 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural evolution under physical and chemical stimuli of metastable Au–Fe nanoalloys obtained by laser ablation in liquid
Andrea
Basagni
a,
Veronica
Torresan
a,
Pasquina
Marzola
b,
Marcela B.
Fernàndez van Raap
 c,
Luca
Nodari
d and
Vincenzo
Amendola
*a
c,
Luca
Nodari
d and
Vincenzo
Amendola
*a
aDepartment of Chemical Sciences, Università di Padova, Via Marzolo 1, I-35131 Padova, Italy. E-mail: vincenzo.amendola@unipd.it
bDepartment of Computer Science, University of Verona, Verona 37134, Italy
cPhysics Institute of La Plata (IFLP-CONICET), Physics Department, Faculty of Exact Sciences, National University of La Plata, La Plata, Argentina
dCNR-ICMATE Institute of Condensed Matter Chemistry and Technologies for Energy, Italian National Research Council, Corso Stati Uniti 4, 35127 Padova, Italy
First published on 24th May 2022
Abstract
Metastable alloy nanoparticles are investigated for their variety of appealing properties exploitable for photonics, magnetism, catalysis and nanobiotechnology. Notably, nanophases out of thermodynamic equilibrium feature a complex “ultrastructure” leading to a dynamic evolution of composition and atomic arrangement in response to physical–chemical stimuli. In this manuscript, metastable Au–Fe alloy nanoparticles were produced by laser ablation in liquid, an emerging versatile synthetic approach for freezing multielement nanosystems in non-equilibrium conditions. The Au–Fe nanoalloys were characterized through electron microscopy, elemental analysis, X-ray diffraction and Mössbauer spectroscopy. The dynamics of the structure of the Au–Fe system was tracked at high temperature under vacuum and atmospheric conditions, evidencing the intrinsic transformative nature of the metastable nanoalloy produced by laser ablation in liquid. This dynamic structure is relevant to possible application in several fields, from photocatalysis to nanomedicine, as demonstrated through an experiment of magnetic resonance imaging in biological fluids.
Introduction
Metastable alloys can be found in multicomponent nanoparticles (NPs) made of thermodynamically immiscible elements, when non-equilibrium synthetic processes are adopted.1–3 This is possible by bringing precursors and educts to high energy states, such that products initially adopt metastable structures, i.e. structures with higher thermodynamic free energy than in the most stable configuration.1,4,5 The resulting metastable compositions are useful to benchmark the current modelling and experimental assessment abilities.4 Besides, the portfolio of functional nanostructures is sensibly widened by the inclusion of metastable phases, enabling a list of applications in several key technological fields such as quantum technologies, sustainability, energy conversion and healthcare.1–4,6–9Metastable phases are often associated with a complex “ultrastructure” due to incomplete element segregation and chemical coordination, leading to glassy regions mixed with ordered crystalline arrangements of atoms.10–13 This “slippery” atomic conformation is difficult to observe even with advanced electron microscopy techniques.13,14 Nonetheless, it is intrinsically connected to the dynamic evolution of the composition, atomic arrangement, and properties of metastable nanoalloys upon response to physical or chemical external stimuli.5,10,15 Hence, in the realm of condensed matter, metastable nanoalloys can be seen as a special type of soft matter, namely of materials that are structurally altered by external physical stimuli of the magnitude of thermal fluctuations.16 The prediction of the equilibrium morphology in metallic NPs has been performed by various computational approaches.17–24 Such an example are Monte Carlo simulations based on semi-empirical potentials derived from the individual potentials calculated for each atom type by density functional theory. However, this approach is limited at a few thousand atoms and on a short timescale compared to most of the real world applications of nanoalloys.18 On the other hand, there is still a gap between modelling abilities and the complex chemistry experienced by nanoalloys under operation. In fact, metastable nanoalloys have a multicomponent nature and are exposed to compositional alterations in response to chemical stimuli.15,25 This is the case, for instance, for the selective reactivity of one element of the alloy with chemical species in the surrounding, typically involving redox reactions. Both thermodynamics and kinetics govern the transient dissolution of a metal during its oxidation, which is relevant for nanoalloy stability in biological environments as well as for the stability of electrocatalysts during operation in fuel cells.26,27
In this study, metastable Au–Fe alloy nanoparticles are produced by laser ablation in liquid (LAL) of a bulk Au–Fe target. LAL proved to be an efficient and versatile synthetic technique for the preparation of nanoalloys.28–32 LAL has the advantages of a self-standing set-up, the absence of chemical precursors and compatibility with the principles of green chemistry.33 Besides, in case of thermodynamically immiscible alloys, laser ablation synthesis is known to create nonequilibrium morphologies, frozen in metastable states representing relative thermodynamic minima.18,33 LAL allowed the synthesis of homogeneous bimetallic NPs in systems showing both miscibility only at high temperature such as Au–Fe and Au–Co systems,34–39 and complete immiscibility at any temperature and even in the liquid state such as the Ag–Fe and Ag–Co systems.11,12,40 For instance, for a transition metal content below 20 at%, the Fe and Co nanoalloys obtained by LAL are structurally stable in the ambient atmosphere and water and exhibited the magnetism expected for the doping of Au and Ag with magnetic centers.34,36,37 The physical and chemical properties of these alloys are interesting for a variety of applications.41 For instance, enhanced electrocatalytic oxygen evolution compared to single-element constituents was observed.35 In the case of photothermal nanostructures such as metal-coated silica core–shells, it was calculated that Au–Fe nanoalloys can convert near-infrared light into heat with an efficiency 90–100% higher than that of pure gold equivalents.42 This is due to the electronic structure of the nanoalloys, which was correlated to their plasmonic response by electron energy loss spectroscopy on individual NPs.43 The Au and Ag nanoalloys exhibit also the local electric field enhancement features suitable for the generation of bright surface-enhanced Raman scattering signals from molecules located at the junction between two NPs.36 Au–Fe NPs behaving as multimodal contrast agents have been developed,15,44 also with a 4-D behaviour leading to spontaneous size reduction and facilitated clearance of the NPs in living organisms.15
However, there is still much to explore about the dynamic evolution of Au-based alloy NPs in typical operating conditions for catalysis, plasmonics and nanomedicine. The comprehension of the time evolution of metastable nanoalloys is the key for enabling new applications and functions where the transformation of the nanomaterial during the process can be a high-value-added feature, which leads to a future generation of dynamic nanotechnologies.6 At the same time, the investigation of alloy formation during LAL promises to shed light on the main parameters required for the successful control of metastable phases.
Here, to obtain information on the formation process of the nanoalloy during the LAL synthesis, Au–Fe NPs have been characterized as obtained and after thermal aging in vacuum or in the ambient atmosphere. The morphological and structural changes are intimately related to the complex composition of the Au–Fe NPs produced by LAL. The dynamics of the Au–Fe nanoalloy structure emerges also in realistic operating conditions, such as by ageing in aqueous solutions of relevance for nanomedicine applications.
The results confirmed the intrinsic transformative nature of the Au–Fe nanoalloy and the importance of using a synthetic approach such as laser ablation in liquid that can freeze multielement nanosystems in non-equilibrium conditions. Overall, the results are relevant to the many fundamental scientific questions and challenges about nanoalloys, such as the evolution under operating conditions, the control and tailoring of nanoalloy formation processes in liquid phases and the determination of transformation and ageing processes of nanoalloys in realistic environments.
