
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCeO2 supported high-valence Fe oxide for highly active and stable water oxidation†
Hongzhi
Liu‡
ab,
Jun
Yu‡
 *a,
Jinghuang
Lin‡
c,
Bin
Feng
c,
Mingzi
Sun
d,
Chen
Qiu
a,
Kun
Qian
e,
Zhichun
Si
b,
Bolong
Huang
*d,
Jean-Jacques
Delaunay
f,
Yuichi
Ikuhara
c and
Shihe
Yang
*ag
*a,
Jinghuang
Lin‡
c,
Bin
Feng
c,
Mingzi
Sun
d,
Chen
Qiu
a,
Kun
Qian
e,
Zhichun
Si
b,
Bolong
Huang
*d,
Jean-Jacques
Delaunay
f,
Yuichi
Ikuhara
c and
Shihe
Yang
*ag
aGuangdong Provincial Key Lab of Nano-Micro Material Research, School of Chemical Biology and Biotechnology, Shenzhen Graduate School, Peking University, Shenzhen, China. E-mail: yu.jun@pku.edu.cn; chsyang@pku.edu.cn
bShenzhen International Graduate School, Tsinghua University, Shenzhen, China
cInstitute of Engineering Innovation, The University of Tokyo, Tokyo, Japan
dDepartment of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong SAR, China. E-mail: bhuang@polyu.edu.hk
eDepartment of Chemistry and Biochemistry, Northern Illinois University, DeKalb, Illinois 60115, USA
fSchool of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
gInstitute of Biomedical Engineering, Shenzhen Bay Laboratory, Shenzhen 518107, China
First published on 24th June 2023
Abstract
Despite the high intrinsic electrocatalytic activity for the oxygen evolution reaction (OER), stable high valence metal ions such as Fe4+ are very difficult to form in oxide catalysts. In this work, by elaborating a novel FeHV/CeO2@NF (nickel foam) catalyst, we stabilized high-valence Fe ions on a CeO2 support and achieved a record low overpotential of 219 mV to reach the current density of 50 mA cm−2. Theoretical calculations revealed that the fluent d–f electron transfer between ultra-small FeOx nanoparticles (US-FeOx) and CeO2 guarantees the robust high valence of surface Fe sites, which enables the optimum adsorption and efficient conversions in the OER process. Meanwhile, the electronic modulations induced by the US-FeOx also improve the site-to-site electron transfer to lower the reaction energy barriers for excellent OER performance. Moreover, the FeHV/CeO2@NF catalyst delivered excellent stability, sustaining a high current density (200 mA cm−2) for over 500 h, and the simple preparation method gave access to a large-area electrode (100 cm2), paving the way for large-scale hydrogen production by water splitting.
Broader contextWater electrolysis-based hydrogen production in a sustainable fashion is one of the most promising ways to meet the global energy demand of a green future. The development of highly active and stable water oxidation catalyst is the key for large-scale application of water electrolysis because of the kinetically sluggish four-electron transfer process of oxygen evolution reaction (OER). Despite the high intrinsic electrocatalytic activity for the OER, stable high valence metal ions such as Fe4+ are very difficult to form in oxide catalysts. Herein we successfully stabilized high-valence Fe ions on Ce4+/3+O2 support and achieved superior OER performance. The novel FeHV/CeO2@NF (nickel foam) catalyst achieved a record low overpotential of 219 mV to reach the current density of 50 mA cm−2 and delivered excellent stability of sustaining a high current density (200 mA cm−2) for over 500 h. Theoretical calculations revealed that the fluent d–f electron transfer between ultra-small FeOx nanoparticles (US-FeOx) and CeO2 guarantees the robust high valence of surface Fe sites, which enables the optimum adsorption and efficient conversions in the OER process. Moreover, the simple preparation method gave access to a large-area catalyst electrode (100 cm2), paving the way for large-scale hydrogen production by water splitting. This work sets the stage for designing and preparing large-area high valence transition metal-based catalysts with high OER activity and high stability for industrial production of clean hydrogen from water splitting. |
Introduction
In order to take on the challenges of imminent energy, environmental and climatic crises caused by burning traditional fossil fuels, the development of renewable clean energy sources such as hydrogen, wind and solar energy has become a top priority agenda. Among possible solutions, water electrolysis-based hydrogen production in a sustainable fashion is the most promising one.1–3 Extensive efforts have been made to tackle the kinetically sluggish four-electron transfer process of the OER, which causes huge energy loss and limits the large-scale application of water electrolysis for hydrogen production.4–7 Many non-noble metal-based electrocatalysts have been developed to reduce the overpotential for water oxidation.8–10 Transition metal-based oxides or hydroxides have displayed good OER performance in alkaline environments yet free of the shortcomings of the scarcity and expensiveness of precious metal-based catalysts,11–13 but their catalytic activity and stability under high current operating conditions need to be further improved.It is widely known that the valence state of transition metals, e.g., Fe ions, is pivotal to the OER activity of transition-metal-based catalysts. The high-valence transition metals have been reported to be the actual OER active sites and shown high OER intrinsic activity.14–18 The Fe3+ ions in the widely applied catalysts, such as NiFe-LDH, need be oxidized to Fe4+ first under applied potentials before they can contribute to the OER process.19–21 It thus appears that direct synthesis of stable Fe4+ based catalysts should reduce the overpotential of the OER. However, given that the typical oxidation states of Fe are +2 and +3, it is very difficult to obtain such stable Fe4+-containing electrocatalysts. As far as we know, Fe4+ has only been reported in ABO3 perovskite structure catalysts; however, that synthesis process was complicated and needed a very high annealing temperature (≥800 °C).22,23
Because of the easy switch between Ce4+ and Ce3+, CeO2 has been widely used as a metal catalyst support to modulate the electronic structure through the so-called strong metal–support interaction (SMSI).24,25 This also applies to metal oxides/hydroxides on a CeO2 support, but the electronic interactions are commonly weaker and could hardly change the valence states of surface metal ions.26,27 It has been reported that the transferred charge between the surface metal and support is inversely related to the metal cluster size,25,28 the electronic interaction between the CeO2 support and metal ions could thus be enhanced by reducing the particle size, making possible the formation of stable Fe4+ on the CeO2 support.
Here, we report a stable FeOx catalyst with a stable high valence of 4+, which shows greatly reduced overpotential for the OER. This was realized by a straightforward electroplating-etching method, whereby electroplated CeO2 coating on a nickel foam (NF) was etched with ferric nitrate solution (Schematic illustration in Fig. 1(a)), installing and stabilizing the high valence Fe4+ in FeOx/CeO2. The Fe4+-containing OER electrocatalyst (named FeHV/CeO2@NF) only requires 238 mV overpotential to deliver 100 mA cm−2 current density in 1 M KOH (pH = 13.6). Also, the OER potential of FeHV/CeO2@NF can be stabilized at about 1.53 V vs. RHE (without iR compensation) to reach a high current density of 200 mA cm−2 for over 500 h. Density functional theory (DFT) calculations uncovered that the effective interfacial d–f electron transfer from the US-FeOx to CeO2 maintains the stable high valence states of Fe sites, which accounts for the high electroactivity for the OER. The optimized electronic structures guarantee the fast conversions of the intermediates on the catalyst, leading to the largely lowered overpotential required for the OER. This advanced catalyst system combined with its simple fabrication method should help to set the stage for the industrialization of sustainable OER catalysts and water-splitting systems.
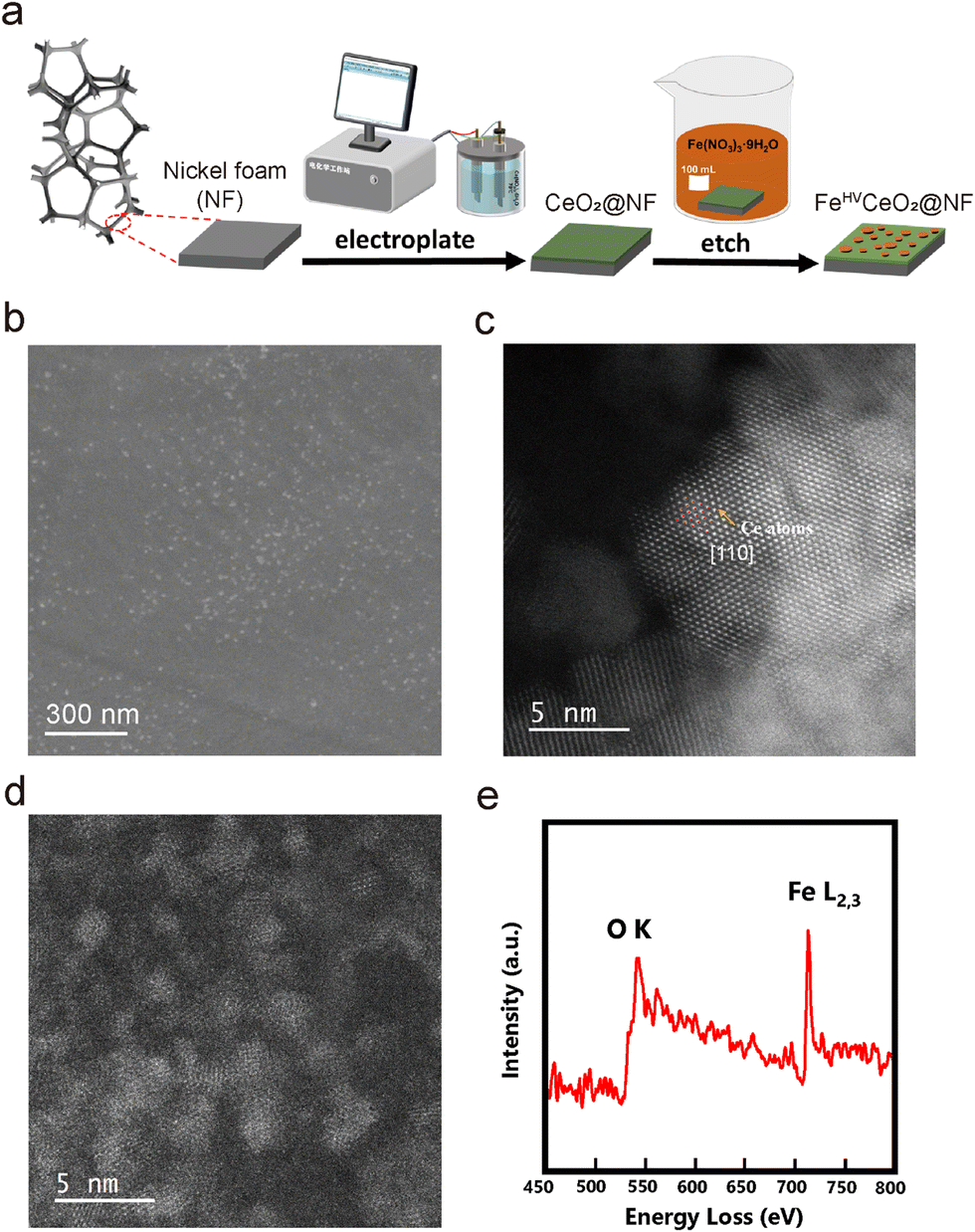 | ||
| Fig. 1 Morphology and structure characterization of the FeHV/CeO2@NF catalyst. (a) Schematic illustration of the synthesis process. (b) SEM image of FeHV/CeO2@NF. (c) and (d) HAADF-STEM image of CeO2 (c) and FeOx particles (d) in FeHV/CeO2@NF. (e) EELS (electron energy loss spectroscopy) spectrum of d. | ||
Synthesis and characterization of high-valence Fe oxide
The scanning electron microscope (SEM) morphology of the CeO2@NF sample is shown in Fig. S1 (ESI†). A uniform electroplated layer, consisting of CeO2 nanoparticles, is formed on the surface of NF. After being etched in ferric nitrate solution, CeO2@NF transforms to FeHV/CeO2@NF and the morphology is shown in Fig. 1(b). The CeO2 layer becomes flat, and there appears dense uniformly dispersed small white spots on the surface of CeO2, which are probably iron oxide nanoparticles. The scanning transmission electron microscopy (STEM)-energy dispersive X-ray spectrometry (EDS) mapping images (Fig. S2, ESI†) prove the uniform distribution of Ce and Fe in FeHV/CeO2@NF. The high-angle annular dark field (HAADF)-STEM images of FeHV/CeO2@NF are shown in Fig. 1(c) and (d). The Ce atomic columns belonging to CeO2 [110] are clearly observed (Fig. 1(c)). In general, the sizes of the FeOx nanoparticles are smaller than 5 nm (Fig. 1(d)), which is confirmed by the atomic force microscope (AFM) images (Fig. S3, ESI†). The electron energy loss spectroscopy (EELS) spectrum taken from Fig. 1(d) is shown in Fig. 1(e), and it proves that the ultra-small nanoparticles are indeed iron oxide. These iron oxide particles are detached from the CeO2 substrate, probably because of the sonication process during TEM sample fabrication. The lattice fringes of both the CeO2 support and the ultra-small FeOx particles with a size of about 2–5 nm are also observed from the HRTEM image of FeHV/CeO2@NF (Fig. S4, ESI†). For the sake of comparison, LFe/CeO2@NF (larger FeOx particles) and Fe@NF (without CeO2 layer) reference samples were also fabricated. The LFe/CeO2@NF sample was synthesized by increasing the concentration of Fe3+ in the etching solution. The morphology of the LFe/CeO2@NF catalyst is shown in Fig. S5 (ESI†), which presents larger iron oxide particles (∼100 nm) than that of the FeHV/CeO2@NF catalyst (Fig. 1(c)). As for the Fe@NF sample, what was partially etched away was the NF substrate in the acidic ferric nitrate solution accompanied by the formation of numerous holes, which is shown in Fig. S6 (ESI†).The X-ray photoelectron spectroscopy (XPS), X-ray absorption fine structure (XAFS) and soft X-ray absorption spectroscopy (SXAS) were carried out to analyze the electronic interaction between CeO2 and Fe in FeHV/CeO2@NF, and the results are shown in Fig. 2. Fig. 2(a) presents the XPS spectra for the Fe 2p region of Fe@NF, LFe/CeO2@NF and FeHV/CeO2@NF. While the peaks of Fe@NF and LFe/CeO2@NF in the range of 710–712 eV correspond to Fe3+, the peak of FeHV/CeO2@NF in the range of 712–714 eV can be assigned to Fe3+ and Fe4+.23,29 The Fe K-edge XANES spectra of the FeHV/CeO2@NF catalyst and Fe2O3 and Fe foil reference samples are shown in Fig. 2(b). Clearly, the Fe absorption edge of FeHV/CeO2@NF shifts towards higher energy (inset of Fig. 2(b)) than that of Fe2O3, which confirms a higher Fe valence state in FeHV/CeO2@NF than +3 in Fe2O3,30,31 and also is in agreement with the XPS result. Further support is from the SXAS shown in Fig. 2(c). The Fe L-edge is divided into two peaks (at 714 eV and 726 eV) corresponding to the L3 and L2 edges. Compared with standard Fe2O3, the Fe L-edge in FeHV/CeO2@NF presents a blue shift, again evidencing a higher Fe valence state than +3 in Fe2O3.32,33Fig. 2(d) shows the Ce 3d XPS spectra, and the total areas of the Ce3+ and Ce4+ peaks are in proportion to their contents.34,35 The Ce3+ content in the FeHV/CeO2@NF catalyst is calculated to be 35%, while the Ce3+ content in CeO2@NF is 31%. The above results indicate an electronic transfer between Fe and Ce in FeHV/CeO2@NF, that is, part of Fe3+ is oxidized to Fe4+ associated with the shift of Ce4+ to Ce3+.
