
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon oxidation with sacrificial anodes to inhibit O2 evolution in membrane-less bioelectrochemical systems for microbial electrosynthesis†
Nils
Rohbohm
a,
Tianran
Sun
ab,
Ramiro
Blasco-Gómez
 ac,
James M.
Byrne
de,
Andreas
Kappler
df and
Largus T.
Angenent
*afghi
ac,
James M.
Byrne
de,
Andreas
Kappler
df and
Largus T.
Angenent
*afghi
aEnvironmental Biotechnology Group, Department of Geosciences, University of Tübingen, Schnarrenbergstr. 94-96, 72076 Tübingen, Germany. E-mail: l.angenent@uni-tuebingen.de
bState Key Lab of Urban and Regional Ecology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China
cLaboratory of Chemical and Environmental Engineering (LEQUiA), Institute of the Environment, University of Girona, Carrer Maria Aurelia Capmany, 69, 17003 Girona, Spain
dGeomicrobiology Group, Department of Geosciences, University of Tübingen, Schnarrenbergstr. 94–96, 72076 Tübingen, Germany
eSchool of Earth Sciences, University of Bristol, Wills Memorial Building, Queens Road, Bristol, BS8 1RJ, UK
fCluster of Excellence: EXC 2124: Controlling Microbes to Fight Infection, Tübingen, 72076, Germany
gAG Angenent, Max Planck Institute for Biology, Max Planck Ring 5, D-72076 Tübingen, Germany
hDepartment of Biological and Chemical Engineering, Aarhus University, Gustav Wieds Vej 10D, 8000 Aarhus C, Denmark
iThe Novo Nordisk Foundation CO2 Research Center (CORC), Aarhus University, Gustav Wieds Vej 10C, 8000 Aarhus C, Denmark
First published on 24th July 2023
Abstract
Microbial electrosynthesis is an emerging biosynthesis technology that produces value-added chemicals and fuels and, at the same time, reduces the environmental carbon footprint. However, constraints, such as low current densities and high inner resistance, disfavor this technology for industrial-scale purposes. The cathode performance has been strongly improved in recent years, while the anode performance has not been given enough attention despite its importance in closing the electric circuit. For traditional water electrolysis, O2 is produced at the anode, which is toxic to the anaerobic autotrophs that engage in microbial electrosynthesis. To overcome O2 toxicity in conventional microbial electrosynthesis, the anode and the cathode chamber have been separated by an ion-exchange membrane to avoid contact between the microbes and O2. However, ion-exchange membranes increase the maintenance costs and compromise the production efficiency by introducing an additional internal resistance. Furthermore, O2 is inevitably transferred to the catholyte due to diffusion and electro-osmotic fluxes that occur within the membrane. Here, we proved the concept of integrating carbon oxidation with sacrificial anodes and microbes to simultaneously inhibit the O2 evolution reaction (OER) and circumvent membrane application, which allows microbial electrosynthesis to proceed in a single chamber. The carbon-based anodes performed carbon oxidation as the alternative reaction to the OER. This enables microbial electrosynthesis to be performed with cell voltages as low as 1.8–2.1 V at 10 A m−2. We utilized Methanothermobacter thermautotrophicus ΔH in a single-chamber Bioelectrochemical system (BES) with the best performing carbon-based anode (i.e., activated-carbon anode with soluble iron) to achieve a maximum cathode-geometric CH4 production rate of 27.3 L m−2 d−1, which is equal to a volumetric methane production rate of 0.11 L L−1 d−1 in our BES, at a coulombic efficiency of 99.4%. In this study, M. thermautotrophicus ΔH was majorly limited by the sulfur source that inhibited electromethanogenesis. However, this proof-of-concept study allows microbial electrosynthesis to be performed more energy-efficiently and can be immediately utilized for research purposes in microbial electrosynthesis.
Broader contextIn microbial electrolysis cells, electric power serves as the driving force for microbial reduction of carbon dioxide at the cathode to produce multi-carbon and electron-dense organic molecules that can serve as fuels and terminal chemical products. This idea is called microbial electrosynthesis and, combined with renewable energy to generate electrical power, makes it an emerging technology for sustainable chemical production. Microbial activity and electrode performance are critical factors in determining the overall efficiency of microbial electrosynthesis. Several studies have focused on modifying the cathode to improve the microbe-electrode interaction and compatibility due to its importance in generating reducing power for microbial electrosynthesis. However, so far, the focus was rarely on the anode performance. Here, we employed a sacrificial carbon-based anode that enabled us to remove the ion-exchange membrane in the electrochemical cell. We proved the concept of a singlechamber bioelectrochemical cell using Methanothermobacter thermautotrophicus ΔH with this sacrificial carbon-based anode. This study shows a different path forward to perform microbial electrosynthesis, especially in the laboratory for basic research. |
Introduction
Globally increasing demands for value-added chemicals and fuels require innovative chemical and biochemical synthesis strategies to proceed in a more cost-efficient and environmentally friendly manner. Microbial electrosynthesis is a bioprocess with bioelectrochemical systems to convert inorganic gases into valuable, multi-carbon compounds to store excessive renewable energy such as wind or solar energy.1,2 This makes it an attractive technology for achieving the goal of a carbon-neutral society. Microbial activity and electrode performance are critical factors in determining the overall efficiency of microbial electrosynthesis. However, the inherent problem of scaling-up microbial electrosynthesis is the currently low current density of the bioelectrochemical system, which results in a considerably lower production rate compared to chemical electrosynthesis that produces the same compounds (Table S1, ESI†). To increase the current density, the electrochemical performance must be increased by, for example, improving the cathode performance. Indeed, several studies have focused on the modification of the cathode to improve microbe–electrode interactions and its biocompatibility.3,4Besides the biocompatibility problems, another troublesome part of bioelectrochemical systems is the ion-exchange membrane. Classically, the anode is separated from the cathode by an ion-exchange membrane to avoid interaction of the O2 evolving at the anode with the strict anaerobic microbes in the cathodic chamber.5 However, applying an ion-exchange membrane in bioelectrochemical systems has several disadvantages. By including an ion-exchange membrane, the internal resistance is increased.6 This will lead to decreased ion transport, which consequently limits electron transfer and reduces the current generation. Ideally, protons are primarily transported from the anodic to the cathodic chamber using a cation exchange membrane to maintain hydrogen production. However, the transfer of other cations, such as sodium or potassium, to the cathode chamber cannot be neglected and reduces the ionic transport number of protons. Consequently, the slower mass transfer rate of protons leads to a pH gradient.7 In addition, the ion-exchange membrane also generates an electro-osmotic flow that allows all ionic and non-ionic solutes, including O2, to pass through membrane into the cathode chamber, thus, making its purpose less effective.8 The pore size of the membrane in addition to O2 permeation, which allows biofilm formation, makes it vulnerable to fouling.9 Finally, the combination of all the disadvantages will result in high maintenance and operating costs.10
The ideal bioelectrochemical system would, therefore, be a single-chamber system in which the ion-exchange membrane has been removed.11 This would improve not only the current generation but also the synthesis efficiency. Cl2 and O2 are the main gases produced on the anode side, with Cl2 being not only a strong oxidizing agent to many elements but also very toxic. While Cl2 can be avoided by removing the chlorine-ions from the anodic chamber, O2 would still be produced without modifications at the anode through water splitting and hinder anaerobic microbes from growing for microbial electrosynthesis to proceed. Thus far, few reports have been published showing membrane-less bioelectrochemical systems, and most reports are about microbial fuel cells and enzymatic fuel cells. Furthermore, most membrane-less, single-chamber systems were operated without O2-sensitive microbes.12,13 Giddings et al.5 designed a membrane-less system for microbial electrosynthesis in which the anode was placed at the boundary of the liquid interface to avoid O2 from mixing. Besides, different patent applications for single-chamber bioelectrochemical systems were filed with setups to capture and remove O2 away from the cathode.14,15 However, none of these innovations had tried to circumvent the O2-evolution reaction (OER) at the anode.
An appealing approach to avoid O2 formation at the anode is carbon-assisted water electrolysis (CAWE) for which carbon is oxidized at the anode instead of the OER in conventional electrolysis.16,17 The classical hydrogen-evolution reaction (HER) is not disturbed and proceeds at CAWE. CAWE can include: (1) the oxidation of carbon-based electrodes, which we refer to as carbon oxidation with sacrificial anodes; and (2) the oxidation of soluble carbon materials (which we did not study) or non-soluble carbon materials in the anolyte. Here, we address carbon oxidation of sacrificial anodes, forming CO2, CO, and oxygenated surface-functional groups. The desired two chemical reactions can be described as follows (eqn (1) and (2)):
| C + 2H2O → CO2 + 4H+ + 4e− E0 = +0.21 V | (1) |
| C + H2O → CO + 2H+ + 2e− E0 = +0.51 V | (2) |
A major advantage of using CAWE instead of conventional water electrolysis with O2 evolution at the anode is the lower operating cell voltage, resulting in lower energy consumption rates. For example, the standard electrode cell potential for the four-electron process (eqn (1)) for CAWE is E0cell,CAWE = −0.21 V, while the conventional water electrolysis reaction requires E0cell,water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) electrolysis = −1.23 V.16 Another advantage of CAWE compared to conventional water electrolysis is the higher carbon-oxidation reaction rate than the OER rate at the anode (exchange current density: j0,CAWE =10−6 Acm−2vs. j0,OER = 10−7 A cm−2).16 Albeit, the carbon-oxidation reaction at the anode is still slower than the HER rate at the cathode (j0,HER = 10−3 A cm−2).18 Finally, a necessary substrate for microbial electrosynthesis is CO2 or CO, which is the desired product of carbon oxidation with sacrificial anodes. Thus, the matching product formation as extra carbon source for the microbes is another pertinent advantage of carbon oxidation with sacrificial anodes for microbial electrosynthesis.
electrolysis = −1.23 V.16 Another advantage of CAWE compared to conventional water electrolysis is the higher carbon-oxidation reaction rate than the OER rate at the anode (exchange current density: j0,CAWE =10−6 Acm−2vs. j0,OER = 10−7 A cm−2).16 Albeit, the carbon-oxidation reaction at the anode is still slower than the HER rate at the cathode (j0,HER = 10−3 A cm−2).18 Finally, a necessary substrate for microbial electrosynthesis is CO2 or CO, which is the desired product of carbon oxidation with sacrificial anodes. Thus, the matching product formation as extra carbon source for the microbes is another pertinent advantage of carbon oxidation with sacrificial anodes for microbial electrosynthesis.