Materials and methods
Au–Fe NPs were synthesized by laser ablation of an Au/Fe 25/75 at% bulk target (99.9% pure, from Mateck GmBH) placed at the bottom of a cell containing 0.04 mg mL−1 PEG-SH (2000 Da, Laysan Bio) in ethanol (HPLC grade, from Sigma-Aldrich). The laser pulses (1064 nm, 6 ns, 50 Hz) were focused at a fluence of 18 J cm−2 by a lens with focal length f = 100 mm. The colloid was then stored at −20 °C overnight and collected by centrifugation at 1000 rcf for 15 min at 5 °C. Finally, the NPs were dried and resuspended in ethanol or in water.UV-visible absorption spectroscopy was performed with a JASCO V770 UV-vis-NIR spectrometer in 2 mm quartz cells. Bright field transmission electron microscopy (TEM) analysis was performed with an FEI Tecnai G2 12 operating at 100 kV and equipped with a TVIPS CCD camera. Room temperature Mössbauer spectra were collected using a conventional constant acceleration spectrometer with a 57Co source, nominal strength 1850 MBq. The hyperfine parameters isomer shift (δ), quadrupole splitting (Δ) or quadrupole shift if magnetic coupling is present (ε), and half linewidth at half maximum (Γ+) were expressed in mm s−1, with the internal magnetic field in Tesla (T), while the relative area (A) in % was obtained by means of standard least-squares minimization techniques. δ is quoted relative to α-Fe foil. Specific magnetization (M) as a function of applied magnetic field (H) at room temperature was obtained using a vibrating sample magnetometer (LakeShore 7404), operated with maximum applied fields of 1.5 T. Magnetic measurements were performed on the lyophilised NP samples previously dispersed in Milli-Q water. The colloid suspension was sealed into a heat-shrinkable tube to prevent sample evaporation and spills.
In situ X-ray diffraction (XRD) patterns were collected on a Bruker D8 Advance Plus diffractometer equipped with a non-ambient chamber (MTC-FURNACE) that allows heating and can be used in a vacuum (3 × 10−3 mbar), inert gas or air. The diffractometer was operated at 40 kV and 40 mA using a Cu Kα radiation source. To trace the structural changes, the samples were prepared by drop-casting on a silicon zero-background plate; this latter was then loaded into the Al2O3 holder. Each measurement step comprised four stages: heating/cooling to the set temperature at a rate of 10 °C min−1; holding for 10 min for temperature stabilization; re-alignment of the sample to compensate for thermal expansion; measuring the symmetric (θ–2θ) diffraction pattern in the 2θ range of 30–70°. The crystallographic phase identification was performed by a search/match procedure using Bruker DIFFRAC.EVA software and the COD database while the diffractograms were analyzed with TOPAS Academic V6 (Bruker AXS). Rietveld refinements were carried out by fitting the background with a Chebychev function, a broad Gaussian peak due to the amorphous phase, and the required phases (Au COD 9008463, Au1−xFex COD 9008463, α-Fe2O3 COD 2101169, Fe-BCC COD 4113936). The thermal expansion at various temperatures was considered in the Rietveld refinement of the lattice parameters by considering the lattice expansion values reported in the literature for Au, α-Fe2O3 and Fe-BCC. The shape of the reflections was modelled through the fundamental parameter approach incorporated in the program, separating the instrumental and the sample contributions. Fit indicators Rwp, Rexp, and GoF (Goodness of Fit) were used to assess the quality of the refined structural models.
Scanning electron microscopy (SEM) was performed with a Zeiss Sigma HD microscope, equipped with a Schottky FEG source, one detector for backscattered electrons and two detectors for secondary electrons (InLens and Everhart Thornley). The microscope is coupled to an EDX detector (from Oxford Instruments, x-act PentaFET Precision) for X-ray microanalysis, working in energy dispersive mode.
Magnetic resonance imaging (MRI) was performed with a Bruker system operating at 7 T (Bruker Biospin, Ettlingen, Germany). In phantom measurements, the samples were dispersed in aqueous solution by serial dilution starting from a solution with an Fe concentration of 2.83 mM. The transversal relaxivity (r2 value) was calculated from the slope of the best fit line of relaxation rates (1/T2) versus iron concentrations. The T2 map phantom images were acquired using a multislice multiecho sequence with the following parameters: TR = 2000 ms, TE = from 6.5 to 170.43 ms, FOV = 55 × 55 mm, matrix size = 128 × 128, slice thickness = 1 mm, number of echoes = 25.
Results and discussion
The Au–Fe NPs were produced by LAL of a bulk Au/Fe 25/75 (atomic%) target in ethanol with ns laser pulses (Fig. 1A). The NPs were analysed without any further purification, except solvent evaporation when required. In this way, all the synthesis products remained in the sample. The UV-vis spectrum of the colloid (Fig. 1B) exhibits an absorption edge in the near UV and no peaks in the visible range ascribable to the surface plasmon resonance of gold NPs. These features are typical of Au–Fe alloys with iron content exceeding 25 at%.42,43 On the other hand, the TEM analysis (Fig. 1B) shows the presence of NPs with size up to tens of nm, in addition to a multitude of ultrafine NPs with size of only a few nm. The NPs are not grouped together into aggregates on the TEM grid, despite solvent evaporation, which suggests the presence of non-crystalline material in between them, typically due to iron oxidation. Indeed, iron oxidation has been systematically observed during the LAL of targets containing iron, even in organic solvents.11,33,38,39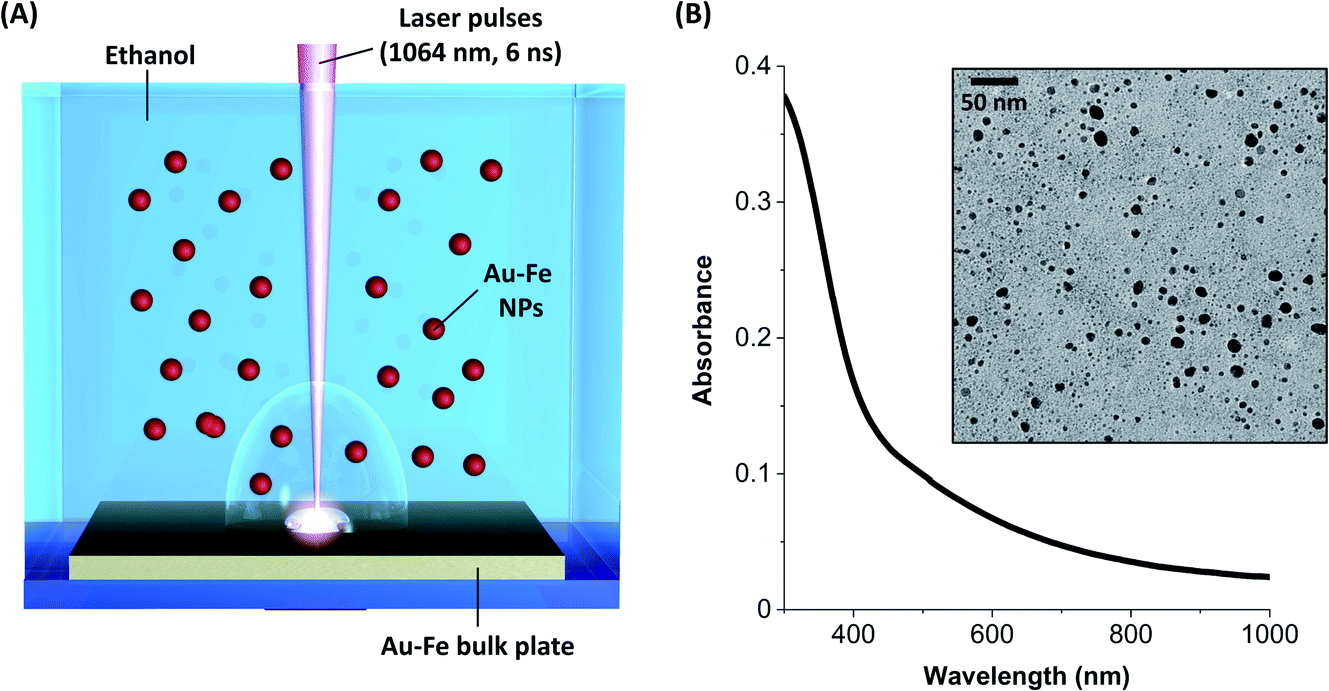 | ||
| Fig. 1 Sketch of LAL synthesis (A), UV-vis spectrum and TEM image of the resulting Au–Fe NPs (B). | ||
Given the complexity of the laser-generated product, Mössbauer spectroscopy is an ideal method to assess the crystalline and chemical state of iron in a sample of NPs. In this case, the room temperature Mössbauer spectrum (Fig. 2A) shows the presence of a broad, poorly resolved sextet, in the −5 mm s−1 to 5 mm s−1 range, together with a well defined, intense doublet, centered at ≈0.30 mm s−1. The relative intensity of lines in the magnetically split component suggests the presence of a relaxation process near TC. This signal could originate from an Au–Fe solid solution. Despite the magnetic component undergoing a magnetic ordering, and therefore the hyperfine parameters being unable to thoroughly describe the system, a fitting is proposed by using Lorentzian lines. The fit consists of three subcomponents: two broad sextets and a doublet. The two sextets (Γ+: 0.44 mm s−1 and Γ+: 0.25 mm s−1), showing an internal field of ≈28 T and ≈21 T respectively, could be representative of an Fe rich alloy. Usually Fe–Au alloys, with an Fe content lower than 20–25 at%, show a complex non-magnetically split absorption at room temperature,45 while a magnetic pattern results upon increasing the Fe load. For instance, Borg and coworkers46 estimated an ordering temperature close to RT when the Fe content is higher than 20 at%, while other authors47,48 reported the existence of magnetically ordered patterns at higher Fe concentration. It is worth observing that the two sextets show positive isomer shifts (≈0.2 mm s−1 for both) suggesting a decrease of the charge density at the nucleus due to the Au neighbors.49 Concerning the intense doublet, the hyperfine parameters are typical of Fe(III) species (δ: 0.32 mm s−1, Δ: 0.84 mm s−1, A: 40%). The presence of Fe(III) species can be reasonably attributed to non-crystalline paramagnetic ferric oxide, as already inferred from TEM analysis, and ferric centers located at the alloy surface as a result of a surface oxidation. The presence of the Au–Fe alloy in a paramagnetic state, that is above the Curie temperature, can be excluded: the shape and the parameters of the doublet do not fit with the literature.45,50
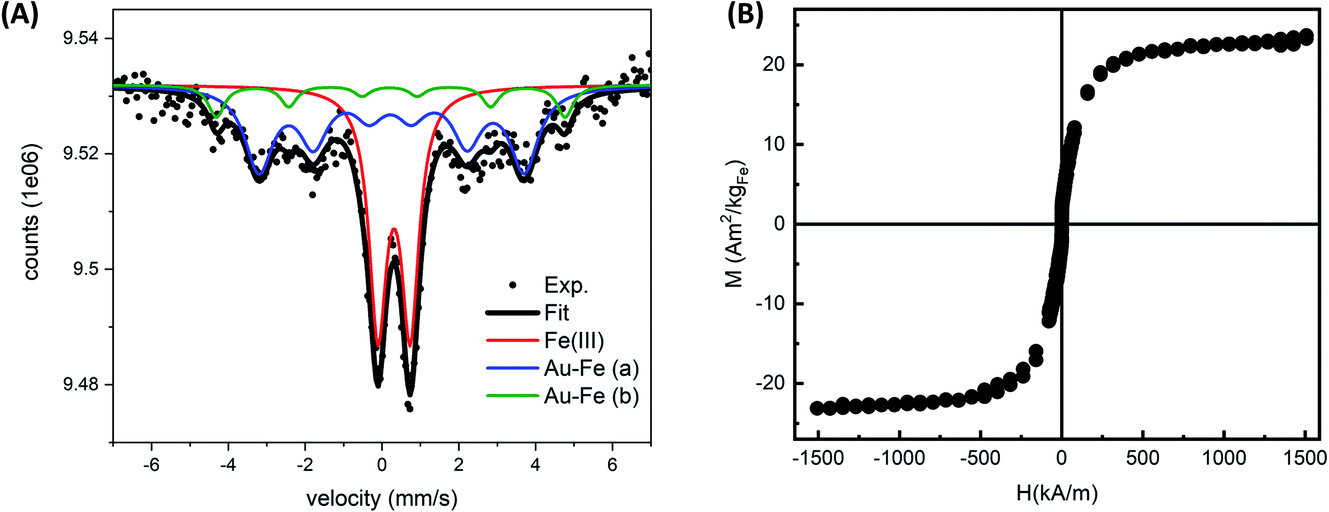 | ||
| Fig. 2 (A) Room temperature Mössbauer spectrum of Au–Fe NP powder (black circles) and fit (black line). Three components were used for the fit: a doublet typical of Fe(III) species (red line) and two broad sextets representative of Fe–Au alloys (blue and green lines). (B) Hysteresis cycles at room temperature obtained by VSM measurements on Au–Fe NPs, after subtraction of the diamagnetic contribution. | ||
To obtain further evidence of the formation of Au–Fe alloy domains near to magnetic order, room temperature magnetometry was performed on the sample resuspended in water and lyophilized (Fig. 2B). The hysteresis cycles evidenced a superparamagnetic behavior of the alloy NPs, indicating that it is above the percolation threshold for ferromagnetism in Au–Fe crystals.34,51 The magnetization at room temperature (M) as a function of the applied magnetic field (H) was fitted using a Langevin function weighted with a log-normal distribution of magnetic moments, resulting in a saturation value of 22.30 ± 0.13 Am2 per kg Fe. This value is sensibly lower than that in pure Fe.34,51 These findings are compatible with a partial dissolution and oxidation of the Au–Fe NPs redispersed in water, but are also expected from the surface effects in NPs, the disorder introduced by alloying with Au and the fraction of non-crystalline ferric oxide identified by Mössbauer spectroscopy.34,52
LAL allows freezing the Au–Fe NPs in a metastable state but the resulting sample contains structural heterogeneities such as the ultrafine NPs and the Fe(III) component, evidenced respectively by TEM and Mössbauer analysis. Furthermore, the mechanism of ejection of the material from the target involves at least two simultaneous processes, which are the expulsion of molten drops and vapours from the target, which is followed by cooling and condensation in the final product.53,54 An experiment of thermal aging can provide further information on the evolution of the ablated material and can help understanding whether it was subjected to rapid cooling by thermalization with the surrounding liquid or interaction with oxygen dissolved in solution. Hence, the structural evolution of the Au–Fe sample at temperatures from 30 to 700 °C in an inert (vacuum) and oxidizing (air) environment was monitored by powder XRD and Rietveld refinement (Fig. 3).