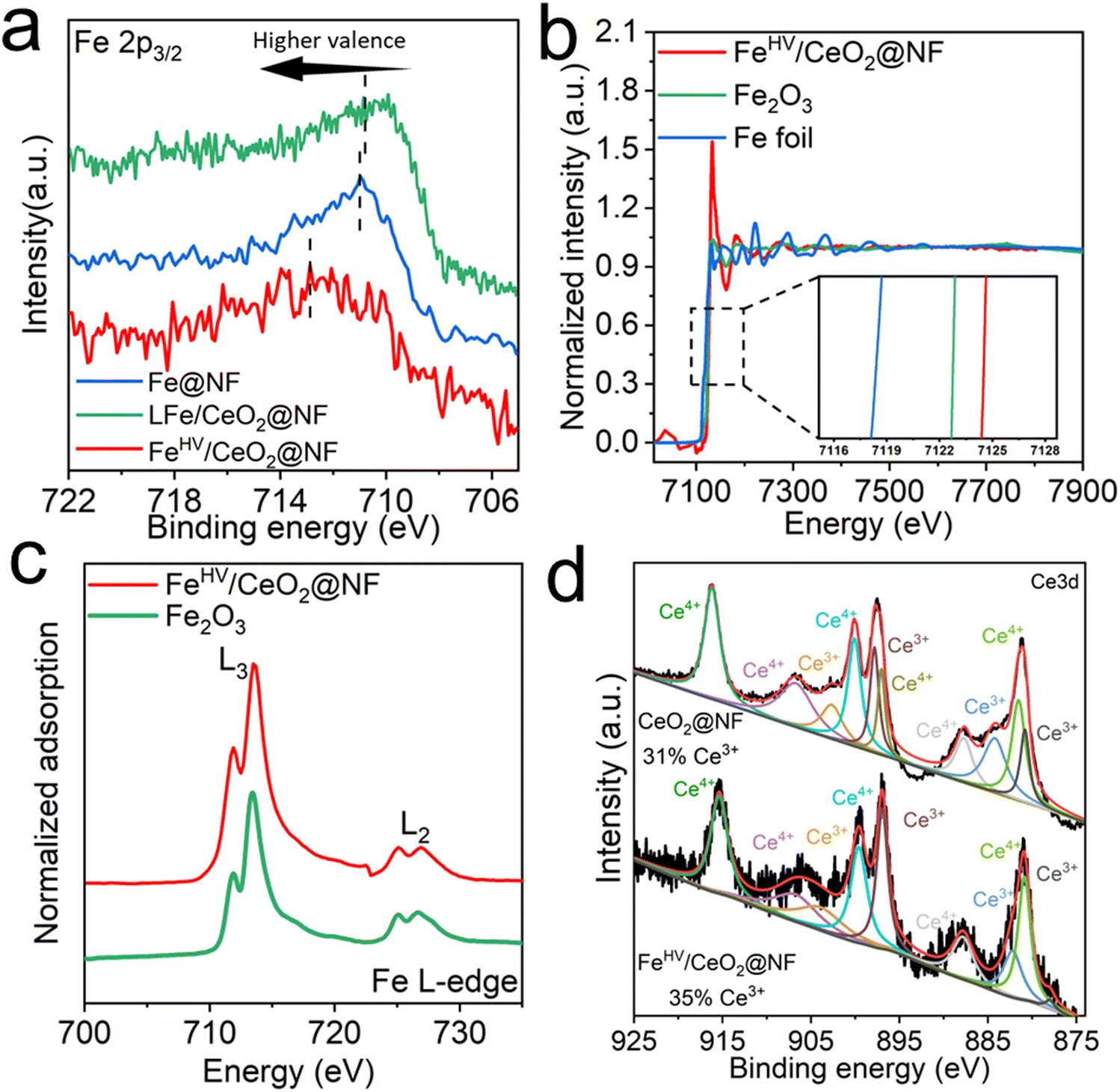 | ||
| Fig. 2 Electronic structure characterization of the catalysts. (a) Fe 2p XPS spectra of the catalysts. (b) XANES of the samples at the Fe K-edge. The inset enlarges the absorption edges and displays the valence changes of Fe in the samples. (c) Soft X-ray absorption spectra (SXAS) of the catalyst and the Fe2O3 control sample at the Fe L-edge. (d) Ce 3d XPS spectra of the catalysts. | ||
Electrochemical performance of FeHV/CeO2@NF
The electrochemical performances of the electrodes were investigated using a three-electrode system in an alkaline environment (1 M KOH, pH = 13.6). As can be seen from Fig. S8 (ESI†), the FeHV/CeO2@NF electrode displays much better OER performance than the Fe@NF, CeO2@NF LFe/CeO2@NF and RuO2@NF electrodes. The faradaic efficiency is calculated to be 98% at the current density of 30 mA cm−2 (with the overpotential as low as 210 mV). To reach the high current densities of 100 mA cm−2 and 200 mA cm−2, the required overpotentials are only 238 mV and 261 mV, respectively. Comparatively speaking, under the overpotential of 238 mV (261 mV), the current density of the FeHV/CeO2@NF electrode is 5.7(2.6), 8.1(4) and 42.5(43.4) times larger than those of the Fe@NF, LFe/CeO2@NF and CeO2@NF electrodes, respectively, as can be seen clearly in Fig. 3(a). The Nyquist plot in Fig. 3(b) shows that the semicircle radius of the FeHV/CeO2@NF electrode is much smaller than that of Fe@NF and LFe/CeO2@NF, indicating the smallest charge transfer resistance of the FeHV/CeO2@NF electrode.36,37 The Tafel plots in Fig. 3(c) show that the FeHV/CeO2@NF electrode has the smallest Tafel slope of 40 mV dec−1, which is beneficial to accelerate the reaction kinetics.38,39 The OER activity comparison between the FeHV/CeO2@NF electrode and recently reported OER electrode26,33,34,40–45 by comparing the overpotential to reach the geometric current density of 30, 50 and 100 mA cm−2 (Fig. 3(d) and Table S1, ESI†) indicates the superior OER activity of the FeHV/CeO2@NF electrode. As shown in Fig. S9 (ESI†), the OER performance of the reference sample on a Cu foam substrate illustrates the positive role of Ni foam.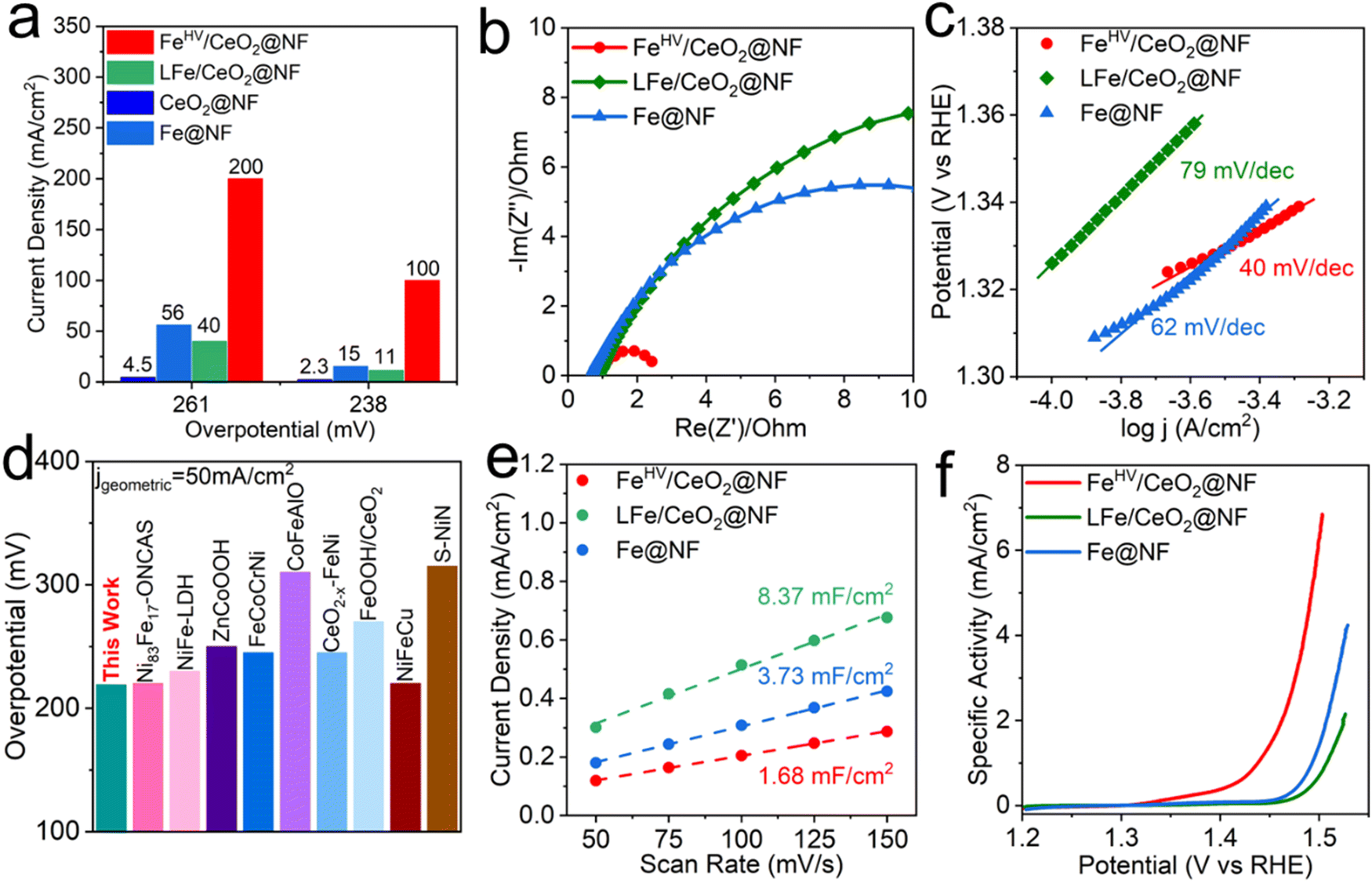 | ||
| Fig. 3 OER performance characterization of the electrodes. (a) Comparison of the current densities achieved by different electrodes at special overpotentials. (b) EIS curves, measured at frequencies from 10−1 to 105 Hz. (c) Tafel slope plots. (d) Activity comparison among OER electrocatalysts26,33,34,40–45 based on the overpotential needed to reach the geometric current density of 50 mA cm−2 (for more details see Table S1, ESI†). (e) Electrochemical double-layer capacitance. The slope of current density at OCP vs. scan rate represents the double-layer capacitance. (f) Polarization curves with the current density normalized by ECSA. | ||
In order to explore whether the high catalytic activity of the FeHV/CeO2@NF electrode comes from the high specific surface area or the high intrinsic activity of the active sites, we conducted cyclic voltammetry (CV) tests on the FeHV/CeO2@NF, LFe/CeO2@NF and Fe@NF electrodes (Fig. S10, ESI†) in the non-Faraday potential range (0.32–0.40 V vs. Hg/HgO) and calculated the electrochemical double-layer capacitance (Cdl) of the three electrodes. The ECSA was calculated according to the formula ECSA = CDL/CS, where a specific capacitance of CS = 0.040 mF cm−2 was used in this work. The calculated ECSA values are shown in Fig. 3(e) and summarized in Table S2 (ESI†), as well as other relevant electrochemistry parameters. The ECSAs of the LFe/CeO2@NF and Fe@NF electrodes are about 5 times and 2.2 times as large as that of FeHV/CeO2@NF, which demonstrates that the high OER activity of the FeHV/CeO2@NF electrode derives from the high intrinsic activity of FeHV active sites considering that the lower ECSA means an electrode has less active sites. The specific activity (jECSA) is further obtained and shown in Fig. 3(f), where it is evident that the FeHV/CeO2@NF electrode has much higher intrinsic OER activity than LFe/CeO2@NF and Fe@NF electrodes. Fig. S11 and Table S3 (ESI†) show the comparison of the specific activity between the FeHV/CeO2@NF electrode and a series of recently reported high OER performance electrodes by comparing the overpotential at an ECSA normalized current density of 1 mA cm−2. The result shows that the FeHV/CeO2@NF electrode has the best OER specific activity among these electrodes.
Formation of high-valence Fe oxide for the OER by DFT calculations
To further reveal the formation mechanism of high-valence Fe oxide (FeOx) and its contributions to the OER performances in Fe/CeO2, DFT calculations have been further introduced to investigate the electronic structures and energetic preferences. For the bonding and anti-bonding orbitals near the Fermi level (EF), the electron-rich feature of the surface US-FeOx is noted (Fig. 4(a)). The electron-rich features indicate a higher electron density of Fe/CeO2 near the EF. Since more electrons locate close to EF, the electron transfer barriers are much lower, suggesting that the electrons are easier to be transferred from the Fe/CeO2 surface in accordance with the higher electroactivity of the Fe sites. Meanwhile, the strong orbital couplings at the interface between US-FeOx and CeO2 result in favorable electronic modulations. In comparison, CeO2 only exhibits an electron-rich feature near the oxygen vacancies while the remaining sites of the surface with dominant contributions from anti-bonding orbitals cannot support the efficient adsorption and conversion of intermediates (Fig. 4(b)). The projected partial density of states (PDOS) have unraveled the electronic structure of Fe/CeO2 (Fig. 4(c)). Fe-3d orbitals have shown the eg–t2g splitting near the EF, where the overlapping with the Ce-4f orbitals supports the potential d–f electron transfer. The detailed electronic contributions of surface Ni sites have been revealed, viz., the Ni-3d orbitals mainly locate near the EF with the dominant peak at EV − 0.85 eV, thereby pulling up the overall electroactivity as well as the electron transfer rate with an improved *OH adsorption energetics and kinetics for optimum OER performance. In addition, the O-2p orbitals have exhibited evident orbital coupling with both Ce-4f and Fe-3d, indicating the small barriers for electron transfer within Fe/CeO2. In comparison, the Ce-4f orbitals are much flattened and O-2p orbitals also become less broadened in CeO2 (Fig. 4(d)). The limited overlap between O-2p and Ce-4f orbitals induces an evident barrier of electron transfer, which results in the significantly increased overpotentials of the OER.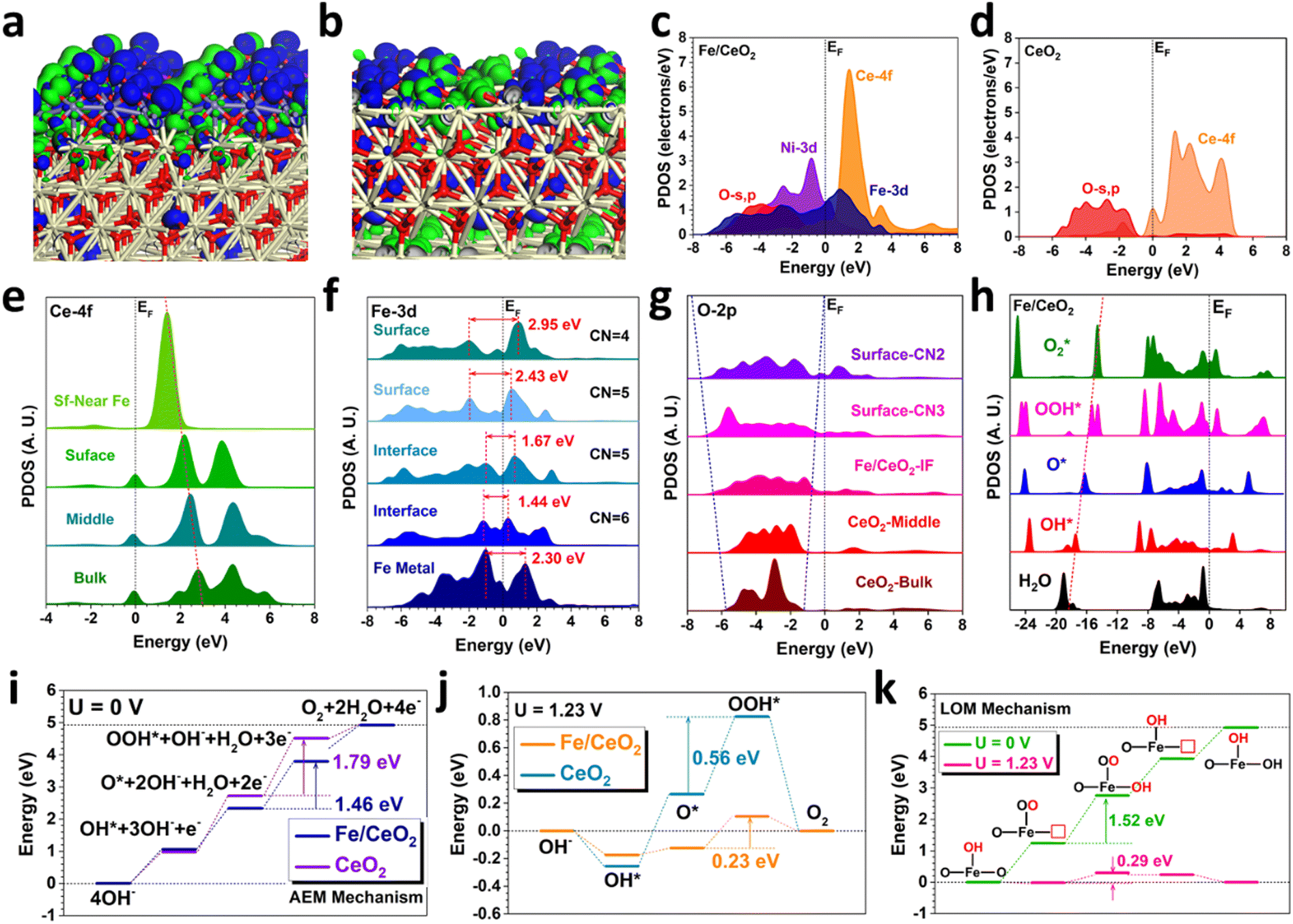 | ||
| Fig. 4 DFT calculations. (a) The 3D contour plot of electronic distribution near the Fermi level of Fe/CeO2. (b) The 3D contour plot of electronic distribution near the Fermi level of CeO2. Yellow balls = Ce, Blue balls = Fe, Purple balls = Ni, and Red balls = O. Blue isosurface = bonding orbitals, and green isosurface = anti-bonding orbitals. (c) The PDOS of Fe/CeO2. (d) The PDOS of CeO2. (e) The site-dependent PDOS of Ce-4f in Fe/CeO2. (f) The site-dependent PDOS of Fe-3d in Fe/CeO2. (g) The site-dependent PDOS of O-2p in Fe/CeO2. (h) The PDOS of key adsorbates during the OER process in Fe/CeO2. (i) The reaction energy changes of the OER process on Fe/CeO2 and CeO2 under U = 0 V through the AEM mechanism. (j) The reaction energy changes of the OER process on Fe/CeO2 and CeO2 under U = 1.23 V through the AEM mechanism. (k) The reaction energy changes of Fe/CeO2 through the LOM mechanism. | ||
In order to interpret the detailed interactions between surface US-FeOx and CeO2, the site-dependent PDOSs have been supplied to illustrate the electronic structure evolution (Fig. 4(e)). We notice a gradual shifting for the empty Ce-4f orbital towards the EF from the bulk to the surface sites near the Fe SNPs, potentially leading to efficient d–f electron transfer from Fe-3d orbitals to Ce-4f orbitals. Thus there appears to be a trend of maintaining the high oxidation states of Fe SNPs induced by the O sites, which is also confirmed by the experimental findings that the surface US-FeOx indeed exists in the high valence states. With the d–f electron transfer, the Fe-3d orbitals display essential contributions to the OER process. Specifically, the Fe sites at the interface show an alleviated eg–t2g splitting of 3d orbitals in Fe/CeO2, benefiting the fast electron transfer from Fe SNPs to CeO2 (Fig. 4(f)). Compared to the interface, however, the surface Fe sites have shown an enlarged eg–t2g splitting. The overall downshifted 3d orbitals of the surface Fe sites significantly enhance the energy band overlap with surface O sites, thus lowering the electron transfer energy costs and further improving the electroactivity of the high valence Fe sites towards the water oxidation process.46,47 Moreover, the O coordination numbers decrease with increasing eg–t2g splitting, especially for the surface Fe sites, supporting the higher electroactivity of Fe sites with high valence states. For the O sites, it is noted that the orbitals become much broadened from the bulk CeO2 to the surface, which considerably enhances their couplings with the Fe-3d orbitals to promote electroactive Fe sites towards the OER (Fig. 4(g)). More importantly, the increased electron density near the Fermi level has improved the electron transfer at the interface. In addition, the PDOSs of the key intermediates have been revealed on the Fe/CeO2 (Fig. 4(h)). Notably, the σ orbitals of O-2p in the key intermediates have shown a highly linear correlation for the OER process, which provides an efficient intermediate conversion process with efficient electron transfer. Such a correlation also supports that the surface US-FeOx are the main active sites that contribute to the superior OER performances in Fe/CeO2.
We next examine the reaction trends of the uphill OER process from the kinetic perspectives regarding both the adsorbate evolution mechanism (AEM) and lattice-oxygen-mediated mechanism (LOM). For the conventional AEM, the highest energy barrier occurs in the rate-determining step (RDS) from O* to OOH*. In comparison, Fe/CeO2 shows an energy barrier of 1.46 eV for the RDS, which is much lower than that with the CeO2 at 1.79 eV and thus guarantees the much-improved OER performance. The overpotential has been estimated by reference to the standard equilibrium potential (U = 1.23 V) (Fig. 4(j)). Although Fe/CeO2 shows an uphill trend from OH* to OOH*, the subtle energy barriers lead to a small overpotential of 0.23 V. In contrast, CeO2 shows overbinding of OH* in the initial reaction process, leading to high barriers for the following conversions to O* and OOH* with a large overpotential of 0.56 V. For the LOM mechanism (Fig. 4(k)), on the other hand, the highest energy barrier of 1.52 eV is located in the *OH adsorption step on the nearby oxygen vacancy sites of Fe. Compared to the RDS in AEM, the LOM presents a higher energy barrier. Using the standard equilibrium potential of 1.23 V as the reference, the overpotential of OER is determined to be 0.29 V, which is higher than that of the AEM. Therefore, we can conclude that the AEM is preferred over the LOM for our OER catalyst, which agrees with the experimental characterizations (Fig. S12, ESI†).
OER stability of FeHV/CeO2@NF
Besides catalytic activity, operational stability is another key parameter to evaluate the performance of a given electrocatalyst, especially for the industrial water splitting production of H2. The chronopotentiometry measurement in 1 M KOH is carried out to evaluate the long-term stability of FeHV/CeO2@NF. In the continuous stability test of 30, 50, 100 and 200 mA cm−2, the potential at every stage remains almost constant and at a high current density of 200 mA cm−2, the potential can be stabilized at about 1.53 V vs. RHE (without iR compensation) for up to 500 h (Fig. 5(a)). By comparing the linear sweep voltammetry (LSV) curves of the electrode before and after the stability test, it can be seen that after the long-term (590 h) and high-current stability test, the overpotential increases by only 16 mV at the current density of 200 mA cm−2 (Fig. 5(b)), indicating that FeHV/CeO2@NF is a super-stable electrode for water oxidation. To assess the valence stability of Fe in the FeHV/CeO2@NF electrocatalyst, XPS, SXAS and Mössbauer spectroscopy were carried out on the samples after a 10 hours OER test at the current density of 30 mA cm−2. As comparatively shown in Fig. S13–S15 (ESI†), the excellent stability of Fe4+ in the FeHV/CeO2@NF electrode is clearly evidenced. The inductively coupled plasma optical emission spectrometer (ICP-OES) is further used to measure the concentration of metal ions in the electrolyte during the water electrolysis of the FeHV/CeO2@NF electrode. As shown in Fig. 5(c), only Fe ions are detected and the content tends to be stable after the 60 h stability test. The excellent stability may derive from the fine and highly dispersed iron oxide nanoparticles, which can strongly combine with ceria and are difficult to fall off during the OER test. Therefore, the FeHV/CeO2@NF electrode behaves steadily for water oxidation. Fig. 5(d) and Table S4 (ESI†) summarize the stability comparison of recently reported efficient oxygen evolution electrodes,26,33–35,41,42,44,45,48–57 and the stability of the FeHV/CeO2@NF electrode is outstanding among them.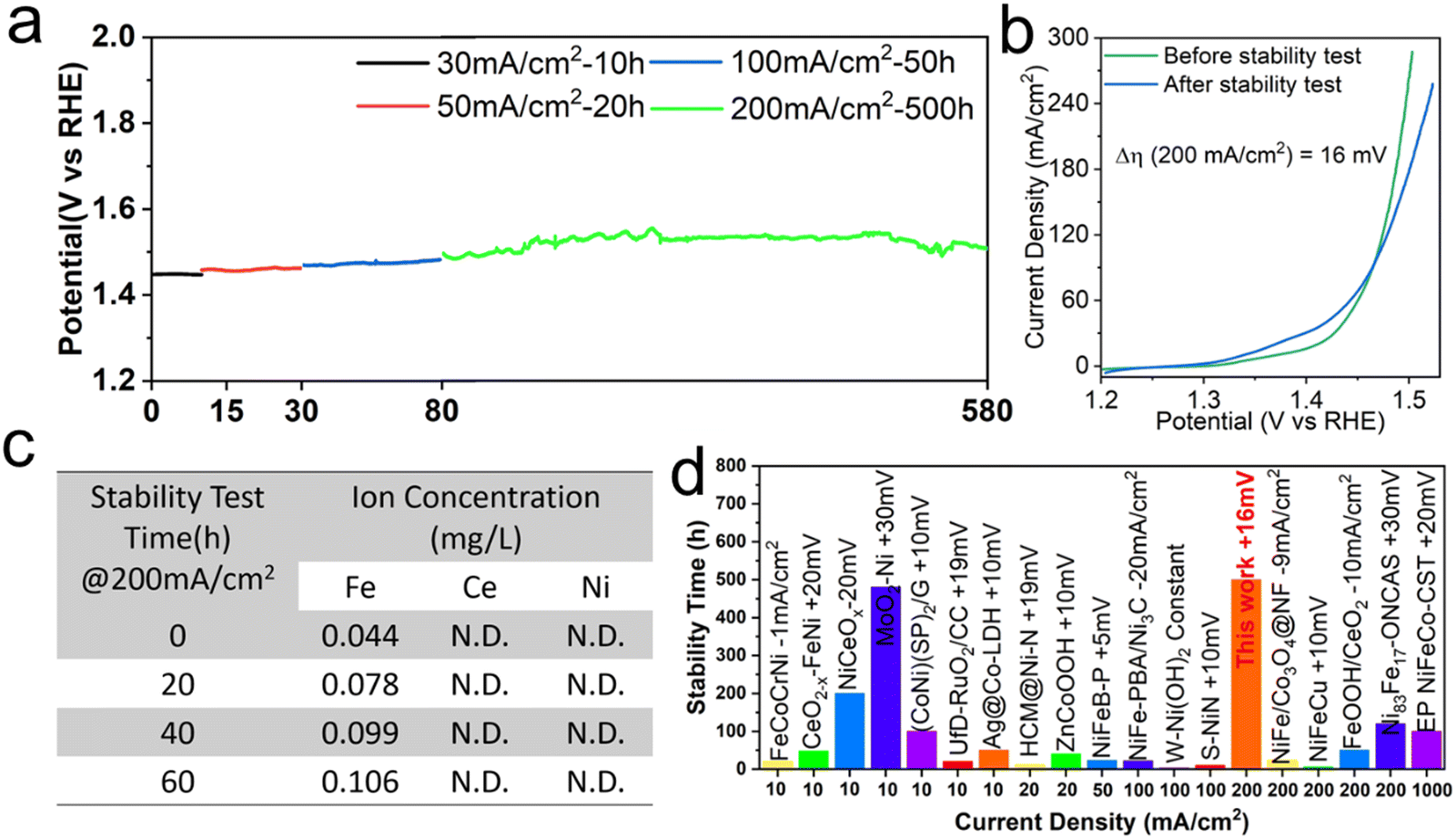 | ||
| Fig. 5 Electrochemical stability characterization of the FeHV/CeO2@NF electrode in 1 M KOH. (a) The chronopotentiometry measurements. (b) Polarization curves before and after the stability test with a scan rate of 10 mV s−1. (c) The ion concentrations of Fe, Ce, and Ni (data acquired by inductive coupled plasma emission spectroscopy) in the electrolyte during the 60 h water electrolysis process. N.D. stands for a value below the measurement limit of 0.005 mg L−1. (d) Summary of the stability comparison26,33–35,41,42,44,45,48–57 of recently reported efficient oxygen evolution electrocatalysts (details in Table S4, ESI†). The electrolyte is 1 M KOH or NaOH. | ||
Large-area fabrication and OER performance under industrial conditions
Last but not least, an industrializable water splitting electrode should be equipped with the ability to scale up its production. Currently, common electrode preparation methods in laboratories often involve complex processes and small electrode sizes, making it difficult to meet the industrial requirements for large-scale production. The electroplating-etching method presented here enables us to prepare a large-area electrode due to the simplicity of the preparation process and the scalability of the required experimental equipment, which is of great significance for the realization of industrialized hydrogen production by electrolysis of water. The large-size FeHV/CeO2@NF electrode with 10 × 10 cm2 effective area is synthesized through the electroplating-etching method, and a photograph is shown in Fig. 6(a). We selectively cut out five 1 × 1 cm2 electrodes (labeled in Fig. 6(a)) from the 10 × 10 cm2 electrode to perform the OER test, and compare their performances with the directly prepared 1 × 1 cm2 electrode (reference sample in Fig. 6(b)). The results show that the performance of these six electrodes remained consistent within a certain fluctuation range (Fig. 6(b)). This proves that the large-area electrode has good uniformity, which in turn confirms the scalability of this preparation method. There is no doubt that this is one small but important step towards the realization of industrialized hydrogen production by electrolysis of water. Finally, the industrial-level (6 M KOH, 60 °C) OER activity and stability tests of the FeHV/CeO2@NF electrode (1 cm2) were conducted. The polarization curve in Fig. S16 (ESI†) shows that the current density can reach 500 mA cm−2 at the overpotential of 270 mV (without iR compensation). As shown in Fig. 6(c), the electrode of FeHV/CeO2@NF could deliver large current densities of 200 mA cm−2 and 300 mA cm−2 at only 1.44 V and 1.47 V vs. RHE (without iR compensation) and remain stable up to 300 h in such a harsh environment, further indicating the potential industrial application.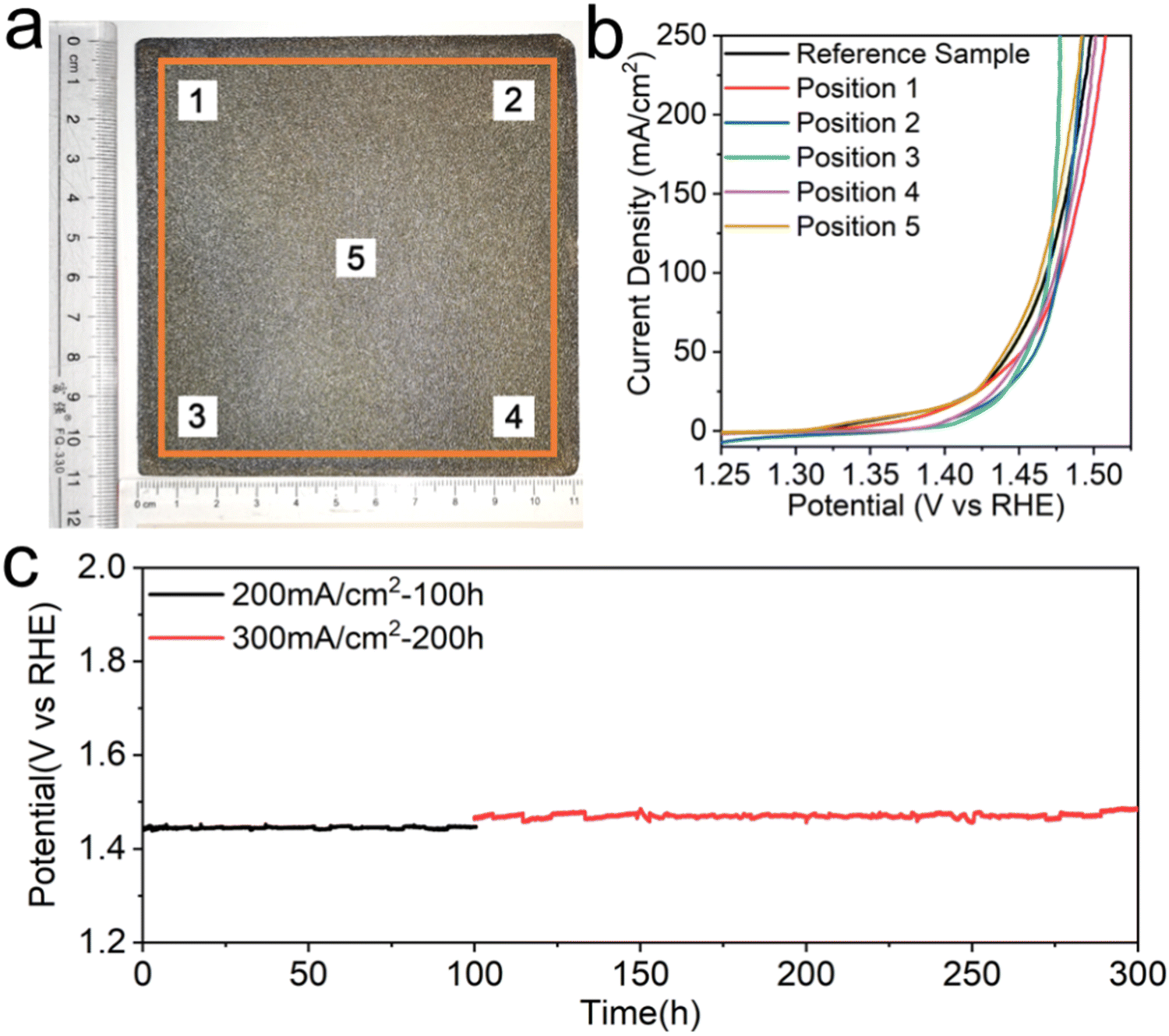 | ||
| Fig. 6 Industrial application of the FeHV/CeO2@NF electrode. (a) Photograph of the large-area (100 cm2) FeHV/CeO2@NF electrode. (b) Polarization curves of the five different positions (1 cm2) in the large-area FeHV/CeO2@NF electrode (the reference sample is directly synthesized with 1 cm2). (c) The chronopotentiometry measurements in 6 M KOH and at 60 °C. | ||
Conclusions
We have demonstrated an efficient and ultra-stable high valence Fe4+-containing OER electrode (named FeHV/CeO2@NF) and its scalable preparation (100 cm2 reported here) by a simple electroplating-etching method. The ultra-small size of the FeOx nanoparticles on the CeO2 support enabled the formation of Fe4+, which has shown high intrinsic activity. Remarkably, the FeHV/CeO2@NF electrode could deliver large current densities of 200 mA cm−2 and 300 mA cm−2 at only 1.44 V and 1.47 V vs. RHE (without iR compensation) and remain stable up to 300 h under harsh industrial conditions (6 M KOH, 60 °C). DFT calculations unraveled the electronic modulations induced by the d–f electron transfer at the interface of Fe/CeO2, which held the US-FeOx in a stabilized high valence Fe4+ state conducive to the OER process. This work provides a guide to designing and preparing large-area high valence transition metal-based electrodes with high OER activity and high stability for industrial production of clean hydrogen from water splitting.Author contributions
Shihe Yang and Jun Yu conceived and supervised the project. Hongzhi Liu conducted the experiments. Mingzi Sun performed the computational study under the supervision of Bolong Huang. Jinghuang Lin performed the STEM analysis with Bin Feng and Yuichi Ikuhara. Kun Qian performed the XAFS characterization. Hongzhi Liu, Jun Yu, Mingzi Sun, Bolong Huang and Shihe Yang wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflicts.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21972006, 22261160370, U2001217), Shenzhen Science and Technology Innovation Commission (KCXFZ20201221173604012), Shenzhen Peacock plan (KQTD2016053015544057), Guangdong Basic and Applied Basic Research Foundation (2021B1515120079), the National Natural Science Foundation of China/Research Grant Council of Hong Kong Joint Research Scheme (N_PolyU502/21), the funding for Projects of Strategic Importance of The Hong Kong Polytechnic University (Project Code: 1-ZE2V), Departmental General Research Fund (Project Code: ZVUL), Shenzhen Fundamental Research Scheme-General Program (JCYJ20220531090807017) and Shenzhen Innovation Fund (JCYJ20220818101018038). A part of this work was supported by “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT). J. L. was supported as a Japan Society for the Promotion of Science (JSPS) fellowship for part of this work. The authors would like to thank NSRL for the synchrotron beamtime. B. H. also thanks the support from Research Centre for Carbon-Strategic Catalysis of The Hong Kong Polytechnic University.References
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 1–13 CrossRef.
- Z. Wang, T. Hisatomi, R. Li, K. Sayama, G. Liu, K. Domen, C. Li and L. Wang, Joule, 2021, 5, 344–359 CrossRef CAS.
- Z. Zhou, Y. Kong, H. Tan, Q. Huang, C. Wang, Z. Pei, H. Wang, Y. Liu, Y. Wang, S. Li, X. Liao, W. Yan and S. Zhao, Adv. Mater., 2022, 34, 2106541 CrossRef CAS PubMed.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- M. Tahir, L. Pan, F. Idress, X. Zhang, L. Wang, J. J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- J. Yu, X. Du, H. Liu, C. Qiu, R. Yu, S. Li, J. Ren and S. Yang, Energy Fuels, 2021, 35, 19000–19011 CrossRef CAS.
- Y. Liu, Y. Wang, S. Zhao and Z. Tang, Small Methods, 2022, 6, 2200773 CrossRef CAS PubMed.
- Y. Yan, S. Liang, X. Wang, M. Zhang, S. M. Hao, X. Cui, Z. Li and Z. Lin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2110036118 CrossRef CAS PubMed.
- T. Wang, Y. He, Y. Liu, F. Guo, X. Li, H. Chen, H. Li and Z. Lin, Nano Energy, 2021, 79, 105487 CrossRef CAS.
- X. Cui, S. Lei, A. C. Wang, L. Gao, Q. Zhang, Y. Yang and Z. Lin, Nano Energy, 2020, 70, 104525 CrossRef CAS.
- H. Sun, Z. Yan, F. Liu, W. Xu, F. Cheng and J. Chen, Adv. Mater., 2020, 32, 1806326 CrossRef CAS PubMed.
- D. Y. Chung, P. P. Lopes, P. F. B. D. Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik, Y. Zhu, S. Seifert, S. Lee, V. R. Stamenkovic and N. M. Markovic, Nat. Energy, 2020, 5, 222–230 CrossRef.
- J. Kang, X. Qiu, Q. Hu, J. Zhong, X. Gao, R. Huang, C. Wan, L. M. Liu, X. Duan and L. Guo, Nat. Catal., 2021, 4, 1050–1058 CrossRef CAS.
- J. Huang, H. Sheng, R. D. Ross, J. Han, X. Wang, B. Song and S. Jin, Nat. Commun., 2021, 12, 3036 CrossRef CAS PubMed.
- J. Wang, S. J. Kim, J. Liu, Y. Gao, S. Choi, J. Han, H. Shin, S. Jo., J. Kim, F. Ciucci, H. Kim, Q. Li, W. Yang, X. Long, S. Yang, S. P. Cho, K. H. Chae, M. G. Kim, H. Kim and J. Lim, Nat. Catal., 2021, 4, 212–222 CrossRef CAS.
- L. Gao, X. Cui, C. D. Sewell, J. Li and Z. Lin, Chem. Soc. Rev., 2021, 50, 8428 RSC.
- S. Zhao, Y. Yang and Z. Tang, Angew. Chem., Int. Ed., 2022, 61, e202110186 CrossRef CAS PubMed.
- S. Zhao, C. Tan, C. T. He, P. An, F. Xie, S. Jiang, Y. Zhu, K. H. Wu, B. Zhang, H. Li, J. Zhang, Y. Chen, S. Liu, J. Dong and Z. Tang, Nat. Energy, 2020, 5, 881–890 CrossRef CAS.