An example of carbon oxidation with carbon-based anodes was shown by Holubowitch et al.,17 who demonstrated an abiotic membrane-less electrochemical cell that was capable of reducing O2 to deoxygenate water. While their main focus was on reducing O2, we identified with their data that either no O2 was produced or O2 production was decreased at the anode and that O2 was electrochemically reduced at the cathode. Carbon oxidation with carbon-based anodes was not only tested abiotically but also for bioelectrochemical systems. Lai et al.19 found a lower O2 content and a higher CO2 content in their anodic compartment using graphite. They concluded that carbon oxidation with the graphite anode was the main cause of the lower O2 formation. A follow-up study for the removal of perchloroethylene in a membrane-less bioelectrochemical system used graphite granules as a single anode to avoid O2 production.20 To our knowledge, carbon oxidation with sacrificial anodes has not been tested for microbial electrosynthesis.
The objective of this work was a proof of concept to show that carbon oxidation can be utilized in a single-chamber bioelectrochemical system to specifically perform microbial electrosynthesis. We used different types of carbon electrodes as anodes and assessed whether O2 formation was inhibited or just decreased. In addition, we tested iron (soluble and coated) materials with carbon electrodes to further circumvent the OER by including a parallel faradaic reaction. After we had studied the feasibility of our carbon electrodes and deciphered the mechanism of the inhibition, we operated our carbon electrodes in bioelectrochemical systems to evaluate their potential for microbial electrosynthesis. Here, we report the successful implementation of carbon oxidation with a sacrificial anode for microbial electrosynthesis using Methanothermobacter thermautotrophicus ΔH, which is an autotrophic and anaerobic microbe, and which is being used in the power-to-gas concept to store renewable electric power into CH4 gas.21
Results and discussion
The minimal required cell potential is lower for carbon oxidation than for O2 evolution at the anode, while the kinetics of the carbon oxidation reaction is higher
For our proof-of-concept process, we used the two carbon-oxidation reactions from CAWE compared to the OER from conventional water electrolysis. The two carbon-oxidation reactions have a theoretically more favorable thermodynamic potential of +0.21 V vs. RHE (reversible hydrogen electrode which depends on the solution pH) for CO2 production (four electrons in eqn (1)) and +0.51 V vs. RHE for CO production (two electrons in eqn (2)), respectively,22 than the OER, with a standard potential of +1.23 V vs. RHE. For conventional water electrolysis with the OER, the overall cell potential would be −1.23 V. However, electrode materials have properties that introduce an overpotential. In the case of carbon electrodes, the overpotential can be up to 500 mV or higher.23To measure the overpotentials (η) for our specific BES for carbon oxidation with carbon-based anodes, OER, and HER, we performed linear sweep voltammetry on the activated carbon (as the anode) and carbon cloth (as the cathode). Linear sweep voltammograms demonstrated that the measured overpotential (|η|= [Eonsetpotential + Ereferenceelectrodecorrection] − [E0 − 0.059 pH]) of carbon oxidation with activated carbon was +1.16 V, while the HER for carbon cloth had an overpotential of +0.190 V (Fig. 1). Thus, the minimum required cell potential for CAWE anodes would be −1.56 V (E0HER − ηHER − [E0carbonoxdiation + ηcarbonoxidation]), which is −1.37 V greater than the theoretical value (E0cell,carbonoxidation = E0HER − E0carbonoxidation = −0.21 V). The OER with the activated carbon had an overpotential of +0.86 V (Fig. 1). Therefore, the minimal required cell potential for conventional water electrolysis would be equivalent to −2.28 V (Ecell,waterelectrolysis = E0HER − ηHER − [E0OER + ηOER]), which is −1.05 V greater than the theoretical value. The considerably less negative overall cell potential for CAWE compared to conventional water electrolysis should lower the energy consumption in our system as well.
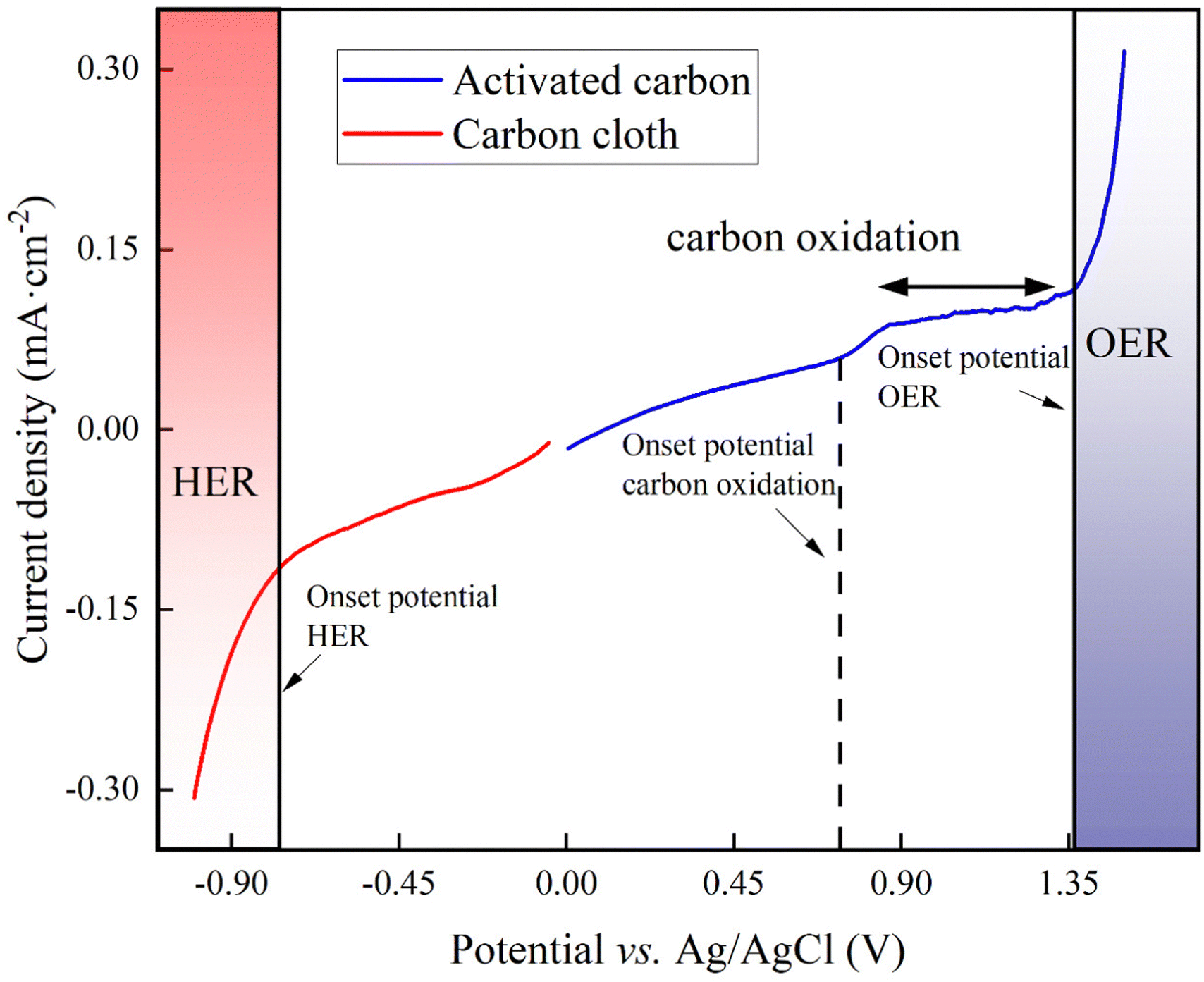 | ||
| Fig. 1 Linear sweep voltammogram of the activated-carbon anode (blue line) and the carbon-cloth cathode (red line) in 66 mM phosphate buffer (pH = 7.2) at a scan rate of 20 mV s−1. | ||
However, besides the cell potentials, the kinetics of the different electrochemical reactions are also important. The linear-sweep voltammogram of activated carbon did not show any pristine carbon-oxidation peaks to compare the kinetic value of the carbon oxidation to the OER (Fig. 1). However, the exchange current density, which is a dimension of the reaction rate, can be deduced from the linear-sweep voltammograms by means of the Butler–Volmer equation (eqn (S1) in Note S1 of the ESI†).24 The exchange current density for carbon oxidation was 10−5 A cm−2 and was one magnitude higher than the OER at 10−6 A cm−2. Therefore, the carbon oxidation reaction with sacrificial anodes is a faster electrochemical reaction than OER.
Carbon-based anodes with and without iron species circumvented the need for a membrane, opening the possibility of a membrane-less system
To identify whether O2 production is inhibited by carbon oxidation, we measured the O2 content of an abiotic, two-electrode electrochemical system for five days. The O2 content measurement was performed abiotically to circumvent using L-cysteine, which would be the preferred sulfur source and reducing agent in our medium for the BES experiments, and which would have removed O2 from the electrolyte. The abiotic experiments were performed in triplicate for two temperature conditions: room temperature and 60 °C, which is the optimal growth temperature of M. thermautotrophicus ΔH. We used a graphite rod, thermally activated graphite felt, carbon cloth, and activated carbon as our carbon anodes for four out of seven treatments and included a platinum anode that was poised at 10 A m−2 in 0.066 M phosphate buffer as the positive control to compare the results of the carbon oxidation with sacrificial anodes to the OER (Fig. 2). | ||
| Fig. 2 O2 concentration in the electrolyte of platinum, graphite-rod, thermally activated graphite felt, carbon-cloth, and activated-carbon anodes over time. Two temperature conditions were included. Two treatments with thermally activated graphite felt and three treatments with activated carbon were included, including iron additions. The error bars show the standard deviation for the average values from triplicate systems: (A) the O2 concentration at room temperature for 180 min; (B) the O2 concentration at room temperature for 5 days; (C) the O2 concentration at 60 °C for 180 min; and (D) the O2 concentration at 60 °C for 5 days. We omitted the data for the first 180 min from panels B and D for clarity reasons. No L-cysteine was added to these abiotic experiments. | ||
We added Fe2+ species in the form of: (1) soluble Fe2+ species (0.1 g L−1 of Fe2+, corresponding to 0.27 g L−1 FeSO4); and (2) coated magnetite for three out of seven treatments to this abiotic O2 evolution study. We added Fe2+ species because abiotic electrochemical systems increased the current density for carbon oxidation with sacrificial anodes without reducing the Coulombic efficiency.25 Soluble Fe2+ is directly reduced at the carbon-based anode to Fe3+ at electrochemical rates that are higher than carbon oxidation and the OER (eqn (3)).
| At the anode: Fe2+ → Fe3+ + e− | (3) |
Therefore, soluble Fe2+ is an ultimate electron donor in addition to the carbon-based anodes when Fe2+ is continuously added to the system. Soluble Fe2+ was added to the thermally activated-graphite-felt and activated-carbon anodes. Magnetite is another biocompatible Fe2+ source. We coated magnetite on the activated carbon to form the anode after performing a magnetite-to-carbon ratio analysis (Note S2 of the ESI†). Magnetite is a mixed-valent Fe(II), Fe(III) magnetic iron mineral that oxidizes to maghemite (γ-Fe2O3), donating one electron (eqn (4)):
| 3Fe3O4 → 4γ-Fe2O3 + Fe2+ + 2e− | (4) |
All carbon-based anodes with and without Fe2+ species showed an inhibitory effect on the O2 production compared to the positive control for the first 180 min at room temperature, with the graphite-rod and carbon-cloth anodes producing more O2 compared to the thermally activated-graphite-felt and activated-carbon anodes (Fig. 2(A)). Thus, a pretreatment to activate the graphite or carbon will permit a faster inhibition of O2 synthesis. We turned off the electrochemical system with the platinum anode after 180 min to avoid accumulating O2 and H2 and creating a Knallgas reaction. For the next 5 days, the accumulated O2 concentration of all the carbon-based anodes was relatively low (<1 ppm), but activated-carbon anodes with and without soluble Fe2+ could sustain a nearly O2-free environment (Fig. 2(B)). However, magnetite-coated activated-carbon anodes showed a higher O2 concentration at room temperature compared to the other activated carbon-based electrodes for unknown reasons (Fig. 2(B)). A possibility is a change of the composition and reactivity of the activated carbon after synthesis of magnetite (pH change from acidic to basic).
In contrast, all the experiments with carbon-based anodes at 60 °C inhibited O2 production during the first 180 min, except for the graphite-rod anode (sky-blue line, Fig. 2(C)). In addition, no O2 was produced at 60 °C for the thermally activated-graphite-felt and activated-carbon anodes with and without soluble Fe2+ and activated-carbon anodes with magnetite (Fig. 2(D)). Because the kinetics of a reaction are directly related to the temperature, the increase in the temperature, therefore, increased the carbon-oxidation rate. Likewise, the kinetics of the OER were also affected by the increase in temperature. However, our results show that no production of O2 occurred and that carbon oxidation was the dominating reaction in our system (Fig. 2(D)).
During the five-day operating period, the O2 concentration in the electrolyte with the graphite-rod anode gradually decreased, suggesting that graphite needs an activation time to oxidize its surface and efficiently perform carbon oxidation (Fig. 2(D)). Indeed, the thermally activated-graphite-felt and activated-carbon anodes did not show oxygen evolution during the first couple of days of the operating period (Fig. 2(D)). The concentrations of CO2 and CO were measured for the two temperature conditions and the seven treatments, but the concentrations were very low for both gases due to the low applied current of up to 1 mA cm−2 and were not further monitored. In summary, the carbon-based anodes circumvented O2 production at 60 °C and were used for further thermophilic BES experiments. Because we completely inhibited O2 evolution at the anode, we can remove the membranes from the bioelectrochemical system to perform microbial electrosynthesis. When comparing these abiotic results to the BES experiments, the abiotic O2 evolution results will be conservative because we added reducing agents only to the microbial medium.
Different carbon materials as the anode affected the CH4 production duration of microbial electrosynthesis, and competitive methane production rates were achieved to other BES studies
In our proof-of-concept BES for microbial electrosynthesis, we opted for the microbe M. thermautotrophicus ΔH due to its high growth rate (doubling time of 120 min) and relatively high CH4 production rate.21 This archaeal strain grows anoxically at 60 °C and shows a resilient behavior in the presence of O2 by having the ability to grow after exposure to air for several hours.26 However, as a strict anaerobic methanogen, it is still sensitive to O2 for longer operating periods, resulting in the absence of growth when prolonged O2 is present. The BES experiments were performed at 60 °C and close to neutral-pH conditions with graphite-rod, thermally activated-graphite-felt, and activated-carbon anodes in an experimental design with six treatments including soluble Fe2+ and magnetite. We set the current at 2 mA and with a 1 × 1 cathode-surface area the cathode-based geometric current density was 1 mA cm−2 (Fig. 3).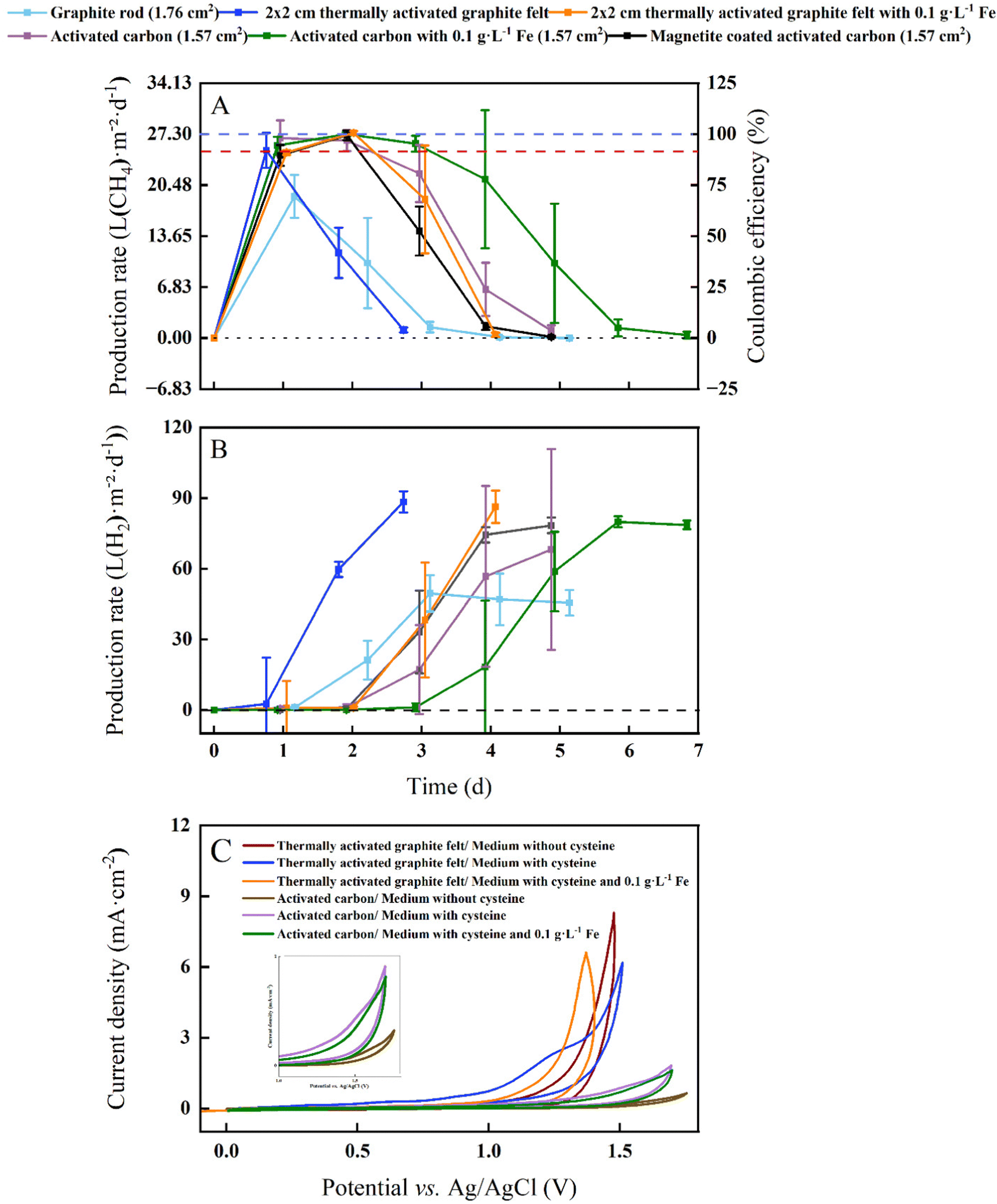 | ||
| Fig. 3 Membrane-less BES performance for electromethanogenesis and cyclic voltammetry analysis with L-cysteine: (A) CH4 production rates corrected to the cathode surface area and Coulombic efficiencies (at I = 10 A m−2); (B) H2 production rate corrected to the cathode surface area during batch experiments over time with the thermophilic archaeon M. thermautotrophicus ΔH. Graphite-rod, thermally activated graphite felt and activated-carbon anodes were included with two treatments and three treatments for thermally activated graphite felt and activated carbon, respectively, including iron additions (soluble iron and magnetite). The error bars show the standard deviation for the average data from triplicate systems. For (A) and (B) Carbon cloth was used as the cathode: (Y-axis 1) cathode-based geometric CH4 production rates and H2 production rates for six treatments; and (Y-axis 2) for (A) Coulombic efficiencies for six treatments. After reaching a maximum H2 production rate, we stopped the current to not accumulate excessive amounts of H2 in our BESs. While the blue-dotted line represents 100% Coulombic efficiency at a 27.30 L m−2 d−1 methane production rate, the red-dotted line shows the empirical cathode-based geometric CH4 production rate of >25 L m−2 d−1 to determine the duration of the optimum performance period; and (C) cyclic voltammograms of thermally activated graphite felt and activated carbon at a scan rate of 5 mV s−1 in MS-medium. In total 6 cyclic voltammogramms were recorded where the MS-medium for each carbon-based electrode contained L-cysteine without soluble Fe2+; L-cysteine with soluble Fe2+; and without L-cysteine and without soluble Fe2+. | ||
M. thermautotrophicus ΔH produced CH4 with CO2 as the sole carbon source during carbon oxidation with sacrificial anodes for all treatments (Fig. 3(A)). After an operating period of one day, the maximum cathode-based geometric CH4 production rates for the BESs with the graphite-rod, the thermally activated graphite felt, the activated-carbon anode were 18.97 ± 2.84, 25.13 ± 2.34, and 26.75 ± 2.38 L m−2 d−1, respectively (Table 1). This resulted in a maximum volumetric CH4 production rate of 0.1 ± 0.001 L L−1 d−1 for the BES with the activated-carbon anode. Coulombic efficiencies of 97.96 ± 8.71% for the BES with the activated-carbon anodes suggest an efficient H2 uptake by the microbes at the cathode (Fig. 3).
| Carbon-based anode | Maximum geometric CH4 production rate in L m−2 d−1 | Maximum volumetric CH4 production rate in L L−1 d−1 | Optimum performance period in d |
|---|---|---|---|
| Graphite rod | 18.97 ± 2.84 | 0.07 ± 0.01 | 0 |
| Thermally activated graphite felt | 25.13 ± 2.34 | 0.10 ± 0.009 | 1 |
| Thermally activated graphite felt with 0.1 g L−1 of Fe2+ | 27.48 ± 0.26 | 0.11 ± 0.001 | 2 |
| Activated carbon | 26.75 ± 2.38 | 0.10 ± 0.009 | 2 |
| Activated carbon with 0.1 g L−1 of Fe2+ | 27.30 ± 0.29 | 0.11 ± 0.001 | 3 |
| Magnetite-coated activated carbon | 27.15 ± 0.65 | 0.11 ± 0.002 | 2 |
By adding soluble Fe2+ to thermally activate-graphite-felt and activated-carbon anodes at 60 °C with M. thermautotrophicus ΔH, we achieved a higher maximum cathode-based geometric CH4 production rate of 27.48 ± 0.26 and 27.30 ± 0.29 L m−2 d−1, respectively, resulting in a volumetric CH4 production rate of 0.11 L L−1 d−1 for both and a higher Coulombic efficiency of 100.61 ± 0.01% and 99.36 ± 0.57% with the added soluble Fe2+ than without (Table 1 and Fig. 3(A)). However, with magnetite-coated activated-carbon anode material, the methane production was lower compared to activated-carbon anode material. The maximum cathode-based geometric CH4 production rate was 27.15 ± 0.65 L m−2 d−1 m with a Coulombic efficiency of 99.40 ± 0.02% (Table 1 and Fig. 3).
To compare the different experimental conditions in our study, we choose an empirical cathode-based geometric CH4 production rate of >25 L m−2 d−1 to determine the duration of the optimum-performance period for each treatment. Under this definition of the optimum performance period, the graphite-rod anode never reached this CH4 production rate, resulting in an optimum performance period of zero (Table 1). We achieved an optimum-performance period of 1 day for the thermally activated-graphite-felt anode. After an operating period of two days, the cathode-based geometric CH4 production rates for the BESs with the activated-carbon anode was 26.44 ± 1.40 L m−2 d−1 after which it decreased, resulting in an optimum-performance period of two days (Table 1). By adding soluble Fe2+ to thermally activated-graphite-felt and activated-carbon anodes, the optimum-performance period was elongated to two and three days at an average cathode-based geometric CH4 production rate of 26.17 ± 0.29 and 26.36 ± 0.84 L m−2 d−1, respectively (Table 1). However, with magnetite, the optimum performance period was shortened to two days at an average cathode-based geometric CH4 production rate of 25.81 ± 1.01 L m−2 d−1, which is one day shorter than for the activated-carbon anodes with soluble Fe2+ (Table 1). For the activated-carbon anodes as an example, the pH remained between 7.3 and 7.8 (Fig. S1, ESI†), and CO2 was never limited (Fig. S2, ESI†).
We achieved a maximum cathode-based geometric CH4 production rate of 27.48 L m−2 d−1 with thermally activated graphite felt and 0.1 g L−1 soluble Fe2+, which was slightly higher than Kracke et al.,4 who achieved a maximum of 26.76 L m−2 d−1 at a cathode-based geometric current density of 1 mA cm−2. The authors utilized a mesophilic methanogen (i.e., Methanococcus maripaludis) in a BES with membranes and a NiMo cathode. However, the reported maximum volumetric CH4 production rate by Kracke et al.4 was 1.38 L L−1 d−1, which was 12.5 times higher than the 0.11 L L−1 d−1 for our study due to a more optimum electrode-surface-to-reactor-volume ratio. An even higher volumetric CH4 production rate of 2.9 ± 1.2 L L−1 d−1 compared to Kracke et al.4 was achieved by Baek et al.,27 using a zero-gap electrochemical cell. Despite its lower geometric CH4 production rate of 18.25 L m−2 d−1 and side-products of acetate and propionate (at a cathode-based geometric current density of 1.74 mA cm−2),27 the zero-gap electrochemical cell has an optimized electrode-surface-to-reactor-volume ratio and has the propensity to increase the production rates for microbial electrosynthesis, including for our system. This promise was already shown by Geppert et al.28 They achieved a higher volumetric CH4 production rate of 12.5 L L−1 d−1 and a cathode-based geometric CH4 production rate of 62.5 L m−2 d−1 by recirculating anolyte and catholytes through a zero-gap design at an applied current density of 3.5 mA cm−2. However, a large amount of biomass was used as an inoculum, making it arguable that all carbon came from CO2.
Noteworthy is the considerably lower volumetric CH4 production rates for all BES studies compared to gas fermentation by sparging H2 and CO2, for which CH4 production rates of 688 L L−1 d−1 (using the standard molar volume of CH4 from 1.28 mol L−1 h−1) had already been achieved in the 1990s.29 It is clear that several breakthroughs are necessary to close the gap between microbial electrosynthesis and gas fermentation for CH4 production. Regardless, our proof-of-concept was successful for which we could produce a cathode-based geometric CH4 production rate of 27.48 L m−2 d−1 with our membrane-less BES with sacrificial anodes.
The duration of methane production in the single-chamber bioelectrochemical system was only a few days due to oxidation of reduced sulfur compounds at the anode and a resulting sulfur limitation
Our abiotic O2 measurements with graphite-rod anodes showed O2 production at 60 °C (Fig. 2(D)), identifying that O2 inhibition to the thermophilic methanogen was a reason for a zero-day optimum-performance period during our BES experiment with graphite-rod anodes (Fig. 3(A)). The thermally activated-graphite-felt anodes and activated-carbon anodes during abiotic experiments exhibited no O2 production after an operating period of five days (Fig. 2(D)), explaining the longer optimum-performance period for these anodes than the graphite-rod anodes (Fig. 3(A)). Still, we could only maintain a longest optimum-performance period of three days for the BES with activated-carbon anodes and soluble Fe2+ (Table 1). By comparing the CH4 production rate (Fig. 3(A)) with the H2 production rate (Fig. 3(B)), we observed different onsets of H2 production after the CH4 production had peaked for graphite-rod, thermally activated-graphite-felt, and activated-carbon anodes (with and without iron). This indicated that the electrochemical system was not limiting (H2 production did not stop). Thus, carbon oxidation had not diminished the available carbon at the anode to maintain a working electrochemical system.This also became clear when we increased the size of the anode surface areas and observed a similar optimum-performance period of one day for each of the BESs with the 1 × 1 cm-, 1 × 2 cm-, and 2 × 2 cm-thermally activated-graphite-felt anodes and with L-cysteine as the sulfur source and reducing agent in our growth medium (Fig. S3, ESI†). While the larger electrode provided a larger surface area for the carbon oxidation to occur and a more stable cell voltage (Fig. S4, ESI†), it did not improve the performance. Such a similar performance showed that, indeed, the carbon material was not limiting but that something else was limiting the methanogens to produce CH4. Without a membrane, L-cysteine oxidizes to L-cystine,30 which may render L-cysteine unavailable as a sulfur source and reducing agent. Therefore, we investigated whether the relatively fast oxidation of reducing sulfur components in the growth medium would diminish our sulfur source for methanogenic growth and activity.
Instead of L-cysteine, we added 0.1 g L−1 Na2S as a sulfur source and reducing agent to our treatment for the BESs to have a measurable sulfur source and a more potent reducing agent to lower the oxygen redox potential (ORP) without harming the microbes.31 We used thermally activated-graphite-felt anodes with 0.1 g L−1 Fe2+ because this resulted in the maximum cathode-based geometric CH4 production rate (Table 1). However, we increased the anode size from 1 × 1 to 2 × 2 cm and also increased the number of methanogenic cells during inoculation by 10 times compared to previous experiments. We compared the treatment with Na2S and soluble Fe2+ to the control with L-cysteine and soluble Fe2+, and obtained a similar: (1) CH4 production pattern; and (2) optimum-performance period of two days compared to previous results (Fig. 3(A)), and between the treatment and the control (Fig. S5A, ESI†).
To determine whether sulfide was oxidized and limiting the methanogens in our BESs, the sulfide concentration of each sampling point was measured for the treatment BESs through a photometric assay. The sulfide concentration decreased rapidly after the first day, proving that sulfide was removed, likely by oxidation (Fig. S5B, ESI†). After three days, sulfide was nearly depleted (Fig. S5B, ESI†). In addition, the ORP value increased from −375 mV to −100 mV showing that the reducing power of sulfide was removed at the same time (Fig. S5C, ESI†). These results suggest that the previous experiments were all limited in sulfur compounds, such that the sulfur source and/or the reducing power became limiting to the methanogens (Table 1).
A potential inhibiting factor for M. thermautotrophicus ΔH is an ORP value that is not negative enough to provide reducing power. In fact, electromethanogenesis would already be inhibited at a minimum ORP of −200 mV.32 Because both sulfur compounds were oxidized during the BES operating conditions, the reducing power and, subsequently, the ORP value for methanogenesis should worsen. To find out whether the depletion of the sulfur source or the increase in the ORP due to the absence of the reducing agent limited the methanogens after oxidation of reduced sulfur compounds, we added another treatment. Besides the Na2S and soluble Fe2+, we added Ti-NTA, L-cysteine, and soluble Fe2+. In fact, we introduced Ti-NTA at the beginning of the experiment and after every sampling point. However, no difference in the cathode-based geometric CH4 production rate was observed between Ti-NTA and the control (L-cysteine) (Fig. S5A, ESI†). In addition, the ORP values were similar throughout the operating period and had crossed the minimum value of −200 mV between Day 1 and Day 3 (Fig. S5C, ESI†). Thus, adding Ti-NTA did not lower the ORP because Ti3+ was also oxidized at the anode, preventing its reducing power from being effective. Therefore, we could not distinguish whether the depletion of the sulfur source or the increase in the ORP due to the absence of the reducing agent limited the methanogens.
Regardless, reduced sulfur compound was the limiting factor of the single-chamber BES. Therefore, the recovery of sulfide in situ after sulfur oxidation is a desirable objective to circumvent the continuous supply of sulfide to the electrochemical system. For a recent study by Izadi et al.,33 sulfide was oxidized at the anode to elemental sulfur after which the sulfide was recovered at the cathode by switching the polarity of the electrodes. Based on their study, we performed a BES experiment using thermally activated graphite felt as both anode and cathode while adding 0.1 g L−1 Na2S as an additional sulfur source. The polarity of the electrodes was switched every 4 h to reduce the elemental sulfur to sulfide. However, our study did not have a desirable outcome because the cathode-based geometric CH4 production rate was lower than in all other BES experiments, possibly due to a stressed environment (Fig. S6A, ESI†). In addition, the sulfide concentration was depleted faster, limiting CH4 production (Fig. S6B, ESI†). We cannot rule out that sulfide was oxidized to higher charged sulfur oxides, such as sulfate or thiosulfate, which are more difficult to reduce back to sulfide, preventing the capability to recover sulfide.
