
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of magnetic nanoparticles with covalently bonded polyacrylic acid for use as forward osmosis draw agents
Irena
Ban
 *a,
Mihael
Drofenik
ab,
Hermina
Bukšek
a,
Irena
Petrinic
a,
Claus
Helix-Nielsen
ac,
Sabina
Vohl
a,
Sašo
Gyergyek
ab and
Janja
Stergar
a
*a,
Mihael
Drofenik
ab,
Hermina
Bukšek
a,
Irena
Petrinic
a,
Claus
Helix-Nielsen
ac,
Sabina
Vohl
a,
Sašo
Gyergyek
ab and
Janja
Stergar
a
aFaculty of Chemistry and Chemical Engineering, University of Maribor, Smetanova 17, SI-2000 Maribor, Slovenia. E-mail: irena.ban@um.si
bDepartment of Materials Synthesis, Jožef Stefan Institute, Jamova cesta 29, SI-1000 Ljubljana, Slovenia
cDepartment of Environmental and Resource Engineering, Technical University of Denmark, Miljøvej 113, 2800 Kgs. Lyngby, Denmark
First published on 28th November 2022
Abstract
Multicoated magnetite (Fe3O4) magnetic nanoparticles (MNPs) with polyacrylic acid (PAA) as a terminal hydrophilic ligand were synthesized and examined for use as a draw solution (DS) agent in forward osmosis (FO). After coating superparamagnetic iron-oxide MNPs with (3-aminopropyl)triethoxysilane (APTES) the carboxyl groups of PAA were bound to APTES amino groups via the crosslinker 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) forming a peptide bond resulting in stable water-soluble particles (MNP@APTES@PAA) with a concentration-normalised osmotic pressure of 1.56 bar L g−1. The MNP@APTES@PAA solution was evaluated as a DS in two FO filtrations with deionized (DI) water as a feed solution (FS): one using freshly prepared MNP@APTES@PAA and one using magnetically recovered (re-concentrated) MNP@APTES@PAA. The resulting MNP@APTES@PAA nanocomposites exhibit good colloidal stability in aqueous solution with a concentration-normalized osmotic pressure of 1.56 bar L g−1. This is 12-fold higher than that in our previous studies of poly-sodium-acrylate coated MNPs and 3-fold higher than that of citric acid coated MNPs. The water recoveries of the two filtrations were 25.7% and 13.6%, respectively, after 2 h of FO filtration time resulting in a DS osmotic pressure of 2.5 bar with a concentration of 4.3 g L−1 and a DS osmotic pressure of 2.6 with a concentration of 3.7 g L−1 respectively.
Water impactThe increased concerns for public health and the environment necessitate developments in the reuse of wastewater. The main contribution to this is the synthesis of inorganic nanoparticles as forward osmosis draw solutions in integrated wastewater treatment processes. |
1. Introduction
In the last decades, the need for drinking water has increased leading to an increased focus on water reuse. Among various methods, forward osmosis (FO) offers potential advantages in recovering clean water from diverse impaired water feed streams.1 FO, also called direct osmosis, uses the osmotic pressure difference between a feed solution (FS) and a draw solution (DS) for driving the FS through a semipermeable membrane. Ideally, the concentrated high osmotic pressure DS extracts water from the dilute low osmotic pressure FS while rejecting all unwanted FS solutes.2 The advantages of FO are low energy use and low membrane fouling propensity.3 However, in order to produce clean water, a second separation step to re-concentrate the DS is needed. Several methods have been investigated including the use of phase-shifting DS agents, membrane distillation and reverse osmosis, and each of these has its advantages and disadvantages.4 Thus, one of the major tasks in FO is choosing an ideal DS with high osmotic pressure and minimum reverse solute flux that can be recovered with minimal energy consumption.Typically used DSs in FO include5 inorganic compounds,6 organic compounds (e.g., polyacrylic acid sodium, methylimidazole-based compounds, hexavalent phosphazene salts, and stimuli-responsive hydrogels),7 functionalized nanoparticles (e.g., magnetic nanoparticles (MNPs)), and Na+-functionalized carbon quantum dots (Na-CQD).8
Recently magnetic nanoparticles (MNPs) have received large attention due to their various advantages, i.e., surface modifiability, magnetic properties, biocompatibility, and ease of manipulation.9 A benefit of MNPs against other possible DSs is facile regeneration by the use of an external magnetic field. In order to secure colloidal stability MNPs are typically surface functionalized using numerous organic grafting agents.10–17 Among them, poly-sodium-acrylate (PSA) and/or polyacrylic acid (PAA) have been reported as efficient osmotic draw agents with high osmotic pressure18–21 but low regeneration capability.22,23 Here, the carboxyl groups of PAA have good coordination and affinity for magnetite nanoparticles preventing their aggregation into larger particles. PAA has a large number of –COOH groups, however, at least one –COOH group must be attached to the surface of the MNPs while the others remain free.10 However, in general for MNPs used as a DS in FO, the choice of coating materials is a critical parameter that is still poorly understood.
For the synthesis of functionalized MNPs, two methods are usually applied: one-pot and two-pot synthesis methods. The one-pot synthesis precipitation of MNPs is accompanied by functionalization of precipitated MNPs. The synthesized average particle size is usually 4–5 nm owing to inhibition of grain growth due to a high concentration of reaction products in situ, i.e., the growth of MNPs is hindered. The one-pot synthesis of functionalized MNPs gives a DS with a relatively high initial osmotic pressure mostly on the account of hydrophilic molecular associates physically bound to the firm chelate bonded monolayer of hydrophilic ligands on the surface of MNPs.24 However, during recycling, DS degradation, i.e., a decrease of the osmotic pressure, may occur primarily due to release of physically bound hydrophilic ligands from the functionalized MNP surface. As these molecules are in equilibrium with those in the carrier liquid they will desorb during the process independent of the suspension concentration. After the release of physically bound ligands, the bonded PAA monolayer remains attached which then determines the final osmotic pressure of the MNP solution. Consequently, ultra-small superparamagnetic MNPs exhibit a relatively large specific surface area with chelate bonded ligands in a monolayer which will induce a large durable osmotic pressure. However, they will exhibit a small operational magnetization, due to their small particle size and a relative low magnetization per MNP, which will limit the exploitation of MNP use for DS application. Thus, Kim et al.25 reported that particles smaller than 11 nm were difficult to separate from the solution even with the application of a strong magnetic field (0–1 T).
On the other hand, two-pot synthesis enables precise engineering of the particle morphology and functionalization, which can facilitate the optimization of DS applicability. In our earlier efforts to produce a poly-sodium-acrylate (PSA) or citric acid (CA) coating on the surface of the synthesized NPs using one-pot synthesis, the ligand PSA or CA was directly attached to MNPs.24,26 One part of PSA was bound to the surface by covalent (chelate) bonds and another part was loosely associated with the PSA–MNP complex and the osmotic pressure of the as-is PSA–MNP was high enough to be used as a DS in FO. UF dialysis revealed a significant amount of loosely bonded PSA ligands, leaving only the firmly bonded ligand molecules on the particle surface. The rather modest osmotic pressure of the dialyzed DS limits the applicability of PSA–MNP as a solute molecule in its current state and additional steric stabilization is needed for its general use as a DS in FO.
The osmotic pressure of the as-is MNP-grafted CA was two-fold higher than that of the as-is PSA–MNP. However, the robustness of CA-MNP-based DSs (i.e., their ability to maintain their osmotic pressure after repeated FO) is still an issue. Our previous work24,26 showed loose bonds which disintegrated after one to two FO cycles, and the osmotic pressure potency was only 0.13–0.51 bar per g L−1 solution. Therefore, further investigations were conducted to establish a stronger (covalent) bond on loosely associated molecules.
Specifically, in order to establish a stronger bond and prevent a decrease of the osmotic pressure after filtration, we applied the synthesis of a DS using two pot synthesis via a carbodiimide (EDC) crosslinker and described a process to obtain MNPs covered with covalently bonded hydrophilic PAA. The major objective of this contribution was the intention to achieve a potent and durable DS without releasing the hydrophilic ligand PAA from the particles’ surface during repeated FO cycles. The MNPs were covered with (3-aminopropyl)triethoxysilane (NH2(CH2)3Si(OC2H5)3 (APTES)) which served as the precursor for the PAA coating using the crosslinker 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC). This crosslinker was activated with carboxylic groups forming a strong covalent bond (peptide bond) between APTES and PAA, forming MNP@APTES@PAA under conditions that stimulated particle stability and PAA solubility.
The MNP@APTES@PAA solutes were assessed as a DS on FO. Specifically, two FO filtrations were performed using freshly prepared and magnetically re-concentrated suspensions of MNP@APTES@PAA particles as a DS and deionized water (DI) in order to test if the functionalized MNPs prevent the release of PAA during the FO process and maintain a stable osmotic pressure.
2. Materials and methods
Iron(II) and iron(III) chloride were purchased from Sigma Aldrich, USA. PAA, ethanol, APTES, and 25% ammonia solution were obtained from Merck Co., USA, while the cross-linker EDC and hydroxysuccinimide (NHS) were obtained from Sigma Aldrich, USA. DI was used for all the reactions and FS. All other chemicals were commercially available analytical grade reagents and were used without further purification. X-ray diffraction patterns were collected by means of a Bruker D2 PHASER 2nd Gen diffractometer (Karlsruhe, Germany). Patterns were collected in the range of 20 to 70° (2θ) with a step size of 0.02θ and a rate of 30 s per step. MNPs were observed with transmission electron microscopy (TEM) (JEOL 2010F, Tokyo, Japan) operated at 200 kV. A dilute colloid was dropped on a holey carbon-coated Cu-grid and left to naturally dry in an atmosphere of air. The particle size was estimated using TEM images of bare magnetite MNPs, using imaging software (DigitalMicrograph, Gatan Inc.). Dynamic light scattering (DLS) and zeta potentials were measured using a Malvern Zetasizer Nano Series, UK, with a 4 mW He–Ne laser running at 633 nm. Analyses were performed on a dilute MNP suspension which afforded multiple scattering that could be considered negligible. The zeta potential was determined using a zeta potential cell. The reflectance Fourier transform infrared spectra with 260 scans and a resolution of 5 cm−1 were obtained using a Shimadzu IRAffinity-1S FTIR spectrometer (PerkinElmer 5000 Inc., Beaconsfield, UK). Thermogravimetric analysis (TGA, TGA/SDTA, 851e Mettler Toledo, Switzerland) of bare and functionalized MNPs was performed in air (up to 900 °C at a heating rate of 10 °C min−1). The weight loss was ascribed to the polymer coating layer on the composite MNPs. The room-temperature magnetization curves were determined with a vibrating sample magnetometer (LakeShore 7304 VSM, Westerville, Ohio, USA). The osmolality of dispersions prepared from MNPs was determined with a freezing point osmometer (Gonotec Osmomat 030, Berlin, Germany) and the osmotic pressure of MNP@APTES@PAA solutions was calculated using the equation in ref. 24. The FO filtration experiments were performed using an AIM™ HFFO module kindly provided by Aquaporin A/S Kgs. Lyngby, Denmark with an effective membrane area of 180 cm2. The batch experiments were performed in cross-flow mode using a double-headed peristaltic pump (Longer Pump® BT100-1, Hebei, China) with a FS and DS circulation of 120.1 mL min−1 in counter-current mode. The active side of the membrane was facing the FS. The mass changes of the FS were monitored continuously every 30 seconds using a digital balance (Ohaus Scout Pro, New Jersey, USA). The specifications of the AIM™ HFFO module and the experimental set-up used in this study were presented in a previous paper.272.1. Synthesis and characterization of magnetite Fe3O4
The preparation of the MNPs proceeded via precipitation of Fe(II) and Fe(III) ions as reported elsewhere;28 in brief: magnetite MNPs were prepared by precipitating Fe(II) and Fe(II) ions (molar ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with ammonia at pH 10 and heating at 80 °C for 60 min. In detail: in a 250 ml three-neck flask 4.30 g FeCl2·4H2O and 11.68 g FeCl3·6H2O were dissolved in 200 ml of DI. When the salts were completely dissolved, the flask was connected to a reflux condenser, and the solution was bubbled in nitrogen for 10 min. Afterward, 45 ml of 25% NH4OH was added (pH = 10.5) and the reaction mixture was stirred and bubbled for 60 min at 80 °C. When the synthesis was finished, MNPs were obtained by placing the suspension on a permanent magnet. MNPs were washed with 100 ml 0.02 M NaCl two times and with DI water two times.
:1) with ammonia at pH 10 and heating at 80 °C for 60 min. In detail: in a 250 ml three-neck flask 4.30 g FeCl2·4H2O and 11.68 g FeCl3·6H2O were dissolved in 200 ml of DI. When the salts were completely dissolved, the flask was connected to a reflux condenser, and the solution was bubbled in nitrogen for 10 min. Afterward, 45 ml of 25% NH4OH was added (pH = 10.5) and the reaction mixture was stirred and bubbled for 60 min at 80 °C. When the synthesis was finished, MNPs were obtained by placing the suspension on a permanent magnet. MNPs were washed with 100 ml 0.02 M NaCl two times and with DI water two times.
XRD analyses of the as-synthesized MNPs showed discrete reflections which were indexed according to the magnetite crystal structure data (JCPDS card (19-0629).29 The average nanoparticle size (τ) was estimated by the Scherrer equation (eqn (1)), from the width of the XRD peaks, and was determined to be 13 nm (Fig. 1(a)). K is the dimensionless shape factor, with a value close to unity (0.9), λ is the X-ray wavelength (nm), β is the line broadening at half the maximum intensity after subtracting the instrumental line broadening (rad), and θ is the Bragg angle (°).
 | (1) |
 | ||
| Fig. 1 a) X-ray diffraction spectrum of the as-prepared MNPs; b) magnetization vs. magnetic field at 293 K. | ||
 | ||
| Fig. 2 TEM image of the as-prepared MNPs. | ||
The results show that the hysteresis of the MNPs cannot be observed due to their superparamagnetic properties. The morphology of the MNPs was examined with TEM. Fig. 2 shows the bare MNPs that were sphere-shaped.
The average size of the magnetite MNPs (12 nm) was confirmed with TEM analysis (Fig. 2). Fig. 3 shows the DLS analysis/results of zeta potential measurements and the corresponding hydrodynamic radii of the particles. The ζ-potential and hydrodynamic radii were measured as a function of the suspension pH.
 | ||
| Fig. 3 Effect of the solution pH on the ζ-potential (black curve) and hydrodynamic radii (red curve) of the as-prepared MNPs. | ||
With increasing pH, the zeta potential decreases, as some of the hydroxyl groups become negatively charged. At pH 5.66, the potential decreases to zero. This is in accordance with the chemical composition of the as-prepared magnetite surface; hydroxyl functional groups on the MNP surface with neutralization allow the pH to reach a value of below 10. The synthesized MNPs exhibit an average size dTEM = 12 nm with an average particle surface area So = 452.4 nm2 and a particle volume Vo = 905.8 nm3. After the functionalization of the magnetite MNPs with precursors based on carboxylic groups, the hydrophilicity for an effective DS was achieved.
3. Results and discussion
3.1. Synthesis and characterization of MNP@APTES
Magnetite MNPs (Fe3O4) were synthesized by the coprecipitation process and stabilized with APTES. APTES binds to the particles via silanol groups, and the amino groups remain free for further functionalization with hydrophilic –COOH groups. To complete the coupling reaction, the crosslinker EDC and N-hydroxysuccinimide (NHS), which stabilizes the intermediate, were added. Through the formation of an amide (–NH–CO–) bond,30 PAA was bound to them to achieve the highest possible osmotic pressure (Fig. 4).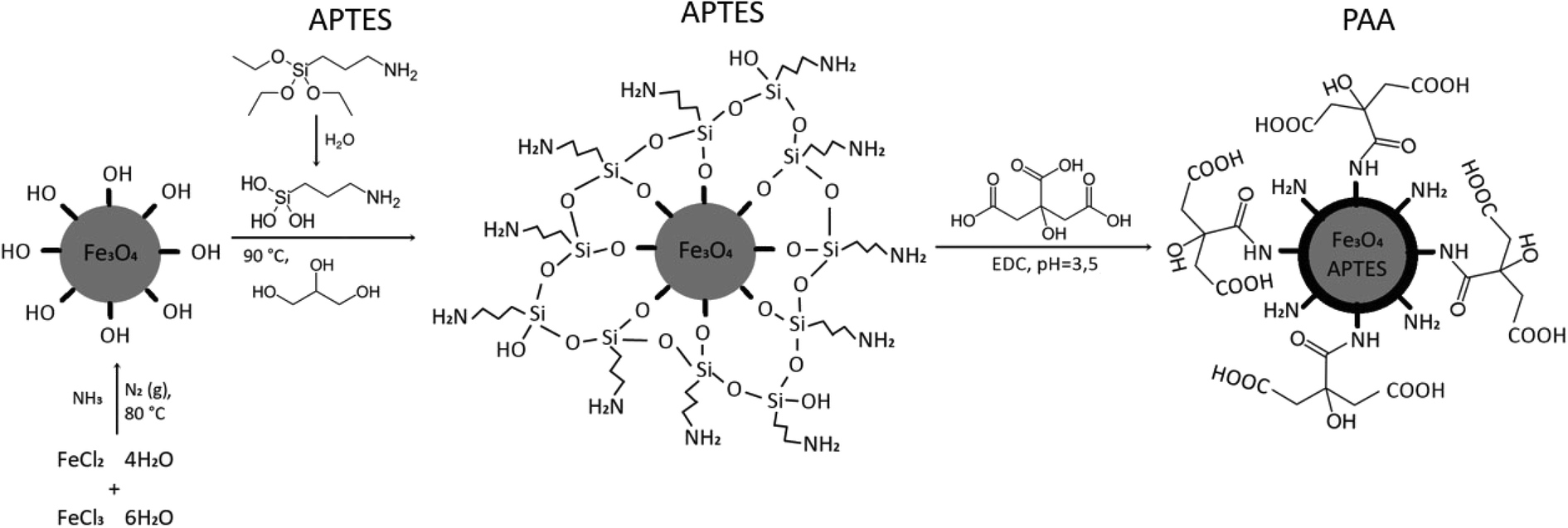 | ||
| Fig. 4 Schematic representation of MNP synthesis and functionalization. | ||
The aqueous solution of the as-synthesized MNPs (Section 3.1.) was mixed with a 10% (v/v) solution of APTES and 40 ml of glycerol and heated to 90 °C for 2 h under a nitrogen atmosphere and constant stirring. After 2 hours the suspension was cooled to room temperature and washed three times with 200 ml DI water, five times with 70 ml methanol, and again three times with 200 ml DI. MNP@APTES was separated by sedimentation using a permanent magnet. The prepared silane-coated MNPs were stored in DI water. Some of them were dried in an oven at 80 °C for additional analyses (Fig. 5).
 | ||
| Fig. 5 Photo of the synthesized functionalized MNPs. | ||
Fig. 6 shows the DLS analysis/results of zeta potential measurements and the corresponding hydrodynamic radii of the MNP@APTES particles. The ζ-potential and hydrodynamic radii were measured as a function of the suspension pH.
 | ||
| Fig. 6 a) Effect of the solution pH on the ζ-potential (black curve) and hydrodynamic radii (red curve) of the MNP@APTES particles and b) the particle size distribution of MNP@APTES particles at pH = 7. | ||
The DLS spectrum shows a representative picture of the surface chemistry of APTES coated MNPs (MNP@APTES). The terminal groups of the MNPs are amino groups. Negative ions, in this case, the OH− ions, are compensating ions to achieve a zero-surface charge. The isoelectric point (IP) of around pH 10 confirms a positively charged surface due to the attached –NH3+ groups. The DLS spectrum (Fig. 6(b)) shows a bimodal particle size distribution at pH 7. The narrow peak at 10 nm represents mostly the distribution of single particles. The average particle size, determined by TEM analysis (bare Fe3O4), is around 12 nm, and the average DLS value of the functionalized particle size is also 10 nm. The peak at 10 nm in the DLS spectrum represents non-agglomerated MNP@APTES composite particles. APTES functionalized MNPs have an organic shell, attached with Si–O bonds to the surface of MNPs, which is not visible in the TEM image. The second peak in the DLS spectrum (Fig. 6(b)) at 150 nm shows the presence of some agglomerates. Here, at pH 7 the electrostatic repulsions govern the suspension properties. At pH 7, where the zeta potential is the highest, some agglomeration in a more mild form still takes place.
In the FTIR spectrum of MNP@APTES (Fig. 7(a)), the peak marked with an arrow is typical for N–H bond stretching, which indicates the presence of –NH2 groups on the MNP surface. The TEM image of MNP@APTES particles (Fig. 7(b)) shows the presence of a typical suspension agglomerate at the IP (pH 10) and this confirms the DLS results.
 | ||
| Fig. 7 a) FTIR spectrum of MNP@APTES with characteristic peaks of –NH2 groups on the surface and b) TEM image of MNP@APTES at pH 10. | ||
The amount of APTES molecules attached to the MNP surface was determined with the data from TGA. The explanation for the TGA curve was complex because Fe3O4 can be oxidized to Fe2O3 at 450 °C; thus the estimated total number of COOH groups was necessary. The important argument to consider magnetite oxidation is according to the oxidation reaction
| Fe3O4 + 1/4O2 → 3/2Fe2O3 | (2) |
 | ||
| Fig. 8 TGA analysis curve of Fe3O4@APTES measured in an air atmosphere. | ||
Considering the process of the ligand APTES – the amount of amine –NH2 groups attached to the MNP surface, the number of active sites on the functionalized MNP surface was estimated as the surface ligand concentration ns using the following equation:
 | (3) |
3.2. Synthesis and characterization of MNP@APTES@PAA
PAA is a good candidate for application as an osmotic draw agent for the functionalization of the MNP@APTES's surface. It possesses a large number of carboxyl groups, which are suitable for attracting water molecules. So, to prepare hydrophilic MNPs PAA (M = 1800 Da) was selected. PAA has been described as an efficient osmotic draw agent for the functionalization of the MNP's surface.30 A large number of hydrophilic carboxylate groups along the polymer chain can serve as a water acceptor and be recognized as a notable hydrophilic solute for the prospective DS. These functional groups may enhance the interaction between water and MNPs, due to free carboxylate groups stretched in water. PAA, attached to coated MNPs in the aqueous solution, may enable dispensability and contribute to an effective DS.For the synthesis of MNP@APTES@PAA, we started the synthesis with stoichiometric amounts of all the constituent components needed to bind links to all, on average, 1579 free –NH2 sites on the particle's surface. The procedure of grafting PAA via hydrophilic COOH ligands, assisted with EDC activation, is achieved with the final step, i.e., the formation of a peptide bond:
| COOH + –NH2 → –CO–NH– + H2O | (4) |
The procedure was as follows: 0.256 g MNP@APTES was suspended in 5 ml of DI. In another 5 ml of DI water PAA (163 mg; n(NH2):n(PAA) = 1:1), 20 mg of EDC (n(EDC):n(COOH from PAA) = 1:1) and 0.02 g sulfo–NHS were added.
The suspension of MNP@APTES was added to the second solution and the pH was increased to 4.5 using 0.1 M NaOH. The solution was stirred at room temperature for 3 hours. After this, MNPs were washed with water several times, 1× with ethanol and afterwards two times with DI water. After each washing, MNPs were settled on a permanent magnet, except after the last washing with DI water (centrifugation: 8000 rpm, 10 min).
The TGA curve above shows a 17.94 wt% weight loss in air (Fig. 9(a)). A magnetite oxidation of 3.3% and a water loss of 2% up to 200 °C were considered in the calculation, and the estimated final weight loss of MNP@APTES@PAA was 19.24%. The TGA curve shows a weight loss of 2% up to 200 °C in the first step and a weight loss of 7.5% up to 300 °C (b.p. of APTES is 217 °C) in the second step. The final weight loss is attributed to the disintegration and oxidation of PAA above 600 °C. From these results, we can also conclude that MNP@APTES molecules were successfully coated with PAA. The total weight loss of the organic material deposited on the MNPs represents 19.24%. From the total weight loss, the amount of APTES loss (11.03%, Fig. 8) is subtracted and the loss is about 8.2%. With a weight loss of 8.2% and using the relation for ns, the number of estimated attached ligand molecules per MNP is about ns = 140 ligands per MNP (452.4 nm2) which is 1 ligand per 3.23 nm2. On the other hand, we have 1579 molecules of APTES per MNP which is 1 ligand per 0.286 nm2. So, the TGA and FTIR analysis showed that the MNPs are covered with 8.2% PAA forming MNP@APTES@PAA with about 140 molecules of PAA attached per particle. This is roughly 9% of all available sites (1579 per average MNP).
 | ||
| Fig. 9 a) TGA of MNP@APTES@PAA and b) FTIR spectra comparison of MNP@APTES (red curve) and MNP@APTES@PAA (black curve). | ||
One of the reasons for the small number of attached PAA molecules at free sites (APTES molecules) is most likely the large difference in molecular weight and thus the much bigger size of PAA molecules. The ratio of the molecular weight is ca. eight (1800 g mol−1/221 g mol−1), which means only a tenth of the occupancy of available free sites on MNPs.
Fig. 9(b) shows a comparison of the FTIR spectra for MNP@APTES (red) and MNP@APTES@PAA (black). The peak marked with an arrow in the black spectrum confirms that MNP@APTES are coated with PAA. The peaks at 1120–1060 cm−1 correspond to the C–O stretching of PAA. The strong absorption peak at 1735 cm−1 is attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode for carboxylate groups, which confirms the successful attachment of PAA on the MNP's surface. It was also observed that the absorbance of COOH peaks relatively decreased after dissociation, indicating that the COOH groups were dissociated into COO− groups.
O stretching mode for carboxylate groups, which confirms the successful attachment of PAA on the MNP's surface. It was also observed that the absorbance of COOH peaks relatively decreased after dissociation, indicating that the COOH groups were dissociated into COO− groups.
The isoelectric point for MNP@APTES@PAA is pH 3.16 (see Fig. 10(a)) which indicates that MNPs were coated with PAA. Fig. 10(b) shows the size distribution of MNP@APTES@PAA particles. The average size of MNP@APTES@PAA agglomerates of MNPs is 250 nm. Specifically, DLS identifies the agglomerates of functionalized particles of an initial average size of uncoated particles which is about 12 nm as examined using the TEM images. Particles are held together by physical attraction forces.
 | ||
| Fig. 10 a) Effect of the solution pH on the ζ-potential (black curve) and hydrodynamic radii (red curve) of MNP@APTES@PAA and b) the particle size distribution of MNP@APTES@PAA particles at pH 7. | ||
3.3. Surface-dissociated nanoparticles of MNP@APTES@PAA
In an aqueous solution at a neutral pH, PAA acts as an anionic polymer since many of the side chains of PAA can lose their protons and obtain a negative charge. This provides the PAA polyelectrolyte with the capability to absorb water which might increase its volume several times. With increasing pH, PAA has the potential to increase the number of PAA dissociated species which is accompanied by the ability to increase the osmotic pressure according to the increase in the number of dissociated particles. The IP of MNP@APTES@PAA is about pH 3. At higher pH > 6, the ionization of the polymer chains releases charges on chains inducing a strong electrostatic repulsion between particular chains, in which stretched polymer chains induce an extended polymer chain conformation, i.e., a brush-like conformation.32Below pH 3.5 PAA molecules lose their charges, and the electrostatic repulsion between chains diminishes, resulting in a collapse of the MNP@APTES@PAA brush configuration. MNPs agglomerate and the precipitation falls immediately out of dispersion.32 Here, steric inter-particle interactions caused by non-charged PAA are not sufficient to balance van der Waals attractive interactions.
This was further explored by measuring the osmotic pressure and DLS spectra of suspensions. The osmotic pressure of the MNP@APTES@PAA suspension as a function of pH was measured and the corresponding diameters were determined by DLS.
It is well-known that the polyelectrolyte analog of PSA being neutral in water must have a much greater dissociation capacity than PAA.22 A higher osmotic pressure and conductivity can be induced in a neutralized polyelectrolyte of PAA solutions resulting from dissociated carboxyl groups and grafted cations. Due to this, one is convinced that neutralization would improve the ability of the suspension to increase the colligative properties. We selected sodium hydroxide to change the pH of the suspension. The alkaline solution of NaOH was added dropwise under mechanical stirring into the MNP solution. The pH was increased, and the constitution of the suspension was examined using DLS, as seen from the diagram in Fig. 11. The osmotic pressure feebly increases with pH values and then notably decreases above pH 6. pH has a strong influence on the structure of the suspension. The average size of agglomerates decreases from 800 nm to 300 nm at pH 6 and is constant up to a high pH value of 11. The presence of an alkali in the solution increases the pH of MNP solutions and enables the dissociation of PAA functional groups from COOH into COO−.
 | ||
| Fig. 11 a) The osmotic pressure of the MNP@APTES@PAA suspension at different pH values and b) sizes of MNPs in the MNP@APTES@PAA suspension at different pH values. | ||
Specifically, a high concentration of dissociated species, i.e., the carboxyl groups, must be a result of dissociation at high pH, which would increase the electrostatic repulsion and decrease the average size of agglomerates and thus must accordingly increase the total amount of species governing the colligative properties. The osmotic pressure depends mostly on the number of charged species present in the DS. Up to pH 6 the osmotic pressure correlates with an increasing number of species due to increasing dissociation of PAA functional groups and due to the decrease of the average size of agglomerates and presumably their larger number. This is consistent with the general properties of osmosis and the mutual correlation between the properties of the solution and pH. However, the further decrease of osmotic pressure despite slight is not consistent with the general principles of osmosis.
The decreasing osmotic pressure must relate to the constitution of the composites in the suspension where the conformation of the hydrophilic ligands must have an impact. One of the options is a possible conformation of PAA chains which might screen the individual grafted cation species so that they do not behave as free independent species. The polyelectrolyte attached to the MNP may show conformational changes at certain concentrations. Simulations with the Monte Carlo (MC) method have shown that the high molecular weight of PAA chains showed more significant conformational changes at high pH values and an increasing ion concentration (high NaOH concentration).33 For PAA with fully charged chains, we can expect folded structures despite being attached to the MNP via APTES. In that case, the grafted cation species cannot be treated as completely free but were screened inside the folded structures and cannot fulfill the expected colligative activity. So, at high pH and consequently a higher ion concentration, the conformation on the attached polyelectrolyte chain can be expected in relation to the MC simulations. This might be a possible reason for the decrease of osmotic pressure by increasing the amount of sodium hydroxide and increasing the pH of the DS.
On the other hand, the stability of the DS osmotic pressure after FO is significant for the use of the DS, which is crucial for applications in FO. From the TGA diagram in Fig. 12, one can see that the weight losses of the composite MNP@APTES@PAA before and after FO are nearly the same. For comparison, the TGA diagram of the as-prepared composite is shown.
 | ||
| Fig. 12 Mass loss of MNP@APTES@PAA (red curve), MNP@APTES@PAA after first FO (blue curve) and MNP@APTES@PAA after second FO (black curve). | ||
From the TGA results, one can see that during the first and second FO processes there is not a substantial loss of attached ligands and that the MNPs remained fully coated with PAA ligands after the first and second FO processes.
The osmotic pressure measurements confirmed the TGA results. After the MNPs were recovered from the magnetic fluid (recycled from DS) – by separation with a permanent magnet –MNP@APTES@PAA was cleaned with water and washed with ethanol to remove possible hydrogen bonding and re-dispersed in water. Depending on the concentration, the osmotic pressure of the recycled DS solution remained constant. In Table 1 we can see that the specific osmotic pressure stayed constant when the composite particles were reused.
| MNP@APTES@PAA | Concentration [g L−1] | Osmotic pressure [bar] | Osmotic pressure/concentration [bar L g−1] | Ref. |
|---|---|---|---|---|
| As-synthesized (before FO) | 5.9 | 9.19 | 1.56 | This work |
| MNP (after first FO), rinsed with EtOH and redispersed in water | 4.2 | 6.77 | 1.61 | This work |
| MNP (after second FO), rinsed with EtOH and redispersed in water | 3.7 | 5.96 | 1.61 | This work |
| *MNP@PSA | 70 | 9 | 0.13 | 26 |
| As-synthesized (before FO) | ||||
| *MNP@CA | 37 | 18.7 | 0.51 | 24 |
| As-synthesized (before FO) | ||||
| *MNP@CA | 14.2 | 4.62 | 0.33 | 24 |
| MNP (after first FO) |
In Table 1 the osmotic pressure is presented in dependence of 2 cycles of DS action. The results show the reproducibility of the MNP@APTES@PAA composite particles as a DS. The osmotic pressures of MNP@PSA and MNP@CA are presented for comparison. As we can see, the parameter osmotic pressure/concentration of MNP@PSA and MNP@CA is much smaller in comparison with MNP@APTES@PAA. The obtained results also confirm our prediction and the aim of our work that firmly bonded ligands will allow for the reuse of composite particles exhibiting a permanent and relatively high osmotic pressure for use in FO applications.
3.4. FO filtrations
Two FO filtrations were performed using freshly synthesized and reused MNP@APTES@PAA as a DS. In Fig. 13(a and b), the results from the water flux versus time and water flux versus recovery, respectively, are presented while in Fig. 13(c and d), the conductivity of the FS and osmotic pressures of the FS and DS for both filtrations are presented. | ||
| Fig. 13 a) Water flux versus time and b) water flux versus recovery during FO processes using freshly synthesized and reused MNP@APTES@PAA as a DS; c) conductivity of the FS and d) osmotic pressures of the FS and DS for both filtrations. | ||
For the first filtration, we used 250 mL of DI water as a FS and 95 mL of 0.6% freshly prepared suspension of MNP@APTES@PAA as a DS. The driving force of the MNP@APTES@PAA suspension was 8.9 bar. During the first filtration, the water flux decreased from a max value of 4.1 LMH to 1.8 LMH in two hours. During that time, 64.1 mL of water (water recovery was 25.7%) passed through the membrane, which corresponds to an increase in the dilution factor of the FS from 1 to 1.7. The osmotic pressure of the FS after two hours increased to 2.1 bar while an increment in conductivity from 1.0 μS cm−1 to 4.9 μS cm−1 was noticed. The osmotic pressure difference between the DS and FS at the end of filtration was 0.4 bar.
The sedimentation process of diluted MNP@APTES@PAA was done with a magnet, followed by a rinsing procedure with EtOH, and then dilution to 200 mL with DI water. The particle recovery from diluted MNP@APTES@PAA was 67.66%. The osmotic pressure of the re-concentrated DS was 6.8 bar, which is lower than the initial osmotic pressure of the MNP@APTES@PAA used in the first run. During recycling, degradation occurs, i.e. there was a decrease of the osmotic pressure, primarily due to the release of physically bound hydrophilic ligands from the functionalized surface. After the release of physically bound ligands, the firmly bonded PAA monolayer remains attached which then determines the final osmotic pressure.
The second filtration was performed using 500 mL of DI water as a FS and 195 mL of 0.4% suspension of MNP@APTES@PAA, reused from the first filtration, as a DS. During FO filtration the water flux decreased from a max value of 3.0 LMH to 1.9 LMH in two hours. During this time, 67.6 mL of water passed through the membrane (water recovery was 13.6%) which corresponded to an increased dilution factor of the DS from the starting value of 1 to 1.3. The FS osmotic pressure increased to 1.1 bar in two hours with a concomitant increment in conductivity from 1.0 μS cm−1 to 2.3 μS cm−1 indicating that some released PAA ligands were present and diffused through the membrane. However, this release is significantly lower than during the first filtration. As a result, the osmotic potential between the FS and DS at the end of filtration was still 1.5 bar. The water recovery from the first filtration was 25.7% and from the second water filtration was 13.6%, after 2 h of filtration time (Fig. 13(b)).
4. Conclusion
The synthesised APTES coated superparamagnetic iron-oxide MNPs form a strong covalent (peptide) bond with carboxyl groups of PAA with the help of an EDC crosslinker. The resulting MNP@APTES@PAA nanocomposites exhibit good colloidal stability in aqueous solution with a concentration-normalized osmotic pressure of 1.56 bar L g−1. This is 12-fold higher than that in our previous studies of PSA coated MNPs and 3-fold higher than that of CA coated MNPs. DLS measurements show the hydrophilic nature and surface chemistry of the nanocomposites. The measurements of the osmotic pressure and size of the nanocomposites at different pH values show that pH has a strong influence on the structure of the suspension. XRD analysis reveals a magnetite crystal structure and TEM analysis shows a spherical (∼12 nm diameter) morphology. Stepwise FTIR, TGA, and IP measurements confirmed the MNP@APTES@PAA structure. The estimated number of ligands per particle (based on TGA analysis) revealed that only about 9% of all available sites (–NH2) of APTES molecules are occupied by PAA molecules (COO–), forming MNP@APTES@PAA. One of the reasons for the small number of bound PAA molecules in “free” sites (APTES molecules) is most likely the large difference in molecular weight of the two ligands.The measured osmotic pressure of MNP@APTES@PAA was 9.19 bar (with a concentration of 5.96 g L −1). We performed two FO filtrations using two different DSs. TGA analyses show that the weight losses of the MNP@APTES@PAA composite before and after FO are nearly the same. The results showed the reproducibility of the MNP@APTES@PAA composite particles as a DS, which confirmed our prediction about the bound ligands.
Author contributions
Conceptualization: M. D., C. H.-N. and I. B.; methodology: S. V., H. B. and I. P.; software: H. B. and S. V.; validation: S. G., I. P., I. B. and C. H.-N.; formal analysis: S. V., H. B. and S. G.; investigation: M. D., S. V., and I. P.; resources: I. B., I. P. and C. H.-N.; data curation; S. V. and H. B.; writing – original draft preparation: M. D., I. B., I. P. and J. S.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was funded by the Slovenian National Agency (grant numbers P3-0036, P2-0006, P2-0089, J1-9169, and J3-1762, L4-1843) and NEPWAT funded by the Novo Nordisk Foundation.References
- D. Johnson, W. Ali, A. Mohammed and N. Hilal, Osmotic's Potential: An Overview of Draw Solutes for Forward Osmosis, Desalination, 2017 DOI:10.1016/j.desal.2017.09.017.
- T. Y. Cath, A. E. Childress and M. Elimelech, Forward osmosis: Principles, applications, and recent developments, J. Membr. Sci., 2006, 281, 70–87 CrossRef CAS.
- N. Akther, A. Sodiq, A. Giwa, S. Daer, H. Arafat and S. Hasan, Recent advancements in forward osmosis desalination: A review, Chem. Eng. J., 2015, 281, 502–522 CrossRef CAS.
- Q. Long, Y. Jia, J. Li, J. Yang, F. Liu and J. Zheng, et al. Recent Advance on Draw Solutes Development in Forward Osmosis, Processes, 2018, 6(9), 165 CrossRef CAS.
- L. Chekli, S. Phuntsho, H. K. Shon, S. Vigneswaran, J. Kandasamy and A. Chanan, A review of draw solutes in forward osmosis process and their use in modern applications, Desalin. Water Treat., 2012, 43(1–3), 167–184 CrossRef CAS.
- M. Elimelech and W. A. Phillip, The Future of Seawater Desalination: Energy, Technology, and the Environment, Science, 2011, 333(6043), 712–717 CrossRef CAS PubMed.
- S. Zhao, L. Zou, C. Y. Tang and D. Mulcahy, Recent developments in forward osmosis: Opportunities and challenges, J. Membr. Sci., 2012, 396, 1–21 CrossRef CAS.
- K. P. Lee, T. C. Arnot and D. Mattia, A review of reverse osmosis membrane materials for desalination—Development to date and future potential, J. Membr. Sci., 2011, 370(1), 1–22 CrossRef CAS.
- T. Alejo, M. Arruebo, V. Carcelen, V. M. Monsalvo and V. Sebastian, Advances in draw solutes for forward osmosis: Hybrid organic-inorganic nanoparticles and conventional solutes, Chem. Eng. J., 2017, 309, 738–752 CrossRef CAS.
- P. Xu, G. M. Zeng, D. L. Huang, C. L. Feng, S. Hu and M. H. Zhao, et al. Use of iron oxide nanomaterials in wastewater treatment: a review, Sci. Total Environ., 2012, 424, 1–10 CrossRef CAS PubMed.
- M. M. Ling, K. Y. Wang and T.-S. Chung, Highly Water-Soluble Magnetic Nanoparticles as Novel Draw Solutes in Forward Osmosis for Water Reuse, Ind. Eng. Chem. Res., 2010, 49, 5869–5876 CrossRef CAS.
- Q. Ge, L. Yang, J. Cai, W. Xu, Q. Chen and M. Liu, Hydroacid magnetic nanoparticles in forward osmosis for seawater desalination and efficient regeneration via integrated magnetic and membrane separations, J. Membr. Sci., 2016, 520 DOI:10.1016/j.memsci.2016.07.033.
- Q. Ge, J. Su, T.-S. Chung and G. Amy, Hydrophilic Superparamagnetic Nanoparticles: Synthesis, Characterization, and Performance in Forward Osmosis Processes, Ind. Eng. Chem. Res., 2011, 50(1), 382–388 CrossRef CAS.
- S. Y. Park, H.-W. Ahn, J. W. Chung and S.-Y. Kwak, Magnetic core-hydrophilic shell nanosphere as stability-enhanced draw solute for forward osmosis (FO) application, Desalination, 2016, 397, 22–29 CrossRef CAS.
- G. Gwak, B. Jung, S. Han and S. Hong, Evaluation of poly (aspartic acid sodium salt) as a draw solute for forward osmosis, Water Res., 2015, 80, 294–305 CrossRef CAS PubMed.
- D. Zhao, S. Chen, P. Wang, Q. Zhao and X. Lu, A Dendrimer-Based Forward Osmosis Draw Solute for Seawater Desalination, Ind. Eng. Chem. Res., 2014, 53(42), 16170–16175 CrossRef CAS.
- Y. Na, S. Yang and S. Lee, Evaluation of citrate-coated magnetic nanoparticles as draw solute for forward osmosis, Desalination, 2014, 347, 34–42 CrossRef CAS.
- H. Bai, Z. Liu and D. D. Sun, Highly water soluble and recovered dextran coated Fe3O4 magnetic nanoparticles for brackish water desalination, Sep. Purif. Technol., 2011, 81, 392–399 CrossRef CAS.
- D. Li and H. Wang, Smart draw agents for emerging forward osmosis application, J. Mater. Chem. A, 2013, 1(45), 14049–14060 RSC.
- Q. Ge, J. Su, G. L. Amy and T. S. Chung, Exploration of polyelectrolytes as draw solutes in forward osmosis processes, Water Res., 2012, 46(4), 1318–1326 CrossRef CAS PubMed.
- M. Ling and T.-S. Chung, Desalination process using super hydrophilic nanoparticles via forward osmosis integrated with ultrafiltration regeneration, Desalination, 2011, 278, 194–202 CrossRef CAS.
- M. M. Ling and T.-S. Chung, Surface-Dissociated Nanoparticle Draw Solutions in Forward Osmosis and the Regeneration in an Integrated Electric Field and Nanofiltration System, Ind. Eng. Chem. Res., 2012, 51(47), 15463–15471 CrossRef CAS.
- C. L. Lin, C. F. Lee and W. Y. Chiu, Preparation and properties of poly(acrylic acid) oligomer stabilized superparamagnetic ferrofluid, J. Colloid Interface Sci., 2005, 291(2), 411–420 CrossRef CAS PubMed.
- I. Petrinic, J. Stergar, H. Bukšek, M. Drofenik, S. Gyergyek and C. Hélix-Nielsen, et al. Superparamagnetic Fe(3)O(4)@CA Nanoparticles and Their Potential as Draw Solution Agents in Forward Osmosis, Nanomaterials, 2021, 11(11), 2965 CrossRef CAS PubMed.
- Y. C. Kim, S. Han and S. Hong, A feasibility study of magnetic separation of magnetic nanoparticle for forward osmosis, Water Sci. Technol., 2011, 64(2), 469–476 CrossRef CAS PubMed.
- I. Ban, S. Markuš, S. Gyergyek, M. Drofenik, J. Korenak and C. Helix-Nielsen, et al. Synthesis of Poly-Sodium-Acrylate (PSA) Coated Magnetic Nanoparticles for Use in Forward Osmosis Draw Solutions, Nanomaterials, 2019, 9(9), 1238 CrossRef CAS PubMed.
- I. Petrinic, H. Bukšek, I. Galambos, R. Gerencsér-Berta, M. Sheldon and C. Helix-Nielsen, Removal of naproxen and diclofenac using an aquaporin hollow fibre forward osmosis module, Desalin. Water Treat., 2020, 192, 415–423 CrossRef CAS.
- M. H. Mashhadizadeh and M. Amoli-Diva, Drug-Carrying Amino Silane Coated Magnetic Nanoparticles as Potential Vehicles for Delivery of Antibiotics, J. Nanomed. Nanotechnol., 2012, 3, 4 Search PubMed.
- R. K. Wardani, K. Dahlan, S. T. Wahyudi and S. G. Sukaryo, Synthesis and characterization of nanoparticle magnetite for biomedical application, AIP Conf. Proc., 2019, 2194(1), 020137 CrossRef.
- M. Wiśniewska, A. Nosal-Wiercińska, I. Ostolska, D. Sternik, P. Nowicki and R. Pietrzak, et al. Nanostructure of Poly(Acrylic Acid) Adsorption Layer on the Surface of Activated Carbon Obtained from Residue After Supercritical Extraction of Hops, Nanoscale Res. Lett., 2017, 12(1), 2–4 CrossRef PubMed.
- J. Sanders and P. Gallagher, Thermomagnetometric evidence of γ-Fe2O3 as an intermediate in the oxidation of magnetite, Thermochim. Acta, 2003, 406, 241–243 CrossRef CAS.
- J. Fresnais, M. Yan, J. Courtois, T. Bostelmann, A. Bée and J. F. Berret, Poly(acrylic acid)-coated iron oxide nanoparticles: quantitative evaluation of the coating properties and applications for the removal of a pollutant dye, J. Colloid Interface Sci., 2013, 395, 24–30 CrossRef CAS PubMed.
- F. Carnal and S. Stoll, Chain stiffness, salt valency, and concentration influences on titration curves of polyelectrolytes: Monte Carlo simulations, J. Chem. Phys., 2011, 134(4), 044909 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2023 |