- B. M. Hunter, N. B. Thompson, A. M. Müller, G. R. Rossman, M. G. Hill, J. R. Winkler and H. B. Gray, Joule, 2018, 2, 747–763 CrossRef CAS.
- A. Elabd and A. Coskun, Chem, 2018, 4, 661–670 CAS.
- J. Y. C. Chen, L. Dang, H. Liang, W. Bi, J. B. Gerken, S. Jin, E. E. Alp and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 15090–15093 CrossRef CAS PubMed.
- Z. Shen, Y. Zhuang, W. Li, X. Huang, F. E. Oropeza, E. J. M. Hensen, J. P. Hofmann, M. Cui, A. Tadich, D. Qi, J. Cheng, J. Li and K. H. L. Zhang, J. Mater. Chem. A, 2020, 8, 4407–4415 RSC.
- S. She, J. Yu, W. Tang, Y. Zhu, Y. Chen, J. Sunarso, W. Zhou and Z. Shao, ACS Appl. Mater. Interfaces, 2018, 10, 11715–11721 CrossRef CAS PubMed.
- C. Sun, H. Li and L. Chen., Energy Environ. Sci., 2012, 5, 8475–8505 RSC.
- Q. Fu and T. Wagner, Surf. Sci. Rep., 2007, 62, 431–498 CrossRef CAS.
- J. X. Feng, S. H. Ye, H. Xu, Y. X. Tong and G. R. Li, Adv. Mater., 2016, 28, 4698–4703 CrossRef CAS PubMed.
- W. Yang, X. Wang, S. Song and H. Zhang, Chem, 2019, 5, 1743–1774 CAS.
- Y. Li, Y. Zhang, K. Qian and W. Huang, ACS Catal., 2022, 12, 1268–1287 CrossRef CAS.
- Y. Zhu, W. Zhou, J. Yu, Y. Chen, M. Liu and Z. Shao, Chem. Mater., 2016, 28, 1691–1697 CrossRef CAS.
- S. Erat, A. Braun, A. Ovalle, C. Piamonteze, Z. Liu, T. Graule and L. J. Gauckler, Appl. Phys. Lett., 2009, 95, 174108 CrossRef.
- T. Tsuyama, T. Matsuda, S. Chakraverty, J. Okamoto, E. Ikenaga, A. Tanaka, T. Mizokawa, H. Y. Hwang, Y. Tokura and H. Wadati, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 115101 CrossRef.
- J. Jiang, F. Sun, S. Zhou, W. Hu, H. Zhang, J. Dong, Z. Jiang, J. Zhao, J. Li, W. Yan and M. Wang, Nat. Commun., 2018, 9, 2885 CrossRef PubMed.
- N. Zhang, X. Feng, D. Rao, X. Deng, L. Cai, B. Qiu, R. Long, Y. Xiong, Y. Lu and Y. Chai, Nat. Commun., 2020, 11, 4066 CrossRef CAS PubMed.
- J. Yu, J. Wang, X. Long, L. Chen, Q. Cao, J. Wang, C. Qiu, J. Lim and S. Yang, Adv. Energy Mater., 2021, 11, 2002731 CrossRef CAS.
- J. Yu, Q. Cao, Y. Li, X. Long, S. Yang, J. K. Clark, M. Nakabayashi, N. Shibata and J. J. Delaunay, ACS Catal., 2019, 9, 1605–1611 CrossRef CAS.
- J. Yu, Q. Cao, B. Feng, C. Li, J. Liu, J. K. Clark and J. J. Delaunay, Nano Res., 2018, 11, 4323–4332 CrossRef CAS.
- J. Yu, Z. Wang, J. Wang, W. Zhong, M. Ju, R. Cai, L. Chen, X. Long and S. Yang, ChemSusChem, 2020, 13, 5273–5279 CrossRef CAS PubMed.
- H. Liu, J. Yu, C. Qiu, R. Yu, S. Li, J. Cheng, J. Wang, Z. Si and S. Yang, Funct. Mater. Lett., 2021, 14, 2130009 CrossRef CAS.
- X. Cui, L. Gao, S. Lei, S. Liang, J. Zhang, C. D. Sewell, W. Xue, Q. Liu, Z. Lin and Y. Yang, Adv. Funct. Mater., 2021, 31, 2009197 CrossRef CAS.
- P. Zhang, L. Li, D. Nordlund, H. Chen, L. Fan, B. Zhang, X. Sheng, Q. Daniel and L. Sun, Nat. Commun., 2018, 9, 381 CrossRef PubMed.
- P. Liu, B. Chen, C. Liang, W. Yao, Y. Cui, S. Hu, P. Zou, H. Zhang, H. J. Fan and C. Yang, Adv. Mater., 2021, 33, 2007377 CrossRef CAS PubMed.
- Z. Qiu, C. W. Tai, G. A. Niklasson and T. Edvinsson, Energy Environ. Sci., 2019, 12, 572 RSC.
- Z. F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 CrossRef CAS.
- T. Wu, S. Sun, J. Song, S. Xi, Y. Du, B. Chen, W. A. Sasangka, H. Liao, C. L. Gan, G. G. Scherer, L. Zeng, H. Wang, H. Li, A. Grimaud and Z. J. Xu, Nat. Catal., 2019, 2, 763–772 CrossRef CAS.
- Y. Hou, M. Qiu, M. G. Kim, P. Liu, G. Nam, T. Zhang, X. Zhuang, B. Yang, J. Cho, M. Chen, C. Yuan, L. Lei and X. Feng, Nat. Commun., 2019, 10, 1392 CrossRef PubMed.
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Science, 2017, 358, 751–756 CrossRef CAS PubMed.
- S. Yagi, I. Yamada, H. Tsukasaki, A. Seno, M. Murakami, H. Fujii, H. Chen, N. Umezawa, H. Abe, N. Nishiyama and S. Mori, Nat. Commun., 2015, 6, 8249 CrossRef PubMed.
- J. Yan, L. Kong, Y. Ji, J. White, Y. Li, J. Zhang, P. An, S. Liu, S. T. Lee and T. Ma, Nat. Commun., 2019, 10, 2149 CrossRef PubMed.
- X. Liu, J. Meng, K. Ni, R. Guo, F. Xia, J. Xie, X. Li, B. Wen, P. Wu, M. Li, J. Wu, X. Wu, L. Mai and D. Zhao, Cell Rep. Phys. Sci., 2020, 1, 100241 CrossRef.
- H. J. Song, H. Yoon, B. Ju, G. H. Lee and D. W. Kim, Adv. Energy Mater., 2018, 8, 1802319 CrossRef.
- R. Ge, L. Li, J. Su, Y. Lin, Z. Tian and L. Chen, Adv. Energy Mater., 2019, 9, 1901313 CrossRef.
- M. Zhang, Y. Zhang, L. Ye, B. Guo and Y. Gong, Appl. Catal., B, 2021, 298, 120601 CrossRef CAS.
- H. Zhang, Y. Liu, T. Chen, J. Zhang, J. Zhang and X. W. D. Lou, Adv. Mater., 2019, 31, 1904548 CrossRef CAS PubMed.
- Y. Kang, Y. Guo, J. Zhao, B. Jiang, J. Guo, Y. Tang, H. Li, V. Malgras, M. A. Amin, H. Nara, Y. Sugahara, Y. Yamauchi and T. Asahi, Small, 2022, 18, 2203411 CrossRef CAS PubMed.
- X. Lin, S. Cao, H. Chen, X. Chen, Z. Wang, S. Zhou, H. Xu, S. Liu, S. Wei and X. Lu, Chem. Eng. J., 2022, 433, 133524 CrossRef CAS.
- J. Lv, L. Wang, R. Li, K. Zhang, D. Zhao, Y. Li, X. Li, X. Huang and G. Wang, ACS Catal., 2021, 11, 14338–14351 CrossRef CAS.
- J. Mo, Y. Ko, Y. S. Yun, J. Huh and J. Cho, Energy Environ. Sci., 2022, 15, 3815–3829 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00113j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |