
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing the activity and selectivity of ppm level copper in gas diffusion electrodes towards CO and CO2 electroreduction†
Xiang
Lyu
 ab,
Jianlin
Li
b,
Tianyu
Zhang
a,
Zhengyuan
Li
a,
In-hui
Hwang
c,
Chengjun
Sun
c,
Charl J.
Jafta
b,
Jun
Yang
b,
Todd J.
Toops
d,
David A.
Cullen
e,
Alexey
Serov
*b and
Jingjie
Wu
*a
ab,
Jianlin
Li
b,
Tianyu
Zhang
a,
Zhengyuan
Li
a,
In-hui
Hwang
c,
Chengjun
Sun
c,
Charl J.
Jafta
b,
Jun
Yang
b,
Todd J.
Toops
d,
David A.
Cullen
e,
Alexey
Serov
*b and
Jingjie
Wu
*a
aDepartment of Chemical and Environmental Engineering, University of Cincinnati, Cincinnati, OH, USA. E-mail: jingjie.wu@uc.edu
bElectrification and Energy Infrastructures Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: serova@ornl.gov
cX-Ray Science Division, Argonne National Laboratory, IL 60439, USA
dBuildings and Transportation Science Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
eCenter for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
First published on 28th November 2022
Abstract
Cu is a unique metal that catalyzes carbon monoxide/carbon dioxide (CO/CO2) to form high-order hydrocarbons and oxygenates through the CO/CO2 reduction reaction (CO/CO2RR) at decent selectivity and productivity. While this has been shown, the limits of the system are still unknown, i.e., the minimum amount of Cu needed for the CO/CO2RR, and the maximum activity that trace amounts of Cu can achieve. Here, we have investigated the activity and selectivity of trace Cu with atomic dispersion over a range of loadings below 2 μg cm−2 and have quantified the mass activity/turnover frequency (TOF) of trace Cu catalysts. A Cu loading of at least 0.042 μg cm−2 initiates the CORR activity with 30% faradaic efficiency (FE) at a partial current density of 102 mA cm−2 forming predominantly CH4. The selectivity moves to C2-based products (CH3COO−, C2H4, and CH3CH2OH) with 70% FE as the Cu loading increases to 0.333 μg cm−2, and increasing the Cu loading to 0.812 μg cm−2 results in a 78% FE to C2 with CH3COO− accounting for 42% of this. The highest mass activity for CH4 reaches 2435 A mg−1 of Cu, corresponding to a TOF of 267 s−1, while C2 activity reaches 584 A mg−1 of Cu, leading to a TOF of 145 s−1. Both TOFs are several orders of magnitude higher than the reported values. Different from the CORR, the CO2RR demands a higher Cu loading and primarily generates C1 (e.g., CO and CH4). Metal impurities can be extended to others that are active towards the CO2RR, such as Zn for CO2-to-CO conversion. Thus, we suggest that the effect of trace metal impurities must be quantified when developing carbon-based metal-free and coordinated single-atom catalysts for the CO/CO2RR in order to avoid overestimating their activity.
Broader contextCarbon dioxide (CO2) removal and utilization have become an essential feature of climate mitigation. The electrochemical CO2 reduction reaction (CO2RR) to form valuable chemicals and fuels utilizing renewable energy is a promising strategy contributing to the carbon-net-zero goal. A catalyst with high activity and selectivity is critical to enabling the large-scale application of the CO2RR. Cu-based catalysts are uniquely known hydrocarbon- and oxygenate-selective metal catalysts. Meanwhile, metal-free carbon-based and metal–nitrogen–carbon (M–N–C with metals of Fe, Ni, and Co) single-atom catalysts have been developed as alternatives to conventional metal catalysts for the CO/CO2 reduction reaction (CO/CO2RR). Since trace Cu on carbon-based supports was reported to show activity towards the CO2RR, the performance overestimation of metal-free and M–N–C catalysts during the CO/CO2RR is a concern. Currently, two questions remain elusive: what is the lowest limit Cu loading to show activity for the CO/CO2RR, and how high is the activity that trace amounts of Cu can achieve? Both questions are critical to scientifically evaluate the intrinsic performance of the catalysts during the CO/CO2RR. In this work, we answered both questions by revealing the activity and selectivity of ppm level copper in gas diffusion electrodes towards the CO/CO2RR. |
Introduction
The electrochemical CO2 reduction reaction (CO2RR) to form valuable chemicals and fuels utilizing renewable energy has attracted remarkable attention, as it is beneficial in producing value-added products while simultaneously consuming CO2 under mild operating conditions.1–4 Nevertheless, the practical cost is still high for large-scale applications partly because of the low selectivity, efficiency, and durability of existing electrocatalysts.5–7 The state-of-the-art electrocatalysts for the CO2RR are metallic.8–11 Ag, Au, and Zn mainly convert CO2 to carbon monoxide (CO),12–15 while Sn and Bi are selective to formate (HCOO−) via a two-electron reduction process in the CO2RR.16–18 Cu stands out as the only known metal catalyst capable of generating high-order products such as C1–C2 hydrocarbons (e.g., CH4 and C2H4) and C2+ oxygenates (e.g., CH3CH2OH, CH3COOH, and C3H7OH,) beyond the 2e− reduction route.19–23 Moreover, Cu is also the unique metal that catalyzes the electrochemical CO reduction reaction (CORR), a critical intermediate reaction in the CO2RR, to form a similar distribution of C2+ hydrocarbons and oxgenates.24,25A trace level of Cu nanoparticles (e.g., 119 ppm) in graphene oxide loaded on a glass carbon substrate exhibited electrochemical activity towards the CO2RR in an H-cell.26 The carbon products were only C1 (CO, CH4, and HCOOH) with a total faradaic efficiency (FE) of about 15% and a total current density of around 5 mA cm−2 at −1.3 V versus reversible hydrogen electrode (vs. RHE throughout the text). Fu et al. prepared atomically dispersed Cu atoms with a low loading of 69 μg cm−2 on nitrogen-rich porous carbon supports and then loaded them onto a carbon paper substrate as gas diffusion electrodes (GDEs).27 They reported ∼60% FE to C2+ products in the CORR under a total current density of 75 mA cm−2 at −0.7 V, while CH4 formation was negligible. Rong et al. synthesized atomically dispersed Cu atoms on a graphdiyne support loaded on a carbon paper substrate to make GDEs as well.28 They observed that the major product of the CORR switched from C1/CH4 (57.3% FE) to C2 (80% FE) at −1 V as the atomically dispersed Cu loading increased from 7.5 to 226 μg cm−2. Those findings indicated that the content of trace Cu on carbon-based supports can substantially influence the electrochemical activity and selectivity in the CO/CO2RR. However, the maximum mass activity and turnover frequency (TOF) in the CO/CO2RR that can be achieved with trace Cu is still unknown. Moreover, understanding how decreasing the Cu loading impacts selectivity in the CO/CO2RR is also unknown.
