
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical weathering of polyurethane microplastics produced complex and dynamic mixtures of dissolved organic chemicals†
Vittorio
Albergamo
 *a,
Wendel
Wohlleben
bc and
Desirée L.
Plata
a
*a,
Wendel
Wohlleben
bc and
Desirée L.
Plata
a
aDepartment of Civil and Environmental Engineering, Massachusetts Institute of Technology, 15 Vassar Street, Cambridge, Massachusetts 02139, USA. E-mail: valberg@mit.edu
bDepartment of Material Physics and Analytics, Advanced Materials Research, BASF SE, Carl-Bosch-Str. 38, 67056 Ludwigshafen, Germany
cDepartment of Experimental Toxicology and Ecology, Advanced Materials Research, BASF SE, Carl-Bosch-Str. 38, 67056 Ludwigshafen, Germany
First published on 11th January 2023
Abstract
Sunlight exposure can naturally mitigate microplastics pollution in the surface ocean, however it results in emissions of dissolved organic carbon (DOC) whose characteristics and fate remain largely unknown. In this work, we investigated the effects of solar radiation on polyether (TPU_Ether) and polyester (TPU_Ester) thermoplastic polyurethane, and on a thermoset polyurethane (PU_Hardened). The microplastics were irradiated with simulated solar light with a UV dose of 350 MJ m−2, which corresponds to roughly 15 months outdoor exposure at 31° N latitude. The particles were characterized using ATR-FTIR and elemental analysis. The DOC released to the aqueous phase was quantified by total organic carbon analysis and characterized by nontarget liquid chromatography coupled to high-resolution mass spectrometry. Polyurethane microplastics were degraded following mechanisms reconcilable with UV photo-oxidation. The carbon mass fraction released to the aqueous phase was 8.5 ± 0.5%, 3.7 ± 0.2%, and 2.8 ± 0.2% for TPU_Ether, TPU_Ester, and PU_Hardened, respectively. The corresponding DOC release rates, expressed as mg carbon per UV dose were 0.023, 0.013, and 0.010 mg MJ−1 for TPU_Ether, TPU_Ester and PU_Hardened, respectively. Roughly three thousand unique by-products were released from photo-weathered TPUs, whereas 540 were detected in the DOC of PU_Hardened. This carbon pool was highly complex and dynamic in terms of physicochemical properties and susceptibility to further photodegradation after dissolution from the particles. Our results show that plastics photodegradation in the ocean requires chemical assessment of the DOC emissions in addition to the analysis of aged microplastics and that polymer chemistry influences the chain scission products.
Environmental significanceThe ubiquitous accumulation of plastic waste in the ocean is a widely acknowledged environmental issue. Despite being durable by design, most synthetic polymers can degrade via photo-oxidative processes initiated by solar ultraviolet radiation, leading to formation of secondary microplastics and releasing chain scission products to the surrounding aqueous environment. In particular, chain scission products can rearrange to complex mixtures of anthropogenic organic chemicals whose bulk properties and fate remain largely unknown. By investigating a set of three different polyurethane microplastics, we showed that polymer chemistry influenced the photo-oxidation mechanisms that took place by the surface of the residual particles and also that the chain scission products in the aqueous phase exhibited different physicochemical properties, rates of accumulation, and susceptibility to further photo-degradation. |
1 Introduction
Approximately 5–13 million metric tons of plastic debris enter the ocean each year as a result of waste mismanagement on land.1–4 Even when designed to be chemically and biologically inert, synthetic polymers fragment into micro- (<5 mm) and nano-size (<100 nm) particles under mechanical forces and when exposed to (photo-)oxidizing agents.5–11 Direct sunlight, particularly in the UV range, induces changes in polymer brittleness, density, size, and surface charge, leading to loss of bulk mechanical properties.12 These changes are the result of oxidative processes initiated by free radicals activated by light in the presence of oxygen,8 which compromise the polymer structure by (i) incorporating oxygen-containing functionalities, (ii) initiating chain scissions that reduce the number-average molecular weight of plastics, and (iii) introducing cross-links.9,12 Specifically, reactive oxygen species activated by light in the UV range initiate the photolytic cleavage of a C–H bond on the polymer backbone to produce a polymer alkyl radical (P˙). Reaction with molecular oxygen leads to formation of unstable polymer peroxy radicals (POO˙), which decompose in hydroxyl radicals (OH˙) and polymer oxy radicals (PO˙) that can decay into chain scission products.13 The reaction terminates when polymer radical species couple mutually or form crosslinks with polymer peroxy radicals.9,13 In particular, chain scissions can release oligomers, monomers and additives to the environment surrounding microplastics, contributing to the pool of dissolved organic carbon (DOC) available to microorganisms with potentially unaccounted consequences on marine ecosystems.9,10,12,14,15 Figures from recent studies indicated that the mass of marine DOC accounts for approximately 600 petagrams of carbon,16 while the contribution of microplastics-derived DOC is estimated at around 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 metric tons per year,17i.e., a 9.5 × 10−9% annual contribution to the reservoir.
000 metric tons per year,17i.e., a 9.5 × 10−9% annual contribution to the reservoir.
Polyurethane (PU) is a family of synthetic copolymers obtained by a condensation reaction of polyisocyanates and polyols to give a urethane linkage (–NH–COO–).18,19 PU is a versatile polymer, as its properties can be easily tailored by changing the polyol and isocyanate precursors and by introducing cross-links.20,21 In 2015, PU ranked as the 7th most produced synthetic polymer with nearly 30 million metric tons fabricated.22 One commercially important class of PUs are thermoplastic elastomers that are not cross-linked and alternate flexible segments, based on long polyester or polyether polyols, and rigid segments, consisting of urethane groups, the reaction products of isocyanates, and hydroxyl functionalities, e.g., from polyols.20,23 Despite the availability of promising recycling technologies,24 landfilling remains the most common way to process an ever-increasing amount of PU-containing waste.18 Although not as abundant as major consumer plastics like polyethylene (PE), polypropylene (PP), and polystyrene (PS), PU microplastics are commonly detected in marine environments, including coastal areas, seafloors and remote areas.10,25–28 If exposed to sunlight, the urethane linkage will photo-oxidize.8,19 Following a characteristic initial yellowing, the homolytic scission of the urethane bond will form free radicals, whose decomposition and recombination ultimately generate short polymer chains, monomers, and low molecular weight photo-oxidized products that partition to the surrounding environment.8,19,29
In this investigation, the effects of solar radiation on PU microplastics were studied in a simulated marine environment. We selected a polyester and a polyether thermoplastic polyurethane elastomer (TPU), both of which are devoid of cross-links, and a cross-linked PU duromer. These polymers were chosen not only because they allowed characterizing the photodegradation behavior of microplastics of the same class in relation to their different chemistries, but also for their commercial and environmental relevance. Changes in microplastics chemistry were investigated using infrared spectroscopy and elemental analysis. The DOC released from polymer photo-oxidation was quantified and subsequently characterized using liquid chromatography coupled to high-resolution mass spectrometry (LC-HRMS) and cheminformatics tools. To our knowledge, this is the first time that PU microplastics photodegradation has been investigated in seawater beyond polymer characterization. Previous research was mainly conducted under dry conditions and heavily focused on the chemistry of residual polymers.29–37 Recently, studies on the characterization of the DOC produced from PP, PE, PS, and polyethylene terephthalate (PET) have shown how nontarget HRMS-based methods can lead to the identification of unknown photo-oxidation products.11,38–41 Confident identification cannot be achieved without reference standards or spectral library match,40,42 and the data reduction of HRMS identification workflows is such that only a minor fraction of the many thousands of detectable signals can be structure-annotated.42–44 While unequivocal identification of chemicals in environmental samples may be crucial, hyphenated HRMS data offer the potential to study bulk characterization parameters that, even if lower on the identification confidence scale,40 can describe a much wider fraction of the carbon pool.45 Therefore, we chose to characterize the DOC with respect to the evolution of number of chemical species generated by photo-oxidation, their masses, degree of saturation, oxidation, and concentration dynamics. Degrees of saturation and oxidation are typically used to describe the bulk properties of DOC in HRMS studies,45 and characterizing their variations alongside changes in number of chemical species, their size, and concentration dynamics can reveal the effects of photo-oxidative processes such as chain scission, oxygen incorporation, and introduction of double bonds.8
2 Experimental
Unless otherwise stated, all data were processed within the R statistical environment (version 4.0.3).462.1 Chemicals and reagents
The chemicals and reagents used for this study were of analytical grade and have been used as received without any further purification. Detailed information about purity and manufacturers is provided in the ESI, Section S1.†2.2 Test materials and sample preparation
Ether-based TPU (TPU_Ester), ester-based TPU (TPU_Ether), and cross-linked PU (PU_Hardened) particles of 350 μm average size were produced at BASF SE (Ludwigshafen, Germany) by means of cryo-milling as described elsewhere.47 Further details including polymer chemistry, density, particle size distribution, and molecular weight distribution are provided in the ESI, Section S2.† All glassware and quartzware were pre-combusted at 450 °C for 10 h. All samples were prepared in triplicate and consisted of 150 mg PU microplastics added to custom-made cylindrical quartz cuvettes of 10.5 × 2 cm size (Technical Glass Products, USA) filled with 30 mL artificial seawater prepared following standard methods,48 where high loadings were used to increase the likelihood of detecting dissolved products. Dark controls were prepared identically but wrapped in three layers of aluminum foil. Blank samples consisted of artificial seawater only. After the irradiation experiments, since PU microplastics were negatively buoyant, the particles were let to settle for approximately 1 h, the supernatant was collected with a 10 mL glass pipette and transferred to a 40 mL glass vial. The microplastic particles were transferred to a 24-well plate with a pre-cleaned, stainless-steel spatula and allowed to dry in a desiccator between 7 and 10 days prior to characterization.2.3 Photo-weathering reactor
Samples were irradiated at 25 °C for 60, 90, and 120 days total using a custom-built solar reactor consisting of a chamber (L × H × D: 120 × 35 × 60 cm) with anodized aluminum reflector walls (Lorin Industries, USA), equipped with a total of ten 40-Watt T12-type light tubes, of which five emitted UV-A (Q-Lab, USA) and five emitted fluorescent light (General Electric, USA). The light tubes were placed at a height of 25 cm from the samples. The peak emission was at 340 nm with a spectral irradiance of 25 μW cm−2 nm−1, as characterized by a Flame miniature spectrometer (Ocean Insight, USA). Irradiance coverage and spectrum in the reactor center are available in the ESI, Section S3 (Fig. S3.1 and S3.2†). After 60, 90, and 120 days in the reactor, the cumulative UV dose values integrated over 300–400 nm range were 175, 262, and 350 MJ m−2, respectively. These doses correspond to roughly 7.5, 11.2, and 15 months outdoor exposure at 31° N latitude. To ensure homogeneous irradiation of the samples, the cuvettes were laid flat on the reactor surface (i.e., perpendicular to the light source). Consequently, it was assumed that the vertical distribution of the UV irradiance across the cuvettes would not vary substantially between samples. All photo-weathered microplastics-containing samples, which were 27 considering 3 polymer chemistries and three replicates per time point, were fit in the center area (60 × 38 cm, highlighted with a red dashed line in Fig. S3.1†) where the irradiance variation was lower than 10%. The first time point was 60 days, corresponding to a UV dose of 175 MJ cm−2, not only to ensure that detectable amounts of DOC could be produced, but also to mimic environmentally relevant conditions in marine environments, as photo-oxidation in aqueous media is slower than under dry conditions.82.4 Attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy
The microplastics were analyzed by Attenuated Total Reflectance-Fourier Transform Infrared Spectroscopy (ATR-FTIR) with an ALPHA II spectrophotometer equipped with a platinum ATR module featuring a monolithic diamond crystal (Bruker Optics, USA). The ATR diamond crystal was coated with approximately 1 mm thickness of test material, which corresponded to roughly 10 mg of microplastics. For each sample, two aliquots were analyzed. Spectra in the region between 4000 and 400 cm−1 were recorded with 32 scans at a resolution of 2 cm−1. Data acquisition, baseline correction and min–max normalization were performed using the software Opus, version 7.8 (Bruker Optics, USA). The carbonyl index (CI), defined as the ratio between the area below the carbonyl (1650–1850 cm−1) and methylene (1420–1500 cm−1) bands, was derived to monitor the progress of photo-oxidation.49 Area under curve values were obtained using the R package pracma.502.5 Elemental analysis
For each sample, aliquots of 1.5–3 mg microplastics were analyzed to determine C, H, O and N content using a PerkinElmer 2400 SERIES II CHNS/O Analyzer (PerkinElmer, USA) at Intertek USA, Inc. (Whitehouse, NJ). The reporting limit was 0.1% mass for all elements.2.6 Total organic carbon
Total Organic Carbon (TOC) analyses were performed using a vario TOC cube configured in liquid mode (Elementar, Germany). From each sample, 15 mL aliquots were transferred to pre-washed (three-times with acidified Milli-Q water) 20 mL polypropylene Luer syringes (VWR, USA), and collected in 40 mL glass vials. Prior to analysis, all samples, blanks and calibrants were acidified with three drops of full-strength (12.1 M) hydrochloric acid and purged with ultrapure air to ensure elimination of inorganic carbon. For quantification, eight calibrants of concentration ranging between 1 and 500 mg L−1 were prepared by diluting a potassium hydrogen phthalate carbon standard (LabChem Inc., USA) in acidified Milli-Q water. The limit of detection and quantification were 0.45 and 0.79 mg L−1, respectively, and the calibration curve is provided in the ESI, Section S4.† The data were used to extrapolate DOC release rates per cumulative UV dose (mg MJ−1) by adapting an approach published elsewhere.51 Details on the extrapolation are provided in the ESI, Section S5.†2.7 LC-HRMS analysis and data processing
Artificial seawater samples from irradiated PU microplastic and associated controls were analyzed by LC-HRMS via injection of a 10 μL aliquot. Separation of the analytes was achieved with an Agilent Infinity 1260 HPLC system consisting of a vacuum degasser, a temperature-controlled autosampler, a column compartment, and a binary pump. The stationary phase was an Agilent ZORBAX Eclipse Plus C18 column (3 × 50 mm, 1.8 μm). The mobile phase consisted of water (A) and methanol (B), both acidified with 0.1% formic acid; the gradient program expressed as A:B was 90:10 at 0 min, 50:50 at 4 min, 5:95 from 17 to 25 min, and 90:10 from 25.1 to 29 min. The flow rate was 0.2 mL min−1. Due to the high salt content of the samples, the first 1.5 min of the analytical run was diverted to waste. Detection was achieved with an Agilent 6545 quadruple time-of-flight HRMS equipped with a Jet Stream electrospray ion source operated in positive mode, adopting an acquisition method previously detailed by Ferrer et al. (2020).52 In brief, full-scan HRMS data (MS1) were acquired for ions in the 100–1000 m/z range at 2 GHz high dynamic range mode. High-resolution tandem mass spectra (MS2) were recorded in data-dependent acquisition in the range of 40–700 m/z in narrow mode with a precursor isolation window of ±1.3 m/z. For each cycle, one MS1 spectrum (0.5 s) over the range 100–1000 Da was recorded, followed by acquisition of MS2 spectra of up to four precursors. Each precursor was fragmented at low (15 eV) and high (30 eV) collision energies. Stability of mass accuracy was guaranteed by tuning prior to each analysis batch and by infusing a reference mass calibration solution containing purine (m/z 121.0509 [M + H]+) and HP-921 [hexakis(1H,1H,3H-tetrafluoro-pentoxy)phosphazene] (m/z 922.0098 [M + H]+). For the analysis, 900 μL aliquots were transferred to 3 mL polypropylene syringes pre-rinsed with acidified Milli-Q water and fitted with 0.2 μm PTFE filters (VWR, USA). Before filtration, 100 μL of a 5 mg L−1 stock solution of adipic acid-13C6 and diethylene glycol-D8 (Cambridge Isotope Laboratories, USA) were added, resulting in an internal standard concentration of 500 μg L−1. The filtrate was collected in 2 mL LC vials and kept at −15 °C prior to analysis.
A nontarget screening workflow was applied to the LC-HRMS data to characterize the DOC. The data files were converted to mzML format using ProteoWizard (version 3.0)53 and imported into R for pre-processing using the open source software platform patRoon.54 The settings used to extract the features, grouping, aligning, filtering and formula annotation are detailed in the ESI (S6†). Peaks detected in the blanks (cf. S6†), in the dark controls and stable carbon isotopes were subtracted from the dataset. Molecular formulae explicable by MS1 and MS2 spectra were considered for further data analysis. When feature annotation resulted in more than one formula, the elemental composition with the lowest mass deviation was selected. To characterize the DOC, formula data were plotted in Van Krevelen (VK) diagrams,45,55 which were enhanced to display variables of interests (e.g., chromatographic retention time and m/z values). In VK diagrams, structural relationships between formula-annotated features can be observed graphically. Vertical lines are representative of hydrogenation/dehydrogenation reactions, where decreasing H/C values indicate introduction of double bonds. Horizontal lines with increasing O/C values indicate the substitution of hydrogen with a hydroxyl group. Based on the linearity equation H/C = −a(O/C) + b, lines with a slope of −1 are characteristic of carboxylic acids, while lines with a slope of −2 indicate a difference in methyl group or aldehyde/ketone group.45 Additionally, different slope values correspond to different homologous alkylation series.39,45,55
Statistical analyses were performed using an unpaired t-test considering a significance level of p < 0.05. Correlation tests were performed using the cor.test() function in R using the Spearman method. Unless stated otherwise, all p-values provided in the Results & discussion section refer to the output of unpaired t-tests.
3 Results & discussion
3.1 Microplastic particles characterization
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations, indicating accumulation of quinone structures in all PU microplastics (Fig. S7.8†). This result is in partial agreement with the literature,32,33,58 as other studies have reported a decreasing 1597 cm−1 band, attributing it to CC stretching vibrations in aromatic rings.29,59 Other notable changes in the irradiated samples were the increasing carbonyl peak at 1704 cm−1 wavenumber in all PU microplastics (Fig. S7.8†), a strong indicator of photo-oxidation;9,19,49 the increasing and broadening 3200–3600 cm−1 wavenumber region in all PU microplastics (Fig. S7.9†), consistent with hydroxyl stretching vibration;35,57 and the decreasing band at 1100 cm−1 in TPU_Ether (Fig. S7.10†), characteristic of C–O stretch in ethers, and reflecting the degradation of ether bonds.29,36,56
C stretching vibrations, indicating accumulation of quinone structures in all PU microplastics (Fig. S7.8†). This result is in partial agreement with the literature,32,33,58 as other studies have reported a decreasing 1597 cm−1 band, attributing it to CC stretching vibrations in aromatic rings.29,59 Other notable changes in the irradiated samples were the increasing carbonyl peak at 1704 cm−1 wavenumber in all PU microplastics (Fig. S7.8†), a strong indicator of photo-oxidation;9,19,49 the increasing and broadening 3200–3600 cm−1 wavenumber region in all PU microplastics (Fig. S7.9†), consistent with hydroxyl stretching vibration;35,57 and the decreasing band at 1100 cm−1 in TPU_Ether (Fig. S7.10†), characteristic of C–O stretch in ethers, and reflecting the degradation of ether bonds.29,36,56
Polymer photo-oxidation corresponded to a consistent increase in carbonyl index (Fig. 1), suggesting gradual accumulation of carbonyl groups on the surface of all PU microplastics.29,49,57,60 In the dark controls, the index value of TPU_Ester (9.8 ± 0.2) was higher than that of TPU_Ether (4.5 ± 0.2) by a factor of two, aligning well with the anticipated chemistry of their respective flexible segments. PU_Hardened displayed the lowest carbonyl index value 3.7 ± 0.2 under dark conditions, indicating a lower concentration of carbonyl oxygen along its surface. After a UV dose of 350 MJ m−2, TPU_Ester, TPU_Ether, and PU_Hardened showed mean index enhancements of approximately 40%, 29%, and 46%, respectively, compared to the dark controls. While TPU_Ester exhibited a steady increase, the mean carbonyl index values of TPU_Ether stabilized by receiving a UV dose of 262 MJ m−2. Presuming that photochemical oxygen consumption did not progress sufficiently to result in oxygen deficiency in the aqueous samples,61 this stabilization could have resulted from a steady state in which carbonyl group formation occurred at the same rate as carbonyl group consumption. This could occur via chain scissions in a process similar to that observed for hydroxyl groups by Bottino et al.62
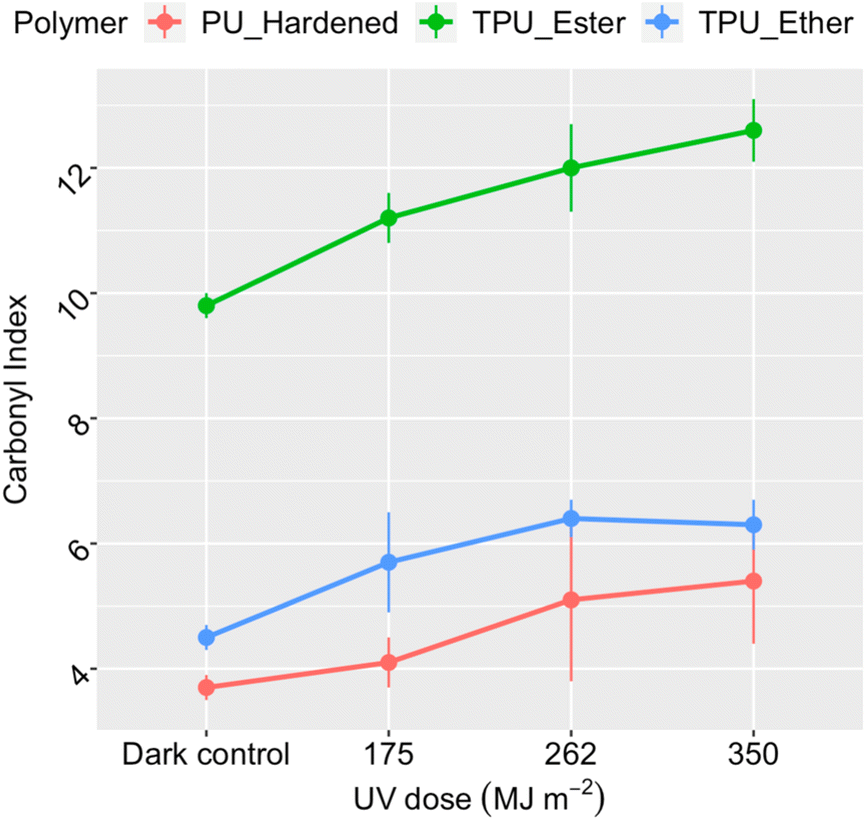 | ||
| Fig. 1 Carbonyl index of the residual polymer as a function of the cumulative UV dose, integrated in the 300–400 nm range, in megajoule per square meter. Data points and error bars represent the mean values and standard deviations, respectively, obtained from the replicate measurements (n = 18 for dark controls and n = 6 for irradiated samples). Invisible error bars are smaller than the data point markers. ATR-FTIR spectra available in the ESI, Section S7.† | ||
3.2 Dissolved organic carbon characterization
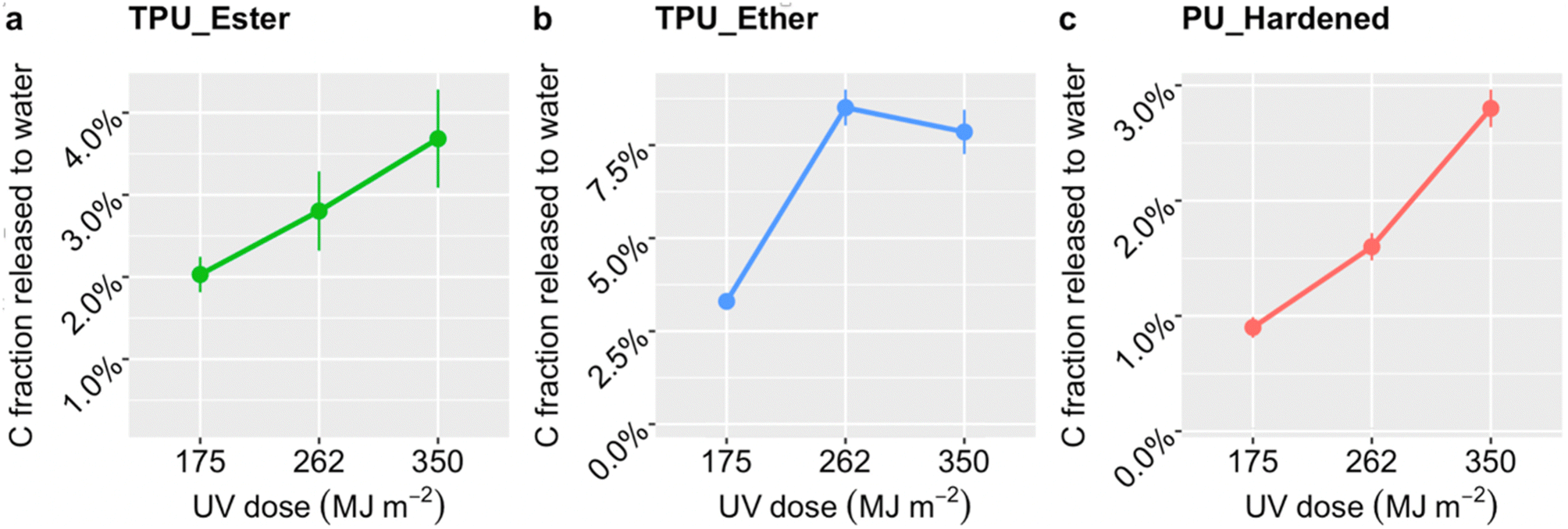 | ||
| Fig. 2 Carbon mass fraction released from TPU_Ester (a), TPU_Ether (b), and PU_Hardened (c) microplastics to the seawater as a function of cumulative UV dose in megajoule per square meter (MJ m−2). The dose values on the x-axis are plotted as factorial variables to improve data visualization. Data points and error bars represent the mean values and standard deviations, respectively, obtained from three replicate samples. Invisible error bars are smaller than the data point markers. | ||
After a UV dose of 175 MJ m−2, TPU_Ester (Fig. 2a) released 2.0 ± 0.2% carbon mass, which increased gradually to 3.7 ± 0.2% with a UV dose of 350 MJ m−2. TPU_Ether (Fig. 2b) released 3.3 ± 0.2% carbon mass with a UV dose of 175 MJ m−2, increasing by nearly a factor of three with a dose of 262 MJ m−2, and remaining substantially unchanged thereafter. The carbon mass released by PU_Hardened (Fig. 2c) increased gradually to 2.8 ± 0.2% by receiving a UV dose of 350 MJ m−2. Overall, the higher degree of carbon release of TPU_Ether aligned well with the greater susceptibility of ether-based TPUs to oxidative processes.19,36,37,47,63 Interestingly, the DOC release profile of TPU_Ether not only stood out for its higher values at all UV doses, but was also consistent with the carbonyl index (Fig. 1), which indicated a stabilization of carbonyl oxygen after a UV dose of 262 MJ m−2. Theoretically, this stabilization could have resulted from a steady state in which the rate of consumption (e.g., via chain scission) equaled the rate of formation of carbonyl oxygen. However, the DOC evidence refuted this possibility, suggesting a prevalence of cross-linking. This was in agreement with findings by Scholz et al.,36 who observed that the solubility of photo-weathered ether-based TPU decreased due to cross-linking while observing a stabilization of molar mass loss. Likewise, Dannoux et al. found that cross-linking competed with chain scission in ether-based TPUs under high-energy radiation.65 The lower release of DOC from PU_Hardened was expected. Cross-linking reduces interchain distances (e.g., via hydrogen or covalent bonds) preventing oxygen diffusion and UV light penetration,66 conferring a de facto resistance to photo-oxidation. Although a direct literature reference on PU_Hardened photodegradation was not found, our results agreed with Lehner et al.,67 who anticipated slow degradation rates for this cross-linked PU. Studies on other cross-linked PUs showed that chemical oxidation produced short chain scission products that reorganized and cross-linked,64 whereas UV photo-oxidation induced only minor chain scissions.68
Carbon release rates were extrapolated from DOC concentrations and UV dose as detailed in the ESI, Section S5.† The corresponding rate values were 0.023, 0.013, and 0.010 mg MJ−1 for TPU_Ether, TPU_Ester and PU_Hardened, respectively. Similar to carbon fraction release profiles (Fig. 2), the release rates aligned with the literature regarding PU chemistry-specific susceptibility to (photo-)oxidative degradation,19,36,37,47,63 with TPU_Ether and PU_Hardened being the polymers that released the most and least DOC per UV dose, respectively. To our knowledge, this is the first time DOC release rates in units of mg MJ−1 are reported for photo-weathered PU microplastics. Wohlleben and Neubauer investigated the release of fragments from (nano)composite flat plates applying different photo-weathering protocols.51 They found that fragments between 2 nm and 10 μm size were released from an ether-based TPU and a crosslinked PU at a rate of 0.69 and 0.17 mg MJ−1, respectively, and that the release from the TPU was slightly less-than-linear. They concluded that while variations of weathering protocol could shift the release rate values, the ranking was mainly influenced by polymer chemistry. It was challenging to draw a thorough comparison between the values obtained by Wohlleben and Neubauer and the present study due to differences in sample forms (flat plates vs. microplastics), exact polymer chemistry, and weathering and sampling protocols. Nevertheless, agreement was found between the rate ranking observed in both studies.
There are limitations to the real-world applicability of the DOC release rates in units of mg MJ−1. Firstly, the assumption of constant particle surface area throughout the experiments may not be appropriate. Chamas et al. incorporated shrinkage of the radius and reduction of surface area in a recent model that describes the degradation of spherical microplastic particles.69 They pointed out that samples with the same composition and mass, but different surface areas can show very different degradation rates and clarified that their model does not account for fragmentation, which abruptly increases the surface area. Secondly, the assumption of constant and homogeneous irradiation may not be appropriate too. In the environment, natural organic matter competes for photons and acts as a scavenger of reactive oxygen species, possibly slowing down polymer photo-oxidation.70 Despite these limitations, we believe that the DOC release rates in units of mg MJ−1 provide an adequate metric to investigate the impact of polymer chemistry on the degree of photodegradation under controlled laboratory conditions.
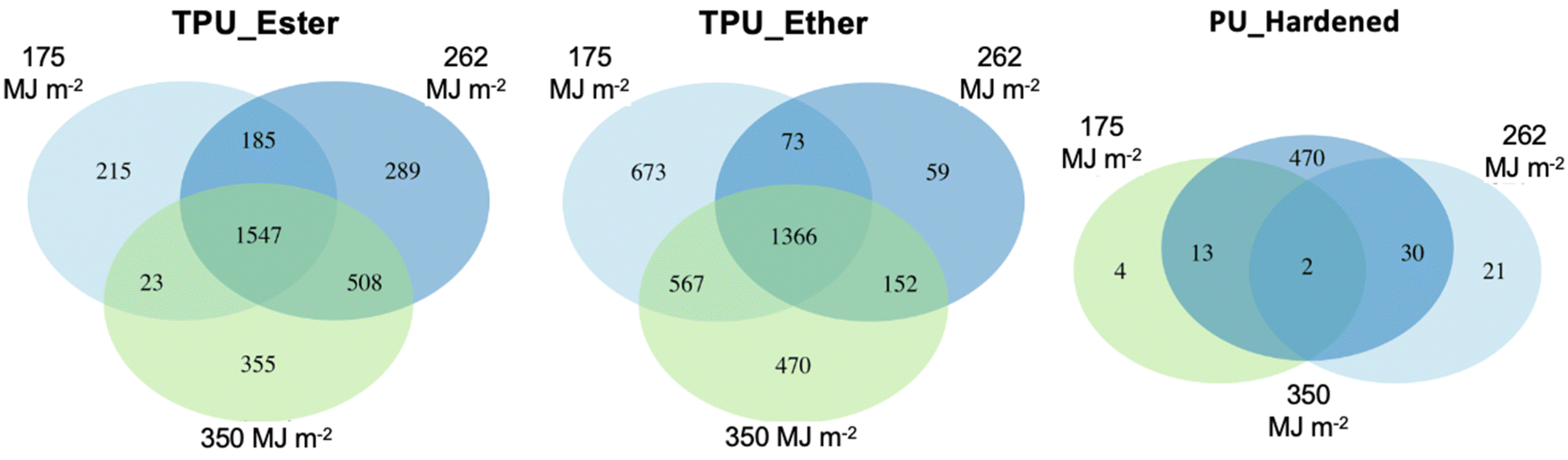 | ||
| Fig. 3 Venn diagrams of feature distribution in TPU_Ester (left), TPU_Ether (center), and PU_Hardened (right) irradiated with a UV dose of 175, 262 and 350 megajoule per square meter (MJ m−2). | ||
The formula-annotated features were visualized in a Van Krevelen (VK) diagram enhanced to display RT information (Fig. 4; surface density plots to facilitate the identification of the most populated regions of the VK diagram are provided in Fig. S9.1†). VK diagrams have been used in recent literature studies to fingerprint microplastic particles,41 and to characterize DOC release from photo-weathered plastic bags.72 However, the visualization approach used for the present investigation is advantageous, as it is coupled with additional plots to visualize the most data-dense areas in the diagrams and (ii) it is enhanced with hyphenated HRMS data of interest, e.g., m/z and RT values, so that DOC physicochemical properties other than elemental ratios can be visually inferred.
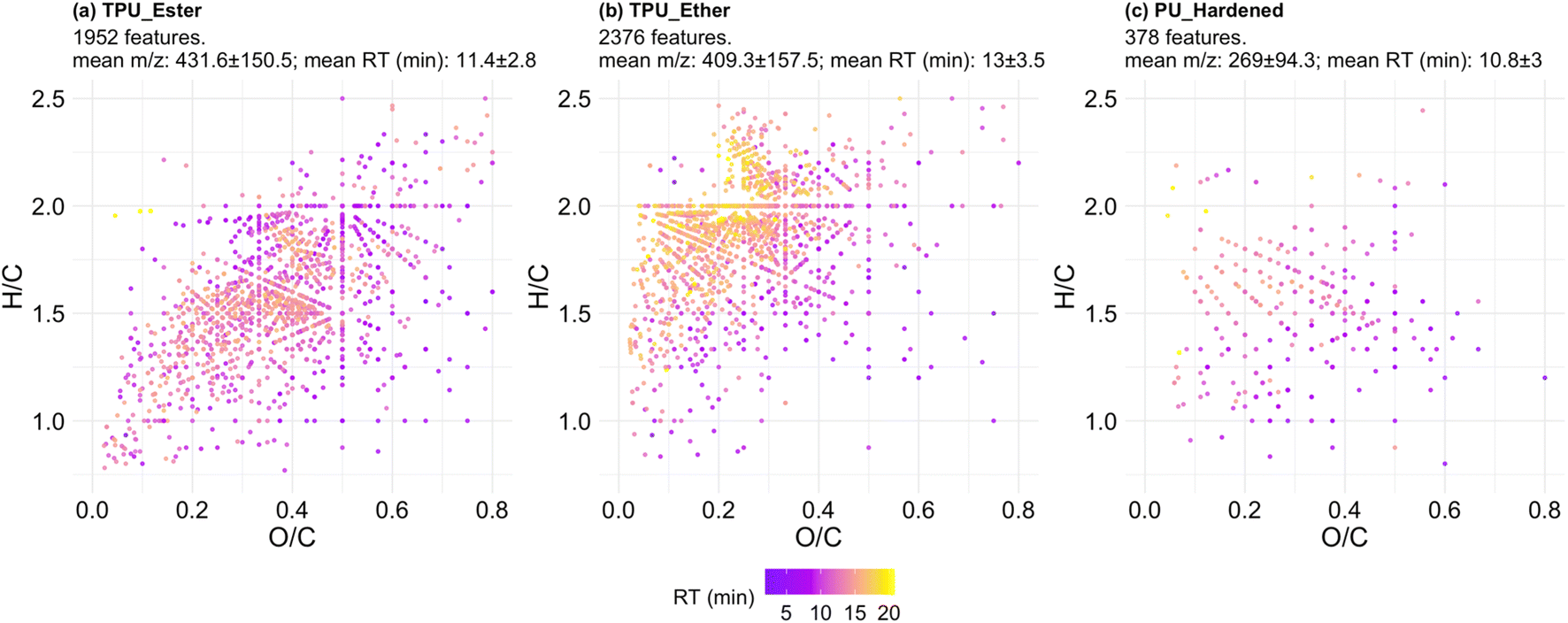 | ||
| Fig. 4 Van Krevelen plots of all formula-annotated features detected in TPU_Ester (a), TPU_Ether (b), and PU_Hardened (c). Each data point represents an annotated formula. The color scale represents chromatographic retention time (RT) in minutes. H/C: hydrogen–carbon ratio; O/C: oxygen–carbon ratio. Number of plotted features, mean and standard deviation of the m/z and RT values are provided in the subtitles. | ||
The features of the TPUs (Fig. 4a and b) differed for: (i) degree of saturation expressed as H/C ratio, which was lower in TPU_Ester; and (ii) oxygen content, expressed as O/C ratio, which was lower in TPU_Ether. These differences aligned with the chemistry of TPU elastomers: ester-based segments contain less hydrogen and more oxygen than ether-based segments of the same chain length due to the carbon to oxygen double bond of ester functional groups. The features in TPU_Ether (Fig. 4b) had the longest RTs of the dataset and populated the VK diagram in an area with high H/C ratios (≥1.5) and low O/C ratios (≤0.5), which is characteristic of aliphatic compounds.39 This was also the most populated area of the VK diagram of TPU_Ether, as shown by the surface density plots (Fig. S9.1†). A dense cluster of 790 late eluting features had H/C and O/C ratios in the range of 1.75–2.5 and 0.2–0.3, respectively, and RT at or beyond 14.4 min. Considering that chromatographic separation was achieved via a reversed-phase mechanism with an octadecyl stationary phase, molecular structures with saturated, long alkyl chains would explain the long retention. This hypothesis was corroborated by the double bond equivalence (DBE) data of TPU_Ether (Fig. 5b), which showed a negligible correlation (Spearman's ρ) between DBE and RT (ρ = 0.041, p-value = 0.047), and a weak correlation between DBE and number of carbon atoms (ρ = 0.29, p-value < 2.2 × 10−16). In contrast, the features of TPU_Ester exhibited a moderate positive correlation between DBE and number of carbon atoms (ρ = 0.58, p-value < 2.2 × 10−16). Based on literature data, UV photo-oxidation of ester-based flexible segments can induce decarboxylation to give alkene group-containing products.19,73 This phenomenon supported the lower H/C (Fig. 4a) and higher DBE values (Fig. 5a) observed in the features of TPU_Ester. The dataset size difference did not allow for a meaningful comparison between PU_Hardened and the TPU elastomers. Moreover, it was challenging to reconcile the dramatically lower number of features produced from the cross-linked PU with its DOC concentrations. A possible explanation is that the chemical species released from PU_Hardened, while fewer, occurred at high concentrations. However, it cannot be excluded that a fraction of the DOC was not detected by HRMS. This would be the case for molecules lacking heteroatoms that could be protonated (e.g., hydrocarbons) or molecules with no affinity for the LC stationary phase that would elute earlier than 1.5 min. The latter case is relevant for small and highly hydrophilic compounds with negative logarithm octanol–water partition coefficient values (logKow). These analytes are typically amenable for LC separation by either Hydrophilic Interaction Liquid Chromatography (HILIC) or normal-phase chromatography. The DBE data (Fig. 5c) showed that the features of PU_Hardened had fewer number of carbon atoms, and, in line with the literature on cross-linked PU degradation,64,68 this indicated that only low molecular weight photo-oxidation products were generated.
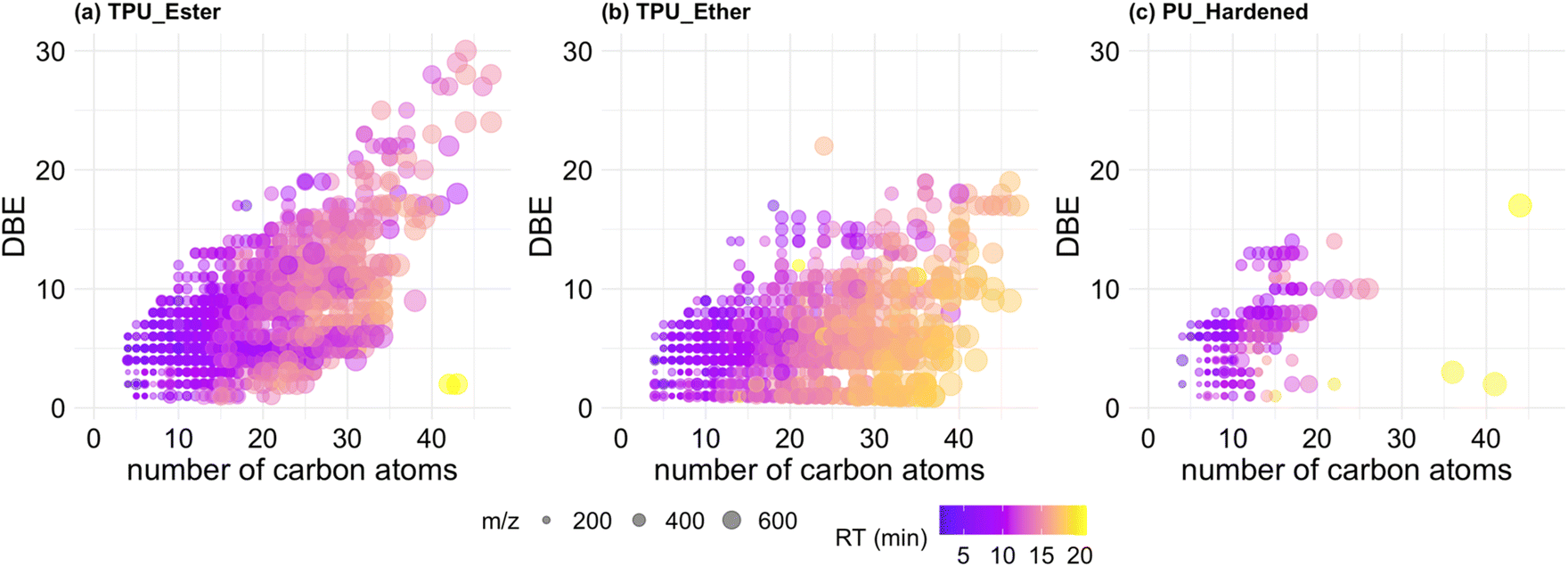 | ||
| Fig. 5 Double-bond-equivalence (DBE) versus number of carbon atoms of all formula-annotated features detected in TPU_Ester (a), TPU_Ether (b), and PU_Hardened (c). Each data point represents an annotated formula. Data point marker size represents feature size expressed as mass-to-charge (m/z) value. The color scale represents feature retention time (RT) in minutes. | ||
The literature indicated that the organic carbon leached from weathered polymers can photodegrade to low molecular weight by-products, partially oxidized DOC and CO2.74,75 To verify whether the features in the DOC exhibited dynamics trends, i.e., they were produced and subsequently photo-degraded, the correlation (Spearman's ρ) between UV dose and normalized feature intensities was derived. This exercise was limited to the formula-annotated features that were detected at all UV dose values, and PU_Hardened was excluded as only 2 features met this criterion. This analysis showed that roughly 80% of features of TPU_Ester had been continuously supplied to the seawater (Fig. S9.2a†), indicating that these chemicals were either recalcitrant to photo-oxidation or their rate of production was higher than their rate of consumption. A different scenario was observed for TPU_Ether, as 61% of the features exhibited a moderate to strong decreasing trend (Fig. S9.2b†). According to the literature, the soft segments of TPU_Ether are very sensitive to photo-oxidation, their degradation is initiated by radical attacks followed by hydrogen abstraction, forming alkoxy radicals and hydroperoxides, and culminating in chain scissions.19,56 Thus, the decreasing intensities of the features of TPU_Ether could be rationalized with the high lability of ether segments to photodegradation, indicating that the chemical species in the DOC pool were, in turn, degraded. This was an interesting finding, as it suggested that a polymer's susceptibility to photodegradation may be conserved in its degradation by-products.
Features that exclusively occurred at a given UV dose, hereafter referred to as unique features, were inspected by plotting the data in VK diagrams enhanced to visualize RT and m/z values (Fig. 6). It is noteworthy that while the unique features may have originated directly from the bulk polymer, these may also be by-products of the (chromophoric fraction of) DOC that were subsequently degraded to undetectable concentrations between UV doses. In TPU_Ester, the number of unique features increased gradually with increasing irradiation. This was accompanied by a significant reduction in mean m/z (p-value < 2.2 × 10−6) and H/C values (p-value = 5.6 × 10−7) at the end of the irradiation experiments, as shown by an unpaired t-test. These observations were in agreement with the literature on photodegradation of ester-based polyurethane,19,29,76 as UV radiation induces photo-oxidation of ester segments with chain scissions through Norrish Type II reactions, which give products with unsaturated chain ends. Surprisingly, and despite the smaller m/z values, the mean RT of the unique features in samples irradiated with a UV dose of 262 MJ m−2 was significantly longer than that of the features detected after a UV dose of 175 MJ m−2 (p-value = 2.9 × 10−15). This indicated a higher degree of hydrophobicity, which would result in longer RT in reversed-phase chromatography.
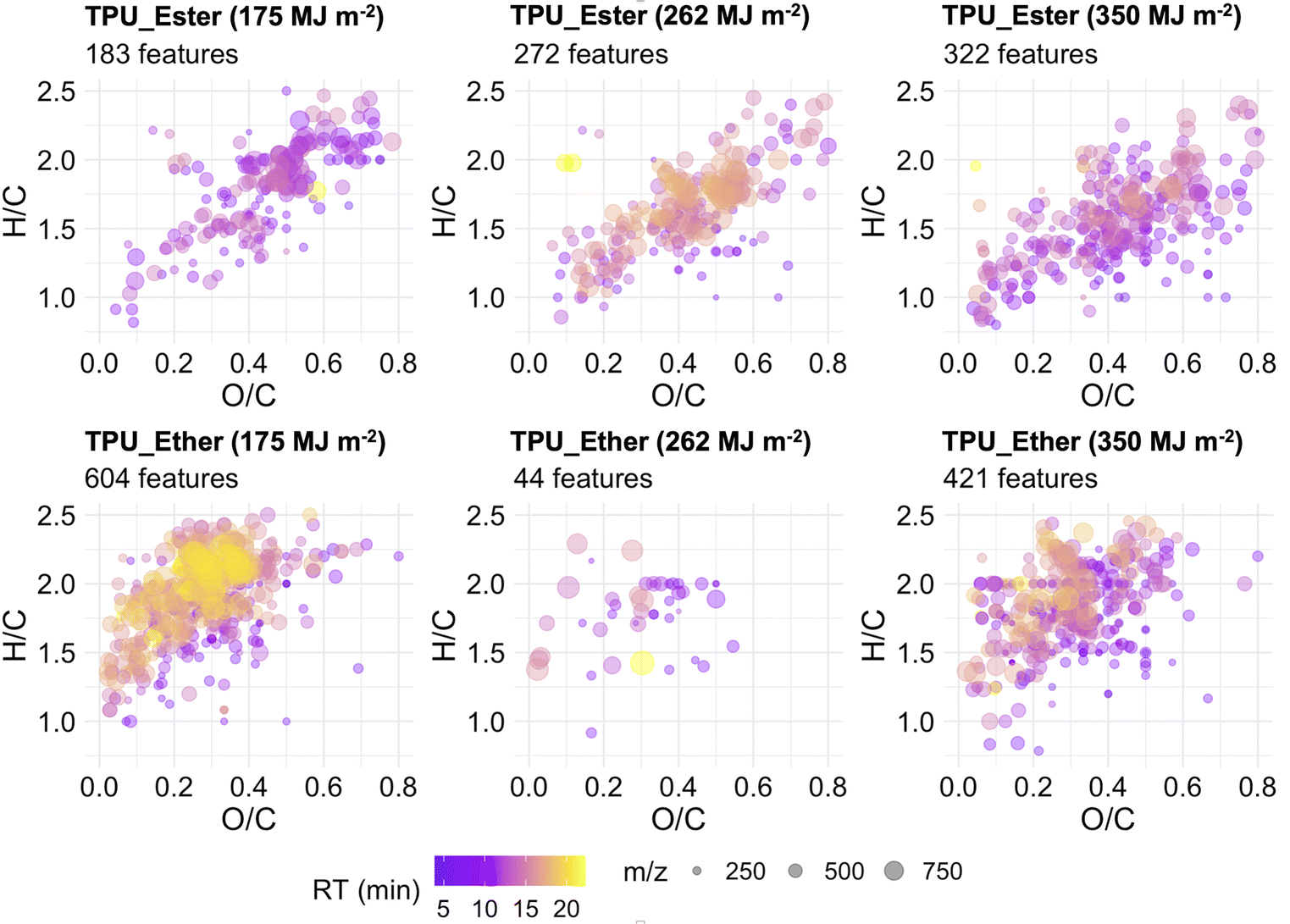 | ||
| Fig. 6 Van Krevelen plots of unique features detected in samples irradiated with a UV dose of 175 MJ m−2 (left), 262 MJ m−2 (middle) and 350 MJ m−2 (right) in TPU_Ester (top) and TPU_Ether (bottom). The data point marker size reflects the feature mass-to-charge (m/z) value, and the color scale represents the feature retention time (RT) in minutes. H/C: hydrogen–carbon ratio; O/C: oxygen–carbon ratio. | ||
The unique features in the DOC of TPU_Ether were 673, 59, and 470 after a cumulative UV dose of 175, 262, and 350 MJ m−2, respectively. It remains unclear why the features uniquely detected after a 262 MJ m−2 UV dose were less abundant by one order of magnitude. The effects of a possibly too aggressive data pre-processing, specifically the replicate abundance filter (cf. ESI Section S6†), were ruled out, as the results did not change after testing less strict filter values (data not shown). Despite the inconsistent trend, it can't be excluded that additional unique features occurred at levels that were below instrumental detection limits, either due to low concentration or low ionization efficiency. Nevertheless, the unique features after a UV dose of 262 MJ m−2 were excluded from further statistical analysis, as the small sample size did not allow for a meaningful comparison with the rest of the TPU_Ether dataset. Significant m/z and RT value reductions (p-value < 2.2 × 10−16) were observed between the unique features detected in the TPU_Ether samples irradiated with a UV dose of 350 MJ m−2 and those detected with 175 MJ m−2. These were accompanied by a moderately significant increase in O/C value (p-value = 0.015), suggesting the introduction of oxygen-containing functional groups such as carbonyl. This was in line with the literature on photodegradation of ether-based polyurethane,19,56 as ether segments are degraded to low molecular weight products with carboxylic end groups.
Considering the entire nontarget screening dataset of the TPUs, it appeared that in TPU_Ester the number of unique features increased along with the intensity trends of the features detected at all UV doses. In TPU_Ether, decreasing number of unique features corresponded to decreasing intensity trends of the features detected at all UV doses. While shifts in m/z values, RT times, and degree of saturation and oxidation were in line with mechanisms reported in the literature, more research is necessary to assess whether the unique features were produced from photo-oxidation of the DOC or from the bulk polymer.
While the present study focused on microplastics' photo-oxidation products, it is noteworthy that 508, 88, and 126 features were detected in the dark controls of TPU_Ester, TPU_Ether, and PU_Hardened, respectively. These features comprised both non-intentionally added substances (NIAS) and additives (IAS) that can leach from polymers regardless of photo-weathering,77 and hydrolytic breakdown products. The substantially higher number of features in the dark controls of TPU_Ester could be expected, as ester-based TPUs undergo hydrolytic degradation.33,35 While these data are provided to the reader, they are not discussed further as they don't fall within the scope of the present work.
4 Conclusions
This investigation showed that (artificial) sunlight can degrade polyurethane microplastics in an aqueous environment via UV photo-oxidation. While offering the potential to mitigate plastic pollution, photo-weathering released an organic carbon pool populated with thousands of degradation products, which were dynamic in terms of accumulation behavior. The progress and degree of photo-weathering, and DOC accumulation behavior appeared to be influenced by polymer chemistry. Our results suggest that a specific polymer's susceptibility to photo-oxidation is conserved in its degradation products, as shown by decreasing intensity trends of the majority of the chemicals detected in the DOC of TPU_Ether. For other polymers, as in the case of TPU_Ester and PU_Hardened, more species of the DOC seemed recalcitrant and thus might have the potential to accumulate in ambient seawater. However, the present study only simulated one specific combination of degradation stresses; other stresses or transport processes contribute to the actual environmental fate of the DOC released from aging plastic. Initiatives such as the Alliance to End Plastic Waste, supported by the recent UN resolution,78 will curb and eventually stop mismanagement of plastic waste. But even the degradation of the plastic waste released to date has the potential to generate more DOC that will at some point reach peak concentration in the oceans, highlighting the need for robust and harmonized analytical strategies to characterize environmental persistence, mobility and ecotoxicology of this highly complex carbon pool. Recent observations that the hazard of aging microplastics may be predominantly attributed to the released DOC add to the relevance of identifying recalcitrant species in the DOC.79 This would extend our understanding of microplastics' environmental fate beyond observations from residual polymer and could support (i) the design of new materials, e.g., exploiting physicochemical properties that allow polymer and by-products to be (bio)degradable, and (ii) regulatory efforts, e.g., by limiting the production and application of polymers that release persistent or toxic DOC.Conflicts of interest
The materials tested here are commercially available products of BASF. W. Wohlleben is employed by BASF. There are no other conflicts of interest to declare.Acknowledgements
This work was supported with funds from BASF. Omar Tantawi is acknowledged for supporting the TOC measurements at MIT's Department of Civil and Environmental Engineering. Mohan Kumar is acknowledged for supporting the LC-HRMS measurements at MIT's Department of Chemistry Instrumentation Facility. Rupert Konradi at BASF's Northeast Research Alliance is thanked for helpful discussions. Three anonymous reviewers are acknowledged for their useful comments.References
- E. MacArthur, Beyond plastic waste, Science, 2017, 358(6365), 843 CrossRef PubMed , https://science.sciencemag.org/content/358/6365/843.
- C. M. Rochman, Microplastics research—from sink to source, Science, 2018, 360(6384), 28–29 CrossRef CAS PubMed.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman and A. Andrady, et al., Plastic waste inputs from land into the ocean, Science, 2015, 347(6223), 768–771 CrossRef CAS PubMed , https://science.sciencemag.org/content/347/6223/768.abstract.
- L. Lebreton, M. Egger and B. Slat, A global mass budget for positively buoyant macroplastic debris in the ocean, Sci. Rep., 2019, 9(1), 12922, DOI:10.1038/s41598-019-49413-5.
- A. Cózar, F. Echevarría, J. I. González-Gordillo, X. Irigoien, B. Úbeda and S. Hernández-León, et al., Plastic debris in the open ocean, Proc. Natl. Acad. Sci., 2014, 111(28), 10239–10244 CrossRef PubMed , https://www.pnas.org/content/111/28/10239.abstract.
- A. L. Andrady, Microplastics in the marine environment, Mar. Pollut. Bull., 2011, 62(8), 1596–1605 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S0025326X11003055.
- Y. K. Song, S. H. Hong, M. Jang, G. M. Han, S. W. Jung and W. J. Shim, Combined Effects of UV Exposure Duration and Mechanical Abrasion on Microplastic Fragmentation by Polymer Type, Environ. Sci. Technol., 2017, 51(8), 4368–4376, DOI:10.1021/acs.est.6b06155.
- J. F. Rabek, Polymer Photodegradation, SPRINGER-SCIENCE+BUSINESS MEDIA, B.V, 1995 Search PubMed.
- E. Yousif and R. Haddad, Photodegradation and photostabilization of polymers, especially polystyrene: review, Springerplus, 2013, 2(1), 398, DOI:10.1186/2193-1801-2-398.
- L. Zhu, S. Zhao, T. B. Bittar, A. Stubbins and D. Li, Photochemical dissolution of buoyant microplastics to dissolved organic carbon: Rates and microbial impacts, J. Hazard. Mater., 2020, 383, 121065 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S0304389419310192.
- B. Gewert, M. Plassmann, O. Sandblom and M. MacLeod, Identification of Chain Scission Products Released to Water by Plastic Exposed to Ultraviolet Light, Environ. Sci. Technol. Lett., 2018, 5(5), 272–276, DOI:10.1021/acs.estlett.8b00119.
- A. L. Andrady, in Persistence of Plastic Litter in the Oceans BT - Marine Anthropogenic Litter, ed. M. Bergmann, L. Gutow and M. Klages, Springer International Publishing, Cham, 2015. pp. 57–72, DOI:10.1007/978-3-319-16510-3_3.
- J. F. Rabek, in Photo-Oxidative Degradation BT - Photodegradation of Polymers: Physical Characteristics and Applications, ed. J. F. Rabek, Springer, Berlin, Heidelberg, 1996, pp. 51–97, DOI:10.1007/978-3-642-80090-0_4.
- L. Hermabessiere, A. Dehaut, I. Paul-Pont, C. Lacroix, R. Jezequel and P. Soudant, et al., Occurrence and effects of plastic additives on marine environments and organisms: A review, Chemosphere, 2017, 182, 781–793 CrossRef CAS PubMed.
- C. Romera-Castillo, M. Pinto, T. M. Langer, X. A. Álvarez-Salgado and G. J. Herndl, Dissolved organic carbon leaching from plastics stimulates microbial activity in the ocean, Nat. Commun., 2018, 9(1), 1430, DOI:10.1038/s41467-018-03798-5.
- M. Fakhraee, L. G. Tarhan, N. J. Planavsky and C. T. Reinhard, A largely invariant marine dissolved organic carbon reservoir across Earth's history, Proc. Natl. Acad. Sci., 2021, 118(40), e2103511118, DOI:10.1073/pnas.2103511118.
- C. Romera-Castillo, S. Birnstiel, X. A. Álvarez-Salgado and M. Sebastián, Aged Plastic Leaching of Dissolved Organic Matter Is Two Orders of Magnitude Higher than Virgin Plastic Leading to a Strong Uplift in Marine Microbial Activity, 2022 Search PubMed.
- A. Kemona and M. Piotrowska, Polyurethane Recycling and Disposal: Methods and Prospects, Polymers, 2020, 12(8), 1752 CrossRef CAS PubMed , https://pubmed.ncbi.nlm.nih.gov/32764494.
- F. Xie, T. Zhang, P. Bryant, V. Kurusingal, J. M. Colwell and B. Laycock, Degradation and stabilization of polyurethane elastomers, Prog. Polym. Sci., 2019, 90, 211–268 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0079670018302995.
- W. Wohlleben, M. W. Meier, S. Vogel, R. Landsiedel, G. Cox and S. Hirth, et al., Elastic CNT–polyurethane nanocomposite: synthesis, performance and assessment of fragments released during use, Nanoscale, 2013, 5(1), 369–380, 10.1039/C2NR32711B.
- J. Datta, P. Kosiorek and M. Włoch, Synthesis, structure and properties of poly(ether-urethane)s synthesized using a tri-functional oxypropylated glycerol as a polyol, J. Therm. Anal. Calorim., 2017, 128(1), 155–167, DOI:10.1007/s10973-016-5928-2.
- R. Geyer, J. R. Jambeck and K. L. Law, Production, use, and fate of all plastics ever made, Sci. Adv., 2017, 3(7), e1700782 CrossRef PubMed , https://advances.sciencemag.org/content/3/7/e1700782.
- S. Laurichesse, C. Huillet and L. Avérous, Original polyols based on organosolv lignin and fatty acids: new bio-based building blocks for segmented polyurethane synthesis, Green Chem., 2014, 16(8), 3958–3970, 10.1039/C4GC00596A.
- A. Loredo-Treviño, G. Gutiérrez-Sánchez, R. Rodríguez-Herrera and C. N. Aguilar, Microbial Enzymes Involved in Polyurethane Biodegradation: A Review, J. Polym. Environ., 2012, 20(1), 258–265, DOI:10.1007/s10924-011-0390-5.
- L. D. K. Kanhai, R. Officer, O. Lyashevska, R. C. Thompson and I. O'Connor, Microplastic abundance, distribution and composition along a latitudinal gradient in the Atlantic Ocean, Mar. Pollut. Bull., 2017, 115(1), 307–314 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S0025326X16310116.
- M. Mistri, M. Scoponi, T. Granata, L. Moruzzi, F. Massara and C. Munari, Types, occurrence and distribution of microplastics in sediments from the northern Tyrrhenian Sea, Mar. Pollut. Bull., 2020, 153, 111016 CrossRef CAS PubMed.
- X. Peng, M. Chen, S. Chen, S. Dasgupta, H. Xu and K. Ta, et al., Microplastics contaminate the deepest part of the world's ocean, Geochem. Perspect. Lett., 2018, 9, 1–5 Search PubMed , http://www.geochemicalperspectivesletters.org/article1829.
- R. Coyle, G. Hardiman and K. O. Driscoll, Microplastics in the marine environment: A review of their sources, distribution processes, uptake and exchange in ecosystems, Case Stud. Chem. Environ. Eng., 2020, 2, 100010 CrossRef , https://www.sciencedirect.com/science/article/pii/S2666016420300086.
- D. Rosu, L. Rosu and C. N. Cascaval, IR-change and yellowing of polyurethane as a result of UV irradiation, Polym. Degrad. Stab., 2009, 94(4), 591–596 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0141391009000135.
- X. D. Chen, Z. Wang, Z. F. Liao, Y. L. Mai and M. Q. Zhang, Roles of anatase and rutile TiO2 nanoparticles in photooxidation of polyurethane, Polym. Test., 2007, 26(2), 202–208 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0142941806001887.
- Y. Wang, H. Wang, X. Li, D. Liu, Y. Jiang, Z. Sun and L. Balan, O3/UV Synergistic Aging of Polyester Polyurethane Film Modified by Composite UV Absorber, J. Nanomater., 2013, 2013, 1–7, DOI:10.1155/2013/169405.
- R. P. Singh, N. S. Tomer and S. V. Bhadraiah, Photo-oxidation studies on polyurethane coating: effect of additives on yellowing of polyurethane, Polym. Degrad. Stab., 2001, 73(3), 443–446 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0141391001001276.
- L. M. Gradinaru, S. Vlad, I. Spiridon and M. Petrescu, Durability of polyurethane membranes in artificial weathering environment, Polym. Test., 2019, 80, 106144 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0142941819312103.
- A. Paberza, L. Stiebra and U. Cabulis, Photodegradation of Polyurethane Foam Obtained from Renewable Resource Pulp Production Byproducts, J. Renewable Mater., 2015, 3(1), 19–27, DOI:10.7569/JRM.2014.634138.
- E. Pellizzi, A. Lattuati-Derieux, B. Lavédrine and H. Cheradame, Degradation of polyurethane ester foam artifacts: Chemical properties, mechanical properties and comparison between accelerated and natural degradation, Polym. Degrad. Stab., 2014, 107, 255–261 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0141391013004230.
- P. Scholz, V. Wachtendorf, U. Panne and S. M. Weidner, Degradation of MDI-based polyether and polyester-polyurethanes in various environments - Effects on molecular mass and crosslinking, Polym. Test., 2019, 77, 105881 CrossRef , https://www.sciencedirect.com/science/article/pii/S0142941819302363.
- E. G. Bajsić and V. Rek, Thermal stability of polyurethane elastomers before and after UV irradiation, J. Appl. Polym. Sci., 2001, 79(5), 864–873, DOI:10.1002/1097-4628(20010131)79:5%3C864::AID-APP110%3E3.0.CO.
- A. Khaled, A. Rivaton, C. Richard, F. Jaber and M. Sleiman, Phototransformation of Plastic Containing Brominated Flame Retardants: Enhanced Fragmentation and Release of Photoproducts to Water and Air, Environ. Sci. Technol., 2018, 52(19), 11123–11131, DOI:10.1021/acs.est.8b03172.
- B. Eyheraguibel, M. Traikia, S. Fontanella, M. Sancelme, S. Bonhomme and D. Fromageot, et al., Characterization of oxidized oligomers from polyethylene films by mass spectrometry and NMR spectroscopy before and after biodegradation by a Rhodococcus rhodochrous strain, Chemosphere, 2017, 184, 366–374 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S0045653517308445.
- E. L. Schymanski, J. Jeon, R. Gulde, K. Fenner, M. Ruff and H. P. Singer, et al., Identifying Small Molecules via High Resolution Mass Spectrometry: Communicating Confidence, Environ. Sci. Technol., 2014, 48(4), 2097–2098, DOI:10.1021/es5002105.
- X. Zhang, A. Mell, F. Li, C. Thaysen, B. Musselman and J. Tice, et al., Rapid fingerprinting of source and environmental microplastics using direct analysis in real time-high resolution mass spectrometry, Anal. Chim. Acta, 2020, 1100, 107–117 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S0003267019314412.
- J. Hollender, E. L. Schymanski, H. P. Singer and P. L. Ferguson, Nontarget Screening with High Resolution Mass Spectrometry in the Environment: Ready to Go?, Environ. Sci. Technol., 2017, 51(20), 11505–11512, DOI:10.1021/acs.est.7b02184.
- V. Albergamo, J. E. Schollée, E. L. Schymanski, R. Helmus, H. Timmer and J. Hollender, et al., Nontarget screening reveals time trends of polar micropollutants in a riverbank filtration system, Environ. Sci. Technol., 2019, 53(13), 7584–7594, DOI:10.1021/acs.est.9b01750.
- A. Lai, R. R. Singh, L. Kovalova, O. Jaeggi, T. Kondić and E. L. Schymanski, Retrospective non-target analysis to support regulatory water monitoring: from masses of interest to recommendations via in silico workflows, Environ. Sci. Eur., 2021, 33(1), 43, DOI:10.1186/s12302-021-00475-1.
- S. Kim, R. W. Kramer and P. G. Hatcher, Graphical Method for Analysis of Ultrahigh-Resolution Broadband Mass Spectra of Natural Organic Matter, the Van Krevelen Diagram, Anal. Chem., 2003, 75(20), 5336–5344, DOI:10.1021/ac034415p.
- R Core Team, R: A Language and Environment for Statistical Computing, Found Stat Comput, Vienna, Austria, 2017, http://https//www.R-project.org/ Search PubMed.
- P. Pfohl, M. Wagner, L. Meyer, P. Domercq, A. Praetorius and T. Hüffer, et al., Environmental Degradation of Microplastics: How to Measure Fragmentation Rates to Secondary Micro- and Nanoplastic Fragments and Dissociation into Dissolved Organics, Environ. Sci. Technol., 2022 Aug 16, 56(16), 11323–11334, DOI:10.1021/acs.est.2c01228.
- ASTM D1141-98(2013), Standard Practice for the Preparation of Substitute Ocean Water, ASTM Int., West Conshohocken, PA, 2013, https://www.astm.org Search PubMed.
- J. Almond, P. Sugumaar, M. N. Wenzel, G. Hill and C. Wallis, Determination of the carbonyl index of polyethylene and polypropylene using specified area under band methodology with ATR-FTIR spectroscopy, e-Polym., 2020, 20(1), 369–381, DOI:10.1515/epoly-2020-0041.
- H. W. Borchers, Pracma: Practical Numerical Math Functions, 2021, https://cran.r-project.org/web/packages/pracma/index.html Search PubMed.
- W. Wohlleben and N. Neubauer, Quantitative rates of release from weathered nanocomposites are determined across 5 orders of magnitude by the matrix, modulated by the embedded nanomaterial, NanoImpact, 2016, 1, 39–45 CrossRef , https://www.sciencedirect.com/science/article/pii/S2452074815300021.
- I. Ferrer, D. L. Sweeney, E. M. Thurman and J. A. Zweigenbaum, Nontargeted Screening of Water Samples Using Data-Dependent Acquisition with Similar Partition Searching, J. Am. Soc. Mass Spectrom., 2020, 31(6), 1189–1204, DOI:10.1021/jasms.0c00031.
- M. C. Chambers, B. Maclean, R. Burke, D. Amodei, D. L. Ruderman and S. Neumann, et al., A cross-platform toolkit for mass spectrometry and proteomics, Nat. Biotechnol., 2012, 30, 918, DOI:10.1038/nbt.2377.
- R. Helmus, T. L. ter Laak, A. P. van Wezel, P. de Voogt and E. L. Schymanski, patRoon: open source software platform for environmental mass spectrometry based non-target screening, J. Cheminform., 2021, 13(1), 1, DOI:10.1186/s13321-020-00477-w.
- V. Krevelen, Graphical-statistical method for the study of structure and reaction processes of coal, Fuel, 1950, 29, 269–284 Search PubMed.
- C. Wilhelm and J.-L. Gardette, Infrared analysis of the photochemical behaviour of segmented polyurethanes: aliphatic poly(ether-urethane)s, Polymer, 1998, 39(24), 5973–5980 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0032386197100659.
- H. Wang, Y. Wang, D. Liu, Z. Sun, H. Wang and T. Jiao, Effects of Additives on Weather-Resistance Properties of Polyurethane Films Exposed to Ultraviolet Radiation and Ozone Atmosphere, J. Nanomater., 2014, 2014, 87343, DOI:10.1155/2014/487343.
- K. Ligier, K. Olejniczak and J. Napiórkowski, Wear of polyethylene and polyurethane elastomers used for components working in natural abrasive environments, Polym. Test., 2021, 100, 107247 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0142941821001975.
- T. Zhang, F. Xie, J. Motuzas, P. Bryant, V. Kurusingal and J. M. Colwell, et al., Early-stage photodegradation of aromatic poly(urethane-urea) elastomers, Polym. Degrad. Stab., 2018, 157, 181–198 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0141391018303008.
- C. Han, A. Zhao, E. Varughese and E. Sahle-Demessie, Evaluating Weathering of Food Packaging Polyethylene-Nano-clay Composites: Release of Nanoparticles and their Impacts, NanoImpact, 2018, 9, 61–71 CrossRef PubMed.
- S. S. Andrews, S. Caron and O. C. Zafiriou, Photochemical oxygen consumption in marine waters: A major sink for colored dissolved organic matter?, Limnol. Oceanogr., 2000, 45(2), 267–277, DOI:10.4319/lo.2000.45.2.0267.
- F. A. Bottino, A. R. Cinquegrani, G. Di Pasquale, L. Leonardi and A. Pollicino, Chemical modifications, mechanical properties and surface photo-oxidation of films of polystyrene (PS), Polym. Test., 2004, 23(4), 405–411 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0142941803001399.
- V. Rek, M. Braver, T. Jocić and E. Govorćin, A contribution to the UV degradation of polyurethanes, Die Angew Makromol Chemie, 1988, 158(1), 247–263, DOI:10.1002/apmc.1988.051580114.
- J. Brzeska, M. Morawska, W. Sikorska, A. Tercjak, M. Kowalczuk and M. Rutkowska, Degradability of cross-linked polyurethanes based on synthetic polyhydroxybutyrate and modified with polylactide, Chem. Pap., 2017, 71(11), 2243–2251, DOI:10.1007/s11696-017-0218-4.
- A. Dannoux, S. Esnouf, B. Amekraz, V. Dauvois and C. Moulin, Degradation mechanism of poly(ether-urethane) Estane® induced by high-energy radiation. II. Oxidation effects, J. Polym. Sci., Part B: Polym. Phys., 2008, 46(9), 861–878, DOI:10.1002/polb.21419.
- M. Xie, W. Luo, H. Guo, L. D. Nghiem, C. Y. Tang and S. R. Gray, Trace organic contaminant rejection by aquaporin forward osmosis membrane: Transport mechanisms and membrane stability, Water Res., 2018, 132, 90–98 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S0043135417310679.
- R. Lehner, W. Wohlleben, D. Septiadi, R. Landsiedel, A. Petri-Fink and B. Rothen-Rutishauser, A novel 3D intestine barrier model to study the immune response upon exposure to microplastics, Arch. Toxicol., 2020, 94(7), 2463–2479, DOI:10.1007/s00204-020-02750-1.
- C. Decker, K. Moussa and T. Bendaikha, Photodegradation of UV-cured coatings II. Polyurethane–acrylate networks, J. Polym. Sci., Part A: Polym. Chem., 1991, 29(5), 739–747, DOI:10.1002/pola.1991.080290516.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum and J. H. Jang, et al., Degradation Rates of Plastics in the Environment, ACS Sustainable Chem. Eng., 2020, 8(9), 3494–3511, DOI:10.1021/acssuschemeng.9b06635.
- X. Wu, P. Liu, Z. Gong, H. Wang, H. Huang and Y. Shi, et al., Humic Acid and Fulvic Acid Hinder Long-Term Weathering of Microplastics in Lake Water, Environ. Sci. Technol., 2021, 55(23), 15810–15820, DOI:10.1021/acs.est.1c04501.
- H. L. Röst, T. Sachsenberg, S. Aiche, C. Bielow, H. Weisser and F. Aicheler, et al., OpenMS: a flexible open-source software platform for mass spectrometry data analysis, Nat. Methods, 2016, 13, 741, DOI:10.1038/nmeth.3959.
- A. N. Walsh, C. M. Reddy, S. F. Niles, A. M. McKenna, C. M. Hansel and C. P. Ward, Plastic Formulation is an Emerging Control of Its Photochemical Fate in the Ocean, Environ. Sci. Technol., 2021, 55(18), 12383–12392, DOI:10.1021/acs.est.1c02272.
- D. Rosu, C. Ciobanu, L. Rosu and C.-A. Teaca, The influence of polychromic light on the surface of MDI based polyurethane elastomer, Appl. Surf. Sci., 2009, 255(23), 9453–9457 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0169433209010563.
- L. A. Kaplan and R. M. Cory, Dissolved Organic Matter in Stream Ecosystems: Forms, Functions, and Fluxes of Watershed Tea, ed. J. B. Jones, Stanley EHBT-SE in a CE, Academic Press, Boston, 2016, ch. 6, pp. 241–320, https://www.sciencedirect.com/science/article/pii/B9780124058903000063 Search PubMed.
- L. Galgani, A. Engel, C. Rossi, A. Donati and S. A. Loiselle, Polystyrene microplastics increase microbial release of marine Chromophoric Dissolved Organic Matter in microcosm experiments, Sci. Rep., 2018, 8(1), 14635, DOI:10.1038/s41598-018-32805-4.
- B. Rånby, Photodegradation and photo-oxidation of synthetic polymers, J. Anal. Appl. Pyrolysis, 1989, 15, 237–247 CrossRef , https://www.sciencedirect.com/science/article/pii/0165237089850375.
- K. Gunaalan, E. Fabbri and M. Capolupo, The hidden threat of plastic leachates: A critical review on their impacts on aquatic organisms, Water Res., 2020, 184, 116170 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S0043135420307077.
- United Nations Environment Assembly of the United Nations Environment Programme. End Plastic Pollution: towards an International Legally Binding Instrument, UNEP/EA.5/L.23/Rev.1, 2022 Search PubMed.
- M. T. Ekvall, I. Gimskog, J. Hua, E. Kelpsiene, M. Lundqvist and T. Cedervall, Size fractionation of high-density polyethylene breakdown nanoplastics reveals different toxic response in Daphnia magna, Sci. Rep., 2022, 12(1), 3109, DOI:10.1038/s41598-022-06991-1.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2em00415a |
| This journal is © The Royal Society of Chemistry 2023 |