DOI:
10.1039/D2EY00029F
(Paper)
EES Catal., 2023,
1, 36-44
Pit-embellished low-valent metal active sites customize CO2 photoreduction to methanol†
Received
26th August 2022
, Accepted 23rd October 2022
First published on 11th November 2022
Abstract
Customizing catalytic reaction pathways by precisely designing the metal active sites and electron–hole separated channels of metal oxides to simultaneously achieve a high yield and selectivity of photocatalytic CO2 reduction to liquid fuel remains a challenge. Herein, we for the first time propose that low-valent tungsten sites favor the formation of key CHO* intermediates for highly selective photocatalytic reduction of CO2 to CH3OH. In situ spectroscopic results and DFT calculations demonstrate that coordinately unsaturated low-valent W sites near the tungsten trioxide (denoted WO3−x) pits serve as catalytic sites and electron capture sites enabling the adsorbed CO2 to selectively form a predominant lower-energy *CHO intermediate instead of *CO, thereby triggering a unique reaction pathway for CO2 reduction to CH3OH. Accordingly, the optimal WO3−x delivers a notable CH3OH selectivity of up to 86% with a high evolution rate of 17 μmol g−1 h−1 under sunlight irradiation. This work highlights how low-valent metal active sites in the surface pits can be controlled at the atomic-level to customize the CO2 reduction reaction (CO2RR) pathway to generate valuable liquid fuels.
Broader context
Infinite sunlight-driven CO2 conversion into value-added alternative fuels under mild conditions has emerged as a promising technology to achieve carbon neutrality and ameliorate future energy feedstock supplies. Metal oxides are very promising and efficient catalysts for the photochemical CO2 reduction to CH3OH in practical catalytic systems. However, the rapid electron–hole recombination rate and lack of intrinsic catalytic sites in photocatalysts still limit their photocatalytic performance. Therefore, developing a suitable photocatalyst with selective active sites for both the activation of stable CO2 molecules and the customization of CO2 reduction products is urgently needed but it remains a challenge. In this tandem experimental–computational work, we propose that the formation of CHO* intermediates at low-valent W sites in the surface pits is the key factor determining selectivity. Furthermore, we demonstrate that the unsaturated W atoms on the WO3−x surface pits are the actual catalytic sites; meanwhile, exposing the low-valent W active site promotes the formation of the *CHO intermediates, thereby facilitating CH3OH generation. This work highlights product selectivity changes induced by precisely designing the electronic structure of the catalyst surface, while providing deep mechanistic insights into customizing CO2 reduction to liquid solar fuels.
|
Introduction
Converting carbon dioxide (CO2) to solar fuels via naturally inexhaustible solar energy is a sustainable strategy to simultaneously ameliorate energy feedstock supplies and climate change.1–4 It is known that the CO2 reduction reaction (CO2RR) process is quite complex and ambiguous, containing the necessary protonation coupled electron transfer to generate various deoxygenated products.5–7 Among the possible CO2RR derivations, the 6H+/6e− reduction of CO2 towards CH3OH, an appealing platform molecule and liquid commodity chemical that can be easily stored and handled, exhibits great potential and economic feasibility for application in industry and organic synthesis, namely ‘methanol economy’.8 In terms of the CO2RR process, the metal (M) sites on the photocatalyst surface preferably bond with the C or O atoms in the adsorbed CO2 through orbital hybridization, resulting in a series of reactive intermediates with the M–C or M–O bond.9–13 In this regard, the catalyst-reactive intermediate interaction is the key factor determining the performance of photocatalysts in practical catalytic systems. To date, notwithstanding that the most research efforts have shown that semiconductor photocatalysts can generate dominantly CH4 or CO products with high activity or selectivity during the CO2RR, only a few reports were on liquid CH3OH production under comparable conditions.14,15 More unfortunately, both the selectivity and yield of photocatalytic CO2-to-CH3OH reduction remain extremely unsatisfactory due to the high thermodynamic stability of linear CO2 molecules, rapid recombination kinetics of photogenerated carriers4,16,17 and multi-electron side reactions with similar reduction potentials.18 As a promising approach to addressing these issues, developing a suitable photocatalyst with both high activity and selectivity for CO2 reduction is urgently desired, while it remains a great challenge.
Given that metal oxides are very promising and efficient catalysts, they have been extensively studied for photocatalytic reduction of CO2 to CH3OH.19 As a classical metal oxide photocatalyst, tungsten trioxide (WO3) seems to be one of the most appropriate candidates to achieve the above goal due to its intriguing advantages such as its beneficial band gap (∼2.6 eV), suitable bandgap edge, strong light harvesting ability, high chemical stability, and excellent photoactivity.13,20,21 Meanwhile, theoretical calculations further indicate that WO3 has good CO2 adsorption and activation abilities, which is the premise of the subsequent reduction reaction.22,23 However, the rapid electron–hole recombination rate and lack of intrinsic catalytic sites in WO3 still limit the full utilization of photogenerated charge carriers to initiate the highly selective reduction of CO2 to CH3OH. As we all know, constructing surface defects on photocatalysts is an effective strategy to introduce coordinatively unsaturated and catalytically active metal sites and regulate electronic structures.24,25 This not only promotes local electron delocalization at unsaturated metal sites on the catalyst surface, but also modulates the redox capability of photogenerated charge carriers, activating adsorbed CO2 molecules and optimizing the reactive intermediates’ energy barrier on sites, thereby altering the product selectivity of CO2 reduction.26–28 Additionally, to further improve the activity of the catalyst, rational morphology design and precise electron–hole separated channel construction for WO3 have also offered an opportunity to enable ultra-high specific surface area and expose more accessible active sites, which is beneficial to improving light harvesting while boosting charge carrier separation and migration in the photocatalytic process.29,30 Nevertheless, as far as we know, the design of highly efficient WO3 photocatalysts with selective active sites and electron–hole separated channels for customization of the CO2RR product is still in its infancy and has not been systematically established yet.
On the basis of the above-mentioned considerations, herein, we developed well-defined low-valent W sites to realize highly efficient electron–hole separation and CO2 activation at single-component WO3−x nanosheet surface pits, thus customizing the CO2RR pathway to generate CH3OH. The optimal WO3−x exhibits an impressive CH3OH evolution rate of 17 μmol g−1 h−1 with a high selectivity of up to 86% and appreciable stability for 5 consecutive runs. More importantly, the underlying mechanism is further revealed by experimental and computational results, showing that the unsaturated W atoms on the WO3−x surface pits are the actual catalytic sites for CO2 activation, and the *CO intermediates adsorbed at the active sites are likely to undergo a more rapid and low-energy-barrier protonation process to generate the key *CHO intermediates rather than desorption for CO generation, thereby leading to accelerated catalytic kinetics for CH3OH production from CO2 photoreduction.
Results and discussion
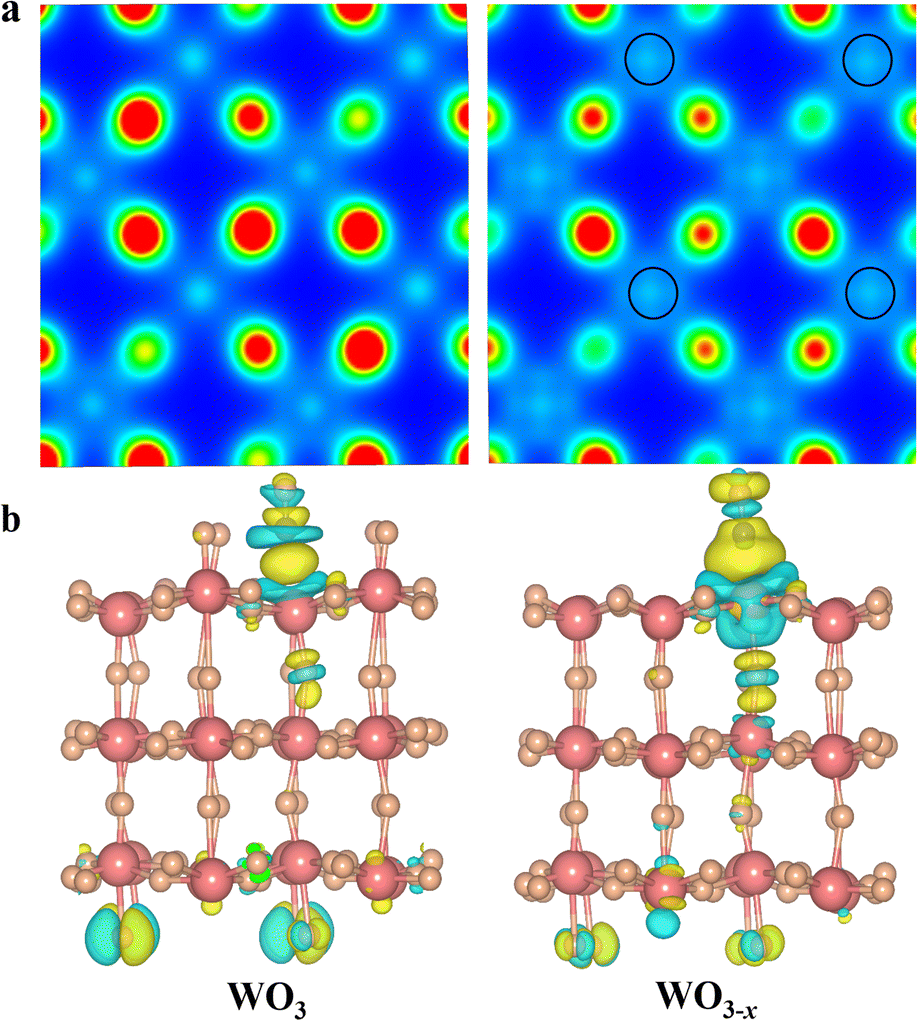
It is widely accepted that the introduction of oxygen vacancies or low-valent metal species could modulate the electronic band structure and create trapping sites to benefit separation of photoinduced charge carriers.31–33 Furthermore, the existence of defects could broaden the light harvesting range and more importantly regulate the local charge density distribution. Thus, we conducted density functional theory (DFT) to check the influence of low-valent W species on the electronic structure of WO3 and WO3−x (see Calculation details in the ESI†).34,35 As depicted in Fig. 1a, the Bader charge distribution of the W atom adjacent to the top oxygen vacancy is 2.79 electrons (2.79 e−) on an optimized WO3−x surface, which is between the charge of W6+ (2.97 e−) in a perfect WO3 and the charge of W4+ (2.27 e−) in WO2. This indicates that the localized electron would transfer to W atoms, resulting in a reduced valence state of around +5.36,37 Interestingly, the low-valent W species on the WO3−x surface exhibits a tremendously enhanced charge density, in which the electron distribution becomes more delocalized, and successively donates its d-orbital electrons to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O antibonding orbital (π*) of adsorbed CO2 molecules to form highly reactive intermediates. In fact, these intermediates are usually regarded as key factors in redefining the selectivity of the final reduction product.37 To further unveil the electron-transfer pathway of CO2 activation on optimized WO3−x surfaces, we applied the charge density difference to track their electron behaviours (Fig. 1b). We found that a crucial *CO intermediate was formed and accumulated on the low-valent WO3−x and perfect WO3 surface.
O antibonding orbital (π*) of adsorbed CO2 molecules to form highly reactive intermediates. In fact, these intermediates are usually regarded as key factors in redefining the selectivity of the final reduction product.37 To further unveil the electron-transfer pathway of CO2 activation on optimized WO3−x surfaces, we applied the charge density difference to track their electron behaviours (Fig. 1b). We found that a crucial *CO intermediate was formed and accumulated on the low-valent WO3−x and perfect WO3 surface.
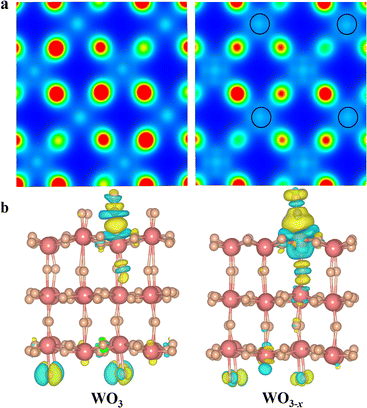 |
| | Fig. 1 (a) Distribution of charge density for the original WO3 (left) and WO3−x (right) plotted from 0 (blue) to 0.035 e− bohr−3 (red). (b) Charge difference diagrams of *CO adsorption on original WO3 (left) and WO3−x (right); blue and yellow surfaces represent charge depletion and accumulation, respectively. | |
In fact, the reaction energy change of this *CO intermediate is a key factor determining the reaction pathway.37,38 In sharp contrast to the perfect WO3 surface, there is a strong electron transfer (0.32 e− lost) from the low-valent W species on WO3−x to the *CO group, and the C–O bond strength is weakened. The weakened C–O bond in the *CO intermediate is stretched from 0.114 to 0.117 nm after introduction of the low-valent W species. This phenomenon indicates that low-valent W species can better stabilize the *CO intermediates and rapidly transfer more electrons to protonate them for production of lower-energy *CHO intermediates during CO2 photoreduction, thereby changing the reaction pathway to generate CH3OH instead of CO.
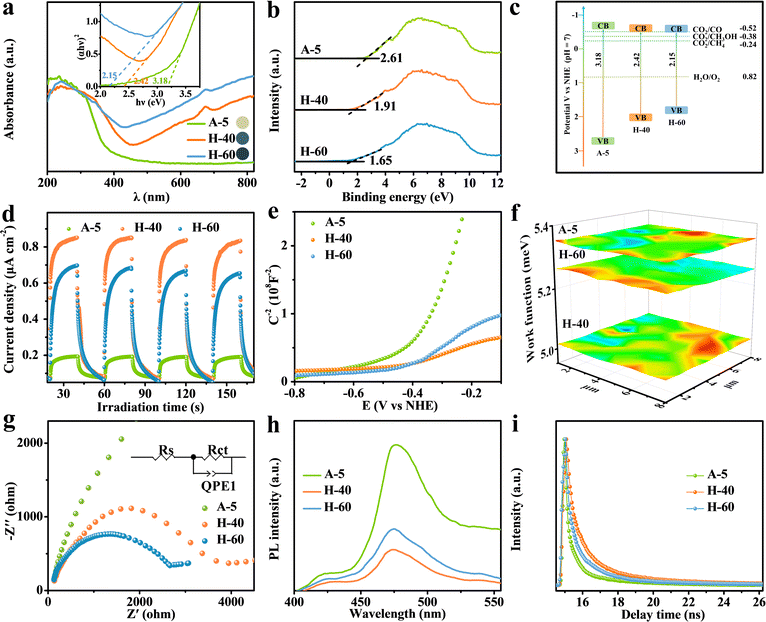
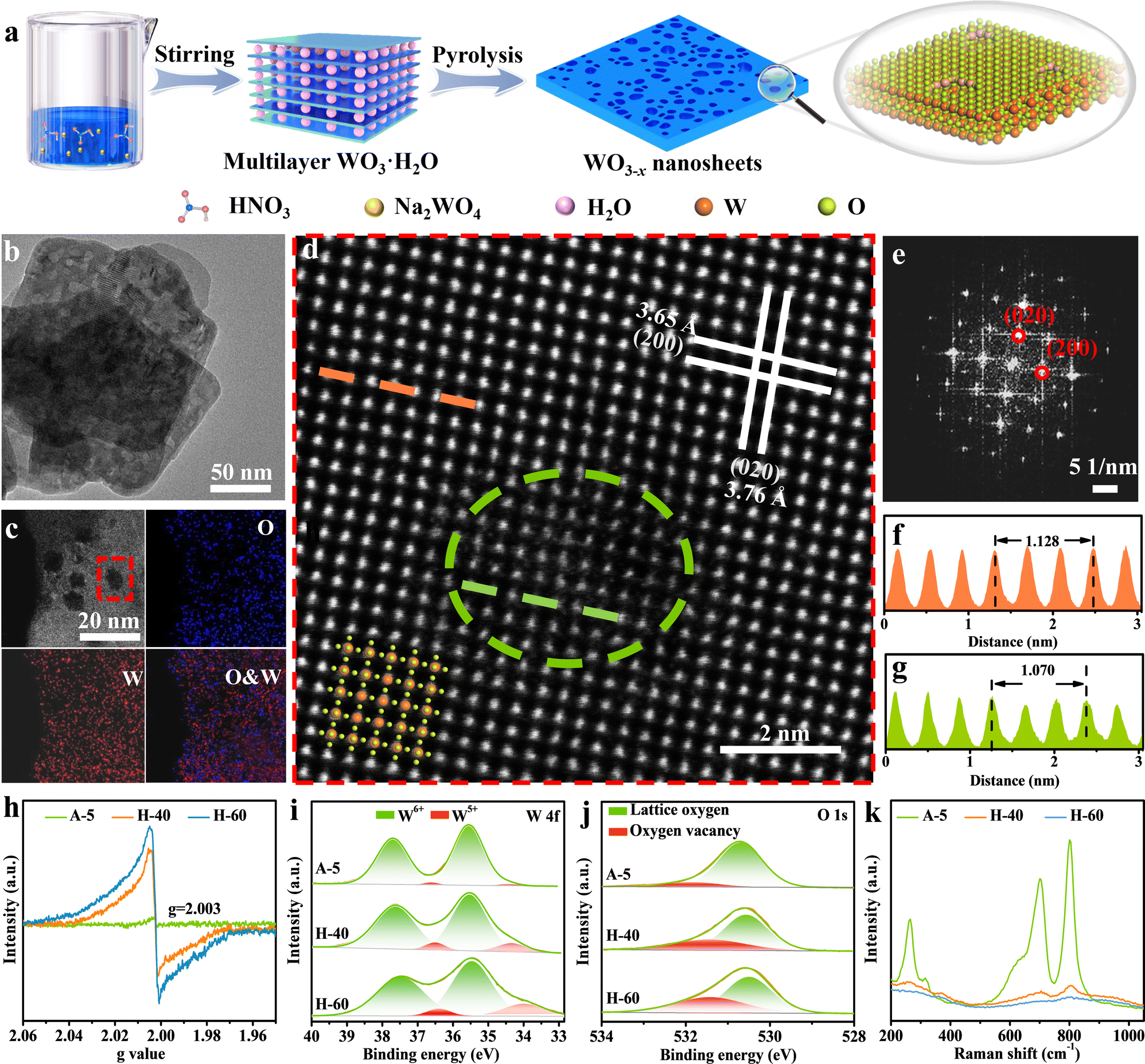
Inspired by the above analysis, we present that creating favourable atomic-level low-valent W species around the pits on the surfaces of WO3−x nanosheets can remarkably improve the performance of photocatalytic CO2 reduction. The preparation of WO3−x nanosheet catalysts via a two-step method is illustrated in Fig. 2a (see the Experimental section in the ESI† for more details). Initially, the WO3·H2O precursor was produced using a wet-chemistry approach by adding sodium tungstate dihydrate into nitric acid solution at a certain concentration. The multilayer WO3·H2O precursor shows a sheet-like morphology and a relatively smooth and flat surface confirmed by the powder X-ray diffraction (XRD) pattern and transmission electron microscopy (TEM) results in Fig. S1 (ESI†). The WO3−x nanosheets with surface pit-decoration and suitable low-valent W sites were subsequently synthesized by an atomic clipping engineering method (Fig. S2, ESI†). The density of low-valent W sites of the WO3−x nanosheets can be adjusted by changing the atmosphere (air or hydrogen) and annealing time. The model catalysts were obtained by calcining the WO3·H2O precursor at 450 °C for 40 and 60 min in hydrogen or calcined at 700 °C for 5 s in air (denoted H-40, H-60, and A-5), respectively. The XRD pattern shown in Fig. S3 (ESI†) for the obtained products could be indexed to the monoclinic tungsten oxide with a distorted ReO3− type unit (JCPDS No. 83-0950). As presented in the Fourier-transform infrared spectroscopy (FT-IR) spectra, the main peaks located at around 743, 811, and 966 cm−1 belong to the O–W–O and O–W stretching modes. This result is in good agreement with the XRD results that all as-prepared samples are the pure monoclinic phase of WO3 (Fig. S4, ESI†). The representative morphology of H-40 is presented in Fig. 2b–d, characterized by TEM and aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). H-40 inherits the two-dimensional sheet-like morphology from the WO3·H2O precursor and contains numerous highly monodisperse pits. We speculate that the formation of surface pits may be due to the loss of crystal water during pyrolysis. The HAADF-STEM and fast Fourier transform (FFT) images in Fig. 2d and e, respectively, confirm the single crystalline structure and the [001] orientation of H-40. The interplanar spacings of 0.376 and 0.365 nm for H-40 can be assigned to (020) and (200) planes, respectively. In addition, the local (020) interplanar spacing (0.357 nm) in the pits is also smaller than its standard value of monoclinic WO3 (0.376 nm), further demonstrating the existence of defects in H-40 (Fig. 2f and g). Elemental mappings (Fig. 2c) further confirm a uniform distribution of W and O elements across H-40, which also agrees well with the XRD results. To further confirm the existence of defective structures, a series of spectroelectrochemistry characterization studies were performed. As shown in Fig. 2h, an obvious electron paramagnetic resonance (EPR) signal at g = 2.003 is observed for A-5, H-40, and H-60, which indicates electrons trapped on metal W species. Furthermore, the stronger EPR signal of H-60 confirms the richer low-valent W species formed during material preparation. This result can be further verified by X-ray photoelectron spectroscopy (XPS). Apparently, the deconvolution of the characteristic W 4f band yields two pairs of peaks, corresponding to the W6+ (located at 37.6 and 35.5 eV) and W5+ (located at 36.5 and 34.3 eV) oxidation states, respectively. Notably, a slightly negative shift of W 4f binding energy and the higher proportion of the peaks at 36.5 and 34.3 eV in H-60, corroborating the existence of more low-valent W5+ (Fig. 2i and Fig. S5, Table S1, ESI†).39–41 This change also affects the electronic structure of oxygen atoms: the H-60 shows the strongest O 1s peak possibly adjacent to the W5+ at 531.4 eV (Fig. 2j).42–44 Furthermore, the Raman bands of H-60 with numerous defective low-valent W5+ sites show peak broadening and a redshift compared to the other counterparts, which may be attributed to phonon softening or enhanced electron–phonon coupling at low-valent W sites. All the above mentioned experimental results support the enrichment of low-valent W sites in H-60 (Fig. 2k).
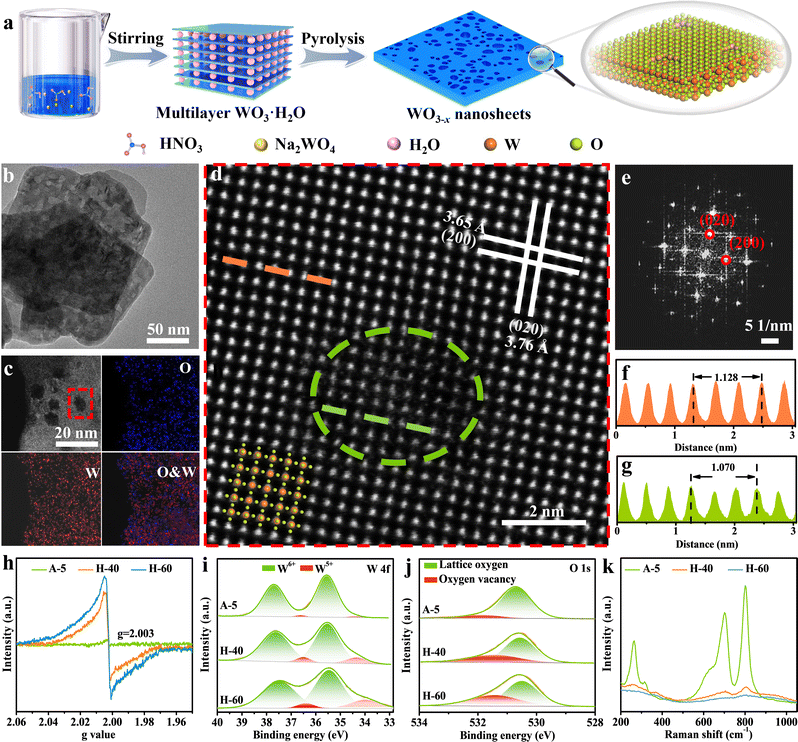 |
| | Fig. 2 (a) Schematic illustration of the synthetic process of 2D WO3−x nanosheets. Characterization of the pit-rich WO3−x nanosheets. (b) TEM image, (c) HAADF-STEM EDX elemental mappings, (d) atomic-resolution HAADF-STEM image of H-40, and (e) the corresponding fast Fourier transform (FFT) electron diffraction pattern along the [001] zone axis. (f and g) Intensity distribution measured from the HAADF-STEM image in the perfect crystal face (orange) and pits (green). (h) EPR spectra, (i) W 4f XPS spectra, (j) O 1s XPS spectra and (k) Raman spectra of A-5, H-40, and H-60. | |