An alternative approach under investigation is the use of metal-free carbon catalysts and metal–nitrogen–carbon (M–N–C with metals of Fe, Ni, and Co). Most carbon and M–N–C catalysts show predominant products of C1 (e.g., CO and CH4) in the CO/CO2RR.29–33 For example, the graphitic-N doped graphene-like carbon catalyst was reported to convert CO2 to CO with a 95% FE and partial current density of 9.07 mA cm−2 at −0.72 V.31 N-doped graphene quantum dots (NGQDs) with functionalized electron-donating groups (e.g., –OH and –NH2) could catalyze CO2 to CH4 with both high selectivity (70% FE) and production rate (partial current density of 200 mA cm−2) at −0.85 V.32 The single Zn atom supported on a microporous N-doped carbon catalyst was reported to catalyze CO2 to CH4 with an 85% FE and partial current density of 31.8 mA cm−2 at −1.8 V (vs. Hg/HgCl2).34 A few carbon catalysts were also reported to form C2 products.35–37 Rigorous evaluation of the activity to the CO/CO2RR of metal-free catalysts should consider the potential effect of trace metal impurities like Cu and Zn because metal impurities will be introduced from reactants/precursors during synthesis of the catalyst and from electrolytes during electrolysis.26,38–43 Therefore, a systematic evaluation of trace metal loading-dependency on GDE was performed as it relates to the CO/CO2RR activity and selectivity.
Herein, to clearly reveal the effect of low levels (ppm) of Cu on the CO/CO2RR performance, trace amounts of Cu with a range of loadings was directly loaded onto carbon paper (CP, Sigracet 39BB gas diffusion layer) via a facile impregnation method. This loading method does not use any additional carbon supports as reported in the literature and subsequently eliminates the effect of the carbon support on the performance. A significant activity towards the CORR is observed on trace Cu-loaded GDE when the loading is higher than 0.042 μg cm−2 (4.9 ppm referred to the electrode mass). Cu loading down to 0.042 μg cm−2 can lead to up to 50% FE of carbon products, with CH4 as the major carbon product, at −0.76 V in the CORR. The primary product switches from C1 to C2 with a total 70% FE at −0.74 V when the Cu loading increases from 0.042 to 0.333 μg cm−2. The CO2RR requires a higher Cu content than the CORR to trigger the activity. Only predominant products of C1 are observed at a Cu loading of 0.812 μg cm−2. The primary product switches from CO to CH4 with the increase in overpotential. These findings indicate that a ppm level of Cu in the electrodes has a significant impact on the CO/CO2RR, especially on the CORR. In addition, this study proposes the importance of the design of Cu catalysts at the atomic scale to maximize the specific activity towards the CO/CO2RR.
Results and discussion
CORR activity over a trace Cu-loaded carbon paper.
The electrocatalytic activity and selectivity towards CO/CO2 reduction were evaluated in a custom flow cell. The geometric area of the CP electrode is 2.8 cm2 (1.67 cm × 1.67 cm), of which 1.0 cm2 (1.0 cm × 1.0 cm) is the active area matching the dimensions of the flow field and the cathode compartment (more details in the Experimental methods of the ESI†). The CP electrodes containing a Cu loading of 0.812 μg cm−2 (denoted as CP–Cu0.812) were first investigated for the CORR. CP–Cu0.812 electrodes exhibit outstanding electrochemical activity to the CORR with the highest