
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFull-spectrum utilization of solar energy for simultaneous CO2 reduction and seawater desalination†
Yuting
Yin‡
 ,
Wenhao
Jing‡
,
Haoran
Qiu‡
,
Feng
Wang
,
Ya
Liu
* and
Liejin
Guo
*
,
Wenhao
Jing‡
,
Haoran
Qiu‡
,
Feng
Wang
,
Ya
Liu
* and
Liejin
Guo
*
International Research Center for Renewable Energy, State Key Laboratory of Multiphase Flow in Power Engineering, Xi’an Jiaotong University, Shaanxi, 710049, China. E-mail: yaliu0112@mail.xjtu.edu.cn; lj-guo@mail.xjtu.edu.cn
First published on 14th July 2023
Abstract
Photocatalytic CO2 reduction, as a highly integrated solar fuel generation technology, cannot efficiently utilize infrared light, resulting in a severe waste of solar energy. Given the fact that bifunctional polyaniline can conduct photocatalysis and promote water evaporation at the same time, integrating the advantages of a gas diffusion layer in mass transfer, we proposed a novel layout of the reacting system. In this constructed three-phase interface, polyaniline exhibited high activity for CO2 reduction, rather than hydrogen evolution. Through quasi-in situ Raman spectroscopy, we found that the introduced copper–cobalt composite oxides would be reduced to cuprous oxide/tricobalt tetroxide once photocatalysis starts. The highest achieved selectivity of ethanol-based C2+ products was >90%. The presence of a greater number of asymmetric adsorption sites between copper and cobalt facilitated C–C coupling, further proved by the density functional theory analysis. For water evaporation, the achieved energy conversion efficiency of solar to vapor was 71.0%. The evaporation was activated via the unabsorbed light by the photocatalyst and the unused light that had been absorbed. The vapor flux was driven across the hydrophobic gas diffusion layer into the flow in the backplate by the temperature gradient between each side of the gas diffusion layer. Subsequently, desalted water was obtained by condensation.
Broader contextDirect solar-driven CO2 reduction to high-value hydrocarbon fuels under mild conditions has been considered a promising technology to tackle carbon neutrality and the energy crisis. However, in photocatalysis, infrared light is usually underutilized in current studies, resulting in serious energy waste. As per our knowledge, this is the first report on comprehensive solar utilization using full-spectrum sunlight to generate multi-carbon products and freshwater simultaneously, optimizing the overall energy conversion process. The self-designed and fabricated bifunctional system embodied the advantages of the gas diffusion layer in mass transfer, which is also rarely seen in previous photocatalytic systems. Additionally, this work provides an in-depth insight into the mechanism of both reactions and their synergistic effect using various characterization methods along with density functional theory analysis. Facilitated C–C coupling was attributed to the asymmetric adsorption sites between copper and cobalt. The key to high solar-to-vapor efficiency was the undamaged amino groups in polyaniline. The process of water evaporation kept water molecules away from the functioning catalytic surface, thus reducing the mass transfer resistance of CO2 and improving the hydrophobicity to avoid the competing reaction of hydrogen evolution. This work is inspiring for the comprehensive and full utilization of solar energy. |
Introduction
Using solar energy to produce hydrocarbon fuels by reducing CO2 is considered to be the ultimate solution to address the energy crisis and decarbonization.1 Since the first discoveries of photocatalytic fuel synthesis in 19722 (Fujishima and Honda's work) and 19783 (M. Halmann's work), this approach has been considered promising in energy and environmental engineering. Unlike photocatalytic reactions, solar water evaporation can use the full-spectrum of energy to produce fresh water.4 The demand for fuels and fresh water exists at the same time in many scenarios, such as ocean voyages, space operations, and Mars exploration. Therefore, it is necessary to build an integrated device that can produce fuels and fresh water at the same time, meanwhile optimizing the use of solar energy. Ultraviolet and visible (UV-vis) light drives solar fuel generation, while infrared light and the unused part of UV-vis light drive water evaporation.There have been several research studies considering comprehensive solar utilization,5,6 some of which are synergistic fuel and fresh water production. In the work of Minmin Gao et al., a H2O–H2 cogeneration system prototype demonstrated ≈4600 μmol m−2 hydrogen production and ≈5000 g m−2 fresh water production in a day under natural light.7 Lihua Han et al. achieved an overall fresh water collection rate of 1.45 kg m−2 h−1 and 21.29 μmol gcat−1 h−1 CO generation rate from CO2 photoreduction in a new double-stage solar catalytic membrane distillation system.8 Hao Liu et al. carried out efficient simultaneous enhanced solar steam generation and CO2 reduction, the highest vapor and CO production rates of which were 3.67 kg m−2 h−1 and 62.55 μmol gcat−1 h−1.9 However, these works were more biased towards seawater desalination in terms of reacting system design. No multi-carbon products were generated. The common configurations of the photocatalytic system nowadays include solid–liquid suspensions, solid–liquid plates, and solid–gas systems,10 in which the mass transfer of reactants is often an obstacle to improving the catalytic performance. In this work, illuminated by the construction of the gas diffusion electrode, the gas diffusion reaction system was designed to enhance the mass transfer of reactants and promote the reaction at the three-phase interface in the gas diffusion layer (GDL).11,12 CO2 was transferred to the reacting interface via the channels of the hydrophobic substrate, instead of the slow diffusion through the liquid phase, which was believed to increase the concentration and further improve the reaction kinetics.13 Besides, the hydrogen evolution reaction was prohibited due to the lack of excess protons (H+) at the hydrophobic catalytic interface macroscopically and microscopically. Notably, in this system, the selectivity to multi-carbon products was higher and no H2 was evolved.
At present, photocatalysts commonly used for photocatalytic CO2 reduction (CO2R) include oxides,14,15 sulfides,16–18 nitrides,19–21etc. Hydrogels and aerogels in 3D porous structures have been researched recently as they possess the ability of modifying the reaction site.22–26 In the meantime, hydrogels with hydrophilic functional groups (–OH, –NH2, –CONH–, –COOH, and –SO3H) are often used in solar vapor generation as they can trap water molecules via hydrogen bonds or electrostatic forces to alter phase change behaviors.27 Polyaniline (PANI) hydrogel can be used as the photocatalyst for CO2R because of its three-dimensional cross-linked structure that facilitates the diffusion of reactants, good visible light response, and good electrical conductivity. At the same time, there are –NH– groups on PANI, which can promote the gasification of water molecules, and thus can purify water while reducing CO2,28 improving the full-spectrum utilization of solar energy. For choosing a cocatalyst, previous investigations have shown that oxygen-bearing copper (OBCu) with densely distributed grain boundaries tends to favor C–C coupling.29,30 In addition, a greater number of asymmetry adsorption sites are often conducive to C–C coupling, leading to the loading of oxygen-bearing cobalt (OBCo).31–33
Inspired by the above, we proposed to conduct CO2R and seawater desalination simultaneously in a self-made gas diffusion system to achieve full-spectrum utilization of solar energy. The PANI hydrogel is prepared in a facile way and further loaded with OBCu–OBCo to improve the solar-to-fuel (STF) efficiency and selectivity to multi-carbon products. In this work, we found out the best layout of the photocatalysts to achieve optimal CO2R and water purification performance. Quasi-in situ Raman spectroscopy and density functional theory (DFT) calculations were carried out to reveal the process of the photocatalytic reaction. The behavior of changed solar to vapor (STV) efficiency was analyzed through multiple characterization methods and calculations as well.
Experimental
Material synthesis
PANI hydrogel was synthesized by facile polymerization of aniline monomer with the help of phytic acid and ammonium persulfate at a low temperature (4 °C). Powder-like PANI aerogel was obtained by performing freeze-drying. OBCu was loaded onto PANI through the chemical reduction of copper(II) acetate monohydrate using sodium borohydride as the reducing agent in an ice bath. OBCo was loaded onto PANI through the deposition of cobalt nitrate hexahydrate under light irradiation while stirring. Further purification of all the photocatalysts for performance tests was conducted by washing with deionized water and freeze-drying several times. Powder-like photocatalysts were sprayed onto carbon paper to fabricate the GDL used in the bifunctional reacting system.Characterization
Surface morphology characterization was performed using an FEI Tecnai G2 F30 transmission electron microscope (TEM) and a JEOL JSM-7800F field emission scanning electron microscope (FE-SEM), in which energy-dispersive X-ray spectroscopy (EDS) analysis was performed using the installed energy-dispersive X-ray detector, OXFORD INCA. The crystal structure analysis was performed using an X-ray diffractometer (XRD), PANalytical X’pert MPD Pro. X-ray photoelectron spectroscopy (XPS) measurements were carried out using a Kratos spectrometer (Axis UltraDLD) with monochromatic Al Kα radiation (hν = 1486.69 eV). The C 1s signal centered at 284.8 eV, and adventitious carbon was used to calibrate the binding energies. The reflectance and transmittance spectra were obtained by ultraviolet-visible spectroscopy (UV-vis) using a double-beam UV4100 UV-vis-NIR spectrophotometer with an integrating sphere detector. The optical bandgap of the semiconductor was calculated by the Tauc plot method using the data of the diffuse reflection spectrum.34 Photoluminescence (PL) spectra were obtained using a PTI QM-4 fluorescence spectrophotometer. Contact angle measurement was carried out on DataPhysics instruments GmbH DCAT 25. Fourier-transform infrared spectra were obtained using a Bruker Vertex 70 FTIR spectrometer using the KBr pellet technique. Infrared images were taken with an infrared thermometer (Fisher Scientific). Quasi-in situ Raman characterization was conducted using a HORIBA LABRAM Soleil high-resolution ultra-sensitive intelligent Raman system. The photocatalysts were sprayed onto carbon paper. Saturated K2CO3 (aq.) acted as the source of the proton. CO2 flowed through the backplate. The structure resembles the one in the activity test. 13C isotopic trace was detected on an Agilent 8860-5977B gas chromatography-mass spectrometer. A NexION 350D, PerkinElmer (PE) Inductively Coupled Plasma Mass Spectrometer (ICP–MS) system was used to determine the concentration of various metal cations (Ca2+, Mg2+, Na+, and K+) in aqueous samples.DFT calculations
All the DFT calculations were carried out with a periodic slab model using the Vienna ab initio simulation program (VASP) with DFT-D3 empirical van der Waals attraction corrections.35–37 The projector augmented-wave (PAW) pseudopotential method was used to describe the nucleus−electron interactions.38 The generalized gradient approximation was used with the PBE exchange–correlation functional. The description of the electron–ion interactions was supported by the projector-augmented wave method. The cut-off energy for the plane-wave basis set was 500 eV. A 3 × 3 × 1 Monkhorst–Pack k-point mesh was used for Brillouin zone integration. The coverage criteria for energies and forces were set to 10−5 eV and 0.02 eV Å−1. The first order Methfessel–Paxton method with a smearing width of 0.1 eV was used. All energies were corrected by the implicit solvent method. Isolated molecules were calculated in boxes of 9 Å × 10 Å × 11 Å using the gamma point with a smearing width of 0.01 eV.Results and discussion
The layout of the bifunctional reacting system
Photocatalysis and seawater desalination were carried out in a self-made gas diffusion system, whose assembly is shown in Fig. 1a and Fig. S1 (ESI†). The reactor consists of one reacting chamber, carbon paper coated with photocatalysts (GDL), and a flow plate. The detailed energy and mass flows in the GDL are exhibited in Fig. 1b. In a typical experiment, once under irradiation, the water body will absorb part of the infrared light to be pre-heated. Then, part of UV-vis light will be absorbed by bifunctional photocatalysts to conduct CO2 reduction occurring at copper–cobalt composite oxides. The unused part of UV-vis and absorbed near-infrared (NIR) light will simultaneously drive water evaporation (detailed deductions considering the energy flow exhibited in the ESI†). In the process, CO2 gas continues to flow through the backplate and diffuse into the saline solution. The pressure difference caused by the temperature gradient between each side of the GDL is the driving force of the vapor flux across the hydrophobic GDL into the CO2 flow.39,40 The outlet gas channel is connected to condensing equipment, in which the purified water vapor is collected in an ice water bath.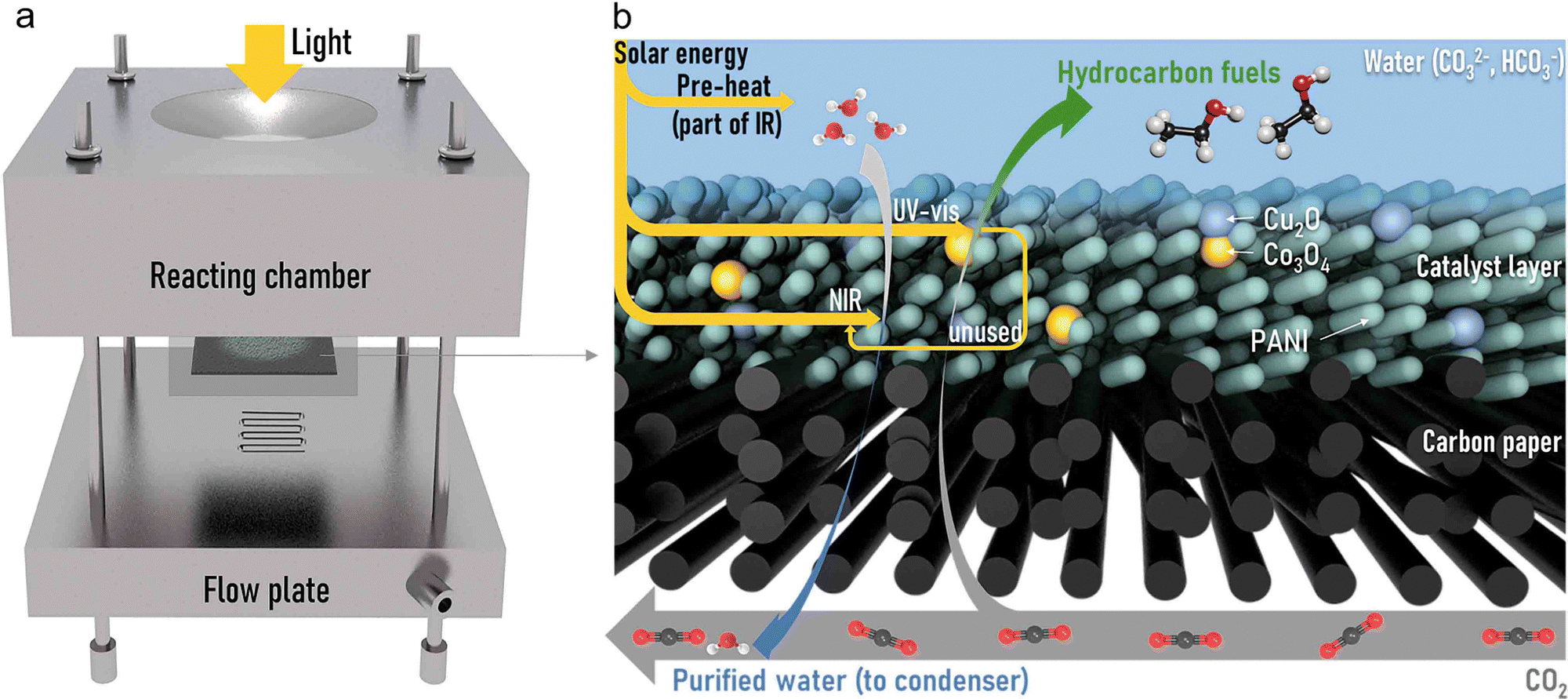 | ||
| Fig. 1 (a) The assembly of the bifunctional gas diffusion system. (b) Energy and mass flow in the GDL. | ||
Structure of the bifunctional hybrid light absorber
PANI-based photocatalysts loaded on carbon paper serve as the bifunctional hybrid light absorber in this work. To illustrate the working principle of this absorber, we must first study its fine composition and structure.
