, C. Moon, P. Liu, S. Bals, S. van Aert, A. Cao, M. Anand, J. K. Nørskov, J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2022, 7, 55–64 CrossRef CAS.
- T. Preocanin, F. Supljika and N. Kallay, J. Colloid Interface Sci., 2009, 337, 501–507 CrossRef CAS.
- M. Smiljanić, U. Petek, M. Bele, F. Ruiz-Zepeda, M. Šala, P. Jovanovič, M. Gaberšček and N. Hodnik, J. Phys. Chem. C, 2021, 125, 635–647 CrossRef.
- B. J. Plowman, B. Sidhureddy, S. V. Sokolov, N. P. Young, A. Chen and R. G. Compton, ChemElectroChem, 2016, 3, 1039–1043 CrossRef CAS.
- X. Li, Q. Chen, I. McCue, J. Snyder, P. Crozier, J. Erlebacher and K. Sieradzki, Nano Lett., 2014, 14, 2569–2577 CrossRef CAS.
- E. N. Saw, V. Grasmik, C. Rurainsky, M. Epple and K. Tschulik, Faraday Discuss., 2016, 193, 327–338 RSC.
- C. A. Starr and D. A. Buttry, ECS Trans., 2014, 58, 19–26 CrossRef.
- C. Rurainsky, A. G. Manjón, F. Hiege, Y.-T. Chen, C. Scheu and K. Tschulik, J. Mater. Chem. A, 2020, 8, 19405–19413 RSC.
- P. S. Yerragopu, S. Hiregoudar, U. Nidoni, K. T. Ramappa, A. G. Sreenivas and S. R. Doddagoudar, Int. Res. J. Pure Appl. Chem., 2020, 37–50 CrossRef.
- A. Arvinte, I.-A. Crudu, F. Doroftei, D. Timpu and M. Pinteala, J. Electroanal. Chem., 2018, 829, 184–193 CrossRef CAS.
- J. Snyder, K. Livi and J. Erlebacher, J. Electrochem. Soc., 2008, 155, C464 CrossRef CAS.
- J. Zhu, Y. Xu, J. Wang, J. Lin, X. Sun and S. Mao, Phys. Chem. Chem. Phys., 2015, 17, 28666–28673 RSC.
- X. Wu, M. El Kazzi and C. Villevieille, J. Electroceram., 2017, 38, 207–214 CrossRef CAS.
- S. Ristig, O. Prymak, K. Loza, M. Gocyla, W. Meyer-Zaika, M. Heggen, D. Raabe and M. Epple, J. Mater. Chem. B, 2015, 3, 4654–4662 RSC.
- V. Petkov, S. Shastri, S. Shan, P. Joseph, J. Luo, C.-J. Zhong, T. Nakamura, Y. Herbani and S. Sato, J. Phys. Chem. C, 2013, 117, 22131–22141 CrossRef CAS.
- N. Alissawi, V. Zaporojtchenko, T. Strunskus, I. Kocabas, V. S. K. Chakravadhanula, L. Kienle, D. Garbe-Schönberg and F. Faupel, Gold Bull., 2013, 46, 3–11 CrossRef CAS.
- S. D. Angelov, S. Koenen, J. Jakobi, H. E. Heissler, M. Alam, K. Schwabe, S. Barcikowski and J. K. Krauss, J. Nanobiotechnol., 2016, 14, 3 CrossRef.
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef.
- C. D. Wagner, W. M. Riggs, L. E. Davis, J. F. Moulder and G. E. Muilenberg, Handbook of X-ray photoelectron spectroscopy, Perkin-Elmer Corporation, MN, USA, 1979 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2fd00092j |
| This journal is © The Royal Society of Chemistry 2023 |