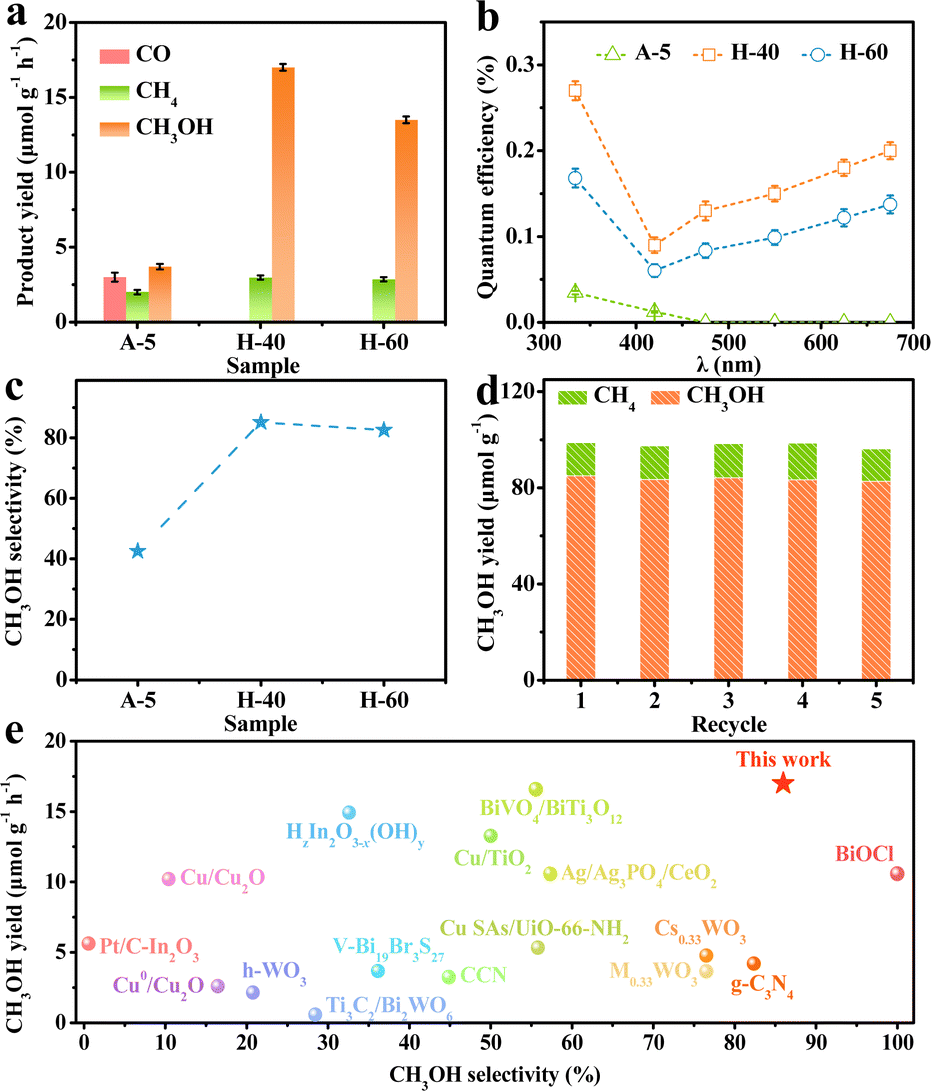
To unravel the influence of unsaturated low-valent W sites on the photocatalytic performance, we probe into the CO2RR properties of the obtained catalysts. The gaseous and liquid products were quantitatively evaluated by gas chromatography and H nuclear magnetic resonance spectroscopy (Fig. S6–S8, ESI†), respectively. As shown in Fig. 3a, the reduction products on the different obtained catalysts upon illumination are CO, CH4, and CH3OH, which gradually accumulated with increasing reaction time. Meanwhile, the 13CO2 isotope labelling experiment and a series of control experiments were operated in Ar or without the sample or in the dark to provide direct proof for the origin of the products (Fig. S9, ESI†), and the molecular ion peaks at m/z 17 and 33 were assigned, respectively, to 13CH4 and 13CH3OH when 13CO2 was used as the reactant, demonstrating that the carbon atoms of the obtained products stemmed from CO2 feedstock instead of any other impurities.45,46 Besides, the production of O2 in the control experiment verified that O2 stemmed from H2O (Fig. S10, ESI†). Notably, H-40 manifests the highest CH3OH production rate of up to 17 μmol g−1 h−1, which is about 4.6 times higher than that of the A-5 material. Furthermore, the quantum efficiency (QE) of the H-40 photocatalyst was measured to be as high as ∼0.2% under monochromatic visible light (675 nm) irradiation.47 The trend of the CH3OH yield agrees well with the photoabsorption of the obtained photocatalysts, demonstrating a high utilization of photogenerated charge carriers (Fig. 3b, 4a and Fig. S11, ESI†). More intriguingly, H-40 achieves the highest CH3OH selectivity of ∼86% among all materials (Fig. 3c). Surprisingly, both the CH3OH yield and selectivity of the achieved cheap H-40 surpass those of most previously reported state-of-the-art photocatalysts (Fig. 3e).38,48–60 To further evaluate its stability, H-40 was repeatedly used in the CO2RR for five cycles and the product yield was tested every 5 h. No obvious variations are observed during measurements, reflecting the high stability of H-40 (Fig. 3d and Fig. S12, ESI†). Besides, XRD, EPR, XPS and TEM measurements of the used H-40 after photocatalysis also support its high structural stability (Fig. S13, S14 and Table S2, ESI†). Clearly, the above results demonstrate that H-40 has high efficiency and selectivity for CO2 photoreduction, which raises the question: how the density of low-valent W sites in WO3−x thermodynamically and kinetically influences the photocatalytic activity of the CO2RR?
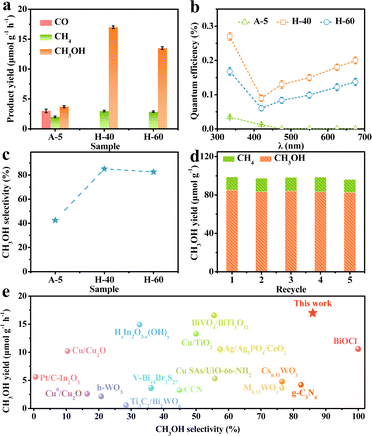 |
| | Fig. 3 (a) Product yields of the CO2RR using the different photocatalysts. (b) QE and (c) CH3OH selectivity values of different samples. (d) Photocatalytic stability of H-40 for five consecutive runs. (e) Comparison of CO2RR performance of WO3−x and other reported photocatalysts. | |
 |
| | Fig. 4 (a) UV-vis absorption spectra (inset: Tauc plots), (b) VB XPS spectra, (c) energy band structure alignments, (d) transient photocurrent responses (0.5 V vs. Ag/AgCl, pH 7), (e) Mott–Schottky plots, (f) SKP maps, (g) EIS spectra, (h) steady-state PL spectra, and (i) time-resolved transient PL decay of A-5, H-40, and H-60, respectively. | |
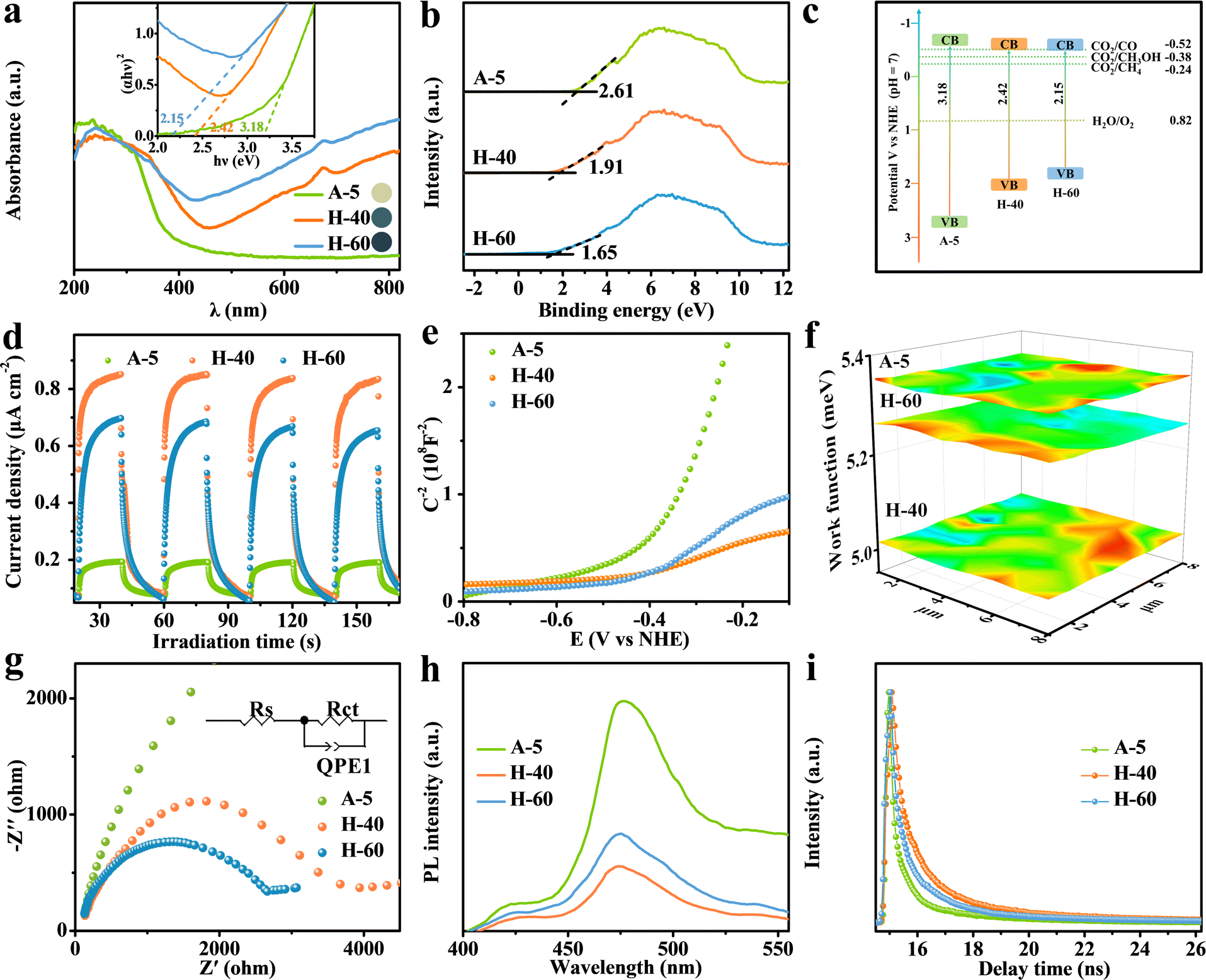
To answer this question, a series of photoelectrochemical characterization studies were conducted to elucidate the excellent CO2RR activity of the obtained photocatalysts. As shown in Fig. 4a, the existence of low-valent W5+ sites results in the formation of abundant internal transition energy levels, which could efficiently narrow the band gap to enhance the visible-light harvesting ability for photocatalytic reactions and tune the sample color range from yellow to dark blue (inset: sample color). The band gaps of A-5, H-40, and H-60 were detected to be 3.18, 2.42, and 2.15 eV, corresponding to their valence band (VB) potentials of 2.61, 1.91, and 1.65 eV (versus normal hydrogen electrode, vs. NHE) (Fig. 4a and b). Compared with the thermodynamic potential (CO2/CH3OH = −0.38 V), the calculated conduction band potential of H-40 is more negative, which is essential to facilitate electron transfer for CO2 conversion into CH3OH (Fig. 4c). Furthermore, the formation of pits on the surface significantly enlarged the surface area, which could provide more accessible catalytic sites to improve the catalytic activity. Notably, the numerous low-valent W sites in H-60 significantly improve the light absorption, while the excessive defect sites on the metal oxide surfaces simultaneously generate more photogenerated charge carrier recombination sites for various chemical reactions, resulting in a decrease in catalytic activity.61,62 Therefore, accurately achieving a suitable density of low-valent W active sites is vital to maximize the photocatalytic activity. This is further verified by the result of the transient photocurrent measurement shown in Fig. 4d, where the enhanced photocurrent of H-40 relative to the other counterparts reveals the promoted generation and separation of photogenerated charge carriers in H-40. The carrier density can be calculated according to the following formula:
| | | Nd = (2/e0εrε0)[d(1/C2)/dE]−1 | (1) |
where
e0 (1.602 × 10
−19 C) is the electron charge,
C is the capacitance of the space charge layer,
Nd is the carrier density,
εr (20 for WO
3) is the dielectric constant, and
ε0 (8.85 × 10
−14 F cm
−1) is the vacuum permittivity.
63 The
Nd value was calculated to be 3.32 × 10
19 cm
−3, approximately 7.8 times higher than the 0.4 × 10
19 cm
−3 for A-5 (
Fig. 4e and Table S5, ESI
†). In addition, the scanning Kelvin probe (SKP) technique was used to probe the work function to determine the electron separation efficiency of the as-prepared photocatalysts.
Fig. 4f shows that H-40 achieves the lowest work function value, causing a higher Fermi level of H-40 in comparison with the other counterparts. This means that the built-in electric field and surface energy band bending are enhanced, indicating that a suitable density of low-valent W
5+ sites is beneficial to improve the efficiency of charge separation. The electrochemical impedance spectroscopy (EIS) further supports the SKP results that H-40 has a lower interfacial charge-transfer resistance resulting from its superior electrical conductivity (
Fig. 4g). Meanwhile, the charge separation efficiencies of A-5, H-40, and H-60 nanosheets were characterized by steady-state photoluminescence (PL) emission spectroscopy; the weaker PL intensity of H-40 reveals efficient inhibition of charge recombination (
Fig. 4h). Furthermore, transient PL emission spectroscopy was employed to determine the specific charge carrier dynamics of the catalysts. The much slower electron decay kinetics of H-40 reveals a distinctly prolonged lifetime of the photogenerated charge carriers by introducing suitably reduced W
5+ sites on H-40 (
Fig. 4i and Table S4, ESI
†).
64–66 All these results provide solid evidence for the proposition that low-valent W
5+ sites on H-40 can efficiently promote the generation and separation of photoinduced charge carries.
Substantially, the reactant (H2O and CO2) adsorption and product desorption on the H-40 surface are crucial during the CO2RR because the activation and protonation of CO2 molecules are involved. As revealed by the N2 adsorption and desorption curves shown in Fig. S15 and S16a (ESI†), moderate pits decorated on H-40 enable a higher surface area, more low-valent W5+ sites, and more electron–hole separation channels, and stabilize more key active species to a certain extent, which can promote the adsorbate on its surface for subsequent surface redox reactions. As expected, H-40 with larger surface area exhibits higher CH3OH activity and selectivity. CO2 temperature-programmed desorption (CO2-TPD) shows a similar adsorption ability to the obtained samples (Fig. S16b, ESI†). The enhanced peaks of H-40 at higher temperatures indicate that the existence of suitable low-valent W5+ sites can dramatically promote the chemisorption ability of CO2. Moreover, this result is further reinforced by the contact angle measurements (Fig. S17, ESI†). Hence, all these results demonstrate that the introduced coordination unsaturated low-valent W5+ sites endow it with higher surface hydrophilicity and enhanced CO2 adsorption capacity.
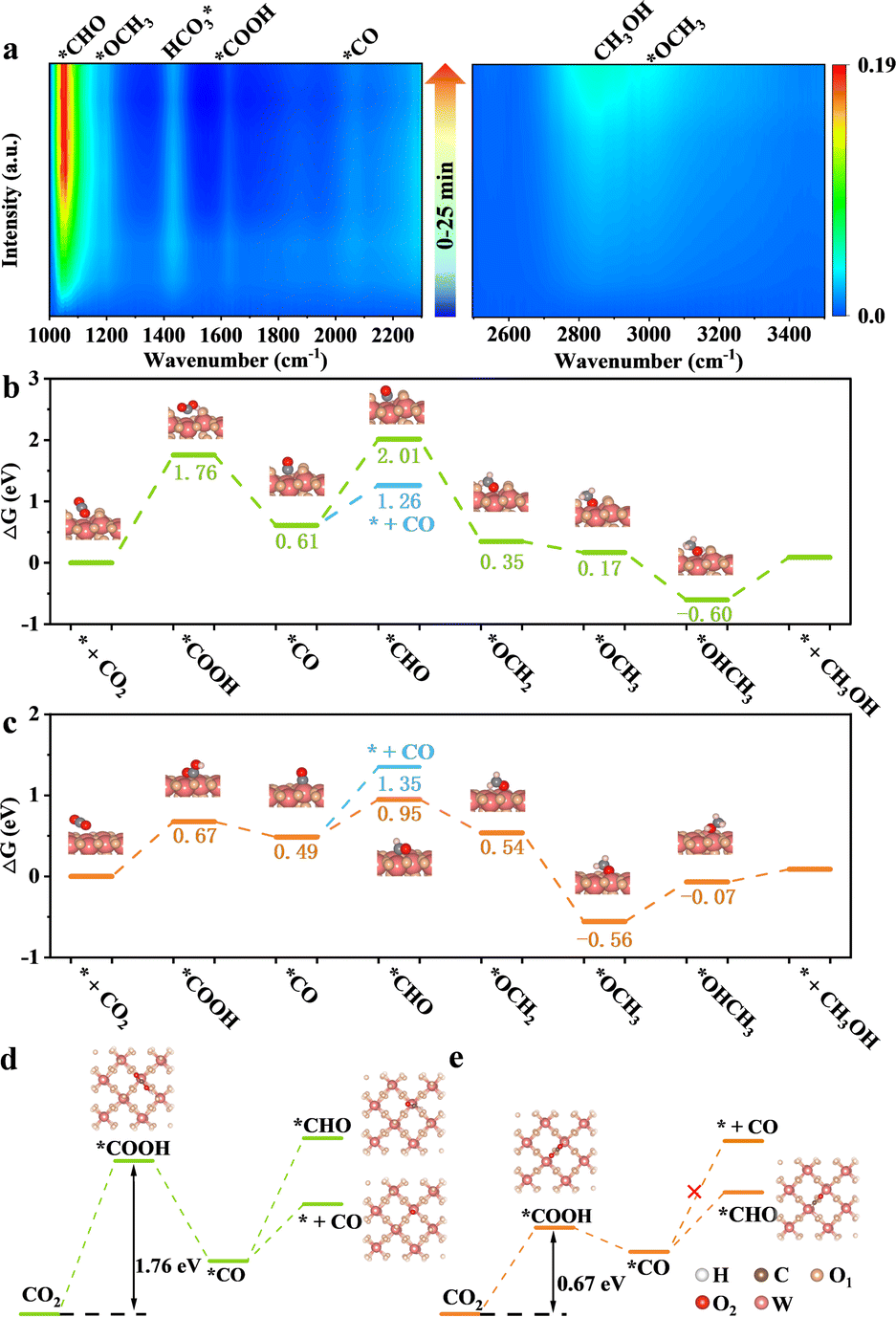
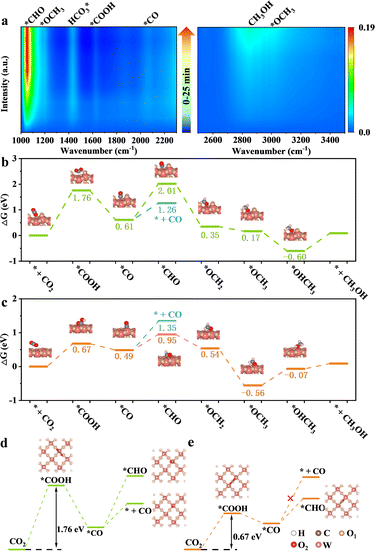
Achieving excellent activity and controllable product selectivity for CO2 photoreduction is generally considered as the bottleneck because of the complicated and sluggish multielectron transfer processes. To probe the evolution of the adsorbed active intermediates on the obtained catalyst surface, in situ FT-IR characterization was performed to track the dynamics process of the CO2RR. As for H-40 (Fig. 5a), the emergence of infrared peaks at 1051, 1430, 1623, and 2065 cm−1 can be assigned to *CHO, 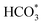 , *COOH, and *CO species, as well as two peaks at 2962 and 1197 cm−1 corresponding to *OCH3 species; in the meantime, the CH3OH peak of the target product at 2850 cm−1 was also observed, consistent with previously reported studies,67–69 and these peak intensities gradually strengthened as a function of irradiation time from 0 to 25 min. Among them, the major preponderant *CHO species is generally regarded as the most significant intermediate during the photocatalytic CO2-to-CH3OH reduction process. It is worth noting that the energy levels of key reactive intermediate species on the catalyst surface can determine whether an easy or complex reaction pathway is present. As expected, the surface coverage of *CHO species on H-40 is much larger relative to other samples, leading to high CH3OH selectivity and a superior yield (Fig. S18, ESI†). Therefore, we deduce that the most likely reaction path for the CO2-to-CH3OH reduction process is outlined below:
, *COOH, and *CO species, as well as two peaks at 2962 and 1197 cm−1 corresponding to *OCH3 species; in the meantime, the CH3OH peak of the target product at 2850 cm−1 was also observed, consistent with previously reported studies,67–69 and these peak intensities gradually strengthened as a function of irradiation time from 0 to 25 min. Among them, the major preponderant *CHO species is generally regarded as the most significant intermediate during the photocatalytic CO2-to-CH3OH reduction process. It is worth noting that the energy levels of key reactive intermediate species on the catalyst surface can determine whether an easy or complex reaction pathway is present. As expected, the surface coverage of *CHO species on H-40 is much larger relative to other samples, leading to high CH3OH selectivity and a superior yield (Fig. S18, ESI†). Therefore, we deduce that the most likely reaction path for the CO2-to-CH3OH reduction process is outlined below:
| | | *CO2 + H+ + e− → *COOH | (R3) |
| | | *COOH + H+ + e− → *CO + H2O | (R4) |
| | | *CHO + H+ + e− → *OCH2 | (R6) |
| | | *OCH2 + H+ + e− → *OCH3 | (R7) |
| | | *OCH3 + H+ + e− → *OHCH3 | (R8) |
where * denotes the active sites of the catalysts.
 |
| | Fig. 5 (a) In situ FT-IR spectra of H-40. Free energy diagrams for reduction of CO2 to CH3OH over (b) WO3 and (c) WO3−x based on DFT calculation as well as the corresponding structure models for every reaction step. Key steps of CO2 photoreduction of (d) WO3 and (e) WO3−x. White, dark brown, light yellow, red and pink balls indicate H, C, O (in WO3), O (in CO2) and W atoms, respectively. | |
To further verify our hypothesis, the remarkable selectivity of the low-valent W5+ mediated WO3 (WO3−x) catalyst was examined by DFT simulation, in which the structural model based on the (001) facet of WO3 was employed. The Gibbs free energy profiles and their optimized reactive intermediate adsorption configurations of each possible step for CO2 reduction to CH3OH on the surface of the perfect WO3 and WO3−x are displayed in Fig. 5b–d. Take the perfect WO3 as an example; the potential-determining step (PDS) is CO2 protonation to generate *COOH with a reaction free-energy-barrier of 1.76 eV. After the introduction of the low-valent W species, this PDS has a lower *COOH formation energy of 0.67 eV, which facilitates the whole reaction. This result indicates that the different adsorption sites likely have a decisive influence on the formation energies of crucial intermediates.33
To clarify this, we performed a precise analysis of the above two models and found a very clear difference in the step of protonation of the *CO intermediate to generate *CHO. Most interestingly, we also find that the *CHO formation energy is higher than the desorption energy of the CO molecules on the perfect WO3, while the CO desorption energy and *CHO formation energy on WO3−x are diametrically opposite. This change might indicate that the low-valent W species induced the redistribution of the photogenerated electrons on the WO3−x surface, thereby efficiently stabilizing and promoting the protonation of *CO to generate *CHO instead of desorbing CO molecules from its surface, which is also supported by the in situ FT-IR results.30,70 Furthermore, the CO-TPD isotherm (Fig. S19, ESI†) confirms the fact that the introduction of suitable low-valent W sites can regulate the CO adsorption and desorption capacity to a certain extent, thereby favouring the subsequent CO protonation and relieving the CO poisoning. This result further suggests that appropriate defect density is critical for customizing liquid products. More importantly, the desorption energy of the CH3OH molecules on the WO3−x surface is much lower than that on the perfect WO3 surface, indicating that suitable low-valent W species are more beneficial for the CH3OH desorption process and efficiently boost its activity and selectivity. The above results demonstrate that the suitable surface pit-embellished low-valent W sites in the WO3−x system could modulate the reaction intermediate's formation energy to change the reaction pathway, which accounts for highly selective photocatalytic CO2 reduction to CH3OH.
Conclusions
In summary, we proposed a general strategy to prepare an extremely efficient WO3−x catalyst and elucidated the origin of its superior CO2RR activity. More specifically, the unique WO3−x nanosheets with abundant surface pits promoted the creation of low-valent W sites to interact with the adsorbed reactant. In situ FT-IR spectra and computational results demonstrate that the delocalized charge near the low-valent W sites not only reduces the reaction energy for CO2 transformation into the key *CHO intermediate, but also regulates the energy level of reaction intermediates to prevent the production of other multi-electron carbon-based products, which is a key characteristic for achieving high CH3OH selectivity. Consequently, a suitable low-valent W concentration enables the H-40 catalyst to show a drastically enhanced yield of CH3OH as well as appreciable stability, which outperformed most previously reported state-of-the-art photocatalysts operated under similar conditions. This work puts forward some inspirations for the rational design of surface electronic structures of photocatalysts and the precise control of the reaction pathways, while providing deep mechanistic insights into customizing the conversion of CO2 into valuable solar fuels.
Author contributions
Y. X. and S. G. conceived and designed the research. W. Z., P. X. Y. and Q. W. carried out the experiments. J. L. Y., Q. Q. L. and M. D. conducted the DFT calculations. K. F. Z., X. W. Z. and Y. Y. participated in the experimental data analyses and scientific discussions. S. G. and K. F. Z. wrote the manuscript. W. Z., M. D., and P. X. Y. contributed equally to this work.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The work was supported by the National Natural Science Foundation of China (grants U1832189, U21A20317, 22288201, 22205002 and 22103001), the research start-up fund from Anhui University (S020118002/060, S020318008/016) and the University Synergy Innovation Program of Anhui Province (GXXT-2020-001). The DFT calculations were performed in the Supercomputing Center of Anhui University and the National Supercomputing Center in Shanghai.
References
- X. Jiang, J. Huang, Z. Bi, W. Ni, G. Gurzadyan, Y. Zhu and Z. Zhang, Adv. Mater., 2022, 34, 2109330 CrossRef CAS PubMed.
- J. W. Wang, L. Jiang, H. H. Huang, Z. Han and G. Ouyang, Nat. Commun., 2021, 12, 4276 CrossRef CAS PubMed.
- Y. Yamazaki, M. Miyaji and O. Ishitani, J. Am. Chem. Soc., 2022, 144, 6640–6660 CrossRef CAS PubMed.
- Y. A. Wu, I. McNulty, C. Liu, K. C. Lau, Q. Liu, A. P. Paulikas, C.-J. Sun, Z. Cai, J. R. Guest, Y. Ren, V. Stamenkovic, L. A. Curtiss, Y. Liu and T. Rajh, Nat. Energy, 2019, 4, 957–968 CrossRef CAS.
- E. Gong, S. Ali, C. B. Hiragond, H. S. Kim, N. S. Powar, D. Kim, H. Kim and S.-I. In, Energy Environ. Sci., 2022, 15, 880–937 RSC.
- Y. Birdja, E. Pérez-Gallent, C. Figueiredo, J. Göttle and T. Koper, Nat. Energy, 2019, 4, 732 CrossRef CAS.
- L. Yuan, M. Y. Qi, Z. R. Tang and Y. J. Xu, Angew. Chem., Int. Ed., 2021, 60, 2–25 CrossRef.
- S. Navarro-Jaén, M. Virginie, J. Bonin, M. Robert, R. Wojcieszak and A. Y. Khodakov, Nat. Rev. Chem., 2021, 5, 564–579 CrossRef.
- Z. Zhang, X. Ding, X. Yang, W. Tu, L. Wang and Z. Zou, EcoMat, 2021, 3, 12078 Search PubMed.
- J. Li, Y. Zhao, M. Xia, H. An, H. Bai, J. Wei, B. Yang and G. Yang, Appl. Catal., B, 2020, 261, 118244 CrossRef CAS.
- R. Yang, J. Duan, P. Dong, Q. Wen, M. Wu, Y. Liu, Y. Liu, H. Li and T. Zhai, Angew. Chem., Int. Ed., 2022, 61, e202116706 CAS.
- Y. Yang, Y. Pan, X. Tu and C. Liu, Nano Energy, 2022, 101, 107613 CrossRef CAS.
- J. Duan, T. Liu, Y. Zhao, R. Yang, Y. Zhao, W. Wang, Y. Liu, H. Li, Y. Li and T. Zhai, Nat. Commun., 2022, 13, 2039 CrossRef CAS PubMed.
- Y. Feng, C. Wang, P. Cui, C. Li, B. Zhang, L. Gan, S. Zhang, X. Zhang, X. Zhou, Z. Sun, K. Wang, Y. Duan, H. Li, K. Zhou, H. Huang, A. Li, C. Zhuang, L. Wang, Z. Zhang and X. Han, Adv. Mater., 2022, 34, 2109074 CrossRef CAS.
- H. Wang, L. Zhang, K. Wang, X. Sun and W. Wang, Appl. Catal., B, 2019, 243, 771–779 CrossRef CAS.
- Z. Zhang, X. Ding, X. Yang, W. Tu, L. Wang and Z. Zou, EcoMat, 2021, 3, 12078 Search PubMed.
- Z. Zhang, J. Liu, J. Wang, Q. Wang, Y. Wang, K. Wang, Z. Wang, M. Gu, Z. Tang, J. Lim, T. Zhao and F. Ciucci, Nat. Commun., 2021, 12, 5235 CrossRef CAS PubMed.
- L. Xiao, X. Xu, Y. Jia, G. Hu, J. Hu, B. Yuan, Y. Yu and G. Zou, Nat. Commun., 2021, 12, 318 CrossRef CAS PubMed.
- K. A. Goulas, A. V. Mironenko, G. R. Jenness, T. Mazal and D. G. Vlachos, Nat. Catal., 2019, 25, 5096–5099 Search PubMed.
- G. Yang, X. Zhu, G. Cheng, R. Chen, J. Xiong, W. Li and Y. Wei, J. Mater. Chem. A, 2021, 9, 22781–22809 RSC.
- H. Zhang, Y. Wang, S. Zuo, W. Zhou, J. Zhang and X. W. D. Lou, J. Am. Chem. Soc., 2021, 143, 2173–2177 CrossRef CAS PubMed.
- J. Yan, T. Wang, G. Wu, W. Dai, N. Guan, L. Li and J. Gong, Adv. Mater., 2015, 27, 1580–1586 CrossRef CAS PubMed.
- Y. Zhao, Z. Han, G. Gao, W. Zhang, Y. Qu, H. Zhu, P. Zhu and G. Wang, Adv. Funct. Mater., 2021, 31, 2104976 CrossRef CAS.
- Y. Sun, Q. Liu, S. Gao, H. Cheng, F. Lei, Z. Sun, Y. Jiang, H. Su, S. Wei and Y. Xie, Nat. Commun., 2013, 4, 2899 CrossRef PubMed.
- B. Zhang, J. Zhang, M. Hua, Q. Wan, Z. Su, X. Tan, L. Liu, F. Zhang, G. Chen, D. Tan, X. Cheng, B. Han, L. Zheng and G. Mo, J. Am. Chem. Soc., 2020, 142, 13606–13613 CrossRef CAS.
- S. Li, L. Meng, W. Tian and L. Li, Adv. Energy Mater., 2022, 32, 2200629 CrossRef.
- Z. Xiao, C. Xie, Y. Wang, R. Chen and S. Wang, J. Energy Chem., 2021, 53, 208–225 CrossRef CAS.
- Z. Yang, T. Sander, J. Gebhardt, T. A. Schaub, J. Schönamsgruber, H. R. Soni, A. Görling, M. Kivala and S. Maier, ACS Nano, 2020, 14, 16887–16896 CrossRef CAS PubMed.
- B. Wu, L. Zhang, B. Jiang, Q. Li, C. Tian, Y. Xie, W. Li and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 4815–4822 CrossRef CAS PubMed.
- Y. Liu, L. Liang, C. Xiao, X. Hua, Z. Li, B. Pan and Y. Xie, Adv. Energy Mater., 2016, 6, 1600437 CrossRef.
- T. W. Kim, Y. Ping, G. A. Galli and K. S. Choi, Nat. Commun., 2015, 6, 8769 CrossRef CAS PubMed.
- T. Yan, N. Li, L. Wang, Q. Liu, A. Jelle, L. Wang, Y. Xu, Y. Liang, Y. Dai, B. Huang, J. You and G. A. Ozin, Energy Environ. Sci., 2020, 13, 3054–3063 RSC.
- L. W. Zhang, L. Wang and Y. F. Zhu, Adv. Funct. Mater., 2007, 17, 3781–3790 CrossRef CAS.
- G. Wang, Z. Chen, T. Wang, D. Wang and J. Mao, Angew. Chem., Int. Ed., 2022, 61, e202210789 CAS.
- G. Moore, S. Howell, M. Brady, X. Xu and K. McNeil, Nat. Commun., 2021, 12, 1–9 CrossRef CAS PubMed.
- Y. Mao, P. Wang, L. Li, Z. Chen, H. Wang, Y. Li and S. Zhan, Angew. Chem., Int. Ed., 2020, 59, 3685–3690 CrossRef CAS PubMed.
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS.
- J. Li, W. Pan, Q. Liu, Z. Chen, Z. Chen, X. Feng and H. Chen, J. Am. Chem. Soc., 2021, 143, 6551–6559 CrossRef CAS PubMed.
- C. Wen, F. Mao, Y. Liu, X. Zhang, H. Fu, L. Zheng, P. Liu and H. Yang, ACS Catal., 2020, 10, 1086–1093 CrossRef CAS.
- M. Sun, R. Gao, J. He, X. Liu, T. Nakajima, X. Zhang and L. Wang, Angew. Chem., Int. Ed., 2021, 60, 17601–17607 CrossRef CAS PubMed.
- Q. He, Y. Zhang, H. Li, Y. Yang, S. Chen, W. Yan, J. Dong, X. Zhang and X. Fan, Small, 2022, 18, 2108034 CrossRef CAS PubMed.
- K. Zhang, L. Yang, Y. Hu, C. Fan, Y. Zhao, L. Bai, Y. Li, F. Shi, J. Liu and W. Xie, Angew. Chem., Int. Ed., 2020, 59, 18003–18009 CrossRef CAS PubMed.
- M. Sun, R. T. Gao, J. He, X. Liu, T. Nakajima, X. Zhang and L. Wang, Angew. Chem., Int. Ed., 2021, 60, 17601–17607 CrossRef CAS PubMed.
- Z. Wei, W. Wang, W. Li, X. Bai, J. Zhao, E. C. M. Tse, D. L. Phillips and Y. Zhu, Angew. Chem., Int. Ed., 2021, 60, 8236–8242 CrossRef CAS PubMed.
- X. Li, W. He, C. Li, B. Song and S. Liu, Appl. Catal., B, 2021, 287, 119934 CrossRef CAS.
- K. Wang, Y. Du, Y. Li, X. Wu, H. Hu, G. Wang, Y. Xiao, S. Chou and G. Zhang, Carbon Energy, 2022 DOI:10.1002/cey2.264.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- J. Albo, M. I. Qadir, M. Samperi, J. A. Fernandes, I. de Pedro and J. Dupont, Chem. Eng. J., 2021, 404, 126643 CrossRef CAS.
- S. Cao, B. Shen, T. Tong, J. Fu and J. Yu, Adv. Funct. Mater., 2018, 28, 1800136 CrossRef.
- H. Guo, J. Ding, S. Wan, Y. Wang and Q. Zhong, Appl. Surf. Sci., 2020, 528, 146943 CrossRef CAS.
- Y. X. Pan, Y. You, S. Xin, Y. Li, G. Fu, Z. Cui, Y. L. Men, F. F. Cao, S. H. Yu and J. B. Goodenough, J. Am. Chem. Soc., 2017, 139, 4123–4129 CrossRef CAS PubMed.
- G. Wang, C. T. He, R. Huang, J. Mao, D. Wang and Y. Li, J. Am. Chem. Soc., 2020, 142, 19339–19345 CrossRef CAS PubMed.
- L. Wang, Y. Wang, Y. Cheng, Z. Liu, Q. Guo, M. N. Ha and Z. Zhao, J. Mater. Chem. A, 2016, 4, 5314–5322 RSC.
- X. Wang, Y. Wang, M. Gao, J. Shen, X. Pu, Z. Zhang, H. Lin and X. Wang, Appl. Catal., B, 2020, 270, 118876 CrossRef CAS.
- X. Wu, Y. Li, G. Zhang, H. Chen, J. Li, K. Wang, Y. Pan, Y. Zhao, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2019, 141, 5267–5274 CrossRef CAS PubMed.
- P. Xia, M. Antonietti, B. Zhu, T. Heil, J. Yu and S. Cao, Adv. Funct. Mater., 2019, 29, 1900093 CrossRef.
- W. Xiong, W. Dai, X. Hu, L. Yang, T. Wang, Y. Qin, X. Luo and J. Zou, Mater. Lett., 2018, 232, 36–39 CrossRef CAS.
- T. Yan, L. Wang, Y. Liang, M. Makaremi, T. E. Wood, Y. Dai, B. Huang, A. A. Jelle, Y. Dong and G. A. Ozin, Nat. Commun., 2019, 10, 2521 CrossRef PubMed.
- Y. Zheng, Z. Duan, R. Liang, R. Lv, C. Wang, Z. Zhang, S. Wan, S. Wang, H. Xiong, C. K. Ngaw, J. Lin and Y. Wang, ChemSusChem, 2022, 15, 216 Search PubMed.
- Y. Zheng, L. Zhang, J. Guan, S. Qian, Z. Zhang, C. K. Ngaw, S. Wan, S. Wang, J. Lin and Y. Wang, ACS Sustainable Chem. Eng., 2021, 9, 1754–1761 CrossRef CAS.
- H. Tan, Z. Zhao, W. Zhu, E. Coker, B. Li, M. Zheng, W. Yu, H. Fan and Z. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 19184–19190 CrossRef CAS PubMed.
- D. Ruan, S. Kim, M. Fujitsuka and T. Majima, Appl. Catal., B, 2018, 238, 638–646 CrossRef CAS.
- M. A. Hossain, R. Al-Gaashani, H. Hamoudi, M. J. Al Marri, I. A. Hussein, A. Belaidi, B. A. Merzougui, F. H. Alharbi and N. Tabet, Mater. Sci. Semicond. Process., 2017, 63, 203–211 CrossRef CAS.
- L. Wang, B. Cheng, L. Zhang and J. Yu, Small, 2021, 17, 2103447 CrossRef CAS PubMed.
- R. Zeng, K. Lian, B. Su, L. Lu, J. Lin, D. Tang, S. Lin and X. Wang, Angew. Chem., Int. Ed., 2021, 60, 25055–25062 CrossRef CAS PubMed.
- J. Wang, E. Kim, D. P. Kumar, A. P. Rangappa, Y. Kim, Y. Zhang and T. K. Kim, Angew. Chem., Int. Ed., 2022, 61, 202113044 Search PubMed.
- S. Bai, T. Li, H. Wang, L. Tan, Y. Zhao and Y.-F. Song, Chem. Eng. J., 2021, 419, 129390 CrossRef CAS.
- S. R. Docherty, N. Phongprueksathat, E. Lam, G. Noh, O. V. Safonova, A. Urakawa and C. Coperet, J. Am. Chem. Soc., 2021, 1, 450–458 CAS.
- W. Wang, Z. Qu, L. Song and Q. Fu, J. Energy Chem., 2020, 47, 18–28 CrossRef.
- H. Wang, D. Yong, S. Chen, S. Jiang, X. Zhang, W. Shao, Q. Zhang, W. Yan, B. Pan and Y. Xie, J. Am. Chem. Soc., 2018, 140, 1760–1766 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence e,
Yu
Yu
a,
Qiquan
Luo
e,
Yu
Yu
a,
Qiquan
Luo
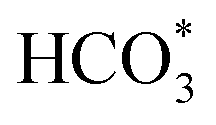 , *COOH, and *CO species, as well as two peaks at 2962 and 1197 cm−1 corresponding to *OCH3 species; in the meantime, the CH3OH peak of the target product at 2850 cm−1 was also observed, consistent with previously reported studies,67–69 and these peak intensities gradually strengthened as a function of irradiation time from 0 to 25 min. Among them, the major preponderant *CHO species is generally regarded as the most significant intermediate during the photocatalytic CO2-to-CH3OH reduction process. It is worth noting that the energy levels of key reactive intermediate species on the catalyst surface can determine whether an easy or complex reaction pathway is present. As expected, the surface coverage of *CHO species on H-40 is much larger relative to other samples, leading to high CH3OH selectivity and a superior yield (Fig. S18, ESI†). Therefore, we deduce that the most likely reaction path for the CO2-to-CH3OH reduction process is outlined below:
, *COOH, and *CO species, as well as two peaks at 2962 and 1197 cm−1 corresponding to *OCH3 species; in the meantime, the CH3OH peak of the target product at 2850 cm−1 was also observed, consistent with previously reported studies,67–69 and these peak intensities gradually strengthened as a function of irradiation time from 0 to 25 min. Among them, the major preponderant *CHO species is generally regarded as the most significant intermediate during the photocatalytic CO2-to-CH3OH reduction process. It is worth noting that the energy levels of key reactive intermediate species on the catalyst surface can determine whether an easy or complex reaction pathway is present. As expected, the surface coverage of *CHO species on H-40 is much larger relative to other samples, leading to high CH3OH selectivity and a superior yield (Fig. S18, ESI†). Therefore, we deduce that the most likely reaction path for the CO2-to-CH3OH reduction process is outlined below: