
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFe–N/C catalysts with tunable mesoporous structures and carbon layer numbers reveal the role of interlayer O2 activation†
Jinwoo
Woo
a,
June Sung
Lim
a,
Taejung
Lim
b,
Du San
Baek
b,
Jae Hyung
Kim
 c,
Jong Hoon
Lee
d,
Hu Young
Jeong
e,
Chang Hyuck
Choi
f and
Sang Hoon
Joo
*b
c,
Jong Hoon
Lee
d,
Hu Young
Jeong
e,
Chang Hyuck
Choi
f and
Sang Hoon
Joo
*b
aSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan, 44919, Republic of Korea
bDepartment of Chemistry, UNIST, Ulsan, 44919, Republic of Korea. E-mail: shjoo@unist.ac.kr
cClean Fuel Research Laboratory, Korea Institute of Energy Research, Daejeon, 34129, Republic of Korea
dUNIST Central Research Facilities, UNIST, Ulsan, 44919, Republic of Korea
eGraduate School of Semiconductor Materials and Devices Engineering, UNIST, Ulsan, 44919, Republic of Korea
fDepartment of Chemistry, Pohang University of Science and Technology (POSTECH), Pohang, 37673, Republic of Korea
First published on 17th November 2022
Abstract
A class of Fe,N-codoped carbon (Fe–N/C) electrocatalysts has made remarkable advances as highly promising non-Pt group metal catalysts for the oxygen reduction reaction (ORR). However, the design of Fe–N/C catalysts whose active site structure and O2 activation mode mimic those of an enzymatic ORR catalyst still remains a challenge. Herein, we report the preparation of mesoporous Fe–N/C catalysts with tunable tube- or rod-like frameworks and carbon layer numbers via solid-state nanocasting of mesoporous silica with an iron–phenanthroline complex. The tube-type Fe–N/C exhibited a larger surface area and active site density than the rod-type Fe–N/C. Unexpectedly, the rod-type Fe–N/C showed superior ORR activity to the tube-type Fe–N/C, with a smaller overpotential, greater turnover frequency (TOF), and lower Tafel slope. Temperature-programmed desorption studies revealed a weaker binding strength of the rod-type Fe–N/C with O2. The rod-type Fe–N/C consisting of multiple carbon layers is likely to activate O2 in the interlayer between the Fe–Nx-containing carbon layers, reminiscent of the enzymatic catalyst, whereas in the tube-type Fe–N/C with 3–4 carbon layers, O2 is mostly activated at a single Fe center on the external carbon layer. As a result, the TOF of the rod-type Fe–N/C exceeded those of reported catalysts. We envisage that our work can provide a new insight into the design of highly active Fe–N/C catalysts.
Broader contextFe–N/C catalysts with atomically dispersed Fe–Nx active sites have been of tremendous interest as a class of promising non-precious metal catalysts for the oxygen reduction reaction (ORR). Although great strides have been made in multiple aspects of Fe–N/C catalysts over the past few years, the realization of a structural analog of the enzymatic ORR catalyst cytochrome c oxidase (CcO), which can cooperatively activate O2 molecules using a bimetallic center, has remained a daunting challenge. In this work, we introduce Fe–N/C catalysts with tunable mesoporous structures and carbon layer numbers, which could serve as model catalysts for revealing the important role of interlayer oxygen activation in boosting ORR activity. We believe that our work provides fundamental and useful guidelines for designing Fe–N/C catalysts for the ORR. |
1. Introduction
In light of ever-increasing climate change, the development of a hydrogen-based energy cycle that can replace the current hydrocarbon-based energy cycle in a carbon-neutral manner is imperative. The overall performance of the hydrogen-based energy cycle critically depends on the efficiency of the electrocatalysts used for the interconversions between H2, O2, and H2O.1–4 For fuel cells that convert chemical energy stored in fuels (e.g., H2) into electrical energy, the development of highly active electrocatalysts that can overcome the sluggish kinetics of the oxygen reduction reaction (ORR) at the cathode is crucial. Despite decades-long efforts, platinum (Pt) or Pt-based alloys are still mainstays as ORR catalysts.5–11 However, they are expensive, scarce, and unevenly distributed.1–4 In this regard, multifaceted efforts have been pursued to develop non-precious metal catalysts (NPMCs) for the ORR.12–24Among the classes of NPMCs, M–N/C catalysts comprising atomically dispersed M–Nx active sites are considered to be the most promising alternatives to Pt-based catalysts, because of their pronounced ORR activity.25–31 In particular, the last few years have witnessed great strides in the diverse aspects of M–N/C-based ORR catalysts. Synthetic advances in M–N/C catalysts that increase the density of accessible M–Nx sites have enabled remarkable enhancement of their ORR activity;32–45 effective suppression of undesirable H2O2 formation has boosted long-term durability;46–49 molecular-level synthetic control and precise speciation of M–Nx sites have helped to identify active sites;50–54 the exploration of non-d-block compositions has uncovered new p- and s-block-element-based M–N/C catalysts;55–58 and the standardization of active site quantification and turnover frequency (TOF) calculations has made the comparison of catalytic activity data from multiple laboratories on fair ground.59–64
One of the outstanding issues in heterogeneous M–N/C catalysts is the realization of a structural analog of the enzymatic ORR catalyst cytochrome c oxidase (CcO). CcO contains a bimetallic center of Fe and Cu and catalyzes the ORR with excellent efficiency under ambient conditions.65,66 Inspired by the structure of CcO, exquisitely designed inorganic homogeneous catalysts, including bifacial porphyrins, have been synthesized.67,68 Enzymatic CcO and its molecular mimics have a bimetallic center that can cooperatively activate the O2 double bond. Considerable efforts have been made to design heterogeneous M–N/C catalysts bearing a bimetallic center, with the aim of mimicking the structural motif of CcO.26,69 However, their syntheses involve a high-temperature pyrolysis step, which makes the realization of such a structure a daunting challenge.
In this work, we developed a simple, solid-state route to Fe–N/C catalysts with tunable mesoporous structures and carbon layer numbers, which were exploited as model catalysts to reveal the important role of interlayer oxygen activation between adjacent Fe,N-containing carbon layers. Nanocasting ordered mesoporous silica with a controlled amount of Fe and N precursors in a dry state, followed by high-temperature pyrolysis and silica template removal, afforded ordered mesoporous Fe,N-containing carbons constructed with a tube-type framework of 3–4 concentric carbon layers or a rod-type framework of ca. 20 carbon layers. The tube-type Fe–N/C exhibited a larger specific surface area and accessible active site density than the rod-type Fe–N/C. Unexpectedly, the rod-type Fe–N/C showed superior ORR activity to the tube-type Fe–N/C, with a lower overpotential, much greater kinetic current density and TOF, and lower Tafel slope. The rod-type Fe–N/C exhibited excellent ORR activity with half-wave potentials of 0.92 and 0.80 V in alkaline and acidic electrolytes, respectively, and its TOF surpassed those of reported Fe–N/C catalysts. The rod-type Fe–N/C showed a weaker oxygen binding strength than the tube-type Fe–N/C as revealed by O2 temperature-programmed desorption studies. The weaker O2 binding strength and lower Tafel slope of the rod-type Fe–N/C suggest that a significant number of O2 molecules are likely to be activated in the interlayer between two adjacent Fe,N-containing carbon layers in this catalyst. In the tube-type Fe–N/C, O2 is expected to be activated mainly on a single Fe center in the outer carbon layers.
2. Results and discussion
Fig. 1a illustrates the preparation scheme for Fe–N/C catalysts. Solid-state nanocasting of hexagonally ordered mesoporous silica (SBA-15) with iron acetate (FeAc) and 1,10-phenanthroline (Phen) as the precursors, followed by high-temperature pyrolysis of the composite and etching of the SBA-15 template, afforded Meso-Fe–N/C_X (where X = weight ratio of the Fe and N precursors to the silica template with the molar ratio of the Fe and N precursors fixed at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) catalysts (see 4. Materials and methods for details). The mixing of Fe2+ ions and Phen in a 1:3 ratio readily generated the Fe(Phen)3 complex in a soluble solvent; however, the complex prepared under wet conditions could generate inactive Fe clusters upon high-temperature pyrolysis in the Fe–N/C catalyst synthesis.70 In turn, we attempted to prepare Fe(Phen)3 under dry conditions in the presence of a silica template, which had a high density of hydrophilic silanol groups on its surface. We hypothesized that silanol groups could facilitate the complex formation. Indeed, Fourier-transform infrared (FT-IR) spectra indicated that, even under solvent-less dry conditions, Fe2+ and Phen formed the Fe(Phen)3 complex in the presence of mesoporous silica. We compared the FT-IR spectra of the mixtures of the precursors (FeAc and Phen) and SBA-15 silica with different mass ratios (denoted Mixture_X) with those of Phen, a mixture of FeAc and Phen prepared under dry conditions, and the Fe(Phen)3 complex prepared in ethanol (Fig. 1b). The FT-IR spectrum of Phen showed a peak at 738 cm−1, which originates from the C–H out-of-plane deformation. It was reported that the position of this peak shifted to a smaller wavenumber when the nitrogen in Phen coordinated with metal ions.71 The Fe(Phen)3 complex generated in ethanol showed a negatively shifted peak at a wavenumber of 725 cm−1, indicating complex formation between Fe2+ and Phen. In contrast, the solid-state mixture of FeAc and Phen exhibited no peak shift in its FT-IR spectrum, indicating that Fe2+ ions and Phen did not bind in the absence of a solvent or silica. The FT-IR spectra of Mixture_X showed that peaks were located between those of Phen and the Fe(Phen)3 complex, suggesting the formation of a complex. A comparison of the FT-IR spectra of the series of Mixture_X samples revealed that, as the amounts of Fe2+ and Phen increased, the peak position gradually shifted to a higher wavenumber.
:3) catalysts (see 4. Materials and methods for details). The mixing of Fe2+ ions and Phen in a 1:3 ratio readily generated the Fe(Phen)3 complex in a soluble solvent; however, the complex prepared under wet conditions could generate inactive Fe clusters upon high-temperature pyrolysis in the Fe–N/C catalyst synthesis.70 In turn, we attempted to prepare Fe(Phen)3 under dry conditions in the presence of a silica template, which had a high density of hydrophilic silanol groups on its surface. We hypothesized that silanol groups could facilitate the complex formation. Indeed, Fourier-transform infrared (FT-IR) spectra indicated that, even under solvent-less dry conditions, Fe2+ and Phen formed the Fe(Phen)3 complex in the presence of mesoporous silica. We compared the FT-IR spectra of the mixtures of the precursors (FeAc and Phen) and SBA-15 silica with different mass ratios (denoted Mixture_X) with those of Phen, a mixture of FeAc and Phen prepared under dry conditions, and the Fe(Phen)3 complex prepared in ethanol (Fig. 1b). The FT-IR spectrum of Phen showed a peak at 738 cm−1, which originates from the C–H out-of-plane deformation. It was reported that the position of this peak shifted to a smaller wavenumber when the nitrogen in Phen coordinated with metal ions.71 The Fe(Phen)3 complex generated in ethanol showed a negatively shifted peak at a wavenumber of 725 cm−1, indicating complex formation between Fe2+ and Phen. In contrast, the solid-state mixture of FeAc and Phen exhibited no peak shift in its FT-IR spectrum, indicating that Fe2+ ions and Phen did not bind in the absence of a solvent or silica. The FT-IR spectra of Mixture_X showed that peaks were located between those of Phen and the Fe(Phen)3 complex, suggesting the formation of a complex. A comparison of the FT-IR spectra of the series of Mixture_X samples revealed that, as the amounts of Fe2+ and Phen increased, the peak position gradually shifted to a higher wavenumber.
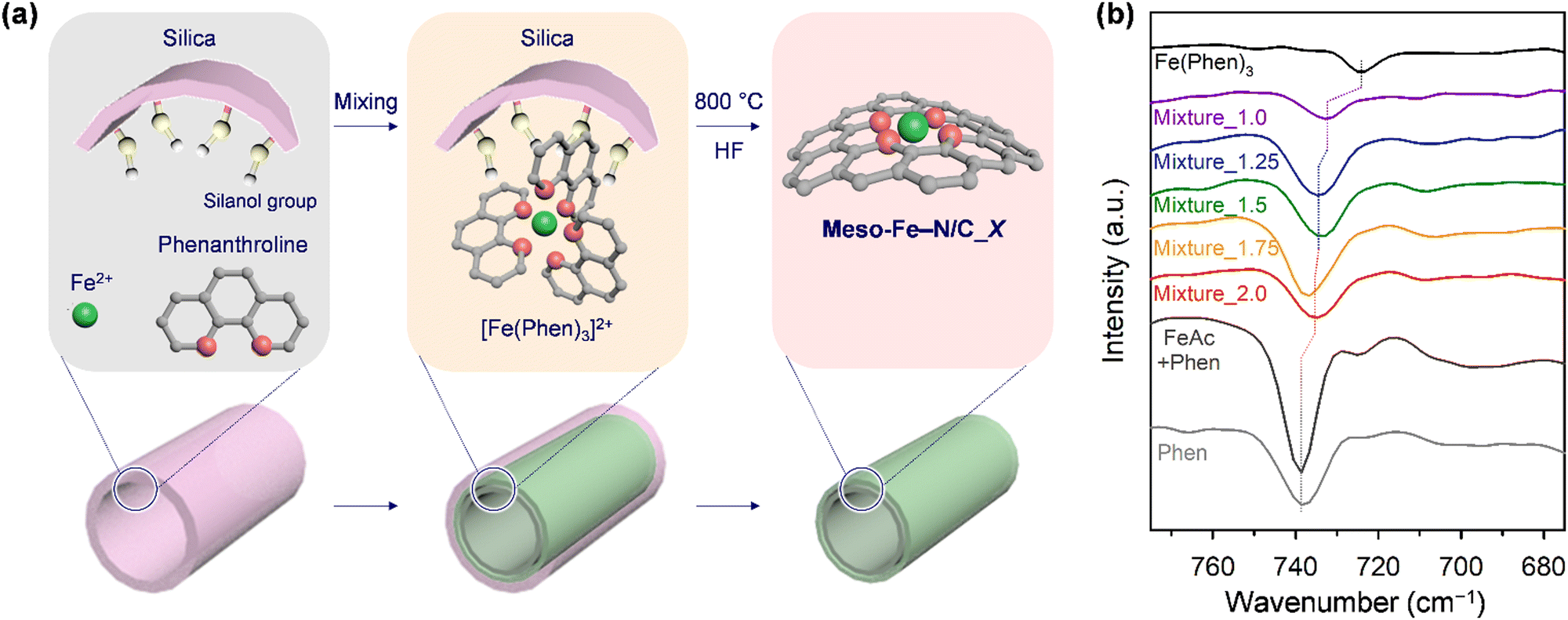 | ||
| Fig. 1 (a) Schematic illustration for the preparation of Meso-Fe–N/C_X catalysts. (b) FT-IR spectra for Mixture_X in the wavelength range of 675–775 cm−1. | ||
High-temperature pyrolysis and silica etching of Mixture_X afforded Meso-Fe–N/C_X samples. Scanning electron microscope (SEM) images of the Meso-Fe–N/C_X catalysts (Fig. S1, ESI†) revealed that they commonly preserved the morphology of the SBA-15 silica template. Small-angle X-ray diffraction (XRD) patterns of Meso-Fe–N/C_X (Fig. S2a, ESI†) exhibited diffraction peaks commensurate with hexagonal p6mm symmetry resulting from the faithful replication of the SBA-15 template. Interestingly, in the series of Meso-Fe–N/C_X catalysts, as the amount of Fe and N precursors decreased, the intensity of the major (100) diffraction peak below 1° gradually decreased, with Meso-Fe–N/C_1.0 exhibiting the lowest peak intensity. The resulting small-angle XRD patterns of Meso-Fe–N/C_2.0 and Meso-Fe–N/C_1.0 are similar to those of CMK-372 and CMK-573 mesoporous carbons, which have solid rod-like and hollow tube-like frameworks, respectively. Wide-angle XRD patterns of Meso-Fe–N/C_X (Fig. S2b, ESI†) showed a broad peak for amorphous carbon (20–30°) along with a peak corresponding to the (002) plane of graphitic carbon (around 26°), without peaks corresponding to the Fe-based crystalline phases. The Meso-Fe–N/C_X catalysts with larger amounts of Fe and N precursors showed a more pronounced peak for graphitic carbon.
The structural differences in the frameworks of Meso-Fe–N/C_X, inferred from their small-angle XRD patterns, could be directly observed in transmission electron microscope (TEM) images (Fig. 2a–d). The TEM image of Meso-Fe–N/C_1.0 (Fig. 2a) clearly shows a hexagonal array of uniform tube-like carbon frameworks and dual mesopores generated inside and between the tubes. The high-resolution TEM (HR-TEM) image (Fig. 2b) indicates that a single tubular structure consists of three to four concentric carbon layers. In contrast, the TEM and HR-TEM images of Meso-Fe–N/C_2.0 (Fig. 2c and d) show that it is constructed with a hexagonal array of uniform rod-like carbon frameworks and has a single mesopore generated between the carbon nanorods. The contrasting pore structures of Meso-Fe–N/C_1.0 and Meso-Fe–N/C_2.0 were further verified by nitrogen adsorption analysis (Fig. 2e, f and Table S1, ESI†). The N2 adsorption–desorption isotherm of Meso-Fe–N/C_1.0, having a tube-like framework (Fig. 2e), showed two distinct capillary condensation steps and hysteresis loops in the relative pressure ranges of 0.4–0.65 and 0.65–0.9, respectively, which are indicative of the generation of dual pores. The corresponding pore size distribution curve in Fig. 2f exhibited two maxima at 4.5 and 10.0 nm. The highly porous nature of Meso-Fe–N/C_1.0 gave rise to a large Brunauer–Emmett–Teller (BET) surface area of 1350 m2 g−1 and pore volume of 1.56 cm3 g−1 (Table S1, ESI†). Meso-Fe–N/C_2.0 with a rod-type framework showed a single hysteresis loop, which was translated as a maximum at 4.5 nm in its pore size distribution curve.
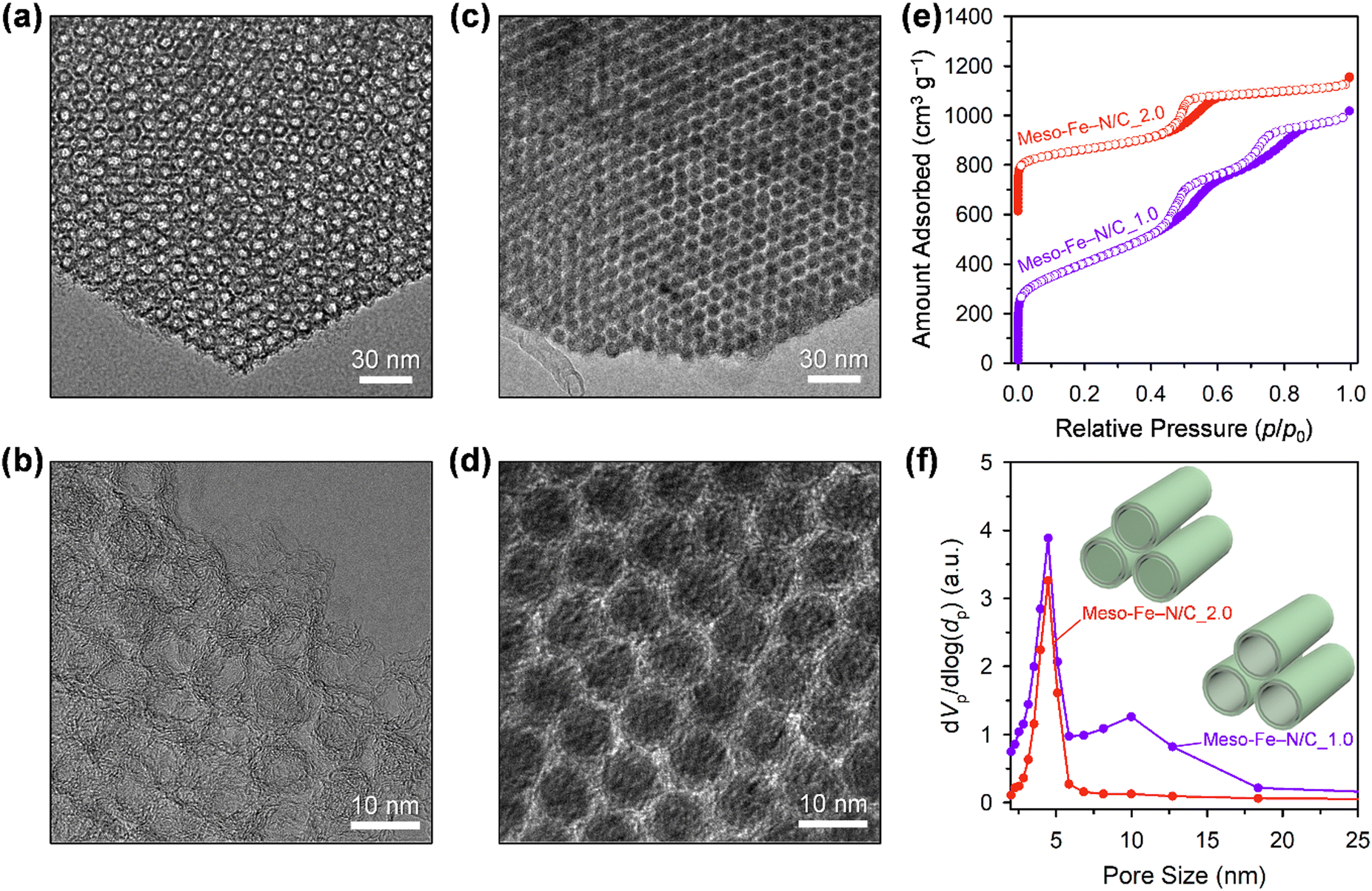 | ||
| Fig. 2 TEM images of (a) Meso-Fe–N/C_1.0 and (c) Meso-Fe–N/C_2.0. High resolution TEM images of (b) Meso-Fe–N/C_1.0 and (d) Meso-Fe–N/C_2.0. (e) N2 adsorption–desorption isotherms of Meso-Fe–N/C_1.0 and Meso-Fe–N/C_2.0 catalysts. The isotherm of the Meso-Fe–N/C_2.0 catalyst was offset by 600 cm3 g−1. (f) Pore size distribution curves obtained from the adsorption branches of the corresponding isotherms with structural models for both catalysts. | ||
A comparison of the pore size distribution curves of all Meso-Fe–N/C_X catalysts (Fig. S3 and Table S1, ESI†) revealed that they commonly had mesopores (4.5 nm) with similar differential pore volumes, which were generated upon the removal of the silica template. Interestingly, for the other mesopore generated inside the carbon tube, as the amounts of Fe and N precursors increased, a gradual decrease in the differential pore volume was observed at a fixed pore size of 10 nm. Hence, Meso-Fe–N/C_1.0 exhibited a maximum pore size of 10 nm with the largest differential pore volume, whereas Meso-Fe–N/C_2.0 exhibited virtually no porosity at this size. The changes in pore size distribution provide critical information on the evolution of the Fe,N-containing carbon structure within the mesopores of SBA-15 silica. At the lowest loading of the Fe and N precursors (Meso-Fe–N/C_1.0), the surface of the silica framework was coated with a few layers of Fe,N-containing carbon to generate uniform tube-like structures. The increased loading of Fe and N precursors resulted in a mix of tube-like and rod-like structures, with a gradual increase in the proportion of the latter. Eventually, at the highest Fe and N precursor loading (Meso-Fe–N/C_2.0), the mesopores of the silica templates were completely filled with rod-like carbon structures. The overall changes in the loading-dependent Fe,N-containing carbon structure formation are presented as structural models in the inset of Fig. S3b (ESI†).
We next investigated the formation of atomically dispersed Fe sites in the Meso-Fe–N/C_X catalysts by a variety of methods. High-angle annular dark-field scanning TEM (HAADF-STEM) images of both Meso-Fe–N/C_1.0 and Meso-Fe–N/C_2.0 catalysts (Fig. 3a and b) show bright dots, which correspond to single atomic Fe species. The electronic and atomic structures of the atomically dispersed Fe sites were investigated by X-ray absorption spectroscopy (XAS) analysis (Fig. 3c and d). Fig. 3c shows the X-ray absorption near-edge structure (XANES) spectra of Meso-Fe–N/C_1.0, Meso-Fe–N/C_2.0, and references. In the pre-edge region (7110–7120 eV), the reference iron(II) phthalocyanine (FePc), with a square planar structure, showed peaks at 7114 (peak A) and 7118 eV (peak B), whereas 5,10,15,20-tetrakis(4-methoxyphenyl)-21H,23H-porphine iron(III) chloride (FeTMPPCl), with a square pyramidal structure, exhibited only peak A. Hence, the presence or absence of peak B can serve as a fingerprint to distinguish the coordination structure of central Fe. Both Meso-Fe–N/C_1.0 and Meso-Fe–N/C_2.0 presented only peak A in the pre-edge region of the XANES spectra, suggesting that they have fifth axial bonding or an off-square planar structure. The peaks at 7132 (peak C) and 7140 eV (peak D) in the edge region can suggest the degree of distortion of the Fe–N4 site with D4h symmetry.74,75 For FePc and FeTMPPCl, the intensity of peak C was lower than that of peak D, suggesting a near-planar structure in both the references. However, the Meso-Fe–N/C_1.0 and Meso-Fe–N/C_2.0 catalysts that had been subjected to high-temperature pyrolysis showed a reversed intensity ratio of peaks C and D, indicating that the Fe atoms were off-centered with distortion. The coordination structure around the Fe center atom was examined using extended X-ray absorption fine structure (EXAFS) analyses (Fig. 3d). The radial distribution function (RDF) of the EXAFS spectrum of the FePc reference showed a major peak at 1.45 Å, which corresponds to the Fe–N coordination, whereas that of the Fe foil presented a peak at 2.18 Å due to Fe–Fe metallic bonding. The EXAFS spectra for both Meso-Fe–N/C_1.0 and Meso-Fe–N/C_2.0 exhibited only a peak near 1.45 Å, indicating the exclusive presence of Fe–N/O bonds. Detailed EXAFS curve fitting results of the two catalysts (Fig. S4 and Table S2, ESI†) indicated that both catalysts have Fe–Nx active sites with similar Fe–N coordination numbers and bond lengths. N 1s X-ray photoelectron spectroscopy (XPS) spectra also confirmed the presence of Fe–Nx species in Meso-Fe–N/C_1.0, Meso-Fe–N/C_2.0, and other Meso-Fe–N/C_X catalysts (Fig. S5a, ESI†). Quantitative analysis of N-containing species (pyridinic N, Fe–Nx, pyrrolic N, graphitic N, and N–O) from the deconvoluted N 1s XPS spectra (Fig. S5a and Table S3, ESI†) revealed that all catalysts have similar distributions of the respective species. In addition, all Meso-Fe–N/C_X catalysts showed similar C 1s (Fig. S5b, ESI†) and O 1s (Fig. S5c, ESI†) spectra. Elemental analyses of the Meso-Fe–N/C_X catalysts by inductively coupled plasma optical emission spectroscopy and using an elemental analyzer (Table S4, ESI†) indicated that all the catalysts had similar contents of Fe and other elements.
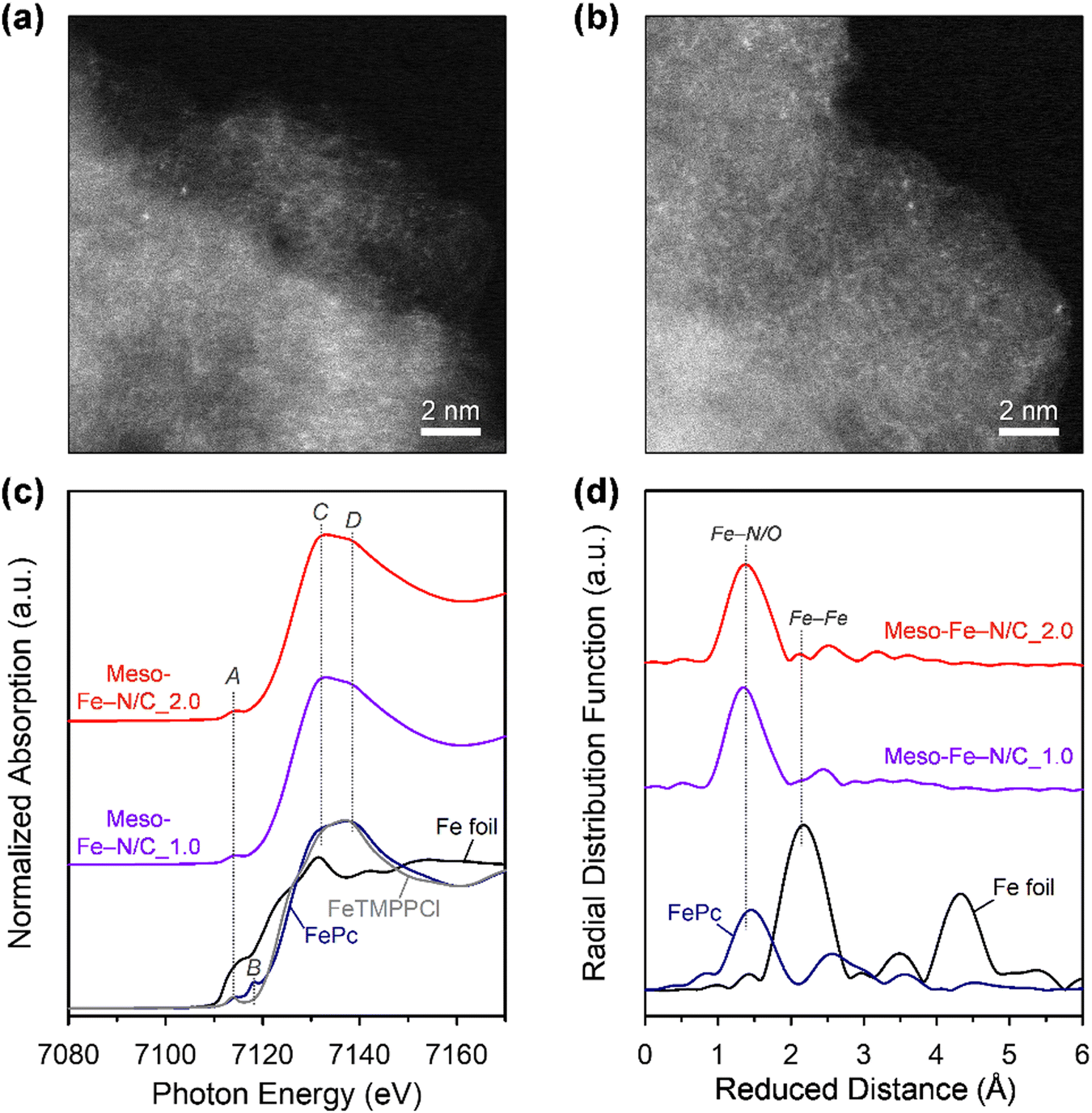 | ||
| Fig. 3 HADDF-STEM images of (a) Meso-Fe–N/C_1.0 and (b) Meso-Fe–N/C_2.0. (c) Fe K-edge XANES spectra for Meso-Fe–N/C_1.0 and Meso-Fe–N/C_2.0, with those of Fe foil (black), FePc (blue), and FeTMPPCl (gray) references. (d) Fourier-transformed EXAFS spectra of Meso-Fe–N/C_1.0 and Meso-Fe–N/C_2.0, with those of the Fe foil and FePc references. | ||
We quantified the catalytically accessible Fe–Nx sites using the CO cryo-adsorption method developed by the Strasser group (Fig. S6, ESI†).27,59–61 This method evaluates a site density under the assumption that one CO molecule adsorbs to one Fe–Nx site at the surface of a catalyst (see 4. Materials and methods for details). The CO uptake amount of Meso-Fe–N/C_1.0 calculated from CO cryo–adsorption data was 77.3 nmol mg−1, which is ca. 35% greater than that of Meso-Fe–N/C_2.0 (57.6 nmol mg−1). The chemisorption results are translated into CO-accessible active surface site densities of 4.7 × 1019 and 3.5 × 1019 sites g−1 for Meso-Fe–N/C_1.0 and Meso-Fe–N/C_2.0, respectively (Fig. 4a and Table 1 and Table S5, ESI†). The site densities of both catalysts are similar to those of the polyaniline-based and ZIF-based Fe–N/C catalysts determined by CO cryo-adsorption.63
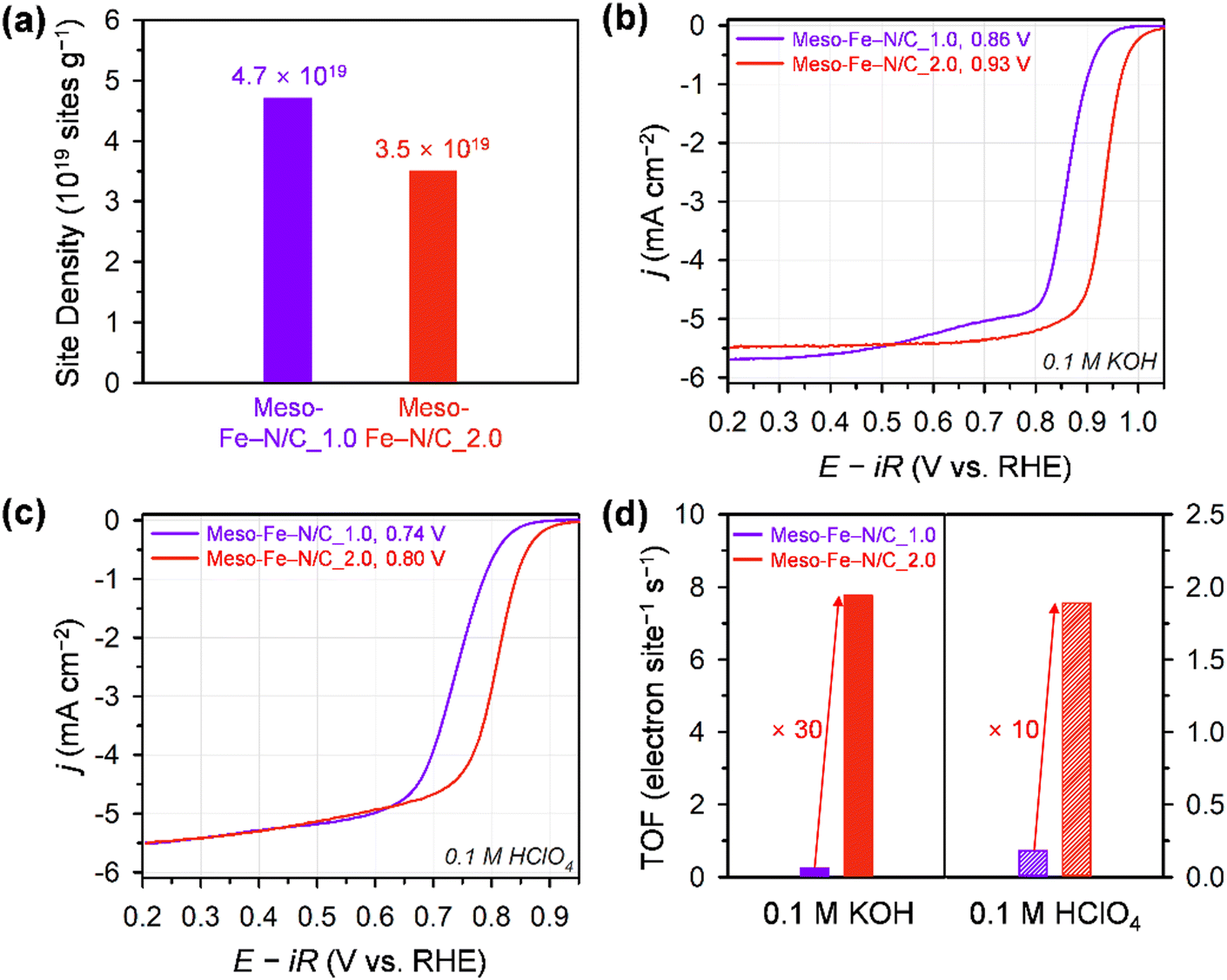 | ||
| Fig. 4 (a) The CO-accessible active site densities of Meso-Fe–N/C_1.0 and Meso-Fe–N/C_2.0 by CO pulse chemisorption at −80 °C. ORR polarization curves of the Meso-Fe–N/C_1.0 and Meso-Fe–N/C_2.0 catalysts in (b) 0.1 M KOH and (c) 0.1 M HClO4. (d) TOF values at 0.9 V (alkaline) and 0.8 V (acid). ORR performances of the catalysts from the RRDE test at a rotating speed of 1600 rpm and scan rate of 5 mV s−1. The catalyst loading on the glassy carbon electrode was 0.6 mg cm−2. | ||
| Sample | Site densitya (sites g−1) | TOF in 0.1 M KOHb (e− site−1 s−1) | TOF in 0.1 M HClO4c (e− site−1 s−1) | T peak (°C) |
|---|---|---|---|---|
| a Active surface site density derived from CO cryo–adsorption data. b Turnover frequency values were calculated at 0.9 V (vs. RHE). c Turnover frequency values were calculated at 0.8 V (vs. RHE). d Peak temperature in O2 TPD. | ||||
| Meso-Fe–N/C_1.0 | 4.7 × 1019 | 0.25 | 0.18 | 65.5 |
| Meso-Fe–N/C_2.0 | 3.5 × 1019 | 7.77 | 1.89 | 58.6 |
We next examined the electrocatalytic performances of Meso-Fe–N/C_X catalysts for the ORR using the rotating ring-disk electrode (RRDE) technique in both alkaline (0.1 M KOH) and acidic (0.1 M HClO4) electrolytes (Fig. 4b, c and Fig. S7, ESI†). The ORR polarization curves of the catalysts in 0.1 M KOH electrolyte (Fig. 4b and Fig. S7a, ESI†) revealed that Meso-Fe–N/C_2.0 exhibited a much higher ORR activity than Meso-Fe–N/C_1.0 with a 70 mV positive shift of the half-wave potential. This ORR activity trend is an unexpected result, given that Meso-Fe–N/C_1.0 has larger surface area and site density than Meso-Fe–N/C_2.0 (vide infra). Both the kinetic current density and mass activity of Meso-Fe–N/C_2.0 at 0.90 V (vs. reversible hydrogen electrode, RHE) are 24 times greater than those of Meso-Fe–N/C_1.0 (Table 1 and Table S5, ESI†). To compare the intrinsic activity of the catalysts, we assessed their TOFs using the number of Fe–Nx sites calculated from the CO cryo-adsorption data (see 4. Materials and methods for details). The TOF of Meso-Fe–N/C_2.0 at 0.9 V (vs. RHE) was 7.77 electron site−1 s−1, which is approximately 31 times higher than that of Meso-Fe–N/C_1.0 (0.25 electron site−1 s−1) (Fig. 4d and Table 1 and Table S5, ESI†). The TOF value of Meso-Fe–N/C_2.0 at 0.9 V (vs. RHE) under alkaline conditions is significantly higher than the values reported in previous studies (Table S6, ESI†).29,60,63 The superior ORR activity of Meso-Fe–N/C_2.0 was further verified by its lower Tafel slope (46 mV dec−1) than that of Meso-Fe–N/C_1.0 (57 mV dec−1) (Fig. S7c, ESI†).
Meso-Fe–N/C_2.0 also exhibited superior ORR activity in acidic media (Fig. 4c and Fig. S7b, ESI†). Meso-Fe–N/C_2.0 showed a half-wave potential of 0.80 V for the ORR in 0.1 M HClO4, which is positively shifted from that of Fe-Phen_1.0 (0.74 V). The superior activity of Meso-Fe–N/C_2.0 was manifested as an approximately 8-fold higher kinetic current density and mass activity, an ∼10 times higher TOF (Table 1 and Table S7, ESI†), and a lower Tafel slope (Fig. S7d, ESI†) than those of Meso-Fe–N/C_1.0. The acidic TOF value of Meso-Fe–N/C_2.0 was compared at 0.8 V (vs. RHE) with those of previously reported Fe–N/C catalysts (Fig. 5).27,31,32,39,55,60,61,63 Notably, the TOF of Meso-Fe–N/C_2.0 under acidic conditions exceeds the reported TOFs of Fe–N/C catalysts evaluated by the cryo-CO adsorption method. Notably, its value is even 1.6 times greater than that of the PANI-CM catalyst,32,59 which is one of the high-performing Fe–N/C catalysts (Table S8, ESI†).
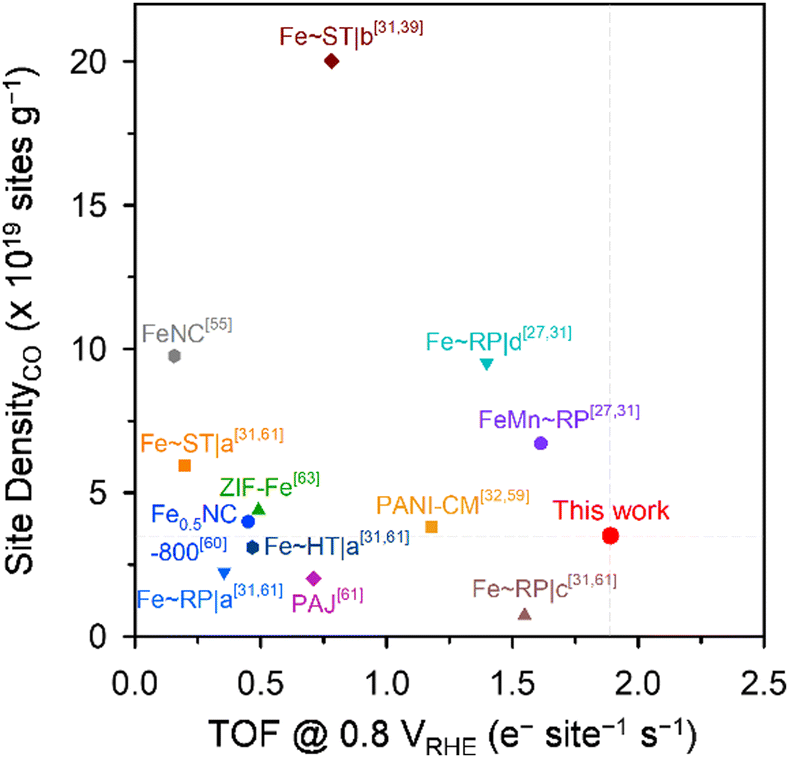 | ||
| Fig. 5 Benchmarking the active site density and TOF (at 0.8 VRHE) of Meso-Fe–N/C_2.0 with reported Fe–N/C catalysts. | ||
We also compared the long-term durability of Meso-Fe–N/C_1.0, Meso-Fe–N/C_2.0, and Pt/C catalysts in 0.1 M KOH (Fig. S8a, ESI†). Both Meso-Fe–N/C_1.0 and Meso-Fe–N/C_2.0 showed excellent long-term durability with a decrease in half-wave potential by only 10 mV after 10000 potential cycles between 0.6 and 1.0 V, whereas Pt/C catalyst underwent a large decrease in catalytic activity with a negative shift of half-wave potential by 60 mV. We also assessed the ORR durability of these catalysts in 0.1 M HClO4 (Fig. S8b, ESI†). The Meso-Fe–N/C_2.0 catalyst showed good long-term durability with a decrease of half-wave potential by 30 mV, which compared favorably with that of Pt/C (20 mV). In contrast, the Meso-Fe–N/C_1.0 catalyst showed a significant decrease in its catalytic activity with a negative shift of half-wave potential by 80 mV, which indicates that thin carbon layers with a high surface area were rapidly deactivated under acidic conditions.
We hypothesized that the unexpected catalytic activity trend between the two catalysts could originate from the difference in the binding energy of oxygen to each catalyst. We performed O2 temperature-programmed desorption (TPD) experiments (Fig. 6 and Fig. S9, ESI†) to qualitatively assess the binding strength of chemisorbed O2 on the Fe sites.76 The TPD spectra presented two peaks at low and high temperatures, corresponding to weakly and strongly adsorbed O2, respectively. Meso-Fe–N/C_2.0 exhibited a lower peak temperature for strongly adsorbed O2 than Meso-Fe–N/C_1.0 (Fig. 6b), indicating the weaker binding strength of Meso-Fe–N/C_2.0 with O2. Meso-Fe–N/C_2.0, consisting of ca. 20 concentric carbon layers, has a significant portion of Fe–Nx sites in the internal carbon layer, whereas Meso-Fe–N/C_1.0, with 3–4 carbon layers, has most of the Fe–Nx species in the external layer. Hence, the difference in the O2 binding energies of the two catalysts can be correlated with the relative ratio of the internal or external Fe–Nx sites. Meso-Fe–N/C_2.0, which has a greater number of internal Fe–Nx species, is more likely to activate O2 cooperatively between two adjacent carbon layers embedding Fe–Nx sites, similar to the active centers of CcO and bifacial porphyrins. As such, its O2 binding strength is weaker than that of Meso-Fe–N/C_1.0, which could be manifested as a lower peak temperature in the O2 TPD spectrum. These results are consistent with the relatively low Tafel slope of Meso-Fe–N/C_2.0. We point out that our TPD and Tafel analyses are in line with the recent theoretical work by Rossmeisl et al., which suggested that the diporphyrin structures would act as promising ORR active sites by promoting a dissociative mechanism that is accessible even at weak binding sites owing to their structural advantages.77
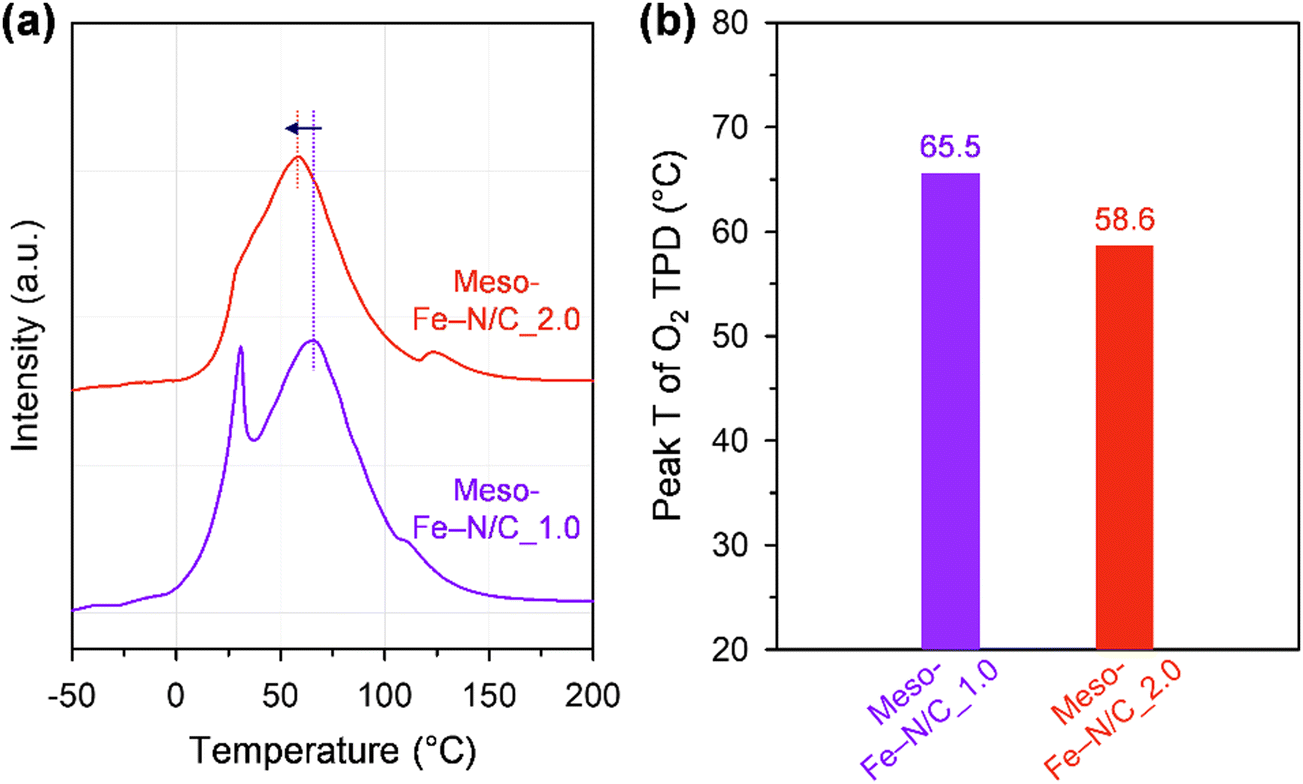 | ||
| Fig. 6 (a) O2 TPD profiles and (b) second O2 desorption peak temperatures of Meso-Fe–N/C_1.0 and Meso-Fe–N/C_2.0. | ||
To further verify the preceding claim experimentally, we synthesized a new catalyst, which was prepared by filling the internal mesopores of Meso-Fe–N/C_1.0 sequentially with an undoped carbon layer and an Fe–Nx site-embedding carbon layer. The resulting Meso-Fe–N_C_Fe catalyst had a rod-type structure, in which the carbon layer was sandwiched between Fe–Nx site-containing carbon layers (Fig. S10a and b, ESI†). The Meso-Fe–N_C_Fe catalyst exhibited intermediate ORR activity between Meso-Fe–N/C_1.0 and Meso-Fe–N/C_2.0, in terms of kinetic current density and mass activity in both alkaline (Fig. S10c and d, ESI†) and acidic (Fig. S10e and f, ESI†) media. This experimental result indicates that the number of Fe–Nx site-containing internal carbon layers plays an important role in enhancing ORR activity.
3. Conclusions
We prepared ordered mesoporous Fe–Nx-containing carbons with controlled framework structures and carbon layer numbers, which could serve as a model system for unraveling the critical role of interlayer O2 activation in boosting ORR activity. The solid-state nanocasting of mesoporous silica with a controlled amount of Fe2+/Phen precursor afforded a tube-like or rod-like framework. Despite the relatively smaller surface area and number of exposed Fe–Nx sites, the rod-type Meso-Fe–N/C_2.0 exhibited superior ORR activity to the tube-type Meso-Fe–N/C_1.0, with a smaller overpotential, greater mass activity and TOF, and lower Tafel slope. Notably, the TOFs in both the alkaline and acidic electrolytes surpassed the reported values. The Tafel analysis and TPD-driven O2 binding strength results indicate that Meso-Fe–N/C_2.0 with multiple carbon layers can have more bifacial-like sites than Meso-Fe–N/C_1.0, which could induce weaker binding strength with O2 and boost the ORR activity. This insight into the role of the Fe–Nx site-embedding carbon layer number in the ORR activity can provide an important guideline for the design of highly active Fe–N/C electrocatalysts.4. Materials and methods
Chemicals
Pluronic P123 (EO20PO70EO20, Mn = 5800), tetraethyl orthosilicate (TEOS, 98%), Phen, and potassium hydroxide (KOH, 99.99%) were purchased from Sigma-Aldrich. FeAc (95%) was purchased from Alfa Aesar. Hydrochloric acid (HCl, 35–37%), ethanol (94.5%), and formic acid (99%) were purchased from Samchun Chemicals. Hydrofluoric acid (HF, 48–51%) was purchased from J. T. Baker. The mesophase pitch was obtained from the Mitsubishi Gas Chemical Company. All chemicals were used as received without further purification.Synthesis of Meso-Fe–N/C_X catalysts
The synthesis of Meso-Fe–N/C_X (where X = weight ratio of the Fe and N precursors to the silica template) was carried out through dry mixing of FeAc, Phen, and mesoporous silica SBA-15. Mesoporous silica SBA-15 with hexagonal symmetry was synthesized following the methods described in previous reports78,79 with some modifications. To prepare Meso-Fe–N/C_X, 1 g of SBA-15 mesoporous silica and the desired amounts of FeAc and Phen (the FeAc to Phen molar ratio was fixed at 1:3) were mixed in an agate mortar for 10 min. For example, for Meso-Fe–N/C_1.0, a total of 1.0 g of Fe and N precursors consisting of 0.243 g of FeAc and 0.757 g of Phen was used. The samples obtained after solid-state impregnation were denoted as Mixture_X. The mixture was heated from room temperature (RT) to 800 °C for 6 h and maintained at that temperature for 3 h under a N2 gas flow (1.0 L min−1). To remove the silica template, the resulting carbon–silica composite was mixed with 1:1 (v/v) EtOH:10% aqueous HF solution, and the slurry was stirred for 30 min, filtered, and washed several times with EtOH. The HF etching process was repeated in the same manner, and the resulting sample was dried at 60 °C to obtain Meso-Fe–N/C_X catalysts.
Synthesis of Meso-Fe–N_C_Fe catalyst
The Meso-Fe–N_C_Fe catalyst was prepared by the sequential formation of an undoped carbon layer and an Fe,N-containing carbon layer inside the mesopores of the pyrolyzed Mixture_1.0 sample and subsequent etching of the SBA-15 template (see Fig. S10b, ESI,† for the structural model of Meso-Fe–N_C_Fe). The Mixture_1.0 sample was pyrolyzed at 800 °C to obtain the Fe–Nx@SBA-15 composite, in which a tubular Fe,N-containing carbon layer was formed on the framework walls of the SBA-15 template. To 0.5 g of the Fe–Nx@SBA-15 composite in an agate mortar, 0.125 g of mesophase pitch and 4 mL of EtOH were added and mixed using a pestle for 10 min. The black slurry was then dried at 60 °C for 8 h. The dried powder was heated to 300 °C at a ramping rate of 1.4 °C min−1 and maintained at that temperature for 4 h under 1 L min−1 N2 flow. The temperature was subsequently increased to 900 °C at a heating rate of 2.5 °C min−1 and maintained for 2 h under N2 flow. The resulting C@Fe–Nx@SBA-15 composite was then dry-mixed with FeAc and Phen to generate an Fe,N-containing carbon layer. The C@Fe–Nx@SBA-15 composite (0.367 g) was mixed with 0.061 g of FeAc and 0.189 g of Phen in an agate mortar for 10 min. The subsequent pyrolysis and HF etching steps were carried out in the same manner as for the Meso-Fe–N/C_X catalysts. The final product was denoted as Meso-Fe–N_C_Fe.Characterization methods
FT-IR spectra were measured using a 670-IR spectrometer (Varian) equipped with a liquid nitrogen-cooled germanium detector in the range of 4000–400 cm−1. SEM images were obtained using an S-4800 scanning electron microscope (Hitachi) operating at 10 kV. TEM and HR-TEM images were acquired using a JEM-2100F TEM (JEOL) instrument at accelerating voltages of 200 and 80 kV, respectively. HAADF-STEM images were obtained using a Titan3 G2 60-300 microscope (FEI) equipped with a double-sided spherical aberration (Cs) corrector operating at an accelerating voltage of 200 kV. XRD patterns were obtained using an X-ray diffractometer (D/MAX2500V/PC, Rigaku) equipped with a Cu Kα radiation source, operated at 40 kV and 200 mA. The XRD patterns of the samples were measured in 2 theta ranges of 0.7–3° and 15–65° at scan rates of 0.5 and 4° min−1, respectively. The textural properties of the samples were analyzed using a nitrogen physisorption analyzer (BELSORP-Max, MicrotracBEL) operated at −196 °C. All the samples were pre-evacuated at 150 °C and 10−2 Pa for 12 h before the measurements. The specific surface areas of the samples were calculated using the BET equation in the relative pressure range of 0.05–0.2, while their pore size distributions were obtained from the adsorption branches of the isotherms using the Barrett–Joyner–Halenda method. XPS measurements were performed using a K-Alpha spectrometer (Thermo Fisher Scientific) equipped with a monochromatic Al-Kα X-ray source (1486.6 eV). The N 1s XPS peak deconvolution was carried out using XPSPeak41 software. A Gaussian–Lorentzian (70:30) mixed function was used to fit the curve after Shirley-type background removal. The Fe content of the samples was analyzed using an inductively coupled plasma optical emission spectrometry analyzer (700-ES, Varian), whereas the carbon, hydrogen, nitrogen, and oxygen contents of the samples were determined using an elemental analyzer (Flash 2000, Thermo Fisher Scientific).
XAS
Fe K-edge XAS spectra were collected at RT at the 6D, 8C, and 10C beamlines of the Pohang Accelerator Laboratory. The storage ring was operated at 3.0 GeV with a beam current of 300 mA in decay mode. The beamline was equipped with a focusing Si(111) double-crystal monochromator that was used to filter the incident photons. The X-ray monochromator was detuned by 30% to remove high-order harmonics and calibrated using standard Fe foil. X-ray intensities were monitored using standard N2-filled ion chambers and an Ar-filled detector. The catalyst powder was pelletized in a sample holder (1 cm wide) to an adequate thickness to obtain a significant transmission signal. Background removal and normalization of the collected XAS data and the Fourier transform of the radial distribution functions were conducted using the Athena software80 with an Rbkg of 1.2 and a Hanning-type window.CO cryo-adsorption
Prior to each CO cryo-adsorption experiment, 0.03 g of a sample was heated to 600 °C at a ramp rate of 10 °C min−1, followed by 15 min holding under He flow to remove pre-adsorbed O2 or H2O molecules.59 Further, CO pulse chemisorption was performed at −80 °C using liquid nitrogen. Ten consecutive 0.5 mL CO pulses at 600 s intervals were passed over the samples, and the CO uptake was monitored and quantified using a thermal conductivity detector. The total amount of adsorbed CO was quantified as the amount adsorbed in the first three CO pulses of a total of ten CO pulses. After He purging for 20 min, five consecutive CO pulse chemisorption experiments were performed to quantify the amount of physisorbed CO. The amount of chemisorbed CO was obtained by subtracting the amount of physisorbed CO from the total amount of adsorbed CO. The active surface site density (SD) was calculated from the CO chemisorption data using the following eqn (1):| SD (sites g−1) = nCO × 10−6 × NA | (1) |
O2 TPD
TPD profiles were collected using a BELCAT II instrument (MicrotracBEL) equipped with a thermal conductivity detector. Prior to each O2 TPD experiment, 0.05 g of sample was degassed at 350 °C for 1 h (ramping rate: 10 °C min−1) under a He flow to remove pre-adsorbed O2 or H2O molecules.76 After degassing, the sample was cooled to −50 °C and O2 adsorption was carried out for 1 h under 10% O2/He gas conditions, followed by He purging for 2 h at −50 °C to remove weakly adsorbed O2. Then, the TPD experiment was performed while heating the sample to 500 °C (ramping rate: 10 °C min−1) under a He flow.Electrochemical measurements
Electrochemical experiments were performed with a bipotentiostat (CHI760E, CH Instruments) and a rotator (AFMSRCE, Pine Research Instrumentation) at RT using a three-electrode electrochemical cell. A Hg/HgO (CHI152, CH Instruments; 1 M KOH filling solution) electrode and a graphite rod were used as the reference and counter electrodes, respectively. The Hg/HgO reference electrode was calibrated with respect to the reversible hydrogen electrode (RHE) scale before use. The RHE calibration was performed in a H2-saturated 0.1 M KOH solution, with a platinum coil as the working electrode and Hg/HgO as the reference electrode. With continuous H2 bubbling, a stable open circuit voltage was obtained within 20 min, which corresponds to the RHE conversion value. A rotating ring-disk electrode (RRDE, AFE7R9GCPT, Pine Research Instrumentation) coated with catalyst ink was used as the working electrode. Prior to every measurement, the RRDE was polished with a 1.0 μm-thick alumina suspension followed by a 0.3 μm-thick suspension to yield a mirror finish. The catalyst ink was prepared by mixing 15 mg of the catalyst powder with 50 μL of H2O, 505 μL of ethanol, and 37.6 μL of Nafion (5 wt%). For the Pt/C catalyst (20 wt% Pt, HiSPEC-3000, Johnson-Matthey), a catalyst ink was prepared by mixing 3.5 mg of Pt/C catalyst with 100 μL of H2O, 1070 μL of ethanol, and 30 μL of Nafion. The prepared ink solution was ultrasonicated until the catalyst was well-dispersed. Additionally, 6 μL of the catalyst ink was pipetted using a micro-pipette onto the glassy carbon disk (0.247 cm2) of the RRDE, resulting in a catalyst loading of 600 μg cm−2 (70 μg cm−2 for Pt/C). A 0.1 M KOH electrolyte was prepared by dissolving high-purity KOH in 18.2 MΩ cm of Millipore water. Before performing linear sweep voltammetry (LSV), the catalyst was cleaned by cycling the potential between 0.05 and 1.2 V (vs. RHE) for 20 cycles at a scan rate of 100 mV s−1 (50 cycles at a scan rate of 500 mV s−1 for Pt/C) in a N2-saturated electrolyte. Subsequently, cyclic voltammetry (CV) was performed in the potential range from 0.05 to 1.2 V for three cycles at a scan rate of 20 mV s−1 (50 mV s−1 for Pt/C). To measure the solution resistance for iR-compensation, electrochemical impedance spectra were obtained at 0.68 V with an AC potential amplitude of 10 mV from 100 kHz to 1 Hz with a rotation speed of 1600 rpm. The LSV polarization curves for the ORR were obtained by sweeping the potential from 1.2 to 0.2 V (from −0.01 V to 1.1 V for Pt/C) at a scan rate of 5 mV s−1 (20 mV s−1 for Pt/C) in an O2-saturated electrolyte at a rotating speed of 1600 rpm. To correct the non-faradaic current (capacitive current) from the LSV curve, the same measurement was performed in a N2-saturated electrolyte. During the potential sweep, the applied potential of the Pt ring was held at 1.3 V (vs. RHE) to measure the 4-electron selectivity. The electron transfer number (n) was calculated using the following eqn (2):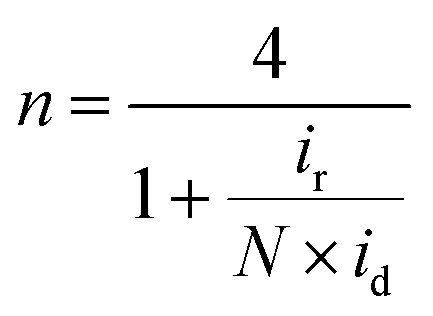 | (2) |
The kinetic current density (jk) was obtained using eqn (3):
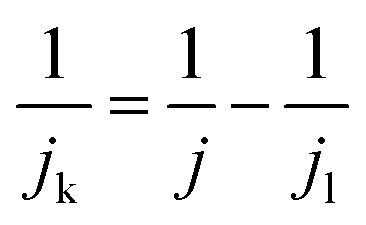 | (3) |
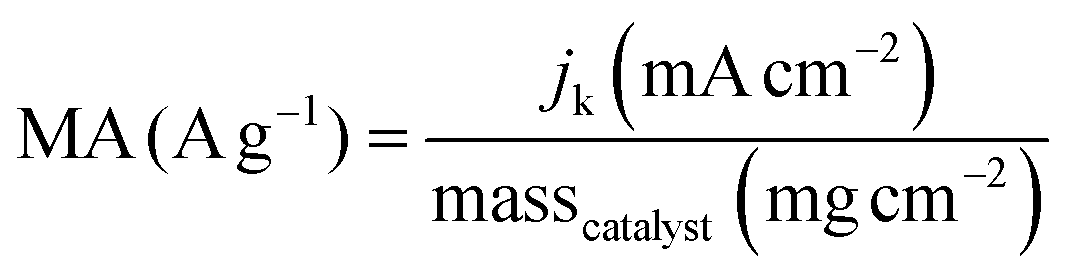 | (4) |
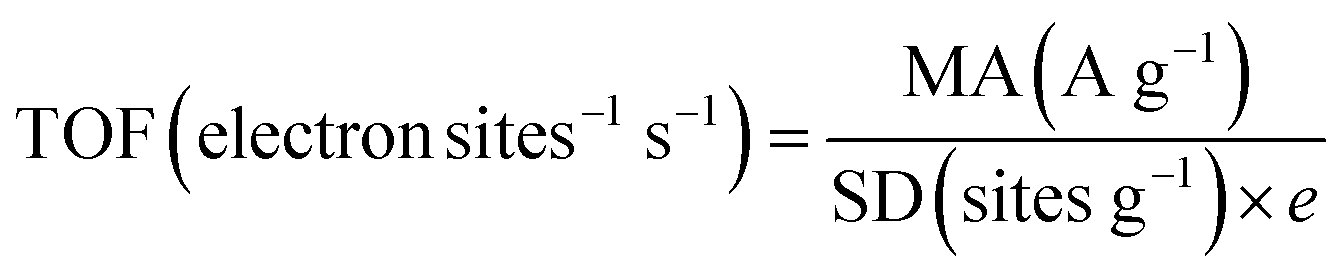 | (5) |
ORR accelerated durability tests of the catalysts were conducted in a N2-saturated 0.1 M KOH solution in the potential range of 0.6–1.0 V (vs. RHE) at a scan rate of 50 mV s−1.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation (NRF) of Korea funded by the Ministry of Science and ICT (MSIT; NRF-2019M3E6A1064521, NRF-2019M3D1A1079306, NRF-2019M1A2A2065614, and NRF-2021R1A2C2007495). The XAS experiment performed at beamlines 6D, 8C, and 10C of the Pohang Accelerator Laboratory was supported in part by the MSIT, POSTECH, and UCRF at UNIST.References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- Z. W. She, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Nat. Mater., 2017, 16, 70–81 CrossRef PubMed.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2017, 16, 57–69 CrossRef CAS PubMed.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- Y. Jiao, Y. Zheng, M. T. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- W. Xia, A. Mahmood, Z. Liang, R. Zou and S. Guo, Angew. Chem., Int. Ed., 2016, 55, 2650–2676 CrossRef CAS PubMed.
- X. X. Wang, M. T. Swihart and G. Wu, Nat. Catal., 2019, 2, 578–589 CrossRef CAS.
- K. Kodama, T. Nagai, A. Kuwaki, R. Jinnouchi and Y. Morimoto, Nat. Nanotechnol., 2021, 16, 140–147 CrossRef CAS PubMed.
- F. Jaouen, E. Proietti, M. Lefèvre, R. Chenitz, J.-P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 RSC.
- G. Wu and P. Zelenay, Acc. Chem. Res., 2013, 46, 1878–1889 CrossRef CAS PubMed.
- Y. J. Sa, J. H. Kim and S. H. Joo, J. Electrochem. Sci. Technol., 2017, 8, 169–182 CrossRef CAS.
- A. A. Gewirth, J. A. Varnell and A. M. DiAscro, Chem. Rev., 2018, 118, 2313–2339 CrossRef CAS PubMed.
- Y. J. Sa, J. Woo and S. H. Joo, Top. Catal., 2018, 61, 1077–1100 CrossRef CAS.
- J. Li and F. Jaouen, Curr. Opin. Electrochem., 2018, 9, 198–206 CrossRef CAS.
- M. Chen, Y. He, J. S. Spendelow and G. Wu, ACS Energy Lett., 2019, 4, 1619–1633 CrossRef CAS.
- U. Martinez, S. K. Babu, E. F. Holby, H. T. Chung, X. Yin and P. Zelenay, Adv. Mater., 2019, 31, 1806545 CrossRef PubMed.
- E. Luo, Y. Chu, J. Liu, Z. Shi, S. Zhu, L. Gong, J. Ge, C. H. Choi, C. Liu and W. Xing, Energy Environ. Sci., 2021, 14, 2158–2185 RSC.
- L. Osmieri, J. Park, D. A. Cullen, P. Zelenay, D. J. Myers and K. C. Neyerlin, Curr. Opin. Electrochem., 2021, 25, 100627 CrossRef CAS.
- J. H. Zagal, S. Specchia and P. Atanassov, Curr. Opin. Electrochem., 2021, 27, 100683 CrossRef CAS.
- S. Specchia, P. Atanassov and J. H. Zagal, Curr. Opin. Electrochem., 2021, 27, 100687 CrossRef CAS.
- J. H. Kim, Y. J. Sa, T. Lim, J. Woo and S. H. Joo, Acc. Chem. Res., 2022, 55, 2672–2684 CrossRef CAS PubMed.
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- E. Proietti, F. Jaouen, M. Lefèvre, N. Larouche, J. Tian, J. Herranz and J.-P. Dodelet, Nat. Commun., 2011, 2, 416 CrossRef PubMed.
- J. Y. Cheon, T. Kim, Y. Choi, H. Y. Jeong, M. G. Kim, Y. J. Sa, J. Kim, Z. Lee, T.-H. Yang, K. Kwon, O. Terasaki, G.-G. Park, R. R. Adzic and S. H. Joo, Sci. Rep., 2013, 3, 2715 CrossRef PubMed.
- N. R. Sahraie, U. I. Kramm, J. Steinberg, Y. Zhang, A. Thomas, T. Reier, J.-P. Paraknowitsch and P. Strasser, Nat. Commun., 2015, 6, 8618 CrossRef CAS PubMed.
- J. Shui, C. Chen, L. Grabstanowicz, D. Zhao and D.-J. Liu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10629–10634 CrossRef CAS PubMed.
- Y. J. Sa, D.-J. Seo, J. Woo, J. T. Lim, J. Y. Cheon, S. Y. Yang, J. M. Lee, D. Kang, T. J. Shin, H. S. Shin, H. Y. Jeong, C. S. Kim, M. G. Kim, T.-Y. Kim and S. H. Joo, J. Am. Chem. Soc., 2016, 138, 15046–15056 CrossRef CAS PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS PubMed.
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang, D. Su, Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS PubMed.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W.-C. Cheong, R. Shen, X. Wen, L. Zheng, A. I. Rykov, S. Cai, H. Tang, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 5422 CrossRef CAS PubMed.
- J. Woo, S. Y. Yang, Y. J. Sa, W.-Y. Choi, M.-H. Lee, H.-W. Lee, T. J. Shin, T.-Y. Kim and S. H. Joo, Chem. Mater., 2018, 30, 6684–6701 CrossRef CAS.
- X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng, W. Yan, H. Wang, M. Xu and J. Shui, Nat. Catal., 2019, 2, 259–268 CrossRef CAS.
- H. Zhang, H. T. Chung, D. A. Cullen, S. Wagner, U. I. Kramm, K. L. More, P. Zelenay and G. Wu, Energy Environ. Sci., 2019, 12, 2548–2558 RSC.
- L. Jiao, R. Zhang, G. Wan, W. Yang, X. Wan, H. Zhou, J. Shui, S.-H. Yu and H.-L. Jiang, Nat. Commun., 2020, 11, 2831 CrossRef CAS PubMed.
- L. Jiao, J. Li, L. L. Richard, Q. Sun, T. Stracensky, E. Liu, M. T. Sougrati, Z. Zhao, F. Yang, S. Zhong, H. Xu, S. Mukerjee, Y. Huang, D. A. Cullen, J. H. Park, M. Ferrandon, D. J. Myers, F. Jaouen and Q. Jia, Nat. Mater., 2021, 20, 1385–1391 CrossRef CAS PubMed.
- H. Adabi, A. Shakouri, N. U. Hassan, J. R. Varcoe, B. Zulevi, A. Serov, J. R. Regalbuto and W. E. Mustain, Nat. Energy, 2021, 6, 834–843 CrossRef CAS.
- F. Xiao, X. Liu, C.-J. Sun, I. Hwang, Q. Wang, Z. Xu, Y. Wang, S. Zhu, H. Wu, Z. Wei, L. Zheng, D. Cheng, M. Gu, G.-L. Xu, K. Amine and M. Shao, Nano Lett., 2021, 21, 3633–3639 CrossRef CAS PubMed.
- X. Xie, L. Peng, H. Yang, G. I. N. Waterhouse, L. Shang and T. Zhang, Adv. Mater., 2021, 33, 2101038 CrossRef CAS PubMed.
- X. Xie, L. Shang, X. Xiong, R. Shi and T. Zhang, Adv. Energy Mater., 2022, 12, 2102688 CrossRef CAS.
- A. Mehmood, M. Gong, F. Jaouen, A. Roy, A. Zitolo, A. Khan, M.-T. Sougrati, M. Primbs, A. M. Bonastre, D. Fongalland, G. Drazic, P. Strasser and A. Kucernak, Nat. Catal., 2022, 5, 311–323 CrossRef CAS.
- G. Chen, Y. An, S. Liu, F. Sun, H. Qi, H. Wu, Y. He, P. Liu, R. Shi, J. Zhang, A. Kuc, U. Kaiser, T. Zhang, T. Heine, G. Wu and X. Feng, Energy Environ. Sci., 2022, 15, 2619–2628 RSC.
- R. Chenitz, U. I. Kramm, M. Lefèvre, V. Glibin, G. Zhang, S. Sun and J.-P. Dodelet, Energy Environ. Sci., 2018, 11, 365–382 RSC.
- C. H. Choi, H.-K. Lim, M. W. Chung, G. Chon, N. R. Sahraie, A. Altin, M.-T. Sourati, L. Stievano, H. S. Oh, E. S. Park, F. Luo, P. Strasser, G. Dražić, K. J. J. Mayrhofer, H. Kim and F. Jaouen, Energy Environ. Sci., 2018, 11, 3176–3182 RSC.
- H. Xie, X. Xie, G. Hu, V. Prabhakaran, S. Saha, L. Gonzalez-Lopez, A. H. Phakatkar, M. Hong, M. Wu, R. Shahbazian-Yassar, V. Ramani, M. I. Al-Sheikhly, D. Jiang, Y. Shao and L. Hu, Nat. Energy, 2022, 7, 281–289 CrossRef CAS.
- S. Liu, C. Li, M. J. Zachman, Y. Zeng, H. Yu, B. Li, M. Wang, J. Braaten, J. Liu, H. M. Meyer III, M. Lucero, A. J. Kropf, E. E. Alp, Q. Gong, Q. Shi, Z. Feng, H. Xu, G. Wang, D. J. Myers, J. Xie, D. A. Cullen, S. Litster and G. Wu, Nat. Energy, 2022, 7, 652–663 CrossRef CAS.
- J. Li, H. Zhang, W. Samarakoon, W. Shan, D. A. Cullen, S. Karakalos, M. Chen, D. Gu, K. L. More, G. Wang, Z. Feng, Z. Wang and G. Wu, Angew. Chem., Int. Ed., 2019, 58, 18971–18980 CrossRef CAS PubMed.
- T. Marshall-Roth, N. J. Libretto, A. T. Wrobel, K. J. Anderton, M. L. Pegis, N. D. Ricke, T. V. Voorhis, J. T. Miller and Y. Surendranath, Nat. Commun., 2020, 11, 5283 CrossRef CAS PubMed.
- J. Li, L. Jiao, E. Wegener, L. L. Richard, E. Liu, A. Zitolo, M. T. Sougrati, S. Mukerjee, Z. Zhao, Y. Huang, F. Yang, S. Zhong, H. Xu, A. J. Kropf, F. Jaouen, D. J. Myers and Q. Jia, J. Am. Chem. Soc., 2020, 142, 1417–1423 CrossRef CAS PubMed.
- J. Li, M. T. Sougrati, A. Zitolo, J. M. Ablett, I. C. Oğuz, T. Mineva, I. Matanovic, P. Atanassov, Y. Huang, I. Zenyuk, A. D. Cicco, K. Kumar, L. Dubau, F. Maillard, G. Dražić and F. Jaouen, Nat. Catal., 2021, 4, 10–19 CrossRef CAS.
- D. Menga, J. L. Low, Y.-S. Li, I. Arčon, B. Koyutürk, F. Wagner, F. Ruiz-Zepeda, M. Gaberšček, B. Paulus and T.-P. Fellinger, J. Am. Chem. Soc., 2021, 143, 18010–18019 CrossRef CAS PubMed.
- F. Luo, A. Roy, L. Silvioli, D. A. Cullen, A. Zitolo, M. T. Sougrati, I. C. Oguz, T. Mineva, D. Teschner, S. Wagner, J. Wen, F. Dionigi, U. I. Kramm, J. Rossmeisl, F. Jaouen and P. Strasser, Nat. Mater., 2020, 19, 1215–1223 CrossRef CAS PubMed.
- S. Liu, Z. Li, C. Wang, W. Tao, M. Huang, M. Zuo, Y. Yang, K. Yang, L. Zhang, S. Chen, P. Xu and Q. Chen, Nat. Commun., 2020, 11, 938 CrossRef CAS PubMed.
- T. Wang, X. Cao, H. Qin, L. Shang, S. Zheng, F. Fang and L. Jiao, Angew. Chem., Int. Ed., 2021, 60, 21237–21241 CrossRef CAS PubMed.
- Z. Lin, H. Huang, L. Cheng, W. Hu, P. Xu, Y. Yang, J. Li, F. Gao, K. Yang, S. Liu, P. Jiang, W. Yan, S. Chen, C. Wang, H. Tong, M. Huang, W. Zheng, H. Wang and Q. Chen, Adv. Mater., 2021, 33, 2107103 CrossRef CAS PubMed.
- N. D. Leonard, S. Wagner, F. Luo, J. Steinberg, W. Ju, N. Weidler, H. Wang, U. I. Kramm and P. Strasser, ACS Catal., 2018, 8, 1640–1647 CrossRef CAS.
- F. Luo, C. H. Choi, M. J. M. Primbs, W. Ju, S. Li, N. D. Leonard, A. Thomas, F. Jaouen and P. Strasser, ACS Catal., 2019, 9, 4841–4852 CrossRef CAS.
- M. Primbs, Y. Sun, A. Roy, D. Malko, A. Mehmood, M.-T. Sougrati, P.-Y. Blanchard, G. Granozzi, T. Kosmala, G. Daniel, P. Atanassov, J. Sharman, C. Durante, A. Kucernak, D. Jones, F. Jaouen and P. Strasser, Energy Environ. Sci., 2020, 13, 2480–2500 RSC.
- G. Bae, H. Kim, H. Choi, P. Jeong, D. H. Kim, H. C. Kwon, K.-S. Lee, M. Choi, H.-S. Oh, F. Jaouen and C. H. Choi, JACS Au, 2021, 1, 586–597 CrossRef CAS PubMed.
- F. Luo, S. Wagner, W. Ju, M. Primbs, S. Li, H. Wang, U. I. Kramm and P. Strasser, J. Am. Chem. Soc., 2022, 144, 13487–13498 CrossRef CAS PubMed.
- R. Z. Snitkoff-Sol, A. Friedman, H. C. Honig, Y. Yurko, A. Kozhushner, M. J. Zachman, P. Zelenay, A. M. Bond and L. Elbaz, Nat. Catal., 2022, 5, 163–170 CrossRef CAS.
- J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 561–588 CrossRef CAS PubMed.
- W. Zhang, W. Lai and R. Cao, Chem. Rev., 2017, 177, 3717–3797 CrossRef PubMed.
- J. P. Collman, N. K. Devaraj, R. A. Decréau, Y. Yang, Y.-L. Yan, W. Ebina, T. A. Eberspacher and C. E. D. Chidsey, Science, 2007, 315, 1565–1568 CrossRef CAS PubMed.
- A. Bhagi-Damodaran, M. A. Michael, Q. Zhu, J. Reed, B. A. Sandoval, E. N. Mirts, S. Chakraborty, P. Moënne-Loccoz, Y. Zhang and Y. Lu, Nat. Chem., 2017, 9, 257–263 CrossRef CAS PubMed.
- J. Wang, W. Liu, G. Luo, Z. Li, C. Zhao, H. Zhang, M. Zhu, Q. Xu, X. Wang, C. Zhao, Y. Qu, Z. Yang, T. Yao, Y. Li, Y. Lin, Y. Wu and Y. Li, Energy Envrion. Sci., 2018, 11, 3375–3379 RSC.
- C. H. Choi, C. Baldizzone, G. Polymeros, E. Pizzutilo, O. Kasian, A. K. Schuppert, N. R. Sahraie, M.-T. Sougrati, K. J. J. Mayrhofer and F. Jaouen, ACS Catal., 2016, 6, 3136–3146 CrossRef CAS.
- X.-X. Qi, N. Ren, S.-L. Xu, J.-J. Zhang, G.-C. Zong, J. Gao, L.-N. Geng, S.-P. Wang and S.-K. Shi, RSC Adv., 2015, 5, 9261–9271 RSC.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712–10713 CrossRef CAS.
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169–172 CrossRef CAS PubMed.
- Q. Jia, N. Ramaswamy, H. Hafiz, U. Tylus, K. Strickland, G. Wu, B. Barbiellini, A. Bansil, E. F. Holby, P. Zelenay and S. Mukerjee, ACS Nano, 2015, 9, 12496–12505 CrossRef CAS PubMed.
- Q. Jia, N. Ramaswamy, U. Tylus, K. Strickland, J. Li, A. Serov, K. Artyushkova, P. Atanassov, J. Anibal, C. Gumeci, S. C. Barton, M.-T. Sougrati, F. Jaouen, B. Halevi and S. Mukerjee, Nano Energy, 2016, 29, 65–82 CrossRef CAS.
- J. H. Kim, D. Shin, J. Lee, D. S. Baek, T. J. Shin, Y.-T. Kim, H. Y. Jeong, J. H. Kwak, H. Kim and S. H. Joo, ACS Nano, 2020, 14, 1990–2001 CrossRef CAS PubMed.
- H. Wan, A. W. Jensen, M. Escudero-Escribano and J. Rossmeisl, ACS Catal., 2020, 10, 5979–5989 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS PubMed.
- M. Kruk, M. Jaroniec, C. H. Ko and R. Ryoo, Chem. Mater., 2000, 12, 1961–1968 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ey00055e |
| This journal is © The Royal Society of Chemistry 2023 |