DOI:
10.1039/D3EY00090G
(Review Article)
EES Catal., 2023,
1, 645-664
Recent advances in electrocatalytic NOx reduction into ammonia
Received
24th April 2023
, Accepted 1st June 2023
First published on 1st June 2023
Abstract
Ammonia (NH3) is an essential ingredient for the production of numerous chemicals which have a wider usage as fertilizers, explosives, and plastics. Currently, NH3 is mainly produced from the conventional Haber–Bosch process, which is energy-consuming and involves the risk of emitting greenhouse gases into the atmosphere. In contrast, electrochemical ammonia synthesis (EAS) from the nitrogen reduction reaction is evolving as a viable solution for sustainable NH3 production under ambient conditions. However, the high N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N dissociation energy and the competitive hydrogen evolution reaction result in an unsatisfactory ammonia yield rate and Faradaic efficiency. In this regard, EAS from reactive nitrogen (NOx) species, especially through the nitric oxide reduction reaction (NORR), could be a sustainable way as it produces valuable NH3 and simultaneously mitigates the gaseous NOx pollutant. Various NORR electrocatalysts have been designed and investigated. The electrocatalytic activity hugely depends on the composition, Gibbs free energy for ‘NO’ or intermediate adsorption on the catalyst surface, and the rate of proton/electron transfer at the solid–liquid–gas interface. Besides, different electrolyte additives have been employed to improve the solubility of NO in aqueous electrolytes. Thus, this review presents an overview of the NORR mechanism, recent advancements in electrocatalysts, and factors influencing the NH3 yield and selectivity. After that, the forthcoming challenges associated with practical realisation of EAS via NORR are discussed.
N dissociation energy and the competitive hydrogen evolution reaction result in an unsatisfactory ammonia yield rate and Faradaic efficiency. In this regard, EAS from reactive nitrogen (NOx) species, especially through the nitric oxide reduction reaction (NORR), could be a sustainable way as it produces valuable NH3 and simultaneously mitigates the gaseous NOx pollutant. Various NORR electrocatalysts have been designed and investigated. The electrocatalytic activity hugely depends on the composition, Gibbs free energy for ‘NO’ or intermediate adsorption on the catalyst surface, and the rate of proton/electron transfer at the solid–liquid–gas interface. Besides, different electrolyte additives have been employed to improve the solubility of NO in aqueous electrolytes. Thus, this review presents an overview of the NORR mechanism, recent advancements in electrocatalysts, and factors influencing the NH3 yield and selectivity. After that, the forthcoming challenges associated with practical realisation of EAS via NORR are discussed.

Harish Reddy Inta
| Harish Reddy Inta received his PhD degree in Chemical Sciences from Indian Institute of Science Education and Research-Kolkata (IISER-K), India in 2022. He is currently working as a postdoctoral researcher at Daegu Gyeongbuk Institute of Science & Technology (DGIST), Daegu, Republic of Korea. His current research interest is on the development of efficient catalysts for electrosynthesis of value-added chemicals such as NH3 and urea from air pollutants (NOx and CO2). |

Dinesh Dhanabal
| Dinesh Dhanabal received his BTech degree in Chemical and Electrochemical Engineering from CSIR-Central Electrochemical Research Institute (CECRI), Karaikudi, India in 2020. He is currently pursuing his Masters’ degree in Energy Science and Engineering at Daegu Gyeongbuk Institute of Science & Technology (DGIST), Dabegu, Republic of Korea, under the supervision of Prof. Sangaraju Shanmugam. His research interest is focussed on development of cost effective and efficient electrocatalysts for the synthesis of value-added chemicals such as NH3 and urea from air pollutants (NOx and CO2). |

Sridhar Sethuram Markandaraj
| Sridhar Sethuram Markandaraj received his BTech degree in Chemical and Electrochemical Engineering from CSIR-Central Electrochemical Research Institute (CECRI), Karaikudi, India in 2020. Recently, he secured his Masters’ degree in Energy Science and Engineering at Daegu Gyeongbuk Institute of Science & Technology (DGIST), Daegu, Republic of Korea, under the supervision of Prof. Sangaraju Shanmugam. His research interests include electrocatalytic nitric oxide reduction to value-added chemicals. |

Sangaraju Shanmugam
| Sangaraju Shanmugam obtained his doctorate in 2004 from the Indian Institute of Technology-Madras (IIT-M), India in the field of heterogeneous catalysis. Thereafter, in 2005, he joined the Department of Chemistry, Bar-Ilan University, Israel as a Postdoctoral Fellow. In 2007, he joined as a JSPS postdoctoral fellow at Waseda University and continued his research in “Catalysis for energy applications”. In 2011, he continued his academic career as an Assistant Professor at Daegu Gyeongbuk Institute of Science & Technology (DGIST), Republic of Korea and was promoted to Tenured Full Professor in 2019. His research interests include cost-effective membranes and catalysts for fuel cell and redox-flow batteries and electrosynthesis of value-added chemicals from atmospheric nitrogen and air pollutants. |
Broader context
Recently, electrochemical ammonia synthesis (EAS) has drawn significant attention for replacing the energy consuming and environmentally polluting Haber–Bosch process. In this context, the electrochemical nitrogen reduction reaction (NRR) is considered to be a viable solution for sustainable NH3 production under ambient conditions. However, the high NN dissociation energy and the competitive hydrogen evolution reaction cause challenges to attain satisfactory ammonia production with good selectivity. Meanwhile, the nitric oxide (NOx) pollutant could be an alternative N-source for EAS as it possesses low N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond dissociation energy and a distinctive reduction potential (0.71 V vs. RHE). To catalyze the electrochemical nitric oxide reduction reaction (NORR), the development of suitable catalysts with good catalytic activity and product (NH3) selectivity is highly desirable. Hence, the scientific community has attempted to design and develop various electrocatalysts and evaluate their performance towards the NORR. The electrocatalytic activity of the materials is found to be dependent on the composition, Gibbs free energy for ‘NO’ or intermediate adsorption on the catalyst surface, and the rate of proton/electron transfer at the solid–liquid–gas interface. Besides, the addition of ‘NO’ complexing agents into an aqueous electrolyte is found to increase the solubility of ‘NO’, thereby enhancing the ammonia production rate. Although the developed catalysts show considerable NH3 yield rates, there is still a necessity to develop efficient and earth-abundant catalysts for practical NH3 production. Hence, we present this Review article to give an overview of the NORR mechanism, recent advancements in the electrocatalysts, and factors influencing the NH3 yield and selectivity. Furthermore, the key issues related to the efficient electroreduction of NO to NH3 are described. O bond dissociation energy and a distinctive reduction potential (0.71 V vs. RHE). To catalyze the electrochemical nitric oxide reduction reaction (NORR), the development of suitable catalysts with good catalytic activity and product (NH3) selectivity is highly desirable. Hence, the scientific community has attempted to design and develop various electrocatalysts and evaluate their performance towards the NORR. The electrocatalytic activity of the materials is found to be dependent on the composition, Gibbs free energy for ‘NO’ or intermediate adsorption on the catalyst surface, and the rate of proton/electron transfer at the solid–liquid–gas interface. Besides, the addition of ‘NO’ complexing agents into an aqueous electrolyte is found to increase the solubility of ‘NO’, thereby enhancing the ammonia production rate. Although the developed catalysts show considerable NH3 yield rates, there is still a necessity to develop efficient and earth-abundant catalysts for practical NH3 production. Hence, we present this Review article to give an overview of the NORR mechanism, recent advancements in the electrocatalysts, and factors influencing the NH3 yield and selectivity. Furthermore, the key issues related to the efficient electroreduction of NO to NH3 are described.
|
Introduction
The thirst for energy keeps expanding with the growing technological society of humankind in this modern era.1 The rise in the human population, urbanization, and industrial activities increases the energy demand worldwide.2–6 This huge energy demand results in the over-exploitation of fossil fuels and causes a surge in carbon and greenhouse gas (GHG) emissions. These emissions trap the thermal radiation in the earth's atmosphere, raise the global temperature, and cause climate change.7 Thus, the energy crisis leads to domino effects related to global warming and accelerated climate change.8
With reference to the U.S. Energy Information Administration (EIA) data in 2021, the transportation sector utilizes around 28% of the total energy consumption of the U.S., which is mainly derived from fossil fuels.9 In the transportation sector, maritime transport controls 90% of global goods trading, accounting for 3.1% of global CO2 emissions.10,11 To mitigate environmental pollution, the Intergovernmental Panel on Climate Change (IPCC) formed a crucial strategy of decarbonizing the transport sector to reach the goals of the Paris climate change agreement to limit global temperature raise within 1.5 °C of pre-industrial levels.12,13 Therefore, the International Maritime Organization (IMO) set several checkpoints to complete the decarbonization of maritime transportation. The IMO GHG strategy sets out to reduce CO2 emission per transport work by up to 40% by 2030 and 70% by 2050 as of 2008 emission levels.14,15 Utilizing zero-carbon fuels in marine transport is the key strategy of IMO to meet the aims of the GHG strategy. Ammonia has attracted everyone's attention as a potential zero-carbon fuel for maritime transport and has been applied in direct ammonia fuel cells and solid oxide fuel cells (SOFCs).16,17 Compared with liquid hydrogen (8.5 MJ L−1), ammonia possesses a high energy density of 12.7 MJ L−1.18 Moreover, storing ammonia at −33 °C with less energy consumption for storage is more convenient, whereas hydrogen needs a cryogenic condition of −235 °C.19,20 With these considerations, many researchers foresee ammonia as a potential decarbonization fuel for heavy transportation like maritime transport.21–24
Besides, ammonia (NH3) is a crucial substance for various industries to produce fertilizers, medicines, refrigerants, explosives, and pesticides. Thus, NH3 is extensively produced from a well-established very old Haber–Bosch Process (HBP).25 However, the ammonia produced by the conventional HBP is not a green commodity. The required hydrogen feedstock for the HBP is exclusively extracted from fossil fuels by steam methane reforming or coal gasification processes. Thus, the HBP consumes 2% of global fossil fuel usage. As the HBP depends on the fossil fuel for the H2 source, it is not a sustainable and greener process. The ammonia production by the energy consuming HBP currently accounts for 2% of global energy consumption and 1.4% of global CO2 emission (carbon emission during gas extraction is excluded).21,26 Therefore, the decarbonization of ammonia production will decarbonize the energy, transportation, and chemical sectors so that the effects of GHG emissions and climate change can be controlled. MacFarlane et al. pointed out that the ammonia industry's decarbonization will occur in three phases, with different technologies.21 As shown in Fig. 1, today the “grey ammonia” is exclusively produced from fossil fuels with the enormous release of CO2 into the environment. Therefore, the first generation will utilize carbon capture and storage technology to capture all the CO2 emissions from HBP (and SMR) to produce “purple ammonia”. The second generation will produce “blue ammonia” or “renewable ammonia”. At this stage, the HBP will be electrified with renewable energy and the hydrogen feedstock will be switched from fossil resources to green hydrogen produced by water electrolysis. Eventually, in the third generation, the HBP will no longer be needed as the renewable energy-assisted electrochemical reduction of the N-source and the H-source from water will be commercialized to produce “sustainable ammonia” or “green-ammonia”.
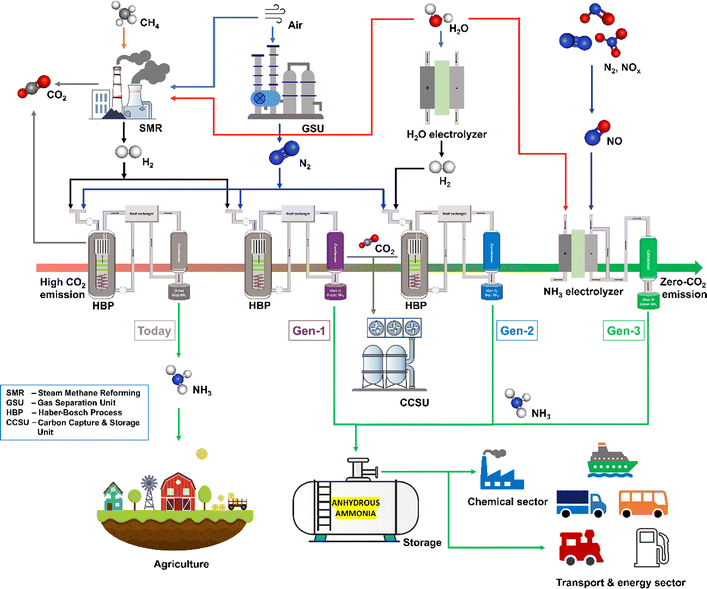 |
| | Fig. 1 Ammonia economy roadmap showing current and projected contributions of the current and Gen 1 (purple), Gen 2 (light blue), and Gen 3 (green) ammonia production technologies. | |
The energy-intensive HBP is carried out under harsh conditions such as high temperature (300–500 °C) and high pressure (150–200 atm). Moreover, each pass in the HBP results in only 15% conversion efficiency and it needs multiple passes to achieve a high conversion efficiency of 97%.27 Therefore, it is a substantial challenge to develop alternative routes to produce NH3 in a sustainable way. Recently, electrochemical NH3 synthesis (EAS) through the nitrogen reduction reaction (NRR) has greatly attracted attention for the production of NH3 under ambient conditions.28,29 The electroreduction of N2 to NH3 only requires protons (H+) from water and the electricity drawn from renewable energy resources, and could be conducted under ambient conditions with no carbon emission.30–34 However, the electrochemical ammonia yield from the NRR is unsatisfactory because of high NN bond dissociation energy, low polarizability, and limited solubility of electrolytes. In addition, the competitive hydrogen evolution reaction (HER) in the NRR potential range (0.148 V vs. RHE) is another bottleneck to attaining high NH3 Faradaic efficiency.30,35 In contrast to N2, nitric oxide (NO) is partly active due to its lower bond energy (NO 607 kJ mol−1, NN 941 kJ mol−1 at 25 °C).36 The electronic structure of NO containing unpaired electrons and the lower energy of its 2π* orbital make it noticeably polar and reactive to undergo electrochemical reactions.37 NO possesses a positive electron affinity (0.024 eV) and a lower ionization potential (9.2 eV) compared to molecular N2 (−1.9 eV and 15.8 eV, respectively), allowing it to be more readily active toward redox reactions.38,39 Moreover, the standard reduction potential for NO to NH3 is about 0.71 V vs. RHE, which is advantageous to attain a high NH3 yield along with high Faradaic efficiency.40,41
Besides, groundwater pollutants (such as NO3− and NO2−) have also been used as reactive nitrogen species for EAS. Meanwhile, this process could be a promising alternative to biological denitrification and physical separation processes for removing nitrate ions from wastewater.42 Despite the electroreduction of nitrogen species that could lead to the formation of various products such as N2, N2O, NH3, and NH2OH, the thermodynamic potentials for NH3 production are as follows:41,43
| | | NO3− + 9H+ + 8e− → NH3 + 3H2O E0 = 0.88 V vs. RHE | (1) |
| | | NO2− + 7H+ + 6e− → NH3 + 2H2O E0 = 0.86 V vs. RHE | (2) |
| | | NO + 5H+ + 5e− → NH3 + H2O E0 = 0.71 V vs. RHE | (3) |
The EAS by the electroreduction of NO
3− and NO
2− is a more energy consuming and complex process compared with the NRR and NO reduction reaction (NORR) owing to the involvement of more number of electrons in the former (
i.e., NO
3− and NO
2− electroreduction requires 8 and 6 electrons, whereas the NORR requires only 5 electrons).
44–49 In addition, NO
3−/NO
2− to NH
3 production proceeds through the formation of ‘NO’ as a key intermediate.
50,51 Besides, NO
3− and NO
2− electroreductions are liquid phase reactions, so it would require additional expenses and systems to separate the electrochemically synthesized NH
3 from the electrolyte. In contrast, EAS through the NORR is advantageous for practical implementation as the NORR can be conducted in the gas phase using a membrane electrode assembly, and the present gas separation and liquefying units in HBP plants can be reused.
On the other hand, mitigating hazardous NOx species from the atmosphere is a substantial challenge as it reacts with atmospheric oxygen and moisture to produce nitrous or nitric acid, which causes acid rain, smog, etc.52 The current technology used to convert hazardous NOx into harmless N2 is selective catalytic reduction (SCR), which uses valuable NH3 and H2 as reductants.53,54 The SCR also requires high temperature and pressure application to reduce NOx to N2, which has no chemical use. Thus, the development of efficient technologies to remove NOx from the atmosphere is a substantial challenge. In this regard, the NORR into NH3, N2, etc., through renewable energy resources such as solar and wind, could be a viable and practical solution. Thus, electrochemical NORR to NH3 production is a kind of “kill two birds with one stone” strategy as it mitigates the NOx pollutant and produces a value-added product. Additionally, as a potential alternative to the traditional HBP, NORR electrolysis will decarbonize the above-mentioned energy, transport, and chemical industries to save the global ecology. However, the practical implementation of this technology is hugely dependent on the development of efficient electrocatalysts that can catalyze the NORR at low overpotentials along with good product selectivity. Hence, this review provides an overview of the NORR mechanism, recent advancements in electrocatalyst development, and factors influencing the NH3 yield and selectivity. In addition, the associated challenges that still need to be addressed for the practical realization of EAS as an alternative to the HBP will be discussed.
Mechanism of NOx reduction
The electrocatalytic NORR process can produce various products such as hydroxylamine (NH2OH), ammonia (NH3), nitrous oxide (N2O), and nitrogen (N2) owing to the multi-electron coupled proton reactions. The thermodynamic potentials for the corresponding products are as follows:| | | NO + 3 (H+ + e−) → NH2OH E0 = 0.38 V vs. RHE | (4) |
| | | NO + 5 (H+ + e−) → NH3 + H2O E0 = 0.71 V vs. RHE | (5) |
| | | 2NO + 2 (H+ + e−) → N2O + H2O E0 = 1.59 V vs. RHE | (6) |
| | | 2NO + 4 (H+ + e−) → N2 + 2H2O E0 = 1.68 V vs. RHE | (7) |
In addition, hydrogen (H2) can be formed at more cathodic potentials (below 0 V vs. RHE). The product selectivity of the NORR is hugely dependent on the catalyst surface, applied potential, ‘NO’ coverage, etc. For instance, the NORR over nanostructured Pd/C has only produced H2, while the PdCu/C catalyst has generated N2 as a main product (∼68%) by forming NH3 and N2H4 as side products.55 Meanwhile, Cu exhibits higher selectivity towards NH3.34 Thus, the chemical composition and the electronic structure of the catalyst are the key to obtaining desirable products. Electrode potential also plays a significant role in product selectivity. Both theoretical and experimental studies on transition metal electrodes manifest that at more positive potentials (>0.3 V) N2O forms as a main product, whereas NH3 can be generated along with H2 at more negative potentials.56 N2 and NH2OH products can be observed at the intermediate potential window. Besides, low ‘NO’ coverage over the catalyst surface preferentially produces single-N products (NH3, NH2OH), while high coverage leads to ‘NO’ dimerization resulting in the N–N coupled product (N2, N2O) formation.57
During the NORR, initially ‘NO’ adsorption on the catalyst surface occurs with different configurations such as N-end, O-end, and side-on. The subsequent electron transfer leads to the formation of various reaction intermediates and products (Fig. 2). The ‘NO’ adsorption to NH3 occurs via either a dissociative or associative pathway. In the dissociative pathway, the first step is splitting the N–O bond on the active site. Then, each adsorbed atom (N* or O*) will be hydrogenated separately to yield NH3 and H2O. In the case of the associative pathway, both ‘N’ and ‘O’ atoms in the NO molecule will be protonated consequently to produce a ‘HxNOHy’ intermediate, which will eventually reduce to NH3. In both routes, the hydrogenation process proceeds via either the Tafel or Heyrovsky-type mechanism. In the former case, prior to the ‘NO’ hydrogenation, the solvated protons will be adsorbed onto the catalyst's surface to from H*, whereas the hydrogenation of ‘NO’ molecules or the intermediates occurs directly on the catalyst surface in the latter case. Based on the type of hydrogenation, the dissociative and associative pathways are further divided into four categories, termed as dissociative-Tafel (D-T), associative-Tafel (A-T), dissociative-Heyrovsky (D-H), and associative-Heyrovsky (H-A) mechanisms.34 Furthermore, the associative mechanisms are classified into distal-O, distal-N, alternating-O, and alternating-N pathways. In the distal pathway, one of the two atoms (N or O) in NO will be completely hydrogenated to produce NH3 or H2O. Thereafter, O* or N* will undergo the hydrogenation process. In contrast, both N and O atoms undergo alternative hydrogenation steps in the alternating pathway.
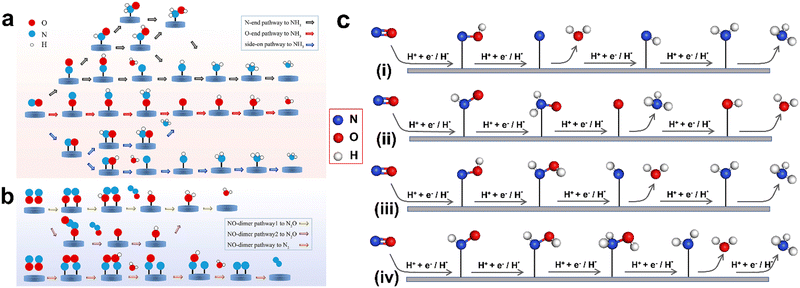 |
| | Fig. 2 (a) Schematic pathways of the NORR process toward NH3 synthesis. (b) Schematic pathways of the NORR process toward N2O/N2 synthesis. Reproduced with permission from ref. 67, copyright 2022, ACS. (c) Scheme of distal-O (i), distal-N (ii), alternating-O (iii), and alternating-N pathways (iv), which are all assigned to the associative mechanism. The H+ + e− and H* represent protons and adsorbed H*, corresponding to the Heyrovsky and Tafel-type hydrogenation, respectively. | |
Due to the involvement of multi-electron transfers, there are numerous varieties of mechanisms, intermediates, and rate-determining steps proposed for the NORR in the literature. The mechanistic studies are initially performed on the Pt surface in an aqueous electrolyte using various techniques such as voltammetry, in situ infrared spectroscopy, and online mass spectroscopy.37,58,59 Earlier, Colucci et al. suggested that continuous NO reduction occurs through the rate-determining step (RDS) of NO* + H+ + e− → NOH*.60 After that, Gootzen et al. demonstrated the involvement of the NO dissociation step (NO* → N* + O*) in the mechanism.61 Meanwhile, the adsorbate reduction (NO adsorbed onto the Pt electrode) and continuous reduction (NO saturated electrolyte) products of the NORR are found to be different. Later, Vooys et al. clarified the mechanistic pathways in both scenarios with the help of kinetic parameters and product selectivity by utilizing the potential, pH, NO surface coverage, and the effect of supporting electrolytes as variables.58 During the adsorbate NO reduction in an acidic medium, NH3 is predominantly formed, whereas the continuous NO reduction yields N2O and NH3 products at above and below 0.4 V, respectively. The similar Tafel slope observed for the first two reduction peaks in adsorbate NO reduction indicates that the first proton-coupled electron transfer is in electrochemical equilibrium, and NH3 will be generated through the N–O bond breaking after the RDS, as given below.
| | | NO* + H+ + e− →HNO* (equilibrium) | (8) |
| | | HNO* + H+ + e− → H2NO* (RDS) | (9) |
| | | H2NO* + 3H+ + 3e− → NH4+ + H2O (fast) | (10) |
At below 0.4 V, the continuous NO reduction also produces the NH
3 in a similar mechanistic pathway. Above 0.4 V, N
2O is produced through the dimerization of NO* with the ‘NO’ present in the solution. In another report, Rosca
et al. proposed that the NORR mechanism on Pt (100) follows the NO → HNO* → NH* + O* → NH
3 + H
2O pathway, in which HNO* → NH* + O* is the RDS.
59 Thereafter, several DFT studies were also carried out to attempt to understand the exact mechanism over the Pt surface.
62–64 However, the conclusive mechanism could not be derived as the kinetic barriers were not considered in these studies.
65
Besides Pt, the NORR mechanism was studied on various transition metals such as Pd, Rh, Ru, Ir, and Au.66 Except Au, all the metals showed high selectivity towards N2O and NH3 at more positive and negative potentials, respectively, like Pt. The similar onset potentials of all metals indicate that the N2O formation occurs through a common mechanism, while the various Tafel slopes manifest the different RDSs over metal surfaces. This could be most likely due to the shift in the occurrence of the first electron-transfer step during the catalytic process. Among all the metals, Pd is found to be the most active catalyst for selective N2 formation via the N2O intermediate. However, the NO to NH3 mechanism over these metals is still unclear.
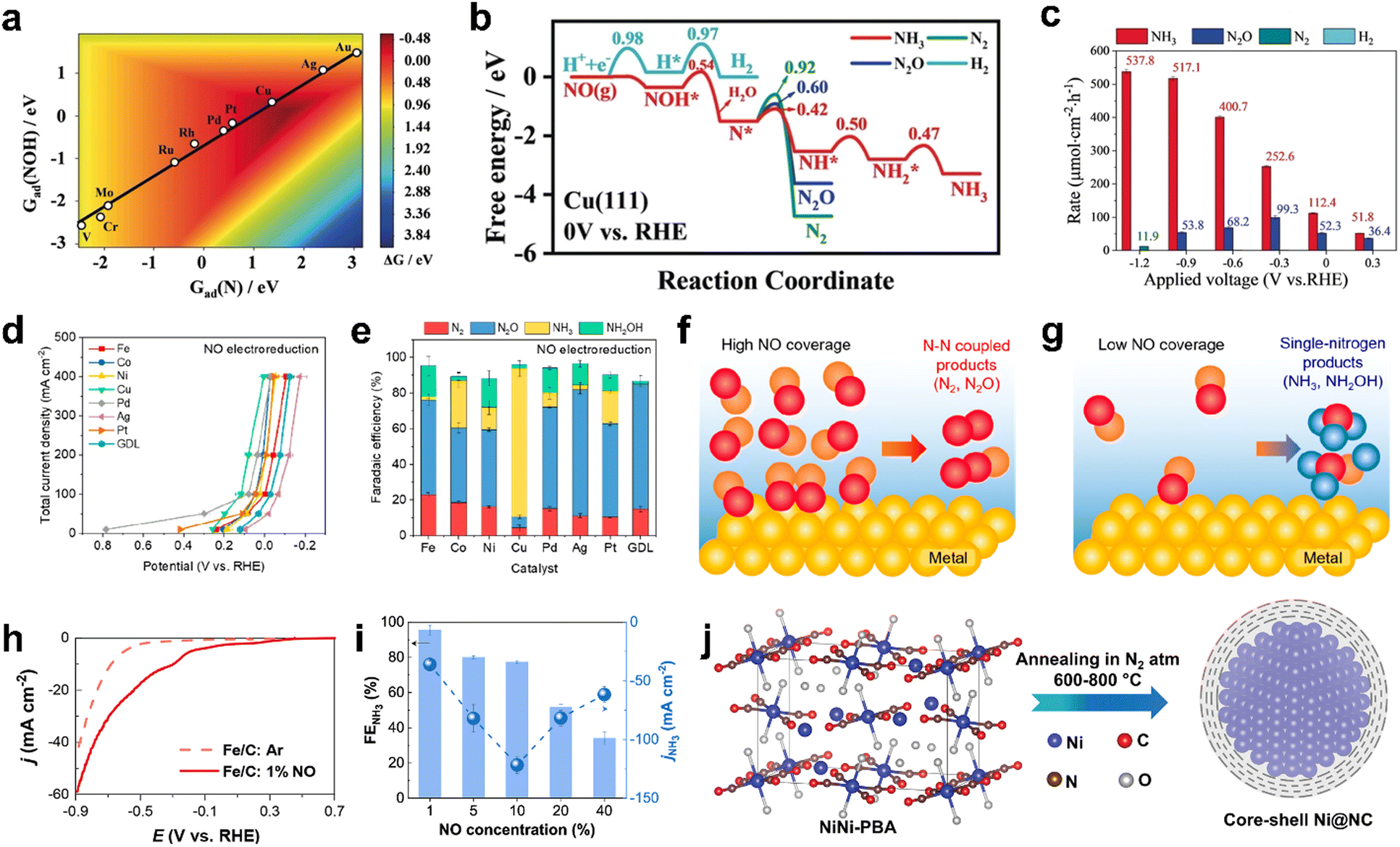
Recently, several DFT studies attempted to correlate the NORR activity of various metals by using the adsorption energies of *NO and *N intermediates as descriptors.43,67,68 Furthermore, to understand the selectivity of NH3 formation over H2, the adsorption energy of *H is also considered. As shown in Fig. 3a, metals present on the left of the vertical line (Pt and Pd) show higher binding ability towards both *NO and *H, leading to dominant H2 production below 0 VRHE. The NH3 formation over the metals located above the horizontal line (Ag and Au) is limited due to the weak binding of *NO. Meanwhile, Cu shows selective binding towards *NO over *H, thereby indicating ideal catalytic behavior towards NH3 formation.43 Furthermore, the limiting potential vs. adsorption energy of *N plot shows that Cu is located on the weak binding side. Cu is predicted to follow the distal-O pathway to produce NH3, and the rate-limiting step is *NO → *NHO (Fig. 3b). The limiting potential of ‘Cu’ can be reduced by adjusting the metal surface with a slightly negative adsorption energy (not higher than −1.5 eV) of *N. Besides, the DFT studies performed on different metal compounds predicted that the NH3 formation occurs through various mechanistic pathways, intermediates, and RDSs.54,69 In transition metal compounds, the significant overlap between the metal d-orbitals and the π orbitals of NO is the primary criterion for the adsorption and activation of NO molecules through the “acceptance–donation” mechanism, i.e., simultaneous depletion and accumulation of electron density in NO orbitals.54 The above studies indicate that the NORR activity and product selectivity are determined by various parameters such as the catalyst surface, operating potential, NO coverage, and hence, the favorable pathways and RDSs over the catalytic materials could differ.
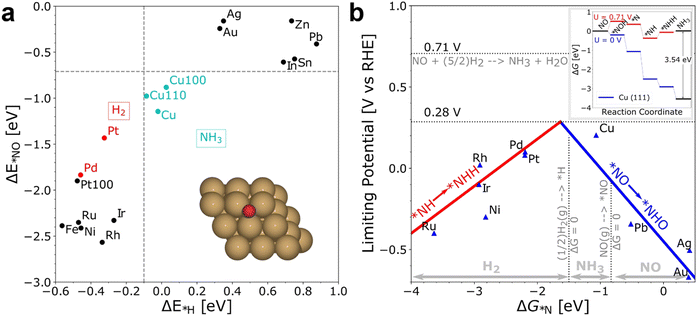 |
| | Fig. 3 (a) The adsorption energies of the intermediates 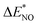 are plotted against are plotted against 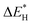 on various metal surfaces. (b) The limiting potential volcano plot for NO reduction to NH3 on various metal surfaces. Reproduced with permission from ref. 43 copyright 2021, Wiley-VCH. on various metal surfaces. (b) The limiting potential volcano plot for NO reduction to NH3 on various metal surfaces. Reproduced with permission from ref. 43 copyright 2021, Wiley-VCH. | |
Electrocatalysts for the NOx reduction reaction (NORR)
Different classes of materials have been developed to investigate their electrocatalytic performance towards the NORR. Based on the chemical composition, the electrode materials can be classified into three categories: precious transition metal-based, non-precious transition metal-based, and p-block element-based electrocatalysts. Meanwhile, several attempts were made to identify the reaction intermediates and unveil the possible reaction mechanism of the NORR over the catalyst surfaces, which will guide the design and production of efficient electrocatalysts with good product selectivity. This section will briefly describe the materials and their catalytic activity in terms of overpotential, selectivity, NH3 yield rates, and Faradaic efficiency (FE). In addition, the efforts toward identifying reaction intermediates to understand the NOx mechanism through computational and experimental studies will be discussed.
1. Precious transition metal-based electrocatalysts
Precious metals such as Pt, Pd, and Ru are renowned for numerous electrocatalytic applications owing to their high electronic conductivity and the ability to bind the various reaction intermediates on their polycrystalline surfaces.70 Despite these metals being found to be active catalysts towards the NORR, the product selectivity is hugely varied as the catalytic surfaces possess different inherent Gibbs free energies for adsorption of NO* and H* intermediates. In the 20th century, the research was extensively focused on the mechanistic investigation of NORR activity over platinum surfaces. In contrast, the recent focus has shifted towards developing efficient electrocatalysts with abundant active sites, optimal Gibbs free energies for adsorption of intermediates to attain high ammonia (NH4+) yield rates, and high FEs at lower overpotentials.
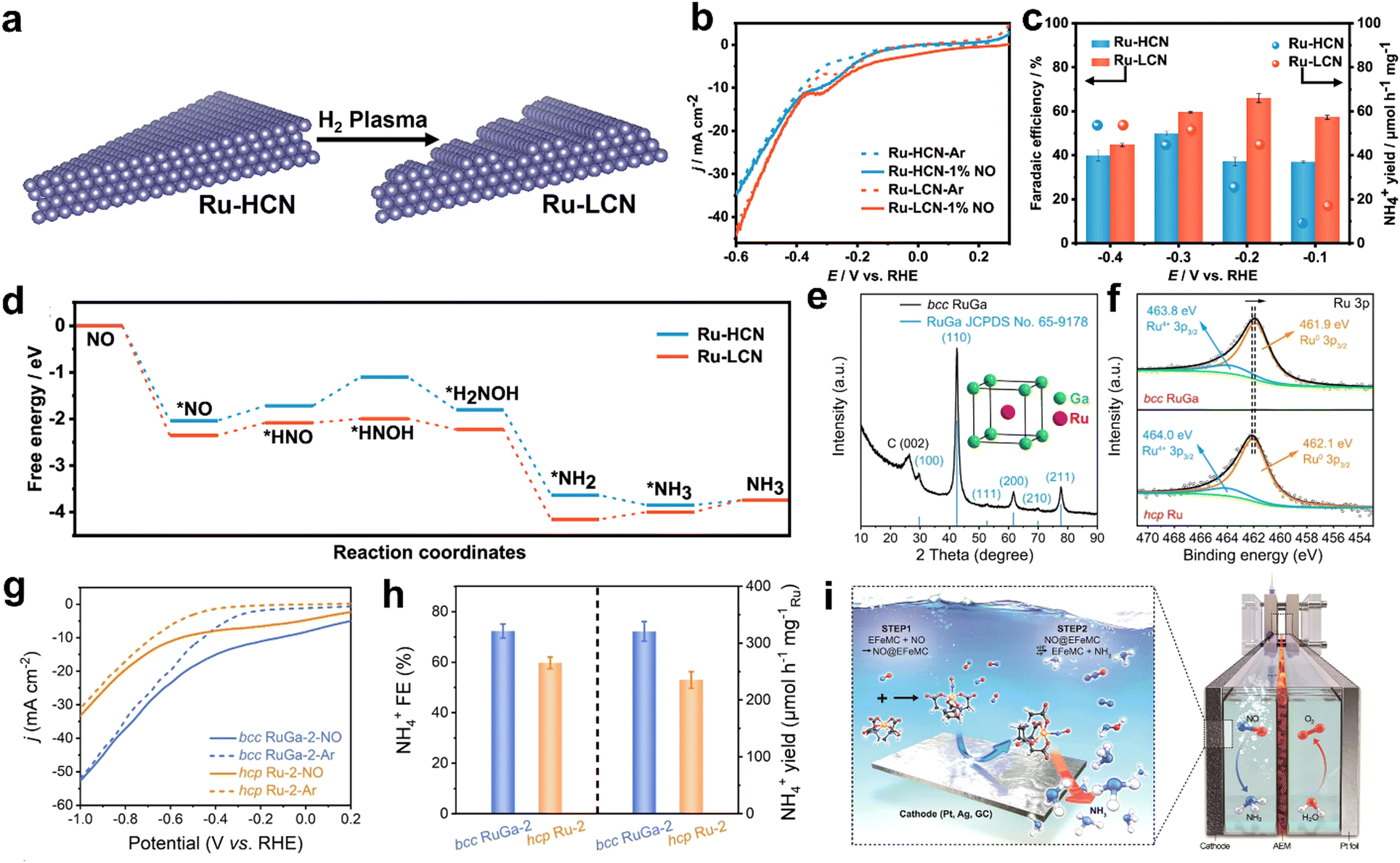
For instance, Yu and co-workers have prepared Ru nanosheets with low coordination numbers (Ru-LCN) through the plasma treatment of Ru nanosheets with a high coordination number (Ru–HCN) (Fig. 4a).71 As shown in Fig. 4b, Ru-LCN possesses enhanced NORR activity as compared to Ru-HCN in 1% NO (99% argon)-saturated 0.5 M Na2SO4 electrolyte. At an optimal potential of −0.2 V vs. RHE, Ru-LCN achieved a high NH4+ yield rate (45.03 μmol h−1 mg−1) and FE (65.96%) in a H-type electrolytic cell (Fig. 4c). Moreover, the FE remains unaltered when the feeding gas is replaced with 99.9% NO instead of 1% NO, which is beneficial to produce NH3 with a wide range of pollutant concentrations. In addition, they demonstrated an improved NH4+ yield rate of up to 84.20 μmol h−1 mg−1 at −0.2 V vs. RHE by constructing a flow electrolyzer (cathode: Ru-LCN, feed gas: 1% NO). The combined in situ measurements and DFT results indicated that the NH3 molecule formation over the Ru surface has occurred through the alternating-N mechanistic pathway. As shown in Fig. 4d, the low coordination number of Ru not only helps in the NO* adsorption onto the Ru surface but also facilitates the subsequent hydrogenation step (PDS) i.e., NO* to HNO*, thereby enhancing the NORR performance. However, the yield rate of Ru-LCN is not sufficiently high to meet the practical ammonia production rate. Recently, Lu et al. observed enhancement in the NORR activity of Ru active sites through the electronic structure optimization by forming an RuGa intermetallic compound (RuGa IMC).72 Furthermore, RuGa nanoparticles were anchored onto the three-dimensional interconnected n-doped reduced graphene oxide nanosheets to improve the reaction kinetics via thermal annealing. Interestingly, the incorporation of Ga into Ru has altered the crystal structure from fcc-Ru to bcc-RuGa in which the isolated Ru atoms are occupied at the center of cubes (Ga atoms occupied at the vertex) (Fig. 4e). Meanwhile, an electron-rich Ru atom has been created through the electron transfer from Ga to Ru in RuGa IMC (Fig. 4f). As shown in Fig. 4g and h, bcc-RuGa exhibits an extraordinary NORR performance (NH4+ yield rate: 320.6 μmol h−1 mg−1Ru and FE: 72.3%), which is superior to that of fcc-Ru (yield rate: 235.4 μmol h−1 mg−1Ru, and FE: 59.8%) in 20% NO-saturated 0.1 M K2SO4 at −0.2 V vs. RHE. In another report, Shi et al. also showed higher intrinsic NORR activity of the Ru0.05Cu0.95 alloy prepared through the coprecipitation of RuxCu1−x(OH)2, followed by an electrochemical reduction step.73 Online differential electrochemical mass spectrometry (DEMS) confirms that ammonia (NH3) is formed over the alloy through the alternating-N mechanistic pathway. Besides, Kim et al. have achieved the selective NH4+ formation using a nanostructured silver (Ag) electrode with nearly 100% FE in a large potential window (0.04 to −0.34 V vs. RHE) by using an EDTA-Fe2+ metal complex (EFeMC) electrolyte that is saturated with ‘NO’.74 They claimed that initially the ‘NO’ molecule gets trapped in the metal complex and subsequent proton-coupled electron transfer from the silver electrode to the metal complex occurs through the outer sphere electron transfer to produce NH3 along with the regeneration of EFeMC (Fig. 4i). During electroreduction, Ag stabilizes the reaction intermediates such as [*(II)-HNO, *(II)-NHOH, and *(II)-NH2OH, where *(II) = EFeMC] through donor–acceptor interaction by binding with the oxygen atoms of intermediates, which are trapped in the metal complex. In addition, the performance and durability of the Ag electrode are tested in a flow cell with the EFeMC electrolyte. In the flow cell, the NH4+ production rate has reached as high as 3.6 mol m2 h−1, along with 100% FE at −0.165 V vs. RHE and the same performance has been maintained for 100 h. Since they have used an additional metal complex in the electrolyte, it is especially important to estimate the economy of the system as compared to the commercial ammonia production cost. The economic analysis suggests that the NH3 production cost through the NORR in the EFeMC-designed electrolyte is 2.5 times higher than the commercial price of ammonia but can be market competitive with an electricity price of $0.03 kW h−1 and with a current density of >125 mA cm−2. Thus, designing efficient electrocatalysts and integrating the electrolyzer with renewable energy sources could be a viable solution for implementing the EFeMC-electrolyte based NORR electrolyzer. In another report, Choi et al. have shown a notable NORR performance from commercial 20 wt% Au supported on Vulcan carbon to produce NH4+ at a relatively lower overpotential (−0.3 V), whereas the electrode is inert towards the NRR.75 Xiong et al. reported improved NORR activity from electrochemically synthesized Au/rGO electrodes. Ferrous citrate (FeIICit) solution was chosen as an electrolyte for the ‘NO’ complexation.76 The NH3 yield of Au/rGO increased linearly with the concentration of FeIICit in the electrolyte, while maintaining the same FE. This indicates that the coordinated NO within the metal complex can only participate in electroreduction, which could solve the limited solubility problem of ‘NO’ in the aqueous electrolyte. Meanwhile, they observed that the FE of NH3 can be maximized up to 98% with the adjustment of electrolyte pH to 1, as the key pronation step of *NOH is more facile in an acidic medium. Furthermore, they hypothesized that selective NH3 formation over the Au surface is due to the stabilization of *N and *NH intermediates. At −0.47 V vs. RHE, an excellent NH4+ yield rate of 438.8 μmol cm−2 h−1 and FE of 93.2% were achieved from 7.6 wt% atomically dispersed iridium (Ir) confined in amorphous MoO3 (Ir/MoO3).77 In this material, the Ir–O5 moiety is found to be the active site for preferential binding of ‘NO’ over ‘H’ and possesses a very low energy barrier (0.01 eV) for the rate determining *NHO → *NHOH step, thereby delivering the high NH3 yield rates and FE. Table 1 summarizes the NORR activity of different precious transition metal-based electrocatalysts.
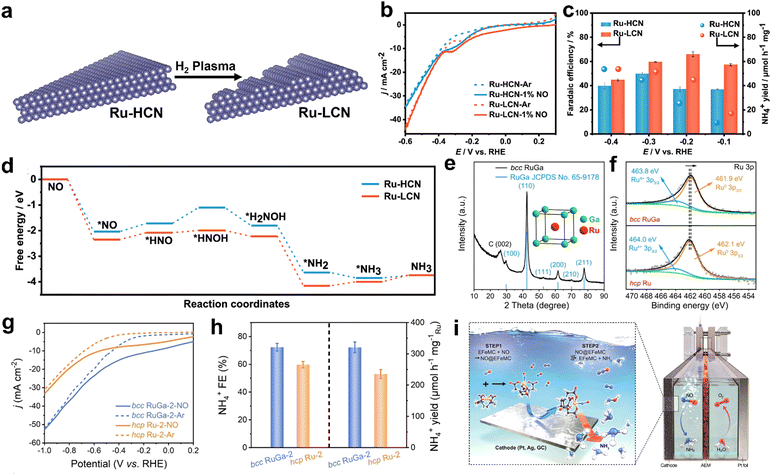 |
| | Fig. 4 (a) Diagram for the preparation of Ru-LCN via plasma treatment on Ru-HCN, (b) LSV curves of Ru-HCN and Ru-LCN at a scan rate of 10 mV s−1 in Ar- and 1% NO-saturated 0.5 M Na2SO4 solution, (c) Faradaic efficiency and yield rate of NH4+ with 1% NO over Ru-HCN and Ru-LCN at each given potential, and (d) reaction Gibbs free energy diagrams of the NORR over Ru-HCN and Ru-LCN on FCC (111) surfaces at 0 V vs. RHE. Reproduced with permission from ref. 71 copyright 2022, ACS. (e) XRD pattern of bcc RuGa IMCs on N-rGO nanosheets, the inset is the crystal structure (unit-cell) model of bcc RuGa IMCs, (f) high-resolution Ru 3p XPS spectra of bcc RuGa IMCs and hcp Ru NPs, (g) LSV curves of bcc RuGa-2 IMCs and hcp Ru-2 NPs in Ar-saturated (dashed lines) or NO-saturated (solid lines) 0.1 M K2SO4 electrolyte, and (h) NH4+ Faradaic efficiencies and NH4+ yield rates over bcc RuGa-2 IMCs and hcp Ru-2 NPs at 0.2 V in NO-saturated 0.1 M K2SO4 electrolyte. Reproduced with permission from ref. 72 copyright 2023, Wiley-VCH. (i) Schematic diagram showing NO capture by EFeMC present in the electrolyte and its electrochemical reduction to ammonia. Reproduced with permission from ref. 74 copyright 2020, ACS. | |
Table 1 Summary of the NORR activity shown by different precious transition metal-based electrocatalysts
| Catalyst |
Synthetic route |
Electrolyte |
NH3 yield rate |
FE (%) |
Potential (vs. RHE) (V) |
Ref. |
| Ru-LCN |
Plasma treatment |
0.5 M Na2SO4 (1% NO) |
45.03 μmol h−1 mg−1 |
65.96 |
−0.2 |
71
|
| RuGa IMCs |
Thermal annealing method |
0.1 M K2SO4 (20% NO) |
320.6 μmol h−1 mg−1Ru |
72.3 |
−0.2 |
72
|
| Ru0.05Cu0.95 |
Coprecipitation–electrochemical reduction |
0.5 M Na2SO4 (20% NO) |
17.68 μmol h−1 cm−2 |
64.9 |
−0.5 |
73
|
| Nanostructured Ag electrode |
Chlorination/dechlorination method |
PBS-EDTA-Fe2+ metal complex (99.9% NO) |
2.29 mol h−1 m−2 |
∼100 |
−0.34 |
74
|
| 20 wt% Au/C |
Commercial |
0.5 M K2SO4 (1 mM NO) |
8.1 ± 0.9 × 102 pmol s−1 cm−2 |
N. A. |
−0.30 |
75
|
| Au/rGO |
Two-step electrochemical reduction |
Citrate–Fe2+ complex (2% NO) |
14.6 μmol h−1 cm−2 |
65.2 |
−0.1 |
76
|
| Ir/MoO3 |
Supercritical CO2 approach |
NO-saturated 0.5M Na2SO4 (99.9% NO) |
439 μmol h−1 cm−2 |
93.2 |
−0.47 |
77
|
2. Non-precious transition metal-based electrocatalysts
(i) Zero valent transition metals.
Despite the high NORR performance of precious metals, their utilization for large-scale NH3 production is hindered by the low abundance and high electrode cost. This demands the exploration of other alternatives for realizing the practical implementation of the NORR to NH3 technology. In this context, Long et al. have performed computational screening over different transition metal catalysts towards NORR by choosing the N* adsorption free energy [Gad(N)] as an activity descriptor.34 Among the chosen transition metals, copper (Cu) is found to be the ideal candidate for selective NH3 production through the NORR (Fig. 5a). In addition, the NH3 formation over the Cu (111) surface is predicted to occur via an associative Heyrovsky distal-O mechanism (AHDO), in which the protonation of NOH* (0.54 eV) is the rate-limiting step. As shown in Fig. 5b, the high energy barriers for N–N, N–NO, and H–H couplings, as compared to N* continual protonation steps over Cu (111), have successfully eliminated the formation of N2, N2O, and H2 side products, thereby leading to high activity and selectivity towards NH3 production. They have validated the above predictions by performing electrocatalytic NORR experiments on copper substrates. A porous Cu foam has shown excellent NORR activity to produce NH3 as a dominant product in a wide potential range from −1.2 to 0.3 V vs. RHE in 0.25 M Li2SO4 (Fig. 5c). A maximum NH3 formation rate of about 517 μmol cm−2 h−1 along with high FE (93.5%) was achieved at −0.9 V vs. RHE and the activity was maintained for 100 hours. In contrast, hardly any NH3 is formed when the Cu foam electrode is subjected to the NRR, thereby indicating the competitiveness of the NORR over the NRR on the Cu surface. Later, Chen et al. used the chemical reduction method for uniform decoration of ultrasmall ‘Cu’ nanoparticles onto the porous TiO2, which was synthesized by the sol–gel method.78 The electrode exhibited a high NH3 yield of 3520.80 μg h−1 mg−1 and FE of 86.49% at −0.3 V vs. RHE in ‘NO’-saturated 0.1 M K2SO4. The improved surface area and porous channels of TiO2/Cu have improved the ‘NO’ molecule adsorption, leading to high NH3 production. Nevertheless, the poor solubility of NOx in electrolytes is another roadblock to further development. This necessitates evaluating the NORR activity of electrocatalysts in a flow cell configuration by supporting them onto gas diffusion electrodes (GDEs) to alleviate the mass transport limitation. Ko et al. have demonstrated the gaseous NOx (NO and N2O) reduction at high reaction rates (400 mA cm−2) over various transition metals deposited on GDEs in a gas-fed three-compartment flow cell.57 In contrast to the previous reports, all the transition metals showed good electrocatalytic performance towards the NORR, while the product selectively is hugely varied (Fig. 5d). The higher activity shown by all metals could be due to the NO-saturated environment in the flow cell (100% NO). Among all the metals, Cu showed excellent selectivity (>80%) towards NH3 from the NORR, whereas other metals, such as Fe, and Pd, have produced N2O as a major product in an alkaline medium (Fig. 5e). Besides, Cu also showed good N2ORR activity and it has selectively produced N2 as a reduction product. In addition, they have studied the effect of NO partial pressure in a flow cell on product selectivity. It was found that irrespective of the electrocatalyst, N–N coupled products (N2 and N2O) and single-N products (NH3 and NH2OH) will be formed at low and high ‘NO’ coverages, respectively, thereby indicating the importance of the NO concentration over the catalyst surface towards product selectivity (Fig. 5f and g). In addition, they showed the pH dependency of the NORR towards NH3 FE over Cu. It could deliver nearly 100% FE at pH = 0.5, as the first proton transfer step of NO (*NO + H+ ↔ *NOH) is the rate determining step (RDS) of NH3. Besides, NH3 production from low concentrated ‘NO’ is of practical significance, especially while using flue gas as a NO source in the flow cell. Cheon et al. also reported excellent NORR activity from zero valent iron (Fe)-incorporated carbon black, which is supported on GDE at low NO concentrations (1–10%).79 As shown in Fig. 5h, Fe/C-supported GDE has achieved a maximum FENH3 of 91% at −0.4 V vs. RHE and jNH3 of 19 mA cm−2 at −0.6 V vs. RHE, respectively, with 1% NO as a feed gas in a GDE cell. In addition, a maximum jNH3 of 122 mA cm−2 was achieved at 10% NO (FE: 77%). A further increase in the NO concentration led to a decrease in jNH3 and FE, owing to the formation of N–N coupled products (N2 and N2O) at high ‘NO’ coverage over the electrode surface (Fig. 5i). The computational calculations revealed that the ammonia formation over the Fe (110) surface occurred through the breaking of the N–O bond in the H2NO intermediate. Although the NH3 yield rate of Fe/C-supported GDE is significantly improved in 0.5 M H2SO4 compared to PBS buffer, the dissolution of Fe in an acidic medium causes a decline in activity during long term NO electrolysis. The above studies show that the NH3 production rate is significantly higher in the acidic medium than in neutral and alkaline electrolytes, owing to the facile protonation steps in the acidic medium. However, transition metals tend to dissolve in acidic environments, which causes catalyst degradation. Hence, it is necessary to protect the catalytic metal centers for durable ammonia production. To resolve this issue, our group attempted to protect catalytic Ni centers with a thin graphitic carbon coating by making a core–shell Ni@NC architecture via one-step pyrolysis of the Ni[Ni(CN)4] PBA precursor at different temperatures from 600 to 800 °C (Fig. 5j).40 Among them, Ni@NC synthesized at 800 °C produced the highest NH3 yield rate of 34.6 μmol cm−2 h−1 with a high FE (72.3%) at a low overpotential of 550 mV (0.16 V vs. RHE) and maintained the catalytic activity for 24 h in 0.1 M HCl. In contrast, bare Ni nanoparticles displayed a moderate NH3 yield rate (8.9 μmol cm−2 h−1) and they were etched away from the electrode surface during the NORR measurements. The improved activity of Ni@NC is attributed to the facile ionic/gaseous transport through the porous multilayer NC shell to the catalytically active Ni core. In addition, the anti-corrosive property of the multilayer NC shell retards the dissolution of the Ni core in an acidic medium, thereby ensuring the stability of Ni@NC. Furthermore, a solar powered NORR/OER electrolyzer (constructed by employing Ni@NC and RuO2 as the cathode and anode, respectively) has achieved a NH3 FE of > 50%, with a solar-to-fuel (STF) efficiency of ≈1.7%. Another work observed a stable NORR performance from N-doped carbon-wrapped nickel nanoparticles supported onto the carbon fiber (NiNC@CF).80 Initially, nanoflake-like Ni-MOF was directly grown onto the CF via a solvothermal method. The subsequent thermal pyrolysis led to the decomposition of organic matter, producing a zero-valent Ni nanoparticle-dispersed porous N-doped carbon microstructure on CF. The resulting electrode achieved a maximum FE of 87% with an NH3 yield rate of 94 mmol cm−2 h−1 at −0.5 V vs. RHE and showed good durability in the PBS electrolyte (pH 7). Meanwhile, the corresponding prototype two-electrode electrolyzer (NORR/OER) has delivered a NH3 yield rate of 27 mmol cm−2 h−1 with the highest FE (80%), at a cell potential of 2.75 V. Wang and coworkers have attained a high NH3 yield of 439.5 μmol cm−2 h−1 (1465.0 μmol h−1 mgcat−1) and an FE of 72.58% at −0.6 V vs. RHE from the hexagonal-close-packed cobalt nanosheets (hcp-Co) prepared through a facile hydrothermal method.81 In contrast, the face-centered cubic phase of the Co nanosheets (fcc-Co) showed inferior activity (NH3 yield: 142.10 μmol cm−2 h−1 and FE: 57.12%), thereby indicating that the crystal structure/phase of the material has a significant
role in the NORR. The DFT studies unveil that hcp-Co activates the adsorbed ‘NO’ molecule through the donation of 0.6 e− through d–π* orbitals and facilitates the rate determining first protonation step (NO* → NOH*) with a low energy barrier of 0.32 eV. Despite both hcp-Co and fcc-Co possessing a similar energy barrier for the RDS, the proton diffusion/shuttling is highly favourable over hcp-Co (−0.80 eV) as compared to fcc-Co (−0.67 eV). Hence, the improved activity of hcp-Co could be attributed to the expedited protonation kinetics during the NORR. Table 2 summarizes the NORR activity of different zero valent non-precious transition metal-based electrocatalysts.
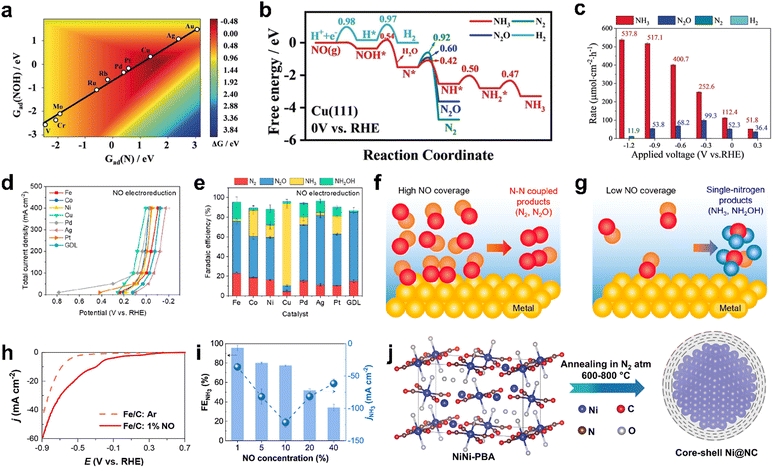 |
| | Fig. 5 (a) A two-dimensional activity map of different metals for ammonia production. All the reaction free energies are shown at 0 V vs. RHE, (b) free-energy diagrams for the HER, NORR to NH3, N2O, and N2 over Cu (111) under 0 V vs. RHE, the kinetic barriers are shown in eV, and (c) reaction rates of the NORR on Cu foam at various potentials. Reproduced with permission from ref. 34 copyright 2020, Wiley-VCH. (d) Total current density vs. potential plots of various metal catalysts towards NO electroreduction performance, (e) Faradaic efficiency of various catalysts at 0.10 ± 0.02 V vs. RHE for NO electroreduction, and (f and g) schematics showing the effect of NO coverage on product selectivity in high and low NO coverages. Reproduced with permission from ref. 57 copyright 2022, ACS. (h) Linear sweep voltammetry scan of the Fe/C-incorporated GDE cell for 1% NO reduction using 0.5 M PBS electrolyte (pH 7), (i) the effect of NO concentration on NH3 faradaic efficiency and NH3 partial current density in a NO electrolyzer operated at −0.6 V vs. RHE and 0.5 M PBS (pH 7). Reproduced with permission from ref. 79 copyright 2022, ACS. (j) Schematic synthesis route of Ni@NC from NiNi-PBA. Reproduced with permission from ref. 40 copyright 2022, Wiley-VCH. | |
Table 2 Summary of the NORR activities shown by different zero valent non-precious transition metal-based electrocatalysts
| Catalyst |
Synthetic route |
Electrolyte |
NH3 yield rate |
FE (%) |
Potential (vs. RHE) (V) |
Ref. |
| Cu foam |
Commercial |
0.25 M Li2SO4 (30 ml min−1) |
517.1 μmol cm−2 h−1 |
93.5 |
−0.9 |
34
|
| TiO2/Cu |
Sol–gel followed by chemical reduction |
0.1 M K2SO4 (10% NO) |
3520.80 μg h−1 mg−1 |
86.49 |
−0.3 |
78
|
| Cu Ti fibers |
Air spraying |
0.05 M Na2SO4 (10% NO) |
400 μmol cm−2 h−1 |
90 |
−0.6 |
82
|
| Copper nanoparticles (GDE) |
Commercial |
0.1 M NaOH + 0.9 M NaClO4 (100% NO) |
1246 μmol cm−2 h−1 |
78 |
0 |
57
|
| Fe/C-incorporated GDE |
Physical mixture of Fe and C |
0.5 M PBS solution (10% NO) |
908 μmol cm−2 h−1 |
77 |
−0.6 |
79
|
| Ni@NC |
Pyrolysis of NiNi-PBA |
0.1 M HCl (99.9% NO) |
34.6 μmol cm−2 h−1 |
72.3 |
0.16 |
40
|
| NiNC@CF |
Solvothermal + annealing |
0.5 M PBS solution (pH 7) (99.9% NO) |
94 mmol cm−2 h−1 |
87 |
−0.5 |
80
|
| hcp-cobalt nanosheets |
Hydrothermal method |
0.1 M Na2SO4 |
439.5 μmol cm−2 h−1 |
72.58 |
−0.6 |
81
|
(ii) Multi-valent transition metal compounds.
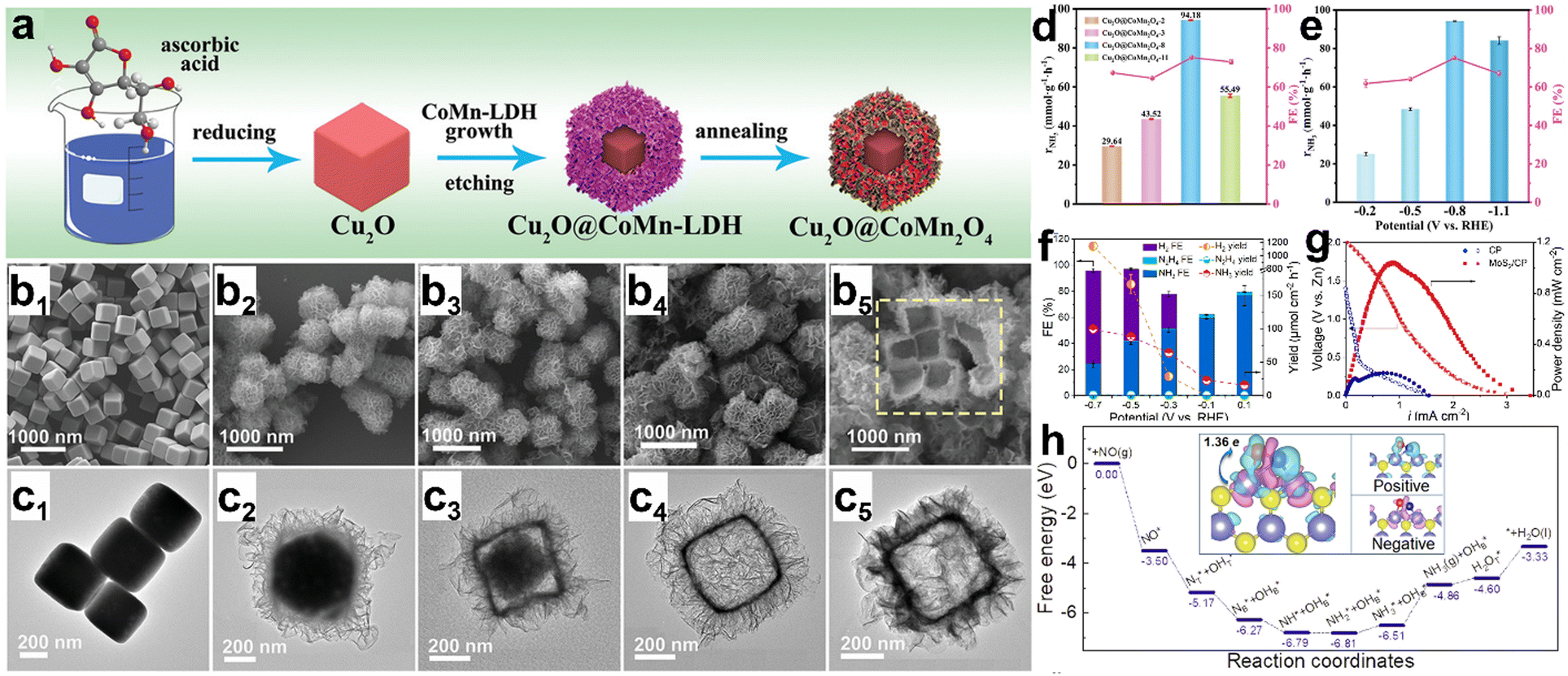
The NORR activity and the NH3 selectivity hugely depend on the electronic structure, morphology, and vacancies of electrocatalysts. In this regard, various metal compounds, such as oxides, sulfides, and phosphides, have been explored and it is found that all the materials can perform NORR activity with high NH3 selectivity. As the biological reduction of NO into N2O is catalyzed by an iron-centered heme group of ‘NO reductase’ Fe-based electrocatalysts are expected to show good ‘NO’ binding and perform the subsequent reduction.83–85 Sun et al. have directly grown a uniform Fe2O3 nanorod array onto a 3-dimensional carbon paper (Fe2O3/CP) and evaluated its NORR activity with diluted NO (10%).86 In addition, to improve the ‘NO’ solubility, they introduced Fe(II)EDTA complex into the 0.1 M Na2SO4 electrolyte. Fe2O3/CP has shown a good NH3 yield of 41.6 μmol h−1 cm−2 (FE: 86.73%) at −0.4 V vs. RHE and maintained the activity for 12 h. Meanwhile, they found that NO3− contamination (resulting from the reaction between NO and O2 followed by the dissolution in water) in the electrolyte causes interference of cathodic currents, thereby hindering the NORR activity judgment of electrodes. Hence, it is necessary to perform NORR in gas-tight (without the aeration of NO) cells to obtain reliable results. Besides, they demonstrated the NH3 production (145.28 mg h−1 mgcat−1) at a high-power density (1.18 mW cm−2) from the Zn–NO battery, which is assembled using Fe2O3/CP and a zinc plate as the cathode and anode, respectively. DFT studies predict that ‘NO’ strongly binds onto the Fe2O3 (104) surface through an “acceptance–donation” mechanism and subsequently gets activated through the 2p* back-donation effect to give rise to NH3 molecule formation. The same group has shown boosted NORR activity using an oxygen vacancy (VO)-rich MnO2 nanowire array (MnO2−x NA) supported on a Ti mesh in 0.2 M Na2SO4.87 The VO in MnO2−x NA is created by annealing the as prepared MnO2 NA in an Ar atmosphere at 350 °C. MnO2−x NA has achieved a high NH3 yield (27.51 × 10−10 mol s−1 cm−2) and FE (82.8%) as compared to pristine MnO2 NA (8.83 × 10−10 mol s−1 cm−2, 44.8%) at −0.7 V vs. RHE, thereby highlighting the advantage of VO in MnO2−x for enhanced NORR activity. DFT calculations reveal that the hydrogenation step of MnO2 (211) is associated with a high energy barrier due to the strong binding (−1.4 eV) of ‘NO’. In contrast, VO in the MnO2−x (211) surface allows the moderate binding (−0.5 eV) of ‘NO’ molecules, which makes the subsequent hydrogenation step more facile. Similarly, an oxygen vacancy-rich TiO2 nanoarray supported on a Ti plate (TiO2−x/TP) has shown superior NORR activity compared to TiO2/TP as the ‘NO’ adsorption is more facile in the former.88 Encouragingly, an open nano array structure of TiO2−x/TP enables the exposure of abundant active sites for generating greater NH3 yields. Wu et al. have achieved an exceptionally high NH3 yield rate of 20 mg h−1 cm2 with nearly unit FE from CoNi(5:5)Ox@Cu at −0.68 V vs. RHE.89 Moreover, the material maintained its NORR activity for 36 h. The in situ Raman spectroscopic and DFT studies concurrently manifest that ‘NO’ most likely adsorbs onto the Co site of CoNi(5:5)Ox@Cu more spontaneously as compared to individual Co3O4@Cu and NiO@Cu. Subsequently, NH3 is selectively produced through the O-end distal pathway, as the energy barriers are significantly high for the formation of other products (N2, H2, N2H4, etc.). Besides, the confinement of reaction intermediates in nanoreactors is proven to be a prudent approach for efficient NORR. Bai et al. have developed a synthetic strategy to prepare hollow Cu2O@CoMn2O4 nanoreactors from a Cu2O nanocube template through the simultaneous etching of Cu2O and growth of CoMn-LDH followed by annealing in Ar (Fig. 6a).90 Meanwhile, they found that the NO adsorption ability linearly increases with nanoreactor internal space, which can be controlled by the etching time (Fig. 6b and c). The void-confinement effect of the Cu2O@CoMn2O4 nanoreactors is beneficial to maintain high reactant concentration and intermediate confinement inside the nanoreactor space and improve the electron transfer efficiency. The optimized material Cu2O@CoMn2O4-8 (etched for 8 min) has shown remarkable NORR activity with a high NH3 production rate of 94.18 mmol g−1 h−1 and an FE of 75.05% at −0.8 V vs. RHE (Fig. 6d and e). Zhang et al. found that 2H-MoS2 flakes grown on graphite felt (MoS2/GF) can efficiently convert NO into NH3 in an acidic electrolyte.54 As shown in Fig. 6f and g, the electrode has shown a maximum NH3 yield rate of 99.6 μmol h−1 cm−2 (FENH3 < 30%) at −0.7 V vs. RHE from the electrocatalytic NORR and the corresponding Zn–NO battery has achieved a power density of 1.04 mW cm−2 (an NH3 yield of 411.8 μg h−1 mgcat−1). DFT studies unveiled that ‘NO’ adsorption on MoS2 (101) occurs through an ‘acceptance–donation mechanism’ and the subsequent activation is enabled by the electron transfer from MoS2 to the anti-bonding orbital of ‘NO’ (Fig. 6h). However, the competitiveness of the HER over the NORR on the MoS2 surface at high cathodic potentials resulted in low FENH3. To suppress the HER on MoS2, Liu's group performed NORR in an ionic liquid (IL)-based electrolyte, which can significantly limit the proton concentration over the electrode surface.91 They regulated the electronic structure of MoS2via phosphorous (P) doping to enhance the NH3 yield. As expected, FENH3 on P-MoS2 nanospheres in 1-butyl-1-methypyrrolidinium tris(pentafluoroethyl)trifluoro phosphate increased up to 70% and achieved an NH3 yield rate of 237.7 μg h−1 mgcat−1 at −0.6 V vs. RHE, which is superior to that of pristine MoS2. Additionally, the computational studies showed that the associated energy barriers from NO adsorption to NH3 formation on the MoS2 surface are significantly lowered after P-doping, which could be the reason for such an enhanced NH3 yield from P-MoS2. Besides doping, creating sulfur vacancies in metal sulfides is also proven to be an effective way to enhance the electrocatalytic NORR performance. The sulfur vacancy-rich CoS1−x nanosheets prepared through the plasma treatment have shown an NH3 yield rate of 44.67 μmol cm−2 h−1 (FE: 54%) at −0.4 V vs. RHE in 0.2 M Na2SO4.92 The ‘S’ vacancy-free CoS counterpart could only display an NH3 yield rate of 27.02 μmol cm−2 h−1 (FE: 37%), as it possesses low active site density (reflected from double layer capacitance (Cdl) measurements) as compared to CoS1−x. In addition, the ‘NO’ activation barrier is found to be relatively lower on CoS1−x (100) facets, which may be attributed to the localization of electrons around the sulfur vacancies.
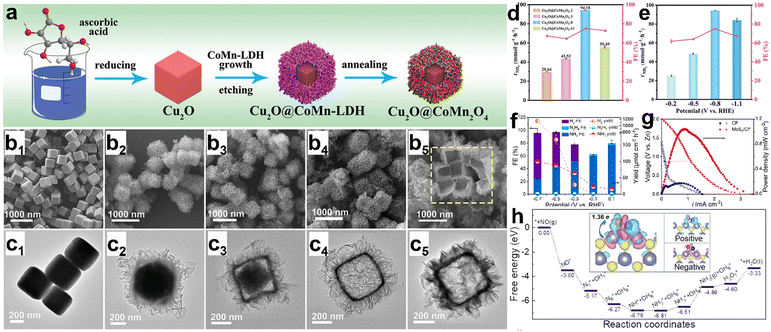 |
| | Fig. 6 (a) Schematic illustration of the preparation of a hollow Cu2O@CoMn2O4−x nanoreactor. SEM images of (b1) Cu2O, (b2) Cu2O@CoMn2O4-2, (b3) Cu2O@CoMn2O4-3, (b4) Cu2O@CoMn2O4-8 and (b5) Cu2O@CoMn2O4−x-11. TEM images of (c1) Cu2O, (c2) Cu2O@CoMn2O4-2, (c3) Cu2O@CoMn2O4-3, (c4) Cu2O@CoMn2O4-8 and (c5) Cu2O@CoMn2O4-11, (d) NH3 production rate and selectivity of Cu2O@CoMn2O4−x, and (e) NH3 production rate and selectivity of Cu2O@CoMn2O4-8. Reproduced with permission from ref. 90 copyright 2022, Wiley-VCH. (f) Product distribution for MoS2/GF at each given potential in a NO-saturated 0.1 M HCl electrolyte containing 0.5 mM FeIISB, (g) polarization and power density plots of CP and MoS2/CP, and (h) free energy landscape for NORR on MoS2 (101). The charge density differences for the adsorbed NO are displayed as insets and cyan (positive) and red (negative) regions represent electron accumulation and loss, respectively. Reproduced with permission from ref. 54 copyright 2021, Wiley-VCH. | |
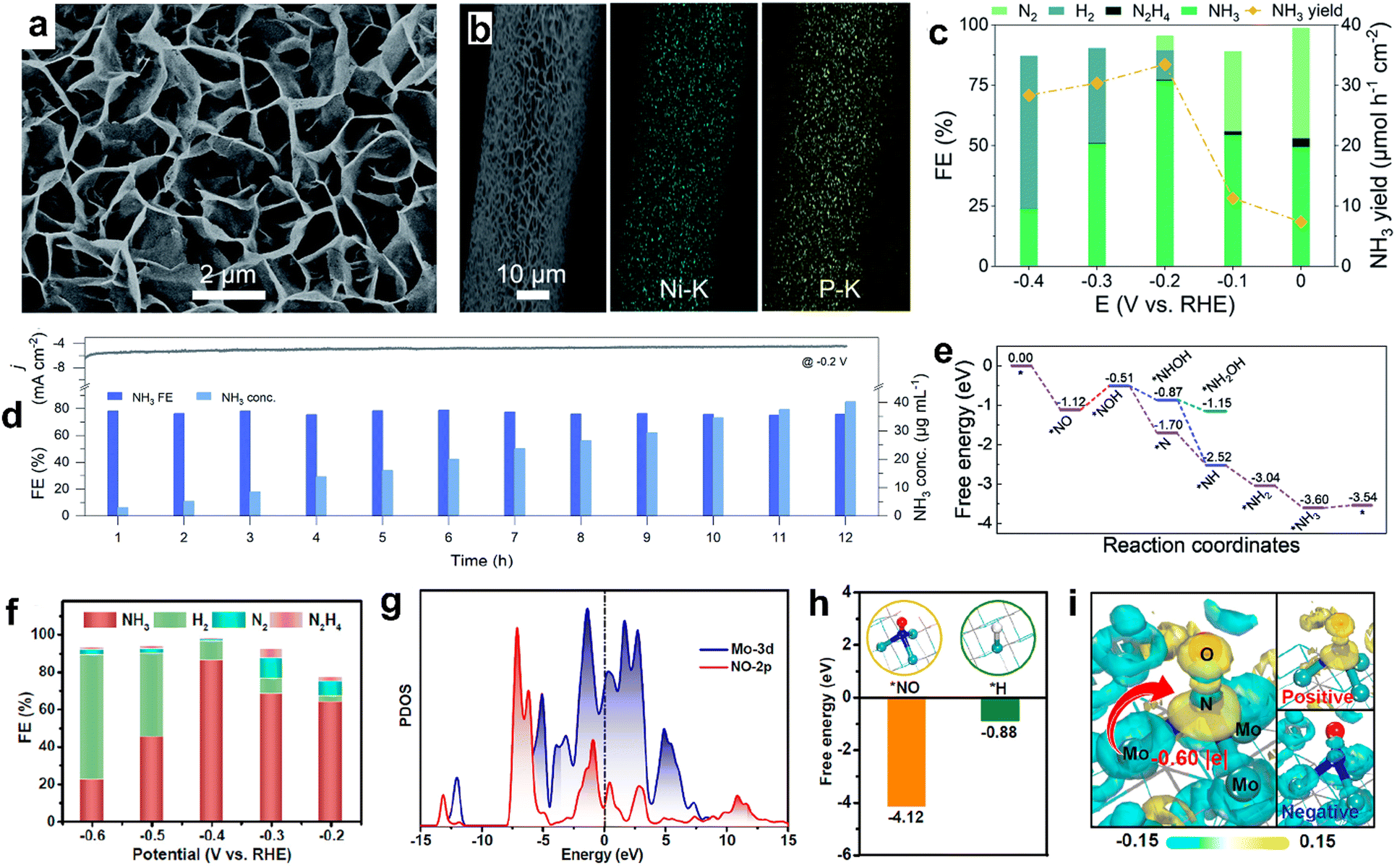
Transition metal phosphides have also shown promising catalytic activity by catalyzing the NORR at relatively low onset potentials (about −0.01 V vs. RHE). However, the excellent NH3 selectivity is limited to low overpotentials as the competitive HER prevails at larger cathodic potentials on metal phosphides. For instance, Mou et al. synthesized a Ni2P nanosheet array supported on carbon paper (Ni2P/CP) through the vapor phase phosphorization of the as prepared Ni(OH)2/CP precursor (Fig. 7a and b).93 As shown in Fig. 7c and d, Ni2P/CC has achieved a maximum NH3 yield of 33.47 μmol h−1 cm−2, high FE up to 77% at −0.2 V vs. RHE in 0.1 M HCl saturated with 10% NO, and maintained good long-term stability for up to 12 h. The assembled Zn–NO battery using Ni2P/CP as the cathode delivered a discharge power density of 1.53 mW cm−2 by producing an ammonia yield of 62.05 μg h−1 mgcat−1. Theoretical calculations indicate that two-way charge transfer occurs between Ni2P (111) and NO, which can simultaneously accumulate and deplete the electron density in the antibonding and bonding orbitals of NO, respectively. Consequently, the N–O bond will elongate and gets weaken on the Ni2P (111) surface to facilitate the successive hydrogenation steps and produces NH3 through the distal pathway (Fig. 7e). Similarly, CoP nanowire and FeP nanorod arrays are also revealed to be excellent candidates for catalyzing the NORR at low overpotentials, and both have achieved high FENH3 (88%) at −0.2 V vs. RHE in Na2SO4 and PBS electrolytes, respectively.94,95 Transition metal carbides are another class of materials predicted to show a strong affinity towards NO adsorption owing to the good orbital overlap between d and p-orbitals of the metal and NO, respectively. This could facilitate the back-donation of electrons from metal centers to NO, activating the NO molecule to produce ammonia. Chu's group developed molybdenum carbide (Mo2C) nanosheets by using methane gas as a carbon source through the carbonization of MoO3 nanosheets at 700 °C.96 As shown in Fig. 7f, Mo2C nanosheets produced the main product NH3 with a yield rate of 122.7 μmol h−1 cm−2 (FENH3: 86.3%) at −0.4 V vs. RHE in Na2SO4 and the same efficiency is retained for continuous 20 h electrolysis. As shown in Fig. 7g–i, DFT studies verify the preferred adsorption of ‘NO’ over the ‘H’ atom on the Mo2C surface and the subsequent electron transfer from the ‘Mo’ center to absorbed NO. Hence, the Mo2C surface has helped to enhance the NORR and delivers high NH3 selectivity by impeding HER kinetics. In another report, Liu and coworkers demonstrated stable NORR performance with high NH3 selectivity from MoC nanocrystals confined in N-doped carbon nanosheets (MoC/NCS).97 Carbon support is found to be beneficial for abundant active site exposure and accelerated electron transport. In addition, the corresponding Zn–NO battery exhibited a peak power density of 1.8 mW cm−2 and a large NH3 yield rate of 782 ± 10 μg h−1 cm−2. Furthermore, the potential determining step on the MoC (111) surface is predicted to be the hydrogenation of *NH, which is associated with a small energy barrier of 0.35 eV, leading to high NH3 selectivity over the other side products (H2, N2, and N2O). Table 3 summarizes the NORR activity of different multi-valent non-precious transition metal compounds.
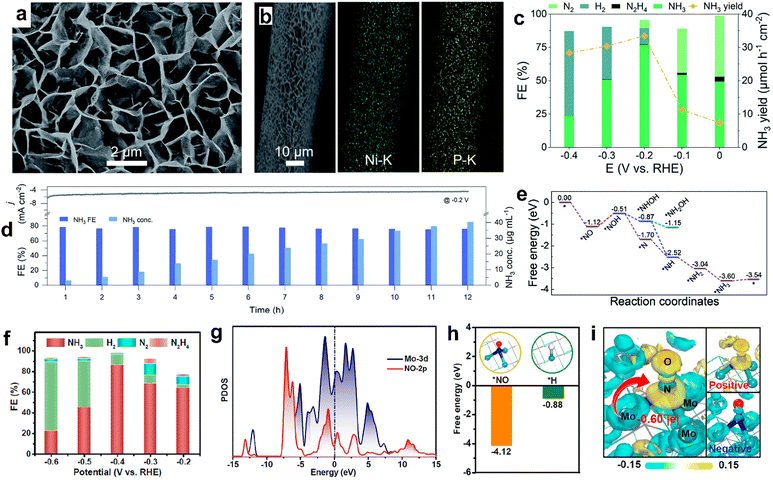 |
| | Fig. 7 (a) SEM image of Ni2P/CP, (b) EDX elemental map of Ni2P/CP, (c) FEs of the major reduction products and NH3 yields for Ni2P/CP, (d) long-term stability tests for continuous generation of NH3 on Ni2P/CP, and (e) free energy diagrams for the NORR on the Ni2P (111) surface along the distal and alternating pathways. Reproduced with permission from ref. 69 copyright 2021, RSC. (f) FEs of Mo2C at various potentials, (g) projected density of states (PDOS) profiles of adsorbed NO and its bonded Mo atoms, (h) comparison of *NO and *H adsorption on Mo2C, and (i) electron density distributions of NO adsorption on Mo2C (001). Reproduced with permission from ref. 96 copyright 2023, ACS. | |
Table 3 Summary of the NORR activity shown by different multi-valent non-precious transition metal compounds
| Catalyst |
Synthetic route |
Electrolyte |
NH3 yield rate |
FE (%) |
Potential (vs. RHE) (V) |
Ref. |
| Fe2O3 nanorods |
Hydrothermal + annealing in Ar |
0.1 M Na2SO4 + 0.5 mM Fe(II)EDTA (10% NO) |
41.6 μmol h−1 cm−2 |
86.73 |
−0.4 |
86
|
| MnO2−x nanowires |
Solvothermal + annealing in Ar |
0.2 M Na2SO4 (10% NO) |
27.51 × 10−10 mol s−1 cm−2 |
82.8 |
−0.7 |
87
|
| TiO2−x nanoarray |
Hydrothermal + H+ exchange + annealing |
0.2 M PBS (10% NO) |
1233.2 μg h−1 cm−2 |
∼65 |
−0.7 |
88
|
| CoNi(5:5)Ox@Cu |
Electrodeposition |
1 M KOH |
20 mg h−1 cm−2 |
∼100 |
−0.68 |
89
|
| NiO nanosheets |
Hydrothermal + calcination in air |
0.1 M Na2SO4 + 0.5 mM Fe(II)EDTA (10% NO) |
2130 μg h−1 cm−2 |
90 |
−0.6 |
98
|
| NiFe LDH nanosheets |
Hydrothermal method |
0.25 M Li2SO4 + 0.5 mM Fe(II)EDTA (99.99% NO) |
112 μmol h−1 cm−2 |
82 |
−0.7 |
99
|
| Cu2O@CoMn2O4 hollow nanoreactors |
CoMn-LDH growth on Cu2O nano-cube + annealing in Ar |
0.1 M Na2SO4 (99% NO) |
94.18 mmol g−1 h−1 |
75 |
−0.8 |
90
|
| MoS2/GF |
Hydrothermal method |
0.1 M HCl + 0.5 mM iron(II) sodium benzoate (10% NO) |
99.6 μmol cm2 h−1 |
<30 |
−0.7 |
54
|
| P-doped MoS2 nanospheres |
Hydrothermal + heat treatment with NaH2PO2 |
0.1 M 1-butyl-1-methypyrrolidinium tris(pentafluoroethyl) trifluorophosphate (99.99% NO) |
237.7 μg h−1 mgcat−1 |
69 |
−0.6 |
91
|
| CoS1−x nanosheet |
Hydrothermal + plasma treatment |
0.2 M Na2SO4 (10% NO) |
44.67 μmol cm−2 h−1 |
53.62 |
−0.4 |
92
|
| Ni2P nanosheets |
Hydrothermal + vapor phase phosphorization |
0.1 M HCl (10% NO) |
33.47 μmol h−1 cm−2 |
76.9 |
−0.2 |
93
|
| CoP nanowires |
Hydrothermal + vapor phase phosphorization |
0.2 M Na2SO4 (10% NO) |
47.22 μmol h−1 cm−2 |
88.3 |
−0.2 |
94
|
| FeP nanorods |
Hydrothermal + vapor phase phosphorization |
0.2 M PBS solution (10% NO) |
85.62 μmol h−1 cm−2 |
88.49 |
−0.2 |
95
|
| Mo2C nanosheets |
Liquid exfoliation of MoO3 + vapor phase carbonization |
0.5 M Na2SO4 (99.99% NO) |
122.7 μmol h−1 cm−2 |
86.3 |
−0.4 |
96
|
| MoC/NCS |
Pyrolysis of a molybdenum complex and melamine |
0.1 M HCl + 0.5 mM iron(II) sodium benzoate (99.99% NO) |
1350 μg h−1 cm−2 |
89 |
−0.8 |
97
|
| Amorphous NiB2 |
Reflux approach |
0.5 M Na2SO4 (99.99% NO) |
167.1 μmol h−1 cm−2 |
90 |
−0.4 |
100
|
| Nanoporous VN film |
Vapor phase nitridation of V2O5 |
0.1 M HCl + 0.5 mM iron(II) sodium benzoate (99.99% NO) |
1.05 × 10−7 mol cm−2 s−1 |
85 |
−0.6 |
101
|
(iii) Transition metal-based single atom catalysts (SACs).
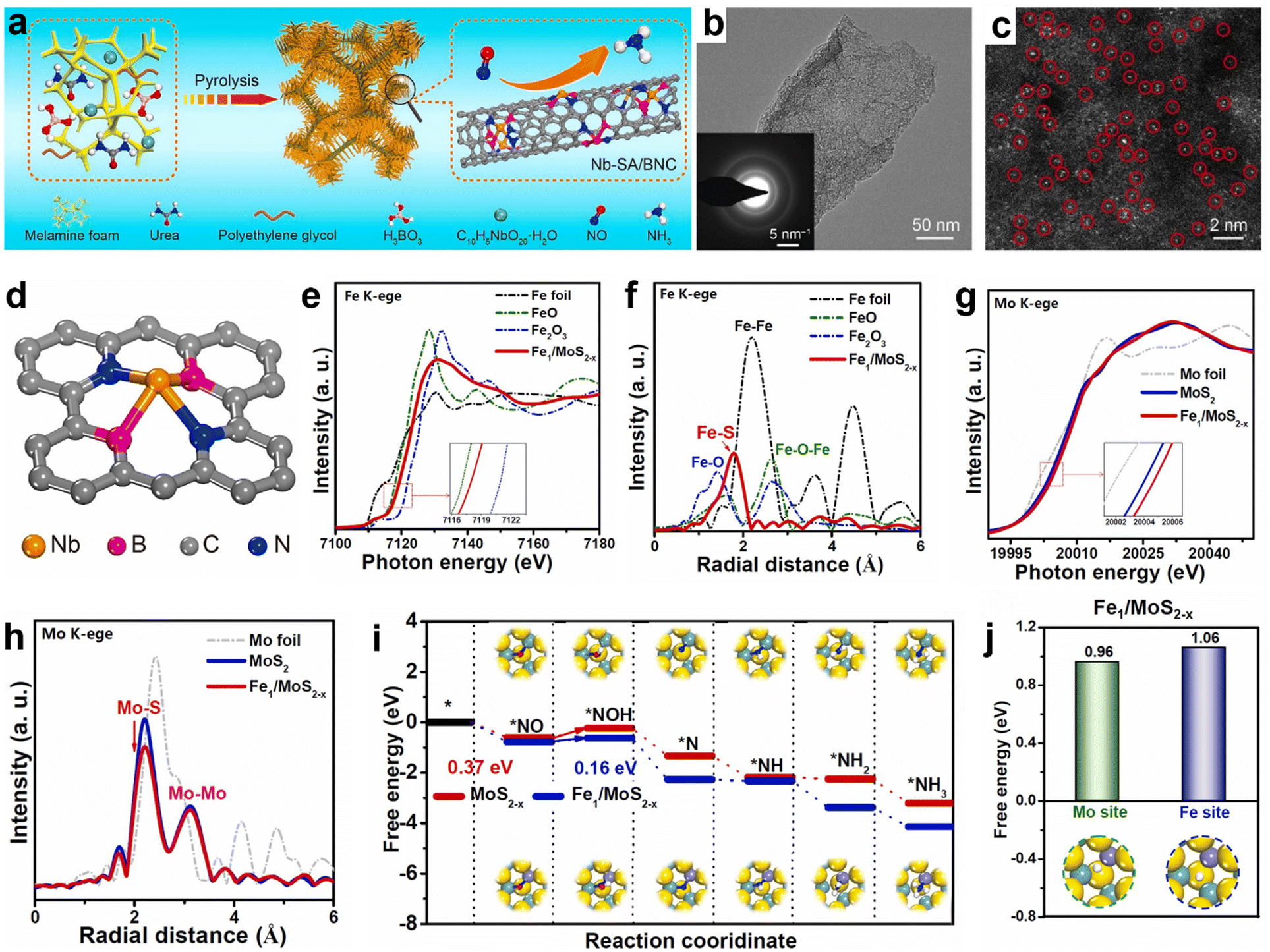
Recently, SACs have evolved as promising materials for various electrocatalytic applications such as the HER and ORR/OER owing to their maximized atom-utilization efficiency.102,103 Meanwhile, their NORR activity and product selectivity are hugely dependent on the coordination environment around the metal center.104,105 For instance, Peng et al. have shown good NORR activity from single-atom Nb sites supported on B, N co-doped carbon nanotubes (Nb-SA/BNC) prepared through the pyrolysis of melamine foam that was soaked in a precursor solution consisting of Nb(V) oxalate hydrate, polyethylene glycol, urea, and boric acid (Fig. 8a).106 The atomic dispersion of Nb atom sites over the amorphous carbon nanotubes is identified using a high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image (Fig. 8b and c). As shown in Fig. 8d, each Nb atom in Nb-SA/BNC is found to be coordinated with two B atoms and two N atoms to create the Nb-B2N2 unit. The ‘NO’ adsorption is predicted to occur on the Nb site through the d–π* orbital overlap and subsequently gets activated through the electron donation from the Nb atom. An appreciable NORR activity along with high NH3 selectivity was noted from atomically dispersed cobalt (Co) single atoms onto the MoS2 basal planes (Co/MoS2).107 It exhibited a maximum NH3 yield and FENH3 of 217.6 μmol h−1 cm−2 and 87.7%, respectively, at −0.5 V vs. RHE and showed excellent durability for 15 h. X-Ray absorption fine structure (EXAFS) analysis confirms the atomic level dispersion of Co atoms and is coordinated with three surface S atoms of MoS2 to form a Co–S3 moiety, which is predicted to be the active site for NO reduction. In addition, they emphasized that during the NORR, the NO bond activates through the electron donation from the Co–S3 moiety, and the consecutive hydrogenation steps occur through the NHO pathway (*NO → *NHO → *NHOH → *NH2OH→ *NH2 → *NH3). Similarly, Cu–S3 moieties of the single-atomic copper supported on MoS2 (Cu/MoS2) also served as efficient catalytic centers for the NORR.105 Meanwhile, the same group has used a combined strategy of atomic doping and vacancy engineering to improve the NORR activity of SACs.108 They have prepared atomically Fe-doped and S-vacancy-rich MoS2 (Fe/MoS2−x) nanoflowers using a facile one-step hydrothermal method and achieved a high NH3 yield rate of 288.2 μmol h−1 cm−2 (FE: 82.5%) at −0.6 V vs. RHE. EXAFS spectra of Fe K-edge and Mo K-edge confirm the existence of atomically distributed Fe dopants coordinated with lattice S atoms and the abundant sulfur vacancies (VS) in Fe/MoS2−x (Fig. 8e–h). In addition, the DFT calculations reveal that the cooperative effect of Fe-doping and VS has facilitated the NO adsorption and activation on Fe–Mo dual sites (Fig. 8i). Furthermore, the high ‘H’ adsorption barrier on Fe–Mo dual sites of Fe/MoS2−x has prohibited HER thereby resulting in high NH3 selectivity (Fig. 8j). Table 4 summarizes the NORR activity of different Transition metal based single-atom catalysts.
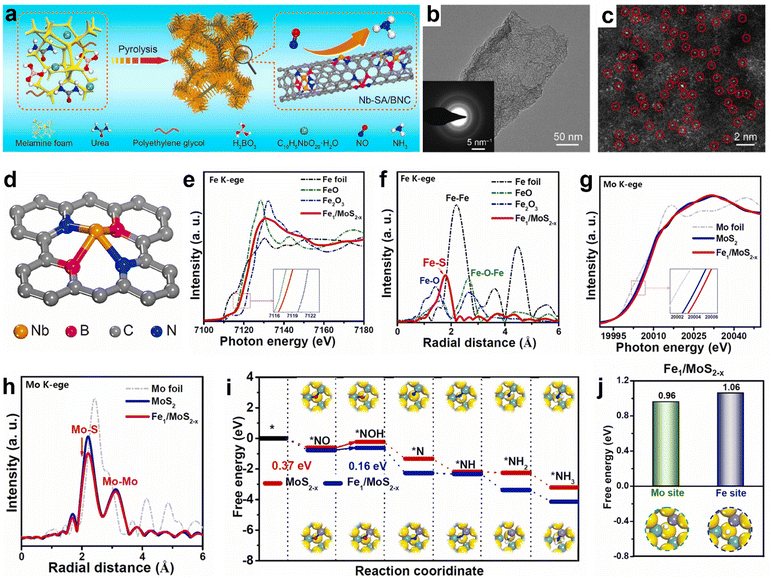 |
| | Fig. 8 (a) Schematic diagram of the synthetic procedure of Nb-SA/BNC, (b) TEM image and SAED pattern of Nb-SA/BNC (inset of Fig. 5b), (c) HAADF-STEM image of Nb-SA/BNC, in which some of the SA Nb sites are highlighted by red circles, and (d) atomic model of Nb-SA/BNC. Reproduced with permission from ref. 106 copyright 2020, Elsevier. Fe K-edge (e) XANES, and (f) EXAFS spectra of Fe/MoS2−x and reference samples of Fe foil, FeO, and Fe2O3. Mo K-edge, (g) XANES and (h) EXAFS spectra of Mo foil, MoS2, and Fe/MoS2−x. (i) Free energy diagrams for the NORR on MoS2−x and Fe/MoS2−x, and (j) free energies of *H adsorption on MoS2 and Fe/MoS2−x. Reproduced with permission from ref. 108 copyright 2023, Elsevier. | |
Table 4 Summary of the NORR activity shown by different transition metal-based single-atom catalysts
| Catalyst |
Synthetic route |
Electrolyte |
NH3 yield rate |
FE (%) |
Potential (vs. RHE) (V) |
Ref. |
| Nb/BNC |
Carbonization in Ar |
0.1 M HCl (100% NO) |
8.2 × 10−8 mol cm−2 s−1 |
77 |
−0.6 |
106
|
| Ce/NHCS |
Impregnation + carbonization |
0.05 M HCl + 0.5 mM iron(II) sodium benzoate (99.99% NO) |
1023 μg h−1 mg−1cat |
91 |
−0.7 |
109
|
| W/MoO3−x |
Supercritical CO2 approach |
0.5 M Na2SO4 (99.99% NO) |
∼ 250 μmol h−1 cm−2 |
91.2 |
−0.4 |
110
|
| Co/MoS2 |
Hydrothermal + impregnation method |
0.5 M Na2SO4 (99.99% NO) |
217.6 μmol h−1 cm−2 |
87.7 |
−0.5 |
107
|
| Cu/MoS2 |
Hydrothermal + impregnation method |
0.5 M Na2SO4 (99.99% NO) |
337.5 μmol h−1 cm−2 |
90.6 |
−0.6 |
105
|
| Fe/MoS2−x |
Hydrothermal method |
0.5 M Na2SO4 (99.99% NO) |
288.2 μmol h−1 cm−2 |
82.5 |
−0.6 |
108
|
3. p-Block element based electrocatalysts
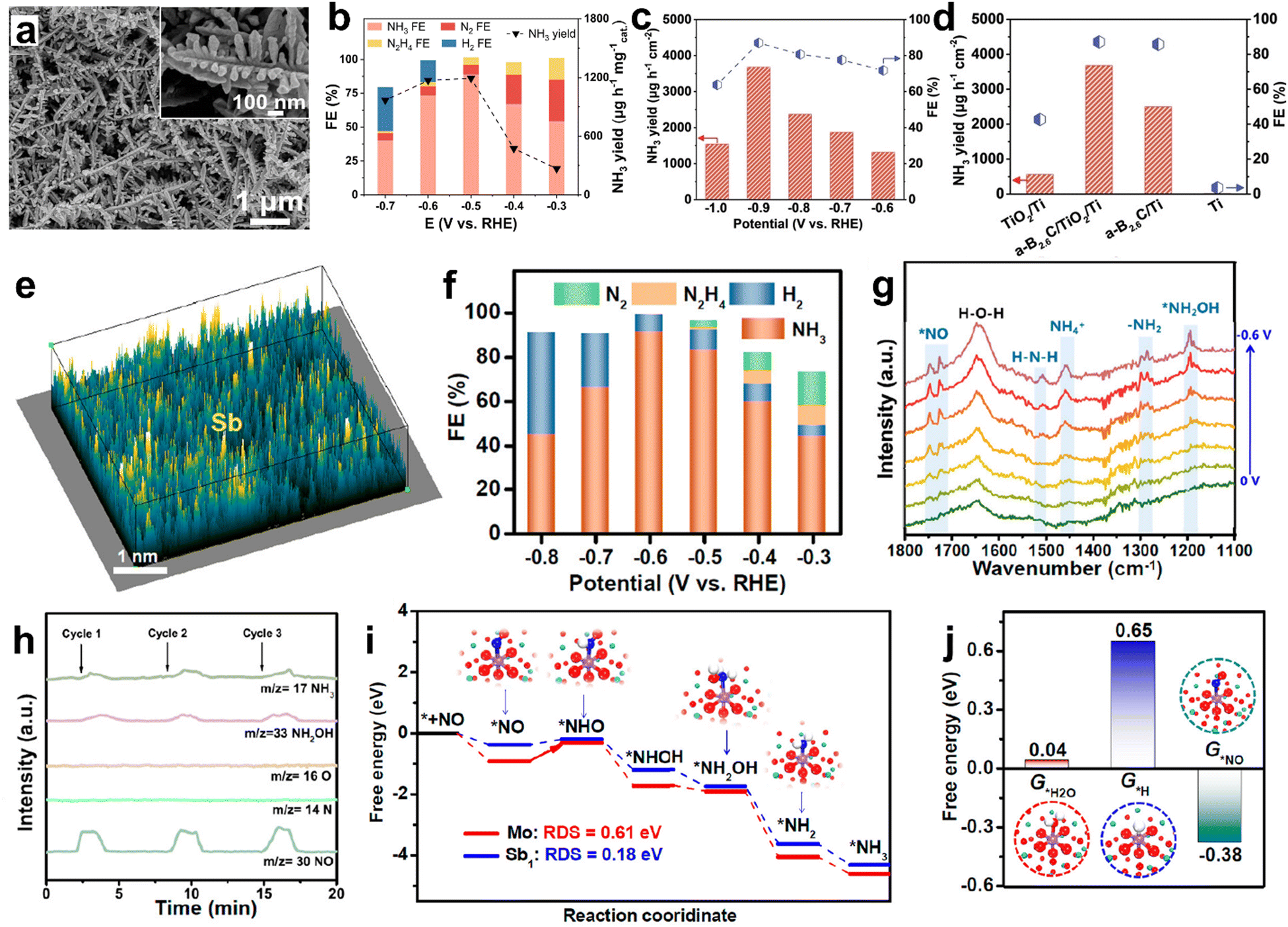
The previous section showed that d-block element-based electrocatalysts show promising NO molecule adsorption and activation owing to their unoccupied d-orbitals. However, the unoccupied d-orbitals are also favorable for ‘H’ atom binding, facilitating the competing HER and hindering the NH3 selectivity in a wide potential window.111,112 In contrast, excellent NH3 selectivity is expected from p-block element-based electrocatalysts as they exclusively bind to ‘NO’ rather than ‘H’. Lin et al. achieved an excellent FENH3 of about 90% and a high NH3 yield rate of 1194 μg h−1 mg−1cat at −0.5 V vs. RHE from Bi nano dendrites (Fig. 9a).111 Moreover, a good FENH3 is maintained in a wide potential window of −0.3 V to −0.6 V. At the same time, the H2 gas is only detectable at high cathodic potentials (Fig. 9b). In addition, the DFT calculations predict that ‘NO’ binds strongly over the Bi (012) surface as compared to the hydrogen atom, and subsequently gets activated through a two-way charge transfer mode thereby resulting in high NH3 FE. Similarly, an outstanding FENH3 of 93% is observed at −0.4 V vs. RHE of Bi nanoparticles decorated onto the carbon nanosheet. The corresponding Zn–NO battery has produced a maximum NH3 yield of 355.6 μg h−1 cm−2 at a discharge current density of 4 mA cm−2. Sulfur vacancy (Vs)-rich SnS2−x also showed an exceptional FENH3 of 90.3% (NH3 yield rate: 78.6 μmol h−1 cm−2) at a large cathodic potential of −0.7 V vs. RHE, as the HER is significantly suppressed over the SnS2−x surface.112 Compared to SnS2−x, the SnS2 counterpart showed an inferior NH3 yield rate and FENH3, thereby highlighting the advantage of the defect engineering strategy towards enhanced NORR. Theoretical computations unraveled that a VS-induced unsaturated Sn site in SnS2−x donates about −0.6 electrons to the adsorbed ‘NO’ to cause the NO bond elongation and activation, while the electron donation is restricted over the Sn site of pristine SnS2. Additionally, the free energies for all protonation steps on SnS2−x are lower than those of SnS2 and the RDS in the former is *NH2 → *NH3 with an uphill of 0.4 eV. Liang et al. have observed the efficient ‘NO’ activation on amorphous B2.6C, which is enabled by the effective electron injection into NO (π2p*) from the B–C bond.113 Given that, the amorphous B2.6C is sputtered onto a Ti plate-supported TiO2 nanobelt array (a-B2.6C@TiO2/Ti) and found that the electrode can deliver a high NH3 yield rate of 3678.6 μg h−1 cm2 and a FENH3 of 87.6% at −0.9 V vs. RHE, which are far higher than those of B2.6C/Ti and TiO2/Ti (Fig. 9c and d). Meanwhile, the electrode maintained the same activity for up to 12 hours for bulk electrolysis. In addition, a maximum NH3 yield of 1125.2 μg h−1 cm−2 was obtained for the corresponding Zn–NO battery at a discharge current of 10 mA cm−2. Metal-free boron phosphide (BP) also delivered a high FE of 83.3% with an NH3 yield rate of 96.6 μmol h−1 cm2 at −0.7 V vs. RHE.114 The DFT calculations indicate that both B and P atoms of BP can synergistically activate ‘NO’ through a strong p–p orbital overlap. In addition, the first hydrogenation step (*NO → *NHO) is found to be the RDS with a low energy barrier of 0.68 eV over BP(111). A promising NORR activity is noted from antimony single atoms confined in amorphous MoO3 (Sb/a-MoO3) nanosheets (Fig. 9e).115 In Sb/a-MoO3, Sb exists in a +3valence state and is surrounded by five O atoms to form Sb1–O5 units, as is confirmed by the Sb K-edge XANES and EXAFS studies. As shown in Fig. 9f, Sb/a-MoO3 has exhibited an excellent FENH3, of about 92%, and a high NH3 yield rate of 273.5 μmol h−1 cm−2 at −0.6 V vs. RHE in 0.5 M Na2SO4. The combined in situ electrochemical studies and theoretical calculations demonstrated that Sb/a-MoO3 prefers to follow an alternating-N pathway (Fig. 9g–i). Nevertheless, the ‘Sb’ site possesses optimal binding free energy for *NO and unfavorable binding free energies for both *H2O and *H, thereby impeding the competing HER (Fig. 9j).
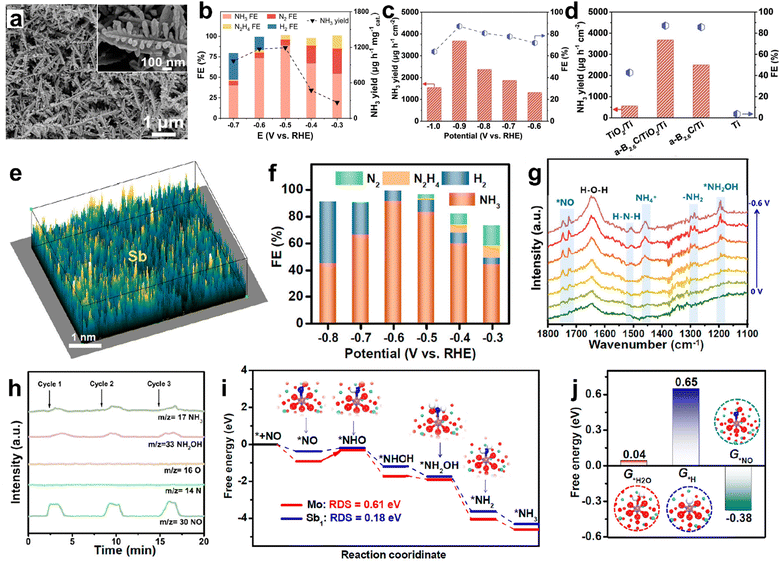 |
| | Fig. 9 (a) SEM image (inset: high-magnification image) of Bi NDs, (b) FEs of the major reductive products and NH3 yields for Bi NDs/CP at each given potential. Reproduced with permission from ref. 111 copyright 2022, Elsevier. (c) FEs and NH3 yields of a-B2.6C@TiO2/Ti at different potentials, (d) performance comparison. Reproduced with permission from ref. 113 copyright 2022, Wiley-VCH. (e) 3D atom image of p-block Sb single atoms confined in amorphous MoO3 (Sb/a-MoO3), (f) FEs of different products at various potentials, (g) potential-dependent in situ FTIR spectrum of Sb/a-MoO3 from 0 to −0.6 V, (h) online DEMS spectrum of Sb/a-MoO3 during the NORR electrolysis at −0.6 V, (i) free energy diagrams of alternating-N pathways on a-MoO3 and Sb/a-MoO3, and (j) binding free energies of *H2O, *H, and *NO on Sb/a-MoO3. Reproduced with permission from ref. 115 copyright 2023, ACS. | |
Meanwhile, atomically isolated, and unsaturated Sb sites located on Sb2S3 also showed a significant NH3 yield rate and excellent FENH3 ∼94% owing to the powerful ‘NO’ activation through the electron donation–back donation phenomenon and the selective ‘NO’ adsorption over H2O/H.116 Besides, metal-free carbon-based materials are also found to be promising candidates toward efficient electroreduction of NO into NH3 in a neutral medium. For instance, a honeycomb like carbon nanofiber coated onto a carbon paper has achieved a high FENH3 and NH3 yield of 88.33%, 22.35 μmol h−1 cm−2, respectively, at −0.6 V vs. RHE and showed good durability for up to 10 h.117 During the NORR, the interconnected nanocavities in the carbon fiber can effectively entrap the NO gas, after that, the electrochemically active –OH functional groups on the fiber can facilitate the NO reduction with a low energy input. Similarly, metal-free g-C3N4 nanosheets deposited onto carbon paper (CNNS/CP) also showed remarkable NORR performance.118 In addition, the regulation of the interface microenvironment around the CNNS/CP electrode through hydrophobic treatment is found to be beneficial for the improved gas–liquid–solid triphasic interface. Therefore, the NO mass transfer and availability over the electrode surface significantly increased, leading to 2 times enhancement in the NH3 production. Table 5 summarizes the NORR activity of different p-block element-based electrocatalysts.
Table 5 Summary of the NORR activity shown by different p-block element-based electrocatalysts
| Catalyst |
Synthetic route |
Electrolyte |
NH3 yield rate |
FE (%) |
Potential (vs. RHE) (V) |
Ref. |
| Bi nanodendrites |
Electrodeposition |
0.1 M Na2SO4 + 0.5 mM Fe(II)EDTA (10% NO) |
1194 μg h−1 mg−1cat |
89.2 |
−0.5 |
111
|
| Bi@C |
Annealing of bismuth citrate |
0.1 M Na2SO4 + 0.5 mM Fe(II)EDTA (10% NO) |
1164.8 μg h−1 mg−1cat |
93 |
−0.5 |
119
|
| SnS2−x |
Hydrothermal + plasma treatment |
0.5 M Na2SO4 (99.99% NO) |
78.6 μmol h−1 cm−2 |
90.3 |
−0.7 |
112
|
| B2.6C@TiO2 |
Magnetron sputtering of B2.6C onto TiO2 |
0.1 M Na2SO4 + 0.5 mM Fe(II)EDTA (10% NO) |
3678.6 μg h−1 cm−2 |
87.6 |
−0.9 |
113
|
| BP |
Vacuum-sealing approach |
0.5 M Na2SO4 (99.99% NO) |
96.6 μmol h−1 cm−2 |
83.3 |
−0.7 |
114
|
| Sb/a-MoO3 |
Supercritical CO2 approach |
0.5 M Na2SO4 (99.99% NO) |
273.5 μmol h−1 cm−2 |
91.7 |
−0.6 |
115
|
| In/a-MoO3 |
Supercritical CO2 approach |
0.5 M Na2SO4 (99.99% NO) |
242.6 μmol h−1 cm−2 |
92.8 |
−0.6 |
120
|
| Sb/Sb2S3 |
Solvothermal method |
0.5 M Na2SO4 (99.99% NO) |
168.6 μmol h−1 cm−2 |
93.7 |
−0.7 |
116
|
| Honeycomb carbon nanofiber |
Electrospinning followed by carbonization |
0.2 M Na2SO4 (10% NO) |
22.35 μmol h−1 cm−2 |
88.3 |
−0.6 |
117
|
| g-C3N4 nanosheets |
Calcination of melamine + ultrasonication |
0.1 M PBS solution (20% NO) |
30.7 μmol h−1 cm−2 |
45.6 |
−0.8 |
118
|
Perspectives and challenges
The research progress indicates that the electrocatalytic NORR has great potential to replace the conventional Haber–Bosch process for NH3 production. However, the practical implementation of the NORR for NH3 production is hindered by several challenges. First, most reported catalysts show high cathodic potentials (overpotentials) and exhibit unsatisfactory NH3 yield rates and FENH3 values. Thus, developing efficient electrocatalysts with high NH3 product selectivity is of paramount interest for reducing the cell potential in a commercial electrolyzer. In this context, strategies to modulate the electronic structure of the electrocatalysts through alloying, doping, and defect engineering could be considered. The operando and computational studies also guide the designing of the catalysts with ideal crystal facets and compositions for selective ‘NO’ adsorption over ‘H’ and the subsequent NORR intermediate stabilization. In addition, the materials can be grown with unique morphologies, high surface area, and ideal porosity to improve the charge transport and active site density.
Second, most of the NORR studies have used expensive 100% NO gas as a source for NH3 production as the solubility of ‘NO’ is poor in aqueous electrolytes (H-type cells). Although the utilization of ‘NO’ complexation agents in the electrolyte seems a promising strategy for attaining high NH3 yield rates from dilute NO (10%), the additional cost of the reagent, recyclability, and the NH3 separation from the electrolyte could be worrisome. In contrast, few studies have demonstrated that NH3 can be produced from dilute NO (1–10% NO) through the gas phase electrolysis using gas diffusion electrodes (GDEs) coated with electrocatalysts in a flow cell configuration. Thus, to understand the feasibility of NH3 synthesis from diluted ‘NO’ or exhaust gas, the NORR performance of the materials must be evaluated in the gas phase (GDE and flow cells) along with ‘NO-saturated electrolytes’ (in H-type cells) by using the diluted ‘NO’ reactant (1–10%). The catalysts should possess ideal Gibbs free energy and significant orbital overlap for ‘NO’ adsorption and the subsequent hydrogenation steps. Also, the gas phase electrolysis of diluted ‘NO’ could eliminate the formation of N–N coupled side products (N2 and N2O).
Third, the NORR measurements must be performed in gas-tight cells to obtain reliable results as there is a high probability of interference of cathodic currents from NO3− contamination. Furthermore, to identify the exact nitrogen source for NH3 formation, 15NO isotopic labelling experiments should be carried out. Despite most of the reports relying only on the colorimetric method for NH3 quantification, the concurrent NMR measurements should be considered for further verification. DFT studies are used extensively to identify the active sites, Gibbs free energies for ‘NO’ and intermediate adsorption, RDS, and probable mechanistic pathways over different catalytic surfaces. However, the most favorable pathway of the NORR to NH3 is still elusive as the results are hugely varied over the different crystalline planes and compounds. This makes it difficult to predict the ideal catalytic surfaces for efficient NORR to NH3. Along with the computational studies, the real-time FTIR and DEMS studies could be beneficial to identify the reaction intermediates and to understand the favorable mechanism.
Fourth, the NORR performance of many reported electrocatalysts was evaluated in neutral and alkaline media. However, to obtain NH3 from commercial PEM electrolyzers, efficient and durable catalysts that could function in an acidic medium are highly desirable. In addition, the durability tests on the developed catalysts must be performed at high NH3 yield rates to unveil their suitability for bulk NH3 production. Apart from the H-type cell, the demonstration of NH3 production from a prototype membrane electrode assembly (MEA) electrolyzer will pave the way for practical NH3 synthesis. Besides, replacing of conventional anodic oxygen evolution reaction in the MEA electrolyzer with NOx/SOx oxidation reactions simultaneously produces the value-added NH3 and nitric acid/sulfuric acid chemicals in the cathodic and anodic compartments, respectively.
In the future, screening of electrocatalysts to selectively produce urea from the co-reduction of NO and CO2 will be of great significance. The sustainable urea generation using the MEA electrolyzer through the injection of the industrial exhaust (flue gas) could be a futuristic goal. With sincere efforts in electrocatalyst development and rigorous evaluation methods, we anticipate that the production of value-added products from pollutants could be realized soon.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Mid-level Research Program through the National Research Foundation of Korea (NRF) grant funded by the Ministry of Education (No. 2021R1A2C2009223).
References
- L. A. White, Am. Anthropol., 1943, 45, 335–356 CrossRef.
- R. Madlener and Y. Sunak, Sustain. Cities Soc., 2011, 1, 45–53 CrossRef.
- S. Franco, V. R. Mandla and K. Ram Mohan Rao, Renewable Sustainable Energy Rev., 2017, 71, 898–907 CrossRef.
- R. Avtar, S. Tripathi, A. K. Aggarwal and P. Kumar, Resources, 2019, 8, 136 CrossRef.
- U. Gierałtowska, R. Asyngier, J. Nakonieczny and R. Salahodjaev, Energies, 2022, 15, 3390 CrossRef.
- A. Javaid, N. Arshed, M. Munir, Z. A. Zakaria, F. S. Alamri, H. Abd El-Wahed Khalifa and U. Hanif, Sustainability, 2022, 14, 669 CrossRef.
- L. Chen, G. Msigwa, M. Yang, A. I. Osman, S. Fawzy, D. W. Rooney and P. S. Yap, Environ. Chem. Lett., 2022, 20, 2277–2310 CrossRef CAS PubMed.
- M. J. B. Kabeyi and O. A. Olanrewaju, Front. Energy Res., 2022, 9, 743114 CrossRef.
- U.S. Energy Information Administration, Monthly Energy Review-03, 2021, https://www.eia.gov/totalenergy/data/monthly/archive/00352103.pdf.
- R. Li, Y. Liu and Q. Wang, Mar. Policy, 2022, 143, 105125 CrossRef.
- O. B. Inal, B. Zincir and C. Deniz, Int. J. Hydrog. Energy, 2022, 47, 19888–19900 CrossRef CAS.
- L. Chen, G. Msigwa, M. Yang, A. I. Osman, S. Fawzy, D. W. Rooney and P. S. Yap, Environ. Chem. Lett., 2022, 20, 2277–2310 CrossRef CAS PubMed.
- C. A. Horowitz, Materials, 2016, 55, 740–755 Search PubMed.
- O. Nisiforou, L. M. Shakou, A. Magou and A. G. Charalambides, Sustainability, 2022, 14, 2185 CrossRef CAS.
- W. S. Sou, T. Goh, X. N. Lee, S. H. Ng and K. H. Chai, Energy Policy, 2022, 170, 113239 CrossRef CAS.
- M. Salimi, M. Hosseinpour and T. N. Borhani, Energies, 2022, 15, 6064 CrossRef CAS.
- T. Capurso, M. Stefanizzi, M. Torresi and S. M. Camporeale, Energy Convers. Manag., 2022, 251, 114898 CrossRef CAS.
- S. Wu, N. Salmon, M. M. J. Li, R. Bañares-Alcántara and S. C. E. Tsang, ACS Energy Lett., 2022, 7, 1021–1033 CrossRef CAS.
- C. Sun, X. Fan, Y. Li, H. Han, J. Zhu, L. Liu and X. Geng, Renew. Energ., 2022, 201, 700–711 CrossRef CAS.
- S. J. Oh, J. H. Yoon, K. S. Jeon and J. J. Choi, J. Mech. Sci. Technol., 2022, 36, 1579–1586 CrossRef.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. Ferrero Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- W. S. Chai, Y. Bao, P. Jin, G. Tang and L. Zhou, Renewable Sustainable Energy Rev., 2021, 147, 11254 CrossRef.
- A. Valera-Medina, F. Amer-Hatem, A. K. Azad, I. C. Dedoussi, M. De Joannon, R. X. Fernandes, P. Glarborg, H. Hashemi, X. He, S. Mashruk, J. McGowan, C. Mounaim-Rouselle, A. Ortiz-Prado, A. Ortiz-Valera, I. Rossetti, B. Shu, M. Yehia, H. Xiao and M. Costa, Energy Fuels, 2021, 35, 6964–7029 CrossRef CAS.
- O. B. Inal, B. Zincir and C. Deniz, Int. J. Hydrog. Energy, 2022, 47, 19888–19900 CrossRef CAS.
- M. Wang, M. A. Khan, I. Mohsin, J. Wicks, A. H. Ip, K. Z. Sumon, C. T. Dinh, E. H. Sargent, I. D. Gates and M. G. Kibria, Energy Environ. Sci., 2021, 14, 2535–2548 RSC.
- X. Liu, A. Elgowainy and M. Wang, Green Chem., 2020, 22, 5751–5761 RSC.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- Q. Liu, T. Xu, Y. Luo, Q. Kong, T. Li, S. Lu, A. A. Alshehri, K. A. Alzahrani and X. Sun, Curr. Opin. Electrochem., 2021, 29, 100766 CrossRef CAS.
- H.-J. Chen, Z.-Q. Xu, S. Sun, Y. Luo, Q. Liu, M. S. Hamdy, Z.-s Feng, X. Sun and Yan Wang, Inorg. Chem. Front., 2022, 9, 4608–4613 RSC.
- D. K. Yesudoss, H. Chun, B. Han and S. Shanmugam, Appl. Catal., B, 2022, 304, 120938 CrossRef CAS.
- B. H. R. Suryanto, K. Matuszek, J. Choi, R. Y. Hodgetts, H.-L. Du, J. M. Bakker, C. S. M. Kang, P. V. Cherepanov, A. N. Simonov and D. R. Macfarlane, Science, 2021, 372, 1187–1191 CrossRef CAS PubMed.
- I. Zhu and T. Getting, Environ. Technol. Rev., 2012, 1, 46–58 CrossRef CAS.
- X. Zhang, Y. Wang, Y. Wang, Y. Guo, X. Xie, Y. Yu and B. Zhang, Chem. Commun., 2022, 58, 2777–2787 RSC.
- J. Long, S. Chen, Y. Zhang, C. Guo, X. Fu, D. Deng and J. Xiao, Angew. Chem., Int. Ed., 2020, 59, 9711–9718 CrossRef CAS PubMed.
- B. H. R. Suryanto, H. L. Du, D. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Nat. Catal., 2019, 2, 290–296 CrossRef CAS.
- Y.-I. Kwon, S. K. Kim, Y. B. Kim, S. J. Son, G. D. Nam, H. J. Park, W.-C. Cho, H. C. Yoon and J. H. Joo, ACS Energy Lett., 2021, 6, 4165–4172 CrossRef CAS.
- A. C. A. De Vooys, G. L. Beltramo, B. Van Riet, J. A. R. Van Veen and M. T. M. Koper, Electrochim. Acta, 2004, 49, 1307–1314 CrossRef CAS.
- S.-C. Kuo, Z. Zhang, S. K. Ross, R. B. Klemm, R. D. Johnson, P. S. Monks, R. P. Thorn and L. J. Stief, J. Phys. Chem. A, 1997, 101, 4035–4041 CrossRef CAS.
- M. W. Siegel, R. J. Celotta, J. L. Hall, J. Levine and R. A. Bennett, Phys. Rev. A, 1972, 6, 607–631 CrossRef CAS.
- S. Sethuram Markandaraj, T. Muthusamy and S. Shanmugam, Adv. Sci., 2022, 9, 2201410 CrossRef CAS PubMed.
- T. Mou, J. Long, T. Frauenheim and J. Xiao, ChemPlusChem, 2021, 86, 1211–1224 CrossRef CAS PubMed.
- M. Duca and M. T. M. Koper, Energy Environ. Sci., 2012, 5, 9726–9742 RSC.
- H. Wan, A. Bagger and J. Rossmeisl, Angew. Chem., Int. Ed., 2021, 60, 21966–21972 CrossRef CAS PubMed.
- X. He, X. Li, X. Fan, J. Li, D. Zhao, L. Zhang, S. Sun, Y. Luo, D. Zheng, L. Xie, A. M. Asiri, Q. Liu and X. Sun, ACS Appl. Nano Mater., 2022, 5, 14246–14250 CrossRef CAS.
- J. Liang, Q. Liu, A. A. Alshehri and X. Sun, Nano Res. Energy, 2022, 1, e9120010 CrossRef.
- Q. Liu, L. Xie, J. Liang, Y. Ren, Y. Wang, L. Zhang, L. Yue, T. Li, Y. Luo, N. Li, B. Tang, Y. Liu, S. Gao, A. A. Alshehri, I. Shakir, P. O. Agboola, Q. Kong, Q. Wang, D. Ma and X. Sun, Small, 2022, 18, 2106961 CrossRef CAS PubMed.
- X. Xu, L. Hu, Z. Li, L. Xie, S. Sun, L. Zhang, J. Li, Y. Luo, X. Yan, M. S. Hamdy, Q. Kong, X. Sun and Q. Liu, Sustain. Energy Fuels, 2022, 6, 4130–4136 RSC.
- H. Wang, F. Zhang, M. Jin, D. Zhao, X. Fan, Z. Li, Y. Luo, D. Zheng, T. Li, Y. Wang, B. Ying, S. Sun, Q. Liu, X. Liu and X. Sun, Mater. Today Phys., 2023, 30, 100944 CrossRef CAS.
- Z. Li, J. Liang, Q. Liu, L. Xie, L. Zhang, Y. Ren, L. Yue, N. Li, B. Tang, A. A. Alshehri, M. S. Hamdy, Y. Luo, Q. Kong and X. Sun, Mater. Today Phys., 2022, 23, 100619 CrossRef CAS.
- H. Wang, J. Huang, J. Cai, Y. Wei, A. Cao, B. Liu and S. Lu, Small Methods, 2023, 2300169 CrossRef PubMed.
- J. Cai, J. Huang, A. Cao, Y. Wei, H. Wang, X. Li, Z. Jiang, G. I. N. Waterhouse, S. Lu and S.-Q. Zang, Appl. Catal., B, 2023, 328, 122473 CrossRef CAS.
- H. Qian, S. Xu, J. Cao, F. Ren, W. Wei, J. Meng and L. Wu, Nat. Sustain., 2021, 4, 417–425 CrossRef.
- L. Han, S. Cai, M. Gao, J. Y. Hasegawa, P. Wang, J. Zhang, L. Shi and D. Zhang, Chem. Rev., 2019, 119, 10916–10976 CrossRef CAS PubMed.
- L. Zhang, J. Liang, Y. Wang, T. Mou, Y. Lin, L. Yue, T. Li, Q. Liu, Y. Luo, N. Li, B. Tang, Y. Liu, S. Gao, A. A. Alshehri, X. Guo, D. Ma and X. Sun, Angew. Chem., Int. Ed., 2021, 60, 25263–25268 CrossRef CAS PubMed.
- J. S.-Hernández, C. R. S.-Ramirez, E. R.-Meneses, M. L.-Trujillo, J.-A. Wang, L. L.-Rojas and A. M.-Robledo, Appl. Catal., B, 2019, 259, 118048 CrossRef.
- J. Long, C. Guo, X. Fu, H. Jing, G. Qin, H. Li and J. Xiao, J. Phys. Chem. Lett., 2021, 12, 6988–6995 CrossRef CAS PubMed.
- B. H. Ko, B. Hasa, H. Shin, Y. Zhao and F. Jiao, J. Am. Chem. Soc., 2022, 144, 1258–1266 CrossRef CAS PubMed.
- A. C. A. De Vooys, M. T. M. Koper, R. A. Van Santen and J. A. R. Van Veen, Electrochim. Acta, 2001, 46, 923–930 CrossRef CAS.
- V. Rosca and M. T. M. Koper, J. Phys. Chem. B, 2005, 109, 16750–16759 CrossRef CAS PubMed.
- J. A. Colucci, M. J. Foral and S. H. Lancer, Electrochim. Acta, 1985, 30, 521–528 CrossRef CAS.
- J. F. E. Gootzen, R. M. Van Hardeveld, W. Visscher, R. A. Van Santen and J. A. R. Van Veen, Red. Trav. Chim. Pays-Bas, 1996, 115, 480–485 CrossRef CAS.
- A. Clayborne, H.-J. Chun, R. B. Rankin and J. Greeley, Angew. Chem., Int. Ed., 2015, 54, 8255–8258 CrossRef CAS PubMed.
- H.-J. Chun, V. Apaja, A. Clayborne, K. Honkala and J. Greeley, ACS Catal., 2017, 7, 3869–3882 CrossRef CAS.
- I. Katsounaros, M. C. Figueiredo, X. Chen, F. Calle-Vallejo and M. T. M. Koper, ACS Catal., 2017, 7, 4660–4667 CrossRef CAS.
- T. Mou, J. Long, T. Frauenheim and J. Xiao, ChemPlusChem, 2021, 86, 1211–1224 CrossRef CAS PubMed.
- A. C. A. De Vooys, M. T. M. Koper, R. A. Van Santen and J. A. R. Van Veen, J. Catal., 2001, 202, 387–394 CrossRef CAS.
- Y. Zang, Q. Wu, S. Wang, B. Huang, Y. Dai and Y. Ma, J. Phys. Chem. Lett., 2022, 13, 527–535 CrossRef CAS PubMed.
- P. Lv, D. Wu, B. He, X. Li, R. Zhu, G. Tang, Z. Lu, D. Ma and Y. Jia, J. Mater. Chem. A, 2022, 10, 9707–9716 RSC.
- T. Mou, J. Liang, Z. Ma, L. Zhang, Y. Lin, T. Li, Q. Liu, Y. Luo, Y. Liu, S. Gao, H. Zhao, A. M. Asiri, D. Ma and X. Sun, J. Mater. Chem. A, 2021, 9, 24268–24275 RSC.
- X. Guo, H. Du, F. Qu and J. Li, J. Mater. Chem. A, 2019, 7, 3531–3543 RSC.
- Y. Li, C. Cheng, S. Han, Y. Huang, X. Du, B. Zhang and Y. Yu, ACS Energy Lett., 2022, 7, 1187–1194 CrossRef CAS.
- H. Zhang, Y. Li, C. Cheng, J. Zhou, P. Yin, H. Wu, Z. Liang, J. Zhang, Q. Yun, A. L. Wang, L. Zhu, B. Zhang, W. Cao, X. Meng, J. Xia, Y. Yu and Q. Lu, Angew. Chem., Int. Ed., 2023, 62, e202213351 CAS.
- J. Shi, C. Wang, R. Yang, F. Chen, N. Meng, Y. Yu and B. Zhang, Sci. China: Chem., 2021, 64, 1493–1497 CrossRef CAS.
- D. Y. Kim, D. Shin, J. Heo, H. Lim, J. A. Lim, H. M. Jeong, B. S. Kim, I. Heo, I. Oh, B. Lee, M. Sharma, H. Lim, H. Kim and Y. Kwon, ACS Energy Lett., 2020, 5, 3647–3656 CrossRef CAS.
- J. Choi, J. Choi, H. L. Du, H. L. Du, C. K. Nguyen, C. K. Nguyen, B. H. R. Suryanto, A. N. Simonov, A. N. Simonov, D. R. MacFarlane and D. R. MacFarlane, ACS Energy Lett., 2020, 5, 2095–2097 CrossRef CAS.
- Y. Xiong, Y. Li, S. Wan, Y. Yu, S. Zhang and Q. Zhong, J. Hazard. Mater., 2022, 430, 128451 CrossRef CAS PubMed.
- K. Chen, G. Wang, Y. Guo, D. Ma and K. Chu, Nano Res., 2023 DOI:10.1007/s12274-023-5556-7.
- L. Chen, W. Sun, Z. Xu, M. Hao, B. Li, X. Liu, J. Ma, L. Wang, C. Li and W. Wang, Ceram. Int., 2022, 48, 21151–21161 CrossRef CAS.
- S. Cheon, W. J. Kim, D. Y. Kim, Y. Kwon and J. I. Han, ACS Energy Lett., 2022, 7, 958–965 CrossRef CAS.
- T. Muthusamy, S. Sethuram Markandaraj and S. Shanmugam, J. Mater. Chem. A, 2022, 10, 6470–6474 RSC.
- D. Wang, Z.-W. Chen, K. Gu, C. Chen, Y. Liu, X. Wei, C. V. Singh and S. Wang, J. Am. Chem. Soc., 2023, 145, 6899–6904 CrossRef CAS PubMed.
- P. M. Krzywda, A. Paradelo Rodríguez, N. E. Benes, B. T. Mei and G. Mul, ChemElectroChem, 2022, 9, e202101273 CAS.
- I. M. Wasser, S. De Vries, P. Moënne-Loccoz, I. Schröder and K. D. Karlin, Chem. Rev., 2002, 102, 1201–1234 CrossRef CAS PubMed.
- N. Lehnert, B. W. Musselman and L. C. Seefeldt, Chem. Soc. Rev., 2021, 50, 3640–3646 RSC.
- D. Malko, A. Kucernak and T. Lopes, J. Am. Chem. Soc., 2016, 138, 16056–16068 CrossRef CAS PubMed.
- J. Liang, H. Chen, T. Mou, L. Zhang, Y. Lin, L. Yue, Y. Luo, Q. Liu, N. Li, A. A. Alshehri, I. Shakir, P. O. Agboola, Y. Wang, B. Tang, D. Ma and X. Sun, J. Mater. Chem. A, 2022, 10, 6454–6462 RSC.