FE of carbon products reaching 83% at −0.64 V (Fig. 1a). The FE of C2 (including CH3COO−, C2H4, and CH3CH2OH) products maintains high (>48%) in a wide potential window from −0.6 to −0.87 V (Fig. 1b), and the highest 78% FE of C2 products is achieved at −0.64 V (Fig. 1b). In contrast, the FE of CH4 only rises to a maximum of 19.1% as the potential is negatively swept to −0.86 V. The partial current densities for both C2 and CH4 (jC2 and jCH4) monotonically increase as the potential is swept cathodically. The jC2 and jCH4 climb to 429 and 170 mA cm−2, respectively, at −0.86 V (Fig. 1c). The total current density of the carbon products with a Cu loading of 0.812 μg cm−2 reaches 599 mA cm−2 at −0.86 V, which is even higher than the Cu nanocube44 and nanosheet45 catalysts with loadings as high as 500 μg cm−2 under similar operating conditions, indicating that the trace Cu on CP exhibits extraordinary electrochemical activity toward the CORR.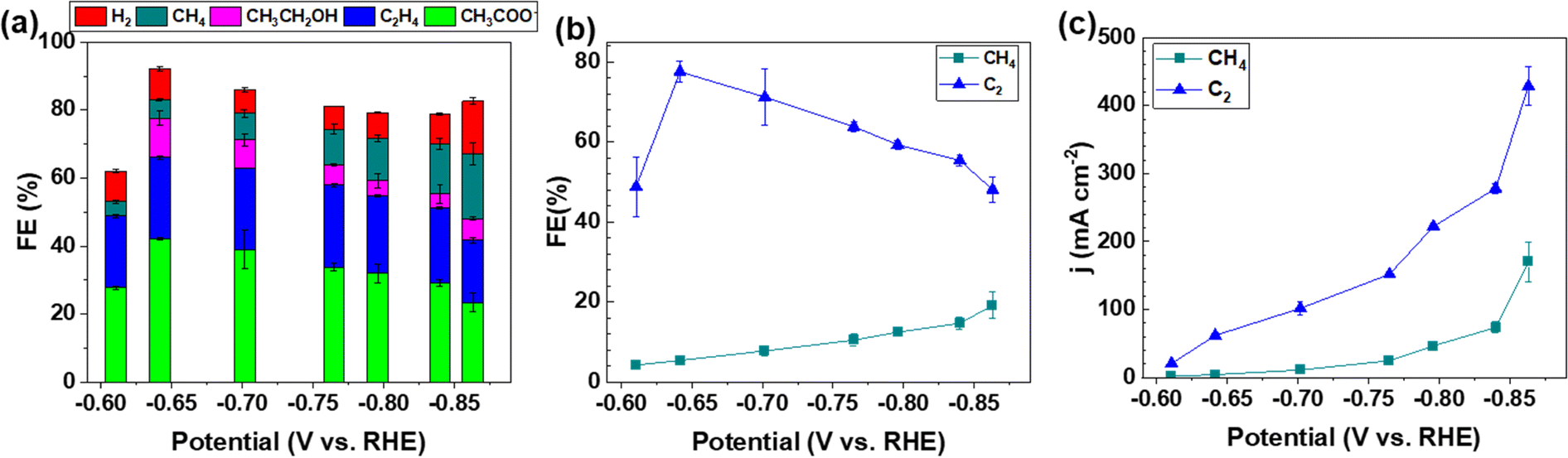 | ||
| Fig. 1 (a) Product distribution during the CORR as a function of the applied potential on the CP–Cu0.812 electrode. (b) FEs of C2 and CH4. (c) The partial current density of C2 and CH4. The error bars represent the standard deviation from the measurements of three independent electrodes. | ||
To systematically investigate the impact of trace Cu loadings on the CORR, we prepared CP electrodes with Cu loadings ranging from 0.016 to 1.53 μg cm−2, as shown in Table S1 (ESI†). Notably, the Cu loading in the as-received pristine CP is 0.016 μg cm−2 (PCP–Cu0.016), and thus, the total Cu content in the prepared CP electrodes includes this Cu impurity in the PCP. The CORR performance of the CP electrodes with different Cu contents is shown in Fig. 2 and Fig. S2 (ESI†). A significant effect on the CORR is observed when the Cu loading exceeds 0.042 μg cm−2 Cu (4.9 ppm Cu in the total electrode), and 50% FE of carbon products with the predominant carbon product of CH4 is obtained at this Cu level. The peak FE of 39% for CH4 is reached with CP–Cu0.070 electrodes. The primary product switches to C2 when the Cu loading further increases to 0.333 μg cm−2, indicating that the C–C coupling reaction is accelerated with the existence of more Cu active sites. The highest C2 FE of 78% is achieved at −0.64 V with a Cu loading of 0.812 μg cm−2. The current density of C2 exhibits a positive correlation with the Cu loading, while the current density of CH4 remains in a similar range with other Cu loadings.
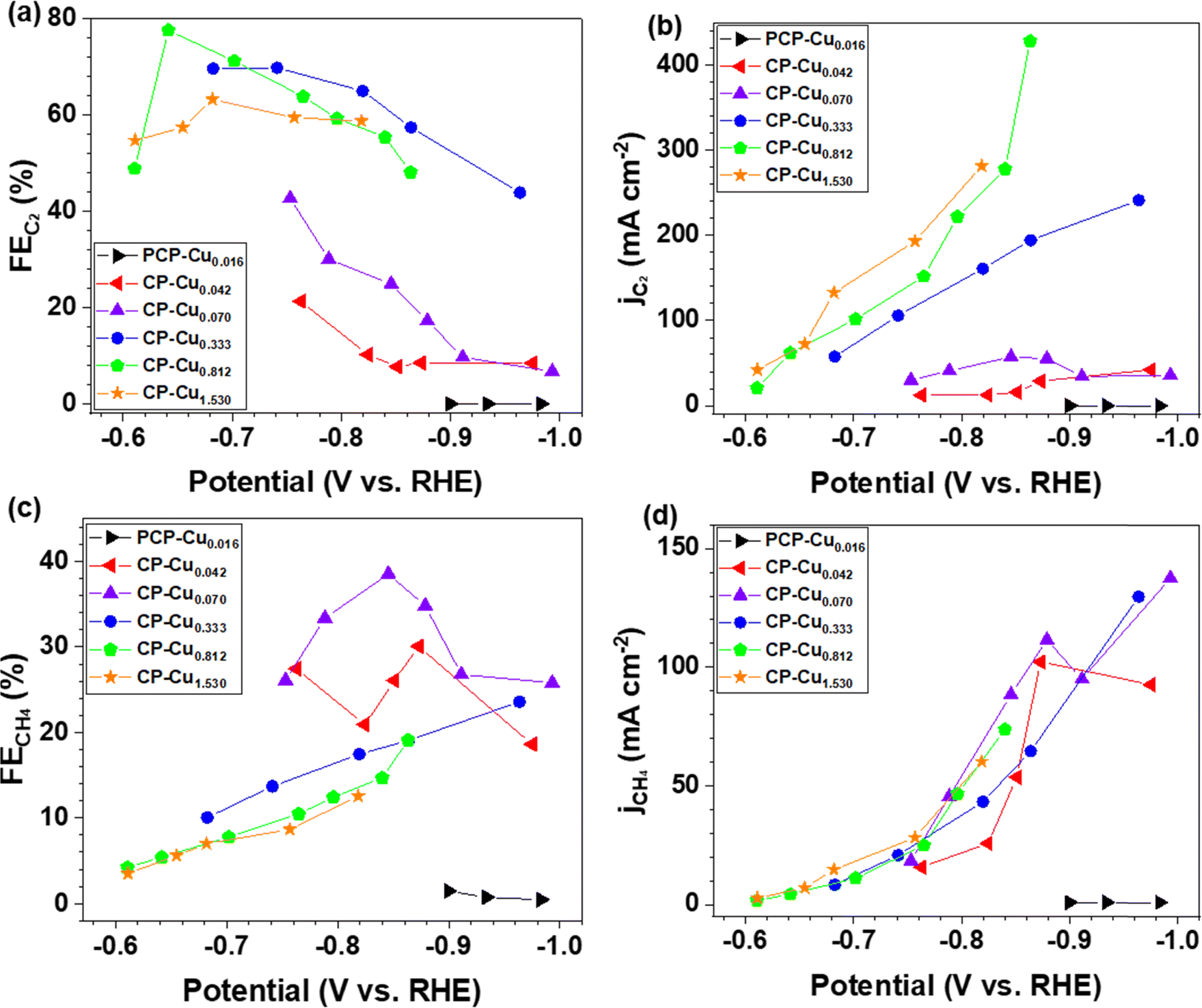 | ||
| Fig. 2 (a) The partial current density and (b) FE of C2 during the CORR as a function of the applied potential using CP–Cu electrodes with various Cu loadings. (c) The partial current density and (d) FE of CH4 as a function of the applied potential using CP–Cu electrodes with various Cu loadings. | ||
The mass activity for C2 reaches 584 A mg−1 of Cu with a FE of 57% at −0.86 V for the CP–Cu0.333 electrodes, which is 2–5 orders of magnitude higher than those reported in the literature (Table S2, ESI†). The mass activity for CH4 rises to 2435 A mg−1 of Cu with a FE of 30.1% at −0.87 V using the CP–Cu0.042 electrodes, which is an increase of 4 orders of magnitude compared with similar Cu/C electrode reported values.12 This is a clear indication that a trace level of Cu can be very active and should always be accounted for in the CORR experiments.