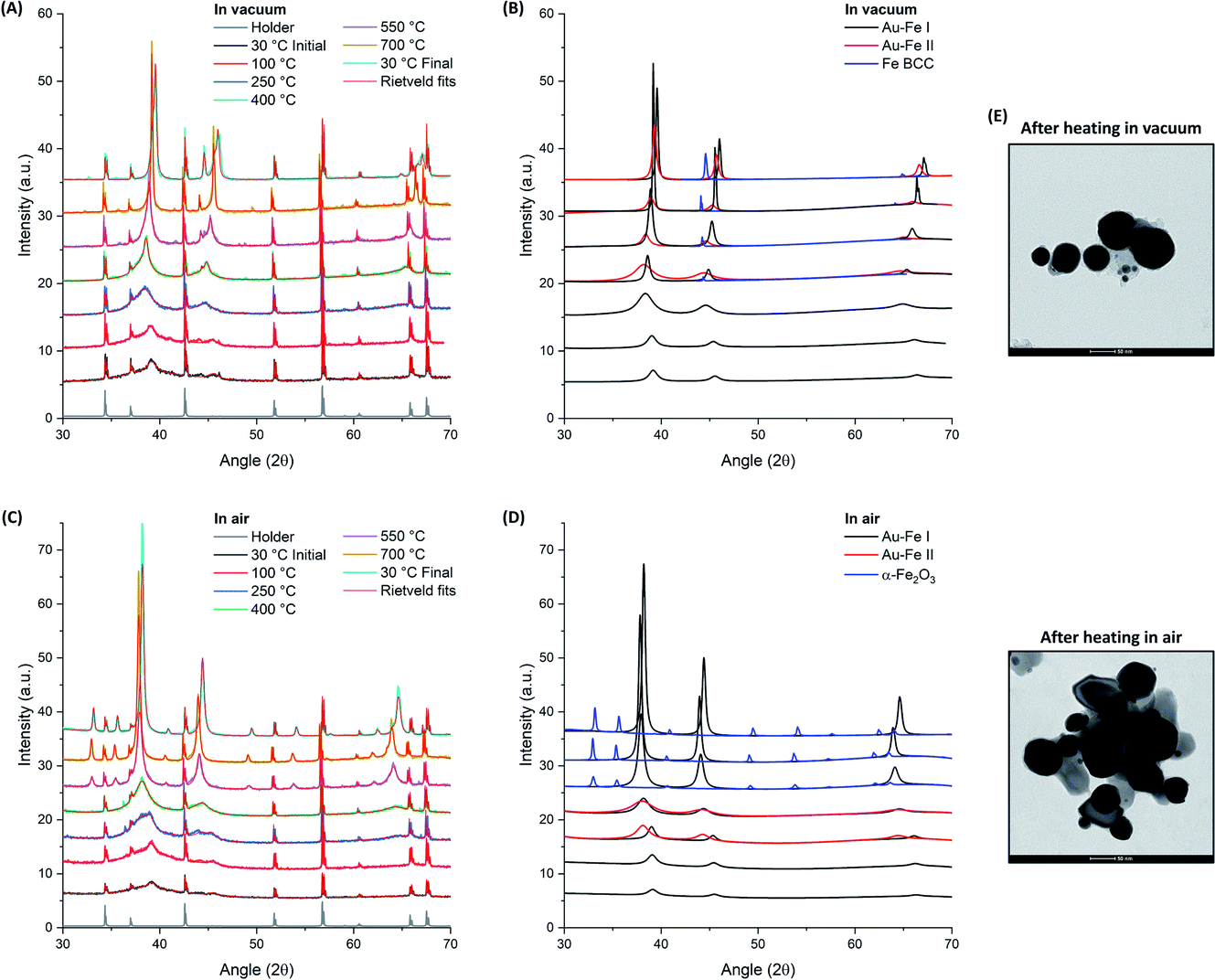 | ||
| Fig. 3 XRD patterns collected on the Au–Fe NP sample heated in vacuum (A and B) or in air (C and D) up to 700 °C. Rietveld refinements are reported as red lines for all patterns and single components are shown in (B) and (D) for the heating in, respectively, vacuum and air. Intensities were not normalized but the spectra were shifted for clarity. (E) TEM images of Au–Fe NPs after the annealing experiment in the two environments. | ||
The Rietveld refinement of the XRD data collected in vacuum at 30 °C (Fig. 3A and B) fully confirms the characteristics previously indicated by UV-vis, TEM and Mössbauer: the reflections correspond to those of the Au FCC cell but with a cell parameter reduced to 3.983 Å (Au–Fe I phase) instead of the 4.079 Å of pure Au (Fig. 4A). This contraction is due to the presence of Fe atoms as substitutional impurities in the Au lattice.34,55 Furthermore, there is an evident background of non-crystalline material, which can be estimated to be 72 wt% (Fig. 4B). The low crystallinity is accompanied by a rather small size of the crystalline domains, estimated at 5.7 ± 0.6 nm from XRD spectrum refinement (Fig. 4C). This is somehow expected considering the composition of the ablated target (Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe 25:75 atomic%) and the immiscibility of the two elements according to their binary phase diagram.56 By increasing the temperature, a clear evolution of the crystalline and amorphous components of the sample is observed, indicative of the annealing process. The degree of crystallinity reaches 100% at 700 °C and remains 100 wt% even after cooling to 30 °C. Sample annealing is accompanied by an increase in the size of the crystalline domains, which reaches 67 ± 9 nm at 700 °C and then stabilizes at 36 ± 6 nm at 30 °C. In the XRD pattern, these features correspond to the remarkable increase of the intensity of FCC peaks (note the intensities in Fig. 3 were not normalized and can be quantitatively compared). The thermal activation of atomic diffusion and the consequent alloying with iron agree with previous experimental observations and theoretical simulations and are explained by the relaxation of structural stress of the disordered phases into metastable states closer to equilibrium, when sufficient thermal energy is applied.17 The crystallization of the amorphous phase begins at 250 °C, when the degree of crystallinity increases from 30 to 69 wt%. This is an indication of a relatively good thermal stability of the material. Notably, the average size of the crystalline domains drops from 5.6 ± 0.6 nm to 3.1 ± 0.3 nm when the temperature increases from 100 °C to 250 °C, indicating the crystallization of the amorphous phase, and then rises to 12.4 ± 0.9 nm at 400 °C. Other important structural changes are observed at 400 °C, because the lattice parameter of the Au–Fe I phase starts to decrease and the Rietveld refinement indicates the appearance of a second phase of the Au–Fe alloy (Au–Fe II) with a higher cell parameter, corresponding to a lower content of Fe (6 at%). These findings and the concomitant detection of Fe-BCC indicate that above 400 °C atomic diffusion between phases plays a dominant role as expected based on the diffusivity calculation of Fe and Au into Au-fcc and Fe-bcc.17 Since the size of the crystalline domains of the Au–Fe II phase is only 2.7 ± 0.6 nm, this component is attributed to a segregation process whereby heterostructures of gold-rich and metallic Fe-rich phases form.
:Fe 25:75 atomic%) and the immiscibility of the two elements according to their binary phase diagram.56 By increasing the temperature, a clear evolution of the crystalline and amorphous components of the sample is observed, indicative of the annealing process. The degree of crystallinity reaches 100% at 700 °C and remains 100 wt% even after cooling to 30 °C. Sample annealing is accompanied by an increase in the size of the crystalline domains, which reaches 67 ± 9 nm at 700 °C and then stabilizes at 36 ± 6 nm at 30 °C. In the XRD pattern, these features correspond to the remarkable increase of the intensity of FCC peaks (note the intensities in Fig. 3 were not normalized and can be quantitatively compared). The thermal activation of atomic diffusion and the consequent alloying with iron agree with previous experimental observations and theoretical simulations and are explained by the relaxation of structural stress of the disordered phases into metastable states closer to equilibrium, when sufficient thermal energy is applied.17 The crystallization of the amorphous phase begins at 250 °C, when the degree of crystallinity increases from 30 to 69 wt%. This is an indication of a relatively good thermal stability of the material. Notably, the average size of the crystalline domains drops from 5.6 ± 0.6 nm to 3.1 ± 0.3 nm when the temperature increases from 100 °C to 250 °C, indicating the crystallization of the amorphous phase, and then rises to 12.4 ± 0.9 nm at 400 °C. Other important structural changes are observed at 400 °C, because the lattice parameter of the Au–Fe I phase starts to decrease and the Rietveld refinement indicates the appearance of a second phase of the Au–Fe alloy (Au–Fe II) with a higher cell parameter, corresponding to a lower content of Fe (6 at%). These findings and the concomitant detection of Fe-BCC indicate that above 400 °C atomic diffusion between phases plays a dominant role as expected based on the diffusivity calculation of Fe and Au into Au-fcc and Fe-bcc.17 Since the size of the crystalline domains of the Au–Fe II phase is only 2.7 ± 0.6 nm, this component is attributed to a segregation process whereby heterostructures of gold-rich and metallic Fe-rich phases form.
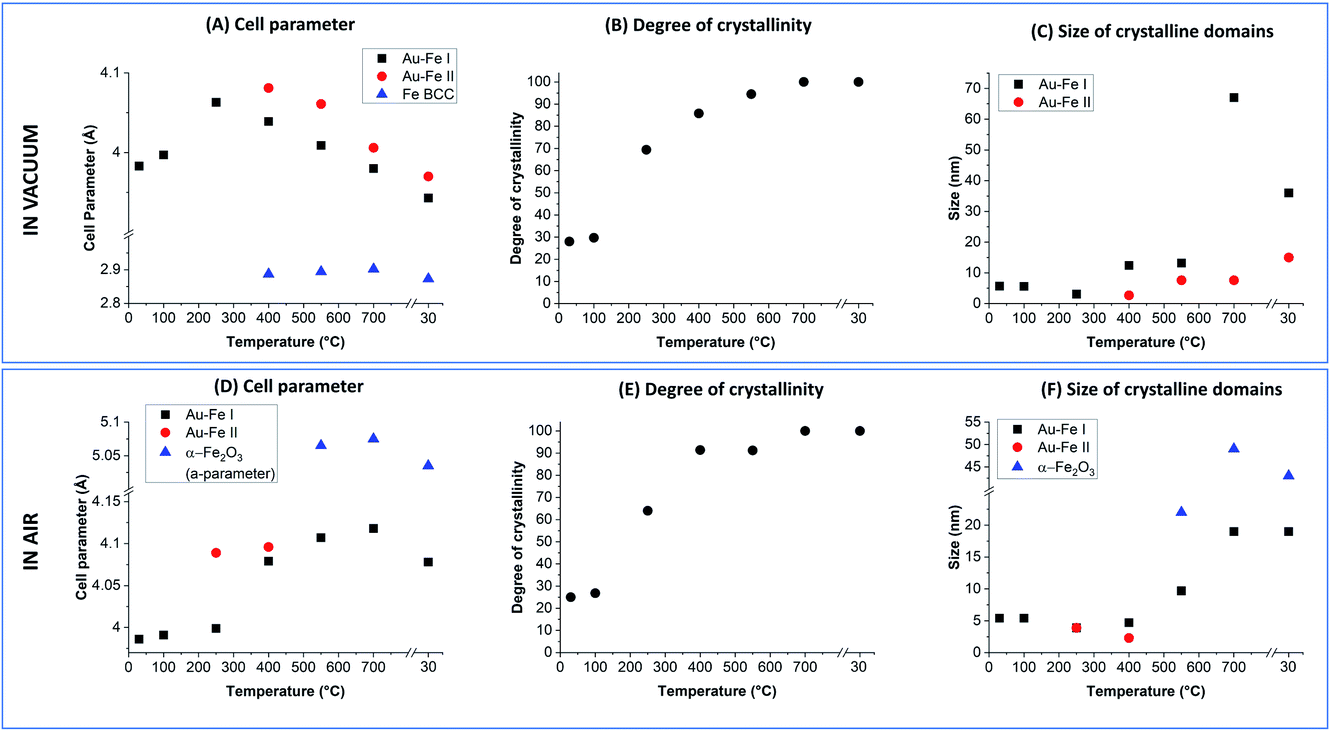 | ||
| Fig. 4 Cell parameters (A and D), degree of crystallinity (B and E) and average size of crystalline domains (C and F) estimated from the Rietveld refinement of the XRD patterns in vacuum and in air. For α-Fe2O3 only the a cell parameter of its trigonal lattice is reported. | ||
The Au–Fe I, Au–Fe II and Fe BCC components remain in the XRD patterns up to 700 °C, even if the cell parameter of the Au–Fe II phase decreases, converging towards that of the Au–Fe I phase. However, a lower size of the crystalline domains (7.6 ± 1 nm) is measured, with the estimated Fe content in the alloy being 43 at% for Au–Fe I and 35 at% for Au–Fe II. This is further evidence that the high temperature entropically and diffusively favours the formation of the Au–Fe alloy at the interface with the Fe BCC crystals. At 30 °C, the three phases maintain the characteristics assumed at 700 °C, apart from the reduction of the size of the crystalline domains of Au–Fe I from 67 ± 9 nm to 36 ± 6 nm, which can be ascribed to the increase of the mixing enthalpy at lower temperature and the consequent tendency for element segregation. It should be noted that the lattice parameter measured for BCC iron (2.873 Å) is slightly larger than the literature value of 2.856 Å,17 suggesting that Au atoms have been partially included in the Fe BCC lattice. TEM images of the Au–Fe NPs after the heating experiment (Fig. 3E) show how the morphology of the sample was sensibly modified by the thermal annealing, with the disappearance of ultrafine NPs and their replacement with NPs of several tens of nm.
SEM analysis on the sample annealed in vacuum (Fig. 5) permitted better identification of the localization of alloy and iron phases indicated by the refinement of the XRD patterns. The annealed sample is composed of a population of NPs with size of tens of nm, appearing homogeneous in the electron micrograph, mixed with NPs of hundreds of nm and inhomogeneous in composition. EDX analysis on the electron bright and opaque regions suggests the segregation of Fe and Au in the submicrometric particles. The morphology of submicrometric particles resembles that of Fe cubes capped with Au prisms, which was observed by thermal annealing in vacuum and modelled by the Barcikowski,18 Langlois57 and Ferrando19 groups. The gold islands grow at the (001) facets of Fe because of the lower interface energy.18,19,57 The precipitation of the Fe BCC from alloy crystals has been observed prevalently for iron content above 50 at%.17,18 Instead, it is not reported for equimolar or Au-rich Au–Fe NPs, where thermal aging resulted in alloying.18 This suggests that NPs with size of hundreds of nm contain significantly more iron than NPs of tens of nm.
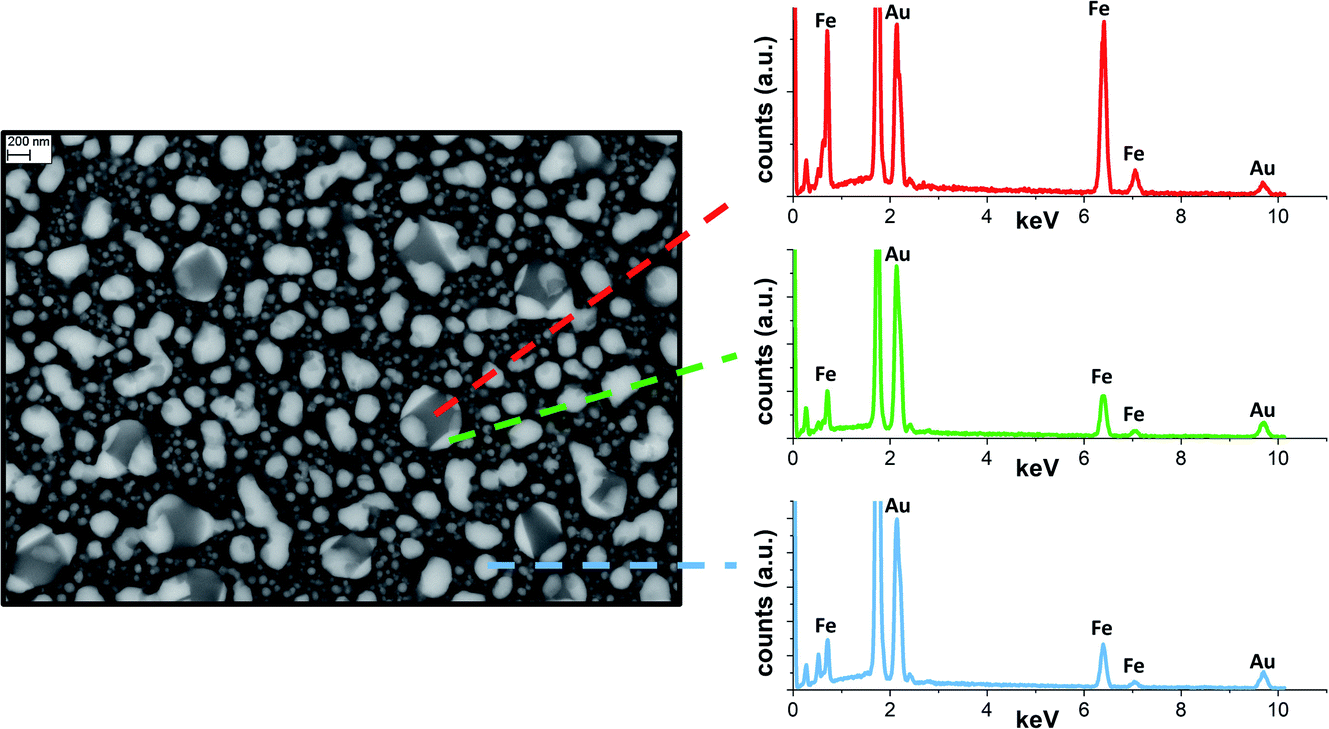 | ||
| Fig. 5 SEM backscattered image of Au–Fe NPs after annealing at 700 °C in vacuum and EDX spectra collected at various points. | ||
For the thermal aging in air, a sharp increase of the degree of crystallinity is again observed when the temperature goes from 100 °C (27 wt%) to 250 °C (64 wt%), reaching 100% crystallinity at 700 °C as well as after cooling back to 30 °C. Besides, also in this case the average size of crystalline domains at 250 °C (3.9 ± 0.6 nm) is smaller than that at 100 °C (5.6 ± 0.6 nm), indicating the beginning of crystallization in the amorphous phase. However, at 400 °C the size of the crystals remains relatively small (4.7 ± 0.6 nm), suggesting that oxidation is hampering the grain coalescence which was possible in vacuum at the same temperature. Importantly, at 250 and 400 °C, a second Au–Fe phase (Au–Fe II) is required for the Rietveld refinement of the XRD patterns, with a cell parameter close to that of pure Au. Also, the cell parameter for the Au–Fe I phase is increased to 4.079 Å, from the initial value of 3.986 Å at 30 °C. All these data point to the oxidation of Fe and its extraction from the Au–Fe alloy, which is definitively confirmed by the XRD spectrum at 500 °C, indicating the presence of α-Fe2O3. Iron oxidation certainly begins at the interface between the Au–Fe NPs and the external environment. Therefore, complete oxidation will first be achieved in small NPs, likely identified as the Au–Fe II phase, as suggested by their large lattice parameter and small size of the crystalline domains estimated by Rietveld analysis.
At 700 °C, as well as after cooling at 30 °C, only pure Au and α-Fe2O3 are detected according to the Rietveld refinement, indicating the complete oxidation of Fe in the sample. As in vacuum, the thermal annealing in air is associated with a remarkable increase of the intensity of the diffraction peaks, as shown in Fig. 3C and D. TEM images of the Au–Fe NPs after the heating experiment in air (Fig. 3E) match with a sample composed of a mixture of Au NPs (more electron-dense) and of Fe oxide NPs (less electron-dense), both with size of tens of nm.
Overall, the picture emerging from the above experiments suggests that laser ablation of the Au–Fe target would lead to the formation of Au–Fe alloy NPs because of the high temperature and rapid cooling of the ablated material, but the presence of oxygen in the liquid solution induces the oxidation of part of the Fe (likely the atomic component) with the formation of a partially oxidized and substantially amorphous material in which the Au–Fe alloy NPs are embedded. In a previous study, this amorphous component was removed by washing in a centrifuge to isolate the crystalline NPs.15 Given that the drops of molten material ejected from the target tend to coalesce, it is likely that a certain degree of oxidation is also present at the interfaces between these grains in the final NPs, which in fact have a defective structure, as previously reported by high resolution TEM.15 This type of ultrastructure can further enhance the reactivity of the laser generated Au–Fe NPs, contributing to their transformative nature, which is known to play an important role in operating conditions. For instance, Au–Fe NPs have been recently considered for nanomedicine applications due to their prominent properties for multimodal imaging, biocompatibility and size reduction in biological fluids.15 Intrigued by this set of features, we exploited the magnetism at room temperature of the Au–Fe NPs to track the transformative behaviour in biological fluids by measuring their performance as contrast agents for magnetic resonance imaging. The sample was kept for 60 days at physiological temperature (37 °C) in foetal calf serum (FCS) containing citrate buffer to reproduce the acidic lysosomal environment (pH of 4.5) which is typical of in vivo operating conditions.58 The MRI contrast ability (Fig. 6A) was assessed by measuring the transverse relaxation time of protons in water in a series of phantoms with variable concentration (Fig. 6B), with a preclinical 7.0 T MRI scanner. The transversal relaxivity (r2) measured at different timepoints shows a clear reduction over time which should be ascribed to the degradation of the alloy in the acidic FCS environment. This is a desirable property for a nanomedicine agent which is supposed to be cleared from the organism after use.15,25 The effect is also qualitatively appreciable from the contrast reduction of the T2-weighted cross-sectional images of phantoms analysed after 30 days (Fig. 6B). The structural modification occurring in the alloy dispersed in aqueous solution at mild acidic conditions (pH 4.5) and 37 °C can also be well monitored with room temperature Mössbauer spectroscopy (Fig. 6C). Despite the high number of counts (≈6.5 × 106) in the spectrum, only a narrow central absorption is well identifiable beyond a weak, and undefined, broad absorption in the range between −4 mm s−1 and 4 mm s−1. No clear evidence of magnetically ordered/relaxing components was detected, suggesting that the Au–Fe alloy lost iron, in agreement with the decrease of r2 measured with MRI. A fit was obtained by means of a single component, a doublet whose parameters can be ascribed to Fe(III) nuclei in a distorted octahedral geometry (δ: 0.47 mm s−1 and Δ: 0.52 mm s−1). According to the literature, these parameters can be assigned to Fe(III) complexes with the citrate ions of the acidic aqueous solution.59
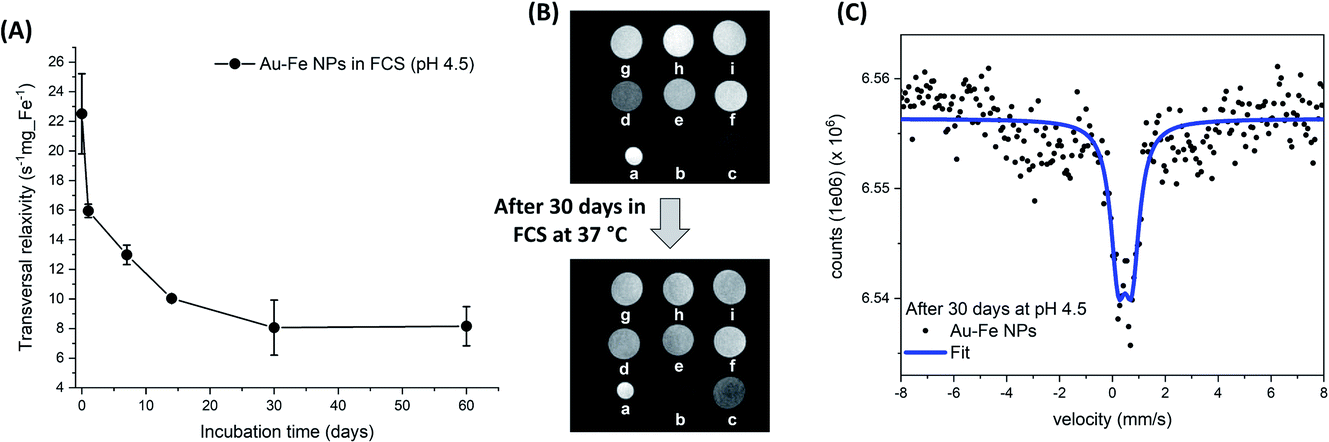 | ||
| Fig. 6 (A) Transversal relaxivity for Au–Fe NPs incubated at 37 °C in FCS at pH 4.5 for up to 60 days. (B) MRI images of phantoms' cross-sections for Au–Fe NP samples at variable dilution (a: 0 mM Fe; b: 2.833; c: 1.417; d: 0.708; e: 0.354; f: 0.177; g: 0.088; h: 0.044; i: 0.022) and at two time points (0 and 30 days). (C) Room temperature Mössbauer spectrum of Au–Fe NPs aged for 30 days at 37 °C in citrate buffer – pH 4.5 (black circles) and fit (blue line). | ||
Conclusions
We reported on the study of laser-generated metastable Au–Fe compounds with the aim of extracting precious information on NP formation and atomic arrangement. This is possible thanks to the modification of morphology and crystalline phase in the metastable nanoalloys and to the different reactivity of elements like Au and Fe with oxygen. To this end, the Au–Fe NPs were exposed to physical (thermal) and chemical (oxidising environment) stimuli. The initial NPs have high crystalline disorder and present a remarkable fraction of oxidised iron. Thermal annealing in vacuum or in air shows that the NPs evolve towards larger nanoparticles of, respectively, the Au–Fe alloy or Au–iron oxide heterostructures. In vacuum above 400 °C, the submicrometric Au–Fe NPs undergo element segregation into the Au–Fe alloy and BCC Fe. Although the exact picture of how nanomaterials form by LAL is still missing some key steps, these results indicate that iron oxidation is crucial in determining the final composition and phase of products. The disordered structure of the Au–Fe NPs plays a role in their stability in biological fluids, facilitating NP dissolution over time as desirable for a nanomedicine agent. Thus, the level of iron oxidation during the laser synthesis emerged as a key parameter for tuning properties such as magnetism, chemical stability, and size distribution of the Au–Fe NPs.The effects of Au–Fe NP ultrastructure on their transformative behaviour represent an intriguing way to realize transformable nanoparticles with dynamic properties. Improving our knowledge of the laser-assisted processes is crucial to identify new and appropriate strategies to obtain full control of the laser-generated products. We expect that, to answer these questions, the development of modern computational techniques up to the long timescales of realistic experiments, and their integration with and the accurate design of experimental investigations, will be mandatory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Alice Busato and Vito Coviello are acknowledged for support with experiments and data collection. The research leading to these results has received funding from the University of Padova P-DiSC grant DYNAMO and from AIRC under MFAG 2021 – ID. 25681 project – P.I. Amendola Vincenzo.References
- M. Aykol, S. S. Dwaraknath, W. Sun and K. A. Persson, Sci. Adv., 2018, 4, eaaq0148 CrossRef PubMed.
- W. Sun, S. T. Dacek, S. P. Ong, G. Hautier, A. Jain, W. D. Richards, A. C. Gamst, K. A. Persson and G. Ceder, Sci. Adv., 2016, 2, e1600225 CrossRef PubMed.
- C. B. Wahl, M. Aykol, J. H. Swisher, J. H. Montoya, S. K. Suram and C. A. Mirkin, Sci. Adv., 2021, 7, eabj5505 CrossRef CAS PubMed.
- J. Hong, J. H. Bae, H. Jo, H. Y. Park, S. Lee, S. J. Hong, H. Chun, M. K. Cho, J. Kim, J. Kim, Y. Son, H. Jin, J. Y. Suh, S. C. Kim, H. K. Roh, K. H. Lee, H. S. Kim, K. Y. Chung, C. W. Yoon, K. Lee, S. H. Kim, J. P. Ahn, H. Baik, G. H. Kim, B. Han, S. Jin, T. Hyeon, J. Park, C. Y. Son, Y. Yang, Y. S. Lee, S. J. Yoo and D. W. Chun, Nature, 2022, 603(7902), 631–636 CrossRef CAS PubMed.
- A. Tymoczko, M. Kamp, C. Rehbock, L. Kienle, E. Cattaruzza, S. Barcikowski and V. Amendola, Nanoscale Horiz., 2019, 4, 1326–1332 RSC.
- V. Amendola, ChemPhysChem, 2021, 22, 622–624 CrossRef CAS PubMed.
- Y. Jin, F. Chen, T. Jin, L. Guo and J. Wang, J. Mater. Chem. A, 2020, 8, 25780–25790 RSC.
- P. S. Cappellari, G. J. Soldano and M. M. Mariscal, J. Phys. Chem. C, 2022, 126, 1742–1750 CrossRef CAS.
- X. Huang, O. Akdim, M. Douthwaite, K. Wang, L. Zhao, R. J. Lewis, S. Pattisson, I. T. Daniel, P. J. Miedziak, G. Shaw, D. J. Morgan, S. M. Althahban, T. E. Davies, Q. He, F. Wang, J. Fu, D. Bethell, S. McIntosh, C. J. Kiely and G. J. Hutchings, Nature, 2022, 603(7900), 271–275 CrossRef CAS.
- Z. Zhang, S. Zhang, Z. Yao, X. Tao, X. Cao, P. Zhang, P. Kuang, Y. Fu, N. H. van Dijk and S. van der Zwaag, Nucl. Instrum. Methods Phys. Res., Sect. B, 2021, 505, 50–57 CrossRef CAS.
- V. Amendola, S. Scaramuzza, S. Agnoli, G. Granozzi, M. Meneghetti, G. Campo, V. Bonanni, F. Pineider, C. Sangregorio, P. Ghigna, S. Fiameni, L. Nodari, S. Polizzi, P. Riello, S. Fiameni and L. Nodari, Nano Res., 2015, 8, 4007–4023 CrossRef CAS.
- A. Guadagnini, S. Agnoli, D. Badocco, P. Pastore, D. Coral, M. B. Fernàndez van Raap, D. Forrer and V. Amendola, J. Colloid Interface Sci., 2021, 585, 267–275 CrossRef CAS PubMed.
- D. Wu, K. Kusada, Y. Nanba, M. Koyama, T. Yamamoto, T. Toriyama, S. Matsumura, O. Seo, I. Gueye, J. Kim, L. S. Rosantha Kumara, O. Sakata, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2022, 144, 3365–3369 CrossRef CAS PubMed.
- A. Ponce, J. A. Aguilar, J. Tate and M. J. Yacamán, Nanoscale Adv., 2021, 3, 311–325 RSC.
- V. Torresan, D. Forrer, A. Guadagnini, D. Badocco, P. Pastore, M. Casarin, A. Selloni, D. Coral, M. Ceolin, M. B. Fernández van Raap, A. Busato, P. Marzola, A. E. Spinelli and V. Amendola, ACS Nano, 2020, 14, 12840–12853 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, 2011 Search PubMed.
- J. Johny, O. Prymak, M. Kamp, F. Calvo, S. H. Kim, A. Tymoczko, A. El-Zoka, C. Rehbock, U. Schürmann, B. Gault, L. Kienle and S. Barcikowski, Nano Res., 2022, 15, 581–592 CrossRef CAS.
- M. Kamp, A. Tymoczko, U. Schürmann, J. Jakobi, C. Rehbock, K. Rätzke, S. Barcikowski and L. Kienle, Cryst. Growth Des., 2018, 18, 5434–5440 CrossRef CAS.
- J. Vernieres, S. Steinhauer, J. Zhao, P. Grammatikopoulos, R. Ferrando, K. Nordlund, F. Djurabekova and M. Sowwan, Adv. Sci., 2019, 6, 1900447 CrossRef.
- J. Pirart, A. Front, D. Rapetti, C. Andreazza-Vignolle, P. Andreazza, C. Mottet and R. Ferrando, Nat. Commun., 2019, 10(1), 1–7 CrossRef CAS PubMed.
- M. Vanzan, R. M. Jones, S. Corni, R. D'Agosta and F. Baletto, ChemPhysChem, 2022, 23, e202200035 CrossRef CAS PubMed.
- P. Ferrari, H. T. Pham, J. Vanbuel, M. T. Nguyen, A. Fielicke and E. Janssens, Chem. Commun., 2021, 57, 9518–9521 RSC.
- J. W. Fagan, K. L. D. M. Weerawardene, A. Cirri, C. M. Aikens and C. J. Johnson, J. Chem. Phys., 2021, 155, 014301 CrossRef CAS.
- C. Zeni, K. Rossi, T. Pavloudis, J. Kioseoglou, S. de Gironcoli, R. E. Palmer and F. Baletto, Nat. Commun., 2021, 12(1), 1–9 CrossRef PubMed.
- V. Amendola, A. Guadagnini, S. Agnoli, D. Badocco, P. Pastore, G. Fracasso, M. Gerosa, F. Vurro, A. Busato and P. Marzola, J. Colloid Interface Sci., 2021, 596, 332–341 CrossRef CAS.
- F. D. Speck, A. Zagalskaya, V. Alexandrov and S. Cherevko, Angew. Chem., Int. Ed., 2021, 60, 13343–13349 CrossRef CAS.
- N. J. Divins, D. Kordus, J. Timoshenko, I. Sinev, I. Zegkinoglou, A. Bergmann, S. W. Chee, S. Widrinna, O. Karslıoğlu, H. Mistry, M. Lopez Luna, J. Q. Zhong, A. S. Hoffman, A. Boubnov, J. A. Boscoboinik, M. Heggen, R. E. Dunin-Borkowski, S. R. Bare and B. R. Cuenya, Nat. Commun., 2021, 12(1), 1–10 CrossRef.
- Z. Lin, J. Yue, L. Liang, B. Tang, B. Liu, L. Ren, Y. Li and L. Jiang, Appl. Surf. Sci., 2020, 504, 144461 CrossRef CAS.
- K. A. Kane, A. C. Reber, S. N. Khanna and M. F. Bertino, Prog. Nat. Sci.: Mater. Int., 2018, 28, 456–463 CrossRef CAS.
- S. Hu, G. Goenaga, C. Melton, T. A. Zawodzinski and D. Mukherjee, Appl. Catal., B, 2016, 182, 286–296 CrossRef CAS.
- M. S. Satya Bharati, B. Chandu and S. V. Rao, RSC Adv., 2019, 9, 1517–1525 RSC.
- V. Torresan, A. Guadagnini, D. Badocco, P. Pastore, G. A. Muñoz Medina, M. B. Fernàndez van Raap, I. Postuma, S. Bortolussi, M. Bekić, M. Čolić, M. Gerosa, A. Busato, P. Marzola and V. Amendola, Adv. Healthcare Mater., 2020, 2001632 Search PubMed.
- V. Amendola, D. Amans, Y. Ishikawa, N. Koshizaki, S. Scirè, G. Compagnini, S. Reichenberger and S. Barcikowski, Chem. – Eur. J., 2020, 26, 9206–9242 CrossRef CAS PubMed.
- V. Amendola, M. Meneghetti, O. M. Bakr, P. Riello, S. Polizzi, S. Fiameni, H. Dalaver, P. Arosio, T. Orlando, C. de Julian Fernandez, F. Pineider, C. Sangregorio and A. Lascialfari, Nanoscale, 2013, 5, 5611–5619 RSC.
- I. Vassalini, L. Borgese, M. Mariz, S. Polizzi, G. Aquilanti, P. Ghigna, A. Sartorel, V. Amendola and I. Alessandri, Angew. Chem., Int. Ed., 2017, 56, 6589–6593 CrossRef CAS.
- V. Amendola, S. Scaramuzza, S. Agnoli, S. Polizzi and M. Meneghetti, Nanoscale, 2014, 6, 1423–1433 RSC.
- A. Guadagnini, S. Agnoli, D. Badocco, P. Pastore, R. Pilot, R. Ravelle-Chapuis, M. B. F. Raap and V. Amendola, ChemPhysChem, 2021, 22, 657–664 CrossRef CAS PubMed.
- V. Amendola, S. Scaramuzza, F. Carraro and E. Cattaruzza, J. Colloid Interface Sci., 2017, 489, 18–27 CrossRef CAS.
- S. Scaramuzza, S. Agnoli and V. Amendola, Phys. Chem. Chem. Phys., 2015, 17, 28076–28087 RSC.
- S. Scaramuzza, D. Badocco, P. Pastore, D. F. Coral, M. B. Fernández van Raap and V. Amendola, ChemPhysChem, 2017, 18, 1026–1034 CrossRef CAS.
- I. Ali, Y. Pan, Y. Lin, Y. Jamil, J. Hu, Z. Gan, J. Chen and Z. Shen, Appl. Phys. A: Mater. Sci. Process., 2021, 127, 1–10 CrossRef.
- V. Amendola, R. Sajia, O. M. M. Maragò, A. Iatì, R. Saija, O. M. M. Maragò, M. A. Iatì, R. Sajia, O. M. M. Maragò and A. Iatì, Nanoscale, 2015, 7, 8782–8792 RSC.
- D. T. L. Alexander, D. Forrer, E. Rossi, E. Lidorikis, S. Agnoli, G. D. Bernasconi, J. Butet, O. J. F. Martin and V. Amendola, Nano Lett., 2019, 19, 5754–5761 CrossRef CAS PubMed.
- V. Amendola, S. Scaramuzza, L. Litti, M. Meneghetti, G. Zuccolotto, A. Rosato, E. Nicolato, P. Marzola, G. Fracasso, C. Anselmi, M. Pinto and M. Colombatti, Small, 2014, 10, 2476–2486 CrossRef CAS.
- G. L. Whittle and S. J. Campbell, J. Phys. F: Met. Phys., 1985, 15, 693 CrossRef CAS.
- R. J. Borg and C. E. Violet, J. Phys. Chem. Solids, 1987, 48, 1239–1244 CrossRef CAS.
- G. Albanese, A. Deriu, J. Moya, E. Angeli, D. Bisero, A. Da Re, F. Ronconi, F. Spizzo, P. Vavassori, M. Baricco and E. Bosco, J. Magn. Magn. Mater., 2004, 272–276, 1545–1546 CrossRef CAS.
- A. K. Mishra, C. Bansal, M. Ghafari, R. Kruk and H. Hahn, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 155452 CrossRef.
- A. Błachowski, K. Ruebenbauer, J. Przewoźnik and J. Zukrowski, J. Alloys Compd., 2008, 458, 96–103 CrossRef.
- B. Window, Phys. Rev. B: Solid State, 1972, 6, 2013 CrossRef CAS.
- B. H. Verbeek and J. A. Mydosh, J. Phys. F: Met. Phys., 1978, 8, L109 CrossRef CAS.
- M. V. Efremova, M. Spasova, M. Heidelmann, I. S. Grebennikov, Z. A. Li, A. S. Garanina, I. O. Tcareva, A. G. Savchenko, M. Farle, N. L. Klyachko, A. G. Majouga and U. Wiedwald, Nanoscale, 2021, 13, 10402–10413 RSC.
- N. A. Inogamov, V. V. Zhakhovsky and V. A. Khokhlov, JETP Lett., 2022, 115, 16–22 CrossRef CAS.
- C.-Y. Shih, M. V. Shugaev, C. Wu and L. V. Zhigilei, Phys. Chem. Chem. Phys., 2020, 22, 7077–7099 RSC.
- S. Pramanik, S. Chattopadhyay, S. Bysakh, A. Mukhopadhyay and G. De, J. Alloys Compd., 2021, 873, 159793 CrossRef CAS.
- H. Okamoto, T. B. Massalski, L. J. Swartzendruber and P. A. Beck, Bull. Alloy Phase Diagrams, 1984, 5, 592–601 CrossRef.
- C. Langlois, P. Benzo, R. Arenal, M. Benoit, J. Nicolai, N. Combe, A. Ponchet and M. J. Casanove, Nano Lett., 2015, 15, 5075–5080 CrossRef CAS PubMed.
- L. Lartigue, D. Alloyeau, J. Kolosnjaj-Tabi, Y. Javed, P. Guardia, A. Riedinger, C. Péchoux, T. Pellegrino, C. Wilhelm and F. Gazeau, ACS Nano, 2013, 7, 3939–3952 CrossRef CAS PubMed.
- A. S. Brar and B. S. Randhawa, Polyhedron, 1984, 3, 169–173 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |