 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mixed-metal nanoparticles: phase transitions and diffusion in Au–VO clusters†
Wolfgang E.
Ernst
 *a,
Maximilian
Lasserus
a,
Daniel
Knez
b,
Ferdinand
Hofer
b and
Andreas W.
Hauser
*a
*a,
Maximilian
Lasserus
a,
Daniel
Knez
b,
Ferdinand
Hofer
b and
Andreas W.
Hauser
*a
aInstitute of Experimental Physics, Graz University of Technology, Graz, Austria. E-mail: wolfgang.ernst@tugraz.at; andreas.hauser@tugraz.at
bInstitute for Electron Microscopy and Nanoanalysis, Graz University of Technology, Graz, Austria
First published on 11th July 2022
Abstract
Nanoparticles with diameters in the range of a few nanometers, consisting of gold and vanadium oxide, are synthesized by sequential doping of cold helium droplets in a molecular beam apparatus and deposited on solid carbon substrates. After surface deposition, the samples are removed and various measurement techniques are applied to characterize the created particles: scanning transmission electron microscopy (STEM) at atomic resolution, temperature dependent STEM and TEM up to 650 °C, energy-dispersive X-ray spectroscopy (EDXS) and electron energy loss spectroscopy (EELS). In previous experiments we have shown that pure V2O5 nanoparticles can be generated by sublimation from the bulk and deposited without affecting their original stoichiometry. Interestingly, our follow-up attempts to create Au@V2O5 core@shell particles do not yield the expected encapsulated structure. Instead, Janus particles of Au and V2O5 with diameters between 10 and 20 nm are identified after deposition. At the interface of the Au and the V2O5 parts we observe an epitaxial-like growth of the vanadium oxide next to the Au structure. To test the temperature stability of these Janus-type particles, the samples are heated in situ during the STEM measurements from room temperature up to 650 °C, where a reduction from V2O5 to V2O3 is followed by a restructuring of the gold atoms to form a Wulff-shaped cluster layer. The temperature dependent dynamic interplay between gold and vanadium oxide in structures of only a few nanometer size is the central topic of this contribution to the Faraday Discussion.
1 Introduction
With numerous potential applications, nanosized metal and metal oxide particles have become increasingly interesting also for fundamental research.1 Prospective applications range from the development of new catalysts and sensors2–7via battery electrode design8 to nano-optical and nano-electronic devices.9,10 Materials based on vanadium oxides have been particularly interesting for catalysis. Due to the different oxidation stages of vanadium, the chemical properties vary strongly, and nanostructured vanadium oxide, prepared in a specific oxidation state, is a highly desired but challenging goal.11 Battery electrodes could be improved by using two-dimensional V2O5 polymorphs as alkali intercalation compounds.12,13 Combinations with gold are considered promising for catalytic oxidation of hydrocarbons or CO14,15 as well as for enhanced sensing properties.16 Significant amounts of clusters can be synthesized from precursor molecules in solution by chemical methods yielding so-called ligand stabilized clusters, see for example ref. 17. These methods have the disadvantage that the influence of the ligand may alter the desired cluster properties. A solvent-free nanoparticle synthesis including some degree of structure and size control has been demonstrated by Palmer and co-workers.18 For the last decade, we have followed an approach to create clusters of atoms or molecules in a cold superfluid helium environment. The method is based on the development of an efficient source for generating superfluid helium droplets in a beam expansion. The droplets can serve as cold traps for the accumulation of other species to form clusters that may be deposited on solid substrates. As we have shown, nanoparticles of virtually any combination of elements can be produced in this way.19 Due to the inert environment of superfluid helium, they are free of any other chemicals or additives. With all processes including the deposition taking place in ultrahigh vacuum (UHV), contaminations of the produced samples are excluded. In this paper, we report the synthesis of mixed gold–vanadium oxide nanoparticles and their deposition on amorphous carbon substrates. The created samples were studied by electron microscopy, energy dispersive X-ray spectroscopy (EDXS), and electron energy loss spectroscopy (EELS). Our focus was set on the temperature dependence of the observed structures and corresponding phase transitions in the range from room temperature to about 650 °C. Prior to this work on the mixed nanoparticles, we have shown that clusters of vanadium pentoxide can be produced in superfluid helium droplets by accumulation of sublimated (V2O5)2 from a hot pick-up oven.20 V2O5 nanoparticles of sizes around 10 nm were clearly identified after deposition at room temperature by scanning transmission electron microscopy (STEM), EELS, and UV-visible absorption spectroscopy.21 This article is structured as follows. First, we provide the details of the helium droplet experiment and our measurement techniques. This section is then followed by a brief overview of our computational attempts to better understand the interactions between gold and vanadium oxide. In Section 4 we present the results of our temperature-dependent studies and discuss the observed structures and phase transitions.2 Experimental details
The experimental setup is depicted in Fig. 1 and described in more detail in ref. 19 and 22. The most important aspects are summarized in the following: highly purified helium is expanded at a stagnation pressure of 20 bar through a 5 μm nozzle, with a temperature of 5.4 K, forming a beam of superfluid He droplets (HeN). The produced droplets have an internal temperature of 0.4 K, which makes them ideal for preserving the structures of dopants and allows to synthesise weakly bound aggregates not achievable otherwise. Due to the He environment chemical interactions can be excluded. The droplet beam is directed through two pick-up zones, where the desired elements are placed in heating crucibles (see sketch in Fig. 1). The crucibles are resistively heated until the vapour pressure of the material is sufficient, such that enough atoms/molecules are picked up by the helium droplets, typically around 10−3 mbar. The superfluidity of the HeN environment enables an unobstructed and frictionless roaming of dopant atoms or molecules. Upon statistical collisions within the HeN, the dopants accumulate and start to form clusters.23 For the described experiment, gold (Ögussa 99.9%) was placed in the first pickup oven and V2O5 (MaTeck 99.9%) powder in the second one. Subsequently, the doped helium beam passes through the UHV chamber with various molecular beam diagnostics like time-of-flight and quadrupole mass spectrometers. It is then terminated on a heatable substrate, suitable for Scanning Transmission Electron Microscopy (STEM) (DENSsolutions Nano-Chip XT carbon) analysis. For the purpose of core–shell cluster creation, we usually take care that the number ratio of core to shell particles is smaller than one to ensure a complete coating of the shell. The dopant numbers cannot be determined quantitatively but we have described a procedure for an estimate based on the shrinkage of the helium droplets after doping.24 | ||
| Fig. 1 Schematics of the three chamber setup ((a) SC, (b) PC, (c) DC) of the HeN beam machine used for the experiments described in this paper, see also ref. 22. The droplet beam produced by the cooled nozzle (1) is shaped by the skimmers (2, 8) and an aperture (9). The droplets can be sequentially doped using different sources (4, 5, 6); a beam flag (7) is used to control the pickup (dashed line indicates closed position). For diagnostics, a TOFMS (10), substrates for ex situ TEM investigations (11), and a quartz crystal microbalance (12) are available on axis, as well as an off-axis QMS (13). The TOFMS is equipped with an electron gun for electron impact ionization (not shown) but also allows photoionization using an external laser. A UHV-transfer system (14) enables the transport of surface deposited samples to external diagnostics without breaking the vacuum. Reproduced from ref. 22 [P. Thaler, A. Volk, D. Knez, F. Lackner, G. Haberfehlner, J. Steurer, M. Schnedlitz and W. E. Ernst, J. Chem. Phys., 2015, 143, 134201], with the permission of AIP Publishing. | ||
After deposition, the samples were taken out of the UHV and kept at ambient conditions for several minutes during transport to the microscope. For STEM analysis, a probe-corrected FEI Titan3 G2 60-300 was used, set up for High-Angle Annular Dark-Field (HAADF) imaging. The electron energy was set to 300 keV and the convergent angle was 19.6 mrad. The predominant Rutherford scattering in this imaging mode results in a contrast which is highly dependent on the atomic number, causing Au to appear brighter than vanadium oxide. Note that a transition between the oxidation states of vanadium oxides is well described in the literature and is induced by several effects, e.g. by electron bombardment and changes in temperature.25–28 Therefore, the exposure time for all STEM measurements in this work is reduced to 2.4 μs (the minimum in this mode), if not stated differently. Elemental maps are obtained by EDX and EELS analyses.
3 Computational approach
Despite the fact that the details of the growth process of mixed-metallic impurities in helium nanodroplets are still an open debate in the community, there is consent and experimental evidence19 that sequential doping is preferably leading to layered structures, which, due to the isotropy of the growth process, translates into e.g. core–shell structures for bimetallic setups. Little is known about scenarios involving metals and metal oxides; however, the substantially different character of the chemical bond in terms of directionality, strength and structural parameters makes any predictions difficult. From the theoretical point of view, a first step is the comparison of binding energies per particle for the two components Au and V2O5. The presence of vanadium(V) oxide is assumed here since it could be confirmed20 as the typical oxide forming in helium droplets after pickup of V2O5 fragments in the helium beam experiment. Note that, in the following computational study, the term ‘unit’ refers to a single Au atom in the case of gold clusters, while it refers to a whole V2O5 unit in the case of the vanadium(V) oxide clusters.Our computational approach for the free-gas oligomers is based on the combination of the GFN-xTB method of Grimme,29 a less expensive method for pre-optimization and structural search based on a tight binding approach, with the application of density functional theory. For the latter part, the ωB97X-V functional,30 a range-separated hybrid GGA density functional with VV10 nonlocal correlation,31 as it is implemented in the Q-Chem program package,32 is used. For O we use the polarized triple-zeta basis set of Weigend and Ahlrichs,33 for V and Au the effective core potential and basis sets of the LANL family.34
We define the binding energy per unit as
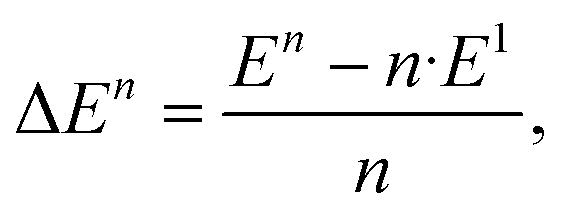 | (1) |
| n | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|
| Aun | −0.95 | −0.95 | −1.29 | −1.43 | −1.69 |
| (V2O5)n | −3.55 | −4.00 | −4.22 | −4.30 | −4.30 |
Table 1 shows an almost converged energy per unit for vanadium(V) oxide at n = 6, while the energy per Au atom is still far from convergence to the bulk value of cohesion energy per particle (3.8 eV experimentally,37 3.1–3.5 eV in theory35,38,39) at this size for the metal cluster. It further reveals a substantial difference in binding energies per unit for the two compounds. It is interesting to compare these values to a representative interaction energy between the two components, e.g. the binding energy of a single Au atom to (V2O5)4, for which we obtain a value of Ei = 3.30 eV with the same computational approach. The corresponding geometry of this mixed structure is depicted in Fig. 2. The Au atom prefers a bridging position between two dangling oxygen atoms. This finding of an interaction energy that is in magnitude higher than the bindingenergy per atom of a small Au cluster but smaller than the energy per unit for vanadium(v) oxidecluster formation, suggests that single Au atoms will prefer direct contact with the vanadium oxideover homonuclear aggregation, while free vanadium oxide fragments should prefer to contribute to the formation of larger vanadium oxide structures. On the other hand, this interaction energy is comparable to the binding energy of a single Au atom in bulk, or, in other words, the cohesion energy per atom of solid gold. This suggests a certain competition for the placement of gold atoms near interfaces of the two components: Au adsorption onto V2O5 units might become energetically more favorable than homonuclear coagulation among gold atoms, if these atoms are only loosely connected to the main gold cluster, for example at edges.
