
Extracellular polymeric substances enhance dissolution and microbial methylation of mercury sulfide minerals†
Zhanhua
Zhang
,
Zhenyu
Zhao
,
Qingxuan
Fang
,
Ruohong
Qiao
and
Tong
Zhang
 *
*
College of Environmental Science and Engineering, Ministry of Education Key Laboratory of Pollution Processes and Environmental Criteria, Tianjin Key Laboratory of Environmental Remediation and Pollution Control, Nankai University, Tianjin 300350, P. R. China. E-mail: zhangtong@nankai.edu.cn; Tel: +86 22-66229516
First published on 22nd November 2022
Abstract
Due to the extremely low solubility, mercury sulfide minerals, as the major environmental mercury sinks, are generally considered to be inert mercury species with minimal bioavailability. Here, we demonstrate that extracellular polymeric substances (EPS), continuously secreted and released by anaerobic methylating bacteria, enhance the dissolution processes of cinnabar (α-HgS) minerals. The enhancing effects of EPS occur to a greater extent in the dissolution of nanoparticulate α-HgS compared to the bulk-scale counterpart. The released EPS–Hg(II) species are available for microbial methylation to produce bioaccumulative neurotoxin, methylmercury. This is probably due to the abundant aromatic proteins in EPS that strongly interact with surface Hg(II) via inner-sphere complexation as well as cation–π interaction. Our study discovers the potential environmental risks of “inert” mercury sulfide minerals in natural microbial habitats, particularly benthic biofilms with abundant microbial EPS, transformed to the severely toxic methylmercury. The mechanistic findings will facilitate an accurate understanding of the interactions between soft and transition metals and microorganism-derived organics, which may dictate the environmental fate and impact of these elements.
Environmental significanceThe naturally occurring transformation of HgS minerals will impact their efficacy as mercury repositories, leading to serious environmental risks. This research draws the attention of the scientific community to microorganism-derived extracellular polymeric substances (EPS) that are widely distributed in natural microbial habitats. EPS, particularly aromatic proteins with abundant functional groups, enhance the dissolution and methylation of α-HgS, which was previously considered the least reactive mercury species. More concerningly, these processes occur to a greater extent at the nano-scale, given that nanominerals and mineral nanoparticles are ubiquitous in the environment. The discovered pathways of EPS-enhanced transformation of minerals may also inform research on the biogeochemistry of other metals. |
Introduction
Mercury sulfide (HgS) minerals, particularly cinnabar (α-HgS), represent the most abundant form of inorganic mercury in the natural environment.1,2 Due to the extremely low solubility of cinnabar with a solubility product of 10−36.8 (HgS(s) + H+ ↔ Hg2+ + HS−, at T = 20 °C and I = 1.0 mol L−1),3 these HgS minerals have been considered as the major environmental mercury sinks that pose few ecological risks. However, recent studies have shown that cinnabar at organic-rich settings may release dissolved mercury species,1,4,5 which at least partly, originated from microbial activities. It's been documented that autotrophic aerobes, such as Acidithiobacillus ferrooxidans, Thiobacillus thioparus, were capable of dissolving HgS minerals through direct oxidation of S(–II) and catalytic oxidation of S(–II) by Fe(III).5–7 Moreover, heterotrophic bacteria, Shewanella oneidensis, in an anaerobic environment were found to enhance the dissolution of HgS minerals, and yet the mechanism for this process remains unknown.1,8It has been widely accepted that the majority of microorganisms in natural habitats occur in biofilms, which serve as the basis of aquatic food chains.9 Microbial biofilms tend to accumulate mercury from the surrounding environment and are deemed to be “hotspots” for mercury methylation.10,11 For example, in rock-hosted biofilms, the concentration of methylmercury (MeHg) accounted for 0.12–22% of the total accumulated mercury, whereas the ratio of MeHg vs. total mercury was as low as 0.10–0.81% in surrounding water.11,12 Microbial biofilms are embedded in self-produced extracellular polymeric substances (EPS), which make up more than 90% of the dry weight of biofilms.13 Previous studies have discovered that EPS mainly consist of proteins and polysaccharides, enriched in aromatic structures and functional groups,14,15 and exhibit relatively high affinities to soft metals.16–18 We recently found that aromatic proteins are the key components of EPS to react with Hg(II), significantly interfering with the precipitation and methylation processes of metacinnabar (β-HgS) nanoparticles.19 Thus, EPS, especially aromatic proteins, may strongly interact with cinnabar, and more concerningly, influence the subsequent bioavailability of this common mercury mineral phase to methylating microorganisms.
In this study, we examined the dissolution and microbial methylation processes of cinnabar minerals in the presence of EPS derived from two model mercury methylating bacteria, Pseudodesulfovibrio mercurii ND132 and Geobacter sulfurreducens PCA. The EPS–Hg(II) interactions were characterized with fluorescence, ultraviolet (UV) absorbance and Raman spectroscopy studies, which were supplemented with quantum chemical calculations. Inner-sphere complexation and cation–π interaction played a critical role in mediating mercury dissolution from cinnabar minerals and the mobilized mercury appeared to be readily available for microbial methylation. These trends are particularly prominent when cinnabar minerals occur at the nano-scale with large specific surface areas. An important implication from our findings is that HgS minerals, which were previously regarded as “inert” mercury species, may cause potential environmental risks in natural microbial habitats enriched with biological macromolecules.
Materials and methods
Extraction and characterization of EPS
P. mercurii ND132 (Cynthia C. Gilmour, Smithsonian Environmental Research Center) and G. sulfurreducens PCA (ATCC 51573) were incubated to extract EPS. The detailed culturing methods of two mercury methylating bacteria are included in the ESI.† A sonication-based method was used for EPS extraction according to Kang et al. (2017).20 Extraction treatments were conducted anaerobically in a glove box (Coy Laboratory Products, U.S.A.). Briefly, the bacterial cells that reached the late-log growth phase were harvested using centrifugation at 4000 rpm for 5 min. Afterward, the cells were washed with and resuspended in deoxygenated phosphate-buffered saline (PBS), consisting of 10 mmol L−1 Na2HPO4 and 2 mmol L−1 KH2PO4 at pH 7.2–7.4. The suspension was sonicated for 8 min in an ice-water bath. Then, the cell suspension was centrifuged and subsequently filtered through 0.22 μm membranes (ANPEL Laboratory Technologies, China) to remove unsettled cells. The filtrate (i.e., aqueous EPS) was stored at −20 °C prior to use.For EPS characterization, total organic carbon (TOC), total carbon (TC), and total nitrogen (TN) contents were determined on a TOC analyzer (vario TOC cube, Elementar, Germany). The total sulfur (TS) content was measured using inductively coupled plasma mass spectrometry (ICP-MS, NexION 2000, PerkinElmer, U.S.A.). Bicinchoninic acid (BCA) assay was used to determine the protein content, and the polysaccharide content was determined by the phenol-sulfuric acid method. The ultraviolet absorption spectra of EPS were obtained on a microplate reader (Spark 10M, Tecan, Switzerland). The specific ultraviolet absorbance at 280 nm (SUVA280) and the slope ratio of the absorption spectra (S275–295/S350–400) was calculated to assess the aromaticity and molecular weight of EPS, respectively.21
Batch experiments of mercury dissolution
Nano-cinnabar and bulk-cinnabar were utilized to conduct dissolution experiments. The detailed synthesis and characterization methods of the cinnabar samples are included in the SI. The background solution, consisting of 0.1 mol L−1 NaNO3 and 10 mmol L−1 PBS (pH 7.2–7.4), was deoxygenated by boiling and then cooled down in a glove box (with 95% N2 and 5% H2) and kept for at least 12 h before use. Cinnabars were added into amber glass vials to reach the solid-to-liquid ratio of 0.5 g L−1, prior to the addition of 15 mg-C L−1 EPS. Experiments without the addition of EPS were performed as a negative control under the same conditions. The amber glass vials sealed with Teflon-lined silicone screw caps were kept at room temperature (23–26 °C) on a rotating mixer (MX-RL-E, DLAB Scientific, China) at a speed of 20 rpm. At each time point, samples were centrifugated at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min, and the dissolved Hg(II) concentrations were analyzed from the supernatant by atomic fluorescence spectrometry (AFS-8500, Beijing Haiguang, China). The efficiency of the centrifugation method was estimated by the control experiments (i.e., cinnabar minerals were added into the amber glass vials without the addition of EPS). The results showed that 98.5% of the added nano-cinnabar and 99.1% of the added bulk-cinnabar were removed from the solution (Fig. S1†). After subtracting the background concentration of mercury, the average dissolution rates of cinnabars were calculated by dividing the increment of dissolved Hg(II) by the incubation time.
000 rpm for 30 min, and the dissolved Hg(II) concentrations were analyzed from the supernatant by atomic fluorescence spectrometry (AFS-8500, Beijing Haiguang, China). The efficiency of the centrifugation method was estimated by the control experiments (i.e., cinnabar minerals were added into the amber glass vials without the addition of EPS). The results showed that 98.5% of the added nano-cinnabar and 99.1% of the added bulk-cinnabar were removed from the solution (Fig. S1†). After subtracting the background concentration of mercury, the average dissolution rates of cinnabars were calculated by dividing the increment of dissolved Hg(II) by the incubation time.
Fluorescence quenching experiments
Three-dimensional excitation-emission matrices (3D-EEM) of EPS samples before and after reacting with cinnabars were determined using fluorescence spectroscopy (F-7100, Hitachi, Japan). The 3D-EEM spectra were collected by scanning the emission spectra (220–500 nm at 2 nm increments) and stepping through the excitation spectra (280–400 nm at 2 nm increments). The scanning speed was adjusted to 12000 nm min−1 with the width of the excitation and emission slit set at 5.0 nm. fluorescence measurements were performed using a 300 nm cutoff filter to minimize the second-order Raleigh scattering. All the EPS samples were diluted three times, and an absorbance-based approach was used to correct for the inner-filter effect.22
A fluorescence titration method was further conducted to evaluate the complexation reaction between Hg(II) and EPS. Five mg-C L−1 EPS solutions buffered with 10 mmol L−1 PBS solution (pH 7.2–7.4) were titrated using Hg(NO3)2 (i.e., the quencher) solution to reach the predetermined Hg(II) concentrations (0–150 μmol L−1). The 3D-EEM spectra were collected under the same experimental condition. Fluorescence quenching data were utilized to generate the modified Stern–Volmer plots of F0/(F0 − F) versus 1/[Q] according to the modified Stern–Volmer equation:18,23
| F0/(F0 − F) = 1/f + 1/(fKqτ0[Q]) = 1/f + 1/(fKSV[Q]) | (1) |
| log[(F0 − F)/F] = logKA + nlog[Q] | (2) |
Characterization of the cation–π interaction
Three aromatic amino acids, including L-Tryptophan (Trp), L-Tyrosine (Tyr), and L-Phenylalanine (Phe) purchased from J&K Scientific Ltd., China, were utilized to analyze the cation–π interaction between Hg(II) and aromatic structures. Ultraviolet (UV) absorption spectra of amino acids before and after reacting with Hg(II) were determined on a microplate reader (Spark 10M, Tecan, Switzerland). All experimental matrices consisted of 10 mmol L−1 PBS solution to maintain the pH at 7.2. Amino acids were added into the experimental matrices to reach the predetermined concentrations (0–100 μmol L−1), prior to the addition of 100 μmol L−1 HgCl2. Ultraviolet difference spectra were obtained by subtracting the background spectra of amino acids from the spectra of amino acids after reacting with Hg(II).A Raman spectrometer (LabRAM HR Evolution, Horiba, Japan) was used for recording the Raman spectra of amino acids in aqueous solution before and after reacting with Hg(II) at an excitation wavelength of 532 nm and an excitation power of 10 mW. An aliquot of HgCl2 stock solution was added to the amino acid solution, containing tryptophan or phenylalanine, to reach the molar ratio of amino acid vs. mercury at 0.5. The difference spectra were obtained by subtracting the background spectra of amino acids with no Hg(II) addition from the spectra of amino acids after reacting with Hg(II).
Quantum chemical calculations
Computational methods were employed to study the cation–π interaction between the aromatic amino acids and mercury. Hg2+ and Hg2+ bound to –SH (i.e., Hg(HS)+) were chosen as typical mercury species. The ground-state geometry structures of the cation–π complexes were optimized using density functional theory (DFT) with the BP86 function in the Gaussian 16 (Revision C.01) software package.27 In order to improve the accuracy for estimating the non-covalent interactions, Grimme's D3BJ dispersion correction was taken into account for all the calculations.28 For geometry structure optimization, the 6-311+G(d,p) basis set was used for C, H, O and N atoms,29 and the def2-TZVP basis set was used for Hg(II).30 The solvation model based on density (SMD) approach was employed to simulate the aqueous environment.31 The electron density difference maps (EDDM) were rendered using the VMD software based on the output files analyzed using the Multiwfn 3.7 software.32,33The specific binding of the model amino acids (i.e., cysteine and tryptophan) on the surface of cinnabar was simulated using DFT calculations. Geometry structure optimizations were conducted in the Vienna Ab initio Simulation Package (VASP)34 within the generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE) formulation.35 The projected augmented wave (PAW) potentials36 were used to describe the ionic cores, and valence electrons were taken into account using a plane wave basis set with a kinetic energy cutoff of 450 eV. Partial occupancies of the Kohn–Sham orbitals were allowed using the Gaussian smearing method with a width of 0.05 eV. The electronic energy was considered self-consistent when the energy change was smaller than 10−4 eV, and a geometry optimization was considered convergent when the force change was smaller than 0.05 eV Å−1. The long-range dispersion forces were described by theDFT-D3 method proposed by Grimme.37 The Brillouin zone was sampled using the surface structures of Gamma-only point sampling, and the supercell (24 Å × 25 Å × 22 Å) applied was quite large for avoiding the image charge interaction of amino acid molecules. The crystal orbital Hamilton population (COHP) was determined using the LOBSTER package38,39 to evaluate the bond strength of the Hg–S bond on the cinnabar surface.
Microbial methylation experiments
The microbial methylation potential of cinnabar minerals in the presence of EPS was assayed using pure cultures of P. mercurii ND132. The bacterial cells were harvested by centrifugation after the cultures were incubated to the late-log phase, and then washed and resuspended with deoxygenated PBS solution, consisting of 10 mmol L−1 Na2HPO4, 2 mmol L−1 KH2PO4, 0.14 mol L−1 NaCl, 3 mmol L−1 KCl, 1 mmol L−1 pyruvate and 1 mmol L−1 fumarate at pH 7.2–7.4, according to Zhang et al. (2019).40 Cinnabars were added into the amber glass vials to reach the solid-to-liquid ratio of 0.5 g L−1, then the washed cells were transferred to the suspension to a final cell density of 2 × 108 cells mL−1 without EPS or with the addition of 15 mg-C L−1 EPS. The active cultures with no cinnabar or EPS addition were included as negative controls (i.e., mercury blank). The sealed amber glass vials were kept at room temperature (23–26 °C) on a rotating mixer at a speed of 20 rpm. Replicate samples were periodically collected and preserved in 0.4% (v/v) HCl (trace-metal grade) at 4 °C for methylmercury (MeHg) analysis. The concentrations of MeHg were determined by distillation, aqueous phase ethylation, gas chromatography, and cold vapor atomic fluorescence spectrometry (Tekran 2700, Tekran, Canada).41 A subset of samples was collected to quantify cell density on a flow cytometer (Accuri C6 Plus, BD Biosciences, U.S.A.).Results and discussion
Enhanced dissolution of cinnabar in the presence of EPS
Bulk and nano-cinnabar were barely soluble at neutral pH, whereas the presence of EPS significantly enhanced the dissolution of two cinnabar minerals (Fig. 1). At the end of the 7 day incubation, EPS derived from P. mercurii ND132 (i.e., EPS(ND132)) and G. sulfurreducens PCA (i.e., EPS(PCA)) resulted in an increased release of dissolved Hg(II) from nano-cinnabar up to 107.24 ± 14.52 nmol g−1 and 42.81 ± 4.87 nmol g−1, respectively, much greater than that of the control group with no EPS addition (i.e., 5.16 ± 1.74 nmol g−1, Fig. 1a). The enhancing effects of EPS were also observed in the dissolution of bulk-cinnabar (Fig. 1b), which occurred to a lower extent compared to nano-cinnabar. Indeed, the average dissolution rates of nano-cinnabar were 21.18 ± 1.48 nmol g−1 d−1 for EPS(ND132) and 9.07 ± 0.93 nmol g−1 d−1 for EPS(PCA), while the average dissolution rates of bulk-cinnabar decreased to 1.52 ± 0.13 nmol g−1 d−1 for EPS(ND132) and 0.94 ± 0.09 nmol g−1 d−1 for EPS(PCA) (Fig. 1c). Overall, the results demonstrated that EPS enhanced the dissolution of cinnabar minerals, which varied with the type of cinnabars and EPS.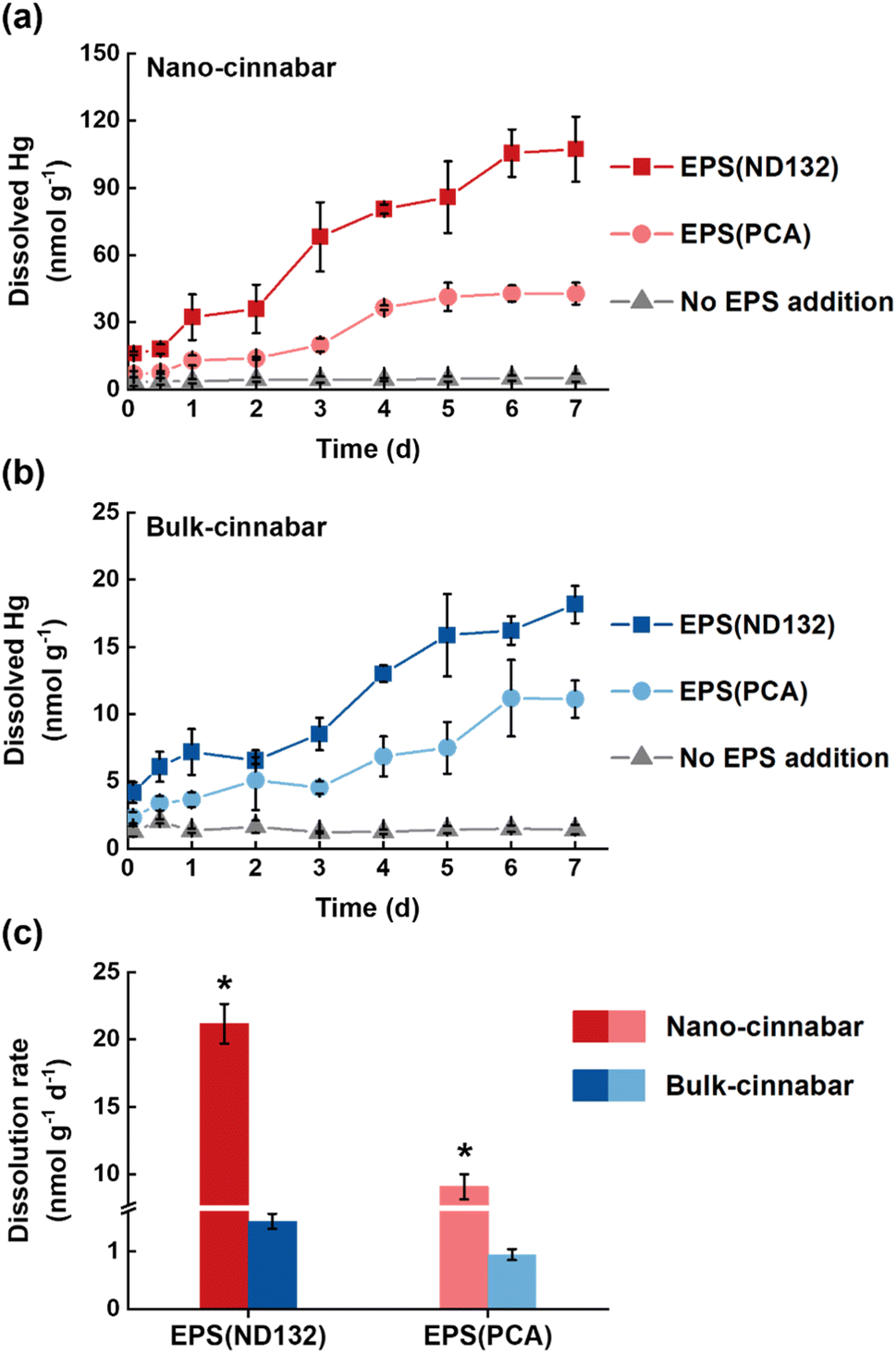 | ||
| Fig. 1 Dissolved mercury from nano-cinnabar (a) and bulk-cinnabar (b) in the presence or absence of EPS, and their average dissolution rates (c). Error bars represent ±1 standard deviation of replicate samples (n = 3). Values that are statistically different (p < 0.01) according to the two-sample t-test are indicated by asterisks. | ||
X-ray diffraction (XRD) patterns clearly showed an α-HgS phase with (011) and (102) as the dominant facets for both cinnabar minerals (Fig. S2†). The crystallite sizes of nano-cinnabar, estimated from the broadening of XRD peaks by the Scherrer formula, were 6–9 nm (Table S1†), consistent with the results of scanning electron microscopy (SEM, Fig. S3†) and transmission electron microscopy (TEM, Fig. S3†). Bulk-scale cinnabar appeared to be irregularly shaped with the size apparently greater than that of nano-cinnabar (Fig. S4†). BET surface area analysis demonstrated that nano-cinnabar exhibited a much larger surface area than bulk-cinnabar (Table S1†), which likely facilitated interactions of EPS with surface Hg(II) and at least in part, contributed to the differences of released dissolved Hg(II) between two cinnabar minerals (Fig. 1). Moreover, EPS(ND132) and EPS(PCA) appeared to enhance dissolution of cinnabars to differential degrees (Fig. 1), pointing to the important role of EPS properties in controlling cinnabar dissolution.
Inner-sphere complexation between EPS and Hg(II)
In comparison with EPS(PCA), EPS derived from P. mercurii ND132 consisted of a higher protein content (Table S2†), suggesting that EPS(ND132) probably contained a larger density of functional groups than EPS(PCA). At the end of the nano-cinnabar dissolution experiment, 4.43 ± 0.78 mg-C L−1 EPS(ND132) were removed from the solution, while 2.12 ± 0.35 mg-C L−1 EPS(PCA) were removed. In the case of bulk-cinnabar, only 0.75 ± 0.12 mg-C L−1 EPS(ND132) and 0.47 ± 0.09 mg-C L−1 EPS(PCA) were removed from the solution. The relatively higher removal of EPS(ND132) indicated that more EPS(ND132) components were sorbed onto the cinnabar surface, which was further confirmed by X-ray photoelectron spectroscopy (XPS) analysis (Table S3 and Fig. S5†). The deconvolution of XPS C 1s signals of nano-cinnabar after the dissolution experiments demonstrated that more carbonaceous fragments of EPS(ND132) (e.g., O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O at 15.7%) were bound onto the cinnabar surface than those of EPS(PCA) (e.g., OC–O at 12.6%). The peak shifting in the XPS spectra validated the complexation between the carboxyl groups of EPS and surface mercury atoms of cinnabar. The carboxyl groups (OC–O) in uncomplexed EPS are commonly at 288.5 eV.42,43 After the interaction, the binding energy of OC–O shifted to larger values by 0.3 eV for EPS(ND132) and 0.2 eV for EPS(PCA). Meanwhile, the Hg 4f5/2 and Hg 4f7/2 shifted to lower values by 0.4 eV for EPS(ND132) and by 0.2 eV for EPS(PCA) (Table S3 and Fig. S5†). These results suggest that a fraction of EPS bound onto the cinnabar surface through inner-sphere complexation with surface mercury atoms, which occurred to a greater degree for EPS(ND132) than for EPS(PCA).
C–O at 15.7%) were bound onto the cinnabar surface than those of EPS(PCA) (e.g., OC–O at 12.6%). The peak shifting in the XPS spectra validated the complexation between the carboxyl groups of EPS and surface mercury atoms of cinnabar. The carboxyl groups (OC–O) in uncomplexed EPS are commonly at 288.5 eV.42,43 After the interaction, the binding energy of OC–O shifted to larger values by 0.3 eV for EPS(ND132) and 0.2 eV for EPS(PCA). Meanwhile, the Hg 4f5/2 and Hg 4f7/2 shifted to lower values by 0.4 eV for EPS(ND132) and by 0.2 eV for EPS(PCA) (Table S3 and Fig. S5†). These results suggest that a fraction of EPS bound onto the cinnabar surface through inner-sphere complexation with surface mercury atoms, which occurred to a greater degree for EPS(ND132) than for EPS(PCA).
What's more, after the dissolution experiments, the SUVA280 values of EPS(ND132) and EPS(PCA) did not decrease upon EPS–HgS interactions (Fig. S6†), a similar phenomenon that Waples et al. (2005)44 also discovered during the dissolution of mercury-containing minerals. Since SUVA280 is a widely used indicator for the aromaticity of natural organic matter,45 this result suggested that the aqueous EPS components were still enriched in aromatic structures compared to the adsorbed components. According to our previous studies, the aromatic components in EPS strongly interact with mercury sulfide via inner-sphere complexation.19 Hence, we hypothesize that aromatic components in EPS may be important organic ligands for Hg(II) that cause the breaking of surface Hg–S bonds and the EPS–Hg(II) species released into the solution. Fluorescence quenching experiments were applied to evaluate the complexation between aromatic components in EPS and Hg(II). Two fluorescence peaks at excitation/emission wavelengths (Ex/Em) of 225/335 nm (peak A) and 278/335 nm (peak B) were identified in the 3D-EEM spectra (Fig. 2a), which were assigned to aromatic protein-like substances according to previous research.18,24,46 The fluorescence intensities of both peak A and peak B markedly diminished after reacting with cinnabar minerals, particularly in the nano-cinnabar samples. The intensity of peak A for EPS(ND132) declined as much as 95.4% and 91.1% upon reactions with nano-cinnabar and bulk-cinnabar, respectively. Further, the biomolecular quenching rate constants (Kq) of EPS(ND132)–Hg and EPS(PCA)–Hg (Fig. 2b and S7†) both exceeded the maximum Kq for diffusion-controlled quenching (2.0 × 1010 L mol−1 s−1),24,26 indicating the occurrence of a static quenching process that may involve EPS–Hg(II) inner-sphere complexation.47,48 The Stern–Volmer quenching constants (KSV), and binding constants (KA) of EPS(ND132)–Hg were consistently larger than those of EPS(PCA)–Hg, suggesting a stronger binding affinity of EPS(ND132) to Hg(II) relative to EPS(PCA) (Fig. 2b and S7†). These results were in accordance with the trends of cinnabar dissolution (Fig. 1). Therefore, inner-sphere complexation between the surface Hg(II) of cinnabar and aromatic proteins of EPS, particularly EPS(ND132), likely enabled the dissolution of cinnabar minerals.
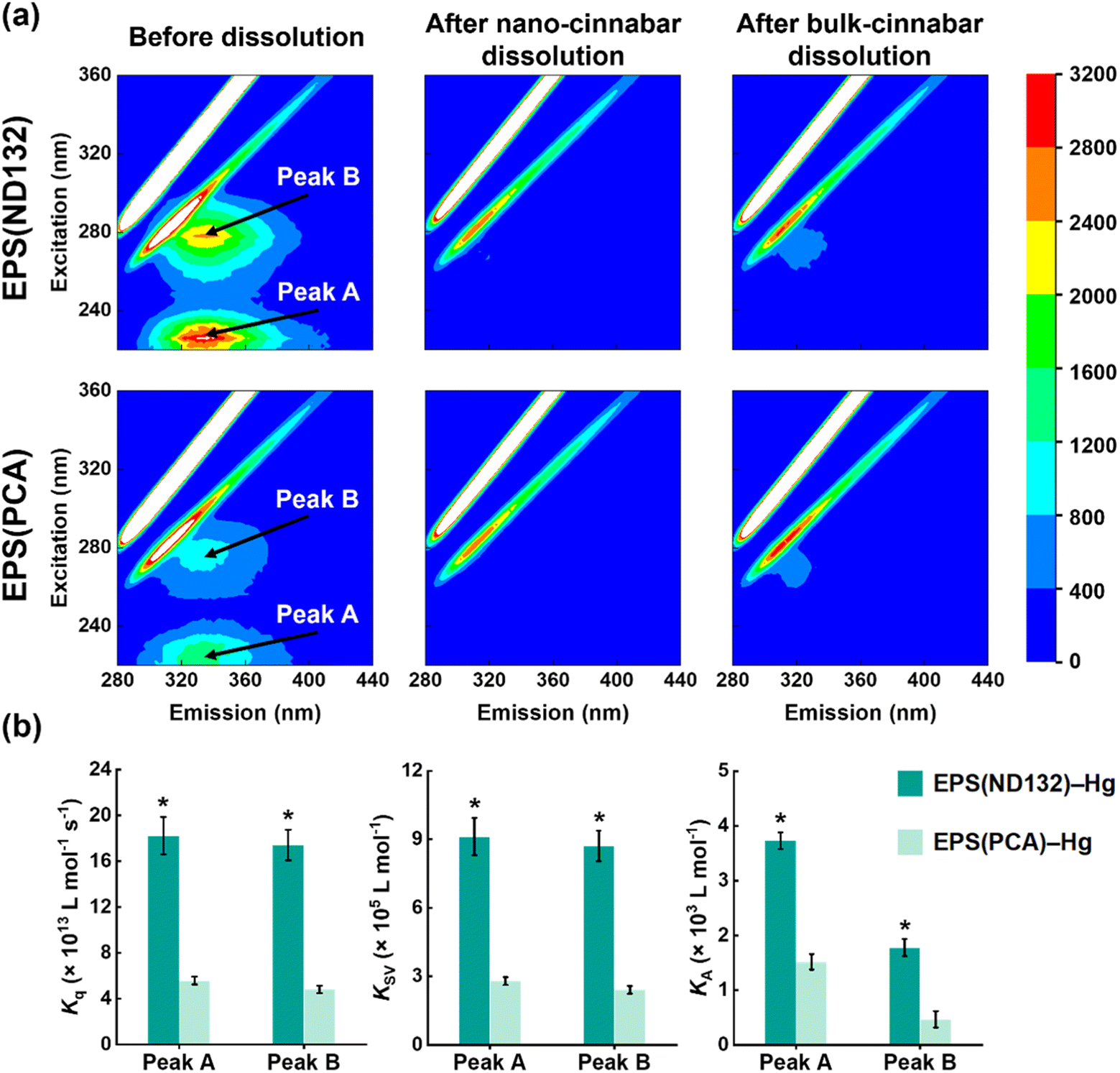 | ||
| Fig. 2 Three-dimensional EEM fluorescence spectra of EPS(ND132) and EPS(PCA) before and after reacting with nano-cinnabar and bulk-cinnabar (a). Biomolecular quenching rate constants (Kq), Stern–Volmer quenching constants (KSV), and binding constants for static quenching (KA) of peak A and peak B in the EEM fluorescence spectra of EPS (b). Error bars represent ±1 standard error of the linear regression for extracting the constant values. Values that are statistically different (p < 0.01) according to the two-sample t-test are indicated by asterisks. | ||
Cation–π interaction between EPS and Hg(II)
EPS(ND132) comprised more aromatic structures, as evidenced by the larger values of SUVA280 for EPS(ND132) compared to that for EPS(PCA) (Table S2†). The above results also indicated the stronger binding affinity of EPS(ND132) to Hg(II) relative to EPS(PCA). However, due to the limitations of the fluorescence quenching methods, the functional groups that bind to Hg(II) in aromatic proteins of EPS remain unclear. Previous studies have discovered that aromatic structures with electron-rich aromatic π-systems may interact with positively charged cations via the cation–π interaction, one of the strongest forms of noncovalent interactions.49 Thus, the cation–π interaction was assessed using experimental and theoretical calculation methods with aromatic amino acids (tryptophan, tyrosine, and phenylalanine, Fig. S8†) as model compounds.50,51 After reacting with Hg(II), ultraviolet difference spectra generated a negative band at 220–225 nm for tryptophan and tyrosine, and a weak positive band at ∼230 nm for phenylalanine (Fig. 3 and S9†). These bands were assigned to the Bb absorption of the benzene ring in aromatic structures, which were suggested to arise from the cation–π interaction between Hg(II) and aromatic amino acids.52,53 The breathing vibration of the benzene/pyrrole ring in tryptophan (i.e., 1015 and 760 cm−1 in Fig. 4a and Table S4†) and the breathing vibration of the benzene ring in phenylalanine (i.e., 1006 cm−1 in Fig. 4b and Table S4†) were prominent in the Raman spectra after reacting with Hg(II), and these Raman bands attributed to the breathing vibration are commonly used as Raman markers of the cation–π interaction.54,55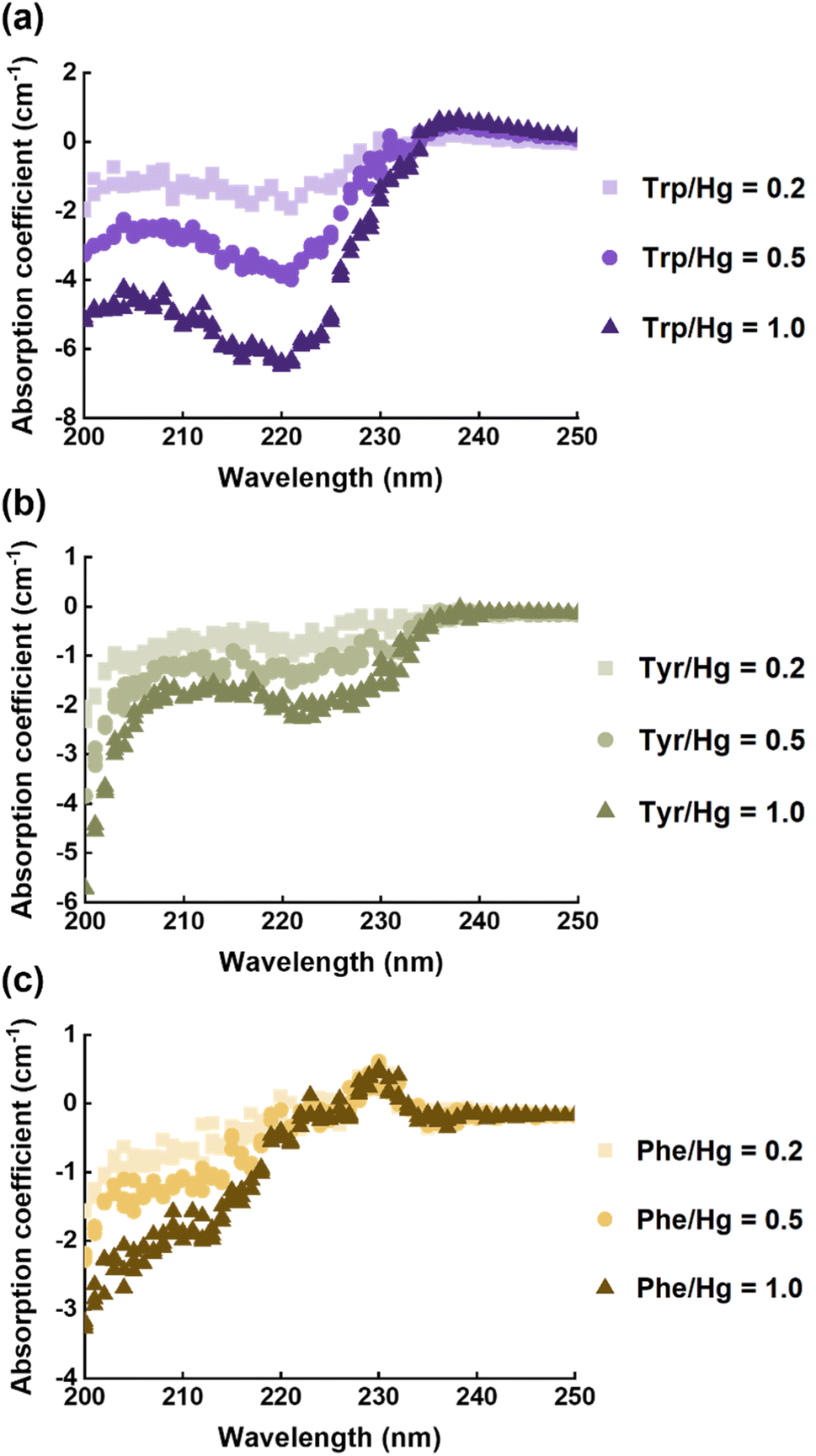 | ||
| Fig. 3 Ultraviolet difference spectra of tryptophan (a), tyrosine (b), and phenylalanine (c) after reacting with mercury at different molar ratios of amino acid vs. mercury. The absorption coefficient (a, cm−1) is converted from the absorbance (A) following equation a = 2.303A/l, where l is the path length (cm). | ||
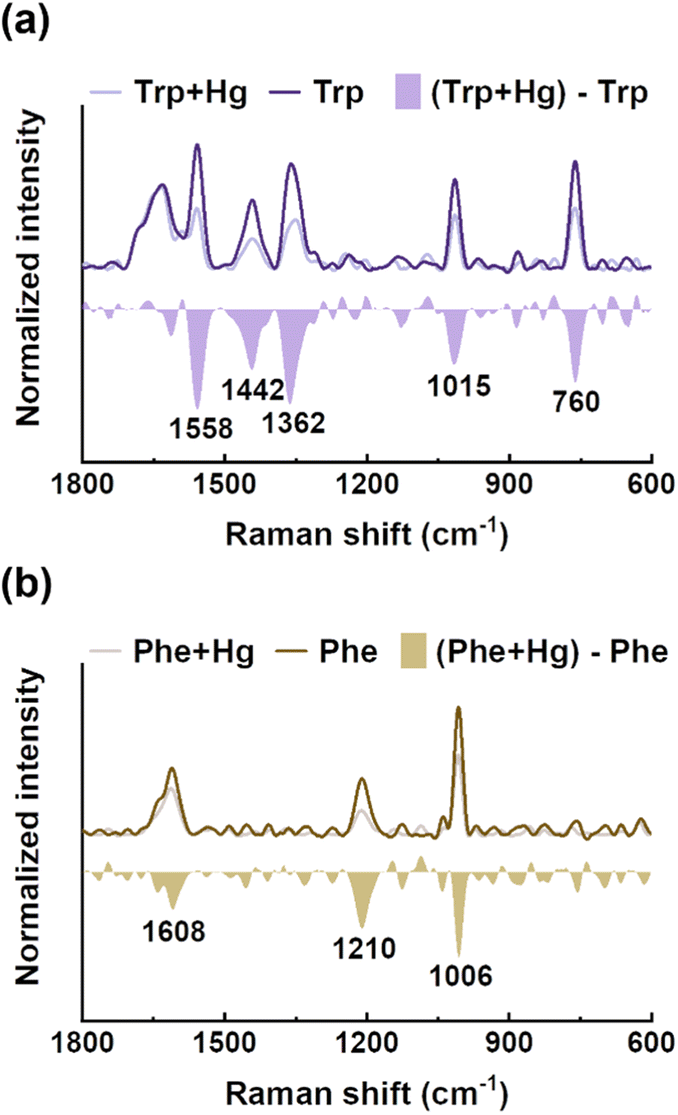 | ||
| Fig. 4 Raman spectra of tryptophan (a) and phenylalanine (b) before and after reacting with mercury, and their difference spectra. | ||
In addition to the spectroscopic evidence, the cation–π interaction between aromatic amino acids and mercury was further confirmed by quantum chemical calculations. The most stable geometry structures of cation–π complexes formed between aromatic structures of the three amino acids and mercury are depicted in Fig. 5 and S10.† Mercury tended to approach the carbon atom on the edge of the aromatic structure to form the πC(η3) conformation (Fig. 5a and S11a†). The distance between mercury and the neighboring carbon atom was 2.24 Å for the indole ring of tryptophan, 2.23 Å for the phenol ring of tyrosine and 2.27 Å for the benzene ring of phenylalanine. When ionic mercury reacted with sulfide to form the mercury sulfide complexes (e.g., Hg(HS)+), the optimized geometry structure favored the πcen(η6) conformation with the Hg(HS)+ above the ring centroid (Fig. S10a and S11b†). The average distance between mercury and the six C atoms in the benzene ring was 3.36 Å for tryptophan, 3.36 Å for tyrosine and 3.38 Å for phenylalanine. Since the electronic configuration of Hg(II) is [Xe]4f145d10 with an unoccupied 6s atomic orbital, the Hg2+ ← π or Hg(HS)+ ← π donation from the aromatic π-systems to the 6s atomic orbital of Hg(II) favor the formation of πC(η3) or πcen(η6) structures.56 These results were supported by the analysis of the electron density difference for the cation–π complexes formed between the aromatic structures of three amino acids and ionic or complexed Hg(II) (Fig. 5b and S10b†). Significant electron transfer occurred from the indole ring of tryptophan, phenol ring of tyrosine and benzene ring of phenylalanine to the ionic or complexed Hg(II), reflecting the nature of the interaction between electron-rich aromatic π-systems and positively charged cations to be the cation–π interaction.56,57
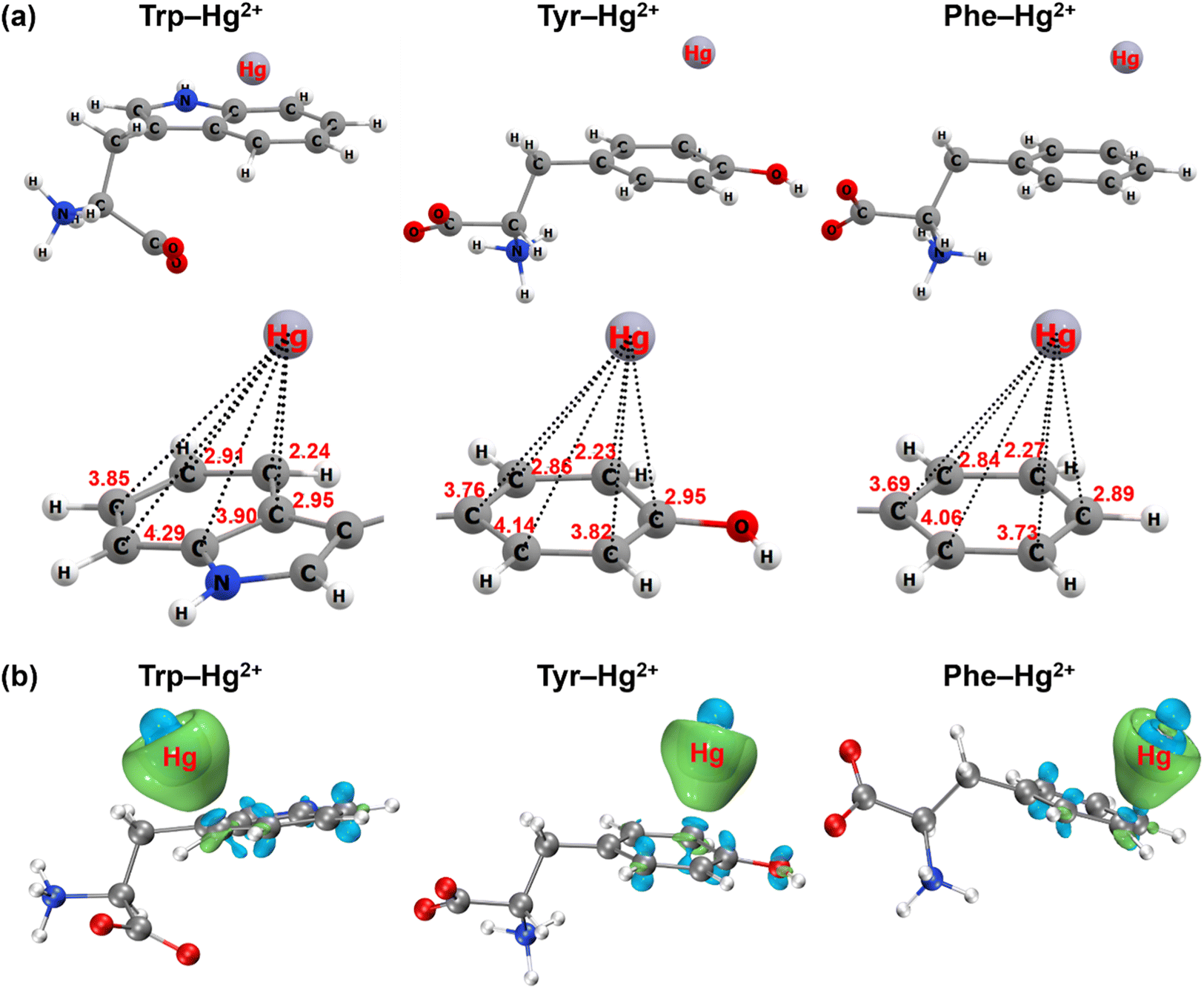 | ||
| Fig. 5 The optimized ground-state geometry structures (a) and electron density difference maps (b) of the cation–π complexes formed between aromatic structures of three amino acids and mercury. The unit of distance between the Hg atom and C atom in (a) is Å. The light green and light blue colors in (b) represent electron accumulation and electron depletion, respectively. The isosurface value (ρ) of the electron density difference is 0.008 au. | ||
Inner-sphere complexation and cation–π interaction induce the dissolution of cinnabar
EPS(ND132) exhibited greater relative nitrogen content (TN/TC) and sulfur content (TS/TC), compared with EPS(PCA) (Table S2†), probably because proteins are the main components in microbial EPS enriched in N- and S-containing functional groups.58,59 The reduced sulfur content of EPS(ND132) was also greater than that of EPS(PCA) given the reduced sulfur tends to be a relatively constant and a high percentage of total organic sulfur.60 Since the reduced sulfur functional groups have the strongest binding affinity toward Hg(II),44,61 cysteine was chosen as the model thiol-containing amino acid to conduct the chemical binding analysis of the Hg–S bond on the cinnabar surface during the dissolution process. The surface models of cinnabar (011) and (102) were established, as these two facets have a relatively low surface energy (i.e., 0.04 and 0.05 J m−2, respectively). However, the atomic configurations of the cinnabar (011) facet became unstable due to the cysteine adsorption-induced distortion, which suggested that the adsorption of cysteine may cause the release of Hg(II) from the cinnabar surface. The calculation results based on the cinnabar (102) facet indicated that cysteine was adsorbed on the surface through the complexation between the surface mercury atom and thiol group (Fig. 6a). In order to quantify the Hg–S bond strength on the cinnabar surface, COHP analysis was employed (Fig. 6c–e). Before the adsorption of cysteine, the integrated COHP (–ICOHP) values of the Hg–S bond on the (102) surface were 0.665 eV, 0.588 eV and 0.063 eV for the Hg–S1, Hg–S2 and Hg–S3 bond, respectively (Fig. 6f). The –ICOHP value decreased after the adsorption of cysteine (Fig. 6f), which suggested that the complexation between the surface mercury atom and thiol group weakened the bond strength, and promoted the breaking of the Hg–S bond on the surface. In addition, Since EPS(ND132) comprised more aromatic components than EPS(PCA) (Table S2†), the cation–π interaction between the surface mercury atom and aromatic structures of EPS may also play an important role in breaking the Hg–S bond on the cinnabar surface. This hypothesis was supported by DFT calculations using tryptophan as the model aromatic amino acid. The indole ring of tryptophan was bound to the surface mercury atom through the cation–π interaction (Fig. 6b), which decreased the –ICOHP value of the Hg–S bond on the cinnabar surface (Fig. 6f). Taken together, the inner-sphere complexation and cation–π interaction between EPS and Hg(II) decreased the Hg–S bond strength of the cinnabar surface, and induced the dissolution of cinnabar.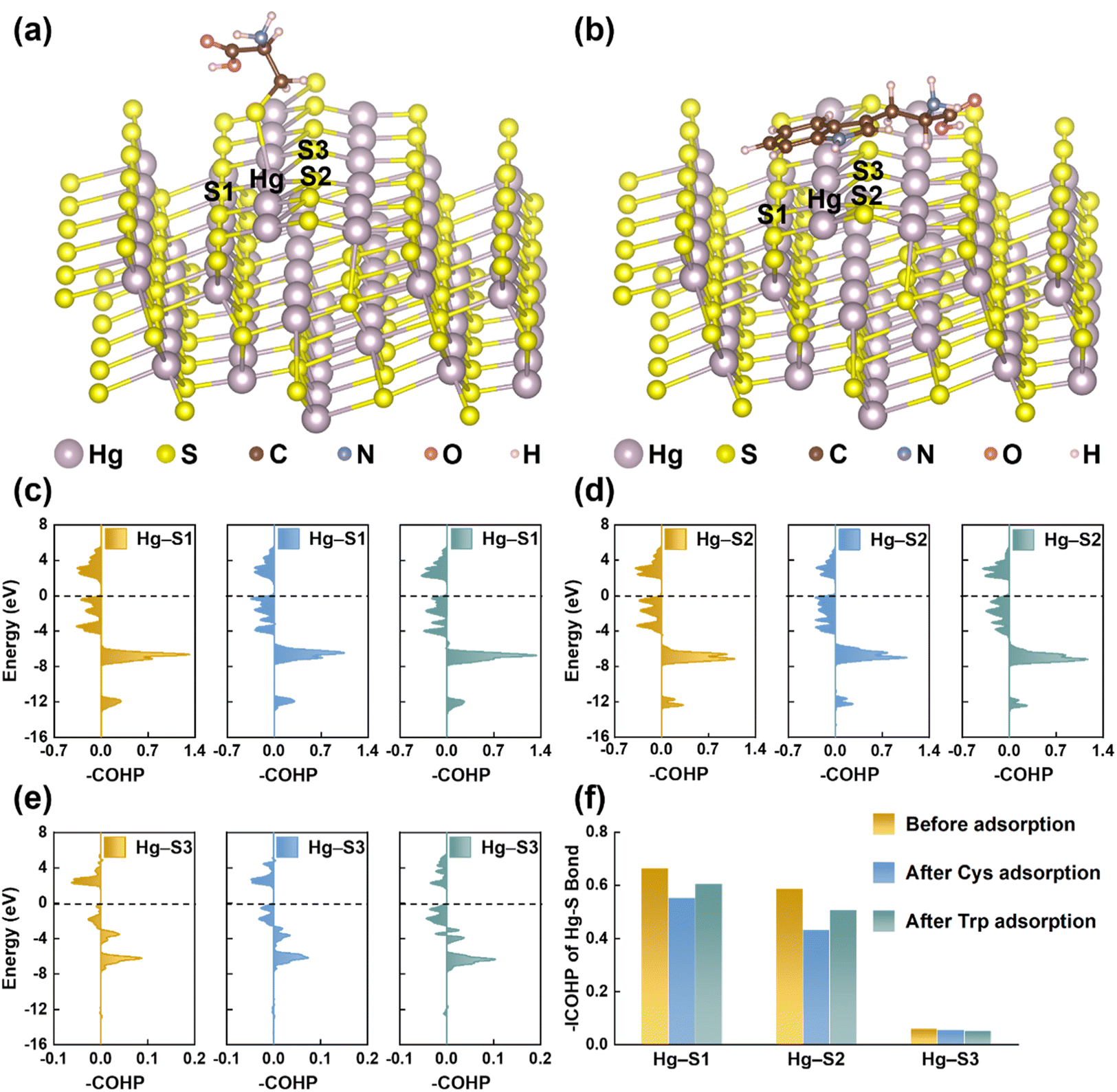 | ||
| Fig. 6 The optimized atomic configurations of the cinnabar (102) facet after cysteine (a) and tryptophan (b) adsorption. The crystal orbital Hamilton population (COHP) analysis of the Hg–S bond at the cinnabar (102) surface (c)–(e) and integrated COHP (ICOHP) values (f) before and after cysteine and tryptophan adsorption. | ||
EPS enhance the microbial methylation of cinnabar
Two cinnabar minerals in the absence and presence of EPS were exposed to P. mercurii ND132, a major microorganism that drives mercury methylation in the anaerobic environment.62 As reported in previous studies, mineral nanoparticles were generally more available for microbial mercury methylation than bulk-scale minerals.63,64 Upon 5 day incubation with the same amount of total inorganic mercury, 2.60 × 10−8 pmol cell−1 and 0.87 × 10−8 pmol cell−1 MeHg were generated in the nano-cinnabar and bulk-cinnabar groups, respectively, in the absence of EPS (Fig. 7). The addition of EPS markedly enhanced the microbial methylation of both cinnabar minerals, with EPS(ND132) displaying a stronger enhancing effect. In the presence of EPS(ND132), MeHg production increased by 3.8 times for nano-cinnabar, and by 3.3 times for bulk-cinnabar, while this increase became 1.9 times for nano-cinnabar and 2.1 times for bulk-cinnabar in the presence of EPS(PCA) (Fig. 7).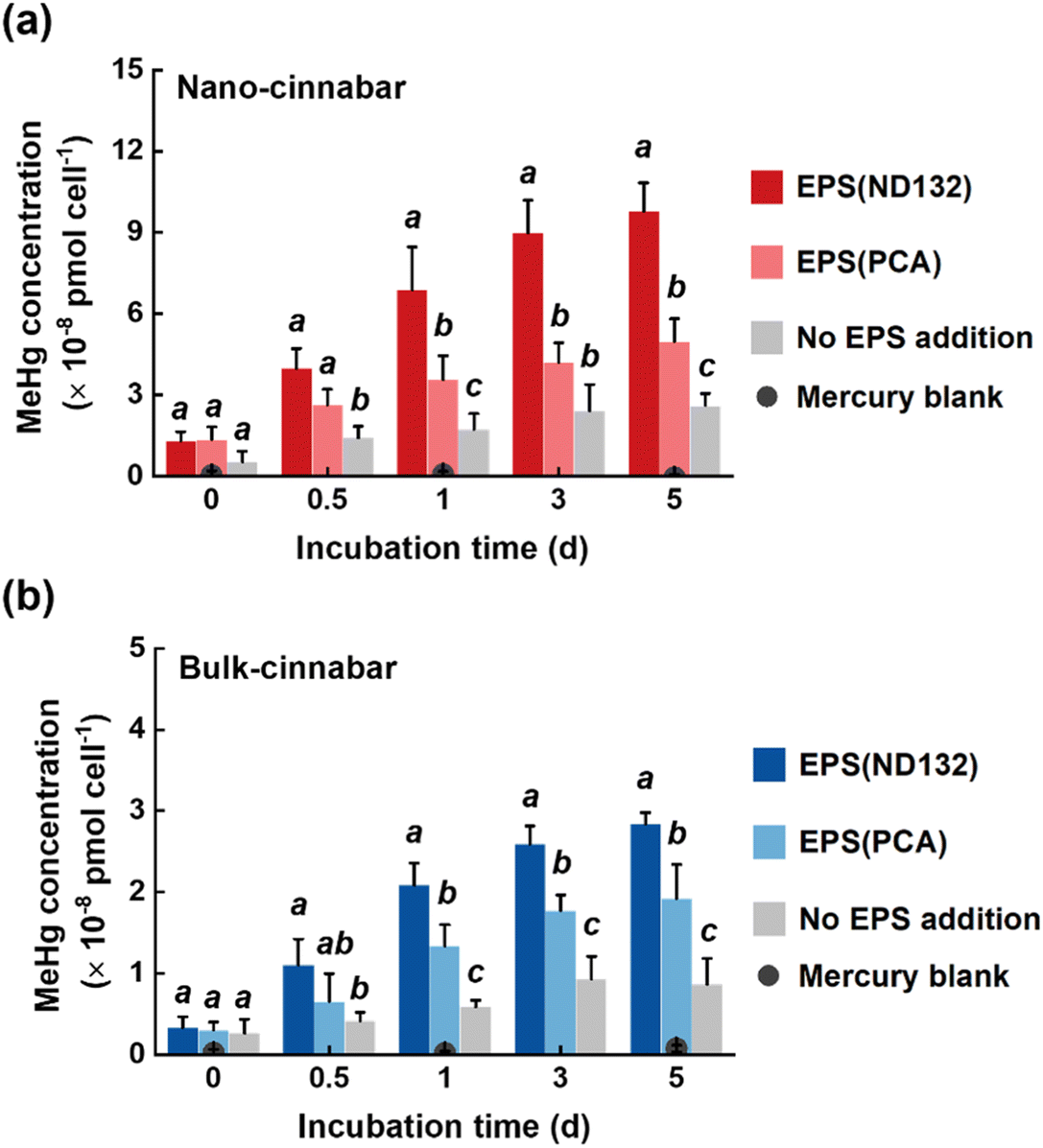 | ||
| Fig. 7 MeHg production from cultures of P. mercurii ND132 after exposure to nano-cinnabar (a) and bulk-cinnabar (b) in the presence or absence of EPS. The concentrations of MeHg were normalized to the cell numbers of P. mercurii ND132. Error bars represent ±1 standard deviation of replicate samples (n = 3–4). Values that are statistically different (p < 0.01) according to the one-way analysis of variance (ANOVA) are indicated by italic lowercase letters. | ||
Compared to mercury-containing minerals, dissolved mercury species are known to exhibit greater methylation potential.64,65 Indeed, the trends of microbial MeHg production (Fig. 7) coincided with the trends of the release of dissolved mercury from cinnabar minerals (Fig. 1) in our study. The EPS-induced enhancement was consistently larger in the mercury dissolution experiments, relative to the mercury methylation experiments. The transport of mercury species across cell membranes prior to intracellular methylation is deemed as an important constraint of MeHg production,66 and it is highly likely that dissolved mercury species bound to different EPS components are not all available or equally available for cellular uptake, which explained the good amount of MeHg produced in the “No EPS addition” samples with mercury dissolution from cinnabar barely discernible.
Conclusions
Our study reveals that EPS derived from mercury methylating bacteria are expected to enhance the dissolution and microbial methylation of cinnabar minerals, particularly at the nano-scale. EPS protein components enriched with soft ligands and aromatic structures play a vital role in dictating the release of mercury from cinnabar, likely through inner-sphere complexation and cation–π interaction. These findings add new perspectives to understanding environmental MeHg contamination by demonstrating a previously unrecognized microbially driven pathway of “inert” cinnabar minerals transformed to the extreme neurotoxin, MeHg. Moreover, EPS-producing microorganisms are widespread in the environment. Soft and transition metals (e.g., Pb, Cd, Cu, Zn, Fe) often occur as insoluble mineral phases, especially as nanoparticulate metal sulfides, in natural microbial habitats.67–69 Therefore, our findings may be extended beyond mercury methylation to deepen the mechanistic understanding of the speciation and transformation of these elements that are either toxins or nutrients.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Key Research and Development Program of China (2019YFC1804403), National Natural Science Foundation of China (22125603, 21976095, 22206089), Tianjin Municipal Science and Technology Bureau (21JCJQJC00060, 20JCZDJC00690), and Fundamental Research Funds for the Central Universities, Nankai University (63211065).References
- Y. Chen, Y. Yin, J. Shi, G. Liu, L. Hu, J. Liu, Y. Cai and G. Jiang, Analytical methods, formation, and dissolution of cinnabar and its impact on environmental cycle of mercury, Crit. Rev. Environ. Sci. Technol., 2018, 47, 2415–2447 CrossRef.
- E. D. Stein, Y. Cohen and A. M. Winer, Environmental distribution and transformation of mercury compounds, Crit. Rev. Environ. Sci. Technol., 1996, 26, 1–43 CrossRef CAS.
- M. Ravichandran, G. R. Aiken, M. M. Reddy and J. N. Ryan, Enhanced dissolution of cinnabar (mercuric sulfide) by dissolved organic matter isolated from the Florida Everglades, Environ. Sci. Technol., 1998, 32, 3305–3311 CrossRef CAS.
- R. Fernández-Martínez, J. Loredo, A. Ordóñez and I. Rucandio, Mercury availability by operationally defined fractionation in granulometric distributions of soils and mine wastes from an abandoned cinnabar mine, Environ. Sci.: Processes Impacts, 2014, 16, 1069–1075 RSC.
- A. D. Jew, S. F. Behrens, J. J. Rytuba, A. Kappler, A. M. Spormann and G. E. Brown, Microbially enhanced dissolution of HgS in an acid mine drainage system in the California Coast Range, Geobiology, 2014, 12, 20–33 CrossRef CAS PubMed.
- A. Vázquez-Rodríguez, C. M. Hansel, T. Zhang, C. Lamborg, C. M. Santelli, S. Webb and S. Brooks, Microbial-and thiosulfate-mediated dissolution of mercury sulfide minerals and transformation to gaseous mercury, Front. Microbiol., 2015, 6, 596 Search PubMed.
- Y. Wang, H. Li, H. Hu, D. Li, Y. Yang and C. Liu, Using biochemical system to improve cinnabar dissolution, Bioresour. Technol., 2013, 132, 1–4 CrossRef CAS PubMed.
- Y. Chen, H. Wang and Y.-B. Si, Research on the bioaccesibility of HgS by Shewanella oneidensis MR-1, Environ. Sci., 2013, 34, 4466–4472 CAS.
- H.-C. Flemming and S. Wuertz, Bacteria and archaea on Earth and their abundance in biofilms, Nat. Rev. Microbiol., 2019, 17, 247–260 CrossRef CAS PubMed.
- P. Dranguet, S. L. Faucheur, C. Cosio and V. I. Slaveykova, Influence of chemical speciation and biofilm composition on mercury accumulation by freshwater biofilms, Environ. Sci.: Processes Impacts, 2017, 19, 38–49 RSC.
- P. Dranguet, S. Le Faucheur and V. I. Slaveykova, Mercury bioavailability, transformations, and effects on freshwater biofilms, Environ. Toxicol. Chem., 2017, 36, 3194–3205 CrossRef CAS PubMed.
- S. Žižek, R. Milačič, N. Kovač, R. Jaćimović, M. J. Toman and M. Horvat, Periphyton as a bioindicator of mercury pollution in a temperate torrential river ecosystem, Chemosphere, 2011, 85, 883–891 CrossRef PubMed.
- H.-C. Flemming and J. Wingender, The biofilm matrix, Nat. Rev. Microbiol., 2010, 8, 623–633 CrossRef CAS PubMed.
- A. W. D. Decho and T. Gutierrez, Microbial extracellular polymeric substances (EPSs) in ocean systems, Front. Microbiol., 2017, 8, 922 CrossRef PubMed.
- G.-P. Sheng, H.-Q. Yu and X.-Y. Li, Extracellular polymeric substances (EPS) of microbial aggregates in biological wastewater treatment systems: A review, Biotechnol. Adv., 2010, 28, 882–894 CrossRef CAS PubMed.
- M. Ueshima, B. R. Ginn, E. A. Haack, J. E. S. Szymanowski and J. B. Fein, Cd adsorption onto Pseudomonas putida in the presence and absence of extracellular polymeric substances, Geochim. Cosmochim. Acta, 2008, 72, 5885–5895 CrossRef CAS.
- L. Wei, Y. Li, D. R. Noguera, N. Zhao, Y. Song, J. Ding, Q. Zhao and F. Cui, Adsorption of Cu2+ and Zn2+ by extracellular polymeric substances (EPS) in different sludges: Effect of EPS fractional polarity on binding mechanism, J. Hazard. Mater., 2017, 321, 473–483 CrossRef CAS PubMed.
- D. Zhang, X. Pan, K. M. G. Mostofa, X. Chen, G. Mu, F. Wu, J. Liu, W. Song, J. Yang, Y. Liu and Q. Fu, Complexation between Hg(II) and biofilm extracellular polymeric substances: An application of fluorescence spectroscopy, J. Hazard. Mater., 2010, 175, 359–365 CrossRef CAS PubMed.
- Z. Zhang, R. Si, J. Lv, Y. Ji, W. Chen, W. Guan, Y. Cui and T. Zhang, Effects of extracellular polymeric substances on the formation and methylation of mercury sulfide nanoparticles, Environ. Sci. Technol., 2020, 54, 8061–8071 CrossRef CAS PubMed.
- F. Kang, X. Qu, P. J. J. Alvarez and D. Zhu, Extracellular saccharide-mediated reduction of Au3+ to gold nanoparticles: New insights for heavy metals biomineralization on microbial surfaces, Environ. Sci. Technol., 2017, 51, 2776–2785 CrossRef CAS PubMed.
- J. R. Helms, A. Stubbins, J. D. Ritchie, E. C. Minor, D. J. Kieber and K. Mopper, Absorption spectral slopes and slope ratios as indicators of molecular weight, source, and photobleaching of chromophoric dissolved organic matter, Limnol. Oceanogr., 2008, 53, 955–969 CrossRef.
- D. N. Kothawala, K. R. Murphy, C. A. Stedmon, G. A. Weyhenmeyer and L. J. Tranvik, Inner filter correction of dissolved organic matter fluorescence, Limnol. Oceanogr.: Methods, 2013, 11, 616–630 CrossRef.
- J. Zhao and D. J. Nelson, Fluorescence study of the interaction of Suwannee River fulvic acid with metal ions and Al3+-metal ion competition, J. Inorg. Biochem., 2005, 99, 383–396 CrossRef CAS PubMed.
- W. Shou, F. Kang and J. Lu, Nature and value of freely dissolved EPS ecosystem services: Insight into molecular coupling mechanisms for regulating metal toxicity, Environ. Sci. Technol., 2018, 52, 457–466 CrossRef CAS PubMed.
- T. L. Hill, in Cooperativity Theory in Biochemistry: Steady-State and Equilibrium Systems, Springer-Verlag, New York, 1985 Search PubMed.
- X. Wang, W. Song, H. Qian, D. Zhang, X. Pan and G. M. Gadd, Stabilizing interaction of exopolymers with nano-Se and impact on mercury immobilization in soil and groundwater, Environ. Sci.: Nano, 2018, 5, 456–466 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C. 01, Gaussian, Inc., Wallingford, CT, U.S.A., 2019 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual molecular dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. P. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- R. Dronskowski and P. E. Bloechl, Crystal orbital Hamilton populations (COHP): Energy-resolved visualization of chemical bonding in solids based on density-functional calculations, J. Phys. Chem., 1993, 97, 8617–8624 CrossRef CAS.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, LOBSTER: A tool to extract chemical bonding from plane-wave based DFT, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
- L. Zhang, S. Wu, L. Zhao, X. Lu, E. M. Pierce and B. Gu, Mercury sorption and desorption on organo-mineral particulates as a source for microbial methylation, Environ. Sci. Technol., 2019, 53, 2426–2433 CrossRef CAS PubMed.
- USEPA, in Method 1630: Methylmercury in Water by Distillation, Aqueous Ethylation, Purge and Trap, and Cold Vapor Atomic Fluorescence Spectrometry, United States Environmental Protection Agency, Washington, DC, 2001 Search PubMed.
- C. Yin, F. Meng and G. Chen, Spectroscopic characterization of extracellular polymeric substances from a mixed culture dominated by ammonia-oxidizing bacteria, Water Res., 2015, 68, 740–749 CrossRef CAS PubMed.
- C. Li, L. Zhou, H. Yang, R. Lv, P. Tian, X. Li, Y. Zhang, Z. Chen and F. Lin, Self-assembled exopolysaccharide nanoparticles for bioremediation and green synthesis of noble metal nanoparticles, ACS Appl. Mater. Interfaces, 2017, 9, 22808–22818 CrossRef CAS PubMed.
- J. S. Waples, K. L. Nagy, G. R. Aiken and J. N. Ryan, Dissolution of cinnabar (HgS) in the presence of natural organic matter, Geochim. Cosmochim. Acta, 2005, 69, 1575–1588 CrossRef CAS.
- J. L. Weishaar, G. R. Aiken, B. A. Bergamaschi, M. S. Fram, R. Fujii and K. Mopper, Evaluation of specific ultraviolet absorbance as an indicator of the chemical composition and reactivity of dissolved organic carbon, Environ. Sci. Technol., 2003, 37, 4702–4708 CrossRef CAS PubMed.
- G.-P. Sheng and H.-Q. Yu, Characterization of extracellular polymeric substances of aerobic and anaerobic sludge using three-dimensional excitation and emission matrix fluorescence spectroscopy, Water Res., 2006, 40, 1233–1239 CrossRef CAS PubMed.
- S. Bi, L. Ding, Y. Tian, D. Song, X. Zhou, X. Liu and H. Zhang, Investigation of the interaction between flavonoids and human serum albumin, J. Mol. Struct., 2004, 703, 37–45 CrossRef CAS.
- Q. Chang, Z. Zhang, Y. Ji, L. Tian, W. Chen and T. Zhang, Natural organic matter facilitates formation and microbial methylation of mercury selenide nanoparticles, Environ. Sci.: Nano, 2021, 8, 67–75 RSC.
- A. S. Mahadevi and G. N. Sastry, Cation-π interaction: Its role and relevance in chemistry, biology, and material science, Chem. Rev., 2013, 113, 2100–2138 CrossRef CAS PubMed.
- J. P. Gallivan and D. A. Dougherty, Cation-π interactions in structural biology, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459–9464 CrossRef CAS PubMed.
- C. Ruan and M. T. Rodgers, Cation-π interactions: Structures and energetics of complexation of Na+ and K+ with the aromatic amino acids, phenylalanine, tyrosine, and tryptophan, J. Am. Chem. Soc., 2004, 126, 14600–14610 CrossRef CAS PubMed.
- M. Ngu-Schwemlein, J. Merle, W. Meeker, K. Risdon-Langdon and T. Nixon, Evaluating the involvement of tryptophan on thiolated peptide-mercury(II) complexes: Cation-pi interactions, Inorg. Chim. Acta, 2020, 506, 119552 CrossRef CAS.
- H. Yorita, K. Otomo, H. Hiramatsu, A. Toyama, T. Miura and H. Takeuchi, Evidence for the cation-π interaction between Cu2+ and tryptophan, J. Am. Chem. Soc., 2008, 130, 15266–15267 CrossRef CAS PubMed.
- D. E. Schlamadinger, J. E. Gable and J. E. Kim, Hydrogen bonding and solvent polarity markers in the UV resonance Raman spectrum of tryptophan: Application to membrane proteins, J. Phys. Chem. B, 2009, 113, 14769–14778 CrossRef CAS PubMed.
- H. Takeuchi, UV Raman markers for structural analysis of aromatic side chains in proteins, Anal. Sci., 2011, 27, 1077 CrossRef CAS PubMed.
- H.-B. Yi, H. M. Lee and K. S. Kim, Interaction of benzene with transition metal cations: Theoretical study of structures, energies, and IR spectra, J. Chem. Theory Comput., 2009, 5, 1709–1717 CrossRef CAS PubMed.
- Ç. A. Demircan and U. Bozkaya, Transition metal cation−π interactions: Complexes formed by Fe2+, Co2+, Ni2+, Cu2+, and Zn2+ binding with benzene molecules, J. Phys. Chem. A, 2017, 121, 6500–6509 CrossRef PubMed.
- F. Qu, H. Liang, J. He, J. Ma, Z. Wang, H. Yu and G. Li, Characterization of dissolved extracellular organic matter (dEOM) and bound extracellular organic matter (bEOM) of Microcystis aeruginosa and their impacts on UF membrane fouling, Water Res., 2012, 46, 2881–2890 CrossRef CAS PubMed.
- P. H. Santschi, C. Xu, K. A. Schwehr, P. Lin, L. Sun, W.-C. Chin, M. Kamalanathan, H. P. Bacosa and A. Quigg, Can the protein/carbohydrate (P/C) ratio of exopolymeric substances (EPS) be used as a proxy for their ‘stickiness’ and aggregation propensity?, Mar. Chem., 2020, 218, 103734 CrossRef CAS.
- A. Manceau and K. L. Nagy, Quantitative analysis of sulfur functional groups in natural organic matter by XANES spectroscopy, Geochim. Cosmochim. Acta, 2012, 99, 206–223 CrossRef CAS.
- D. Riccardi, H.-B. Guo, J. M. Parks, B. Gu, A. O. Summers, S. M. Miller, L. Liang and J. C. Smith, Why mercury prefers soft ligands, J. Phys. Chem. Lett., 2013, 4, 2317–2322 CrossRef CAS.
- C. C. Gilmour, D. A. Elias, A. M. Kucken, S. D. Brown, A. V. Palumbo, C. W. Schadt and J. D. Wall, Sulfate-reducing bacterium Desulfovibrio desulfuricans ND132 as a model for understanding bacterial mercury methylation, Appl. Environ. Microbiol., 2011, 77, 3938–3951 CrossRef CAS PubMed.
- L. Tian, W. Guan, Y. Ji, X. He, W. Chen, P. J. Alvarez and T. Zhang, Microbial methylation potential of mercury sulfide particles dictated by surface structure, Nat. Geosci., 2021, 14, 409–416 CrossRef CAS.
- T. Zhang, B. Kim, C. m. Levard, B. C. Reinsch, G. V. Lowry, M. A. Deshusses and H. Hsu-Kim, Methylation of mercury by bacteria exposed to dissolved, nanoparticulate, and microparticulate mercuric sulfides, Environ. Sci. Technol., 2012, 46, 6950–6958 CrossRef CAS PubMed.
- S. Jonsson, U. Skyllberg, M. B. Nilsson, P.-O. Westlund, A. Shchukarev, E. Lundberg and E. Björn, Mercury methylation rates for geochemically relevant HgII species in sediments, Environ. Sci. Technol., 2012, 46, 11653–11659 CrossRef CAS PubMed.
- H. Hsu-Kim, K. H. Kucharzyk, T. Zhang and M. A. Deshusses, Mechanisms regulating mercury bioavailability for methylating microorganisms in the aquatic environment: a critical review, Environ. Sci. Technol., 2013, 47, 2441–2456 CrossRef CAS PubMed.
- P.-I. Chou, D.-Q. Ng, I. C. Li and Y.-P. Lin, Effects of dissolved oxygen, pH, salinity and humic acid on the release of metal ions from PbS, CuS and ZnS during a simulated storm event, Sci. Total Environ., 2018, 624, 1401–1410 CrossRef CAS PubMed.
- G. Du Laing, E. Meers, M. Dewispelaere, B. Vandecasteele, J. Rinklebe, F. M. G. Tack and M. G. Verloo, Heavy metal mobility in intertidal sediments of the Scheldt estuary: Field monitoring, Sci. Total Environ., 2009, 407, 2919–2930 CrossRef CAS PubMed.
- J. Morse and G. Luther III, Chemical influences on trace metal-sulfide interactions in anoxic sediments, Geochim. Cosmochim. Acta, 1999, 63, 3373–3378 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2em00394e |
| This journal is © The Royal Society of Chemistry 2023 |