A control experiment was carried out to evaluate the effect of the CP treatment with the solvent mixture of DI water and IPA but without additional CuCl2 on the CORR. After exposure to the solution, the Cu loading declines from 0.016 to 0.006 μg cm−2, indicating the Cu contamination in the PCP can be partially dissolved in the solvent mixture. The electrochemical results of the CP–Cu0.006 electrodes are shown in Fig. S3 (ESI†). Only trace CH4 is produced and H2 predominates, which is a similar performance to PCP–Cu0.016. Another control experiment was performed to investigate the effect of solvent on the Cu impregnation, where the highest concentration of 5 mM of CuCl2 in DI water was used. The performance of this control electrode for the CORR is displayed in Fig. S4 (ESI†). The HER exclusively occurs under current densities from 107 to 321 mA cm−2, and only trace amounts of CH4 with a FE < 2.2% are detected. This finding suggests that the Cu ions in pure DI water could not be loaded onto the CP substrate, as the CP surface is highly hydrophobic. Fig. S5 (ESI†) shows contact angle measurements on CP with pure DI water and a mixture of DI water/IPA (v/v: 1/1). The contact angle decreases from 169 to 42 °C when replacing the pure DI water with the solvent mixture.
The product distribution of the CORR as a function of the trace Cu loading is further plotted in Fig. 3. The FEs of each CORR product under different Cu loadings are compared at the current density of around 130 mA cm−2 (Fig. 3a). The peak FE of 33% for CH4 is achieved with 0.070 μg cm−2 Cu loading. The CH3COO− is predominant with a peak FE of 41% at a Cu loading of 0.333 μg cm−2. The results indicate that the trace level of Cu is already very active towards the CORR to form CH3COO−. The FE ratio of C1/C2 is 1.1 with the CP–Cu0.070 electrode, and it drops to 0.11 with the CP–Cu0.812 electrode, indicating the selectivity varies by 10 times after 0.742 μg cm−2 Cu is added. Fig. 3b depicts the product distribution of the CORR at around 330 mA cm−2 as the trace Cu loading increases, and a similar trend is observed with 130 mA cm−2. The notable difference is that the FE of C2 decreases while the FE of C1 increases slightly at a higher current density and overpotential, as the high overpotential prefers CH4 formation.46–48
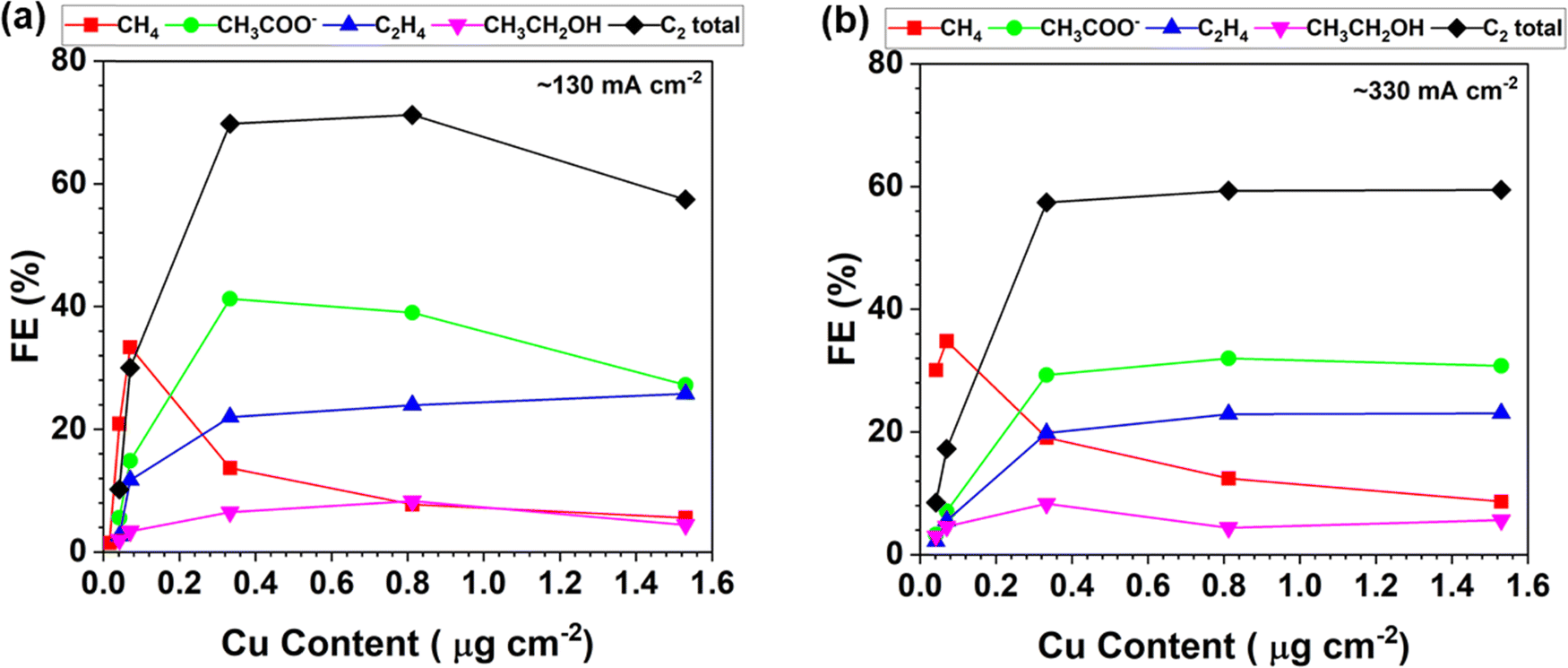 | ||
| Fig. 3 Product distribution of the CORR as a function of the Cu loading in the electrodes at the current density of around 130 mA cm−2 (a) and 330 mA cm−2 (b). | ||
The performance of CO2RR with different Cu loadings in carbon paper
The electrochemical activity of trace Cu on CP was also evaluated for the CO2RR (Fig. S6, ESI†). The low (CP–Cu0.070) and high (CP–Cu0.812) Cu loading electrodes were chosen for the CO2RR since these electrodes exhibit the predominant selectivity towards C1 and C2 in the CORR, respectively. The highest FE of carbon products with the CP–Cu0.070 electrode in the CO2RR is 62%, lower than in the CORR (83%), indicating that the CO2RR is less sensitive to trace Cu than the CORR. It is worth pointing out that trace Cu favors formation of C1 products in the CO2RR, which is different from C2 products being favored in the CORR. The predominant product is CO at low potentials, and it transitions to CH4 when the potential is swept more negatively for both CP–Cu0.070 and CP–Cu0.812 electrodes. The peak FE of CO is 26% appearing at −0.57 V for the CP–Cu0.070 electrode while the maximum FE of CH4 is 24% at −0.67 V. The CP–Cu0.812 electrode shows the highest FE of CO, 43%, at −0.55 V and 38% FE of CH4 is achieved at −0.83 V. The CP–Cu0.070 electrodes possess mass activities of 155 and 375 A mg−1 Cu for CO and CH4, respectively, which are 2–3 orders of magnitude higher than those reported in the literature (Table S2, ESI†). This finding indicates that Cu impurities as low as 0.070 μg cm−2 (8.2 ppm relative to the total electrode) can significantly affect the performance of carbon catalysts for C1 (e.g., CO and CH4) generation. Our previous work found that functionalized N-doped graphene quantum dots (NGQDs) could catalyze CO2 to CH4 with a high selectivity of 70% FE at −0.85 V, and the Cu impurity on electrodes is 0.056 μg cm−2.32 We observed that the highest FE of CH4 is 24% at −0.67 V for 0.070 μg cm−2 Cu and 14% at −0.81 V for 0.042 μg cm−2 Cu (Fig. S6, ESI†). Compared to trace Cu only, the functionalized NGQDs requires a more negative potential to reach the maximum FE of C2H4. The facts indicate that both functionalized NGQDs and trace Cu may contribute to CH4 formation in the CO2RR, even though functionalized NGQDs play a more significant role.The trace metal effect on the CO/CO2RR can be extended to other metal impurities, such as Ag, Au, Zn, Sn, and Bi. To verify this hypothesis, we loaded trace Zn (∼1.6 μg cm−2) onto the CP by impregnating CP in a 5 mM ZnCl2 solution (2 mL, H2O(v)/IPA(v) = 1/1 solvent) for 1 h. The electrochemical activity of trace Zn to the CO2RR is shown in Fig. S7 (ESI†). The FE of CO is higher than 60% in the wide testing potential range of −0.65 to −1.08 V, and the total current density reaches 300 mA cm−2 at −1.08 V, indicating that the trace Zn on the CP exhibits outstanding activity for CO generation. M–N–C catalysts are commonly employed as single-atom metal electrocatalysts for catalyzing CO2 to CO with high FEs.41–43 However, to the best of our knowledge, the metal impurities, such as Au, Ag, and Zn, were never determined and evaluated with those M–N–C catalysts, which may result in the overestimation of M–N–C catalyst performance.
Chemical structure of Cu
In order to correlate the chemical structure and oxidation status of Cu with the electrochemical activity towards the CORR/CO2RR, the trace Cu structure was characterized by a combination of electron microscopy and spectroscopy. The microstructure of the CP–Cu0.812 post-electrode was characterized by scanning electron microscopy (SEM), as shown in Fig. S8 (ESI†). No aggregate of Cu at the micron size was observed. The corresponding energy dispersive X-ray spectroscopy (EDS) elemental mapping is shown in Fig. S9 (ESI†). It is well known that characterization of ppm level Cu in carbon materials is a challenge. The Cu structure on the CP–Cu0.812 post-electrode was determined further with aberration-corrected scanning transmission electron microscope (AC-STEM). No metal nanoparticles or clusters were found, and only a few Cu single atoms were observed after many attempts, as shown in Fig. 4a and b.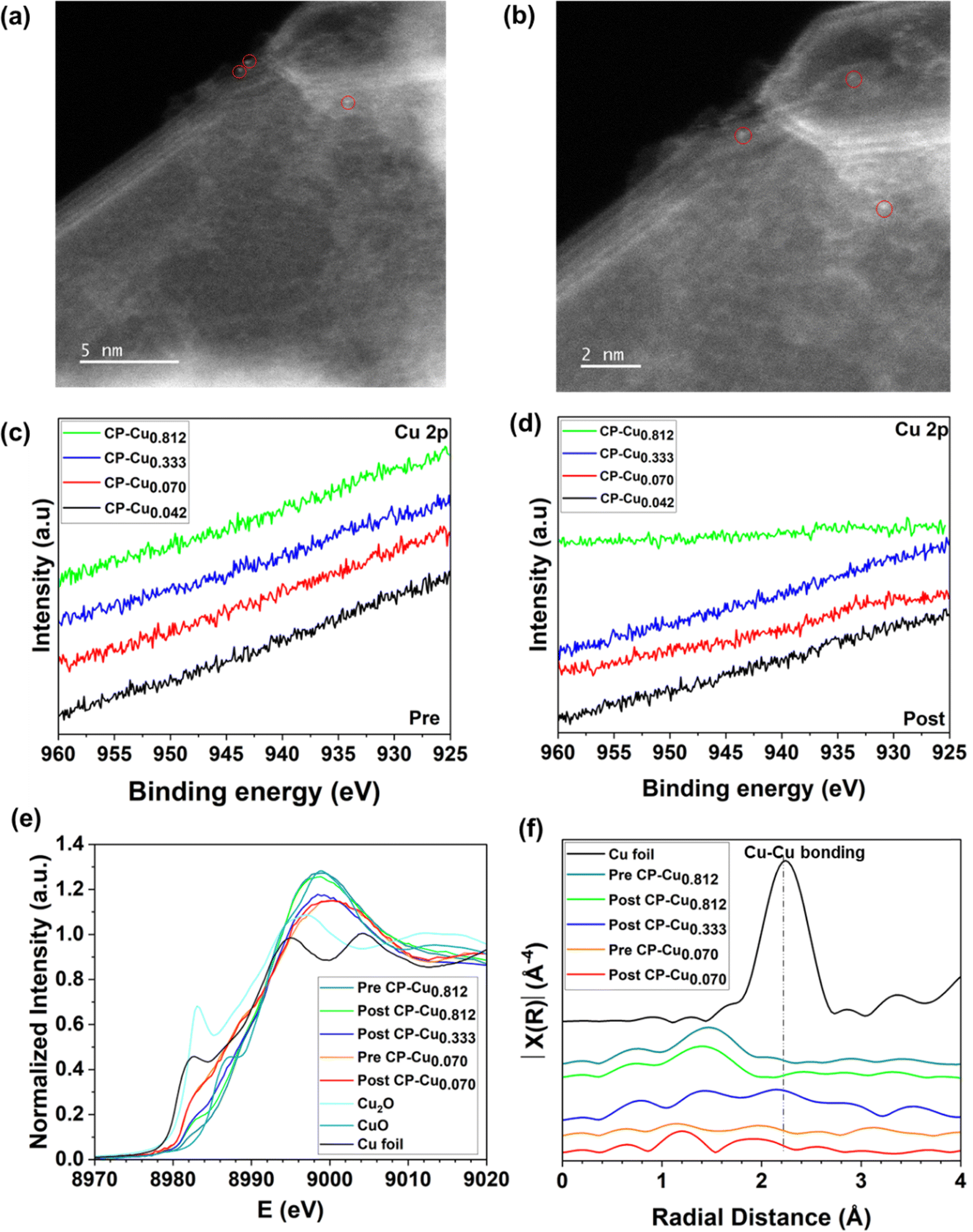 | ||
| Fig. 4 Structural characterization of the CP–Cu electrodes. (a and b) AC-HAADF-STEM images of single atom Cu in the CP–Cu0.812 post-electrode after the CORR. (c) High-resolution XPS spectra of Cu 2p of the pre-experiment CP–Cu electrodes. (d) High-resolution XPS spectra of Cu 2p of the post-experiment CP–Cu electrodes in the CORR. (e) Cu K-edge normalized XANES spectra as a function of incident photon energy (E) for the CP–Cu electrodes. (f) Fourier transform of k3-weighted EXAFS in R space of the CP–Cu electrodes. | ||
Fig. 4c and d show the high-resolution X-ray photoelectron spectroscopy (XPS) spectra of Cu 2p of pre- and post-electrodes, respectively. No notable peaks of Cu 2p are visible on any electrodes due to the ultralow Cu loading. X-ray absorption spectroscopy (XAS) was employed to further investigate the chemical structure of Cu species at the atomic scale. Fig. 4e presents the X-ray absorption near edge structure (XANES) spectra of the Cu K-edge for CP–Cu0.070 (pre- and post-), CP–Cu0.333 (post-), and CP–Cu0.812 (pre- and post-) electrodes. The Cu shows a higher energy absorption edge at higher Cu loadings, suggesting an increase of Cu valence with an increase of Cu loading. The possible reason is that the high-loading Cu species are more susceptible to being oxidized by oxygen in the air due to the impregnation method. Compared to the pre-electrodes of CP–Cu0.812, the post-electrode shows lower Cu energy due to the reduction of Cu in the CORR. The energy absorption from all electrodes is in the range between CuO and Cu2O, indicating that Cu in the post-reaction electrodes has a chemical state of Cuδ+ (1 < δ < 2). Fig. 4f displays the Cu Fourier-transformed k3-weighted extended X-ray absorption fine structure (EXAFS) in R space. Only a notable peak at ca. 1.5 Å, which is associated with Cu–C/O scattering, is observed in both the pre-and post-electrodes of CP–Cu0.812.28,49,50 No Cu–Cu bonding peak is seen for any electrodes, which confirms that the Cu species are atomically dispersed on CP. According to the analysis above, we conclude that the high electrochemical activity towards the CO/CO2RR comes from a trace level of atomically dispersed Cu with a chemical state of Cuδ+ (1 < δ < 2).
Since the Cu species are atomically dispersed in the CP, the TOF of trace amounts of Cu catalysts for the CO/CO2RR were calculated assuming the existence of single atom Cu. For the CP–Cu0.333 electrodes, the TOF for the CO-to-C2 products conversion reaches 145 s−1 at −0.86 V, which is 4 orders of magnitude higher than those reported in the literature (Table S3, ESI†). For the CP–Cu0.042 electrodes, the TOF for the CO-to-CH4 conversion achieves 267 s−1 at −0.87 V, which has increased by 2 orders of magnitude compared with reported values in the literature (Table S3, ESI†).
Proposed mechanism for the CO/CO2RR in the trace Cu loaded CPs
We attempted to propose the reaction mechanism of the CO/CO2RR on trace Cu loaded CPs based on documented theoretical investigations and our experimental observations. At a very low Cu loading in the CP (<0.333 μg cm−2), the primary product in the CORR is CH4, while it switches to C2 when the Cu loading is higher than 0.333 μg cm−2. The predominant product switches from C1 to C2 with an increased Cu loading in the CP, while the Cu species remain atomically dispersed in the CP. The density of the Cu single atom is hypothesized to be responsible for the switch of the predominant product in the CORR. A low density of Cu loading prefers the C1 products, while a high density of Cu loading favors the C2 products. It was reported that the single-site Cu can facilitate the protonation of CO* to CHO* and then form C1 products due to the strong chemisorption of H* and the high partial pressure of CO in the CORR.28 The lack of Cu pair active sites makes C1 the primary product. The C–C coupling is accelerated with the increase of atomic Cu density.27 The C–C coupling reaction can proceed via either the Eley–Rideal mechanism or the Langmuir–Hinshelwood mechanism. The Eley–Rideal mechanism is supposed to be predominant in the CORR due to the high partial pressure of CO.Distinguished from the CORR, the main product is only C1 (e.g., CO and CH4) in the CO2RR with trace amounts of Cu, indicating that the C–C coupling in the CO2RR is more challenging than that in the CORR. One possible reason is that it is difficult for the C–C coupling to occur by the Eley–Rideal mechanism due to the low pressure of CO under the CO2RR conditions. The other possible reason is that the CO2 is more inert than CO, and the adoption of CO2 on the Cu species is much more difficult than for CO.
Conclusions
We revealed the activity and selectivity of trace amounts of Cu with various loadings towards the CO/CO2RR by simply impregnating Cu onto CP. An extraordinary mass specific activity and selectivity toward the CORR is observed with 50% FE of carbon products including the predominant CH4 when the Cu loading is higher than 0.042 μg cm−2. The predominant product switches from CH4 to C2 with an increase of Cu to 0.333 μg cm−2. When further increasing the Cu loading to 0.812 μg cm−2, the overall highest FE of C2 achieves 78%, of which the major product of CH3COO− accounts for 42%. The record mass activities for CH4 and C2 reach 2435 and 584 A mg−1 of Cu, corresponding to TOFs of 267 and 145 s−1, respectively. Different from the CORR, a higher loading of Cu is required to trigger the CO2RR. Moreover, trace Cu primarily catalyzes the CO2RR to form C1 (e.g., CO and CH4) even when the Cu loading reaches 0.812 μg cm−2, indicating trace Cu is not in favor of C–C coupling in the CO2RR. The effect of trace metal impurities can be extended to others that are active towards the CO2RR, such as Zn for CO2-to-CO conversion. We suggest that the effect of trace metal impurities must be quantified when developing metal-free and M–N–C catalysts for the CO/CO2RR.Disclaimer
This manuscript has been co-authored by UT-Battelle, LLC, under contract DE-AC05-00OR22725 with the US Department of Energy (DOE). The US government retains and the publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for US government purposes. The DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (https://energy.gov/downloads/doe-public-access-plan).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Office of Fossil Energy and Carbon Management of the U.S. Department of Energy under Award Number DE-FE0031919. A portion of this work was funded by ORNL's Laboratory Directed Research and Development (LDRD) Seed Money program. This research used resources of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, and is supported by the U.S. DOE under Contract No. DE-AC02-06CH11357, and the Canadian Light Source and its funding partners.References
- J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Nat. Catal., 2022, 5, 268–276 CrossRef CAS.
- Z. Han, D. Han, Z. Chen, J. Gao, G. Jiang, X. Wang, S. Lyu, Y. Guo, C. Geng and L. Yin, Nat. Commun., 2022, 13, 1–10 Search PubMed.
- J. Timoshenko, A. Bergmann, C. Rettenmaier, A. Herzog, R. M. Arán-Ais, H. S. Jeon, F. T. Haase, U. Hejral, P. Grosse and S. Kühl, Nat. Catal., 2022, 5, 259–267 CrossRef CAS.
- T. Zhang, J. C. Bui, Z. Li, A. T. Bell, A. Z. Weber and J. Wu, Nat. Catal., 2022, 5, 202–211 CrossRef CAS.
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani and X. Wang, Science, 2020, 367, 661–666 CrossRef PubMed.
- Z. Li, Y. Fang, J. Zhang, T. Zhang, J. D. Jimenez, S. D. Senanayake, V. Shanov, S. Yang and J. Wu, J. Mater. Chem. A, 2021, 9, 19932–19939 RSC.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li and Y. Wang, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- P. Saha, S. Amanullah and A. Dey, Acc. Chem. Res., 2022, 11214–11217 Search PubMed.
- W. Choi, S. Park, W. Jung, D. H. Won, J. Na and Y. J. Hwang, ACS Energy Lett., 2022, 7, 939–945 CrossRef CAS.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave and E. B. Duoss, Nat. Energy, 2022, 1–14 Search PubMed.
- Y. Li, A. Ozden, W. R. Leow, P. Ou, J. E. Huang, Y. Wang, K. Bertens, Y. Xu, Y. Liu and C. Roy, Nat. Catal., 2022, 5, 185–192 CrossRef CAS.
- J. L. Hitt, Y. G. C. Li, S. S. Tao, Z. F. Yan, Y. Gao, S. J. L. Billinge and T. E. Mallouk, Nat. Commun., 2021, 12, 1114 CrossRef CAS PubMed.
- A. K. Ummireddi, S. K. Sharma and R. G. S. Pala, J. Catal., 2022, 213–221 CrossRef CAS.
- C. Li, H. Xiong, M. He, B. Xu and Q. Lu, ACS Catal., 2021, 11, 12029–12037 CrossRef CAS.
- T. Zhang, X. Li, Y. Qiu, P. Su, W. Xu, H. Zhong and H. Zhang, J. Catal., 2018, 357, 154–162 CrossRef.
- Z. Wu, H. Wu, W. Cai, Z. Wen, B. Jia, L. Wang, W. Jin and T. Ma, Angew. Chem., 2021, 133, 12662–12667 CrossRef.
- F. Cheng, X. Zhang, K. Mu, X. Ma, M. Jiao, Z. Wang, P. Limpachanangkul, B. Chalermsinsuwan, Y. Gao and Y. Li, Energy Technol., 2021, 9, 2000799 CrossRef CAS.
- J. Li, J. Li, X. Liu, J. Chen, P. Tian, S. Dai, M. Zhu and Y.-F. Han, Appl. Catal., B, 2021, 298, 120581 CrossRef CAS.
- R. O. Yang, J. Y. Duan, P. P. Dong, Q. L. Wen, M. Wu, Y. W. Liu, Y. Liu, H. Q. Li and T. Y. Zhai, Angew. Chem., Int. Ed., 2022, 21 DOI:10.1002/anie.202116706.
- G. D. Shi, Y. L. Xie, L. L. Du, X. L. Fu, X. J. Chen, W. J. Xie, T. B. Lu, M. J. Yuan and M. Wang, Angew. Chem., Int. Ed., 2022, 61 DOI:10.1002/anie.202203569.
- Y. B. Ma, J. L. Yu, M. Z. Sun, B. Chen, X. C. Zhou, C. L. Ye, Z. Q. Guan, W. H. Guo, G. Wang, S. Y. Lu, D. S. Xia, Y. H. Wang, Z. He, L. Zheng, Q. B. Yun, L. Q. Wang, J. W. Zhou, P. Y. Lu, J. W. Yin, Y. F. Zhao, Z. B. Luo, L. Zhai, L. W. Liao, Z. L. Zhu, R. Q. Ye, Y. Chen, Y. Lu, S. B. Xi, B. L. Huang, C. S. Lee and Z. X. Fan, Adv. Mater., 2022, 34, 2110607 CrossRef CAS PubMed.
- Y. Y. Liu, H. L. Zhu, Z. H. Zhao, N. Y. Huang, P. Q. Liao and X. M. Chen, ACS Catal., 2022, 12, 2749–2755 CrossRef CAS.
- T. Y. Zhang, J. C. Bui, Z. Y. Li, A. T. Bell, A. Z. Weber and J. J. Wu, Nat. Catal., 2022, 5, 202–211 CrossRef CAS.
- X. Chang, A. Malkani, X. Yang and B. Xu, J. Am. Chem. Soc., 2020, 142, 2975–2983 CrossRef CAS PubMed.
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2021, 50, 12897–12914 RSC.
- Y. Lum, Y. Kwon, P. Lobaccaro, L. Chen, E. L. Clark, A. T. Bell and J. W. Ager, ACS Catal., 2016, 6, 202–209 CrossRef CAS.
- X. Fu, Y. Wang, H. Shen, Y. Yu, F. Xu, G. Zhou, W. Xie, R. Qin, C. Dun and C.-W. Pao, Mater. Today Phys., 2021, 19, 100418 CrossRef CAS.
- W. Rong, H. Zou, W. Zang, S. Xi, S. Wei, B. Long, J. Hu, Y. Ji and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 466–472 CrossRef CAS PubMed.
- H. Yang, Y. Wu, Q. Lin, L. Fan, X. Chai, Q. Zhang, J. Liu, C. He and Z. Lin, Angew. Chem., 2018, 130, 15702–15706 CrossRef.
- J. Zhao, Z. Chen and J. Zhao, J. Mater. Chem. A, 2019, 7, 4026–4035 RSC.
- J. Li, W.-Y. Zan, H. Kang, Z. Dong, X. Zhang, Y. Lin, Y.-W. Mu, F. Zhang, X.-M. Zhang and J. Gu, Appl. Catal., B, 2021, 298, 120510 CrossRef CAS.
- T. Zhang, W. Li, K. Huang, H. Guo, Z. Li, Y. Fang, R. M. Yadav, V. Shanov, P. M. Ajayan and L. Wang, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed.
- R. M. Yadav, Z. Li, T. Zhang, O. Sahin, S. Roy, G. Gao, H. Guo, R. Vajtai, L. Wang and P. M. Ajayan, Adv. Mater., 2022, 34, 2105690 CrossRef CAS PubMed.
- L. Han, S. Song, M. Liu, S. Yao, Z. Liang, H. Cheng, Z. Ren, W. Liu, R. Lin, G. Qi, X. Liu, Q. Wu, J. Luo and H. L. Xin, J. Am. Chem. Soc., 2020, 142, 12563–12567 CrossRef CAS PubMed.
- J. Wu, S. Ma, J. Sun, J. I. Gold, C. Tiwary, B. Kim, L. Zhu, N. Chopra, I. N. Odeh and R. Vajtai, Nat. Commun., 2016, 7, 1–6 Search PubMed.
- Y. M. Liu, S. Chen, X. Quan and H. T. Yu, J. Am. Chem. Soc., 2015, 137, 11631–11636 CrossRef CAS PubMed.
- Y. M. Liu, Y. J. Zhang, K. Cheng, X. Quan, X. F. Fan, Y. Su, S. Chen, H. M. Zhao, Y. B. Zhang, H. T. Yu and M. R. Hoffmann, Angew. Chem., Int. Ed., 2017, 56, 15607–15611 CrossRef CAS PubMed.
- C. H. A. Wong, Z. Sofer, M. Kubešová, J. Kučera, S. Matějková and M. Pumera, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13774–13779 CrossRef CAS PubMed.
- G. Dong, M. Fang, H. Wang, S. Yip, H.-Y. Cheung, F. Wang, C.-Y. Wong, S. T. Chu and J. C. Ho, J. Mater. Chem. A, 2015, 3, 13080–13086 RSC.
- A. Wuttig and Y. Surendranath, ACS Catal., 2015, 5, 4479–4484 CrossRef CAS.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, 2001848 CrossRef CAS PubMed.
- A. S. Varela, W. Ju, A. Bagger, P. Franco, J. Rossmeisl and P. Strasser, ACS Catal., 2019, 9, 7270–7284 CrossRef CAS.
- A. S. Varela, W. Ju and P. Strasser, Adv. Energy Mater., 2018, 8, 1703614 CrossRef.
- P. Zhu, C. Xia, C.-Y. Liu, K. Jiang, G. Gao, X. Zhang, Y. Xia, Y. Lei, H. N. Alshareef and T. P. Senftle, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 2010868118 CrossRef PubMed.
- W. Luc, X. Fu, J. Shi, J.-J. Lv, M. Jouny, B. H. Ko, Y. Xu, Q. Tu, X. Hu, J. Wu, Q. Yue, Y. Liu, F. Jiao and Y. Kang, Nat. Catal., 2019, 2, 423–430 CrossRef CAS.
- L. L. Zhuo, P. Chen, K. Zheng, X. W. Zhang, J. X. Wu, D. Y. Lin, S. Y. Liu, Z. S. Wang, J. Y. Liu and D. D. Zhou, Angew. Chem., 2022, 134, e202204967 CrossRef.
- S. Y. Lee, H. Jung, N.-K. Kim, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2018, 140, 8681–8689 CrossRef CAS PubMed.
- A. Eilert, F. S. Roberts, D. Friebel and A. Nilsson, J. Phys. Chem. Lett., 2016, 7, 1466–1470 CrossRef CAS PubMed.
- F. Huang, Y. Deng, Y. Chen, X. Cai, M. Peng, Z. Jia, J. Xie, D. Xiao, X. Wen and N. Wang, Nat. Commun., 2019, 10, 1–7 CrossRef PubMed.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean and R. E. Winans, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ey00071g |
| This journal is © The Royal Society of Chemistry 2023 |