- Z. Li, Z. Ma, J. Liang, Y. Ren, T. Li, S. Xu, Q. Liu, N. Li, B. Tang, Y. Liu, S. Gao, A. A. Alshehri, D. Ma, Y. Luo, Q. Wu and X. Sun, Mater. Today Phys., 2022, 22, 100586 CrossRef CAS.
- Z. Li, Q. Zhou, J. Liang, L. Zhang, X. Fan, D. Zhao, Z. Cai, J. Li, D. Zheng, X. He, Y. Luo, Y. Wang, B. Ying, H. Yan, S. Sun, J. Zhang, A. A. Alshehri, F. Gong, Y. Zheng and X. Sun, Small, 2023, 19, 2300291 CrossRef CAS PubMed.
- A. Wu, J. Lv, X. Xuan, J. Zhang, A. Cao, M. Wang, X.-Y. Wu, Q. Liu, Y. Zhong, W. Sun, Q. Ye, Y. Peng, X. Lin, Z. Qi, S. Zhu, Q. Huang, X. Li, H. B. Wu and J. Yan, Adv. Energy Mater., 2023, 13, 2204231 CrossRef CAS.
- C. Bai, S. Fan, X. Li, Z. Niu, J. Wang, Z. Liu and D. Zhang, Adv. Funct. Mater., 2022, 32, 2205569 CrossRef CAS.
- T. Wei, H. Bao, X. Wang, S. Zhang, Q. Liu, J. Luo and X. Liu, ChemCatChem, 2023, 15, e202201411 CrossRef CAS.
- L. Zhang, Q. Zhou, J. Liang, L. Yue, T. Li, Y. Luo, Q. Liu, N. Li, B. Tang, F. Gong, X. Guo and X. Sun, Inorg. Chem., 2022, 61, 8096–8102 CrossRef CAS PubMed.
- T. Mou, J. Liang, Z. Ma, L. Zhang, Y. Lin, T. Li, Q. Liu, Y. Luo, Y. Liu, S. Gao, H. Zhao, A. M. Asiri, D. Ma and X. Sun, J. Mater. Chem. A, 2021, 9, 24268–24275 RSC.
- J. Liang, W. F. Hu, B. Song, T. Mou, L. Zhang, Y. Luo, Q. Liu, A. A. Alshehri, M. S. Hamdy, L. M. Yang and X. Sun, Inorg. Chem. Front., 2022, 9, 1366–1372 RSC.
- J. Liang, Q. Zhou, T. Mou, H. Chen, L. Yue, Y. Luo, Q. Liu, M. S. Hamdy, A. A. Alshehri, F. Gong and X. Sun, Nano Res., 2022, 15, 4008–4013 CrossRef CAS.
- K. Chen, P. Shen, N. Zhang, D. Ma and K. Chu, Inorg. Chem., 2023, 62, 653–658 CrossRef CAS PubMed.
- G. Meng, M. Jin, T. Wei, Q. Liu, S. Zhang, X. Peng, J. Luo and X. Liu, Nano Res., 2022, 15, 8890–8896 CrossRef CAS.
- P. Liu, J. Liang, J. Wang, L. Zhang, J. Li, L. Yue, Y. Ren, T. Li, Y. Luo, N. Li, B. Tang, Q. Liu, A. M. Asiri, Q. Kong and X. Sun, Chem. Commun., 2021, 57, 13562–13565 RSC.
- G. Meng, T. Wei, W. Liu, W. Li, S. Zhang, W. Liu, Q. Liu, H. Bao, J. Luo and X. Liu, Chem. Commun., 2022, 58, 8097–8100 RSC.
- K. Chen, Y. Tian, Y. Li, Y. Liu and K. Chu, J. Mater. Chem. A, 2023, 11, 7409–7414 RSC.
- D. Qi, F. Lv, T. Wei, M. Jin, G. Meng, S. Zhang, Q. Liu, W. Liu, D. Ma, M. S. Hamdy, J. Luo and X. Liu, Nano Res. Energy, 2022, 1, e9120010 CrossRef.
- J. Xi, H. S. Jung, Y. Xu, F. Xiao, J. W. Bae and S. Wang, Adv. Funct. Mater., 2021, 31, 2008318 CrossRef CAS.
- F. D. Speck, J. H. Kim, G. Bae, S. H. Joo, K. J. J. Mayrhofer, C. H. Choi and S. Cherevko, JACS Au, 2021, 1, 1086–1100 CrossRef CAS PubMed.
- D. H. Kim, S. Ringe, H. Kim, S. Kim, B. Kim, G. Bae, H.-S. Oh, F. Jaouen, W. Kim, H. Kim and C. H. Choi, Nat. Commun., 2021, 12, 1856 CrossRef CAS PubMed.
- K. Chen, G. Zhang, X. Li, X. Zhao and K. Chu, Nano Res., 2023, 16, 5857–5863 CrossRef CAS.
- X. Peng, Y. Mi, H. Bao, Y. Liu, D. Qi, Y. Qiu, L. Zhuo, S. Zhao, J. Sun, X. Tang, J. Luo and X. Liu, Nano Energy, 2020, 78, 105321 CrossRef CAS.
- X. Li, K. Chen, X. Lu, D. Ma and K. Chu, Chem. Eng. J., 2023, 454, 140333 CrossRef CAS.
- K. Chen, J. Wang, J. Kang, X. Lu, X. Zhao and K. Chu, Appl. Catal., B, 2023, 324, 122241 CrossRef CAS.
- W. Zhang, X. Qin, T. Wei, Q. Liu, J. Luo and X. Liu, J. Colloid Interface Sci., 2023, 638, 650–657 CrossRef CAS PubMed.
- K. Chen, J. Wang, H. Zhang, D. Ma and K. Chu, Nano Lett., 2023, 23, 1735–1742 CrossRef CAS PubMed.
- Y. Lin, J. Liang, H. Li, L. Zhang, T. Mou, T. Li, L. Yue, Y. Ji, Q. Liu, Y. Luo, N. Li, B. Tang, Q. Wu, M. S. Hamdy, D. Ma and X. Sun, Mater. Today Phys., 2022, 22, 100611 CrossRef CAS.
- X. Li, G. Zhang, P. Shen, X. Zhao and K. Chu, Inorg. Chem. Front., 2022, 10, 280–287 RSC.
- J. Liang, P. Liu, Q. Li, T. Li, L. Yue, Y. Luo, Q. Liu, N. Li, B. Tang, A. A. Alshehri, I. Shakir, P. O. Agboola, C. Sun and X. Sun, Angew. Chem., Int. Ed., 2022, 61, e202202087 CAS.
- G. Zhang, Y. Wan, H. Zhao, Y. Guo and K. Chu, Dalton Trans., 2023, 52, 6248–6253 RSC.
- K. Chen, Y. Zhang, J. Xiang, X. Zhao, X. Li and K. Chu, ACS Energy Lett., 2023, 8, 1281–1288 CrossRef CAS.
- K. Chen, Y. Zhang, W. Du, Y. Guo and K. Chu, Inorg. Chem. Front., 2023, 10, 2708–2715 RSC.
- L. Ouyang, Q. Zhou, J. Liang, L. Zhang, L. Yue, Z. Li, J. Li, Y. Luo, Q. Liu, N. Li, B. Tang, A. Ali Alshehri, F. Gong and X. Sun, J. Colloid Interface Sci., 2022, 616, 261–267 CrossRef CAS PubMed.
- K. Li, Z. Shi, L. Wang, W. Wang, Y. Y. Liu, H. Cheng, Y. Yang and L. Zhang, J. Hazard. Mater., 2023, 448, 130890 CrossRef CAS PubMed.
- Q. Liu, Y. Lin, L. Yue, J. Liang, L. Zhang, T. Li, Y. Luo, M. Liu, J. You, A. A. Alshehri, Q. Kong and X. Sun, Nano Res., 2022, 15, 5032–5037 CrossRef CAS.
- K. Chen, N. Zhang, F. Wang, J. Kang and K. Chu, J. Mater. Chem. A, 2023, 11, 6814–6819 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Dinesh
Dhanabal
,
Dinesh
Dhanabal




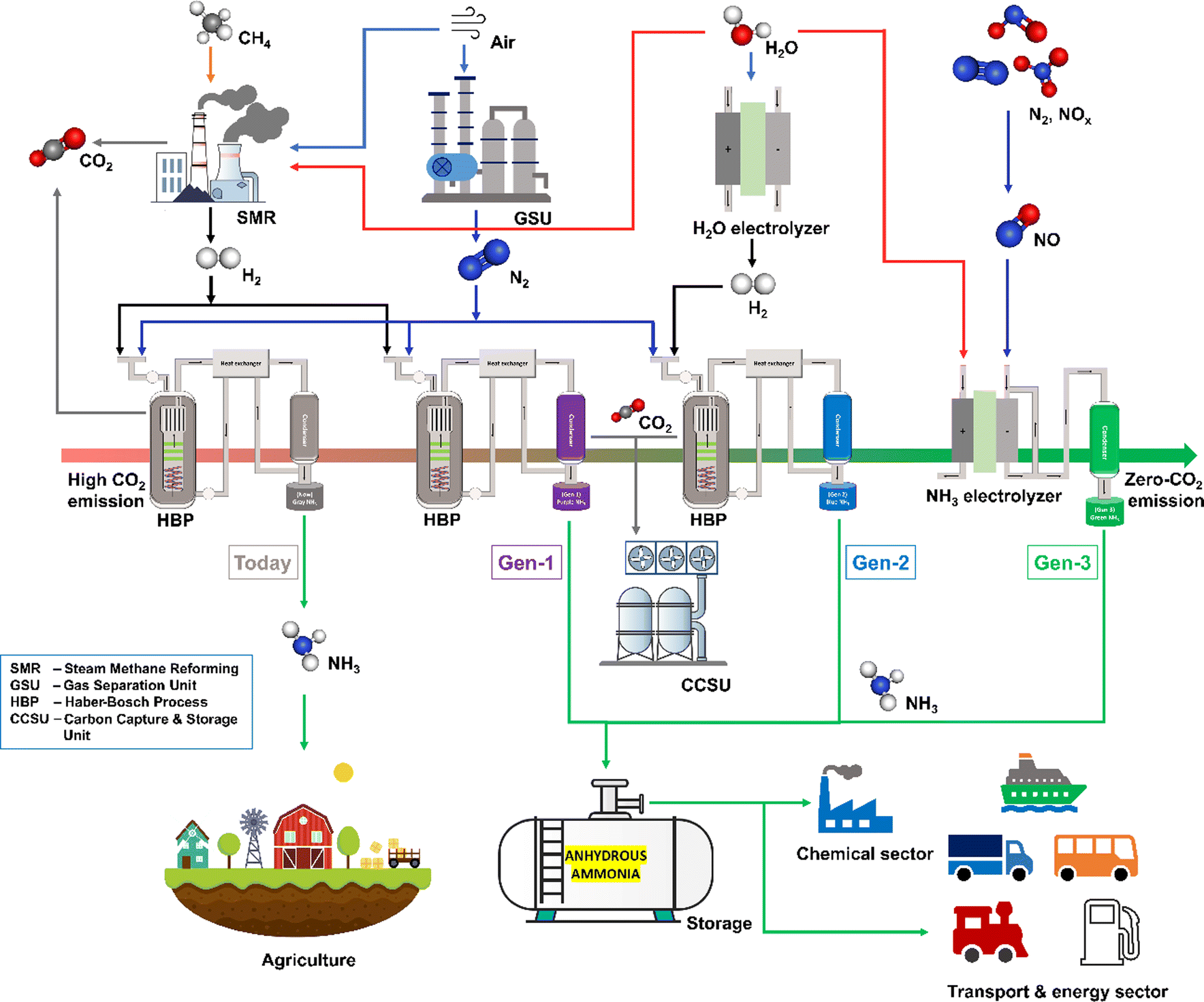
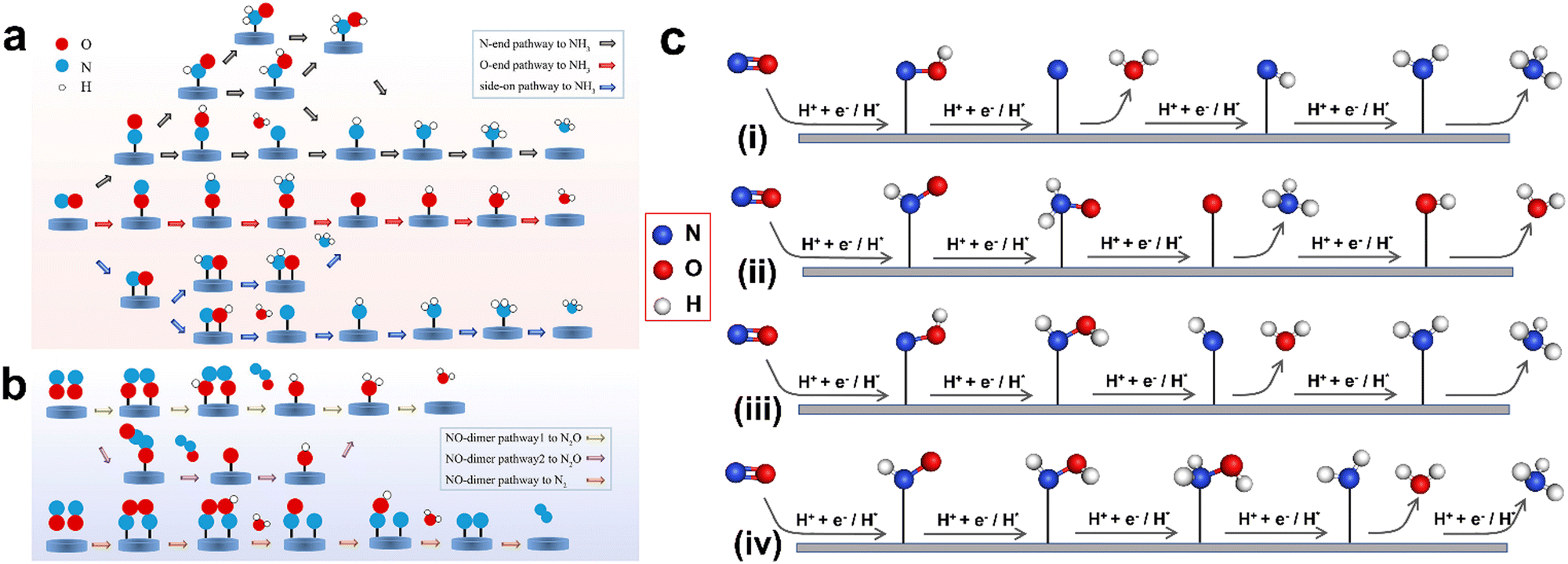
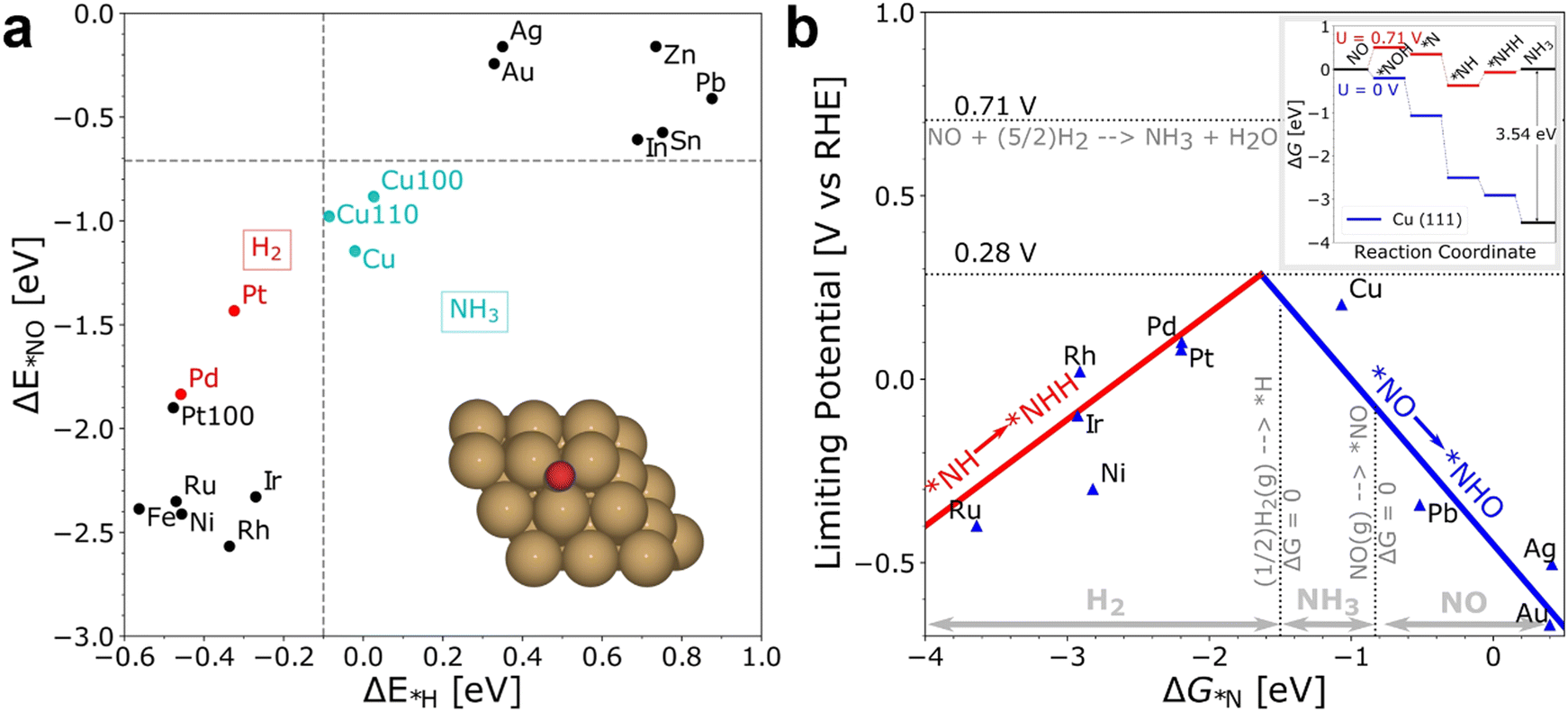
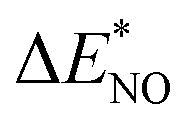 are plotted against
are plotted against 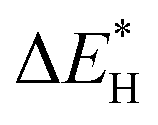 on various metal surfaces. (b) The limiting potential volcano plot for NO reduction to NH3 on various metal surfaces. Reproduced with permission from ref. 43 copyright 2021, Wiley-VCH.
on various metal surfaces. (b) The limiting potential volcano plot for NO reduction to NH3 on various metal surfaces. Reproduced with permission from ref. 43 copyright 2021, Wiley-VCH.