
Enhanced oxygen activation on an atomically dispersed Au catalyst with dual active sites for room-temperature formaldehyde oxidation†
Huixia
Li
a,
Shaofan
Fang
b,
Guimin
Jiang
a and
Zuotai
Zhang
 *a
*a
aSchool of Environmental Science and Engineering, Guangdong Provincial Key Laboratory of Soil and Ground Water Pollution Control, Southern University of Science and Technology, Shenzhen 518055, People's Republic of China. E-mail: zhangzt@sustech.edu.cn
bInternational Collaborative Laboratory of 2D Materials for Optoelectronics Science and Technology of Ministry of Education, Institute of Microscale Optoelectronics, Shenzhen University, Shenzhen 518060, P. R. China
First published on 25th October 2022
Abstract
Formaldehyde (HCHO) is known to be a hazardous indoor air pollutant. Noble metal catalysts offer a feasible approach for indoor HCHO decomposition. Oxygen activation via a noble metal such as gold plays a critical role in realizing the total HCHO oxidation, but then it is vague how different-sized noble metals function synergistically on oxygen activation. Here, an Au/CeO2 catalyst with atomically dispersed Au (Au/CeO2-550) was successfully prepared through a thermal aging treatment, and a synergistic mechanism for the HCHO oxidation reaction involving co-loading of single Au atoms and Au nanoclusters on CeO2 was revealed. Strikingly, the HCHO oxidation performance of the obtained Au/CeO2-550 catalyst at room temperature was significantly greater than that of other Au/CeO2 catalysts, which was attributed to a concerted catalysis mechanism of the individual Au atoms and nanoclusters. The surface oxygen species of CeO2 activated by single Au atoms can promptly transform HCHO into dioxymethylene species, while Au nanoclusters well adsorb and activate molecular oxygen to oxidize the dioxymethylene into formate, which is further oxidized into carbonates, and the carbonates are eventually decomposed into CO2 and H2O. This synergy results in enhanced HCHO oxidation at surface sites between neighboring single Au atoms and Au nanoclusters.
Environmental significanceWith the development of society, people spend more and more time indoors, and indoor air quality has been an increasingly more important issue affecting human health. Formaldehyde (HCHO) is extremely toxic and universal in indoor environments with a variety of emission sources, and regarded as a priority carcinogenic compound. Therefore, it is vital to remove indoor HCHO in order to meet human health needs. To date, catalytic oxidation at room temperature is the most promising approach to indoor HCHO removal because of the high efficiency and zero energy input, as enabled by well-designed catalysts. Thus, in this study, we prepared an atomically dispersed Au catalyst with dual active sites and found a new synergistic oxygen activation pathway and reaction mechanism for realizing room-temperature HCHO oxidation. |
1. Introduction
HCHO is extensively regarded as a hazardous indoor air pollutant, and it is primarily emitted from used building and decoration materials.1–3 Prolonged and sustained exposure to HCHO poses a significant threat to human health, and it can cause many serious health problems, such as cancer, skin irritation, and respiratory diseases.4,5 Catalytic oxidation, due to its high efficiency and zero energy input, is of great significance for air cleaning in an enclosed environment.6–8 Hence, developing effective catalysts to remove indoor-air HCHO, as well as understanding the catalytic reaction mechanism, is significant to conform to the air-quality requirements and reduce public health risks.Rational design of catalysts with active sites for several complex elementary reaction steps can significantly improve the catalytic performance in many chemical processes.9,10 In general, among catalyst systems with multistep reactions, pure single-site catalysts could show poor catalytic performance as they may only activate one of several elementary reaction steps.11 Hence, combining multi-active sites into one catalyst is an effective method to accelerate catalytic reaction processes via synergistic effects involving various active sites.12–14 However, constructing catalysts with multiple active sites and studying the corresponding catalytic redox reaction mechanisms remain challenging. Therefore, including a range of active metal species within a catalyst may offer the chance to construct atomic catalysts with multiple active sites, leading to efficient charge transfers during multistep reactions.15,16 For example, Pt-GT-1 was found to be a highly efficient catalyst owing to the presence of both single Pt atoms and Pt clusters, which synergistically enhanced the conductivity to facilitate the hydrogen evolution reaction (HER).17 Meanwhile, the group of Ding Ma demonstrated that the (Pt1–Ptn)/α-MoC structure has high mass-specific activity and a good anti-oxidation capability that prevents deactivation during the water–gas shift (WGS) reaction.18 For the HCHO catalytic oxidation, noble metal catalysts have also shown excellent performance, owing to efficient O2 activation, and numerous studies on the exploitation of supported noble metal catalysts with higher dispersion and reduced loading have been reported.19–22 Supported Au catalysts generally possess superior catalytic activities because Au can excellently activate surface oxygen species of metal oxide supports to achieve outstanding HCHO oxidation performance.23–25 Besides, some single-atom Au catalysts have also demonstrated good catalytic performance for HCHO oxidation, because Au atoms can activate surface lattice oxygen and promote migration of bulk lattice oxygen, thus generating surface active oxygen species for HCHO oxidation.26 Therefore, the single Au atoms and Au nanoparticles both play positive roles in the HCHO oxidation process, but the definite interaction mechanisms have not been clarified for the case in which the two species are integrated within one catalyst. In particular, it is not clear how individual Au atoms and Au nanoclusters can function synergistically to activate oxygen in the HCHO catalytic oxidation.
Herein, we report the use of a thermal aging method to fabricate an atomically dispersed Au catalyst supported on CeO2 nanorods for catalyzing the HCHO oxidation reaction. Our aim was to clarify the nature of the synergistic effect of single Au atoms and Au nanoclusters in activating oxygen. Impressively, the Au/CeO2 sample calcined at 550 °C, with Au nanoclusters and single Au atoms, displays a room-temperature HCHO oxidation activity that is outstanding being much superior to those of thermally aged Au/CeO2 catalysts. We further examined the mobility and reactivity of the surface oxygen species on the catalysts and O2 activation, to explain the synergistic mechanisms involving the single Au atoms and Au nanoclusters in the HCHO oxidation reaction. The result of this study may help to advance the field of efficient supported dual-site noble metal catalyst design, leading to the elimination of HCHO and other harmful pollutants from air.
2. Results and discussion
2.1 Morphology, structure, and component analysis
A schematic illustration for the preparation of the Au/CeO2-550 catalyst is displayed in Fig. 1a. Briefly, all the precursors were well mixed and then hydrothermally heated at 120 °C to obtain CeO2 nanorod supports. Subsequently, the CeO2 support was impregnated with the Au precursor solution without any change to its macroscopic shape, to obtain the Au/CeO2 sample, which was then calcined at a temperature of 550 °C (Fig. 1a). Details of the synthetic process are described in the Experimental section (ESI†). For characterization, first, the morphology and atom arrangement within the prepared Au/CeO2-550 catalyst were observed carefully. From the HAADF-STEM images shown in Fig. 1b and c, it is apparent that the morphology of the CeO2 supports remained unchanged after Au loading. Moreover, the analysis of the enlarged high magnification HAADF-STEM image and the Fourier transform image of region II (Fig. 1d and e) revealed that the crystalline CeO2 support of the Au/CeO2-550 catalyst had a d-spacing of 0.31 nm, corresponding to the (111) surface (insets in Fig. 1d and e). Note that Au nanoparticles are not observed on the Au/CeO2-550 surface shown in Fig. 1c, but several individual well-separated Au atoms are seen on the surface of the CeO2 nanorods shown in Fig. 1f. The larger-scale HAADF-STEM image and the corresponding EDS mapping images of the Au/CeO2-550 catalyst in Fig. S2 (ESI†) also indicate no Au aggregation, with Au atoms apparently homogenously dispersed over the CeO2 nanorods. To further confirm the above observation, the enlarged high magnification HAADF-STEM image (Fig. 1g), energy-dispersive X-ray spectroscopy (EDS) mapping images (Fig. 1h), and the representative AC HAADF-STEM image (Fig. 1i) of the Au/CeO2-550 catalyst were obtained. The chemical element distributions in the Au/CeO2-550 sample clearly show that the Au species are highly dispersed on the CeO2 nanorods (Fig. 1h). In fact, the Au species are presented in the form of Au nanoclusters and single Au atoms. The Au atoms were doped into the CeO2 lattice (Fig. 1i). The Au atoms of Au/CeO2-550 at the interface of nanocluster/support migrated from the Au nanoclusters, steadied by the oxygen coordination at defect sites in the CeO2 lattice, as previous literature reported.27 However, the Au species exhibit different sizes on the surface of the CeO2 support because of the thermal treatment at different temperatures, as shown in Fig. S3.†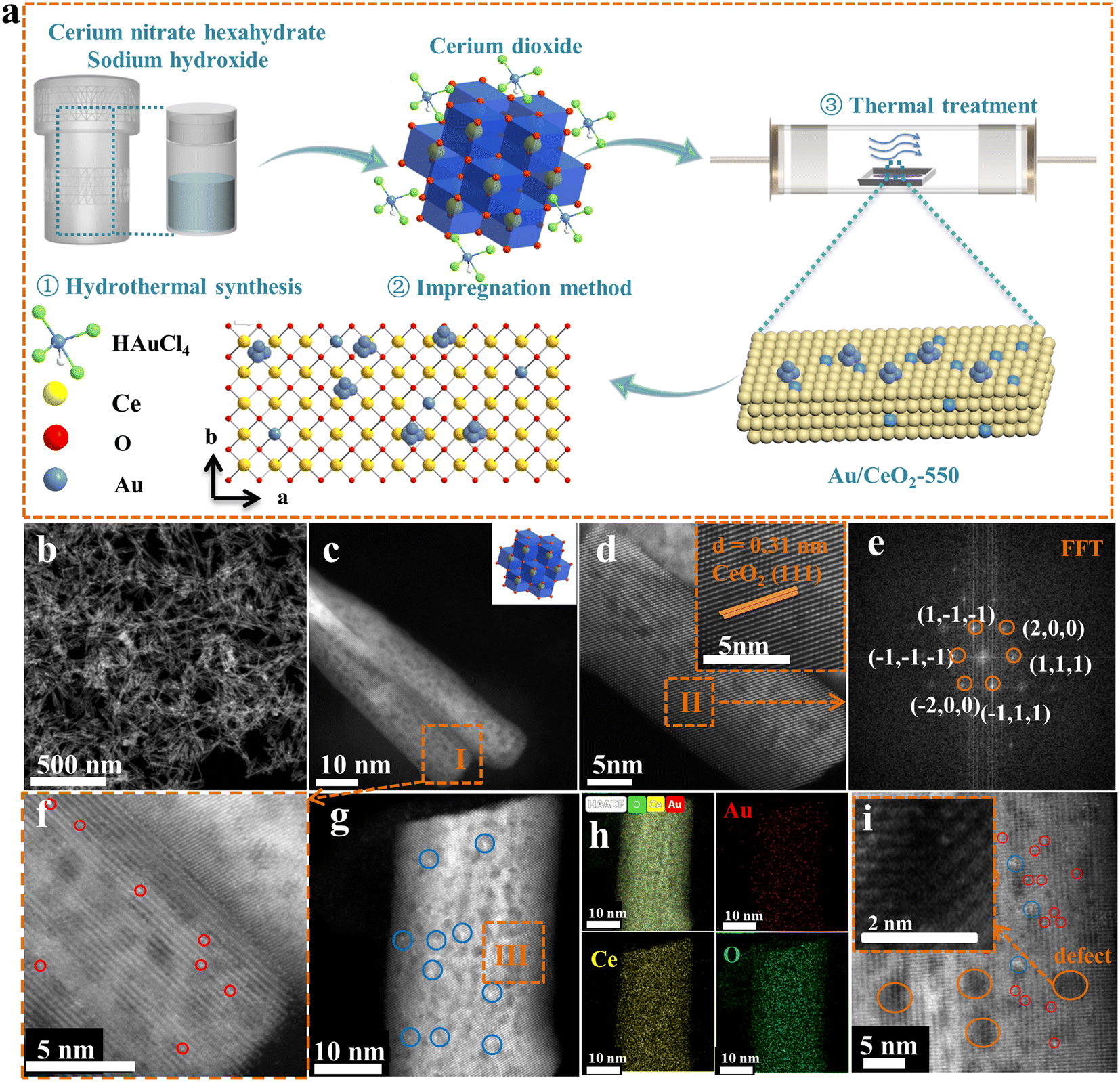 | ||
| Fig. 1 Preparation and characterization of the representative sample Au/CeO2-550. (a) Schematic illustration of the fabrication process. (b) Low-magnification HAADF-STEM image. (c) High magnification HAADF-STEM image and inset: the corresponding structure diagram. (d) Enlarged high magnification HAADF-STEM image and inset: the corresponding lattice spacing image. (e) FFT result of the area outlined by the rectangle overlaid on panel (d). (f) Representative AC HAADF-STEM image of the area indicated by the overlaid dashed line in panel (c). (g) Enlarged high magnification HAADF-STEM image and (h) energy-dispersive X-ray spectroscopy (EDS) mapping images of the same area. (i) Representative AC HAADF-STEM image (the overlaid red, blue, and orange circles indicate the location of single Au atoms, Au nanoclusters, and oxygen defects, respectively) and inset: the corresponding oxygen defect image. | ||
The phase structures and compositions of the prepared composites were further identified by X-ray diffraction (XRD) and Raman spectroscopy. A strong CeO2 diffraction peak (PDF No. 34-0394) is seen in the XRD pattern of each catalyst (Fig. 2a).28 A comparison of the XRD patterns of the samples revealed that the highest diffraction peak, at 28.5° (corresponding to the (111) lattice plane of CeO2 nanorods), became stronger and was dramatically shifted. This indicates that Au loading led to some Au cations being successfully doped into the CeO2 lattice. Specifically, the diffraction peak of the (111) lattice plane for Au/CeO2 was shifted to a lower angle compared with that of CeO2, suggesting that the cell parameter change may be caused by ceria doping with Au+ and Ce3+, which is expected because the size of the Ce4+ cations (Ce4+: 0.097 nm)29 is smaller than those of Au+ (Au+: 0.137 nm) and Ce3+ (Ce3+: 0.114 nm). In contrast, doping with Au3+ (cation size: 0.070 nm) results in decreased ceria cell parameters, leading to increased diffraction peak angles, as was observed for the Au/CeO2-350 and Au/CeO2-550 catalysts. In addition, for Au/CeO2-750, the cell parameters are similar to those for CeO2, which may be because Au was partially segregated and formed larger Au and Au2O3 nanoparticles. Evidence of such a segregated Au (111) phase was observed in the diffractogram of the Au/CeO2-750 catalyst but not in the diffractograms of the other catalysts and Au3O2 nanoparticles were observed in the HRTEM image (Fig. S4†). This finding is consistent with the above-discussed electron microscopy results for the Au/CeO2, Au/CeO2-350 and Au/CeO2-550 catalysts (Fig. 1, S2 and S3†).
 | ||
| Fig. 2 (a) The XRD patterns (left) and expanded regions of the XRD patterns in the range of 27–30° (right) of CeO2, Au/CeO2, Au/CeO2-350, Au/CeO2-550 and Au/CeO2-750 catalysts (the asterisk represents the Au diffraction peak). (b) Raman spectra of the same catalysts. | ||
Raman spectroscopy could investigate the structural properties of metal oxidation materials because of its high sensitivity to M–O bond arrangement.30Fig. 2b presents the Raman spectra of the CeO2, Au/CeO2, Au/CeO2-350, Au/CeO2-550 and Au/CeO2-750 catalysts. A prominent band at ∼455.5 cm−1, seen in the spectrum of pure CeO2, was assigned to the F2g Raman-active mode of fluorite-structured CeO2, confirming our XRD observations.31 This band can be assigned to the breathing mode of oxygen around Ce4+ cations.32 The shift of this band position offers information about the introduction of foreign cations into the ceria lattice. A shift of the F2g band toward lower values was observed when foreign cations with sizes larger than Ce4+ were inserted into the lattice, while a shift toward higher values was observed when smaller cations were present in the lattice.33 Therefore, the lower values of the F2g band Raman shifts observed for our catalysts suggest the presence of large cations. Intriguingly, the F2g band of Au/CeO2 was shifted to lower wavenumbers (442.6 cm−1), whereas the F2g bands of Au/CeO2-350 (460.9 cm−1), Au/CeO2-550 (461.7 cm−1) and Au/CeO2-750 (459.4 cm−1) were shifted to higher wavenumbers compared to that of CeO2 (455.5 cm−1). This F2g band shift trend resembles the observed XRD peak shift trend, which can be explained by considering the incorporation of Au ions into the CeO2 lattice.
Based on the above analyses, doping is confirmed for the Au/CeO2 composites with heterostructure interfaces and CeO2-induced interfacial Auδ+ site enrichment. The greatest amount of Au3+ doping within the parent ceria framework was achieved in the Au/CeO2-550 catalyst.
To further examine the Au valence state information on the surface and the surface compositions of the as-prepared catalysts, Au 4f and O 1s XPS analyses were performed (Fig. 3). The Au 4f spectra contain two principal peaks: a high binding energy peak (Au 4f5/2) and a low binding energy peak (Au 4f7/2). These two peaks can be further subdivided into three main peaks for the above samples, as shown in Fig. 3a. The Au 4f7/2 peaks at binding energies of 83.2–83.7 eV were assigned to Au0, while the signals at higher binding energies in the regions of 84.4–85.0 eV and 86.2–86.6 eV corresponded to Au+ and Au3+, respectively.34 According to the deconvolution calculation results displayed in Fig. 3c, the Au3+ content in the Au/CeO2-550 sample (32.2%) was much greater than those in the Au/CeO2, Au/CeO2-350, and Au/CeO2-750 samples. In addition, the Au/CeO2-550 catalyst contained the lowest Au0 content (28.9%). Meanwhile, the Au/CeO2-750 catalyst was found to contain a high surface Au0 content of 60.4%. The TEM results (Fig. S3†) indicate that the Au nanoparticle size is larger in the sample than that in the other catalyst samples. Additionally, it is observed that the Au 4f7/2 binding energy for Au/CeO2, Au/CeO2-350, and Au/CeO2-550 was shifted higher than that of the Au/CeO2-750 sample, which might reflect the differences in the coordination number of the Au particles of different sizes, the electronic state of Au, and the interaction between Au and the support.35 These results are consistent with the above-described HAADF-STEM (Fig. 1), XRD (Fig. 2a), and Raman (Fig. 2b) characterization. According to previous research,5 the existence of Auδ+ on the surface of supports results in greater oxygen species activation during oxidation reactions, which explains the excellent HCHO oxidation activity of the Au/CeO2-550 catalyst.
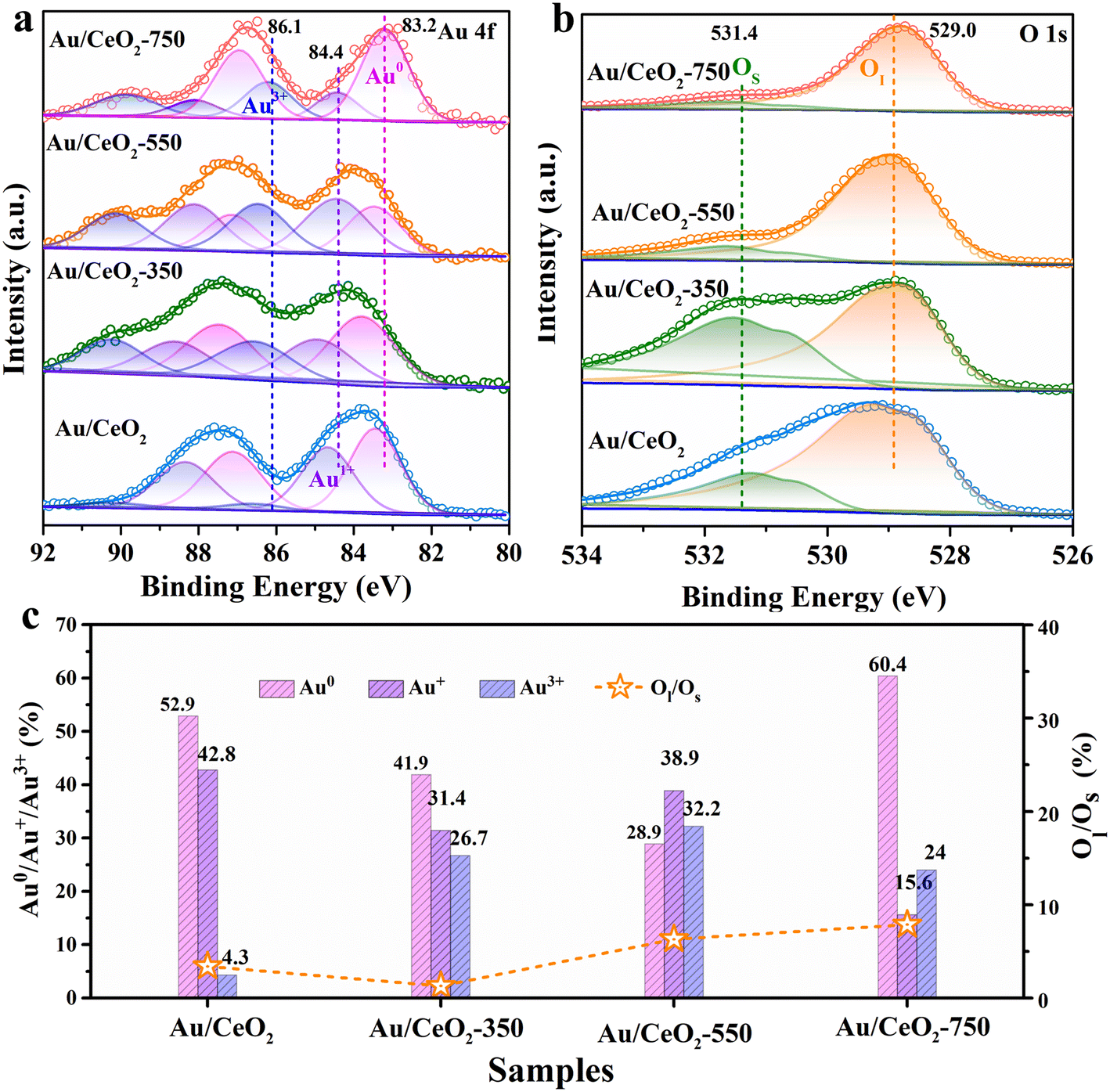 | ||
| Fig. 3 (a) XPS Au 4f and (b) O 1s spectra of the catalysts. (c) Au0/Au+/Au3+ species content and the ratio of lattice oxygen to surface oxygen (Ol/Os) of each catalyst as obtained via XPS fitting. | ||
All the O 1s XPS spectra in Fig. 3b contain two peaks, a sharp one at 529.0 eV and a broad one at 531.4 eV, which are attributed to surface lattice oxygen (Ol) and surface adsorbed oxygen (Os), respectively.36 A comparison of the areas of fitted peak areas revealed the ratios and relative contents of different oxygen species. As shown in Fig. 3c, the Ol/Os ratios of the Au/CeO2-550 and Au/CeO2-750 catalysts were higher than those of the other two catalysts. HCHO oxidation over the CeO2-based catalysts followed the Mars–van-Krevelen mechanism, in which Ol plays a key role. Furthermore, the Auδ+ species is considered to be an active site for substrate activation, so in the presence of a larger Auδ+ content, a higher content of surface lattice oxygen can be activated to achieve excellent HCHO oxidation performance.
2.2 Optimizing oxygen activation and reduction ability
The surface reactive oxygen species and reducibility of the CeO2, Au/CeO2, Au/CeO2-350, Au/CeO2-550 and Au/CeO2-750 catalysts were assessed by O2-TPD and H2-TPR. To further characterize the oxygen desorption behaviors, surface reactive oxygen species, and lattice oxygen mobility of the samples, O2-TPD measurements were carried out (Fig. 4a). Usually, bulk oxygen vacancies are produced from adsorbed surface oxygen species via the subsequent route: O2 (ads) → O2− (ads) → O− (ads) → O2− (ads/lattice).37–39 O2 (ads), i.e. physisorption oxygen, can be removed by argon purification as a pretreatment.40 Surface-adsorbed peroxy species (O2−) and monatomic species (O−), corresponding to the surface defects, desorbed in the temperature ranging from 30 to 250 °C and 280 to 340 °C, respectively.41 O2− was found to be the surface or bulk lattice oxygen species at temperatures above 350 °C.42 Three different desorption oxygen species were presented for CeO2, Au/CeO2, Au/CeO2-350, Au/CeO2-550 and Au/CeO2-750. In order to verify that the desorbed peaks are from oxygen species of the above catalysts during O2-TPD measurement, we analyzed the desorbed gas by mass spectrometry for Au/CeO2-550 (Fig. S5†). The analysis results show that there are no other impurity gas desorption i.e. CO2, H2 and CO except for oxygen, indicating that the impurity gas on the catalyst surface has been completely removed by pretreatment. Thus, the peak at below 200 °C corresponded to the surface adsorbed peroxy species O2− (ads), and that centered at 200–450 °C corresponded to O− and surface lattice oxygen. The bulk lattice oxygen was desorbed at a temperature above 500 °C. Note that for Au/CeO2-550, a larger desorption peak was observed at lower temperatures, and the desorption peaks were much larger below 200 °C, indicating that Au/CeO2-550 has more abundant and more easily migrating surface-adsorbed oxygen species, owing to the synergistic Au single-atom and nanocluster activation on CeO2.43 The peak in the range of 210–350 °C corresponds to the surface adsorbed monatomic species, O− (ads), and that centered at 350–500 °C is surface lattice oxygen. The bulk lattice oxygen desorption peak of Au/CeO2-550 appeared at an unexpectedly lower temperature and was much wider and more intense than those of CeO2, Au/CeO2, Au/CeO2-350 and Au/CeO2-750, suggesting that single-Au-atom loading can remarkably advance the mobility of lattice oxygen, on account of Au doping within the CeO2 lattice.37 Considering the temperature range of the HCHO oxidation reaction, chemisorbed surface oxygen species (O2− and O−) and surface lattice oxygen are expected to be the principal active oxygen species. Thus, the synergistic enhancement of oxygen activation of single Au atoms and Au nanoclusters is likely to be the main reason for the outstanding catalytic performance of this catalyst in the HCHO oxidation reaction.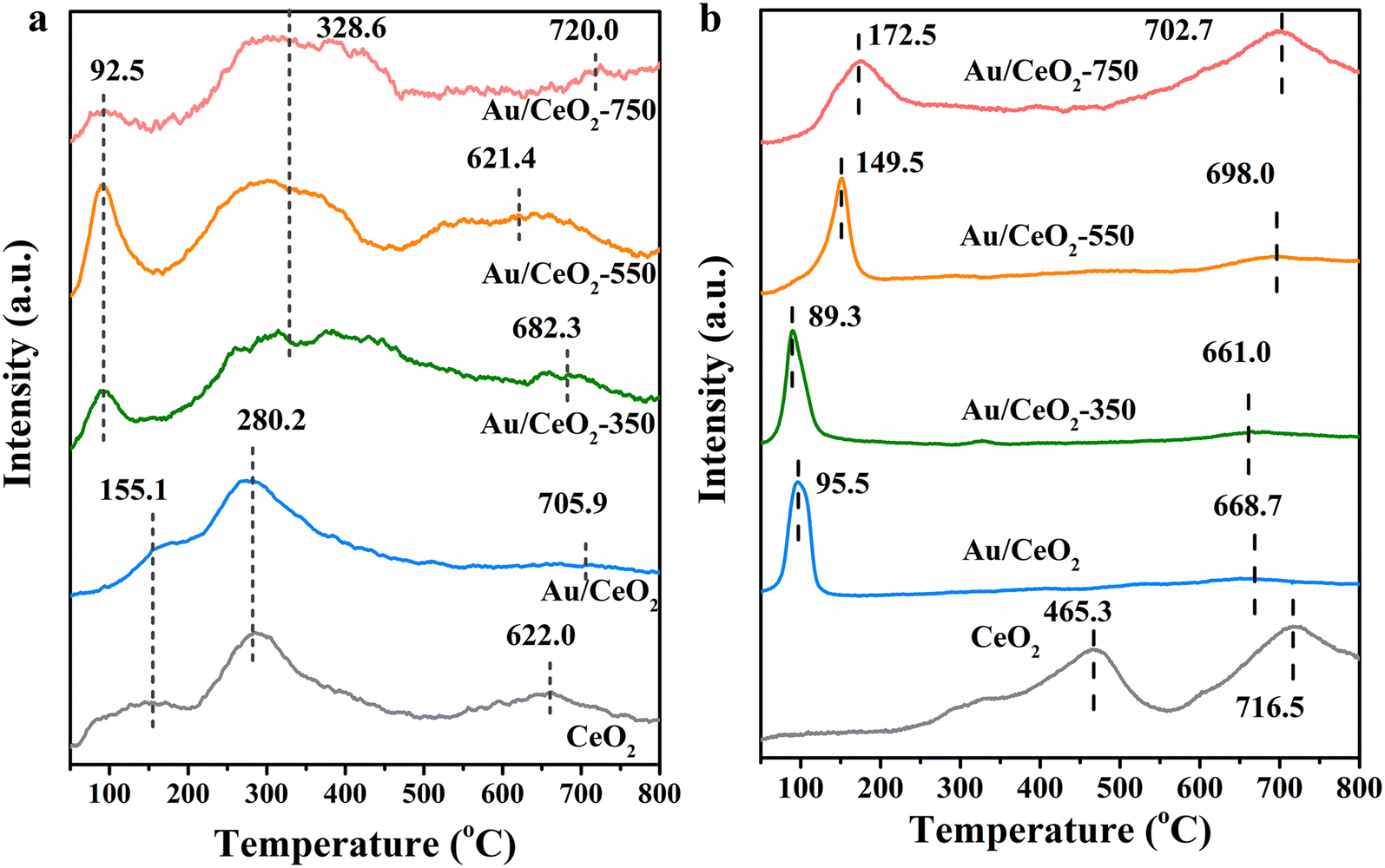 | ||
| Fig. 4 (a) O2-TPD and (b) H2-TPR profiles of all the catalysts. | ||
H2-TPR profiles allow identification of the different redox species in the samples. H2-TPR profiles of the prepared samples are presented in Fig. 4b. Two primary H2-consumption peaks are apparent in each profile. For the CeO2 sample, one of these peaks (465.3 °C) belongs to the reduction of surface capping oxygen and OH groups in ceria, and the other peak is assigned to the reduction of bulk oxygen in ceria.44 Comparing the H2-TPR results for the Au/CeO2 samples with that for the CeO2 reference, it is apparent that the addition of gold significantly modified the reduction behavior. In particular, the two reduction peaks were shifted to much lower temperatures. This phenomenon indicates that gold increases the reducibility of surface oxygen on CeO2 and facilitates the oxygen transfer across the solid–gas interface during the reaction. The variation in the reduction temperatures among the Au/CeO2, Au/CeO2-350, Au/CeO2-550 and Au/CeO2-750 samples could be because of the different calcination temperatures. With increasing calcination temperature, the first reduction peak shifted to a higher temperature indicating the increasing difficulty for reducing Auδ+ and the hydrogen consumption is also relatively low (Table S2†). The reduction peak observed for Au/CeO2-750 shifts to a higher temperature because of the strong Au–CeO2 interaction.45 However, too small surface area of the catalyst calcined at too high temperature (750 °C) (Table S1†) would lead to serious aggregation of Au nanoparticles and thus cause the catalyst to have low activity during HCHO oxidation. Only those catalysts with a moderate interaction, such as that calcined at 550 °C, showed stable activity of HCHO oxidation above room temperature.46,47 This further justified the claim that a moderate metal–support interaction is necessary to ensure the high activity and stability of the Au/CeO2-550 catalyst for HCHO oxidation.
The results discussed above indicate that the combined effects of single Au atoms and Au nanoclusters can dramatically enhance the activation of surface lattice oxygen. The surface oxygen species in the vicinity of both single Au atoms and Au nanoclusters exhibited greater mobility and reducibility, which are especially crucial for catalyzing HCHO oxidation.
2.3 Catalytic performance for HCHO oxidation
The catalytic activities of the samples were investigated for the oxidation of HCHO to CO2 and H2O. The temperature dependence of the HCHO oxidation from room temperature to 280 °C is presented in Fig. 5a. As expected from the above-described data analyses, the catalytic activities of Au/CeO2, Au/CeO2-350, Au/CeO2-550, and Au/CeO2-750 were found to be superior to that of pure CeO2 (Fig. S7†), as evidenced by lower HCHO conversion temperatures. Only 2 wt% Au loading enhances the activity considerably, indicating that Au is the active substance. The temperature at which the conversion of HCHO to CO2 and H2O was 100%, T100, was close to room temperature for Au/CeO2-550, far lower than that for the other Au-based catalysts (Au/CeO2, Au/CeO2-350, and Au/CeO2-750). In addition, although the Au/CeO2-350 catalyst reached 100% HCHO conversion at 100 °C, the conversion of Au/CeO2-350 tends to significantly degrade at high temperatures (>200 °C). To further investigate the cause of this performance degradation, we used CO as the probe molecule to detect the desorption capacities of Au/CeO2-350 and Au/CeO2-550 via in situ DRIFTS as shown in Fig. S8.† The intensity of Au0–CO and Auδ+–CO bands of the Au/CeO2-350 catalyst was weaker than that of the Au/CeO2-550 catalyst, which indicating that the adsorbed CO of the Au/CeO2-350 catalyst was lost much more slowly (Fig. S8c and d†). Thus, the Au/CeO2-350 catalyst possesses a weak gas-desorption capacity, which may hinder effective gas adsorption. The Au/CeO2-550 catalyst maintained a high HCHO conversion over a wide temperature range, and the operation-temperature window for 100% conversion, was larger than those of the other catalysts in this study (Fig. 5a) as well as those of other previously reported Au-based catalysts (Table S3†). Temperature-dependent TOFs and reaction rates were calculated for the catalysts, as shown in Fig. 5b and c, which decrease in the following order: Au/CeO2-550 > Au/CeO2-350 > Au/CeO2-750 > Au/CeO2 at 100 °C. Apparent activation energy values were estimated for the Au/CeO2, Au/CeO2-350, Au/CeO2-550, and Au/CeO2-750 catalysts as 8.64, 8.30, 7.75 and 19.27 kJ mol−1, respectively (Fig. 5d). Notably, the apparent activation energy of Au/CeO2-550 is the lowest among the given catalysts, indicating that the catalyst might have different active sites and that HCHO oxidation followed different reaction pathways. Therefore, for the Au/CeO2-550 catalyst, it is highly possible that the substitution of Au3+ into CeO2 and the Au nanoclusters enhances oxygen activation ability, increasing the ΔT100 value. In addition, the XRD patterns after the HCHO oxidation test revealed the stable nature of the Au/CeO2-550 catalyst (Fig. S9†). To further evaluate the potential application in indoor environments, the catalytic performance of Au/CeO2-550 was tested in HCHO oxidation for 100 h under HCHO oxidation reaction conditions (Fig. S10†); the HCHO conversion remains to be 100% for 84 h. After the 84 h, the HCHO conversion gradually declines from 100% to 79.3% under the HCHO oxidation reaction conditions.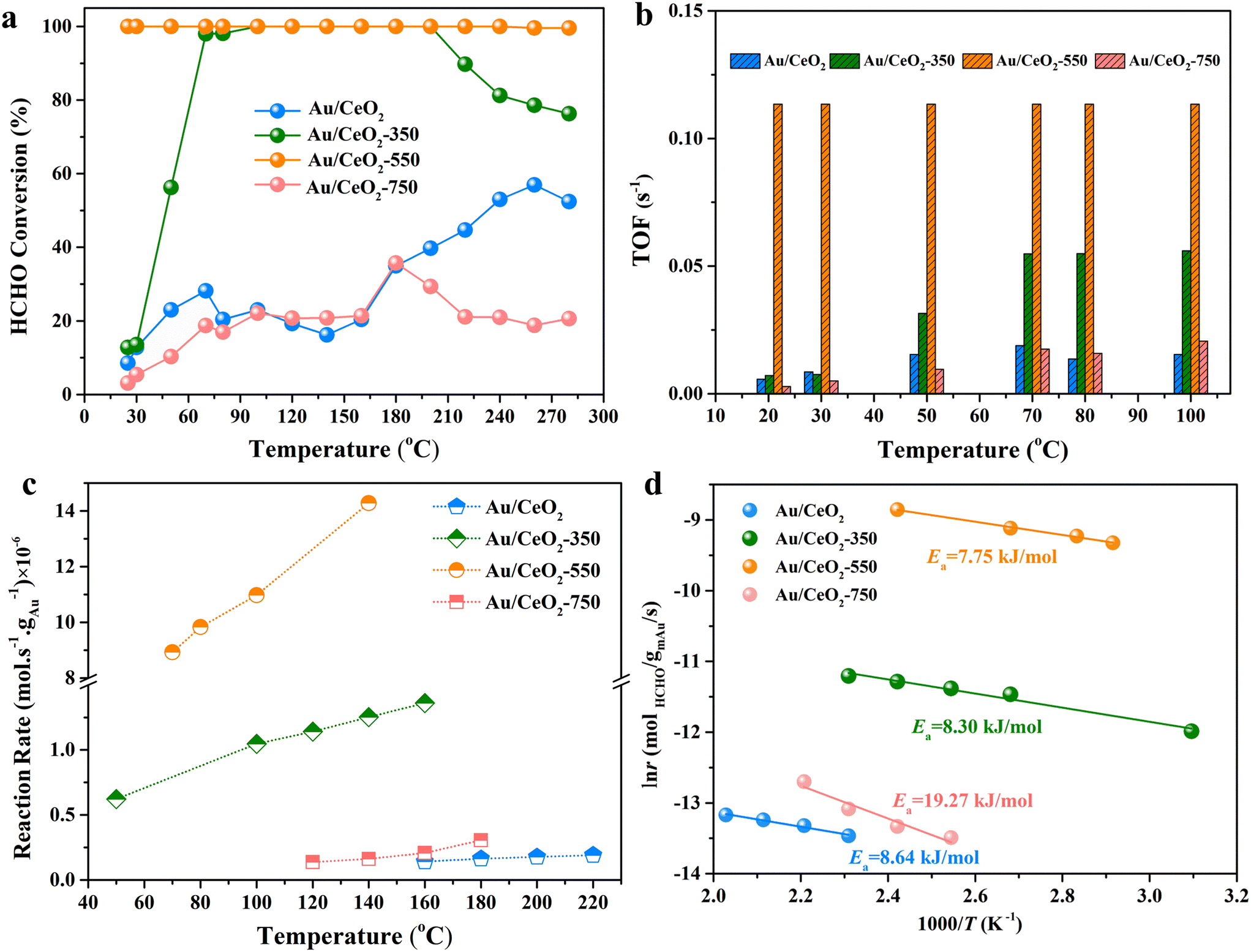 | ||
| Fig. 5 (a) Temperature dependence of HCHO conversions on the Au/CeO2, Au/CeO2-350, Au/CeO2-550 and Au/CeO2-750 catalysts. (b) Comparison of TOFs of the catalysts for HCHO oxidation at various temperatures. (c) Reaction rates normalized by the surface area at various temperatures for each catalyst. (d) Arrhenius plots for HCHO oxidation using each catalyst. | ||
2.4 Identifying active sites and reaction mechanism underlying the outstanding HCHO oxidation performance
To further identify the active sites and understand the mechanism underlying the enhancement of the HCHO oxidation performance of the Au/CeO2-550 catalyst, DRIFTS measurements were performed under HCHO oxidation reaction conditions. The DRIFTS data for the samples, acquired at temperatures in the range of 30–280 °C, are displayed in Fig. 6, S11 and S12.† In the spectra of Au/CeO2-550, for example (Fig. 6), it is apparent that the intensity of the characteristic hydroxyl bands (3532 cm−1) gradually decreased with temperature.48 This indicates that rapid transformation of HCHO after its adsorption on the catalyst occurs, and that OH species participate in this transformation process. The bands centered at approximately 2932 cm−1, 2846 cm−1, (1566 cm−1 and 1548 cm−1), and (1371 cm−1 and 1356 cm−1) were attributed to the formate CH symmetric stretching (νs), CH asymmetric stretching (νas), COO asymmetric stretching (νas) and COO symmetric stretching (νs), respectively.49–52 The most intense IR-active vibration of CO2 in the gas phase occurred at approximately 2333 cm−1, as observed in the middle part in Fig. 6.53 The band at 1471 cm−1 could be assigned tentatively to the CH bending vibration of dioxymethylene (DOM),53 and the band appearing at 1310 cm−1 to the ν(OCO) stretching of carbonate species.54 However, for the CeO2 catalyst, only weak formate bands were observed (Fig. S11†). This result reflects the adsorption of HCHO on the CeO2 surface rather than on Au. The Au/CeO2-550 catalyst enabled the total conversion of HCHO to CO2 and H2O end products via the formation of formate, DOM, and carbonate intermediates, while the CeO2 catalysts failed to convert the intermediates to end products. Because the conversion of formate, DOM and carbonate intermediates to CO2 and H2O needs the participation of active O species; these results confirm that Au provides active sites for O2 activation.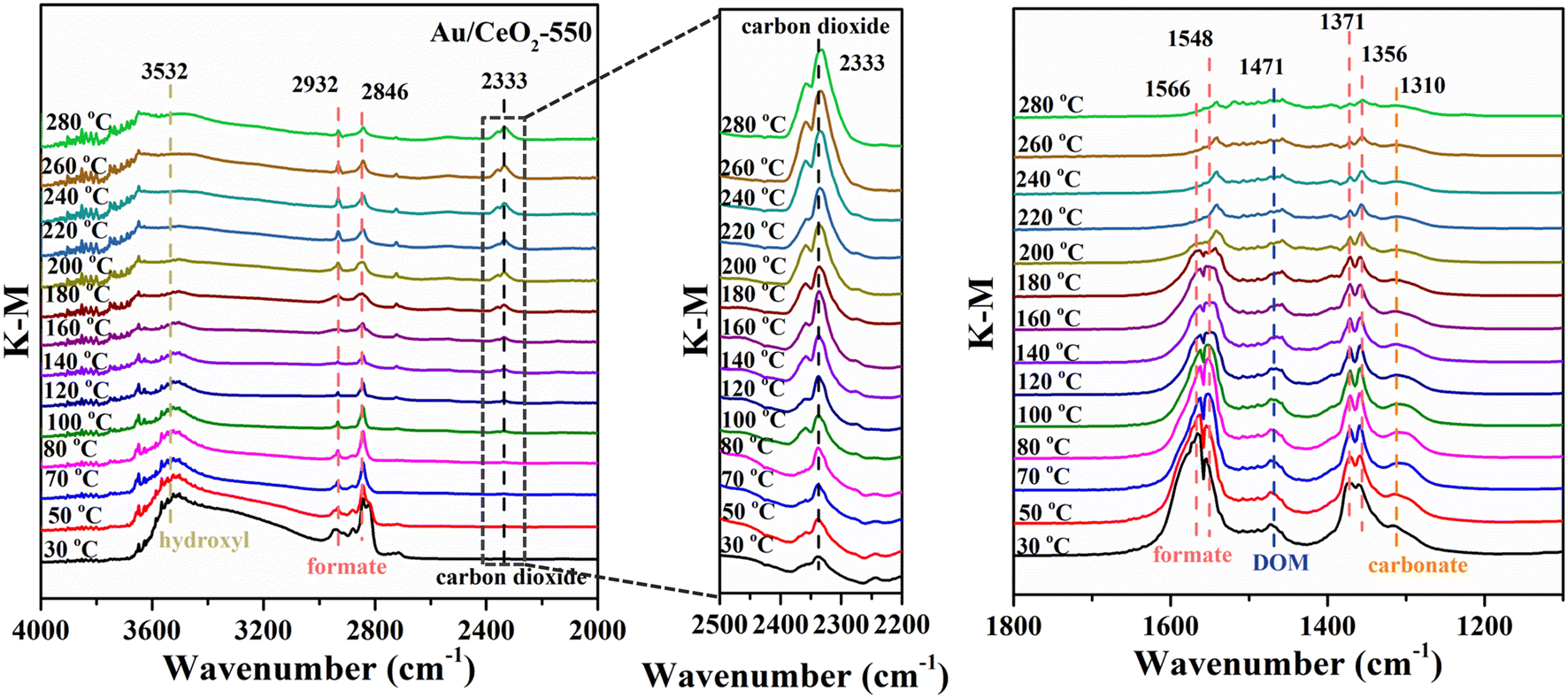 | ||
| Fig. 6 In situ DRIFTS results for the Au/CeO2-550 catalyst at various temperatures during HCHO oxidation over different wavenumber regions and the expanded regions of the in situ DRIFTS patterns in the range of 2500–2200 cm−1. | ||
Notably, the reaction intermediates produced during oxidation using the different catalysts are apparently different. For the Au/CeO2 and Au/CeO2-350 samples, the main reaction intermediates during the HCHO oxidation reaction were formate and dioxymethylene (DOM), as shown in Fig. S12a and b.† As the temperature was increased, the formate and DOM band intensities decreased, indicating that the two species take part in the HCHO oxidation reaction. Nevertheless, the reaction intermediates for oxidation using Au/CeO2-750 were mainly formate and carbonate species (Fig. S12c†). Thus, two different types of reaction intermediates were formed. The formate, DOM, and carbonate species appeared at the same time for the all the catalyst. However, for the Au/CeO2-550 catalyst, as the temperature increased, the formate, DOM and carbonate band intensities decreased, while the intensity of carbon dioxide vibration increased, demonstrating that HCHO molecules were converted into CO2 and H2O via different reaction routes for the Au/CeO2-550 catalyst. In the in situ DRIFTS experiment, the flowing HCHO is 220 ppm, the catalyst amount is just about 2 mg less than that (50 mg) in the catalytic performance test. Under the condition of abundant HCHO, the vibration intensity of CO2 may be affected by the reaction rate.55 According to the enlarged region of CO2 in Fig. 6, the vibration of CO2 for Au/CeO2-550 at low temperatures with a low reaction rate is not stronger than that at high temperature with a high reaction rate. Furthermore, in situ DRIFTS spectra were obtained during a study of the time dependence of HCHO adsorption, He purging, and the O2 oxidation process for the Au/CeO2-550 catalyst (Fig. S13†). It is noteworthy that strong formate, DOM and carbonate bands appeared upon the adsorption of HCHO on the Au/CeO2-550 catalyst, even though no oxygen was present in the feed stream, suggesting that HCHO reacted with surface oxygen species activated by individual Au atoms. As time progressed during the He purging, the formate, DOM and carbonate band intensities remained basically unchanged, demonstrating that the generation and desorption of the products reached a dynamic balance during the reaction. The formate band intensity evidently increased during O2 oxidation, whereas the DOM and methoxy vibration bands became quite weak, which might suggest that the latter species were converted into formate. Moreover, a slight increase in the carbonate band intensities was observed, which might suggest that formate was subsequently converted into carbonate. In addition, the bands at ∼2371 cm−1 and 2337 cm−1 are assigned to carbon dioxide (Fig. S13b†). These peaks of carbon dioxide appeared under an HCHO atmosphere. The intensity of carbon dioxide did not change significantly after the He treatment. The possible reason for this phenomenon is that HCHO can react with surface reactive oxygen on the CeO2 support. Subsequently, the intensity of carbon dioxide gradually became strong when O2 was injected. After O2 purging for 60 min, the peak intensity of CO2 was low indicating that Au/CeO2-550 would favor a faster desorption of CO2 through which the HCHO oxidation is accelerated. Active oxygen atoms, generated by adsorbed O2 and activated by Au (mainly in the metallic form), are critical in the transformation of DOM to formate.48 Therefore, O2 adsorbed on the Au nanocluster surface could be well activated to participate in the subsequent reactions.
These results suggest that the excellent oxygen activation ability of Au/CeO2-550 can be attributed to a synergistic effect of dual active sites, leading to a faster intermediate species formation rate, and thus an enhanced catalytic HCHO oxidation performance was obtained.
Using the results of DRIFTS, HAADF-STEM, XPS, H2-TPR, O2-TPD and activity analysis, and supported by previously reported data, a probable HCHO oxidation reaction mechanism has been recommended, as summarized in Fig. 7. The intermediate products of the samples are mainly divided into three categories depending on the Au/CeO2 calcination temperature: (I) DOM and formate; (II) formate, DOM and carbonate; (III) formate and carbonate.
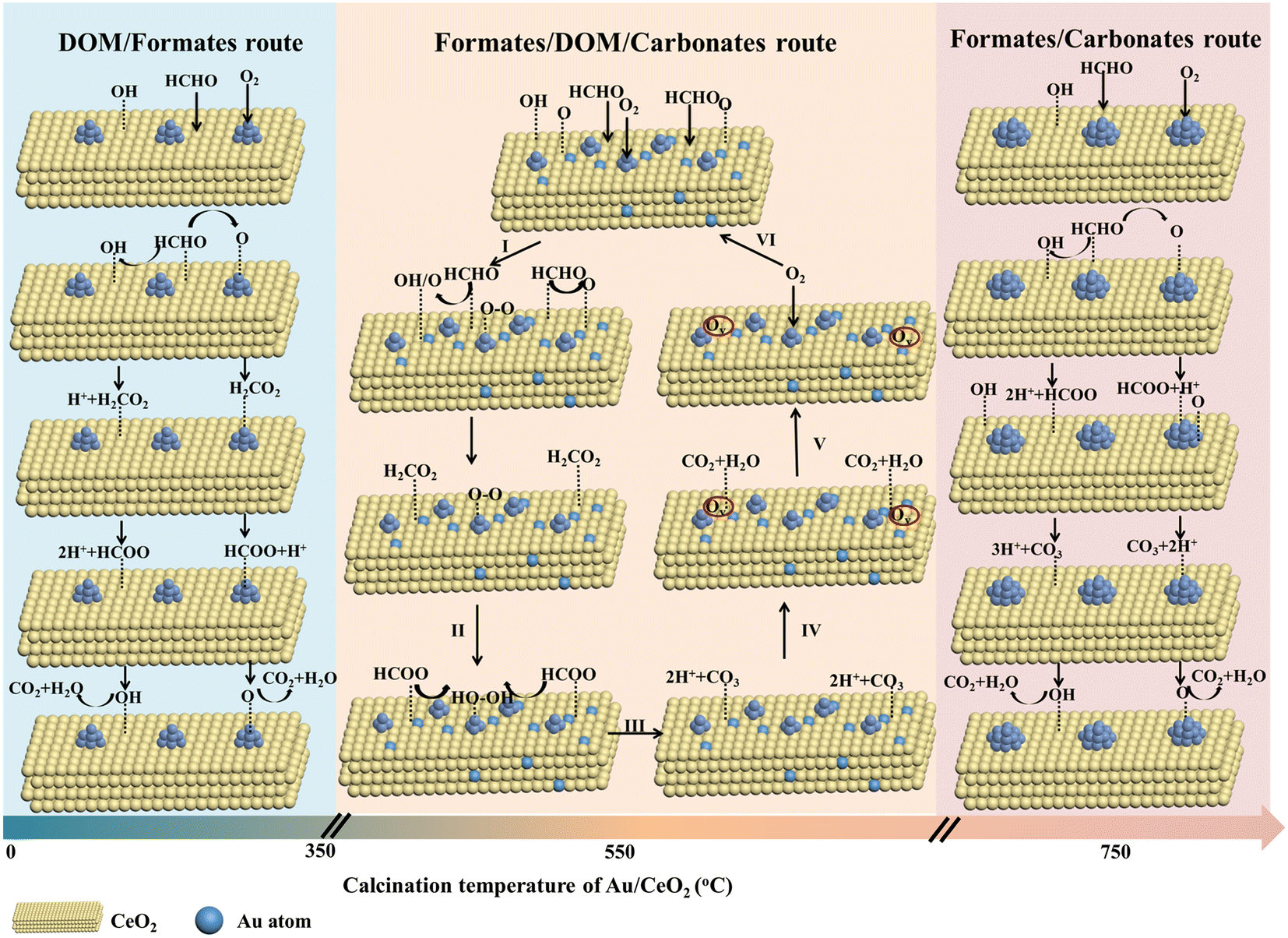 | ||
| Fig. 7 Proposed mechanism of the HCHO oxidation reaction on the Au/CeO2 catalysts calcined at different temperatures. | ||
For the Au/CeO2 and Au/CeO2-350 catalysts, DOM and formate are the reaction intermediates. HCHO was first adsorbed on the surface of CeO2. Certain enriched Auδ+ species were present on the surface and interface (Au/CeO2: 47.1% and Au/CeO2-350: 58.1%), as shown in Fig. 3, and multiple adsorption sites for oxygen activation. The reaction steps involve the activation of oxygen/hydroxyl on CeO2, and activated oxygen/hydroxyl on the support surface to react with the adsorbed HCHO, leading to the formation of DOM. The DOM species are further converted to formate, which was then decomposed into CO2 and H2O. The oxygen activation ability (Fig. 4a) strongly influenced the reaction, and for this reason Au/CeO2 exhibited a poorer catalytic performance than Au/CeO2-350.
In the case of Au/CeO2-750, the formation of formate contains hydroxyl groups and activated oxygen on the CeO2 surface. Thus, the rate of the subsequent transformation into carbonate, CO2 and H2O should be mainly dependent on the surface OH concentration, oxygen activation and Au nanoparticle sizes, which will explain the significant difference in the catalytic performance for this sample compared to the Au/CeO2 and Au/CeO2-350 samples.
The in situ DRIFTS results indicated that the formation of formate, DOM and carbonate occurred on the main pathway for the production of gaseous CO2 on Au/CeO2-550 catalyst with Au nanoclusters and single Au atoms. At the single Au sites, the HCHO molecules reacted with the surface oxygen species of CeO2 activated by individual Au atoms to form DOM. At the same time, O2 molecules were adsorbed onto Au nanoclusters and activated (step I). Then, DOM can be further oxidized by O2 to generate formate (step II). Subsequently, intermediate formate species were further oxidized to unstable carbonate by the active oxygen species generated on Au nanoclusters (step III). The carbonate rapidly decomposed into harmless CO2 and H2O products (step IV). Next, the oxygen vacancies (Vo) were generated with the adsorbed CO2 and H2O, which rapidly desorbed from the surface (step V). Finally, the expended activated surface oxygen species are refilled by O2 adsorption on oxygen vacancies to regenerate the catalyst (step VI).
In the above process, optimizing the Au distribution is beneficial for increasing the density of synergistic active sites while ensuring that the material costs remain as low as possible. In addition, enhancing oxygen activation and the transfer of active oxygen species on the catalyst surface opens a new avenue for improving HCHO oxidation catalytic performance.
3. Conclusion
In summary, we showed that CeO2-supported Au catalysts with both Au nanoclusters and single Au atoms exhibited enhanced HCHO oxidation performance at room temperature. In addition, it was found that the overall HCHO conversion to CO2 involved several reaction intermediates, including DOM, formates, and carbonates, as well as diverse active oxygen species, such as OH and O activated by Au atoms and O2 activated by Au nanoclusters, on the CeO2 surface. The synergistic effect of single Au atoms and Au nanoclusters resulted in a dramatic increase in the oxidation of DOM and formate, and accelerated the decomposition of carbonate, which is particularly important in defining the overall HCHO oxidation kinetics. The single Au atoms facilitated the generation of surface reactive oxygen, which rapidly oxidized HCHO to DOM. Meanwhile, Au nanoclusters could adsorb and constantly provide active O2, which efficiently oxidizes DOM and formate. The results of this study provide a clearer understanding of the possible synergistic mechanism involving oxygen activation at individual Au atoms and Au nanoclusters that facilitates efficient HCHO oxidation. More generally, this study produced insights that will assist in the design of highly efficient atomically dispersed catalysts for the removal of indoor volatile organic compounds (VOCs).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Science Fund for Distinguished Young Scholars (Grant 52225407). The authors are grateful for the financial support from the Shenzhen Science and Technology Innovation Committee (JCYJ20200109141437586 and KCXFZ20211020174805008). The authors thank the Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme 2018.References
- R. Maddalena, M. Russell, D. P. Sullivan and M. G. Apte, Formaldehyde and other volatile organic chemical emissions in four FEMA temporary housing units, Environ. Sci. Technol., 2009, 43, 5626–5632 CrossRef CAS PubMed.
- S. Huang, X. Zhu, B. Cheng, J. Yu and C. Jiang, Flexible nickel foam decorated with Pt/NiO nanoflakes with oxygen vacancies for enhanced catalytic formaldehyde oxidation at room temperature, Environ. Sci.: Nano, 2017, 4, 2215–2224 RSC.
- Y. Wang, J. Ye, C. Jiang, Y. Le, B. Cheng and J. Yu, Hierarchical NiMn2O4/rGO composite nanosheets decorated with Pt for low-temperature formaldehyde oxidation, Environ. Sci.: Nano, 2020, 7, 198–209 RSC.
- B. Hileman, Formaldehyde: Assessing the risk, Environ. Sci. Technol., 1984, 18, 216A–221A CrossRef.
- J. Chen, D. Yan, Z. Xu, X. Chen, X. Chen, W. Xu, H. Jia and J. Chen, A Novel Redox Precipitation to Synthesize Au-Doped alpha-MnO2 with High Dispersion toward Low-Temperature Oxidation of Formaldehyde, Environ. Sci. Technol., 2018, 52, 4728–4737 CrossRef CAS PubMed.
- C. Zhang and H. He, A comparative study of TiO2 supported noble metal catalysts for the oxidation of formaldehyde at room temperature, Catal. Today, 2007, 126, 345–350 CrossRef CAS.
- K. Li, J. Ji, M. He and H. Huang, Complete oxidation of formaldehyde over a Pd/CeO2 catalyst at room temperature: tunable active oxygen species content by non-thermal plasma activation, Catal. Sci. Technol., 2020, 10, 6257–6265 RSC.
- H. Xie, X. Chen, C. Zhang, Z. Lao, X. Liu, X. Xie, R. Semiat and Z. Zhong, Identifying the Fe3Mn3O8 phase as a superior catalyst for low-temperature catalytic oxidation of formaldehyde in air, Environ. Sci.: Nano, 2022, 9, 767–780 RSC.
- D. Cao, H. Xu and D. Cheng, Construction of Defect-Rich RhCu Nanotubes with Highly Active Rh3Cu1 Alloy Phase for Overall Water Splitting in All pH Values, Adv. Energy Mater., 2020, 10, 1903038 CrossRef CAS.
- K. Chen, K. Liu, P. An, H. Li, Y. Lin, J. Hu, C. Jia, J. Fu, H. Li, H. Liu, Z. Lin, W. Li, J. Li, Y. R. Lu, T. S. Chan, N. Zhang and M. Liu, Iron phthalocyanine with coordination induced electronic localization to boost oxygen reduction reaction, Nat. Commun., 2020, 11, 4173 CrossRef CAS PubMed.
- T. Kou, S. Wang and Y. Li, Perspective on High-Rate Alkaline Water Splitting, ACS Mater. Lett., 2021, 3, 224–234 CrossRef CAS.
- H. Sun, J. He, Z. Hu, C.-T. Chen, W. Zhou and Z. Shao, Multi-active sites derived from a single/double perovskite hybrid for highly efficient water oxidation, Electrochim. Acta, 2019, 299, 926–932 CrossRef CAS.
- M. Song, Y. Wu, C. Xu, X. Wang and Y. Su, Synergistic effects of multi-active sites in silver modified Bi degrees -BiVO4 toward efficient reduction of aromatic nitrobenzene, J. Hazard. Mater., 2019, 368, 530–540 CrossRef CAS PubMed.
- P. Sun, S. Zhai, J. Chen, J. Yuan, Z. Wu and X. Weng, Development of a multi-active center catalyst in mediating the catalytic destruction of chloroaromatic pollutants: A combined experimental and theoretical study, Appl. Catal., B, 2020, 272, 119015 CrossRef CAS.
- S. Ye, F. Luo, T. Xu, P. Zhang, H. Shi, S. Qin, J. Wu, C. He, X. Ouyang, Q. Zhang, J. Liu and X. Sun, Boosting the alkaline hydrogen evolution of Ru nanoclusters anchored on B/N–doped graphene by accelerating water dissociation, Nano Energy, 2020, 68, 104301 CrossRef CAS.
- X. Zhang, G. Cui, H. Feng, L. Chen, H. Wang, B. Wang, X. Zhang, L. Zheng, S. Hong and M. Wei, Platinum-copper single atom alloy catalysts with high performance towards glycerol hydrogenolysis, Nat. Commun., 2019, 10, 5812 CrossRef CAS PubMed.
- J. N. Tiwari, S. Sultan, C. W. Myung, T. Yoon, N. Li, M. Ha, A. M. Harzandi, H. J. Park, D. Y. Kim, S. S. Chandrasekaran, W. G. Lee, V. Vij, H. Kang, T. J. Shin, H. S. Shin, G. Lee, Z. Lee and K. S. Kim, Multicomponent electrocatalyst with ultralow Pt loading and high hydrogen evolution activity, Nat. Energy, 2018, 3, 773–782 CrossRef CAS.
- X. Zhang, M. Zhang, Y. Deng, M. Xu, L. Artiglia, W. Wen, R. Gao, B. Chen, S. Yao, X. Zhang, M. Peng, J. Yan, A. Li, Z. Jiang, X. Gao, S. Cao, C. Yang, A. J. Kropf, J. Shi, J. Xie, M. Bi, J. A. van Bokhoven, Y. W. Li, X. Wen, M. Flytzani-Stephanopoulos, C. Shi, W. Zhou and D. Ma, A stable low-temperature H2-production catalyst by crowding Pt on alpha-MoC, Nature, 2021, 589, 396–401 CrossRef CAS PubMed.
- Y. Wu, M. Song, Z. Chai, J. Huang and X. Wang, Integrating an Ag0–Ag+ mediated Ag2Ta4O11/Ag8(Nb0.5Ta0.5)26O69 heterojunction to quickly decontaminate indoor gaseous formaldehyde under indoor temperature, humidity and sunlight irradiation conditions, Environ. Sci.: Nano, 2020, 7, 1831–1840 RSC.
- Z. Zhang, G. He, Y. Li, C. Zhang, J. Ma and H. He, Effect of Hydroxyl Groups on Metal Anchoring and Formaldehyde Oxidation Performance of Pt/Al2O3, Environ. Sci. Technol., 2022, 56, 10916–10924 CrossRef CAS.
- S. Zhu, J. Zheng, S. Xin and L. Nie, Preparation of flexible Pt/TiO2/γ-Al2O3 nanofiber paper for room-temperature HCHO oxidation and particulate filtration, Chem. Eng. J., 2022, 427, 130951 CrossRef CAS.
- J. Liu, W. Chen, T. He, Y. Fang, Z. Zhong, X. Wang, Z. Li and Y. Song, Lewis base sites of non-oxide supports boost oxygen absorption and activation over supported Pt catalysts, RSC Adv., 2022, 12, 12537–12543 RSC.
- C. Ma, D. Wang, W. Xue, B. Dou, H. Wang and Z. Hao, Investigation of formaldehyde oxidation over Co3O4-CeO2 and Au/Co3O4-CeO2 catalysts at room temperature: effective removal and determination of reaction mechanism, Environ. Sci. Technol., 2011, 45, 3628–3634 CrossRef CAS.
- J. Xu, Z. Qu, Y. Wang and B. Huang, HCHO oxidation over highly dispersed Au nanoparticles supported on mesoporous silica with superior activity and stability, Catal. Today, 2019, 327, 210–219 CrossRef CAS.
- J. Qu, D. Chen, N. Li, Q. Xu, H. Li, J. He and J. Lu, 3D Gold-Modified Cerium and Cobalt Oxide Catalyst on a Graphene Aerogel for Highly Efficient Catalytic Formaldehyde Oxidation, Small, 2019, 15, e1804415 Search PubMed.
- J. Chen, M. Jiang, J. Chen, W. Xu and H. Jia, Selective immobilization of single-atom Au on cerium dioxide for low-temperature removal of C1 gaseous contaminants, J. Hazard. Mater., 2020, 392, 122511 CrossRef CAS PubMed.
- S. Chen, S. Li, R. You, Z. Guo, F. Wang, G. Li, W. Yuan, B. Zhu, Y. Gao, Z. Zhang, H. Yang and Y. Wang, Elucidation of Active Sites for CH4 Catalytic Oxidation over Pd/CeO2 Via Tailoring Metal–Support Interactions, ACS Catal., 2021, 11, 5666–5677 CrossRef CAS.
- X.-L. Wang, X.-P. Fu, W.-W. Wang, C. Ma, R. Si and C.-J. Jia, Effect of Structural Evolution of Gold Species Supported on Ceria in Catalyzing CO Oxidation, J. Phys. Chem. C, 2019, 123, 9001–9012 CrossRef CAS.
- S. Putla, M. H. Amin, B. M. Reddy, A. Nafady, K. A. Al Farhan and S. K. Bhargava, MnOx Nanoparticle-Dispersed CeO2 Nanocubes: A Remarkable Heteronanostructured System with Unusual Structural Characteristics and Superior Catalytic Performance, ACS Appl. Mater. Interfaces, 2015, 7, 16525–16535 CrossRef CAS.
- P. Sudarsanam, B. Mallesham, P. S. Reddy, D. Großmann, W. Grünert and B. M. Reddy, Nano-Au/CeO2 catalysts for CO oxidation: Influence of dopants (Fe, La and Zr) on the physicochemical properties and catalytic activity, Appl. Catal., B, 2014, 144, 900–908 CrossRef CAS.
- Y. Kang, M. Sun and A. Li, Studies of the Catalytic Oxidation of CO Over Ag/CeO2 Catalyst, Catal. Lett., 2012, 142, 1498–1504 CrossRef CAS.
- D. Jampaiah, S. J. Ippolito, Y. M. Sabri, B. M. Reddy and S. K. Bhargava, Highly efficient nanosized Mn and Fe codoped ceria-based solid solutions for elemental mercury removal at low flue gas temperatures, Catal. Sci. Technol., 2015, 5, 2913–2924 RSC.
- D. Mukherjee, B. G. Rao and B. M. Reddy, CO and soot oxidation activity of doped ceria: Influence of dopants, Appl. Catal., B, 2016, 197, 105–115 CrossRef CAS.
- H.-F. Li, N. Zhang, P. Chen, M.-F. Luo and J.-Q. Lu, High surface area Au/CeO2 catalysts for low temperature formaldehyde oxidation, Appl. Catal., B, 2011, 110, 279–285 CrossRef CAS.
- N. S. Portillo-Vélez and R. Zanella, Comparative study of transition metal (Mn, Fe or Co) catalysts supported on titania: Effect of Au nanoparticles addition towards CO oxidation and soot combustion reactions, Chem. Eng. J., 2020, 385, 123848 CrossRef.
- S. Ren, W. Liang, Q. Li and Y. Zhu, Effect of Pd/Ce loading on the performance of Pd-Ce/gamma-Al2O3 catalysts for toluene abatement, Chemosphere, 2020, 251, 126382 CrossRef CAS.
- Y. Huang, K. Ye, H. Li, W. Fan, F. Zhao, Y. Zhang and H. Ji, A highly durable catalyst based on CoxMn3–xO4 nanosheets for low-temperature formaldehyde oxidation, Nano Res., 2016, 9, 3881–3892 CrossRef CAS.
- H. Zhou, Q. Wu, B. Qi and G. Feng, Facile One-Step Flame Synthesis of La1−xSrxMnO3 Nanoparticles for CO Catalytic Oxidation, J. Chem., 2021, 2021, 1–11 Search PubMed.
- Y.-H. Quan, C. Miao, T. Li, N. Wang, M.-M. Wu, N. Zhang, J.-X. Zhao and J. Ren, Effect of preparation methods on the structure and catalytic performance of CeO2 for toluene combustion, J. Fuel Chem. Technol., 2021, 49, 211–219 CrossRef.
- Q. Liu, L.-C. Wang, M. Chen, Y. Cao, H.-Y. He and K.-N. Fan, Dry citrate-precursor synthesized nanocrystalline cobalt oxide as highly active catalyst for total oxidation of propane, J. Catal., 2009, 263, 104–113 CrossRef CAS.
- Z. Fei, S. He, L. Li, W. Ji and C. T. Au, Morphology-directed synthesis of Co3O4 nanotubes based on modified Kirkendall effect and its application in CH4 combustion, Chem. Commun., 2012, 48, 853–855 RSC.
- J. Ye, M. Zhou, Y. Le, B. Cheng and J. Yu, Three-dimensional carbon foam supported MnO2/Pt for rapid capture and catalytic oxidation of formaldehyde at room temperature, Appl. Catal., B, 2020, 267, 118689 CrossRef CAS.
- C. Zhang, Y. Li, Y. Wang and H. He, Sodium-promoted Pd/TiO2 for catalytic oxidation of formaldehyde at ambient temperature, Environ. Sci. Technol., 2014, 48, 5816–5822 CrossRef CAS PubMed.
- D. Valechha, S. K. Megarajan, A. Al-Fatesh, H. Jiang and N. Labhasetwar, Low Temperature CO Oxidation Over a Novel Nano-Structured, Mesoporous CeO2 Supported Au Catalyst, Catal. Lett., 2018, 149, 127–140 CrossRef.
- H. Peng, C. Rao, N. Zhang, X. Wang, W. Liu, W. Mao, L. Han, P. Zhang and S. Dai, Confined Ultrathin Pd-Ce Nanowires with Outstanding Moisture and SO2 Tolerance in Methane Combustion, Angew. Chem., Int. Ed., 2018, 57, 8953–8957 CrossRef CAS.
- D. Cui, J. Liu, J. Yu, J. Yue, F. Su and G. Xu, Necessity of moderate metal-support interaction in Ni/Al2O3 for syngas methanation at high temperatures, RSC Adv., 2015, 5, 10187–10196 RSC.
- Y. Wang, S. Zhu, J. Lu, J. Liu, Y. Zhao, S. He, Y. Zhao, H. Lu and Y. Luo, Boosting hydrogen production from steam reforming of glycerol via constructing moderate metal-support interaction in Ni@Al2O3 catalyst, Fuel, 2022, 324, 124583 CrossRef CAS.
- S. Li, C. I. Ezugwu, S. Zhang, Y. Xiong and S. Liu, Co-doped MgAl-LDHs nanosheets supported Au nanoparticles for complete catalytic oxidation of HCHO at room temperature, Appl. Surf. Sci., 2019, 487, 260–271 CrossRef CAS.
- Y. Li, C. Zhang, J. Ma, M. Chen, H. Deng and H. He, High temperature reduction dramatically promotes Pd/TiO2 catalyst for ambient formaldehyde oxidation, Appl. Catal., B, 2017, 217, 560–569 CrossRef CAS.
- Y. Li, X. Chen, C. Wang, C. Zhang and H. He, Sodium Enhances Ir/TiO2 Activity for Catalytic Oxidation of Formaldehyde at Ambient Temperature, ACS Catal., 2018, 8, 11377–11385 CrossRef CAS.
- C. Ma, C. Yang, B. Wang, C. Chen, F. Wang, X. Yao and M. Song, Effects of H2O on HCHO and CO oxidation at room-temperature catalyzed by MCo2O4 (M=Mn, Ce and Cu) materials, Appl. Catal., B, 2019, 254, 76–85 CrossRef CAS.
- X. Qin, X. Chen, M. Chen, J. Zhang, H. He and C. Zhang, Highly efficient Ru/CeO2 catalysts for formaldehyde oxidation at low temperature and the mechanistic study, Catal. Sci. Technol., 2021, 11, 1914–1921 RSC.
- L. Zhu, J. Wang, S. Rong, H. Wang and P. Zhang, Cerium modified birnessite-type MnO2 for gaseous formaldehyde oxidation at low temperature, Appl. Catal., B, 2017, 211, 212–221 CrossRef CAS.
- X. Sun, J. Lin, H. Guan, L. Li, L. Sun, Y. Wang, S. Miao, Y. Su and X. Wang, Complete oxidation of formaldehyde over TiO2 supported subnanometer Rh catalyst at ambient temperature, Appl. Catal., B, 2018, 226, 575–584 CrossRef CAS.
- T. Yang, Y. Huo, Y. Liu, Z. Rui and H. Ji, Efficient formaldehyde oxidation over nickel hydroxide promoted Pt/γ-Al2O3 with a low Pt content, Appl. Catal., B, 2017, 200, 543–551 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2en00805j |
| This journal is © The Royal Society of Chemistry 2023 |