Soluble iron elongated the optimum-performance period for reasons that are related to sulfide oxidation
We performed cyclic voltammetry (CV) measurements with two carbon materials at the anode in an abiotic experiment with 6 treatments of which with and without L-cysteine and with and without soluble Fe2+ were included (Fig. 3(C)). We were interested in observing the onset potential of the oxidation of reduced sulfur compounds during the forward electrochemical scan (5 mV s−1). The thermally activated graphite felt with L-cysteine and without soluble Fe2+, showed the earliest onset potential of the six cyclic voltammograms and an electrochemical shoulder with a mid-point potential of +1.25 V vs. Ag/AgCl (Fig. 3(C)). The resulting faster oxidation of L-cysteine rate, explains why the optimum-performance period for the BES was the shortest between the thermally activated graphite felt and activated carbon anode treatments (one day in Table 1). Adding soluble Fe2+ to the thermally activated graphite felt anode with L-cysteine clearly shifted the onset potential to a higher voltage (Fig. 3(C)), reducing the oxidation of L-cysteine rate in our BES, and explaining the longer optimum-performance period (two days in Table 1). The no-L-cysteine control (i.e., no-sulfur control) shifted the onset potential further to a higher voltage, which was anticipated (Fig. 3(C)).Even though the current densities for the three cyclic voltammograms with activated carbon were approximately four times lower than with thermally activated graphite felt (Fig. 3(C)), we observed the same order of first onset potentials (L-cysteine without soluble Fe2+; L-cysteine with soluble Fe2+; and without L-cysteine and without soluble Fe2+ [control]). We found a mid-point potential of +1.5 V vs. Ag/AgCl with the activated carbon with L-cysteine and without soluble Fe2+ (Fig. 1(C)). The higher mid-point potential of L-cysteine oxidation (1.5 vs. 1.25 V) and the lower current densities explain why the activated carbon with L-cysteine but without iron showed a longer optimum-performance period for the BES compared to the thermally activated graphite felt (two days vs. 1 day in Table 1) – the slower L-cysteine oxidation rate elongated the growth period of methanogens. The addition of soluble Fe2+ to the activated carbon with L-cysteine, showed the same result as with thermally activated graphite felt: (1) a slower onset potential; and (2) a higher mid-point potential (1.6 V vs. Ag/AgCl in Fig. 3(C)), explaining the higher optimum-performance period (three days in Table 1). Thus, the addition of soluble Fe2+, reduced the L-cysteine oxidation rate.
We suspect that the formation of iron sulfide (FeS) in the electrolyte and/or at the anode slowed the oxidation of reduced sulfur compounds, allowing the methanogens a longer growth period before the sulfur source became limiting. Indeed, the activated carbon-based anodes with soluble Fe2+ showed a decreased overall iron concentration throughout time (Fig. S7, ESI†). The initial iron concentration was 0.1 g L−1, continuously decreasing until it stabilized after three days (Fig. S7, ESI†). In addition, we observed the precipitation of FeS, which persisted as a black precipitate. The removal of sulfide through FeS is a desired outcome in water treatment.34 Fortunately, it has been shown to benefit M. thermautotrophicus ΔH by improving CH4 production after adding Fe.35 Here, the formation of FeS benefited the microbe by lowering the oxidation of reduced sulfur compounds rate. However, FeS is still redox active with sulfide oxidation as an ultimate result, explaining that the maximum optimum-performance period was still only three days (Table 1).
Magnetite, which is an alternative redox mediator to soluble iron, shortened the optimum performance period
The cyclic voltammogram of synthesized magnetite nanoparticles that were immobilized on an Au electrode featured two peaks with a peak splitting of +0.28 V, representing the oxidative peak at −0.12 V vs. Ag/AgCl and the reductive peak at −0.41 V vs. Ag/AgCl (Fig. 4(A)). The comparison of the standard potential of both magnetite-oxidation reactions with the literature exhibited that the two-electron oxidation of magnetite to maghemite (eqn (4)) occurred for our system.36 Roberts et al.37 had shown that magnetite is prone to develop this irreversible reaction, which would have a negative impact on long-duration BES experiments. Indeed, the cyclic voltammograms of bulk magnetite showed decreasing reaction rates with increasing cycles (Fig. 4(B)), which can be explained by the formation of maghemite (eqn (4)).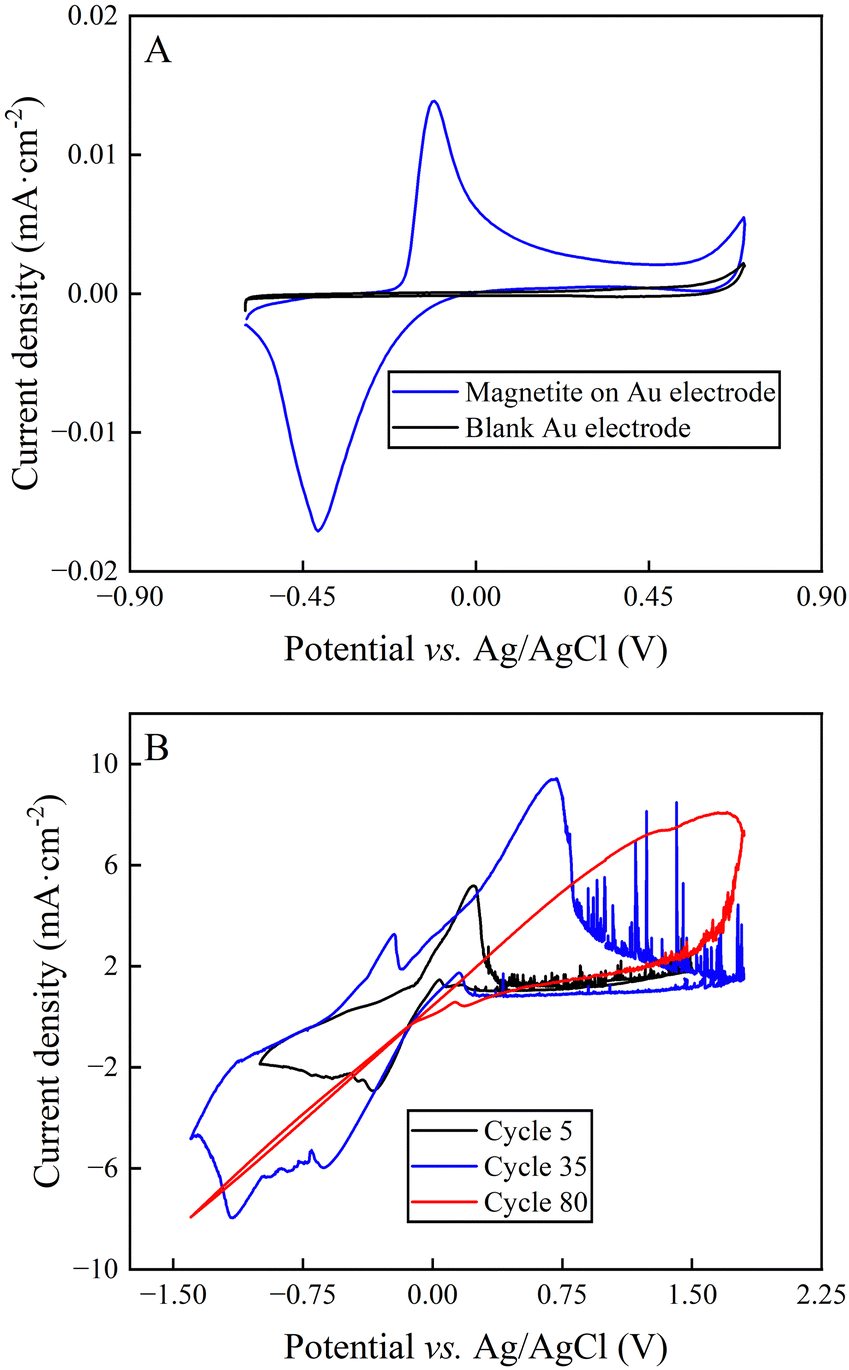 | ||
| Fig. 4 Electrochemical analysis of magnetite: (A) cyclic voltammogram of magnetite nanoparticles that were drop-casted on an Au electrode at a scan rate of 20 mV s−1 in 66 mM phosphate buffer; and (B) cyclic voltammograms of bulk magnetite at cycle 5, 35, and 80 at a scan rate of 20 mV s−1 in 50 mM PBS. | ||
To quantify the reaction rates, Tafel slopes were calculated from bulk magnetite during different cycles of the cyclic voltammogram (ESI,† Note S1 and Fig. 4(B)). Bulk magnetite electrodes achieved 187 mV dec−1 at cycle 5. It transitioned to 617 mV dec−1 and 1310 mV dec−1 at cycles 35 and 80, respectively (Fig. 4(B)). The formation of maghemite increased the inner resistance of the anode material to decrease its reaction rate by forming a resistor-type cyclic voltammogram (Fig. 4(B)), which likely would constrain the BES. This is one possible reason why we observed the shorter optimum performance period of two days vs. three days for magnetite-coated activated carbon anodes and activated-carbon anodes, respectively. However, more work would be necessary to exclude other possible reasons.
When sulfur source limitations are overcome, carbon at the anode would become a limiting factor
The oxidation and the removal of the carbon anode itself due to its sacrificing nature, would likely be the next liming factor of our BES due to a lack of accessible carbon to be oxidized. Our proof-of-concept BESs used relatively small geometric anode surfaces of 1.57 cm2. For our calculation, we used the ideal reaction of donating four electrons and producing CO2 during carbon oxidation (eqn (1)). To calculate the radius change (Δr) during the two-day optimum performance period of the activated carbon anode (as one example anode), we first calculated the volume (V) of carbon that was consumed (eqn (5)–(8)), which was equivalent to 1.10 × 10−2 cm3 (8.95 × 10−4 mol carbon), using the equation of the Coulombic efficiency.4 The exposed surface of the activated carbon anode corresponded to a halved cylinder so that the volume after the optimum performance period and its corresponding radius change would be: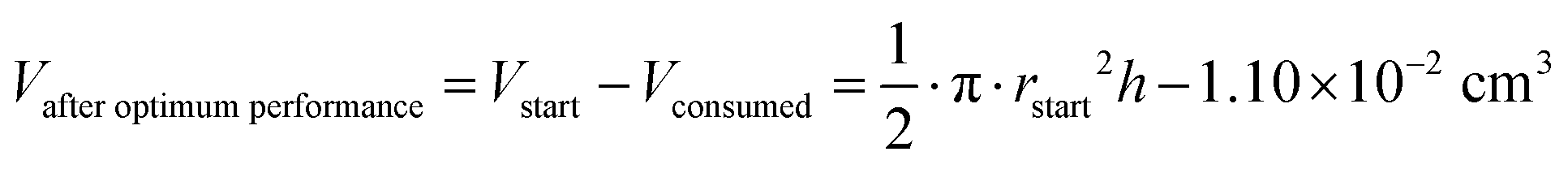 | (5) |
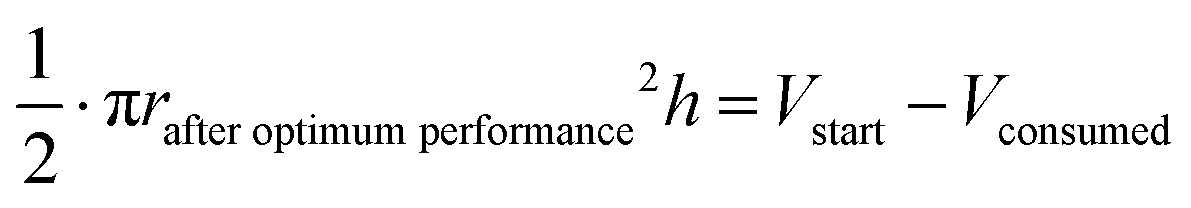 | (6) |
 | (7) |
| Δr = rstart − rafteroptimumperformance ≈ 34 μm | (8) |
With rstart = 0.475 cm and h = 2.175 cm. The relatively small change in the radius of 34 μm shows that the accessible carbon did not limit the system's performance yet. We could not determine when the carbon would become limiting due to the sulfur source limitations. If this occurs, the accessibility of carbon should be increased. This can be achieved by adding more carbon after increasing the ratio of anode surface area to reactor volume or improving the reactivity of the carbon. In addition, anodes could be replaced periodically.
A final limitation would be the impact of carbon corrosion on the CH4 production rates
We observed that the activated carbon-based electrodes started to disintegrate, and small carbon particles were found in suspension (Fig. S8, ESI†) that might impact the CH4 production rate during long-term operating conditions (considerably longer than three days). We refer to this as carbon corrosion, which could impact the CH4 production rate mostly negatively due to: (1) the exfoliation of carbon material at the anode, limiting carbon oxidation; and (2) the toxicity of a tar-like solution to methanogens. On the other hand, there is a mechanism known from abiotic carbon oxidation systems that could improve CH4 production, which we refer to as indirect carbon oxidation by Fe3+ (described below). Therefore, we examined carbon corrosion by investigating the surface and the bulk of the anodes with FTIR to track the change of functional groups exhibited at the carbon material. We used the activated-carbon anodes from our abiotic experiments during the measurements of the liquid O2 concentration (no soluble iron was added).Several oxidized functional groups from the initial activated carbon material, such as ketones and phenol groups, were identified by the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching bond at 1700–1800 cm−1 and –OH stretching bonds at 3300–3600 cm−1, respectively (Fig. S9, ESI†). The absorbance peak at 2500–3000 cm−1 represented the C–H stretching bond of the paraffin oil, which we had added to produce the anodes. The larger absorbance of –OH bonds suggested an alkaline chemical activation of the carbon from the manufacturer. Almost all the –OH stretches disappeared after five days of anode oxidation. The phenol functional group can act as a catalyst for carbon oxidation and facilitate the CO2 reaction at the anode. Once the functional group was consumed, the electrode exfoliated in small carbon particles.
O stretching bond at 1700–1800 cm−1 and –OH stretching bonds at 3300–3600 cm−1, respectively (Fig. S9, ESI†). The absorbance peak at 2500–3000 cm−1 represented the C–H stretching bond of the paraffin oil, which we had added to produce the anodes. The larger absorbance of –OH bonds suggested an alkaline chemical activation of the carbon from the manufacturer. Almost all the –OH stretches disappeared after five days of anode oxidation. The phenol functional group can act as a catalyst for carbon oxidation and facilitate the CO2 reaction at the anode. Once the functional group was consumed, the electrode exfoliated in small carbon particles.
While small concentrations of carbon particles in solution might not have a big influence, higher concentrations have been shown to influence methanogenesis. Cavalcante et al.38 identified that adding conductive material of carbon particles to a culture of methanogens enhanced methanogenesis. However, inhibition of methanogenesis occurred even at lower carbon material concentrations when the inoculation was performed at a relatively low cell density. In addition, Umetsu et al.39 showed that M. thermautotrophicus ΔH adsorbed on –OH and –COOH functional groups and prevented hydrogen or mineral uptake, inhibiting methanogenesis.
We also found that carbon corrosion and the reaction of the released carbon with Fe3+ (eqn (9)) could explain another stimulation of the CH4 production rate besides: (1) reducing the oxidation of reduced sulfur compounds rate due to FeS formation; and (2) the presence of conductive particles. Iron would need to be present in our membrane-less BES for this other stimulation, and we refer to this mechanism as indirect carbon oxidation by Fe3+. The continuous addition of soluble Fe2+ stimulated abiotic carbon oxidation systems because Fe2+ is directly reduced at the carbon-based anode to Fe3+ (eqn (3)). However, even without continuous soluble Fe2+ addition, a current increase can be anticipated due to the circular nature of the electron-mediating process, with the ultimate electron donor coming from the carbon-based anode material than the continuously added Fe2+. This is due to indirect carbon oxidation by Fe3+ using carbon in the solution that originated from carbon corrosion at the anode (Fig. 5). Indirect carbon oxidation reduces Fe3+ back to Fe2+ in the solution, while carbon is oxidized to CO2 (eqn (9) and Fig. 5). The reactions of the electron-mediating process can be described as follows:
| C + 4Fe3+ + 2H2O → CO2 + 4Fe2+ + 4H+ | (9) |
 | ||
| Fig. 5 Overview of the reactions that occur at the anode and cathode during the carbon oxidation using thermally activated graphite felt and activated carbon anodes with 0.1 g L−1 of Fe2+. Water splitting takes place at the cathode to produce H2. The main reaction at the anode is carbon oxidation, followed by the oxidation of Fe2+ and indirect carbon oxidation by the formed Fe3+. For more information, see section on the impact of carbon corrosion on the CH4 production rates. | ||
Because the reduction and oxidation of soluble iron species occur at the anode and with carbon in solution in the electrolyte, respectively, there is a continuous reformation of Fe2+ to maintain a higher current density for the electrochemical cell. An advantage of the circular nature of this electron-mediating process is that Fe3+ is oxidized in the electrolyte with carbon and not at the cathode, which, as a competing process, would have reduced the Coulombic efficiency for methane production.
We do not know whether carbon corrosion would have positive or negative effects on the CH4 production rate and the duration of the optimum-performance period for our BES. We would first need to solve the reduced sulfur compound oxidation problem. Likely, there may be an initial positive effect when carbon corrosion is only starting to occur, but when more and more carbon particles accumulate, we may observe a negative effect of carbon corrosion. Therefore, measures should be taken to reduce the negative effects of carbon corrosion on the anode activity and microbial activity to lengthen the optimum performance period of anode oxidation with sacrificial anodes.
Carbon oxidation with sacrificial anodes for single-chamber BES is ready for research purposes within the field of microbial electrosynthesis
We showed that implementing carbon oxidation with sacrificial anodes in a single-chamber BES can perform microbial electrosynthesis. We cultivated M. thermautotrophicus ΔH with in situ produced H2 at the cathode under constant pH and temperature conditions to produce CH4 for an optimum performance period of three days. The growth behavior followed a similar trend for all BESs until the amount of sulfur source limited the CH4 production rates. Our maximum cathode-based geometric CH4 production rate was already compatible with other studies due to similarly set current densities.4 Further optimizations of our system should not only increase the geometric CH4 production rate but also prolong the production days to several weeks. With the relatively low current densities of microbial electrosynthesis, our system is already useful for research purposes because the sulfur source will not become limiting that fast with lower current densities. Our work can also inform the earlier studies with a single-chamber BES in the field of perchlorate removal.20 We advise the authors to replace the graphite anode materials with activated-carbon anode materials and to add soluble iron to increase the performance.Before the industrial application of carbon oxidation with sacrificial anodes in BES for microbial electrosynthesis can commence, several breakthroughs are required
Our system would have a considerably lower theoretical power consumption than membrane-based two-compartment cells due to the lower potential to operate the BES (Fig. S4 and S10, ESI†). After optimizing the single-chamber BES and making it continuous, a direct comparison between the production rates and power consumption rates should be performed with a conventional membrane-based cell for microbial electrosynthesis. Another advantage of the single-chamber cell compared to a membrane-based cell would be the lower capital costs due to the absence of ion-exchange membranes, albeit a full techno-economic analysis would be necessary to be conclusive. These advantages make our system a desirable opportunity for scale-up. However, several problems must be overcome.We must prolong the optimum performance period to several months rather than a few days before scale-up can commence. We observed the limiting factor of sulfur source availability in our membrane-less BES, which had a promising current density. Utilizing a microbe that is not reliant on a redox-active sulfur compound as either a sulfur source for growth or to provide reducing power would be one route to overcome the utilization of a single-chamber BES. One possibility is to grow the microbes with thiosulfate; however, a considerably slower growth rate may be problematic. If this problem can be overcome, we predict two additional limitations for our membrane-less BES: (1) the availability of accessible carbon; and (2) carbon corrosion. Because carbon gets consumed over time, innovative BESs must be designed to refill the consumed carbon at the anode.
Undoubtedly, carbon corrosion would inhibit the growth of microbial cells during operating periods of several weeks, depending on the current densities. In addition, carbon corrosion would introduce a higher content of –OH, –CO, and –COOH functional groups, increasing the charge transfer resistance of the carbon material and decreasing the electrochemical performance.40 Consequently, the carbon anode decreases its conductivity and current density, while deteriorating. Ideally, carbon corrosion is avoided by efficiently transforming carbon into CO2 or CO. Studies to mitigate carbon corrosion currently tend to use either expensive catalysts to protect the carbon material or structurally stronger carbon, such as graphene or carbon nanotubes.41 In our case, excessive carbon corrosion has to be avoided, and a continuous system rather than our batch system could remove the by-products. Furthermore, mesoporous carbon would enlarge the surface area and the porosity, allowing more reaction sites for carbon oxidation rather than carbon corrosion.
Conclusion
We have proven the concept that microbial electrosynthesis is possible in a BES without membranes by utilizing carbon oxidation with sacrificial anodes. Our BES produced CH4 with an oxygen-sensitive thermophilic methanogen at a cathode-based geometric current density of 1 mA cm−2 for 3 days after which reduced sulfur compounds became limiting in the growth medium due to sulfur oxidation at the anode. The system is ready for microbial electrosynthesis experiments with relatively low current densities to perform basic research. However, for industrial applications, several breakthroughs are necessary to overcome limitations at the anode, such as sulfur oxidation, refilling of carbon, and protecting against carbon corrosion.Experimental section
Chemicals, strain, and medium
All chemicals were used as received unless otherwise noted in the text. These chemicals were of technical or analytical grade and were purchased from Sigma-Aldrich (St. Louis, USA), Alfa Aeser (Haverhill, USA), Merck (Darmstadt, Germany), Thermo-Fischer (Waltham, USA), VWR (Radnor, USA), and Carl Roth (Karlsruhe, Germany). The strain M. thermautotrophicus ΔH (DSM 1053) was obtained from the DSMZ (Braunschweig, Germany). A pre-culture was grown at 60 °C in 100 mL serum bottles with 20 mL of modified anaerobic MS-Medium31 containing (per liter): 6.77 g NaHCO3; 0.17 g K2HPO4; 0.235 g (NH4)2SO4; 0.09 g MgSO4; 0.06 g CaCl2 × 2 H2O; 0.23 g KH2PO4; 1 mL trace elements (10×); 1 mL of 4 M H2SO4; 0.5 g L-cysteine hydrochloride. The trace-element solution was a 10× stock solution containing (per 100 mL): 0.2 g nitrilotriacetic acid; 3 g MgSO4 × 7 H2O; 0.5 g MnSO4; 0.3 g FeSO4 × 7 H2O; 0.11 g CoSO4 × 7 H2O; 0.068 g CaCO3; 0.18 g ZnSO4; 0.01 g CuSO4 × 5 H2O; 0.018 g KAl(SO4)2 × 12 H2O; 0.01 g H3BO3; 0.01 g Na2MoO4 × 2 H2O; 0.48 g (NH4)2Ni(SO4)2 × 6 H2O; 0.01 g Na2WO4 × 2 H2O; 0.01 g Na2SeO4. The trace-element solution was adjusted to a pH value of 1 with H2SO4. The MS-Medium was buffered only in the bioelectrochemical system by adding 2.5 M 3-(N-morpholino)propanesulfonic acid (MOPS) at pH = 7.2 for a final media concentration of 25 mM. 25 mM Ti-NTA was prepared according to Moench et al.42 All solutions were sterilized either through a sterile filter or autoclaved and made anaerobic by sparging with N2 or N2/CO2.Preparation of carbon-based electrodes
The activated carbon served not only as active material but also as a conductive and surface enlarging material and was used as purchased (∼100 mesh particle size (∼150 μm), DARCO®, Cabot Norit Americas Inc, Marshall, USA). The activated carbon and paraffin oil (Sigma-Aldrich) was homogeneously mixed in a mortar at 70%:30% to form a paste.43 The resulting paste was pelleted into a self-made electrode holder and cured in an 80 °C vacuum oven for 4 h. Activated-carbon-associated magnetite was synthesized through a co-precipitation method by mixing activated carbon with 2 M FeCl3 and 1 M FeCl2.44 Then, 20% NH4OH was slowly dropped into the solution until pH 9. The solution was stirred for 1 h to complete the synthesis of magnetite. The precipitate was centrifuged, cleaned with deionized water until pH 7, and dried in a vacuum oven at 80 °C for 4 h. Different amounts of activated carbon to magnetite were used during the synthesis ranging from 0% to 60% of activated carbon. In this study, activated carbon that was co-precipitated with magnetite is called magnetite-coated activated carbon (ESI†). The 60% activated carbon and 40% maghemite mixture was synthesized by adding 60% activated carbon and 40% magnetite mixture into an aerated oven at 80 °C for 24 h.Measurement of dissolved O2
The O2 measurements were carried out in a self-designed five-neck round flask. To measure the dissolved O2, an O2-sensitive foil (0–100% O2, PreSens Precision sensing, Regensburg, Germany) was glued at the mid-height of the flask. The electrochemical cell was operated as a two-electrode setup with a 1 × 1 cm carbon cloth electrode as the cathode. A platinum wire, a graphite rod, a thermally activated graphite felt, a carbon cloth, activated carbon, and activated carbon that was coated with magnetite were used as anodes. Thermally activated graphite felt and activated carbon anodes were used with addition of soluble iron. The measurements were carried out in 0.066 M phosphate buffer at room temperature and 60 °C. The phosphate solution was buffered at 7.2 to mimic the pH of the MS medium used in the bioelectrochemical system. Before each experiment, the cells were purged with N2 gas for 1 h to secure an O2-free solution.Sample analyses
The bioelectrochemical systems were sampled once per day. The gas compositions of the bioelectrochemical systems were sampled with a 500 μL gastight syringe (Hamilton, Reno, USA) and analyzed using a gas chromatograph (SRI GC, Torrance, USA). The gas chromatograph used a Haysep D column (length 3 m, outer diameter 1/8”, SRI GC) and a Molsieve 13X column (length 3 m, outer diameter 1/8”, SRI GC). It was equipped with a thermal coupled detector and a flame ionization detector to measure N2, O2, H2, and CO2, and CH4, respectively. N2 and H2 were used as carrier gas. Coulombic efficiencies for CH4 were calculated according to Kracke et al.4 Cell growth was measured by cell counting with a light microscope (X41, Olympus, Shinjuku, Japan). The anodes were collected after the experiment and stored anoxically at −20 °C for further analyses. The outer layer and the bulk of magnetite particles were analyzed through a sequential extraction. First, the outer layer of magnetite particles was dissolved by acidifying with 1 M HCl for 1 h. It was followed by a complete dissolution of the magnetite particle by acidifying with 6 M HCl for 24 h. The iron content from the liquid samples and the oxidation state of iron in the magnetite electrodes were further analyzed via the Ferrozine assay.44,45 Sulfide concentrations were determined photometrically based on a modified version of Cline and Gilboa-Garber.46,47 Sulfide samples were stabilized with Zn(ac)2 and stored at −20 °C. The diamine reagent (N,N-dimethyl-p-phenylenediamine sulfate salt and FeCl3) was mixed with the stabilized sample at a 1:1 ratio and kept in the dark for 30 min for the reagents to react. The samples were measured at 565 nm. The ORP was measured with an ORP electrode (Hanna Instruments, Vöhringen, Germany) connected to an ORP controller (Alpha pH 800, Eutech Instruments, Singapore).
Electrochemical system
All electrochemical measurements (cyclic voltammetry, linear sweep voltammetry, and chronopotentiometry) were conducted in a five-neck round flask with a working volume of 50 mL. The electrochemical cells were operated as a two-electrode or a three-electrode setup and controlled via a potentiostat (VMP3, Bio-Logic, Grenoble, France). An Ag/AgCl (3 M KCl, 0.210 V vs. SHE) in-house-made reference electrode was used for the three-electrode setup. All measurements with a reference electrode were ohmic-drop corrected and were determined by potentiostatic electrochemical-impedance spectroscopy. Ohmic-drop-corrected potentials were calculated following the method of Sun et al.48 The reaction kinetics of carbon and magnetite were calculated with the Butler–Volmer equation, using linear sweep voltammetry and cyclic voltammetry measurements (ESI†). The 1 × 1 cm carbon cloth electrode (PANEX 30-PW06, Zoltek, St. Louis, USA), thermally activated graphite felt (Sigracell, SGL carbon, Bonn, Germany) and the platinum wire (Bioanalytical Systems, Inc., West Lafayette, USA) were used as cathodes, and the carbon-based electrode, graphite rod (Mersen, Suhl, Germany), carbon cloth (Zoltek), thermally activated graphite felt (SGL carbon), the carbon paste electrode holder (Bio-Logic), and the gold electrode (Bio-Logic) were used as anodes depending on their intended application, which is described in the results part. A titanium wire (Sigma Aldrich, St. Louis, USA) was threaded through the carbon cloth and thermally activated graphite felt, and glued with carbon conductive cement (Thermo-Fischer, Waltham, USA) for better conductivity. The temperature was either room temperature or controlled by a water bath at 60 °C (ICC basic IB R RO 15 eco, IKA, Staufen im Breisgau, Germany). The supporting electrolyte for the abiotic experiments was phosphate buffer (0.066 M) or phosphate buffered saline (0.05 M), with the pH buffered at 7.2. Before each abiotic electrochemical experiment, a high-purity N2 gas was introduced to remove O2. The bioelectrochemical systems were either autoclaved or chemically sterilized before the beginning of each experiment. The reactors were filled with 50 mL MS-Medium, were made anaerobic by sparging with 80:20 (v/v) N2/CO2 through a sterile filter, and were continuously stirred with a magnetic stir bar. The reactors were operated in batch mode and were fed for 5 min with pure CO2 as the sole carbon source prior to the experiment and every day for 1 min after liquid sampling to exchange the headspace. After feeding, an overpressure of 80 mbar of CO2 was kept in the headspace. The electrochemical cells were submerged in a water bath to prevent heat loss for optimal growth of M. thermautotrophicus ΔH at 60 °C.
Immobilization of magnetite
Magnetite was immobilized by dispersing magnetite nanoparticles onto an Au electrode (Biologic) to test its electrochemical behavior. The protocol is an adjusted version from Sun et al.48 Briefly, ten milligrams of the active powder and 40 μL of Nafion were mixed with 960 μL 70% ethanol and sonicated for 15 min. Then, 10 μL were drop casted onto the surface of the Au electrode and dried at room temperature. To avoid iron from oxidizing, the preparations for the drop casted magnetite were performed inside a glovebox (mBraun, Garching, Germany).SEM, EDX, X-ray diffraction, and FTIR
A 50 μL aliquot of 0.1% poly-L-lysine solution was placed onto a glass slide and was dried for 1 h in a 60 °C incubator. The samples were placed onto a poly-L-lysine coated glass slide that was attached to aluminum stubs, using carbon adhesive tabs. The sample was coated with a 10 nm deposition of platinum, using a BAL-TEC™ SCD 005 sputter coater (Leica biosystems, Nussloch, Germany). The purpose of the coating was to reduce any charging effects during scanning electron microscope (SEM) analysis. All images were taken in secondary-electron (SE) mode using a Zeiss Crossbeam 550L focused ion beam (FIB) SEM (Zeiss, Wetzlar, Germany), which was operated with an acceleration voltage of 2 kV. The SEM was equipped with an Oxford Instrument energy dispersive spectrometry detector (Ultim Max, Oxford Instrument, Abingdon, United Kingdom). Elemental maps of iron and carbon were obtained along with corresponding spot analysis.X-ray diffraction (XRD) was carried out on dry material using Bruker's D8 Discover GADDS XRD2 micro-diffractometer that was equipped with a Co-anode (Cu Kα radiation, λ = 0.154 nm) at parameters of 30 kV/30 mA (Billerica, USA). The total measurement time was 240 s at two detector positions, which were 15° and 40°. Fourier transform infrared (FTIR) spectroscopy was performed on a Vertex 80v equipped with an RT-DLaTGS detector (Bruker, Billerica, USA). 0.5 mg of sample was mixed homogenously with 250 mg KBr (FTIR grade, Carl Roth, Karlsruhe, Germany) and compressed at 10T to form a pellet. Samples were scanned 32 times in the range of 400–4500 cm−1.
Author contributions
L. T. A., T. S., and R. B.-G. initiated the work. N. R., T. S., and L. T. A. designed the experiments. N. R. performed the laboratory experiments and analyzed the data. L. T. A. and T. S. supervised the project. N. R. and L. T. A. wrote the manuscript. All edited the paper and approved the final version.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, EBiotech, SPP2240; L.T.A.) – project number 445506379, the Alexander von Humboldt Foundation in the framework of the Alexander von Humboldt Professorship (L. T. A.), and The Novo Nordisk Foundation CO2 Research Center with grant number NNF21SA0072700 (L. T. A.). L. T. A. and A. K. are also grateful to funding from the Deutsche Forschungsgemeinschaft under Germany's Excellence Strategy – EXC 2124 – 390838134. R. B.-G. was supported by an FPI grant (BES-2015-074229) from the Spanish Ministry of Economy and Competitiveness (MINECO) within the research project CTQ2014-53718R. The authors gratefully acknowledge the Tübingen Structural Microscopy Core Facility (Funded by the Federal Ministry of Education and Research (BMBF) and the Baden-Württemberg Ministry of Science as part of the Excellence Strategy of the German Federal and State Governments) for their support and assistance in this work. The authors also thank the German Research Foundation DFG (INST 37/1027-1 FUGG) for the financial support provided for the acquisition of the cryogenic focused ion beam scanning electron microscope. We thank Prof. Dr Uwe Schröder (University of Greifswald) for helpful and insightful discussions.Notes and references
- K. P. Nevin, T. L. Woodard, A. E. Franks, Z. M. Summers and D. R. Lovley, mBio, 2010, 1, e00103 CrossRef PubMed.
- K. Rabaey and R. A. Rozendal, Nat. Rev. Microbiol., 2010, 8, 706–716 CrossRef CAS PubMed.
- P.-L. Tremblay, L. T. Angenent and T. Zhang, Trends Biotechnol., 2017, 35, 360–371 CrossRef CAS PubMed.
- F. Kracke, J. S. Deutzmann, W. Gu and A. M. Spormann, Green Chem., 2020, 22, 6194–6203 RSC.
- C. G. S. Giddings, K. P. Nevin, T. Woodward, D. R. Lovley and C. S. Butler, Front. Microbiol., 2015, 6, 468 Search PubMed.
- P. Długołęcki, P. Ogonowski, S. J. Metz, M. Saakes, K. Nijmeijer and M. Wessling, J. Membr. Sci., 2010, 349, 369–379 CrossRef.
- R. A. Rozendal, H. V. M. Hamelers and C. J. N. Buisman, Environ. Sci. Technol., 2006, 40, 5206–5211 CrossRef CAS PubMed.
- K. J. Kim, A. G. Fane, M. Nystrom, A. Pihlajamaki, W. R. Bowen and H. Mukhtar, J. Membr. Sci., 1996, 116, 149–159 CrossRef CAS.
- L. Jourdin, S. M. T. Raes, C. J. N. Buisman and D. P. B. T. B. Strik, Front. Energy Res., 2018, 6, 7 CrossRef.
- J. J. Fornero, M. Rosenbaum and L. T. Angenent, Electroanalysis, 2010, 22, 832–843 CrossRef CAS.
- N. Mano, F. Mao and A. Heller, ChemBioChem, 2004, 5, 1703–1705 CrossRef CAS PubMed.
- D. Call and B. E. Logan, Environ. Sci. Technol., 2008, 42, 3401–3406 CrossRef CAS PubMed.
- S. Cheng, D. Xing, D. F. Call and B. E. Logan, Environ. Sci. Technol., 2009, 43, 3953–3958 CrossRef CAS PubMed.
- M. Kumar, S. Sandipam, S. K. Puri, S. S. V. Ramakumar, L. Diels and L. Bouwan, European Pat., EP38319832020, 2020 Search PubMed.
- D. R. Lovley and K. P. Nevin, USA Pat., 9856449, 2012 Search PubMed.
- H. Ju, S. Badwal and S. Giddey, Appl. Energy, 2018, 231, 502–533 CrossRef CAS.
- N. E. Holubowitch, A. Omosebi, X. Gao, J. Landon and K. Liu, Electrochim. Acta, 2019, 297, 163–172 CrossRef CAS.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- A. Lai, F. Aulenta, M. Mingazzini, M. T. Palumbo, M. P. Papini, R. Verdini and M. Majone, Chemosphere, 2017, 169, 351–360 CrossRef CAS PubMed.
- M. Zeppilli, E. Dell’Armi, L. Cristiani, M. Petrangeli Papini and M. Majone, Water, 2019, 11, 2579 CrossRef CAS.
- M. R. Martin, J. J. Fornero, R. Stark, L. Mets and L. T. Angenent, Archaea, 2013, 2013, 157529 CrossRef PubMed.
- S. G. Ji, H. Kim, W. H. Lee, H.-S. Oh and C. H. Choi, J. Mater. Chem. A, 2021, 9, 19834–19839 RSC.
- Y. Yi, G. Weinberg, M. Prenzel, M. Greiner, S. Heumann, S. Becker and R. Schlögl, Catal. Today, 2017, 295, 32–40 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, Hoboken, NJ, USA, 2nd edn, 2000 Search PubMed.
- R. Yin, Y. Zhao, S. Lu, H. Wang, W. Cao and Q. Fan, Electrochim. Acta, 2009, 55, 46–51 CrossRef CAS.
- A. Kiener and T. Leisinger, Syst. Appl. Microbiol., 1983, 4, 305–312 CrossRef CAS PubMed.
- G. Baek, R. Rossi, P. E. Saikaly and B. E. Logan, Water Res., 2022, 219, 118597 CrossRef CAS PubMed.
- F. Geppert, D. Liu, E. Weidner and A. ter Heijne, Int. J. Hydrogen Energy, 2019, 44, 21464–21469 CrossRef CAS.
- N. Nishimura, S. Kitaura, A. Mimura and Y. Takahara, J. Ferment. Bioeng., 1992, 73, 477–480 CrossRef CAS.
- M. Zhou, J. Ding, L.-P. Guo and Q.-K. Shang, Anal. Chem., 2007, 79, 5328–5335 CrossRef CAS PubMed.
- C. Fink, S. Beblawy, A. M. Enkerlin, L. Mühling, L. T. Angenent and B. Molitor, mBio, 2021, 12, e02766 CrossRef CAS PubMed.
- S. Hirano, N. Matsumoto, M. Morita, K. Sasaki and N. Ohmura, Lett. Appl. Microbiol., 2013, 56, 315–321 CrossRef CAS PubMed.
- P. Izadi, M. N. Gey, N. Schlüter and U. Schröder, iScience, 2021, 24, 102822 CrossRef CAS PubMed.
- H.-W. Lin, C. Kustermans, E. Vaiopoulou, A. Prévoteau, K. Rabaey, Z. Yuan and I. Pikaar, Water Res., 2017, 118, 114–120 CrossRef CAS PubMed.
- J. Cheng, H. Dong, H. Zhang, L. Yuan, H. Li, L. Yue, J. Hua and J. Zhou, Bioresour. Technol., 2020, 314, 123799 CrossRef CAS PubMed.
- M. J. Bradley and P. G. Tratnyek, ACS Earth Space Chem., 2019, 3, 688–699 CrossRef CAS.
- J. J. P. Roberts, J. A. Westgard, L. M. Cooper and R. W. Murray, J. Am. Chem. Soc., 2014, 136, 10783–10789 CrossRef CAS PubMed.
- W. A. Cavalcante, T. A. Gehring and M. Zaiat, Chem. Eng. J., 2021, 415, 128882 CrossRef CAS.
- M. Umetsu, T. Sunouchi, Y. Fukuda, H. Takahashi and C. Tada, Archaea, 2020, 2020, 9432803 CrossRef PubMed.
- X. Liu, W. Xu, D. Zheng, Z. Li, Y. Zeng and X. Lu, J. Mater. Chem. A, 2020, 8, 17938–17950 RSC.
- J. Zhao, Z. Tu and S. H. Chan, J. Power Sources, 2021, 488, 229434 CrossRef CAS.
- T. T. Moench and J. G. Zeikus, J. Microbiol. Methods, 1983, 1, 199–202 CrossRef CAS.
- Y. Zhang, F. Li, X. Liu, J. Lu and G. Zhang, Electrochim. Acta, 2017, 242, 165–172 CrossRef CAS.
- J. Sorwat, A. Mellage, A. Kappler and J. M. Byrne, J. Hazard. Mater., 2020, 400, 123139 CrossRef CAS PubMed.
- L. L. Stookey, Anal. Chem., 1970, 42, 779–781 CrossRef CAS.
- J. D. Cline, Limnol. Oceanogr., 1969, 14, 454–458 CrossRef CAS.
- N. Gilboa-Garber, Anal. Biochem., 1971, 43, 129–133 CrossRef CAS PubMed.
- T. Sun, B. D. A. Levin, J. J. L. Guzman, A. Enders, D. A. Muller, L. T. Angenent and J. Lehmann, Nat. Commun., 2017, 8, 14873 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00141e |
| This journal is © The Royal Society of Chemistry 2023 |