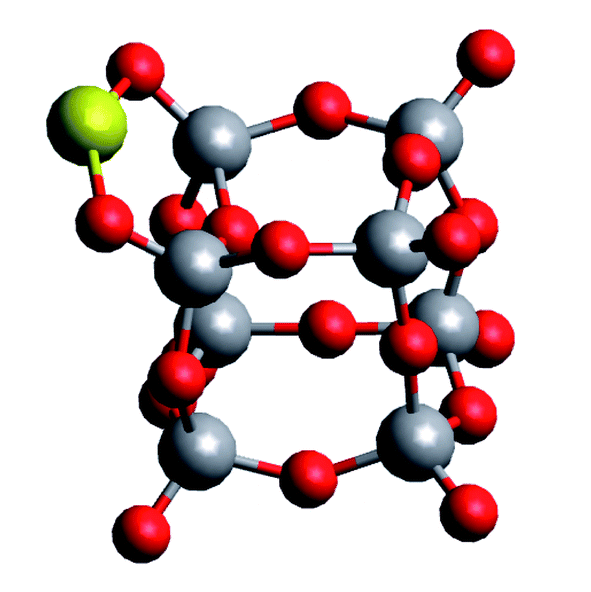 | ||
| Fig. 2 Adsorption of a single Au atom onto a (V2O5)4 cluster, calculated with the ωB97X-V density functional. | ||
In the process of our mixed cluster formation, this kind of competition should only represent an exception. As observed in all our preceding experiments for the generation of clusters of two materials in helium droplets, the formation of a cluster of thousands of atoms of the material picked up in the first cell has been finished in the millisecond flight time before the second material is added. This means that in the current case, a gold cluster of about 2000 atoms has aggregated when V2O5 units, presumable in the form of dimers,20 are added one by one. In this case, the situation seems to amount to a question about ‘nano-wetting’ of the gold cluster surface by (V2O5)2. Still comparing with our previous experience about core–shell formation, we would expect a similar result for the newly formed Au–V2O5 particles. While we cannot give precise numbers of our dopant particles, our experimental procedure always aims at a relation of the number of shell particles to core particles larger than one to ensure a full coating. From the theoretical side, calculations of binding energies of V2O5 molecules and/or dimers to gold cluster surfaces or at least to Au(111) would be desirable but are not available to our knowledge. Observations by the Freund group40 during the experimental creation of thin V2O5 films on Au(111) indicated island formation due to coalescence at preferred locations. Although their experiments differ from our procedures, there remains the question whether our coating by subsequent doping will work as in the case of two metals or result in nano-islands.
The above calculations for individual units will become relevant when the dynamics after cluster deposition and the thermal and electron beam treatment have started. Then situations may occur where diffusion or migration of individual gold atoms leads to the described bond competition.
4 Experimental results and discussion
Fig. 3 shows the results of the analytical STEM investigations of the particles. The high angle annular dark field images in Fig. 3a and b provide a contrast which depends on the atomic number. Therefore, bright regions exhibit very small Au clusters with sizes between 2 and 5 nm, the medium gray regions are the vanadium oxide deposits. In addition, an EELS and EDX-spectrum image has been recorded in parallel41 on the structure surrounded by the green frame in Fig. 3a. The magnified HAADF image of this area is given in Fig. 3b. The EDX elemental map of gold is displayed in Fig. 3c using the signal of the Au M line at 2.2 keV, thus confirming that the bright particles are indeed Au clusters. In Fig. 3d we have the EELS elemental map showing the vanadium distribution recorded with the V-L2,3 ionization edge which correlates with the medium gray regions in the HAADF image of Fig. 3b thus highlighting the vanadium oxide phase. Due to the overlapping of the V-L2,3 edge with the O K edge it was not possible to record oxygen maps using the O K edge.42 Although the near edge-fine structures (ELNES) of the EELS signal has been successfully employed for the identification of vanadium oxides and their oxidation states in previous experiments on vanadium oxide particles as well as thin films,43 we could not employ this method for the identification of the vanadium oxidation state in our STEM measurements. In order to avoid particle damage and to prevent electron beam induced reduction, we have to use a low dose electron beam and extremely small exposure times for all STEM measurements in this work. We reduced the exposure time to 2.4 μs which is the minimum in this mode. Therefore, we have a rather weak EELS signal and a lower spatial and energy resolution compared to other published spectra.42,43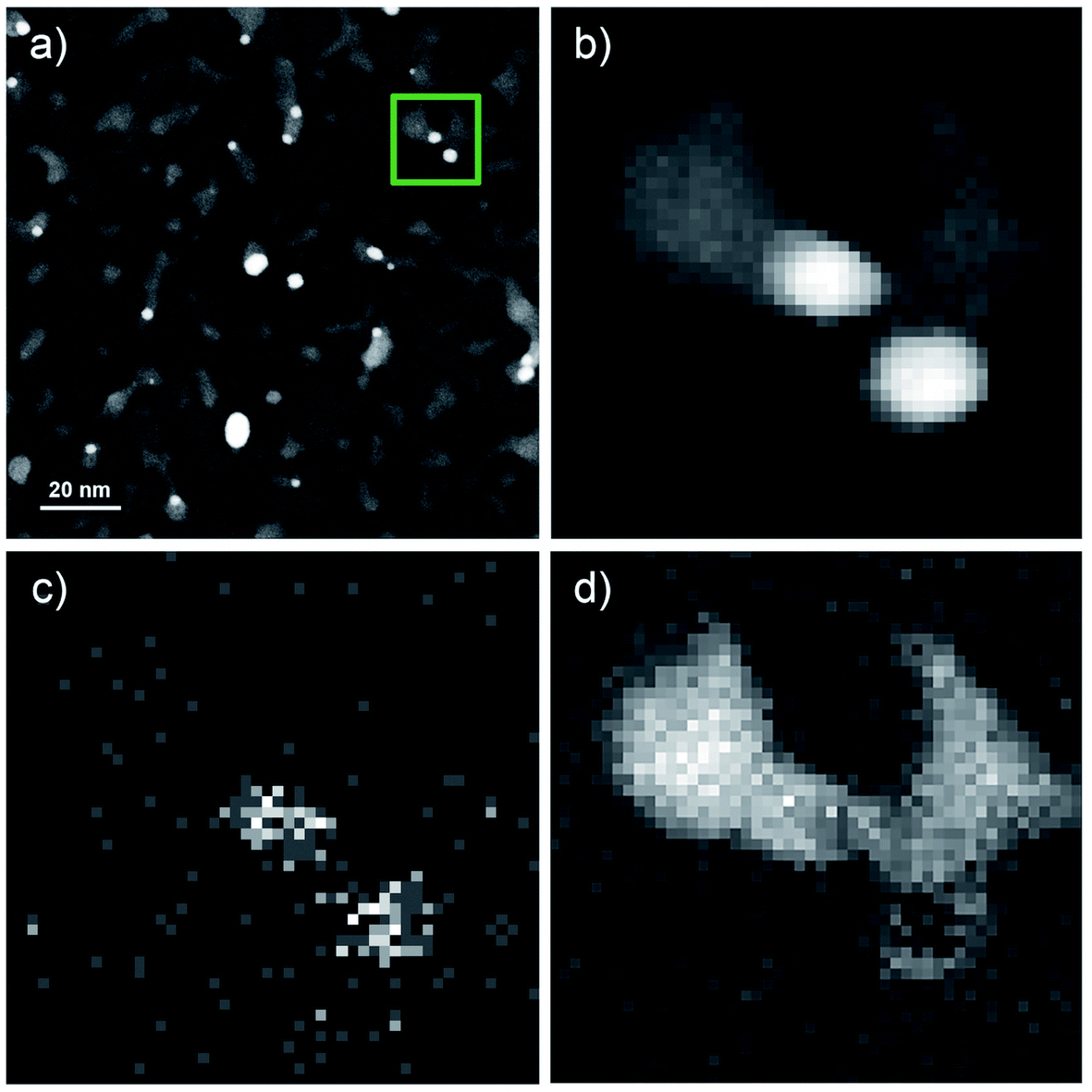 | ||
| Fig. 3 Overview of STEM HAADF images of V2O5/Au structures. (b) HAADF region for the measured EELS and EDX spectroscopy. (c) Spatially resolved EDX analysis based on the Au Mα peak at 2.1 keV. (d) Spatially resolved elemental EELS analysis using the signal of the V L-edge. (b), (c), and (d) are magnified images of the green marked area in (a), a square 20 nm x 20 nm. | ||
Nonetheless, our previous measurements of vanadium oxide nanoparticles alone under the same conditions and on the same substrates, were shown to be pure V2O5 nanostructures,21 therefore we identify these clusters as Au–V2O5. When inspecting the overview image in Fig. 3a, the Au share of each structure does not seem to be encapsulated by the larger V2O5 part of any structure. Our previous two-component metal cluster aggregation had yielded core@shell structures such as Au@Ag, Co@Au, Ni@Au, Fe@Au, and Ag@ZnO.44–49 Therefore, we expected an Au core surrounded by a V2O5 shell, but in the current images, most Au clusters are connected to larger vanadium structures at their tail, while a complete coating is never observed.
A representative Au–V2O5 cluster is depicted in Fig. 4. The bright lower part of the structure can be assigned as Au and the darker part on top as V2O5 portion. Within both structures the lattice plane distances are visible as periodic lines. Only planes that are oriented parallel to the incoming electron beam can be clearly identified. Some portions of the image appear washed-out and do not allow lattice structure assignments due to tilted angles with respect to the beam direction. Intensity plots for the areas in the black rectangle, assigned as thin Au layer, and the blue marked area, assigned as vanadium oxide, are shown in the right-hand part of Fig. 4. Literature values for cubic Au(111) and orthorhombic V2O5(211) are 2.36 Å and 2.48 Å, respectively, which lets us choose these assignments in reasonable agreement within the spatial resolution of 0.1 Å of the microscope. In our work on pure vanadium oxide depositions from the helium droplet source,21 corresponding STEM images of V2O5 clusters exhibited (020), (011), and (200) lattice portions with 2.2 Å, 2.7 Å, and 5.7 Å lattice distances, respectively. We have no control of the nanocrystal orientation. Furthermore, the contrast of the vanadium oxide part is not high enough for 3D imaging, a technique which we had demonstrated for Au@Ag core@shell clusters with our instrument before.50 As it also turns out, the Au–V2O5 particles alter their shape during longer electron beam exposure as previously shown for other transition metal oxide particles which have been also prepared by the helium droplet method.51 This makes a reconstruction of a 3D image from sequentially taken exposures unreliable. In the past, V2O5 films have been grown on plane Au(111) surfaces by oxidation of a previously deposited V layer with a sufficiently high pressure of O2.28 In our experiments, primarily V2O5 dimers are added one by one to a gold cluster core grown inside the droplets before,20 without the need for subsequent oxidation. As shown in a model calculation,52 the kinetic energy of the impact of the droplets on the substrate surface is small compared to the binding energy between the atoms and molecules of the grown cluster. Therefore, we believe that the observed V2O5 portions represent regular nanocrystals with typical literature lattice constants.
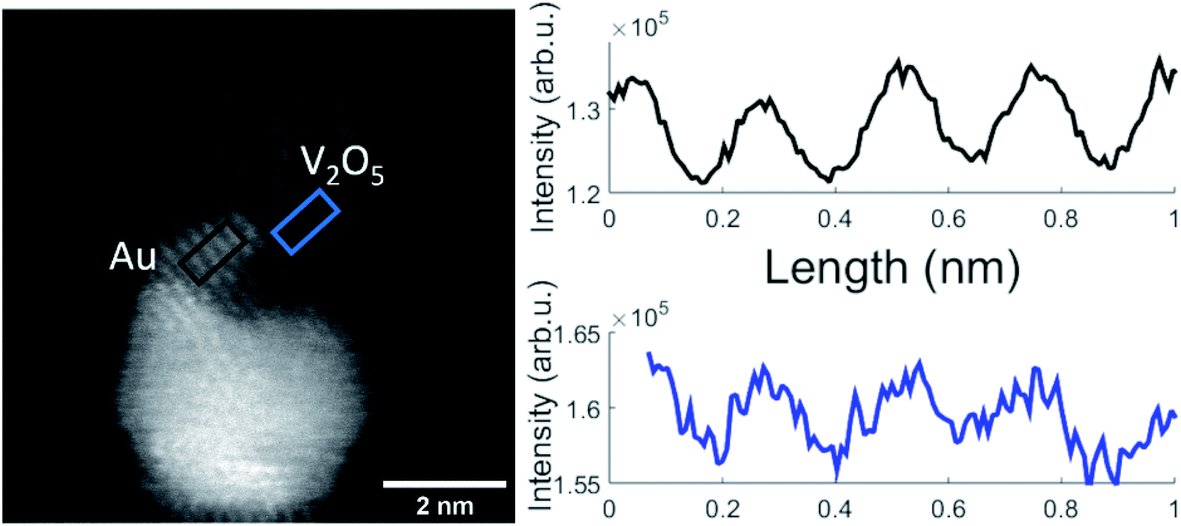 | ||
| Fig. 4 STEM HAADF images of a V2O5/Au structure (left) and the resulting lattice distances of the nanostructure extracted from contrast scans (right). Here the V2O5 part of the Janus-type particle (blue rectangle) exhibits a lattice spacing corresponding to a V2O5(211) orientation (2.48 Å literature value) which is close to the Au(111) lattice plane separation of 2.36 Å as it is retrieved from the black rectangle. | ||
For further understanding of this Au/V2O5 nanosystem, in situ heating was performed with a MEMS-based heating chip during the STEM measurement.53 A Janus-type particle of gold and vanadium oxide was monitored upon heating and appeared to maintain the described Au and V2O5 lattice parts stably up to 300 °C. Changes were observed when the samples were further heated in situ up to a temperature of 650 °C during the STEM measurements. In Fig. 5 several subsequently taken images of a structure at 650 °C are shown. Note that for each image within this figure the temperature was kept constant at 650 °C, and every image represents approximately one second of collection time. Therefore, 80 pictures means that approximately 80 seconds have passed after the first image was produced at this high temperature. For the calculation of the total electron illumination dose of the nanoparticle area, the algorithm described in ref. 51 was used. According to this estimate, the cluster had already been exposed to 3.8 × 107 electrons when the first image was taken, and for every 80 seconds of additional exposure the same number of electrons per second can be assumed if the projected area is approximately constant. In the 237 s from the beginning to the end of this sequence, a significant change is observed and a hexagonal shape seems to evolve with a partial coating of the gold portion with vanadium oxide.
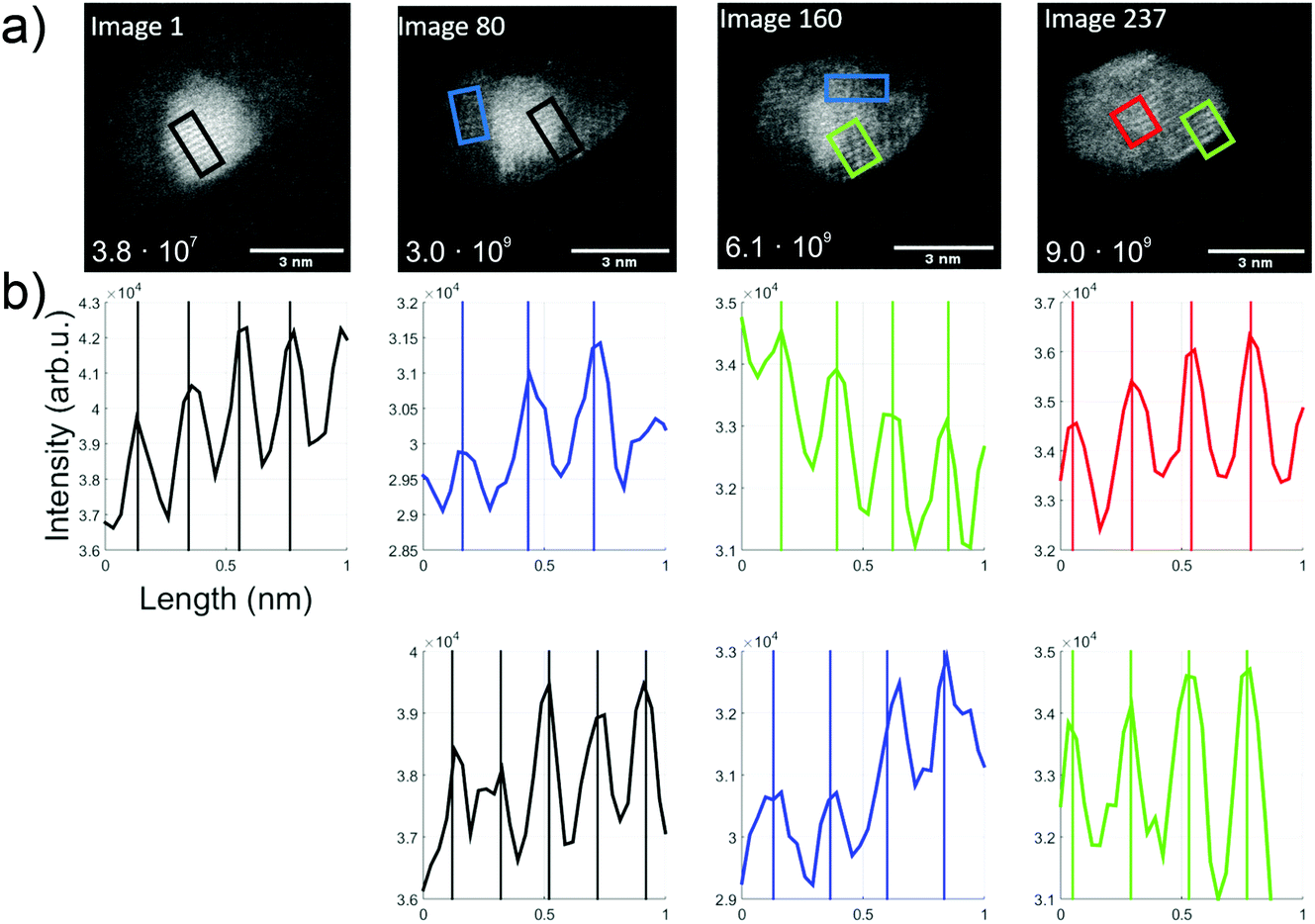 | ||
| Fig. 5 Overview over the thermal kinetics in an Au/V2O3 cluster at constant 650 °C. (a) A time series of TEM images (video provided in the ESI†). The number in the left lower corner indicates the estimated dosage in units of total electrons per cluster area. (b) Intensity profiles, extracted from the images directly above, and printed in matching colors to indicate the corresponding rectangular areas in the TEM images (length about 1 nm). Periodicities are determined via fits. | ||
The first image shows the typical Au fcc lattice in (200) orientation with a plane distance of 2.1 Å, while the second image shows V2O3(104) lattice planes with 2.7 Å (blue) and Au(200) planes with 2.1 Å. In the third image, probed by two rectangular selections (green and blue), a lattice distance of 2.3 Å can be identified which indicates the V2O3(006) plane. In the last image, the formation of 120° angles seems to start, and a spacing of 2.4 Å is measured within the red and green rectangles, which could refer to the Au(111) and/or V2O3(111) plane.
With the lattice plane separations of V2O5 and V2O3 being such that one can find orientations for either one of them with the determined values, an unambiguous assignment is difficult. For example, 2.7 Å distance can be assigned to V2O3(104) but also to V2O5(011). We assume that our vanadium oxide samples show the same thermal reduction behaviour as observed by other groups who report a reduction from V2O5 to V2O3 in TEM studies for temperatures above 600 °C. Su and Schlögl have suggested a transition from V2O5via VO2 to V2O3, where V2O3 remained at 600 °C (ref. 25) and Ramana et al. did not observe any reduced phase below 450 °C. At 500 °C they determined a phase which contained mainly V2O5 and additional, smaller amounts of V2O3 and V4O9.27 After heating beyond 600 °C only V2O3 remained. Both groups reported an ongoing reduction, predicting that V2O3 prevails at temperatures above 600 °C. For this reason, we assign the measured lattice plane separations in the bright areas to Au and the others to V2O3. The image sequence indicates dynamics that may be induced by the electron beam exposure, from image 1 with already more enclosure of the gold part by vanadium oxide (in comparison to the room temperature picture in Fig. 3), to a change of the gold cluster shape with some hexagonal boundaries and a more pronounced vanadium oxide coating.
In order to follow the described dynamics further, we produced a movie sequence on a different cluster which we started after a visual state similar to image 237, maintaining the same electron dose at constant 650 °C for another 250 seconds. Fig. 6 shows a number of images from this movie where the length scale is shown in image 1. Beyond image 50 a magnification is applied in order to make the structure changes more visible. Already in image 1, the structural feature of 120 degree angles becomes more emphasized. 50 s later (image 50), the brighter 120 degree edges indicate some gold diffusion which then increases to cover the edges such that we see a primary gold core with the typical Au(111) lattice plane separation of 2.36 Å, a vanadium oxide part surrounding the gold, perhaps enclosing it entirely, and a gold monolayer outside due to diffusion. The image sequence indicates a significant impact of our electron dose of roughly 107 electrons per second on the diffusion processes towards a partial phase segregation of the gold and vanadium oxide parts. The 120° edges suggest a particular change of the gold cluster shape. According to ref. 54, metal nanoparticles with diameters in the range of 15 nm or below, favour a so-called Wulff shape for energy reasons. A gold cluster would then arrange as a truncated octahedron with 6 square and 8 hexagon surfaces and with 36 edges and 24 vertices (point group Oh, sphericity 0.9099). While such interpretation is somewhat speculative, it may provide an explanation for the structural change upon the impact of thermal and electron beam exposure. In our previous temperature studies of metal core@shell nanoparticles including our model calculations, we have seen different diffusion processes. In the case of Au@Ag and Ag@Au,44 we observe nanoalloys at high temperature; in Ni@Au, nickel diffuses through the gold protective cover,45 as does Fe in Fe@Au.46 Among the iron triad combined with a gold shell, increased temperature tends to promote gold diffusion in Co@Au such that the formerly spherical Au shell becomes thinner on one side of the Co core and thicker on the other.48,55 So far, this is just a qualitative comparison. The binding energies of a gold atom to a gold cluster and a (V2O5)2 unit to a V2O5 cluster20 are comparable with about 3.5 to 4 eV, but the Au–VO system is much more complex and the reduction from V2O5 to V2O3 – perhaps even through intermediate oxidation states – will influence the diffusion dynamics significantly.
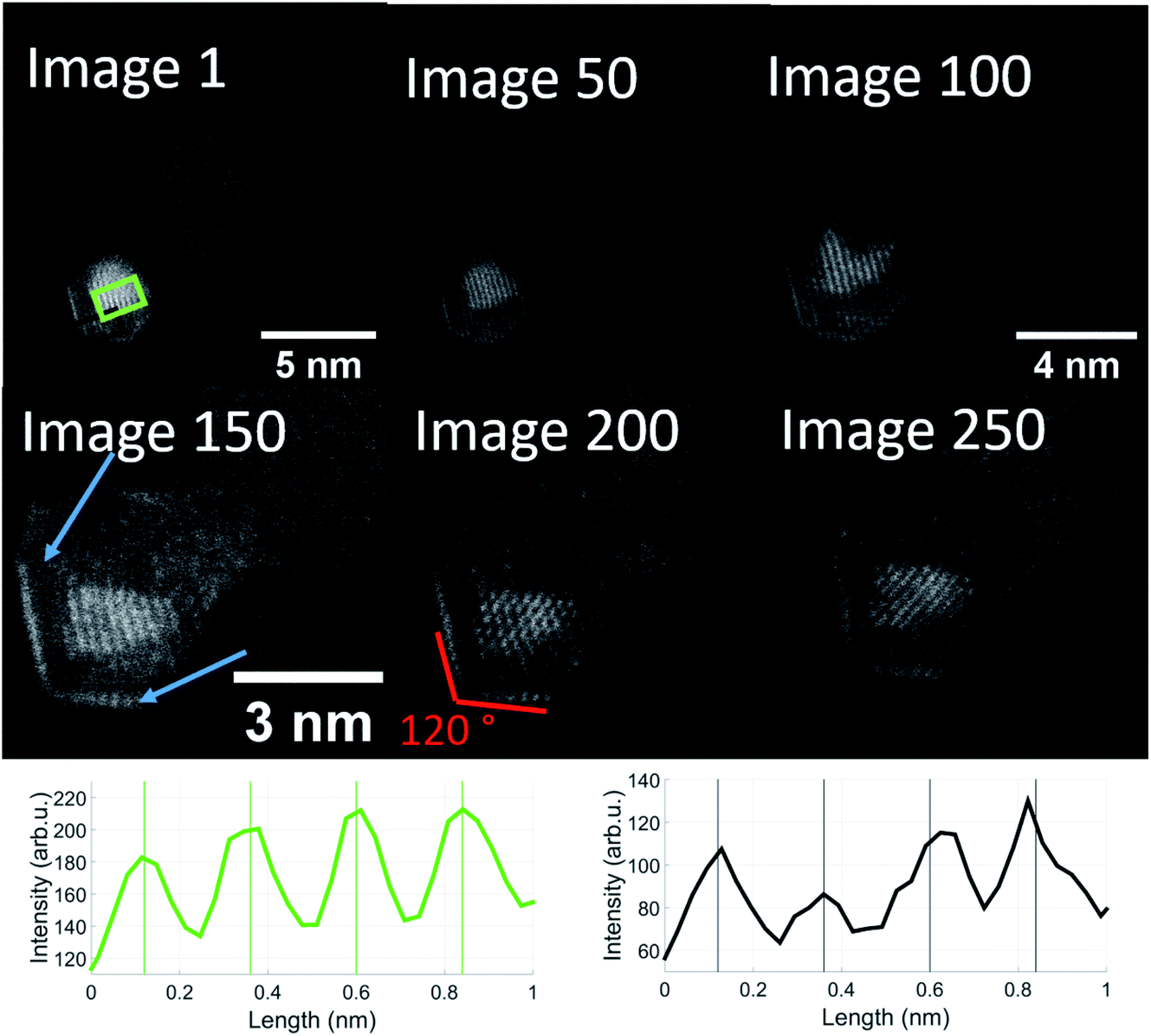 | ||
| Fig. 6 Sequential images taken from the same Au/V2O3 nanostructure at a temperature of 650 °C. Two distinct features can be observed: the Au atoms form a monolayer on the surface of the structure, marked by blue arrows and additionally, these layers as well as the gold core form an angle of exactly 120 degrees. The images were taken from a video which is included in the ESI.† The magnification was increased after image 50 and again after image 100. Note that the enhanced diffusion of the Au atoms has already started in image 1. The onset of a monolayer can be observed in the left corner. The lattice plane distances in the green and black rectangles are close to 2.4 Å, similar to image 237 of Fig. 5, with the difference that the gold and vanadium oxide parts seem more strongly separated, i.e. they presumably correspond to Au(111) (green) and V2O3(111) (black). | ||
5 Summary
Gold–vanadium oxide clusters of about 10 nm diameter were grown inside superfluid helium nanodroplets in a molecular beam expansion and deposited on amorphous carbon substrates. The samples were subsequently studied by scanning transmission electron microscopy, electron energy loss spectroscopy, and energy-dispersive X-ray spectroscopy over a temperature range from room temperature to 650 °C. At 650 °C, the diffusion dynamics inside individual clusters on the surface was monitored via STEM imaging. Several interesting features and processes were observed. In contrast to our metal core@shell helium droplet assisted cluster production, the Au–VO nanostructures exhibit rather a Janus type particle character. Thanks to our high resolution instrument, lattice planes could be identified for the nanocrystals and assigned. At room temperature, V2O5 cluster structures were assigned similar to our earlier investigations.21 Above 600 °C, a reduction to V2O3 was assumed due to other studies on VO films,26–28,43 and corroborated by our lattice plane distance measurements on the deposited structures.Continued electron beam exposure at 650 °C led to a restructuring of the whole nanoparticle towards a hexagonal shape, which we interpret as the formation of an Au Wulff-shaped cluster with a surrounding vanadium oxide layer and an Au-rich surface layer.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to thank all previous co-workers on the cluster project who contributed to the set-up of the lab, especially Philipp Thaler, Alexander Volk, Florian Lackner, Martin Schnedlitz, and the electron microscopy group around Gerald Kothleitner. The work was supported by the Austrian Science Foundation FWF in cooperation with the Christian Doppler Research Association (CDG) under Grant No. PIR8-N34 and is embedded in the NAWI cooperation of the universities in Graz. Further funding was received from the European Union's Horizon 2020 research program under Grant Agreement No. 823717-ESTEEM3.References
- E. Roduner, Chem. Soc. Rev., 2006, 35, 583–592 RSC.
- E. Jimenez-Izal, B. C. Gates and A. N. Alexandrova, Phys. Today, 2019, 72, 38–43 CrossRef CAS.
- Q. Zhang, I. Lee, J. B. Joo, F. Zaera and Y. Yin, Acc. Chem. Res., 2013, 46, 1816–1824 CrossRef CAS PubMed.
- D. Astruc, Chem. Rev., 2020, 120, 461–463 CrossRef CAS PubMed.
- X.-N. Li, L.-N. Wang, L.-H. Mou and S.-G. He, J. Phys. Chem. A, 2019, 123, 9257–9267 CrossRef CAS PubMed.
- M. Hu, J. Chen, Z.-Y. Li, L. Au, G. V. Hartland, X. Li, M. Marquez and Y. Xia, Chem. Soc. Rev., 2006, 35, 1084–1094 RSC.
- N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. R. Lytton-Jean, M. S. Han and C. A. Mirkin, Science, 2006, 312, 1027–1030 CrossRef CAS PubMed.
- F. Wan and Z. Niu, Angew. Chem., Int. Ed., 2019, 58, 16358–16367 CrossRef CAS PubMed.
- A. Rogov, Y. Mugnier and L. Bonacina, J. Opt., 2015, 17, 033001 CrossRef.
- M. Osada and T. Sasaki, J. Mater. Chem., 2009, 19, 2503–2511 RSC.
- M. Liu, B. Su, Y. Tang, X. Jiang and A. Yu, Adv. Energy Mater., 2017, 7, 1700885 CrossRef.
- R. Baddour-Hadjean, M. Safrany Renard, N. Emery, L. Huynh, M. Le and J. Pereira-Ramos, Electrochim. Acta, 2018, 270, 129–137 CrossRef CAS.
- R. Baddour-Hadjean, M. Safrany Renard and J. Pereira-Ramos, Acta Mater., 2019, 165, 183–191 CrossRef CAS.
- D. Andreeva, R. Nedyalkova, L. Ilieva and M. Abrashev, Appl. Catal., B, 2004, 52, 157–165 CrossRef CAS.
- R. Camposeco and R. Zanella, Catal. Today, 2022, 392–393, 49–59 CrossRef CAS.
- X. Yang, W. Wang, C. Wang, H. Xie, H. Fu, X. An, X. Jiang and A. Yu, Powder Technol., 2018, 339, 408–418 CrossRef CAS.
- G. Schmid and L. F. Chi, Adv. Mater., 1998, 10, 515–526 CrossRef CAS.
- R. E. Palmer, R. Cai and J. Vernieres, Acc. Chem. Res., 2018, 51, 2296–2304 CrossRef CAS PubMed.
- W. E. Ernst and A. W. Hauser, Phys. Chem. Chem. Phys., 2021, 23, 7553–7574 RSC.
- M. Lasserus, M. Schnedlitz, R. Messner, F. Lackner, W. E. Ernst and A. W. Hauser, Chem. Sci., 2019, 10, 3473–3480 RSC.
- M. Lasserus, D. Knez, F. Lackner, M. Schnedlitz, R. Messner, D. Schennach, G. Kothleitner, F. Hofer, A. W. Hauser and W. E. Ernst, Phys. Chem. Chem. Phys., 2019, 21, 21104–21108 RSC.
- P. Thaler, A. Volk, D. Knez, F. Lackner, G. Haberfehlner, J. Steurer, M. Schnedlitz and W. E. Ernst, J. Chem. Phys., 2015, 143, 134201 CrossRef.
- A. W. Hauser, A. Volk, P. Thaler and W. E. Ernst, Phys. Chem. Chem. Phys., 2015, 17, 10805–10812 RSC.
- A. Volk, P. Thaler, D. Knez, A. W. Hauser, J. Steurer, W. Grogger, F. Hofer and W. E. Ernst, Phys. Chem. Chem. Phys., 2016, 18, 1451–1459 RSC.
- D. S. Su, M. Wieske, E. Beckmann, A. Blume, G. Mestl and R. Schlögl, Catal. Lett., 2001, 75, 81–86 CrossRef CAS.
- D. S. Su and R. Schlögl, Catal. Lett., 2002, 83, 115–119 CrossRef CAS.
- C. Ramana, S. Utsunomiya, R. Ewing and U. Becker, Solid State Commun., 2006, 137, 645–649 CrossRef CAS.
- S. Guimond, D. Göbke, Y. Romanyshyn, J. M. Sturm, M. Naschitzki, H. Kuhlenbeck and H.-J. Freund, J. Phys. Chem. C, 2008, 112, 12363–12373 CrossRef CAS.
- S. Grimme, C. Bannwarth and P. Shushkov, J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef CAS.
- N. Mardirossian and M. Head-Gordon, Phys. Chem. Chem. Phys., 2014, 16, 9904–9924 RSC.
- N. Mardirossian and M. Head-Gordon, J. Chem. Phys., 2014, 140, 18A527 CrossRef PubMed.
- Y. Shao, et al. , Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- L. E. Roy, P. J. Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029–1031 CrossRef CAS PubMed.
- B. Assadollahzadeh and P. Schwerdtfeger, J. Chem. Phys., 2009, 131, 064306 CrossRef.
- L. Rincon, A. Hasmy, M. Marquez and C. Gonzalez, Chem. Phys. Lett., 2011, 503, 171–175 CrossRef CAS.
- C. Kittel, Introduction to Solid State Physics, Wiley, 8th edn, 2004 Search PubMed.
- N. Takeuchi, C. T. Chan and K. M. Ho, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 1565–1570 CrossRef CAS.
- G. Heimel, L. Romaner, J.-L. Brédas and E. Zojer, Surf. Sci., 2006, 600, 4548–4562 CrossRef CAS.
- S. Guimond, J. M. Sturm, D. Göbke, Y. Romanyshyn, M. Naschitzki, H. Kuhlenbeck and H.-J. Freund, J. Phys. Chem. C, 2008, 112, 11835–11846 CrossRef CAS.
- G. Kothleitner, M. J. Neish, N. R. Lugg, S. D. Findlay, W. Grogger, F. Hofer and L. J. Allen, Phys. Rev. Lett., 2014, 112, 085501 CrossRef.
- D. Su, H. Zandbergen, P. Tiemeijer, G. Kothleitner, M. Hävecker, C. Hébert, A. Knop-Gericke, B. Freitag, F. Hofer and R. Schlögl, Micron, 2003, 34, 235–238 CrossRef CAS.
- C. Hébert, M. Willinger, D. S. Su, P. Pongratz, P. Schattschneider and R. Schlögl, Eur. Phys. J. B, 2002, 28, 407–414 CrossRef.
- M. Lasserus, M. Schnedlitz, D. Knez, R. Messner, A. Schiffmann, F. Lackner, A. W. Hauser, F. Hofer and W. E. Ernst, Nanoscale, 2018, 10, 2017–2024 RSC.
- M. Schnedlitz, M. Lasserus, R. Meyer, D. Knez, F. Hofer, W. E. Ernst and A. W. Hauser, Chem. Mater., 2018, 30, 1113–1120 CrossRef CAS.
- M. Lasserus, D. Knez, M. Schnedlitz, A. W. Hauser, F. Hofer and W. E. Ernst, Nanoscale Adv., 2019, 1, 2276–2283 RSC.
- M. Schnedlitz, R. Fernandez-Perea, D. Knez, M. Lasserus, A. Schiffmann, F. Hofer, A. W. Hauser, M. P. de Lara-Castells and W. E. Ernst, J. Phys. Chem. C, 2019, 123, 20037–20043 CrossRef CAS.
- M. Schnedlitz, D. Knez, M. Lasserus, F. Hofer, R. Fernández-Perea, A. W. Hauser, M. P. de Lara-Castells and W. E. Ernst, J. Phys. Chem. C, 2020, 124, 16680–16688 CrossRef CAS.
- A. Schiffmann, T. Jauk, D. Knez, H. Fitzek, F. Hofer, F. Lackner and W. E. Ernst, Nano Res., 2020, 13, 2979–2986 CrossRef CAS.
- G. Haberfehlner, P. Thaler, D. Knez, A. Volk, F. Hofer, W. E. Ernst and G. Kothleitner, Nat. Commun., 2015, 6, 8779 CrossRef CAS PubMed.
- D. Knez, M. Schnedlitz, M. Lasserus, A. Schiffmann, W. E. Ernst and F. Hofer, Ultramicroscopy, 2018, 192, 69–79 CrossRef CAS PubMed.
- P. Thaler, A. Volk, M. Ratschek, M. Koch and W. E. Ernst, J. Chem. Phys., 2014, 140, 044326 CrossRef PubMed.
- R. Krisper, J. Lammer, Y. Pivak, E. Fisslthaler and W. Grogger, Ultramicroscopy, 2022, 234, 113461 CrossRef CAS.
- G. D. Barmparis, Z. Lodziana, N. Lopez and I. N. Remediakis, Beilstein J. Nanotechnol., 2015, 6, 361–368 CrossRef PubMed.
- D. Knez, M. Schnedlitz, M. Lasserus, A. W. Hauser, W. E. Ernst, F. Hofer and G. Kothleitner, Appl. Phys. Lett., 2019, 115, 123103 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: TEM movie of the structural evolution of an Au–V2O3 cluster as a function of time at 650 °C under constant electron beam exposure. See https://doi.org/10.1039/d2fd00089j |
| This journal is © The Royal Society of Chemistry 2023 |