Fig. 2a and Fig. S2 (ESI†) show the typical SEM image of OBCu–OBCo–PANI loaded on carbon paper from the cross-section and front view. OBCu–OBCo–PANI evenly covers the surface of the carbon paper with an approximate thickness of 40 μm. We can observe that OBCu–OBCo–PANI possesses a granular structure with an axial length of nearly 70 nm as well as a large porosity overall as shown in Fig. 2b. Besides, according to Fig. S4 (ESI†), the morphology did not change after the reaction. The element mapping in the insets of Fig. S5 (ESI†) confirms the successful loading of OBCu and OBCo onto PANI. As in Fig. 2c and d, metal oxides are loaded on PANI in the form of island-like distribution; the interplanar spacing measurement reveals typical pitch values of 3.73, 2.46, and 1.96 Å, corresponding to the (008) crystal plane of CoO2, the (111) crystal plane of Cu2O, and the (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 12) crystal plane of CuO respectively. The TEM SAED patterns in Fig. S3 (ESI†) also confirm the existence of these primary planes.
12) crystal plane of CuO respectively. The TEM SAED patterns in Fig. S3 (ESI†) also confirm the existence of these primary planes.
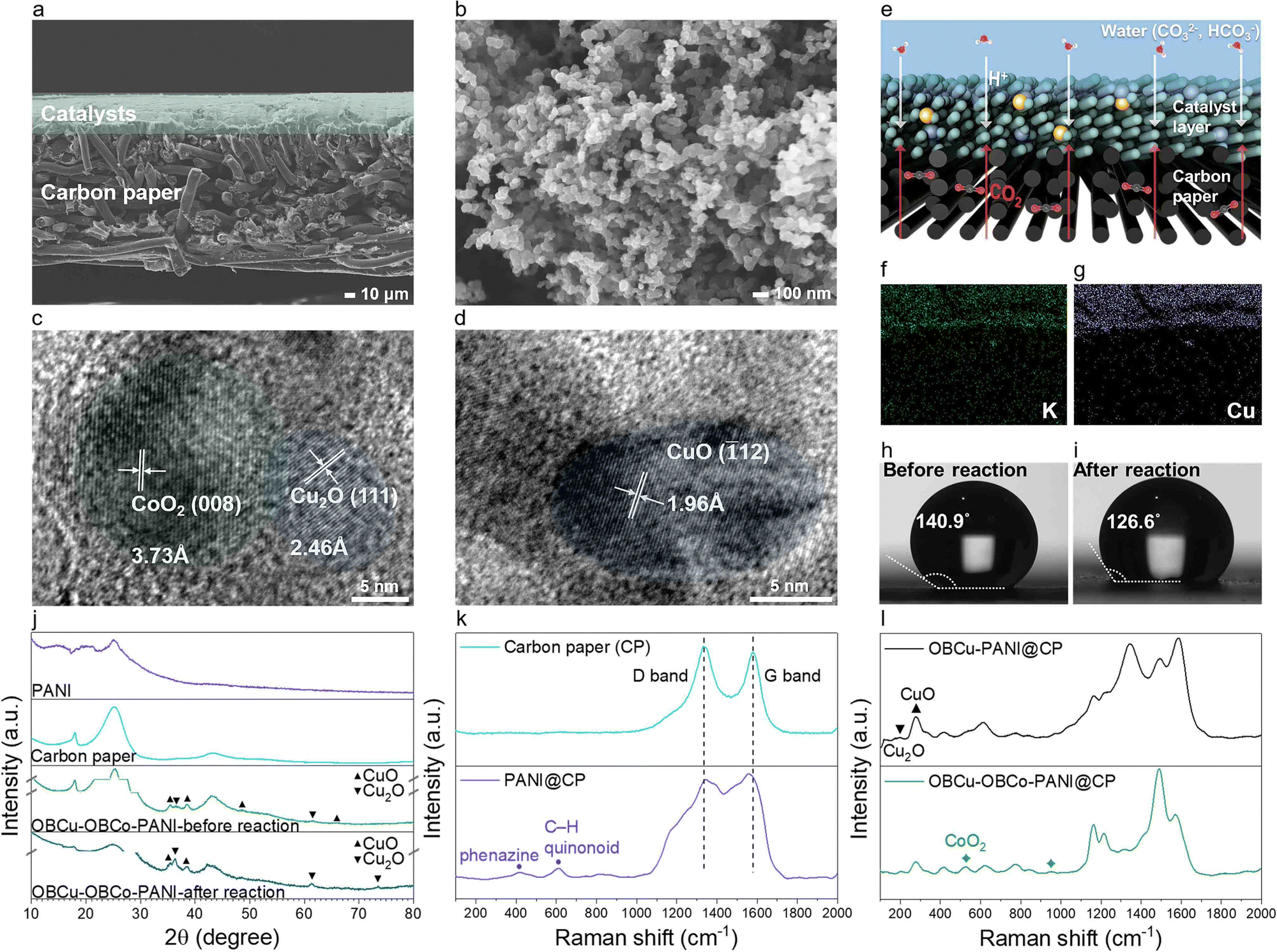 | ||
| Fig. 2 (a) and (b) SEM images of OBCu–OBCo–PANI spayed on carbon paper. (c) and (d) TEM images of OBCu–OBCo–PANI. (e) The macro illustration of the three-phase interface. (f) and (g) The element mapping of cross-section catalysts on carbon paper after reacting in a high concentration K2CO3 (aq.) (0.22 M). (h) and (i) Contact angle measurement of wet catalysts on carbon paper before and after reaction. (j) XRD spectra of PANI, carbon paper, and OBCu–OBCo–PANI on carbon paper before and after the reaction. (k) and (l) Raman spectra of carbon paper, PANI, OBCu–PANI, and OBCu–OBCo–PANI on carbon paper, respectively. | ||
XRD was further carried out to confirm the existence of photocatalysts as in Fig. 2j. The diffraction peaks at 2θ = 16.0°, 20.3°, and 25.3° are all characteristic diffraction peaks of PANI, corresponding to periodicity paralleling of the polymer chains.41 The broad peak is due to the thinness of the material. Comparing the spectra of OBCu–OBCo–PANI before and after the reaction, interestingly, part of the copper oxide was converted to cuprous oxide, as the diffraction peaks belonging to CuO disappear at 2θ = 48.7° and 65.7°, while the peak belonging to Cu2O appears at 2θ = 73.6°. However, the exact existence needs to be assessed by in situ characterization during the reaction as the catalysts will undergo valence change once exposed to air. The peaks of OBCo are not recognized because the content is little and the distribution is dispersed; beyond that, the peak positions of OBCo overlap with copper oxides. The weakening of diffraction signals’ intensity after the reaction is considered to be related to the longitudinal swelling of the material. Besides, we obtained the Raman spectra of carbon paper, PANI, OBCu–PANI, and OBCu–OBCo–PANI which are shown in Fig. 2k and l. Carbon paper has typical D and G bands located at 1335.3 cm−1 and 1577.1 cm−1, respectively. When PANI is added, the peaks at 416.1 cm−1 and 611.5 cm−1 appear, which are ascribed to phenazine-like segments and out-of-plane C–H deformation of the quinonoid ring in PANI, respectively.42,43 The peak at 213.2 cm−1 is assigned to Cu2O and the peak at 276.6 cm−1 is assigned to CuO.44 The loading of CoO2 gives rise to the peaks at 525.2 cm−1 and 950.5 cm−1.
As for the dynamic structure of the light absorber during the reaction, we deem that a macro three-phase interface exists in the GDL resembling the one in Fig. 2e. CO2 is directly transferred through the hydrophobic substrate, the carbon paper, to the photocatalysts. Saturated potassium carbonate solution provides the protons needed in the evolution of hydrocarbon products, and prevents carbon dioxide from dissolving in the aqueous solution instead of participating in the reaction. To give a visual representation of the three-phase interface, we used high concentration K+ to exhibit the position of liquid in the reaction. Comparing Fig. 2f and g, the liquid overlaps the photocatalysts, while the carbon paper remains anhydrous. Thus, the existence of the interface is proved. To understand the nature of the local reacting surface more clearly, we photographed the contact angles of water on typical surfaces as in Fig. 2h, i, and Fig. S6 (ESI†). The contact angle is 151.3° on carbon paper. On wet catalysts, the angle decreases to 140.9° and further decreases to 126.6° after the reaction. The reacting surface is always hydrophobic during the reaction, which ensures the stable existence of the three-phase interface microscopically and further promotes CO2R, hindering the hydrogen evolution reaction. Moreover, the evaporated water can flow through the light absorber into the outlet airway unimpeded because the substrate will not be immersed in water during the reaction. The controlled experiment was set in a solid–liquid suspension reacting system, immersing PANI particles in saturated K2CO3 (aq.), and the selectivity to hydrogen was as high as 89.97%, as shown in Fig. S22 (ESI†).
Performance of CO2R and real-time changes of catalysts in the process
Fig. 3a and b show the photocatalytic activity and selectivity of the loading of various catalysts, respectively. Products were all in the liquid phase dispersed in very few species with no hydrogen generation. With the step-by-step loading of metal oxides, the selectivity for C2+ products increased correspondingly. OBCu–OBCo–PANI had the best selectivity for C2H5OH, up to 93.6%, with a generation rate of 158.7 μmol h−1 g−1. An apparent quantum yield of 3.3% at 410 nm was achieved without sacrificial agents. With the OBCu and OBCo loading, the evaporation was curbed, which was attributed to the destruction of the hydrophilic group (amino groups) in PANI. To address the problem and optimize the CO2R performance simultaneously, we designed the tandem structure, with PANI beneath and OBCu–OBCo–PANI on top receiving the irradiation first, half-thickness of the usual. A high generation rate of 368.9 μmol h−1 g−1 towards HCOOH was obtained. This light absorber in this tandem layout exhibited fine bifunctionality.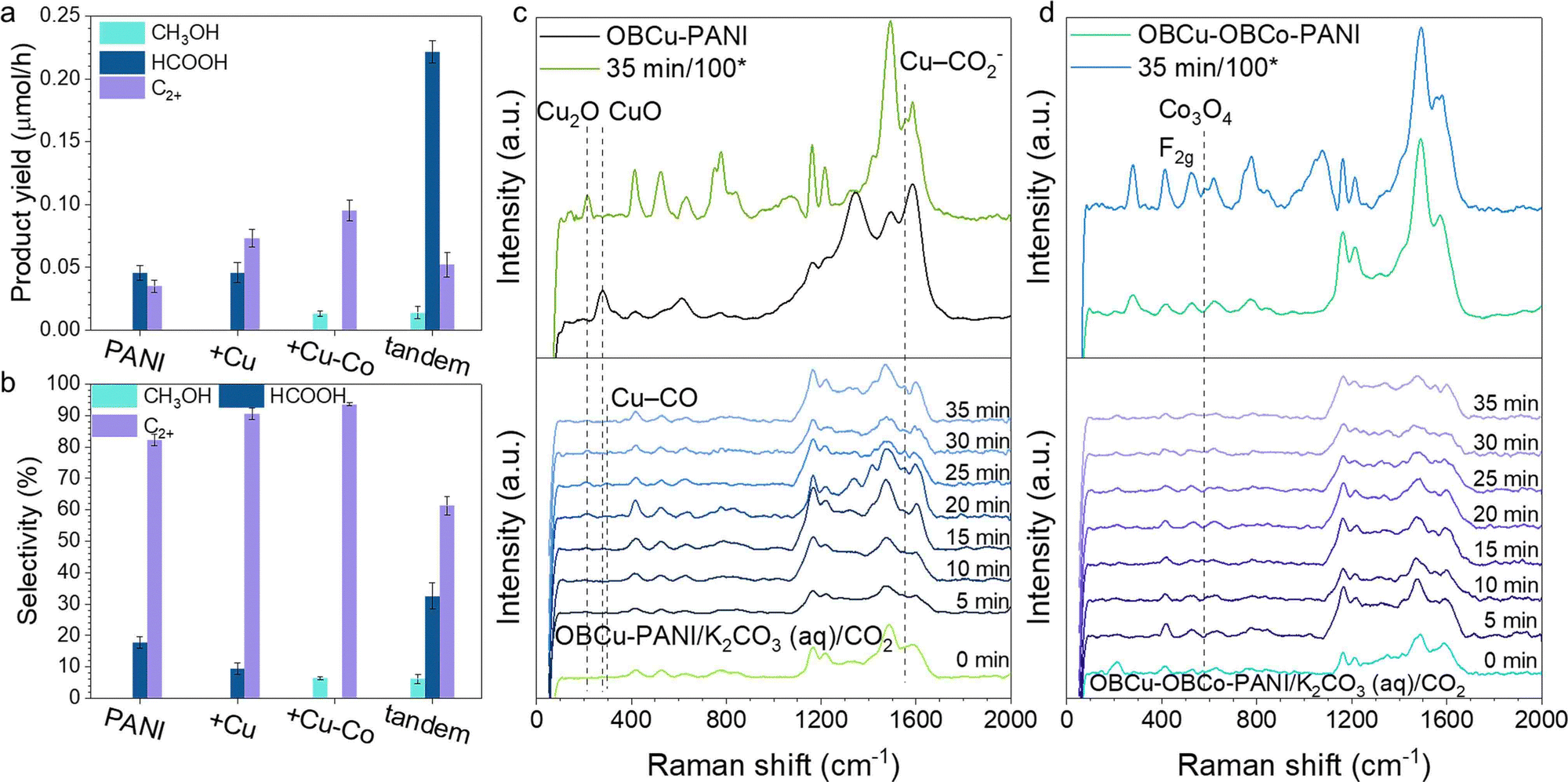 | ||
| Fig. 3 (a) and (b) The product yield and selectivity of PANI, OBCu–PANI (+Cu), OBCu–OBCo–PANI (+Cu–Co), and the tandem structure, with PANI beneath and OBCu–OBCo–PANI on top receiving the light first, half-thickness (tandem). (c) Quasi-in situ Raman spectra of OBCu–PANI during CO2R. (d) Quasi-in situ Raman spectra of OBCu–OBCo–PANI during CO2R. | ||
To gain an in-depth view of the CO2R process, quasi-in situ Raman spectroscopy was conducted to reveal the change of photocatalysts and the existence of intermediates. Fig. S8 (ESI†) shows the unchanged structure of PANI during the CO2R. The addition of K2CO3 (aq.) will bring about several peaks in the Raman spectrum. However, the adding of CO2 flow causes no change compared with the one already immersed in K2CO3 (aq.). It is worth noting that PANI does not change after 2 h of reaction. Fig. 3c shows the change of oxide-bearing copper during the CO2R. The intensity of the peak at 213.2 cm−1 increases as time goes by during the reaction, whereas, the intensity of the peak at 276.6 cm−1 decreases and disappears. It is revealed that CuO is reduced to Cu2O under irradiation, which is in line with the results of XRD. Moreover, we used a higher power lens (100*) to observe the photocatalysts after 35 min of reaction. It turns out that CuO is converted to Cu2O completely. Thus, the functional photocatalyst should mainly be Cu2O–PANI during the CO2R. The emerging peak at 1552.5 cm−1 belongs to CO2− intermediates coordinated with Cu, specifically, Cu–C and Cu–O are co-coordinated on Cu.45–47 The associated peak of Cu–CO frustrated rotation is found to appear at 295.7 cm−1,48 which demonstrates the reaction process and verifies the adsorption site. Fig. 3d shows the change of CoO2 during the CO2R. The typical peaks at 525.2 cm−1 and 950.5 cm−1 overlap with the peaks of K2CO3 (aq.), so no valid information could be acquired. However, the emerging peak at 576.6 cm−1 is associated with the F2g symmetry of Co3O4 (Co2+(Co3+)2O2−4).49,50 Therefore, we deduced that CoO2 was reduced during the reaction. Copper and cobalt both underwent valence drop once the CO2R started; in other words, the photo-induced electrons would flow to the metal sites. Meanwhile, the patterns of PANI remained the same. Thus, it was assumed that the Cu/Co sites were where CO2R took place. This was also confirmed by the detected adsorbed intermediates on copper sites and the enhanced CO2R performance adding OBCo.
Mechanism study of the improved performance for CO2R
Intending to enhance the explication of experimental observations, particularly the generative mechanism of the C2+ product, we meticulously employed DFT as a means of conducting our investigation. The Cu2O–PANI model is comprised of two parts. Firstly, the minimal repeating unit of PANI molecules is placed on the surface of Cu2O, which consists of a benzene ring with a reducing end (–NH) and an oxidizing end (–N).51,52 Our findings indicate that the presence of water molecules near the catalytic surface greatly hinders the transport of CO2 molecules. By calculating the activation energy barrier for CO2 transfer from the solution to the surface with varying amounts of water molecules (Fig. 4a), we found that accumulation of water on the surface significantly raises the transport barrier and reduces the efficiency of the overall reaction. Thus, the evaporation of water is beneficial to CO2R. Subsequently, we introduced Co3O4 clusters in the model, as shown in Fig. 4c. CO is known to be the most important intermediate in producing multi-carbon products, thus we first calculated the free energy profile of CO intermediate formation, shown in Fig. 4b.53–55 The greatest difference is the activation of CO2 molecules, where the oxidizing end N atom of PANI can co-adsorb CO2 molecules with the surface, greatly reducing the first step activation energy, which is about 0.8 eV. The subsequent steps have a small difference, but it is noteworthy that CO molecules tend to proceed to the subsequent reaction rather than dissociating from the surface, regardless of the presence of PANI. However, PANI clearly enhances this effect, thereby providing a necessary condition for the subsequent C–C bonding step. We then investigated the C–C coupling reaction and discovered that the addition of Co3O4 greatly influenced the process. The results are shown in Fig. 4d. It reduced the activation energy of the C–C coupling by 0.28 eV due to its weaker adsorption of CO compared to the stronger adsorption by Cu2O. The asymmetrical adsorption sites facilitated the C–C coupling (Fig. 4c), combined with our previous calculation that the CO produced on the surface was not easily displaced, the likelihood of successful C–C coupling was greatly enhanced. Finally, we calculated the reaction energy barrier for C2–C or 3C coupling, and it is worth mentioning that the calculated C3 synthesis path in this work is through the *CH2CH–*CO coupling to produce *CH2CHCO (Fig. 4c), which is currently the most stable C3 generation path.56,57 The calculation results shown in Fig. 4e indicate that the C3 coupling energy barrier on Cu2O–Co3O4–PANI is only 0.82 eV, far lower than 1.21 eV on Cu2O–PANI.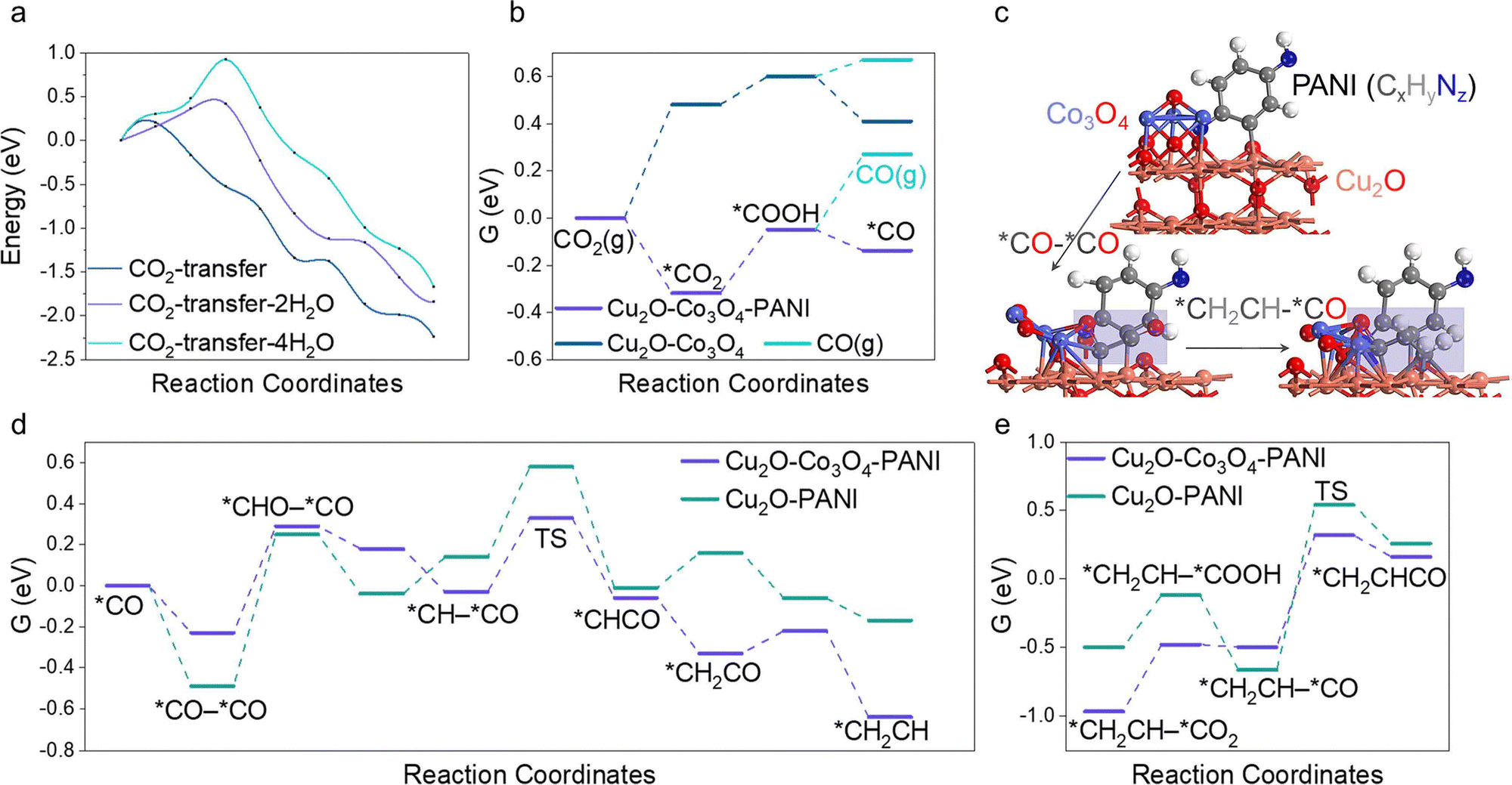 | ||
| Fig. 4 (a) The activation energy diagram calculation by nudged elastic band (NEB) for the transfer of CO2 molecules from the solution to the surface with different numbers of H2O molecules. (b) Free energy diagram for the formation of *CO and CO(g). (c) The structure of Cu2O–Co3O4–PANI and the key intermediates *CO–*CO/*CH2CH–*CO on it. (d) Free energy diagram for the formation of C2 intermediates. (e) Free energy diagram for the formation of the key intermediates for C3 production. | ||
Performance of water purification and factors in STV efficiency improvement
As in Fig. 5c, pristine PANI possesses the best water evaporation capacity, the pure water generating rate of which is 1.13 kg m−2 h−1, and the corresponding energy conversion efficiency is 71.04%. With the OBCu and OBCo loading, the evaporation was curbed, which was attributed to the destruction of the hydrophilic group (amino groups) in PANI. We designed a tandem structure as mentioned before, and the water evaporation capacity was well retained. The generation rate of pure water is 1.03 kg m−2 h−1 and the energy conversion efficiency is 65.12%. Moreover, under the illumination of 940 nm infrared light only, the STV efficiency is 31.5%, far lower than that under full-spectrum illumination, which indicates that CO2R has a synergistic effect with water evaporation. To verify the effectiveness of water evaporation, we used inductively coupled plasma mass spectrometry (ICP-MS) to measure the concentration of potassium ions in the reactor and condenser after the reaction as shown in Fig. 5a and Table S1 (ESI†). The concentration of K+ remaining in the reactor is approximately 104 times that in the purified liquid. There is nearly no trace of impurity ions (0.413 μg mL−1) in the condensing equipment. CO2R and seawater desalination were also carried out simultaneously to test the feasibility of this approach. Using the tandem structure catalysts, only C2H5OH and HCOOH were generated, with a 91.6% selectivity and 696.7 μmol h−1 g−1 yield towards HCOOH. The STV efficiency is 63.0%. The desalination results are exhibited in Fig. 5b and Table S2 (ESI†). The concentration of ions remaining in the reactor is approximately 10 times that in the purified liquid. The complex composition of seawater caused the outcome of the reaction to be less than expected.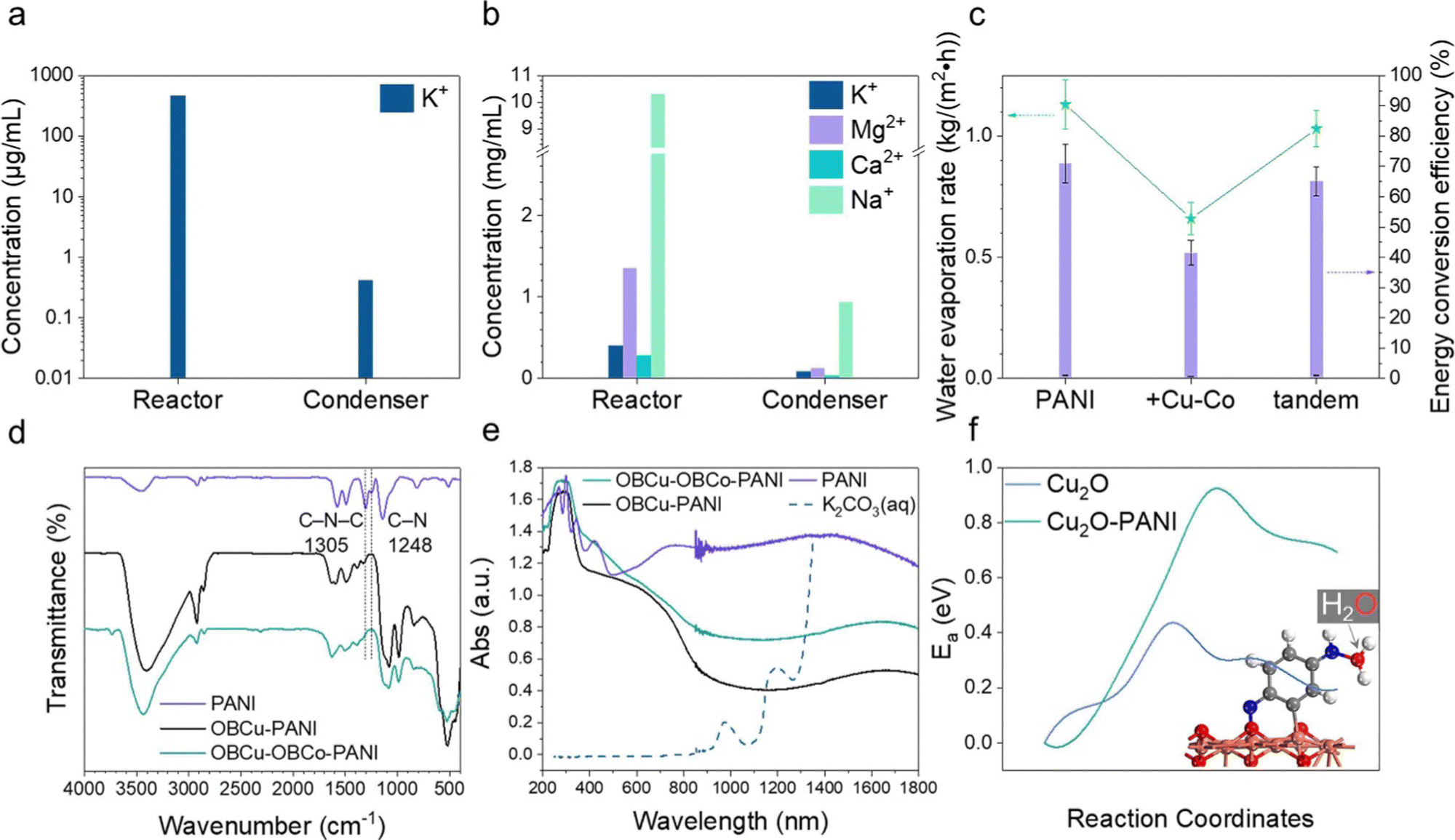 | ||
| Fig. 5 (a) The ion concentration after water purification (K2CO3 (aq.)) in the reactor and condenser. (b) The ion concentrations after seawater desalination in the reactor and condenser. (c) Water evaporation rate and corresponding energy conversion efficiency of PANI, OBCu–OBCo–PANI (+Cu–Co), and the tandem structure, with PANI beneath and OBCu–OBCo–PANI on top receiving the light first, half-thickness (tandem). (d) FTIR spectra of PANI, OBCu–PANI, and OBCu–OBCo–PANI. (e) UV-vis spectra of PANI, OBCu–-PANI, OBCu–OBCo–PANI, and potassium carbonate solution. (f) The activation energy diagram calculation by NEB for the transfer of H2O molecules from the solution to the catalytic surface. | ||
To show the role of the catalysts in water evaporation more vividly, we used infrared imaging to show the temperature distribution of the illuminated surface as in Fig. S13a and b (ESI†). Before the light irradiation, the temperature of the surface was almost uniform, and even the boundary of the carbon paper could not be distinguished. After 40 min of light irradiation, the temperature of the surface stabilized. The shape of PANI on the carbon paper is prominent in Fig. S13b (ESI†) because it is about 9 °C warmer than the surface of the surrounding water. Hence, it is proved that the existence of PANI-based catalysts on carbon paper has a stimulating effect on water evaporation. To uncover the reasons for the different performances of PANI and PANI loaded with metal oxides on the molecular level, the FTIR spectra of the photocatalysts are recorded and analyzed as in Fig. 5d. The peaks at 1574 and 1487 cm−1 are attributed to the stretching vibrations of the quinone ring and benzenoid ring, respectively.41,58–60 Comparing the spectra, the main structure of PANI is not damaged after loading the metal oxides. The obvious blueshift is due to the interaction between PANI and the constituents of the formed metal oxides.61 The peaks at 1305 cm−1 and 1248 cm−1 are related to the C–N–C and C–N stretching modes, respectively. The peak intensity decreases and further disappears as the metal oxides are loaded, indicating damage to the amino groups. We further used UV-vis spectroscopy to analyze the light absorption properties of different PANI-based catalysts and the potassium carbonate solution. The spectrum of K2CO3 (aq.) in Fig. 5e is cut off after 1350 nm due to drastic fluctuation caused by the nearly total absorption of light. The absorption of infrared light above 1400 nm by the liquid body is 100% as the transmittance value is 0 as shown in Fig. S15 (ESI†). It is worth noting that in the near-infrared range (800 nm–1300 nm), the light absorption of catalysts is generally greater than the potassium carbonate solution, especially PANI, thus the higher temperature and corresponding enhanced water molecular energy are mainly attributed to the catalyst-induced absorption in this relatively high-energy region of the solar spectrum. In addition, we found in the UV-vis spectra that the light absorption of PANI is much better than that of OBCu–PANI and OBCu–OBCo–PANI in the near-infrared range, which is in line with the water evaporation test result. The near-infrared region (780–2500 nm) absorption is formed by the superposition of double-, sum-, and difference-frequency of the fundamental frequency in the mid-infrared region of the relatively high-energy chemical bonds in organic substances (mainly C–H, O–H, and N–H).62 And we have already found the damage of amino groups, which are believed to alter phase change behaviors of water, as metal oxides are loaded in the analysis of FTIR spectra. To further confirm this finding, XPS was conducted to provide the change of the N 1s line after the loading of metal oxides. Compared with Fig. S16a (ESI†), after loading OBCu, there is only baseline fluctuation with no characteristic peaks in Fig. S16b (ESI†), which verifies the damage of amino groups. Besides, with the help of the DFT investigation, we confirmed that water molecules tend to be captured by the reducing end of PANI (–NH), forming a bond between the O and N atoms (inset in Fig. 5f). Based on this, we calculated the influence of PANI on the transport of water molecules and the transition state calculation showed that the presence of PANI increases the transfer activation energy of water molecules moving from the solution to the catalytic surface by over 0.5 eV as in Fig. 5f. In other words, PANI can effectively keep water away from the catalytic surface, thereby indirectly facilitating the transport of CO2 molecules and enhancing the CO2R performance. In conclusion, we found that PANI has the functions of (1) adsorbing water molecules through –NH as proven by performance tests and characterization and (2) increasing the hydrophobicity of the catalytic surface to suppress the hydrogen evolution reaction.
Conclusions
In summary, we facilely synthesized OBCu/OBCo loaded PANI as a bifunctional material for simultaneous CO2 photoreduction and seawater desalination to achieve the full-spectrum utilization of solar energy as shown in Fig. 6. The reaction was carried out in a self-designed gas diffusion system to enhance the kinetics and suppress the hydrogen evolution reaction. A high selectivity for C2H5OH, up to 93.6%, with a generation rate of 158.7 μmol h−1 g−1, was achieved. The apparent quantum yield was 3.3% at 410 nm without sacrificial agents. During the reaction, OBCu and OBCo will be reduced to Cu2O and Co3O4 because of the transferred photoinduced electrons proven by quasi-in situ Raman spectra. The asymmetrical adsorption sites of the different metal oxides facilitated the C–C coupling and promoted the 3C coupling process. Undamaged –NH in PANI was deemed to be the key to water evaporation. Pristine PANI possessed the best water evaporation capacity, with an STV efficiency of 71.0%. Thus, the tandem structure, with PANI beneath and OBCu–OBCo–PANI on top, exhibited the best bifunctional performance. The avoided accumulation of water on the catalytic surface significantly increased the hydrophobicity of the catalytic surface while decreasing the transport barrier of CO2. Meanwhile, a full-spectrum irradiation reaction had higher STV efficiency than that of only infrared irradiation. This research sheds light on the development of efficient and sustainable photocatalytic systems with synergistic applications in environmental remediation and energy conversion.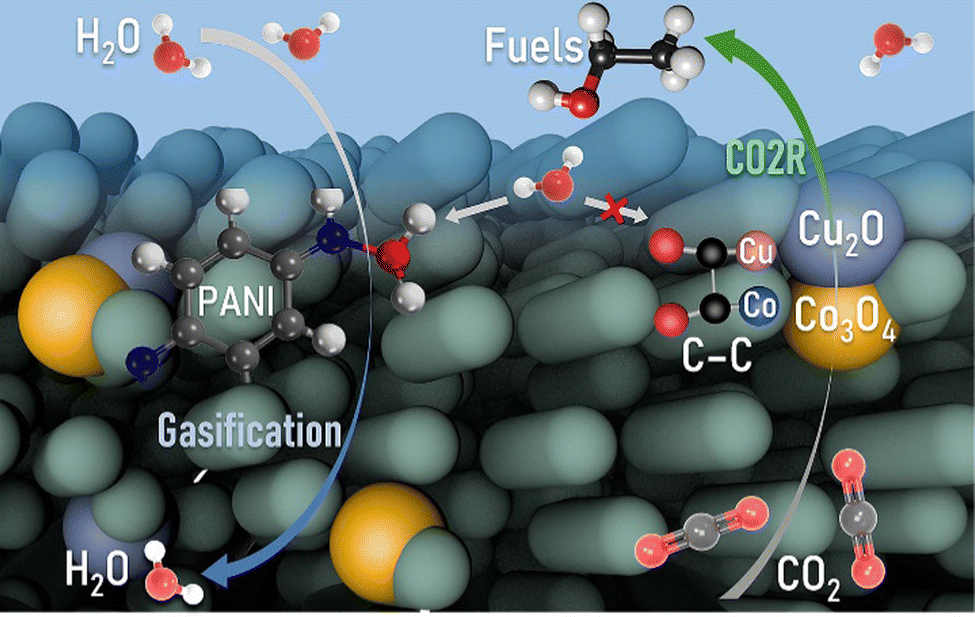 | ||
| Fig. 6 Mechanism of the synergistic CO2 photoreduction and seawater desalination reaction on OBCu–OBCo–PANI. | ||
Author contributions
Y. Y.: writing – original draft, writing – review & editing, conceptualization, investigation, and formal analysis; W. J.: writing – original draft, formal analysis, and software; H. Q.: methodology and conceptualization; F. W.: funding acquisition, project administration, and resources; Y. L.: supervision, conceptualization, writing – review & editing, and funding acquisition; L. G.: supervision, conceptualization, project administration, resources, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (51888103, 51906199) and the Fundamental Research Funds for the Central Universities.References
- A. Wagner, C. D. Sahm and E. Reisner, Nat. Catal., 2020, 3, 775–786 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- M. Halmann, Nature, 1978, 275, 115–116 CrossRef CAS.
- Y. Guo, L. S. de Vasconcelos, N. Manohar, J. Geng, K. P. Johnston and G. Yu, Angew. Chem., Int. Ed., 2022, 61, e202114074 CAS.
- T. Ding and G. W. Ho, Joule, 2021, 5, 1639–1641 CrossRef.
- W. Wang, Y. Shi, C. Zhang, S. Hong, L. Shi, J. Chang, R. Li, Y. Jin, C. Ong, S. Zhuo and P. Wang, Nat. Commun., 2019, 10, 3012 CrossRef PubMed.
- M. Gao, C. K. Peh, L. Zhu, G. Yilmaz and G. W. Ho, Adv. Energy Mater., 2020, 10, 2000925 CrossRef CAS.
- L. Han, J. Mao, A.-Q. Xie, Y. Liang, L. Zhu and S. Chen, Sep. Purif. Technol., 2023, 309, 123003 CrossRef CAS.
- H. Liu, H.-G. Ye, M. Gao, Q. Li, Z. Liu, A.-Q. Xie, L. Zhu, G. W. Ho and S. Chen, Adv. Sci., 2021, 8, 2101232 CrossRef CAS PubMed.
- Y. Liu, F. Wang, Z. Jiao, S. Bai, H. Qiu and L. Guo, Electrochem. Energy Rev., 2022, 5, 5 CrossRef CAS.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Phys. Chem. Chem. Phys., 2018, 20, 16973–16984 RSC.
- L. Han, W. Zhou and C. Xiang, ACS Energy Lett., 2018, 3, 855–860 CrossRef CAS.
- X. Sheng, Z. Liu, R. Zeng, L. Chen, X. Feng and L. Jiang, J. Am. Chem. Soc., 2017, 139, 12402–12405 CrossRef CAS PubMed.
- G. Xi, S. Ouyang, P. Li, J. Ye, Q. Ma, N. Su, H. Bai and C. Wang, Angew. Chem., Int. Ed., 2012, 51, 2395–2399 CrossRef CAS PubMed.
- S. Gong, Y. Niu, X. Liu, C. Xu, D. Xiong, Z. Tu, M. Xu and Z. Chen, Appl. Catal., B, 2022, 316, 121663 CrossRef CAS.
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS.
- F. Wang, F. Huang, F. Yu, X. Kang, Q. Wang and Y. Liu, Cell Rep. Phys. Sci., 2023, 4, 101450 CrossRef CAS.
- Y. Wu, Q. Chen, J. Zhu, K. Zheng, M. Wu, M. Fan, W. Yan, J. Hu, J. Zhu, Y. Pan, X. Jiao, Y. Sun and Y. Xie, Angew. Chem., Int. Ed., 2023, 62, e202301075 CrossRef CAS PubMed.
- Y. Wang, X. Liu, X. Han, R. Godin, J. Chen, W. Zhou, C. Jiang, J. F. Thompson, K. B. Mustafa, S. A. Shevlin, J. R. Durrant, Z. Guo and J. Tang, Nat. Commun., 2020, 11, 2531 CrossRef CAS PubMed.
- P. Huang, J. Huang, S. A. Pantovich, A. D. Carl, T. G. Fenton, C. A. Caputo, R. L. Grimm, A. I. Frenkel and G. Li, J. Am. Chem. Soc., 2018, 140, 16042–16047 CrossRef CAS PubMed.
- W. Xie, K. Li, X.-H. Liu, X. Zhang and H. Huang, Adv. Mater., 2023, 35, 2208132 CrossRef CAS PubMed.
- F. Rechberger and M. Niederberger, Mater. Horiz., 2017, 4, 1115–1121 RSC.
- Q. Han, B. Wang, Y. Zhao, C. Hu and L. Qu, Angew. Chem., Int. Ed., 2015, 54, 11433–11437 CrossRef CAS PubMed.
- J. Puskelova, L. Baia, A. Vulpoi, M. Baia, M. Antoniadou, V. Dracopoulos, E. Stathatos, K. Gabor, Z. Pap, V. Danciu and P. Lianos, Chem. Eng. J., 2014, 242, 96–101 CrossRef CAS.
- Z. Jiang, X. Zhang, G. Yang, Z. Yuan, X. Ji, F. Kong, B. Huang, D. D. Dionysiou and J. Chen, Chem. Eng. J., 2019, 373, 814–820 CrossRef CAS.
- X. Zhao, M. Xu, X. Song, W. Zhou, X. Liu, H. Wang and P. Huo, Chem. Eng. J., 2022, 446, 137034 CrossRef CAS.
- F. Zhao, Y. Guo, X. Zhou, W. Shi and G. Yu, Nat. Rev. Mater., 2020, 5, 388–401 CrossRef.
- Y. Guo, J. Bae, Z. Fang, P. Li, F. Zhao and G. Yu, Chem. Rev., 2020, 120, 7642–7707 CrossRef CAS PubMed.
- H. Jung, S. Y. Lee, C. W. Lee, M. K. Cho, D. H. Won, C. Kim, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2019, 141, 4624–4633 CrossRef CAS PubMed.
- W. Zhang, C. Huang, Q. Xiao, L. Yu, L. Shuai, P. An, J. Zhang, M. Qiu, Z. Ren and Y. Yu, J. Am. Chem. Soc., 2020, 142, 11417–11427 CrossRef CAS PubMed.
- X. Wang, Z. Wang, T.-T. Zhuang, C.-T. Dinh, J. Li, D.-H. Nam, F. Li, C.-W. Huang, C.-S. Tan, Z. Chen, M. Chi, C. M. Gabardo, A. Seifitokaldani, P. Todorović, A. Proppe, Y. Pang, A. R. Kirmani, Y. Wang, A. H. Ip, L. J. Richter, B. Scheffel, A. Xu, S.-C. Lo, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Commun., 2019, 10, 5186 CrossRef PubMed.
- H. Shen, Y. Wang, T. Chakraborty, G. Zhou, C. Wang, X. Fu, Y. Wang, J. Zhang, C. Li, F. Xu, L. Cao, T. Mueller and C. Wang, ACS Catal., 2022, 12, 5275–5283 CrossRef CAS.
- P. Wang, H. Yang, C. Tang, Y. Wu, Y. Zheng, T. Cheng, K. Davey, X. Huang and S.-Z. Qiao, Nat. Commun., 2022, 13, 3754 CrossRef CAS PubMed.
- F. Wooten, Optical properties of solids, Academic Press, Inc., New York, USA, 1972 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- A. Deshmukh, C. Boo, V. Karanikola, S. Lin, A. P. Straub, T. Tong, D. M. Warsinger and M. Elimelech, Energy Environ. Sci., 2018, 11, 1177–1196 RSC.
- J. Wu, K. R. Zodrow, P. B. Szemraj and Q. Li, J. Mater. Chem. A, 2017, 5, 23712–23719 RSC.
- Y. Liu, J. Cui, Y. Liang, W. An, H. Wang, L. Liu, J. Hu and W. Cui, Appl. Surf. Sci., 2020, 509, 145296 CrossRef CAS.
- E. Elanthamilan, A. Sathiyan, S. Rajkumar, E. J. Sheryl and J. P. Merlin, Sustainable Energy Fuels, 2018, 2, 811–819 RSC.
- L. Wang, Q. Yao, H. Bi, F. Huang, Q. Wang and L. Chen, J. Mater. Chem. A, 2015, 3, 7086–7092 RSC.
- H. Li, P. Wei, D. Gao and G. Wang, Curr. Opin. Green Sustainable Chem., 2022, 34, 100589 CrossRef CAS.
- Z. Pan, K. Wang, K. Ye, Y. Wang, H.-Y. Su, B. Hu, J. Xiao, T. Yu, Y. Wang and S. Song, ACS Catal., 2020, 10, 3871–3880 CrossRef CAS.
- I. V. Chernyshova, P. Somasundaran and S. Ponnurangam, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E9261–E9270 CrossRef CAS PubMed.
- Z.-Z. Niu, F.-Y. Gao, X.-L. Zhang, P.-P. Yang, R. Liu, L.-P. Chi, Z.-Z. Wu, S. Qin, X. Yu and M.-R. Gao, J. Am. Chem. Soc., 2021, 143, 8011–8021 CrossRef CAS PubMed.
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2021, 4, 20–27 CrossRef CAS.
- S. Zhao, F. Hu and J. Li, ACS Catal., 2016, 6, 3433–3441 CrossRef CAS.
- X. Hongyan, D. Jiangtao, H. Zhenyin, G. Libo, Z. Qiang, T. Jun, Z. Binzhen and X. Chenyang, Cryst. Res. Technol., 2016, 51, 123–128 CrossRef.
- G. P. Oliveira, B. H. Barboza and A. Batagin-Neto, Comput. Theor. Chem., 2022, 1207, 113526 CrossRef CAS.
- J.-X. Feng, S.-Y. Tong, Y.-X. Tong and G.-R. Li, J. Am. Chem. Soc., 2018, 140, 5118–5126 CrossRef CAS PubMed.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2020, 10, 1754–1768 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- A. R. Woldu, Z. Huang, P. Zhao, L. Hu and D. Astruc, Coord. Chem. Rev., 2022, 454, 214340 CrossRef CAS.
- W. Pei, S. Zhou, J. Zhao, X. Xu, Y. Du and S. X. Dou, Nano Energy, 2020, 76, 105049 CrossRef CAS.
- M. T. Tang, H.-J. Peng, J. H. Stenlid and F. Abild-Pedersen, J. Phys. Chem. C, 2021, 125, 26437–26447 CrossRef CAS.
- R. Wang, M. Han, Q. Zhao, Z. Ren, X. Guo, C. Xu, N. Hu and L. Lu, Sci. Rep., 2017, 7, 44562 CrossRef CAS PubMed.
- K. M. R. Karim, M. Tarek, S. M. Sarkar, R. Mouras, H. R. Ong, H. Abdullah, C. K. Cheng and M. M. R. Khan, Int. J. Hydrogen Energy, 2021, 46, 24709–24720 CrossRef CAS.
- W. Zheng, S. Nayak, W. Yuan, Z. Zeng, X. Hong, K. A. Vincent and S. C. E. Tsang, Chem. Commun., 2016, 52, 13901–13904 RSC.
- M. Shaban, M. Rabia, A. M. A. El-Sayed, A. Ahmed and S. Sayed, Sci. Rep., 2017, 7, 14100 CrossRef PubMed.
- W. Kaye, Spectrochim. Acta, 1954, 6, 257 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthesis procedure; supplements about the bifunctional CO2R and water evaporation system; determination of efficiencies and selectivity; supplements about the characterization of the bifunctional hybrid light absorber; supplements about the performance of catalysts; and details about DFT calculations. See DOI: https://doi.org/10.1039/d3ey00098b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |