
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Boosting the activity of PdAg alloy nanoparticles during H2 production from formic acid induced by CrOx as an inorganic interface modifier†
Kohsuke
Mori
 *ab,
Tatsuya
Fujita
a and
Hiromi
Yamashita
*ab
*ab,
Tatsuya
Fujita
a and
Hiromi
Yamashita
*ab
aDivision of Materials and Manufacturing Science, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: mori@mat.eng.osaka-u.ac.jp; yamashita@mat.eng.osaka-u.ac.jp
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University, Suita, Osaka 565-0871, Japan
First published on 29th November 2022
Abstract
Interfacial modification of PdAg nanoparticles supported on mesoporous carbon (MSC) functionalized with weakly basic phenylamine groups was performed using an amorphous CrOx phase. The resulting PdAgCr/amine-MSC catalyst was found to promote the efficient dehydrogenation of formic acid (HCOOH) serving as a liquid organic hydrogen carrier. A maximum turnover frequency of 6898 h−1 (based on the mass of Pd employed) was achieved, which was 1.6 times larger than the value previously obtained using a PdAg catalyst. Physicochemical characterization and density functional theory calculations indicated that electronic interactions with the CrOx phase induced a significant electronic gap. This effect, in turn, generated unique ensemble sites on the PdAg nanoparticle surfaces at which electron-deficient Agδ+ and electron-rich Pdδ− atoms were adjacent. Kinetic analyses and theoretical investigations demonstrated that O–H bond dissociation was assisted by amine groups on the support surface. Enhanced C–H bond dissociation and H2 desorption resulting from the cooperative action at the ensemble sites were found to play a crucial role in increasing the catalytic activity. The present system is easily synthesized, does not require additives, can be recycled and effectively suppresses CO formation, meaning that this material is a suitable candidate for practical applications in fuel cells.
Broader contextFormic acid (FA, HCOOH) has emerged as one of the most promising hydrogen storage compounds because it is non-flammable, relatively nontoxic, and contains 4.4 wt% (53.4 g L−1) hydrogen. There have been extensive studies in developing catalysts targeted for efficient H2 delivery. Our own group has demonstrated that the dispersed PdAg nanoparticles supported on phenylamine-functionalized mesoporous carbon display an extremely high turnover frequency (TOF) of 5638 h−1 during the dehydrogenation of FA, and this work has provided advanced insights into the architecture of catalytically-active sites for this target reaction (ACS Catal., 2018, 8, 2277). In this study, interfacial modification of PdAg nanoparticles was performed using highly dispersed CrOx. The resulting ternary PdAgCr/amine-MSC is proven to display a maximum TOF of 6898 h−1, which is 1.6 times larger than that with our previously reported binary catalyst. On the basis of kinetic and theoretical investigations, we demonstrated that the significant enhancement of activity is attributed to the interplay of unique ensemble sites on the PdAg nanoparticle surface, where electron-deficient Agδ+ and electron-rich Pdδ− are adjacent. We are sure that these findings are of sufficiently immediate interest to a general chemistry research readership. |
1. Introduction
Hydrogen (H2) has a high gravimetric energy density of 33.3 kW h kg−1, which is three times the value for gasoline, and produces only water as a by-product when converted into energy in an internal combustion engine or a fuel cell.1,2 Hence, H2 is a potentially important energy source. However, the development of safe and efficient methodologies for the storage and transportation of hydrogen gas is required to realize a sustainable hydrogen-based economy.3,4 Conventional storage technologies, including pressurization and/or cryogenic liquefaction, suffer from a tradeoff relationship between storage density and efficiency.5 As such, chemical hydrogen storage in liquid organic hydrogen carriers (LOHCs) may be a viable alternative to physical hydrogen storage.6–8 In an LOHC system, hydrogen is stored by converting a hydrogen-lean compound into its hydrogen-rich counterpart via a catalytic hydrogenation reaction, while hydrogen is released by dehydrogenation of the hydrogen-rich compound. There are several LOHC systems, including cyclohexane–benzene, methylcyclohexane–toluene, decalin–naphthalene and perhydro-N-ethylcarbazole–N-ethyl carbazole, that have been recognized as suitable candidates.9–13Formic acid (FA, HCOOH) has emerged as a prominent LOHC component over the last decade. FA is of interest because of its high hydrogen content (4.4 wt%; 53.4 g L−1), low toxicity and low flammability (flashpoint of 69 °C, which is much higher than the values for methanol (12 °C) and gasoline (−40 °C)) under ambient conditions.14,15 The use of FA could allow economical CO2-mediated hydrogen storage energy cycling based on the regeneration of FA through the hydrogenation of CO2 with H2.16 The selective dehydrogenation of FA is favored thermodynamically (ΔG = −48.4 kJ mol−1), which is necessary to ensure that pure H2 is generated without the formation of CO and H2O via the competitive dehydration pathway (ΔG = −28.5 kJ mol−1). This factor is especially important because the CO byproduct is toxic to Pt-based fuel-cell catalysts. For all these reasons, numerous investigations have been conducted in attempts to identify catalysts, both homogeneous and heterogeneous, capable of promoting FA dehydrogenation under mild conditions.17–23
To ensure that practical systems are developed, current research has focused on the use of reliable heterogeneous catalysts such as Au, Pt and especially Pd, which exhibits excellent catalytic activity. Alloying these elements with additional metals that possess superior resistance to CO poisoning is another promising strategy.24–28 Our group previously developed binary nanoparticle (NP) catalysts based on Pd combined with coinage metals such as Au, Ag or Cu that exhibited superior activity compared with monometallic catalysts.29–31 The synergistic effect provided by these materials is attributed to the electronic activation of Pd species by charge transfer resulting from the different work functions of the two metals. This prior work suggested possible design strategies for producing catalytically active sites promoting the dehydrogenation of FA.
Research regarding the nano-engineering of catalysts for efficient dehydrogenation of FA has indicated that precise tuning of the NP architecture at the angstrom level is required to provide the appropriate activity and selectivity. Controlling particle size can provide large surface-area-to-volume ratios and also optimize the relative proportions of low- and high-coordination surface atoms within regular arrangements of the NPs.32,33 The catalytic behavior of NPs is also affected by morphology due to differences in the preferential exposure of particular crystallographic planes.34 Many supports, including zeolites, silica, carbon and metal–organic frameworks, have been investigated in attempts to obtain strong metal–support interactions, with frequent reports of unique synergistic effects at the interfaces between NPs and supports.35–41 Surface modification with organic molecules can also inhibit the agglomeration of NPs, thus allowing control over the adsorption kinetics of reactants or intermediates through specific geometric phenomena or metal–ligand electronic effects.42,43
We have previously reported that the PdCuCr ternary NPs within a macroreticular basic resin that possesses –N(CH3)2 functional groups are responsible for the efficient production of H2 from formic acid, which is attributed to the stabilization effect of surface Cr clusters and a synergistic effect to boost the C–H bond dissociation step.44 Inspired by our previously obtained insight, the present study utilized a highly dispersed amorphous CrOx phase as an inorganic interface modifier to alter the electronic state of PdAg NPs supported on mesoporous carbon (MSC) functionalized with a weakly basic phenylamine. Unlike organic ligands, which exhibit Lewis basicity, metal centers connected with oxide (O2−) groups in CrOx are positively charged, while those in the periphery of the Cr3+ atoms are highly negatively charged. Thus, the modification of these materials with CrOx improves catalytic activity during dehydrogenation of FA compared with a pure binary PdAg catalyst. These electronic interactions generate unique ensemble sites at which electron-deficient Agδ+ and electron-rich Pdδ− atoms are adjacent, and ultimately lead to a rate-determining step involving the dissociation of C–H bonds in FA, as demonstrated by kinetics analyses and density functional theory (DFT) calculations.
2. Results and discussion
2.1. Characterization of catalysts
The synthetic procedure employed to produce catalysts comprising PdAgCr supported on phenylamine-functionalized MSC (denoted herein as PdAgCr/amine-MSC) is illustrated in Scheme 1. In this procedure, the MSC was treated with an aqueous nitric acid solution, which resulted in the formation of surface O-based functional groups such as –COOH and –OH (Fig. S1A, ESI†). The surface of the acid-treated MSC was subsequently functionalized with p-phenylenediamine to afford amine-MSC. During this step, the surface O-based functional groups reacted with the amine groups to generate amide and C–N bonds. The concentration of amine groups grafted onto the MSC was determined to be approximately 0.57 mmol g−1 based on CHN elemental analysis (Table S1, ESI†), in good agreement with the value of 0.53 mmol g−1 obtained from thermogravimetric analysis (Fig. S1B, ESI†). Pd, Ag and Cr were deposited by mixing the amine-MSC support with aqueous solutions containing Pd(NO3)2, AgNO3 and Cr(NO3)3·9H2O. Finally, each sample was reduced using NaBH4, affording PdAgCr/amine-MSC. | ||
| Scheme 1 Schematic illustration of the synthesis of the PdAgCr-supported amine-MSC catalyst. | ||
The N2 adsorption–desorption isotherm for each specimen displayed a characteristic type IV curve with sharp capillary condensation steps and an H1-type hysteresis loop over the relative pressure (p/p0) range of 0.6–0.9, indicating the presence of ordered and well-preserved cylindrical mesopores (Fig. S2, ESI†). These results confirmed that the original pore structure of the MSC was maintained despite the addition of the modifier and metals. The Brunauer–Emmett–Teller surface areas (SBET) and pore volumes (Vp) calculated from the N2 adsorption–desorption isotherms are summarized in Table S2 (ESI†). After amine functionalization to generate amine-MSC (SBET = 153 m2 g−1, Vp = 0.39 cm3 g−1), both SBET and Vp decreased compared with the values for the original MSC (SBET = 320 m2 g−1, Vp = 0.60 cm3 g−1). A further slight decrease was observed after deposition of the metals (SBET = 104 m2 g−1, Vp = 0.24 cm3 g−1).
Fig. 1 presents a high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of PdAgCr/amine-MSC, together with energy dispersive X-ray spectroscopy (EDX) maps. The NPs had a mean diameter (dave) of 1.5 nm and were well dispersed (Fig. 1a). This value was comparable with that obtained for PdAg/amine-MSC (dave = 1.2 nm) and Pd/amine-MSC (dave = 1.8 nm) specimens, as shown in Fig. S3 and S4 (ESI†). However, a comparison of the particle sizes clearly demonstrated that the addition of the second and third metals resulted in the formation of smaller NPs. Energy-dispersive X-ray spectroscopy maps indicated that the bright particles were composed of Pd, Ag and Cr atoms that are highly dispersed throughout the amine-functionalized material (Fig. 1b–e).
 | ||
| Fig. 1 (a) HAADF-STEM image of PdAgCr/amine-MSC. EDX mapping of (b) Pd, (c) Ag, (d) Cr, and (e) N over the region shown in (a). | ||
The electronic state of each metal was investigated by X-ray photoelectron spectroscopy (XPS). In the case of Pd/amine-MSC, the Pd 3d5/2 and 3d3/2 electron binding energy values were 335.9 and 338.0 eV, and 341.0 and 343.3 eV, respectively. The peaks at lower binding energies were assigned to the Pd0 state, while the higher ones were attributed to Pd2+ (Fig. 2A). The presence of Pd2+ was presumably due to the oxidation of the surface Pd NPs by CrOx or strong interaction with surface amine groups. The Pd 3d peaks generated by PdAg/amine-MSC were shifted to lower binding energies by 0.4 eV compared with those obtained from the corresponding monometallic Pd catalyst. This shift was attributed to the ability of Ag atoms to donate electrons to Pd atoms upon alloying, as a consequence of the net difference in the ionization potentials of the two elements (Ag: 7.57 eV, Pd: 8.34 eV). In contrast, the addition of Cr atoms shifted the Pd 3d peaks toward higher binding energies compared with PdAg/amine-MSC. No differences were observed in the Ag 3d XPS spectra acquired from PdAg/amine-MSC and PdAgCr/amine-MSC (Fig. 2B), suggesting that the presence of Ag+ was minimal. The Cr 2p spectra could be deconvoluted into two components (Fig. 2C), comprising a main peak at approximately 577.5 eV assigned to Cr3+ and a small peak at 580 eV attributed to Cr6+. These results indicate that the Ag atoms were preferentially located in the core regions of the NPs, while Pd atoms were typically situated on NP surfaces and were partially oxidized by the amorphous chromium oxide (CrOx) and/or chromium hydroxide (Cr(OH)3).
 | ||
| Fig. 2 (A) Pd 3d XPS spectra of (a) PdAgCr/amine-MSC, (b) PdAg/amine-MSC and (c) Pd/amine-MSC; (B) Ag 3d XPS spectra of (a) PdAgCr/amine-MSC and (b) PdAg/amine-MSC; and (C) Cr 2p XPS spectrum of PdAgCr/amine-MSC. | ||
X-ray absorption analyses were also performed to elucidate the local structures of the specimens. The shapes of the Pd K-edge X-ray absorption near-edge structure (XANES) spectra of the three Pd-based samples all differed from that of Pd foil but resembled the spectrum of PdO (Fig. 3A). The Fourier-transform extended X-ray absorption fine structure (FT-EXAFS) spectra of all materials exhibited two peaks associated with Pd–O(N) and contiguous metallic Pd–Pd bonds, appearing at 1.6 and 2.6 Å, respectively (Fig. 3B), with no significant shift from the Pd–Pd distance in Pd foil. The first peak, at 1.6 Å, is believed to be partly due to Pd–N bonds originating from interactions with the amine-MSC support. The inverse FT of PdAgCr/amine-MSC was well fitted using Pd–O(N) and Pd–Pd shells with a coordination number (CN) and an interatomic distance (R) of CN = 4.7 and R = 2.00 Å and CN = 6.8 and R = 2.74 Å, respectively (Table 1 and Fig. S5A, ESI†). Assuming that the contribution of Pd–N for all samples is almost the same since the mean diameter for all samples is comparable, the ratio of the contribution of Pd–O bonds to that of Pd–Pd bonds (CNPd–O/CNPd–Pd = 0.69) in the case of PdAgCr/amine-MSC was larger than those for PdAg/amine-MSC (0.52) and Pd/amine-MSC (0.52) (Table 1). These results are in good agreement with the Pd 3d XPS data and suggest that the Pd was partially oxidized following modification with CrOx and/or Cr(OH)3.
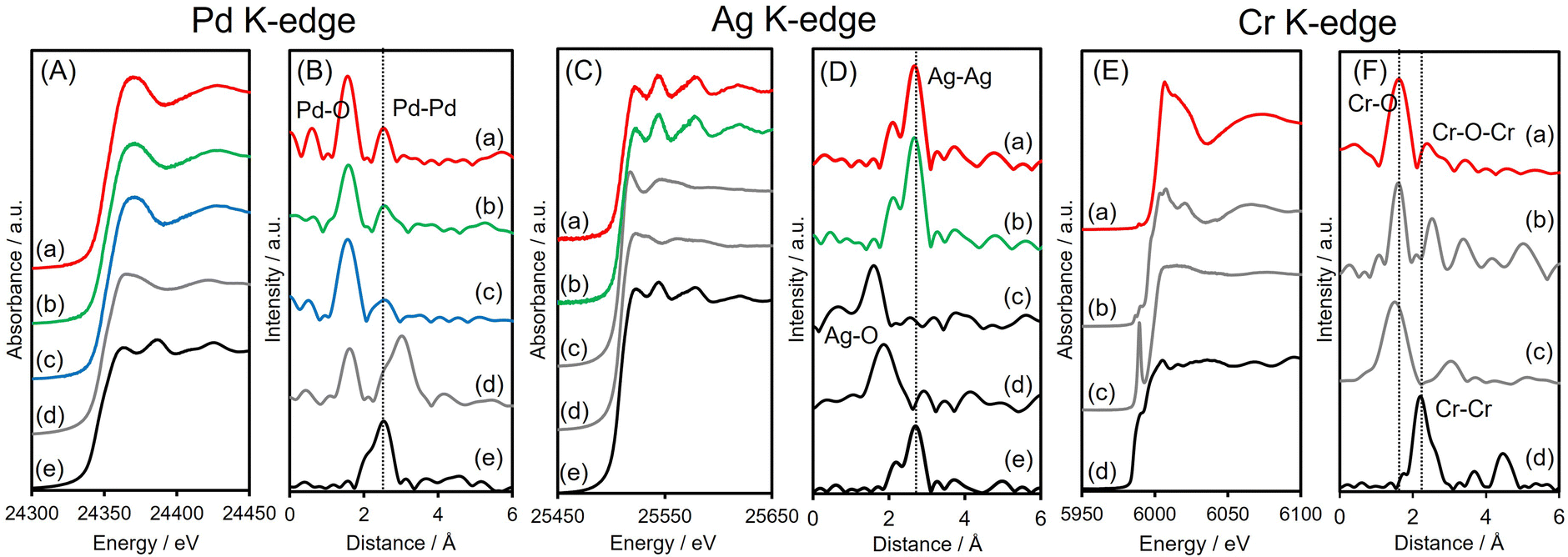 | ||
| Fig. 3 (A) Pd K-edge XANES and (B) FT-EXAFS spectra of (a) PdAgCr/amine-MSC, (b) PdAg/amine-MSC, (c) Pd/amine-MSC, (d) PdO and (e) Pd foil; (C) Ag K-edge XANES and (D) FT-EXAFS spectra of (a) PdAgCr/amine-MSC, (b) PdAg/amine-MSC, (c) Ag2O, (d) AgNO3 and (e) Ag foil; and (E) Cr K-edge XANES and (F) FT-EXAFS spectra of (a) PdAgCr/amine-MSC, (b) Cr2O3, (b) CrO3, and (c) Cr foil. | ||
| K-edge | Shell | CN | R/Å | σ 2 | |
|---|---|---|---|---|---|
| PdAgCr/amine-MSC | Pd | Pd–O(N) | 4.7 | 2.00 | 0.0067 |
| Pd–Pd | 6.8 | 2.74 | 0.0065 | ||
| Ag | Ag–Ag | 11.3 | 2.89 | 0.0077 | |
| Cr | Cr–O | 4.6 | 2.01 | 0.0032 | |
| PdAg/amine-MSC | Pd | Pd–O(N) | 4.8 | 2.02 | 0.0089 |
| Pd–Pd | 9.3 | 2.76 | 0.015 | ||
| Ag | Ag–Ag | 11.3 | 2.82 | 0.0073 | |
| Pd/amine-MSC | Pd | Pd–O(N) | 5.0 | 2.01 | 0.0078 |
| Pd–Pd | 9.6 | 2.73 | 0.015 | ||
The Ag K-edge XANES spectra obtained from PdAgCr/amine-MSC and PdAg/amine-MSC were similar to that of Ag foil and their FT-EXAFS spectra showed a single intense peak ascribed to contiguous Ag–Ag bonds at approximately 2.6–2.8 Å without any noticeable evidence of Ag–O bonds (Fig. 3C and D). This peak is shifted to a slightly shorter interatomic distance compared with that for pure Ag foil, indicating the presence of Pd–Ag bonds. The inverse FT was well fitted using Ag–Ag shells with CN = 11.3 and R = 2.89 Å for PdAgCr/amine-MSC, and CN = 11.3 and R = 2.82 Å for PdAg/amine-MSC, indicating that Ag formed metallic NPs in both samples (Table 1 and Fig. S5B, ESI†). These results are in agreement with the Ag 3d XPS data.
The Cr K-edge XANES spectrum of CrO3 displayed an intense pre-edge peak at 5992 eV which is characteristic of terminal Cr6+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in a tetrahedral coordination (Fig. 3E).44,45 The XANES spectrum obtained for PdAgCr/amine-MSC was different from that for CrO3 but resembled that for Cr2O3, which contains Cr3+. The edge energy for PdAgCr/amine-MSC was higher than that for Cr foil, confirming that the Cr in this sample was in the +3 oxidation state. The length of the first Cr–O bond in PdAgCr/amine-MSC as determined from its FT-EXAFS spectrum was comparable to that in Cr2O3 but longer than that in CrO3. These data suggest the absence of CrO double bonds in PdAgCr/amine-MSC (Fig. 3F). A curve-fitting analysis also confirmed that the interatomic distance for the first Cr–O bond was 2.01 Å, in conjunction with a CN of 4.6 (Table 1 and Fig. S5C, ESI†). Moreover, the intensity of the peak associated with the second coordination sphere for PdAgCr/amine-MSC was significantly weaker that the intensity obtained for Cr2O3. In the case of the PdAgCr specimen, this peak was attributable to contiguous Cr–O–M (M: Cr or Pd) bonds because of the absence of evidence for Ag–O bonds in the Ag K-edge FT-EXAFS spectrum. The reduced intensity in this region was ascribed to the formation of small, structurally disordered nanoclusters. No evidence for metallic bonds, which were detectable at approximately 2.3 Å in the case of Cr foil, was observed. Because the reduction potentials for Cr3+ ions (E°(Cr3+/Cr2+) = −0.42 V, E°(Cr2+/Cr0) = −0.90 V vs. NHE) are more negative than those for Pd2+ and Ag+ ions (E°(Pd2+/Pt0) = +0.99 V, E°(Ag+/Ag0) = +0.80 V vs. NHE), the reduction of Cr3+ ions was inhibited to a greater extent than for Pd2+ and Ag+ ions, and Cr ions preferentially constitute extremely small clusters.
O bonds in a tetrahedral coordination (Fig. 3E).44,45 The XANES spectrum obtained for PdAgCr/amine-MSC was different from that for CrO3 but resembled that for Cr2O3, which contains Cr3+. The edge energy for PdAgCr/amine-MSC was higher than that for Cr foil, confirming that the Cr in this sample was in the +3 oxidation state. The length of the first Cr–O bond in PdAgCr/amine-MSC as determined from its FT-EXAFS spectrum was comparable to that in Cr2O3 but longer than that in CrO3. These data suggest the absence of CrO double bonds in PdAgCr/amine-MSC (Fig. 3F). A curve-fitting analysis also confirmed that the interatomic distance for the first Cr–O bond was 2.01 Å, in conjunction with a CN of 4.6 (Table 1 and Fig. S5C, ESI†). Moreover, the intensity of the peak associated with the second coordination sphere for PdAgCr/amine-MSC was significantly weaker that the intensity obtained for Cr2O3. In the case of the PdAgCr specimen, this peak was attributable to contiguous Cr–O–M (M: Cr or Pd) bonds because of the absence of evidence for Ag–O bonds in the Ag K-edge FT-EXAFS spectrum. The reduced intensity in this region was ascribed to the formation of small, structurally disordered nanoclusters. No evidence for metallic bonds, which were detectable at approximately 2.3 Å in the case of Cr foil, was observed. Because the reduction potentials for Cr3+ ions (E°(Cr3+/Cr2+) = −0.42 V, E°(Cr2+/Cr0) = −0.90 V vs. NHE) are more negative than those for Pd2+ and Ag+ ions (E°(Pd2+/Pt0) = +0.99 V, E°(Ag+/Ag0) = +0.80 V vs. NHE), the reduction of Cr3+ ions was inhibited to a greater extent than for Pd2+ and Ag+ ions, and Cr ions preferentially constitute extremely small clusters.
Based on these data, a reasonable structural model for PdAgCr/amine-MSC was devised. In this model, Ag atoms are preferentially located in the core regions of the NPs, while Pd atoms tend to be situated in the shells. In addition, the amorphous CrOx phase including (Cr(OH)3) is partially located on the surfaces of these Pd-rich shells as a result of the formation of Pd–O–Cr bonds, as illustrated in Scheme 1. This configuration is based on the EXAFS analysis showing that the Pd atoms in PdAgCr/amine-MSC were partially oxidized, either because of the interaction with Cr atoms or the coordination with N atoms in the surface amine groups by the exposure to the surface.
The effect of the CrOx phase on the electronic state of the Pd and Ag atoms was assessed by calculating atomic charges using DFT. These calculations were based on supercell slab models consisting of 5 × 5 surface unit cells including PdAg(111) interacting with Cr2O3 clusters together with pristine PdAg(111). The optimized structures are shown in Fig. 4A, in which the colors of Pd atoms indicate the calculated Mulliken atomic charges. These investigations indicated that the integration of Pd with Ag induced charge transfer from Ag atoms to Pd atoms to produce electron-rich Pdδ− and electron-deficient Agδ+. In addition, Pd atoms in the vicinity of the Cr atoms in Cr2O3 were determined to have a large negative charge as a consequence of electron transfer from the Cr atoms. In contrast, Ag atoms bound to the O atoms of Cr2O3 were highly positive, suggesting electron transfer from the Ag atoms to the O atoms. These electronic interactions generated unique ensemble sites and a significant electronic gap based on the adjacent electron-deficient Agδ+ and electron-rich Pdδ−. Density of states (DOS) calculations showed that the d-band center of PdAg (111) associated with Cr2O3 was located at −2.69 eV and so had a similar energy to that of −2.60 eV for PdAg (111). However, this value was more negative than that of −1.99 eV for Pd (111) (Fig. 4B). These findings confirmed that modification with CrOx did not change the electronic structure of the catalyst in the bulk but rather caused uneven charge distribution on the surface of the material.
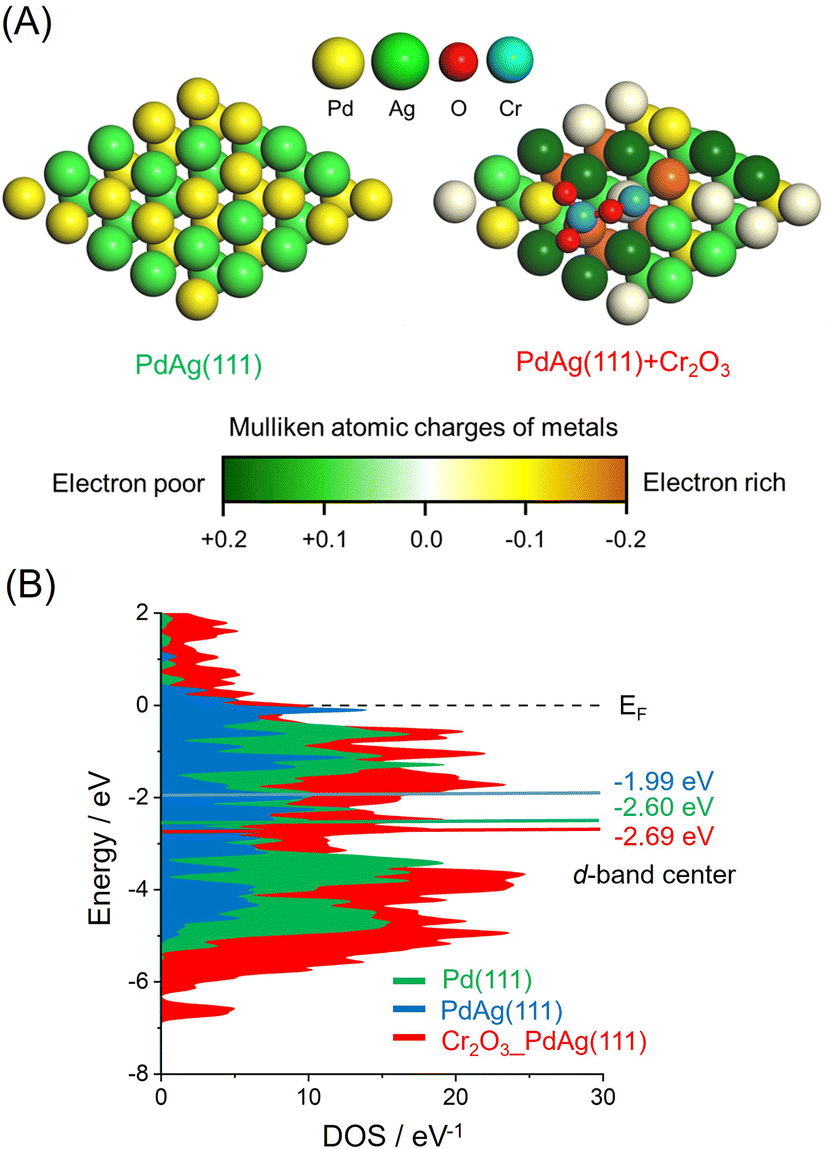 | ||
| Fig. 4 (A) The optimized structure of pristine PdAg(111) and PdAg(111) associated with one stoichiometric Cr2O3 cluster with a color overlay of representative Mulliken atomic charge determined using density functional theory calculations. (B) Calculated DOS plots. | ||
2.2. Catalytic activity during FA dehydrogenation
Dehydrogenation of FA was conducted in an aqueous solution containing HCOOH and HCOONa at a molar ratio of 9/1. No induction period was observed during periodic monitoring of the reaction (Fig. 5A), and H2 and CO2 were generated in a molar ratio of almost 1, indicating complete dehydrogenation of the FA. PdAgCr/amine-MSC displayed the highest activity among the various catalysts and exhibited substantially higher performance than the monometallic Pd and binary PdAg materials.46 Using the optimal FA concentration (Fig. S6, ESI†), a turnover frequency (TOF) of 6898 h−1 (based on the mass of Pd employed) was achieved, which was 1.6 times larger than the value for the PdAg catalyst. The catalytic activity attained by PdAgCr/MSC without amine modification was quite low, confirming that the amine functional group plays a crucial role in determining the elementary steps in the catalytic dehydrogenation of FA, especially in the O–H bond dissociation step, as discussed elsewhere.29,31,46 In addition, the reaction essentially did not occur when using pure Ag or Cr. The addition of Mn, Ni or Co was found to slightly increase the catalytic activity, while the presence of Fe, Zn, Ga or Cu reduced the dehydrogenation performance (Fig. 5B). These results indicate that a significant synergistic effect was obtained from the ternary NPs. The effect of the composition of the PdAgCr catalyst was investigated by simply varying the initial molar ratio of the metal precursors. Fig. 5C shows the effect of the Cr mole fraction relative to the combined moles of Pd and Ag on the Pd-based TOF and indicates that the activity was maximized at a Cr mole fraction of 0.5. This volcano type activity relationship verified that the unusual synergistic effect observed in the present work originated from the combination of the three metals and also demonstrated that an excess of CrOx can suppress access to the active Pd centers. | ||
| Fig. 5 (A) Time course of the reaction in the dehydrogenation of FA using various amine-MSC supported catalysts. (B) Effect of the addition of third metals to the Pd-based catalysts. (C) TOF versus mole fraction of Cr for the PdAg/amine-MSC catalyst. (D) Arrhenius plots in the dehydrogenation of FA. | ||
The Arrhenius plot in Fig. 5D demonstrates that the activation energy (Ea) for the dehydrogenation of FA decreased significantly with increasing number of catalyst components, with values of 22.3 kJ mol−1 for PdAgCr/amine-MSC, 31.0 kJ mol−1 for PdAg/amine-MSC and 35.1 kJ mol−1 for Pd/amine-MSC. These results indicate that modification with CrOx boosted the reaction rate in the elementary step during the dehydrogenation of FA. Notably, PdAg/Cr2O3 prepared using the same impregnation method followed by NaBH4 reduction showed almost no catalytic activity, indicating that Cr2O3 itself does not act as an efficient support. The mean particle size determined by TEM analysis was 7.5 nm, which is larger than that of PdAgCr/amine-MSC (Fig. S7, ESI†). The Pd 3d peaks in XPS analysis generated by PdAg/Cr2O3 shifted to higher binding energies by 1 eV compared with those obtained from PdAgCr/amine-MSC (Fig. S8A, ESI†). Similarly, the shift toward higher binding energies by 1 eV can be observed in Ag 3d XPS spectra (Fig. S8B, ESI†). These shifts were attributed to the ability of Cr2O3 to withdraw electrons from PdAg alloy NPs, as a consequence of the relatively low electronegativity among metal oxides.47
It should be noted that the present catalytic system efficiently suppressed the undesirable formation of CO via the reaction HCOOH → H2O + CO. No CO was detected by the gas chromatography analysis method (the detection limit is approximately less than 2 ppm). This concentration of CO was significantly lower than that generated during conventional gas reforming from methanol, ethanol and methane, and meets the requirement provided in the PEMFC standard, meaning a CO concentration below 10 ppm.48 Upon completion of the reaction, the heterogeneous catalyst was readily separated from the reaction system and analysis of the filtrate by inductively coupled plasma confirmed that leaching of the metals from the catalyst was negligible. From these results it may be deduced that the reaction occurred on the solid surfaces of the material rather than in solution. The recovered catalyst could be recycled at least three times while retaining 90% of its original activity (Fig. S9, ESI†). TEM observations showed that the particle size remained virtually unchanged after the reaction without the formation of large agglomerated NPs (Fig. S10, ESI†). XAFS analyses also indicated a lack of structural changes in the K-edge spectrum obtained from the recovered catalyst relative to that for the fresh material (Fig. S11, ESI†). Unfortunately, the catalytic activity decreased to almost half of its original activity after the fourth recycling experiment. A critical reason for the decreased activity is unclear so far. Moreover, the maximum turnover frequency achieved in this study is 6898 h−1, which is comparable to or higher than those of other more recently reported catalytic systems,49–52 but still lower than those of the most pronounced catalytic systems.53,54 Thus, the further improvement of activity as well as the durability of the present catalytic system is now under investigation.
In a preliminary experiment, the activity of PdAgCr/amine-MSC was evaluated in the hydrogenation of CO2 with H2 to give FA using a 1.0 M aqueous NaHCO3 solution at pH = 8.5 under a total pressure of 2.0 MPa (H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2 = 1:1, volume ratio) at 100 °C. PdAgCr/amine-MSC showed a TON of 893 after 24 h with >99% selectivity, which is larger than those obtained with PdAg/amine-MSC (TON = 830) and Pd/amine-MSC (TON = 310), indicating the synergistic effect between Pd and Ag as well as the modification with CrOx. It can be concluded that unique ensemble sites on the PdAg nanoparticle surfaces, at which electron-deficient Agδ+ and electron-rich Pdδ− atoms were adjacent, are also crucial for attaining high activity during hydrogenation of CO2 with H2. Thus, the electron-deficient Agδ+ facilitates the adsorption of HCO3− ions, whereas the electron-rich Pdδ− accelerates not only H2 dissociation but also the attack of dissociated H atoms on C atoms in HCO3− ions.55
:CO2 = 1:1, volume ratio) at 100 °C. PdAgCr/amine-MSC showed a TON of 893 after 24 h with >99% selectivity, which is larger than those obtained with PdAg/amine-MSC (TON = 830) and Pd/amine-MSC (TON = 310), indicating the synergistic effect between Pd and Ag as well as the modification with CrOx. It can be concluded that unique ensemble sites on the PdAg nanoparticle surfaces, at which electron-deficient Agδ+ and electron-rich Pdδ− atoms were adjacent, are also crucial for attaining high activity during hydrogenation of CO2 with H2. Thus, the electron-deficient Agδ+ facilitates the adsorption of HCO3− ions, whereas the electron-rich Pdδ− accelerates not only H2 dissociation but also the attack of dissociated H atoms on C atoms in HCO3− ions.55
2.3. FA dehydrogenation mechanism
The dehydrogenation reaction proceeds exclusively to afford H2 and CO2 in a molar ratio of 1:1. The presence of amine functional groups on supports significantly boosted the activity. The addition of HCOONa to the reaction mixture increased H2 productivity, indicating the participation of the Pd–formate species as a reaction intermediate. Upon consideration of these preliminary insights as well as the results of previous studies, a possible reaction mechanism for the dehydrogenation of FA assisted by weakly basic surface –NH2 functional groups in the vicinity of metal NPs was devised and is presented in Fig. 6A.31,46,56–59 In this process, the –NH2 groups act as proton scavengers to promote the dissociation of O–H bonds in FA molecules and generate metal-formate species along with –NH3+ groups (Step 1). Each formate intermediate subsequently undergoes isomerization (Step 2), followed by C–H bond dissociation, to produce CO2 and a metal hydride (Step 3). Finally, the hydride reacts with a neighboring –NH3+ group to form molecular hydrogen (Step 4) with the simultaneous regeneration of the initial active species.
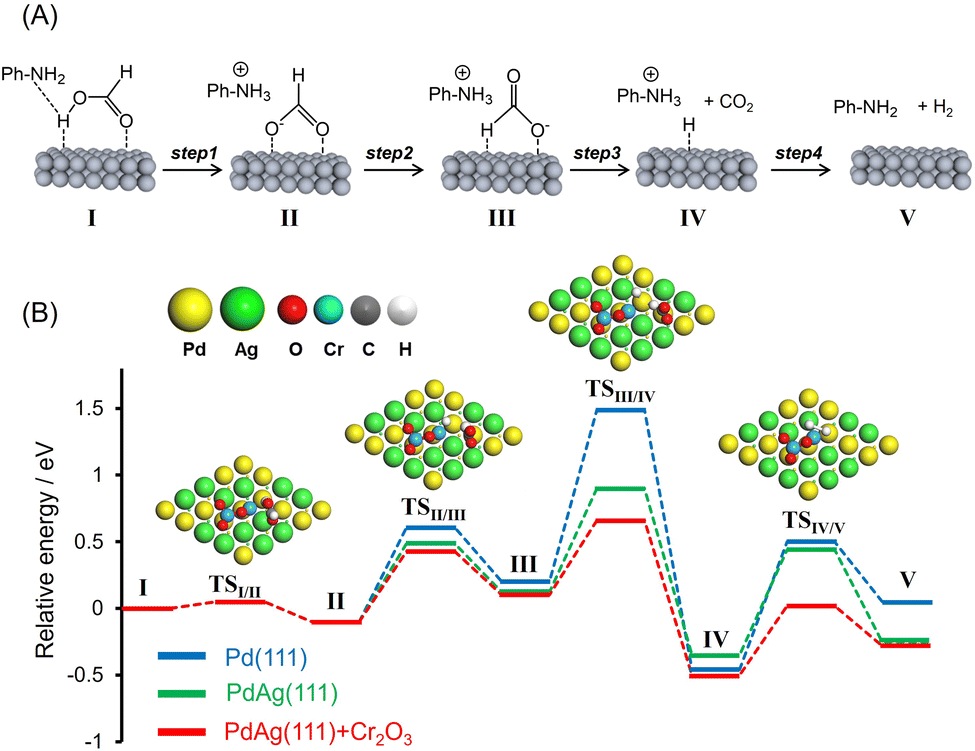 | ||
| Fig. 6 (A) Possible reaction mechanism and (B) potential energy profiles for dehydrogenation of formic acid calculated using pristine Pd(111), PdAg(111), and PdAg(111) associated with one stoichiometric Cr2O3 cluster in the absence of amine molecules. The representative TS structures obtained in the calculation of PdAg(111) + Cr2O3 are shown. | ||
It is likely that FA molecules will encounter amine groups in the vicinity of the NPs prior to adsorption on the surface of the catalyst, as a result of the acid–base interaction. The present DFT calculations demonstrate that the optimized structure of the resulting acid–base pair has a cis-M(O)–M(O–C)-bridged configuration (M = Pd or Ag) in which the carbonyl oxygen and non-carbonyl oxygen interact with Ag and Pd atoms, respectively. The O–H bond length (1.075 Å) in this optimized configuration is larger than the value of 0.985 Å for free trans-HCOOH, suggesting that the O–H bond of the FA undergoes a spontaneous dissociation without any transition state when an incoming HCOOH molecule is coupled with a phenylamine group. Kinetic isotope effect (KIE) studies using HCOOH and HCOOD show that the kH/kD ratios obtained from competitive reactions using the Pd-based catalysts supported on amine-MSC were quite small (approximately 1) compared with that obtained using PdAgCr/MSC without the amine modification (Table 3, entries 1–3 vs. 4). These results provide clear evidence that the addition of –NH2 groups promoted cleavage of the O–H bonds in the FA.
To better understand the role of interfacial CrOx modification in catalyzing the FA dehydrogenation, potential energy profiles were generated using DFT calculations employing Pd(111), PdAg(111) and PdAg(111) slab models together with a Cr2O3 cluster in the absence of amine molecules (Fig. 6B). The calculated activation energies (Ea) are summarized in Table 2. The trans-M(O)–M(H–O)-bridged configuration (I, M = Pd or Ag) was determined to be the most energy favorable HCOOH adsorption structure. In the case of PdAg(111) and PdAg(111) models associated with a Cr2O3 cluster, the adsorbed molecule undergoes O–H bond dissociation and forms HCOO− as an intermediate (II) via transition state TSI/II with an energy barrier of 0.05 eV (Step 1). Subsequently, this species is isomerized to a trans-M(H)–Pd(O)-bridged HCOOH configuration (III) via transition state TSII/III with an energy barrier of 0.53 eV (Step 2). Following this, C–H bond scission occurs via TSIII/IV to form CO2 and a Pd–H species with an energy barrier of 0.56 eV (Step 3). Finally, the catalytic cycle is completed with the release of H2via TSIV/V with an energy barrier of 0.52 eV (Step 4).
| Model | Step 1 (I → II) | Step 2 (II → III) | Step 3 (III → IV) | Step 4 (IV → V) |
|---|---|---|---|---|
| O–H bond dissociation | Isomerization of formate | C–H bond dissociation | H2 desorption | |
| Pd(111) | 0.05 | 0.71 | 1.29 | 0.96 |
| PdAg(111) | 0.05 | 0.49 | 0.78 | 0.79 |
| PdAg(111) + Cr2O3 | 0.05 | 0.53 | 0.56 | 0.52 |
In the case of the Pd(111) and PdAg(111) models, the dissociation of the O–H bond occurs with a similar barrier to that obtained with the PdAg(111) model associated with a Cr2O3 cluster. In sharp contrast, the activation energies for Steps 2, 3 and 4 were determined to increase in the order of PdAg(111) + Cr2O3 < PdAg(111) < Pd(111). The lower energy barriers determined for the PdAg(111) model associated with a Cr2O3 cluster as compared with the Pd(111) and PdAg(111) models confirmed the positive effect of the modification with Cr2O3 in terms of boosting the FA dehydrogenation reaction. Even though this effect is evident with regard to several reaction steps, the effect on C–H bond dissociation was most pronounced (Step 3). This was also determined to be the rate-determining step in the case of pristine Pd(111).
These results agree with the data obtained from a KIE analysis performed using HCOOH and DCOOH. The kH/kD ratio found for PdAgCr/amine-MSC was 1.03, which was smaller than those observed for the monometallic Pd (2.20) and binary PdAg samples (1.37) (Table 3, entries 5–7). These results are consistent with the order of catalytic activity and indicate that the PdAg and PdAgCr catalysts facilitated C–H bond dissociation of the metal-formate intermediate. One possible reason for the improved catalytic activity of the binary PdAg catalyst could be the formation of an electron-rich Pdδ− species based on energy transfer from Ag to Pd atoms, as confirmed by the XPS analysis (Fig. 2B) and DOS calculations (Fig. 4). In this system, the metal-formate intermediate generated over electron-rich Pdδ− species was stabilized by significant back-donation from the metal to the formate that, in turn, facilitated selective C–H bond dissociation via a bidentate configuration.27 The enhancement of the rate-determining C–H bond dissociation step in the case of the PdAgCr catalyst can be explained by considering the electronic state of reaction intermediate III in Fig. 6A. The DFT-optimized configurations are summarized in Fig. 7. In this scenario, the H atom of HCOO− absorbed at an electron-deficient Agδ+ site over PdAg(111) interacting with a Cr2O3 cluster is more positively charged because of the reduced distance of 2.37 Å compared with the values for Pd(111) (3.29 Å) and PdAg (111) (2.42 Å). Accordingly, the electronic charge of the H atom becomes negative, accompanied by elongation of the C–H bond, which is then more likely to undergo dissociation.
| Entry | Catalyst | Formic acid | k H/kD |
|---|---|---|---|
| 1 | Pd/amine-MSC | HCOOH | 1.14 |
| HCOOD | |||
| 2 | PdAg/amine-MSC | HCOOH | 1.07 |
| HCOOD | |||
| 3 | PdAgCr/amine-MSC | HCOOH | 1.00 |
| HCOOD | |||
| 4 | PdAgCr/MSC | HCOOH | 1.67 |
| HCOOD | |||
| 5 | Pd/amine-MSC | HCOOH | 2.20 |
| DCOOH | |||
| 6 | PdAg/amine-MSC | HCOOH | 1.37 |
| DCOOH | |||
| 7 | PdAgCr/amine-MSC | HCOOH | 1.03 |
| DCOOH |
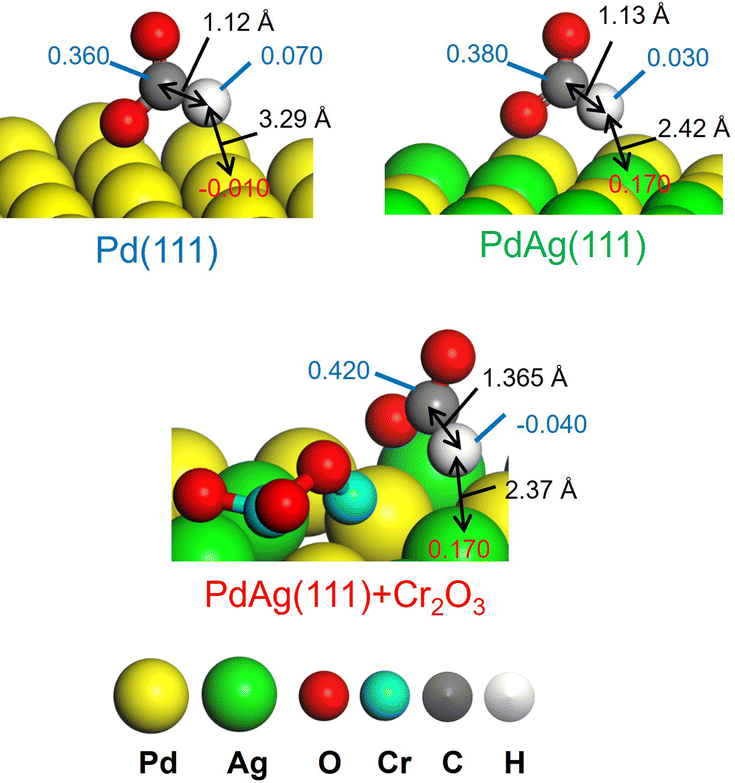 | ||
| Fig. 7 Representative Mulliken atomic charges as determined by DFT calculations in reaction intermediate III for Pd(111), PdAg(111), and PdAg(111) associated with the Cr2O3 cluster. | ||
The synergistic effect involved in promoting the final H2 desorption step was also assessed (Step 4). Reactions performed with a flow of H2 and D2 through the catalyst indicated that the formation rate for HD increased in the order of monometallic Pd < binary PdAg < ternary PdAgCr (Fig. S12, ESI†), in agreement with the lower reaction barriers for the H2 desorption step calculated using DFT (Table 2). It has been suggested that the energy of an adsorbed atom is related to the intrinsic electronic properties of the metallic particles on the catalyst. Specifically, a greater downward shift of the d-band center from the Fermi level of a metal increases the possibility that anti-bonding states will be formed, leading to weaker binding of the adsorbed atom.60 Thus, the binary PdAg catalyst exhibited a lower activation energy for H2 desorption than the monometallic Pd catalyst because the d-band center of PdAg(111) is deeper than that of Pd(111) (Fig. 4B). The participation of multiple interactions induced by the modification with CrOx in this process was also apparent. The dissociated hydride species at electron-rich Pdδ− sites were determined to be negatively charged, while those at electron-deficient Agδ+ sites were found to be positive. These hydrides having different charges generated at sites provided by CrOx modification are believed to have increased the production of hydrogen molecules.
3. Conclusions
This work increased the activity of a binary PdAg catalyst during dehydrogenation of FA based on interfacial modification with a highly dispersed amorphous CrOx phase. The combined effects of the weakly basic amine groups on the material surface and the synergistic catalysis provided by PdAgCr NPs explain the improved activity of this catalyst. The basic amine groups on the surface of the support promoted the dissociation of O–H bonds in FA molecules, while the CrOx phase enhanced the rate-determining C–H bond dissociation step of the metal-formate intermediate as well as the H2 desorption step. These effects resulted from the formation of unique ensemble sites involving adjacent electron-deficient Agδ+ and electron-rich Pdδ− atoms. These findings provide vital information that will assist in the future design of catalytically active centers for dehydrogenation of FA. They also emphasize the importance of interface design for further improvement of catalytic performance as a means of realizing environmentally-friendly hydrogen release systems.4. Experimental section
4.1. Materials
Acetone, nitric acid (HNO3), p-phenylenediamine, AgNO3, Cr(NO3)3·9H2O, sodium hydroxide (NaOH), sodium borohydride (NaBH4), formic acid (FA) and sodium formate (SF) were purchased from Nakalai Tesque, Inc. Pd(II) nitrate solution was obtained from Wako Pure Chemical Ind. Co., Ltd. Mesoporous carbon (MSC, average pore diameter: 100 ± 10 Å) was obtained from Aldrich Chemical Co. All commercially available compounds were used as received. Distilled water was employed as the reaction solvent.4.2. Synthesis of amine functionalized MSC (amine-MSC)
The detailed procedure is described elsewhere.46 Briefly, the MSC (1.0 g) was mixed with aqueous nitric acid solution (60%, 50 mL) at 80 °C for 5 h. The sample was collected by filtration, washed with distilled water, and dried under vacuum overnight. The acid treated MSC (1.0 g) was added into 500 mL of distilled water and sonicated for 2 h. The aqueous dispersion solution was mixed with acetone (50 mL) containing p-phenylenediamine (PDA, 3.24 g) and stirred at room temperature for 48 h. The resultant sample was separated by filtration and washed with acetone and distilled water until no color was confirmed in the filtrate, followed by drying under vacuum overnight.4.3. Synthesis of the PdAgCr supported amine-MSC catalyst (PdAgCr/amine-MSC)
The amine-MSC (0.3 g) was mixed with an aqueous solution (80 mL) containing Pd(NO3)2 (0.606 mL, Pd 47 mM), AgNO3 (2.85 mL, 10 mM), and Cr(NO3)3·9H2O (1.425 mL, 10 mM). After mixing, NaOH aqueous solution (4.2 ml, 1M) was added to adjust the pH around 10 and stirred for 30 min. Subsequently, the sample was reduced by NaBH4 under an Ar atmosphere, collected by filtration, and dried under vacuum overnight, giving PdAgCr/amine-MSC (Pd 1 wt%, Pd:Ag:Cr = 1:1:0.5). Monometallic Pd, binary PdAg, and ternary PdAgM (M = Mn, Co, Ga, Ni, Zn, Fe, and Cu) supported on amine-MSC catalysts were also prepared by the same method. Metal loadings were determined using inductively coupled plasma (ICP) analysis.
4.4. Characterization
Powder XRD patterns, BET surface area, ICM-ES, XPS spectra, and TEM images were acquired according to the procedure described previously.46 Quantification of the loading amount of amine functionality in samples was performed by two methods. One is the CHN elemental analysis using a Micro Corder JM-10 (J-Science Lab Co., Ltd). Another is thermogravimetric (TG) analysis using a Rigaku Thermo Plus EVO2 TG8121 system from room temperature to 700 °C at a heating rate of 10 °C min−1 in air flow (10 mL min−1). STEM images and elemental maps were obtained using a JEOL-ARM 200F apparatus equipped with a Kvex energy-dispersive X-ray detector (JED-2300T) operating at 200 kV. Pd, Ag, and Cr K-edge XAFS spectra were recorded using a fluorescence yield collection technique with a Si(111) monochromator at the 01B1 beamline station in the SPring-8 facility, JASRI, Harima, Japan (proposal no. 2020A0523 and 2020A1062). Data reduction was performed using the REX2000 software program (Rigaku). To obtain radial structure functions, Fourier transformation of the k3-weighted normalized EXAFS data was carried out over the range 3.0 Å < k/Å−1 < 12 Å. The H2–D2 exchange reaction was monitored using a BELCAT II system (MicrotracBEL Corp.). After pretreatment under He flow at 323 K for 30 min, the H2–D2 exchange reaction was performed at 323 K using 25%H2/He and 25%D2/He at a flow rate of 25 mL min−1. The products (H2, HD and D2) were analyzed with an online mass spectrometer. The rate constant was determined from the produced HD amount.4.5. DFT calculations
All DFT calculations were performed with the CASTEP program in the Materials Studio 17.2 software package.61,62 The generalized gradient approximation exchange–correlation functional proposed by Perdew, Burke, and Ernzerhof was combined with the double numerical basis set plus polarization functions. A supercell slab consisting of a surface 5 × 5 unit cell with three atomic (111) surface layers was adopted. For the calculation, periodic boundary conditions were applied to the slabs. The slab was separated by a vacuum space with a height of 20 Å. The reactant atoms and their adjacent atoms were relaxed during geometry optimizations, and the other layers were fixed at the corresponding bulk positions. Transition states (TSs) were determined using the nudged elastic band method, and the activation energy was defined by the energy difference between the TS and reactant.4.6. General procedure for the catalytic dehydrogenation of formic acid
The catalyst was placed in a reaction vessel (30 mL) equipped with a reflux condenser and a gas burette. After purging with N2, FA:SF = 9:1 aqueous solution (1 M, 10 mL) was added to the reaction vessel and reacted at 75 °C with magnetic stirring. TOF values [h−1] were determined according to the equation:| TOF = PatmVH2/RTNPdt |
4.7. General procedure for the catalytic hydrogenation of CO2
CO2 hydrogenation to FA was conducted with a batch reactor system (stainless steel autoclave (60 mL)) using a catalyst (50 mg) and 1.0 M aqueous NaHCO3 solution (10 mL) under a total pressure of 2.0 MPa (H2:CO2 = 1:1, volume ratio) at 100 °C. After 24 h, disodium succinate (external standard) was added and the yield of formic acid was determined by HPLC using a Shimadzu HPLC instrument equipped with a Bio-Rad Aminex HPX-87H Ion Exclusion Column (300 mm × 7.8 mm). 5 mm H2SO4 (0.500 mL min−1) was used as a mobile phase. TON values were determined by dividing the quantity of FA produced after 24 h by the moles of total Pd.
Author contributions
K. M. supervised the project, performed all the calculations and wrote the manuscript. T. F. performed the catalyst preparation, characterization, and catalytic reactions. H. Y. helped supervise the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present work was financially supported by the Iwatani Naoji Foundation. Part of this work was also supported by the TOYOTA Mobility Foundation (TMF) and Kakenhi Grant-in-Aid for Transformative Research Areas (B) (No. 21B206). A part of TEM experiment was carried out by using a facility in the Research Center for Ultra-High Voltage Electron Microscopy, Osaka University. The synchrotron radiation experiments for XAFS measurements were performed at the BL01B1 beamline in SPring-8 with the approval from JASRI (2021B1096 and 2022A1076).References
- M. A. Rosen and S. Koohi-Fayegh, Energy Ecol. Environ., 2016, 1, 10–29 CrossRef.
- J. O. M. Bockris, Science, 1972, 176, 1323 CrossRef CAS PubMed.
- S. Park, J. M. Vohs and R. J. Gorte, Nature, 2000, 404, 265–267 CrossRef CAS PubMed.
- R. von Helmolt and U. Eberle, J. Power Sources, 2007, 165, 833–843 CrossRef CAS.
- M. Felderhoff, C. Weidenthaler, R. von Helmolt and U. Eberle, Phys. Chem. Chem. Phys., 2007, 9, 2643–2653 RSC.
- P. T. Aakko-Saksa, C. Cook, J. Kiviaho and T. Repo, J. Power Sources, 2018, 396, 803–823 CrossRef CAS.
- P. M. Modisha, C. N. M. Ouma, R. Garidzirai, P. Wasserscheid and D. Bessarabov, Energy Fuels, 2019, 33, 2778–2796 CrossRef CAS.
- G. Sievi, D. Geburtig, T. Skeledzic, A. Bösmann, P. Preuster, O. Brummel, F. Waidhas, M. A. Montero, P. Khanipour, I. Katsounaros, J. Libuda, K. J. J. Mayrhofer and P. Wasserscheid, Energy Environ. Sci., 2019, 12, 2305–2314 RSC.
- L. Chen, P. Verma, K. Hou, Z. Qi, S. Zhang, Y.-S. Liu, J. Guo, V. Stavila, M. D. Allendorf, L. Zheng, M. Salmeron, D. Prendergast, G. A. Somorjai and J. Su, Nat. Commun., 2022, 13, 1092 CrossRef CAS PubMed.
- Y. Okada, E. Sasaki, E. Watanabe, S. Hyodo and H. Nishijima, Int. J. Hydrogen Energy, 2006, 31, 1348–1356 CrossRef CAS.
- E. Gianotti, M. Taillades-Jacquin, J. Rozière and D. J. Jones, ACS Catal., 2018, 8, 4660–4680 CrossRef CAS.
- T. W. Kim, H. Jeong, J. H. Baik and Y.-W. Suh, Chem. Lett., 2022, 51, 239–255 CrossRef CAS.
- J. Oh, K. Jeong, T. W. Kim, H. Kwon, J. W. Han, J. H. Park and Y.-W. Suh, ChemSusChem, 2018, 11, 661–665 CrossRef CAS PubMed.
- J. Eppinger and K.-W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC.
- A. K. Singh, S. Singh and A. Kumar, Catal. Sci. Technol., 2016, 6, 12–40 RSC.
- B. Wang, S. Yang, Z. Yu, T. Zhang and S. Liu, Mater. Today Commun., 2022, 31, 103617 CrossRef CAS.
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Chem. Soc. Rev., 2016, 45, 3954–3988 RSC.
- E. Doustkhah, M. Hasani, Y. Ide and M. H. N. Assadi, ACS Appl. Nano Mater., 2020, 3, 22–43 CrossRef CAS.
- N. Onishi, M. Iguchi, X. Yang, R. Kanega, H. Kawanami, Q. Xu and Y. Himeda, Adv. Energy Mater., 2019, 9, 1801275 CrossRef.
- R. Xu, W. Lu, S. Toan, Z. Zhou, C. K. Russell, Z. Sun and Z. Sun, J. Mater. Chem. A, 2021, 9, 24241–24260 RSC.
- M. Younas, M. Rezakazemi, M. S. Arbab, J. Shah and W. U. Rehman, Int. J. Hydrogen Energy, 2022, 47, 11694–11724 CrossRef CAS.
- C. Guan, Y. Pan, T. Zhang, M. J. Ajitha and K.-W. Huang, Chem. – Asian J., 2020, 15, 937–946 CrossRef CAS PubMed.
- M. Ojeda and E. Iglesia, Angew. Chem., Int. Ed., 2009, 48, 4800–4803 CrossRef CAS PubMed.
- Z.-L. Wang, J.-M. Yan, Y. Ping, H.-L. Wang, W.-T. Zheng and Q. Jiang, Angew. Chem., Int. Ed., 2013, 52, 4406–4409 CrossRef CAS PubMed.
- Y.-L. Qin, J. Wang, F.-Z. Meng, L.-M. Wang and X.-B. Zhang, Chem. Commun., 2013, 49, 10028–10030 RSC.
- K. Tedsree, T. Li, S. Jones, C. W. A. Chan, K. M. K. Yu, P. A. J. Bagot, E. A. Marquis, G. D. W. Smith and S. C. E. Tsang, Nat. Nanotechnol., 2011, 6, 302–307 CrossRef CAS PubMed.
- W.-Y. Yu, G. M. Mullen, D. W. Flaherty and C. B. Mullins, J. Am. Chem. Soc., 2014, 136, 11070–11078 CrossRef CAS PubMed.
- K. Mori, M. Dojo and H. Yamashita, ACS Catal., 2013, 3, 1114–1119 CrossRef CAS.
- M. Wen, K. Mori, Y. Kuwahara and H. Yamashita, ACS Energy Lett., 2017, 2, 1–7 CrossRef CAS.
- K. Mori, H. Tanaka, M. Dojo, K. Yoshizawa and H. Yamashita, Chem. – Eur. J., 2015, 21, 12085–12092 CrossRef CAS PubMed.
- J. Li, W. Chen, H. Zhao, X. Zheng, L. Wu, H. Pan, J. Zhu, Y. Chen and J. Lu, J. Catal., 2017, 352, 371–381 CrossRef CAS.
- M. Navlani-García, K. Mori, A. Nozaki, Y. Kuwahara and H. Yamashita, ChemistrySelect, 2016, 1, 1879–1886 CrossRef.
- W. Wang, T. He, X. Liu, W. He, H. Cong, Y. Shen, L. Yan, X. Zhang, J. Zhang and X. Zhou, ACS Appl. Mater. Interfaces, 2016, 8, 20839–20848 CrossRef CAS PubMed.
- N. Wang, Q. Sun, R. Bai, X. Li, G. Guo and J. Yu, J. Am. Chem. Soc., 2016, 138, 7484–7487 CrossRef CAS PubMed.
- M. Navlani-García, K. Mori, Y. Kuwahara and H. Yamashita, NPG Asia Mater., 2018, 10, 277–292 CrossRef.
- S.-T. Gao, W. Liu, C. Feng, N.-Z. Shang and C. Wang, Catal. Sci. Tech., 2016, 6, 869–874 RSC.
- M. Martis, K. Mori, K. Fujiwara, W.-S. Ahn and H. Yamashita, J. Phys. Chem. C, 2013, 117, 22805–22810 CrossRef CAS.
- J. Cheng, X. Gu, P. Liu, T. Wang and H. Su, J. Mater. Chem. A, 2016, 4, 16645–16652 RSC.
- M. Yadav, T. Akita, N. Tsumori and Q. Xu, J. Mater. Chem., 2012, 22, 12582–12586 RSC.
- Y.-J. Chou, H.-C. Ku, C.-C. Chien, C. Hu and W.-Y. Yu, Catal. Sci. Technol., 2020, 10, 7883–7893 RSC.
- Z.-L. Wang, J.-M. Yan, H.-L. Wang, Y. Ping and Q. Jiang, Sci. Rep., 2012, 2, 598 CrossRef PubMed.
- Z.-L. Wang, H.-L. Wang, J.-M. Yan, Y. Ping, O. Song-II, S.-J. Li and Q. Jiang, Chem. Commun., 2014, 50, 2732–2734 RSC.
- K. Mori, K. Naka, S. Masuda, K. Miyawaki and H. Yamashita, ChemCatChem, 2017, 9, 3456–3462 CrossRef CAS.
- T. Shishido, K. Shimamura, K. Teramura and T. Tanaka, Catal. Today, 2012, 185, 151–156 CrossRef CAS.
- S. Masuda, K. Mori, Y. Futamura and H. Yamashita, ACS Catal., 2018, 8, 2277–2285 CrossRef CAS.
- J.-R. Ares-Fernández and K.-F. Aguey-Zinsou, Catalysts, 2012, 2, 330–343 CrossRef.
- G. A. Deluga, J. R. Salge, L. D. Schmidt and X. E. Verykios, Science, 2004, 303, 993–997 CrossRef CAS PubMed.
- A. Zhang, J. Xia, Q. Yao and Z.-H. Lu, Appl. Catal., B, 2022, 309, 121278 CrossRef CAS.
- M. Navlani-García, D. Salinas-Torres, F. D. Vázquez-Álvarez and D. Cazorla-Amorós, Catal. Today, 2022, 397–399, 428–435 CrossRef.
- W. Zou, Y. Liu, C. Song, H. Lin, H. Huang, W. Ye, R. Lu and S. Zhang, Int. J. Hydrogen Energy, 2022, 47, 28518–28529 CrossRef CAS.
- M. Deng, J. Ma, C. Yang, T. Cao, M. Yao, F. Liu, H. Chen and X. Wang, Mater. Today Chem., 2022, 24, 101001 CrossRef CAS.
- Y. Cui, M. Zhao, Y. Zou, J. Zhang, J. Han, Z. Wang and Q. Jiang, J. Energy Chem., 2022, 68, 556–563 CrossRef CAS.
- Q.-L. Zhu, F.-Z. Song, Q.-J. Wang, N. Tsumori, Y. Himeda, T. Autrey and Q. Xu, J. Mater. Chem. A, 2018, 6, 5544–5549 RSC.
- K. Mori, H. Hata and H. Yamashita, Appl. Catal., B, 2023, 320, 122022 CrossRef CAS.
- F.-Z. Song, Q.-L. Zhu, N. Tsumori and Q. Xu, ACS Catal., 2015, 5, 5141–5144 CrossRef CAS.
- Q.-Y. Bi, X.-L. Du, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, J. Am. Chem. Soc., 2012, 134, 8926–8933 CrossRef CAS PubMed.
- K. Mori, Y. Futamura, S. Masuda, H. Kobayashi and H. Yamashita, Nat. Commun., 2019, 10, 4094 CrossRef PubMed.
- K. Mori, S. Masuda, H. Tanaka, K. Yoshizawa, M. Che and H. Yamashita, Chem. Commun., 2017, 53, 4677 RSC.
- B. Hammer and J. K. Norskov, Nature, 1995, 376, 238–240 CrossRef CAS.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045–1097 CrossRef CAS.
- V. Milman, B. Winkler, J. A. White, C. J. Pickard, M. C. Payne, E. V. Akhmatskaya and R. H. Nobes, Int. J. Quantum Chem., 2000, 77, 895–910 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ey00049k |
| This journal is © The Royal Society of Chemistry 2023 |