
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A submersible probe with in-line calibration and a symmetrical reference element for continuous direct nitrate concentration measurements†
Tara
Forrest
,
Thomas
Cherubini
,
Stéphane
Jeanneret
,
Elena
Zdrachek
 ,
Polyxeni
Damala
and
Eric
Bakker
*
,
Polyxeni
Damala
and
Eric
Bakker
*
Department of Inorganic and Analytical Chemistry, University of Geneva, Quai Ernest-Ansermet 30, CH-1211 Geneva, Switzerland. E-mail: eric.bakker@unige.ch
First published on 22nd December 2022
Abstract
Current methods to monitor nitrate levels in freshwater systems are outdated because they require expensive equipment and manpower. Punctual sampling on the field or at a fixed measuring station is still the accepted monitoring procedure and fails to provide real-time estimation of nitrate levels. Continuous information is of crucial importance to evaluate the health of natural aquatic systems, which can strongly suffer from a nitrogen imbalance. We present here a nitrate-selective potentiometric probe to measure the analyte continuously without requiring maintenance or high-power consumption. Owing to a simple design where the sensors are located directly in contact with the sample, the need for constant pump usage is eliminated, requiring just 0.7 mW power per day instead of 184 mW per day and per pump. It is estimated that with this power consumption, the setup can easily run for more than 97 h on four simple Li-ion batteries. A simple in-line one-point calibration step was implemented to allow for drift correction. At the same time, a symmetrical design was used involving a second nitrate probe as a reference electrode placed in the calibrant compartment. This, combined with an in situ calibration step, allows one to quantify nitrate ion concentrations directly, instead of yielding activities. The dependence on ion activity was removed by using the analysed sample spiked with nitrate as the calibrant. This results in essentially the same activity coefficients and additionally reduces junction potentials to a fraction of a millivolt. In addition, a symmetrical reference element served to compensate for fluctuations caused by environmental factors (temperature, convection, etc.) to achieve improved stability and signal reproducibility compared to a traditional Ag/AgCl based reference electrode. The final prototype was deployed in the Arve River in Geneva for 75 h without requiring any intervention. The nitrate levels measured using the symmetrical reference element over this period were estimated at 44.0 ± 3.5 M and agreed well with the values obtained with ion chromatography (38.2 ± 2.1 μM) used as the reference method. Thanks to a modular sensing head the potentiometric sensors can be easily exchanged, making it possible to quantify other types of analytes and leading the way to a new monitoring strategy.
Environmental significanceThe effects of excessive amounts of different forms of N (e.g. NH4 and NO3) in water range from human health impacts to eutrophication (which affects dissolved O2, T and pH). The European Union adopted a motion which sets out the obligation to monitor eutrophication risks and report the results every 4 years. Currently constant monitoring points are lacking due to low spatial density and frequency of monitoring. The observations point out that the monitoring network should be strengthened and a different strategy must be envisioned. We propose here a nitrate-selective potentiometric probe that directly measures concentration continuously, has low maintenance and does not require a large power source. This proposed system could pave the way to replacing manual sampling for better nitrate level estimations. |
Introduction
Nitrogen plays an essential role in plant and crop growth, promoting photosynthesis, which is essential for a healthy ecosystem. Although nitrogen is naturally present in the atmosphere, plants mostly absorb it through the soil. In aquatic environments, nitrate is the main source of nitrogen and originates from the conversion of nitrite by bacteria. The aquatic environmental balance of nitrate is crucial as excess nitrogen will lead to extreme algae growth, resulting in oxygen depletion and eutrophication. Excessive nitrate levels are often found in streams crossing rural areas, where nitrate-based fertilisers are still routinely used for agricultural purposes. This is generally the result of leaching into groundwater or surface runoff. Indeed, to enhance plant growth and therefore productivity, nitrate fertilisers have been used routinely in agriculture all over the world.In 1991, the European Commission (EC) established a decree aiming to reduce nitrate pollution and prohibit further pollution.1 Since this directive, the use of nitrate-based fertilisers has been more adequately regulated and systematic water quality monitoring has been implemented. This trend can also be seen here locally in Lake Geneva, where the nitrate levels have been either constant or decreasing since 1991.2 Although it seems that the general tendency leans towards a decrease in nitrate levels and an increase in the general water quality, the monitoring process is not uniform and still rather tedious. The current strategy relies on fixed sampling stations and punctual sampling at strategic sites, which both require manpower and expensive measurement techniques, making it impossible to ensure a constant monitoring of nitrate levels. Monitoring nitrate runoff continuously is especially important due to the rapid multiplication of algae that generally are the cause for eutrophication. This fragile ecological equilibrium can be quickly impacted if no actions are taken swiftly.
Several methods are generally used to monitor nitrate in freshwater systems, such as ion chromatography,3,4 colorimetric assays,5–7 spectrophotometry8–10 and use of ion-selective electrodes.11–14 Ion chromatography is highly selective and sensitive and a well-established laboratory technique that allows nitrate quantification to the ppb level. Unfortunately, the equipment is pricy and bulky and therefore not applicable for in situ measurements, although some studies have been performed to make it portable.4 Colorimetric assays to determine nitrate date back to the 19th century6 and are mostly based on Griess test after subsequent reduction of nitrates into nitrites. Currently Griess reagent test kits are commercially available and this methodology is still at the core of modern research.7 Although these tests have proven to be reliable, they require reagent mixing by the user, which is incompatible with direct in situ measurements. Also using the reduction to nitrite, this time with a cadmium column, followed by azo-dye spectrophotometric detection, continuous analysers have been developed.15,16 The constant usage of pumps to deliver reagents makes these systems unfit for field measurements where no power source is available. Spectrophotometric water analysis of nitrate is possible because of its absorbance in the UV range but limited to freshwater owing to chloride interference. Continuous and on-site measurements are possible with this technique when coupled with a flow system.10 Long-term measurements in this case are not practical due to high power consumption because of the necessary and constant use of a peristaltic pump and UV spectrophotometer.
The use of ion-selective electrodes to quantify the nitrate levels in water dates back to the 1970s11 and was then based on classical liquid-contact electrodes where a Ag/AgCl reference element is placed on the backside of a polymeric PVC-based membrane in a solution of fixed nitrate and chloride concentrations. Over the years, attempts to miniaturise these sensors and apply them to field measurements have been made,12 but although the stability of these sensors proves to be good at high analyte concentrations (>10−2 M), the lifetime and E0 value reproducibility were poor at lower concentrations13 (∼10−4 M) which are in the environmentally relevant range. Developing ion-selective electrodes that have a long lifetime has always been and remains a fundamental challenge. The focus is often only set on maintaining14 a Nernstian slope and sufficiently low detection limit during the analysis period to ensure an adequate response. Unfortunately, less attention is drawn to maintaining a stable E0 value during the development of new sensors because correcting a long-term potential drift can be compensated by regular calibration. This maintenance step is recommended daily with commercially available ready-made nitrate sensors.22 Having to perform a calibration daily adds a complication, which is incompatible with independent long-term monitoring.
In the last few decades, special attention has been drawn to replacing liquid-contact electrodes with solid-contact electrodes23 where no inner filling solution, which must be punctually replaced, is required. The implementation of a transducing layer between the conducting element and the polymeric membrane increases the stability of the signal and gives more control over potential stability. Such sensors have already been applied to the determination of nitrate levels in environmental samples, but in most cases, measurements are performed on collected samples and not made in situ.24,25 An important characteristic of potentiometric sensors is that they are designed to measure activity rather than concentration, which may be a drawback in environmental analysis because the reporting of concentrations is prevalent. Converting activity to concentration requires one to know the total ionic strength of the sample which is not easily available in situ. Recently, however, direct access to concentration measurements with potentiometric sensors has been achieved for cations.26 It used a symmetrical design, with two different indicator electrodes measured against each other, in which the activity coefficients for the analyte ion and the ion measured at the reference electrode changed similarly with ionic strength. Another approach for direct concentration measurements for anions has been developed based on a pulstrode protocol.27
Submersible potentiometric probes28,29 are by nature designed to perform measurements directly in the field and have been used previously in environmental studies,17,18,30–32 but they require a pump to drive the sample towards the measuring site. To enable constant monitoring, the pump must run continually, which would lead to unreasonable power consumption and difficulties in deployment because of power source availability. We present here a simple nitrate-selective submersible potentiometric probe to perform constant monitoring in an independent manner. For this purpose, solid-contact nitrate-selective PVC based electrodes have been selected. The chosen transducing layer polymer, PEDOT-C14, was based on previous work on nitrate sensors performed by our group.33 This choice was also supported by the observation that electrodes prepared with PEDOT-C14 exhibited more mechanical stability than those prepared with carbon nanotubes as seen in previous work.30 This was attributed to the layer-by-layer deposition procedure of carbon nanotubes23 that promoted membrane fall-off. In this new design, the probes are located in a small recess and are in direct contact with the sample allowing constant monitoring without the requirement of any additional power. A small peristaltic pump has been programmed to run briefly at predetermined time intervals to perform a one-step calibration that is used to correct underlying drifts, that would otherwise bias the signal, and allow direct estimation of the concentration of the analyte of interest. The implementation of this automated process cancels the need for external calibration and reduces the necessity for maintenance.
Following a recent research finding by our group,34–36 a new reference element based on the principle of electrochemical symmetry was also tested and implemented to reduce the temperature influence on the sensor signal. In this case, both reference and measuring electrodes are of the same nature (PVC nitrate-selective membrane) as opposed to an asymmetrical system where a plasticised membrane is measured against an all-solid-state reference, Ag/AgCl in most cases. Although lacking stability over time, the Ag/AgCl reference electrode was chosen for comparison purposes as it is currently the gold standard in the field. It also offers miniaturisation possibilities that are often desired in submersible probes. Traditional reference electrodes, such as classical double junction electrodes, exhibit a higher stability and do not require a Cl− background but are known to be bulky, which is incompatible with the reduced available space in submersible probes. In an asymmetrical setup, outside influences such as temperature can affect the E0 value of the reference and measuring electrode differently, resulting in unwanted drifts and deviations. With an automated one-point calibration occurring 2–3 times a day and a sensing principle based on open circuit potential, the power consumption of the probe remains low and allows independent measurements to be carried out continuously on simple batteries. Longer deployment periods could be envisioned thanks to solar panels in order to have fewer power limitations. After initial testing in a laboratory with both artificial and natural freshwater samples, the prototype was deployed in the Arve River in Geneva for testing under real conditions. The obtained results from the field campaigns were cross correlated with traditional reference measurement methods for nitrate. Comparisons between the proposed system and other types of submersible probes for freshwater sensing can be found in Table 1.
| Principle | Application | LOD | Characteristics | Reference |
|---|---|---|---|---|
| Pump driven fluidic system with an ISE | on site | 20 μM | High energy consumption due to pumps | 17 |
| Pump driven fluidic system with an ISE | in line | 1.6 μM | High energy consumption and manual weekly calibration | 18 |
| Gravity-fed fluidic system with an ISE | in situ | 7 μM | Non-submersible probe | 19 |
| Commercial multiparameter probe (YSI) with an ISE | in situ | 0.2 μM | Matrix effects, limited to 15 m depth | 20 |
| ISE with a solar powered antenna | in situ | 7.5 μM | Non-submersible probe | 21 |
| Low power consumption submersible probe with an ISE | in situ | 6 μM | Submersible, no matrix effects low power consumption, and automated recalibration | This work |
Experimental
Reagents and materials
All aqueous solutions were prepared in deionised water (>18 MΩ cm). Sodium nitrate (≥99.0%, NaNO3), sodium chloride (≥99.5%, NaCl), poly(vinyl chloride) (Selectophore™, PVC), bis(2-ethylhexyl) phthalate (Selectophore™, DEHP), tridodecylmethylammonium nitrate (≥99.0%, TDMAN), tetradodecylammonium tetrakis(4-chlorophenyl)borate (Selectophore™, ETH 500) and tetrahydrofuran (≥99.5%, Selectophore™, THF) were purchased from Sigma-Aldrich. Potassium tetrakis(pentafluorophenyl)borate (≥97%, KTPFPB) was purchased from Alfa Aesar. Analytical grade acetonitrile (≥99.5%, ACN) was purchased from Fischer Scientific. EDOT-C14 was synthesised in-house according to ref. 37.Laboratory electrochemical equipment
Glassy carbon (GC) electrodes were fabricated in-house using a PEEK tube (L × OD × ID = 10 ft × 1/8 in × 0.055 in) as the body with a GC rod glued inside (Ø 1.00 ± 0.05 mm). These electrodes were approximately 25 mm long. Before use they were polished using different diamond powder suspensions (Ø 6−3−1−0.25 μM). Electropolymerisation of the transducing layer was performed in a three electrode system with a PGSTSAT 101 (Metrohm Autolab, B.V., Utrecht, The Netherlands) controlled by the Nova 1.8 software. A platinum electrode was used as a quasi-reference with a glassy carbon rod as the counter electrode. Preliminary potentiometric measurements in the laboratory were performed using a high impedance input 16-channel EMF monitor (Lawson Laboratories, Inc., Malvern, PA) to record the signal with double-junction Ag/AgCl/3 M KCl/1 M LiOAc (Metrohm, Switzerland) as the reference electrode. Ion chromatography was used to determine the concentrations of nitrate anions. The eluent solution consisted of 1 mM NaHCO3, 3.2 mM Na2CO3 and 50 mM H2SO4 for regeneration of the suppressor (pressure = 7.0 mPa, flow = 0.7 mL min−1 and temperature = 30 °C). A 0.2 μm cellulose acetate syringe filter (VWR International GmbH) was used to filter the sample prior to injection into the ion chromatograph. The pH of the sample was determined using an 826 pH lab station (Metrohm) at room temperature (21 °C). Flame atomic absorption spectroscopy (AAS, Varian SpectrAA 240 FC) was used to determine the concentrations of Ca2+ and Mg2+ and atomic emission spectroscopy (AES, Varian SpectrAA 240 FC) was used to quantify K+ and Na+. All samples were acidified to pH ∼2.0 with HCl prior to AAS or AES measurements. The concentration of HCO3− was determined by potentiometric titration. The raw sample was diluted with a known volume of 0.1 M NaOH and then titrated with 0.1 M HCl using a pH-sensitive glass electrode from Metrohm. For light influence measurements, a photo studio light box (PULUZ Technology, Shenzhen, China) equipped with modulable LED lights (max intensity of 1690 LM) was used.Field electrochemical equipment
Potentiometric measurements on the field were performed using an EmStat Pico module (Palm Sens, Houten, The Netherlands) located inside the probe. This whole module is designed around an STM32L476 microcontroller running on an operating system called Chibios. The probe is equipped with a PT1000 temperature probe using a MAX31865 chip. The data is stored internally on a microSD card located on the main board (Fig. S1†). To perform time-based measurements, an integrated clock with a CR2032 battery is mounted internally. To perform the calibration step, two small KMPP peristaltic pumps (Kamoer Fluid Tech, Shanghai) were added to the sensing head. The pumps are controlled by a Grove I2C Motor (TB6612) module that can control two pumps. The pumps and the main board are powered by 4 Li-ion batteries (3500 mA h–3.6 V). The main reference electrode was a simple Ag/AgCl coated wire (Metrohm AG, Switzerland).Electrode preparation
The polymerisation solution used to generate the transducing layer was made of 0.01 M of EDOT-C14 and 0.03 M of KTPFPB in acetonitrile as previously reported in ref. 33. The PEDOT-C14 films were generated on the polished GC electrode by dynamic electropolymerisation (cyclic voltammetry), from −1.1 V to 1.6 V at a scan rate of 0.1 V s−1 for 2 cycles. The transducing layer was then prepolarised by applying a constant potential to a solution of 0.03 M of KTPFPB in acetonitrile to reach an oxidation ratio of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 between PEDOT-C140/PEDOT-C14+.36 The prepolarisation potential was chosen as the potential corresponding to the oxidation peak potential. The electrodes were then air-dried for 30 min. The membrane cocktail was prepared by dissolving 32.35 mg of PVC, 65.77 mg of DEHP, 0.60 mg of TDMAN (10 mmol kg−1) and 1.73 mg of ETH500 (15 mmol kg−1) in 1 mL THF. A total amount of 25 μL (5 times 5 μL) of membrane cocktail was drop cast onto the electrode body and left to dry overnight. The membrane deposition was performed sequentially to avoid overflowing due to the small diameter of the electrode. Prior to analysis, the sensors were conditioned in 1 mM NaNO3 overnight.
:50 between PEDOT-C140/PEDOT-C14+.36 The prepolarisation potential was chosen as the potential corresponding to the oxidation peak potential. The electrodes were then air-dried for 30 min. The membrane cocktail was prepared by dissolving 32.35 mg of PVC, 65.77 mg of DEHP, 0.60 mg of TDMAN (10 mmol kg−1) and 1.73 mg of ETH500 (15 mmol kg−1) in 1 mL THF. A total amount of 25 μL (5 times 5 μL) of membrane cocktail was drop cast onto the electrode body and left to dry overnight. The membrane deposition was performed sequentially to avoid overflowing due to the small diameter of the electrode. Prior to analysis, the sensors were conditioned in 1 mM NaNO3 overnight.
Before any experiments, the functionality of the electrodes was tested by calibrating them with NaNO3 solutions from 10−7 M to 10−2 M in deionised water. The response slope as well as the lower detection limit were recorded as they are needed for further calculations. All activity coefficients were calculated according to ref. 38. To test the reliability of the electrodes under different conditions, the light and pH influence on the potentiometric signal was evaluated. It can be seen in Fig. S2A† that the signal remains constant no matter the light intensity and in Fig. S2B† that the signal remains stable when changing the pH of the solution from 6 to 11, especially in the range that is environmentally relevant (from pH 6.5 to 9.0). To confirm the applicability of these nitrate ion-selective electrodes for measurements over several days, a water layer test was conducted (see Fig. S3†) and showed no water formation between the transducer layer and the ion-selective membrane. The selectivity of these membranes was also tested to ensure the reliability of the measurements on real samples. This experiment was performed according to the MSSM.39 It can be seen in Fig. S4 and Table S1† that the proposed ion-selective electrodes are discriminating against sulfate and chloride, which are the main anions that might interfere in freshwater samples.
Probe design
The entire probe (Fig. 1A) was built and designed in-house. The white casing is made of Delrin tubes (Ø 10 cm) closed by transparent custom lids made of poly(methyl methacrylate) (PMMA). All lids are equipped with fluorinated, carbon-based rubber (FKM) O-rings to make the tubes watertight. A close-up of one of these lids, with the O-ring, can be seen in Fig. 1B, as it acts as the sensing head. The two compartments of the probe were separated on purpose to protect the electronics located in the top compartment in case of a leak in the bottom part, containing the pumps and sensors. The cables connecting both compartments are enclosed in PMMA tubes sealed with silicone rubber to ensure watertightness. The total length of the instrument is 1.0 m (0.92 m without the top connector). Because of the air located inside both tubes when closed, the instrument had to be fixed on a stainless-steel bar (l = 1.4 m) acting as a deadweight. To protect the sensors from foreign objects and perform the calibration step during the immersion in real samples, a piece of metallic mesh was mounted at the edge of the dome-shaped recess where the electrodes are located. The solution used during the calibration step is placed into a soft plastic bag and connected through an inlet located between the two tubes. | ||
| Fig. 1 (A) Full view of the probe with (from bottom to top) the first compartment containing the sensors (behind a piece of protective metallic mesh), cabling and pumps. The calibrant inlet is located in the middle part with the cables running from the first to the second compartment that encloses the main board. An outlet to connect the transmission cable is located on top, close to the anchoring point. (B) Zoomed view of the empty sensing head, where the ion-selective electrodes will be located in a dome-shaped recess. A temperature probe is placed on the side. | ||
A schematic representation of the sensing head can be seen in Fig. 2. The sensing head is divided into two parts: a modified T-junction that contains the reference elements and sensing dome, located in contact with the sample. The top channel, called the calibrant inlet, is used to deliver the calibrant to the ion-selective electrodes during the calibration step. This is achieved using a small peristaltic pump that is activated punctually. Both references, located in the top part of the modified T-junction, will only ever be in contact with the calibrant solution thanks to the smaller diameter connection, separating both parts of the T-junction. The use of a channel exhibiting a smaller diameter prevents mixing between the calibrant and sample during the measuring periods. A small drain channel is located after the recess and can be used to aspirate solution from the sensing dome, for example in case an air bubble gets stuck inside (which will generate potential drifts). The small diameter connection between both parts of the modified T-junction is designed to ensure that when the drain channel is used, solution is pumped selectively from the sensing dome and not from the top part where the reference electrodes are located. The ion-selective electrodes are located at the bottom of the sensing head in a small dome-shaped recess. The dome has a diameter of 12.64 mm for a maximal depth of 3 mm. A schematic side-cut of the dome with its housing can be seen in Fig. S5.† It is possible to fit six measuring electrodes in the sensing dome. This can be useful to obtain intra-electrode reproducibility or measure more than one type of analyte. This geometry was chosen amongst others to have no sharp edges that would impair equal diffusion of the sample in the recess. The sensing dome is closed using metallic mesh (30 μm mesh size-stainless-steel) that shields the electrodes from outer foreign objects but still allows solution to flow freely in and out.
 | ||
| Fig. 2 Scheme of the sensing head. Calibrant solution flows from the top trough the inlet. Both reference electrodes are located in the first chamber of the modified T-junction. A drain channel is present in the second chamber to remove potential bubbles in the sensing dome. The ion-selective electrodes (ISEs) are located all around the sensing dome. | ||
Calibration and measurement principle
The scheme presented in Fig. 3 illustrates the principle of the in-line calibration protocol. During the initial sample measurement phase, the sample fills the dome-shaped recess and the ISEs measure the sample. Both reference electrodes located on top are in contact with the calibrant solution. To avoid significant junction potentials between the calibrant and sample, the calibration solution was prepared by spiking the sample with a small amount of nitrate. With this procedure, the matrix and overall ionic strength are the same in both cases. Due to the high ionic strength of freshwater samples, small concentration variations of ions do not influence the junction potential significantly and the calibrant does not have to be exchanged during the testing period. Thanks to the small channel connecting both parts of the cell, mixing can be neglected. Since mixing never occurs and the references are in a solution of fixed concentrations, it is possible to use an additional ion-selective electrode as a symmetrical reference element. The principle of electrochemical symmetry has been recently investigated by our group34 and is based on the observation that ion-selective electrodes of the same batch always tend to experience similar drifts. It has been shown in this study that traditional reference elements such as Ag/AgCl wires do not experience temperature influence in the same way as ion-selective electrodes. | ||
| Fig. 3 Principle of in-line automated calibration. (A) Initial phase where the calibrant is measured. (B) Calibration phase where the calibrant solution is actively pushed to the sensing dome. (C) Reequilibration phase where the sample gradually fills the sensing dome back. | ||
This difference in temperature influence is of great importance for a probe that is meant for measurements outside the laboratory, where temperature gradients during experiments can be relatively large, especially in surface waters during day and night cycles. In a follow up study,35 it has also been demonstrated that other types of drifts, occurring for example at a transducer–membrane interface or because of water uptake, are also comparable between ion-selective electrodes of the same batch. With these considerations in mind, by measuring one electrode against another of the same type (located in a solution of a fixed concentration), these drifts should cancel themselves out and lead to higher stability and reproducibility.
During the sample measurement, the whole system is passive and renewal of solution in the sensing dome is achieved by natural water motion or buoyancy. During the calibration phase (Fig. 3B), a small pump is activated and the calibration solution is pushed towards the sensing dome. In this configuration, all electrodes are now measuring the same solution. Due to the small volume of the sensing dome, less than a minute is needed to fully replace the sensing dome solution. It has also been seen that the metallic mesh placed in front of the electrodes for protection helps slow down the diffusion of the calibration solution into the bulk, therefore rendering the system more efficient and allowing for a shorter calibration period. This is a significant advantage in terms of power consumption.
During the reequilibration period, the pump is stopped and the sample gradually fills the dome again to go back to its initial state. In field campaigns, the probe is always anchored to a boat or a buoy, hence it is never static. This natural movement will help the reequilibration process and speed it up. The whole calibration procedure can be preprogramed into the probe and run at fixed time intervals (Fig. S6†). The duration of a cycle can be adjusted to space out the number of calibrations to fit the experimental needs. Since the concentration of the calibrant solution remains constant, the potential value corresponding to the calibrant should remain stable throughout cycles and is therefore a good control parameter towards undesired drifts. In addition to this, the calibration step is also used to estimate the concentration of the real sample with better accuracy.
When performing a calibration in a laboratory setup, one is able to obtain a calibration curve that will normally adhere to the theoretical Nernst equation. It is well known that this equation correlates the measured potential with the activity of the ion of interest. During field measurements, estimating the activity of the sample is of limited interest since the ionic strength (aI = γ·cI) has to be known to compute the concentration of the ion of interest. Determining the total ionic strength requires a concentration estimation of all major ionic species. Because this involves benchtop instrumentation it defeats the purpose of direct on-site measurements. In the case presented here, since the calibrant is based on the sample, it can be assumed that the ionic strength is the same in both cases. It is possible to use this information to obtain the concentration of the sample based on the Nikolsky–Eisenmann equations of both solutions:
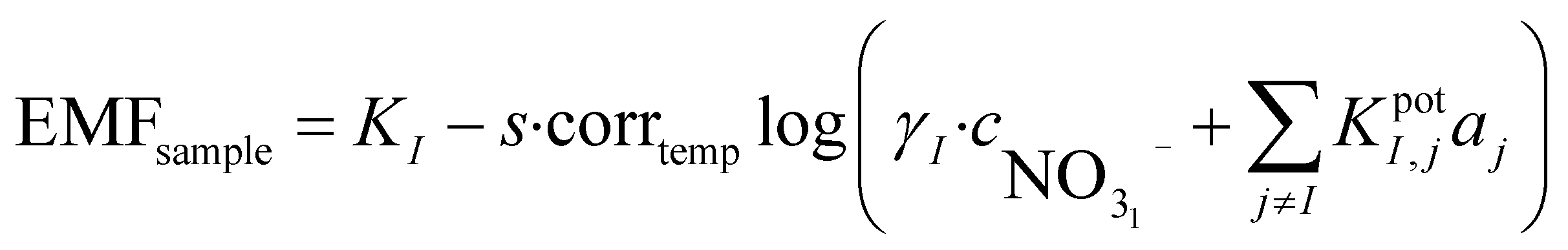 | (1) |
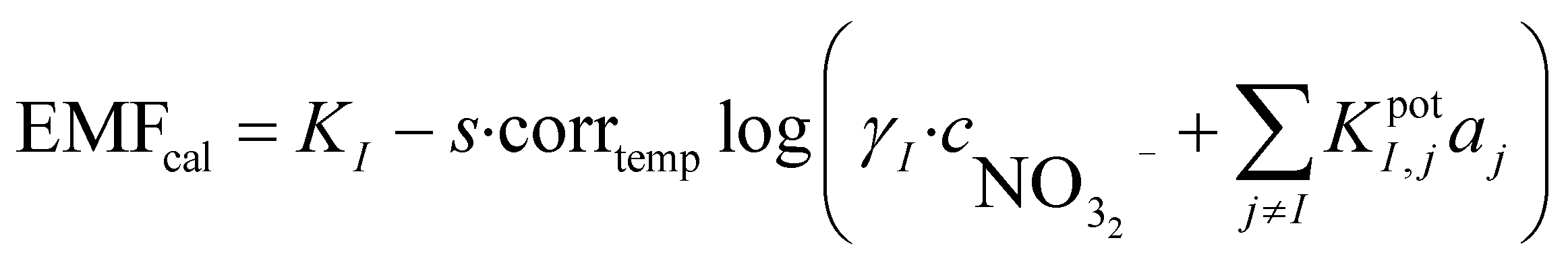 | (2) |
 is the concentration of nitrate in the sample,
is the concentration of nitrate in the sample,  is the concentration of nitrate in the calibrant and KI,jpotaj is the selectivity coefficient. It can be assumed that γI is the same in both cases since the ionic strength of both solutions is very similar. The lower activity-based detection limit determined during initial laboratory testing of the electrode can be converted into a concentration-based detection limit and used instead of the selectivity coefficient:
is the concentration of nitrate in the calibrant and KI,jpotaj is the selectivity coefficient. It can be assumed that γI is the same in both cases since the ionic strength of both solutions is very similar. The lower activity-based detection limit determined during initial laboratory testing of the electrode can be converted into a concentration-based detection limit and used instead of the selectivity coefficient: | (3) |
If eqn (2) is subtracted from eqn (1), the difference potential ΔEMF is obtained. This allows the removal of the parameter KI which is the same in both cases owing to the similar composition of the sample and calibrant. The activity coefficient is now present everywhere in the logarithmic part of eqn (4) and therefore also cancels out:
 | (4) |
It should be emphasised here that this assumption is only possible because both samples have very similar ionic strengths. As mentioned before, the calibrant is prepared on-site by spiking a nitrate aliquot in a known volume of sample. The concentration of nitrate in both solutions can now be correlated:
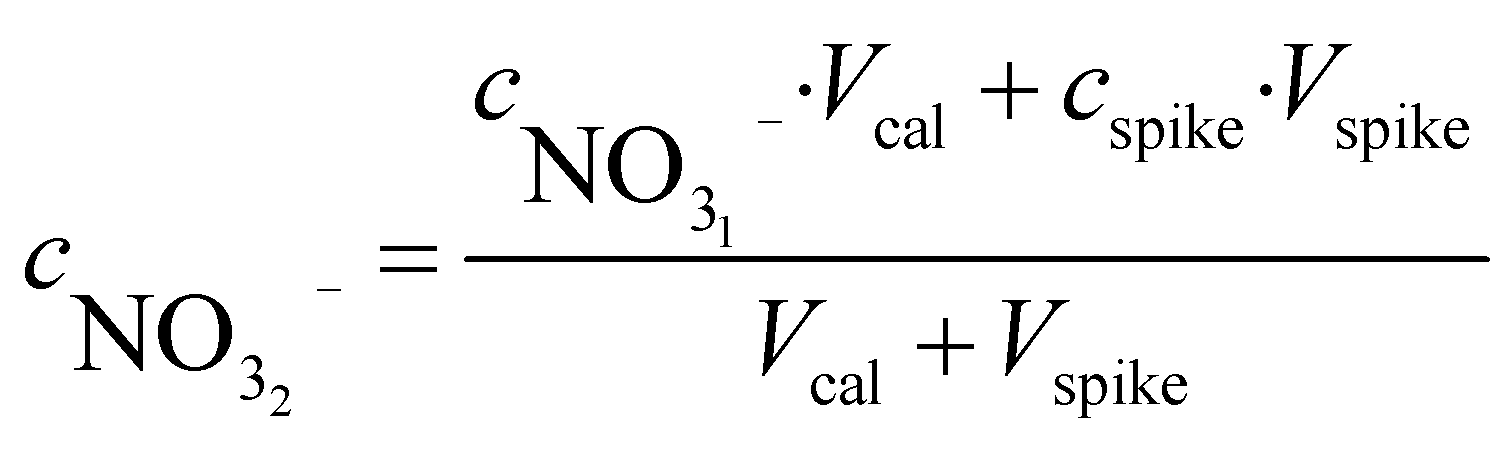 | (5) |
Finally, the temperature correction on the slope can also be adjusted, by knowing at what temperature the laboratory calibration was performed initially:
 | (6) |
If eqn (1)–(6) are now combined, the unknown nitrate concentration in the sample can be calculated using the following equation:
 | (7) |
This makes it now possible to have direct access to the concentration (not activity) of the medium, without having to estimate the total ionic strength, which is impossible during on-site measurements.
Results and discussion
During the development phase of the sensing head, it was realised that its design plays a significant role in achieving a successful one-point calibration step. The critical point was the height of the sensing dome (Fig. S5†) that had to be carefully adjusted. In earlier designs, a smaller dome was tested with a height lower than 2 mm. In this case, unfortunately, a full replacement of the solution during the calibration was never achieved. The calibrant was directly diluted in the sample and a stable signal for the calibrant was never obtained. The optimal dome height was determined to be 3 mm. Attempts with a higher dome (10 mm) were also undertaken but it was seen that the reequilibration phase (Fig. 3C) was difficult in that case as calibrant solution would stay trapped at the top of the dome, creating a dead volume that was never replaced with the sample. The final design had the recess closed by a metal mesh barrier to shield the ion-selective membranes from foreign objects and prevent excessive dilution of the calibrant in the sample during the calibration step. Several materials, such as a dialysis membrane, a cellulose membrane and metallic mesh were considered during the development phase. Although conclusive results were obtained with the cellulose membrane, the material was discarded owing to its poor mechanical robustness. The dialysis membrane proved to be much more robust, but its charged surface40 and low porosity were a challenge for solution renewal, especially after the calibration step. The metallic mesh was kept in the final configuration owing to its high mechanical stability and larger pore size.Tests mimicking a real environment were conducted in a fish tank equipped with a water circulating system to reproduce the natural flow of a river. The tank was filled with a solution containing 0.5 mM NaCl/0.5 mM NaNO3 and the calibrant was set to 0.5 mM NaCl/2.0 mM NaNO3. As seen in Fig. 4, the sample was first measured and then periodically the pump was activated to push the calibrant (containing a higher concentration of nitrate ions) into the sensing dome as depicted in Fig. 3. Due to the different concentrations of nitrate anions in the two solutions the potential value drops according to the Nernst equation when the calibrant solution is pushed towards the dome. Once a stable potential value was obtained for the calibrant, the pump was stopped and the signal was found to gradually return to the initial potential value corresponding to the sample. The rate at which the initial potential is recovered can be adjusted by changing the mesh size. By comparing Fig. 4A and B it can be noticed that with the larger mesh size, the baseline is reached faster than with the smaller sized mesh. More cycles could be performed in 2000 s with the 74 μm mesh size due to a faster reequilibration time. Having different mesh sizes is also an advantage for field testing where it can be adapted depending on the turbulences present in the medium. A smaller mesh size may be more suited in the case of a very turbulent sample to achieve the calibration step, while a larger mesh size might be advantageous in more stagnant environments to not impair the reequilibration process after the calibration step.
 | ||
| Fig. 4 (A) Reversibility test with a piece of 30 μm stainless steel mesh closing the dome. (B) Reversibility test with a piece of 74 μm stainless steel mesh closing the dome. The top red line represents the potential value of the sample (0.5 mM NaNO3) and lower red line represents the potential value of the calibrant (2.0 mM NaNO3). Both solutions contained a background of 0.5 mM NaCl since a Ag/AgCl reference was used. | ||
The potential jump that was observed in both cases (∼−38.0 mV) was consistent with theoretical calculations based on the Nernst equation, taking into account the junction potential. In this case, since solutions of low ionic strength were used, the junction potential had to be considered. The classical Henderson equation was used to estimate the junction potential and all ion mobility values were taken from ref. 41. The liquid junction potential was calculated according to Henderson's eqn (8).
 | (8) |
With a calculated potential difference of −39.0 mV, it can be concluded that the solution exchange during the calibration process is optimal and that the proposed geometry of the sensing head works as expected for the one-point calibration.
To avoid high junction potentials (up to 5 mV) when working with solutions of low ionic strength, follow-up experiments using the same setup were conducted in real water samples sourced from the Rhône River in Geneva. To confirm the small junction potential between two solutions of higher ionic strength, the concentrations of all major cations (K+, Na+, Ca2+ and Mg2+) and anions (NO3−, Cl−, HCO3− and SO42−) were estimated using reference methods specified in the experimental section. The individual concentrations of each ion can be found in Table S2.† The calibrant used in this case was the same Rhône River water sample but spiked with NaNO3. The concentration of nitrate in the calibrant was 155.3 μM compared to 35.3 μM in the sample. The resulting junction potential calculated between the two solutions was lower than 0.3 mV and can be neglected considering that the expected potential shift between both solutions corresponds to −38.0 mV.
The results from the tank-based tests with a real sample are presented in Fig. 5. In this case, a piece of fine mesh (30 μm) was mounted to have the same conditions that would be further used in the field campaign. In addition to testing the one-point calibration protocol, the symmetrical reference element was also implemented. Since the experiment is performed in a controlled environment, the temperature fluctuations are minimal; therefore their influence on both references should not be seen. In both cases, the reversibility is satisfactory and the signal corresponding to the sample is recovered in minimal time. It can be noticed that the time trace corresponding to the response versus the Ag/AgCl reference exhibits more noise and perturbations, especially during the calibration step. This can be explained by the fact that when the pump is activated, the electrodes are subjected to a stronger flow than during the passive sample measurement, causing a small perturbation. Since this flow is similar in the modified T-junction, where the symmetrical reference is located, and the sensing dome, when measured against each other, this interference cancels out, leading to a smoother signal. The potential values recorded for the sample exhibit a smaller standard deviation when measured against the symmetrical reference (2.7 ± 0.2 mV) element than that exhibited when measured against the Ag/AgCl reference (34.2 ± 0.6 mV). The same conclusion can be drawn for the potential values corresponding to the calibrant (−0.6 ± 0.6 mV for the classical system versus −34.3 ± 0.3 mV for the symmetrical system). The observed potential shift between both solutions is closer to the theoretical value with the symmetrical system (−37.0 ± 0.3 mV) compared to the classical system (−34.8 ± 0.6 mV). Cycle by cycle data extracted from Fig. 5 can be found in Table S3† for the symmetrical reference and Table S4† for the Ag/agCl reference element. Both tests performed in the laboratory showed that the one-point calibration is successful and sufficiently accurate to enable one to estimate the nitrate concentration of an unknown sample with eqn (7), without having to use traditional measurement methods for nitrate that are not well suited for field work. This accuracy is reinforced by the use of the symmetrical reference element that helps cancel out small perturbations or drift, seen when using a traditional Ag/AgCl reference element.
 | ||
| Fig. 5 Nitrate potentiometric response in the Rhône River water sample. Reversibility tests over 7 cycles alternating between the raw sample and spiked sample. the pink trace is the potentiometric response versus the Ag/AgCl reference electrode and red trace is the potentiometric response versus the symmetrical nitrate reference. The temperature time trace is in cyan. A 30 μm mesh size was used to mimic the field conditions. For each trace, the highest potential refers to the sample (35.3 μM) and the lowest potential refers to the calibrant (155.3 μM). | ||
Field measurements were performed in the Arve River in spring 2022. The probe was anchored with a rope to a platform close to the shore. During the testing period the smaller mesh size was always used (30 μm) due to a turbulent flow which is typical for this time of the year, due to snow melting in the French Alps where the Arve has its source. The probe was immersed during the day and preprogramed to perform a calibration every 8 h. The details of the calibration cycle can be found in Fig. S6 and Table S5† During each cycle, the pump was activated for a total time of a 120 s and the signal related to the calibrant was recorded at the end. No measurements were taken during the 5 min following the calibration routine to leave some time for the calibrant to dissipate from the dome and be exchanged with the sample again. Knowing the flow rate of the pump (2.6 mL min−1), 15.6 mL of calibrant was consumed every day assuming 3 calibrations are performed. With the 500 mL calibrant bag that was mounted, the solution would have to be renewed only every 30 days. The results generated by the probe were stored internally on an SD card as a text file. These files were then processed with Mathematica to generate time traces and convert potential readings into nitrate concentrations with eqn (7).
Performing 3 calibrations a day means that the pump is activated for a total of 360 s per day. Knowing that the pump requires 50 mA at 3.7 V, the daily power consumption is just 0.77 mW. If the pump was running continuously, as it is classically done in submersible probes, the daily required power would be 185 mW. The main board included in the instrument needs 100 mA at 5 V to run at full power. If needed, the main board can go into standby where it will only need 20 μA at 5 V. Assuming the board runs at full power continuously, the daily required power may be estimated at 0.5 W. With this information, it is assumed that the probe can run for at least 97 h on 4 Li-ion batteries. To remove the need for battery exchange, it is calculated that a 43 × 55 cm solar panel would be sufficient to power the device with only 2 h of direct sunlight a day.
Fig. 6A shows the potentiometric time trace of a nitrate-selective electrode versus the Ag/AgCl reference, showing a drifting signal over time. Over the 75 h of the testing period the overall potential change was estimated at around 30.0 mV (from ∼30.0 mV at t = 0 h to ∼0.0 mV at t = 75 h). Fig. 6B, which represents the potentiometric time trace of the nitrate-selective electrode used as the reference relative to the same Ag/AgCl electrode, shows that this drift is similar. Since both reference electrodes are in a solution of fixed concentration, one can be sure that the observed potential shift is a drift and not a response towards nitrate. The intense drift seen in the first few hours after immersion may originate from the low water temperature, below 10 °C during the testing period. To correct for the drift seen in both cases, the symmetrical reference was used to generate Fig. 6C. Both graphs presented in Fig. 6A and B are raw potentiometric data, where no temperature correction is applied. Applying a temperature correction here to generate Fig. 6C is not needed as it would be cancelled out mathematically. The temperature correction was applied when calculating the concentration of nitrate as seen in Fig. 7. The drift is now compensated for and the observed signal exhibits an increased stability. The spikes, corresponding to the calibration events, are now more reproducible and the overall signal change over the entire testing period is now approximately 6.0 mV.
 | ||
| Fig. 6 (A) Potentiometric response of a nitrate-selective electrode versus the Ag/AgCl electrode. The 30 μm mesh size was used due to the strong flow of the river. (B) Potentiometric response of a symmetrical nitrate-selective reference electrode versus Ag/AgCl. (C) Potentiometric response of the nitrate-selective electrode versus the symmetrical reference. Graphs (A) and (C) represent the same measuring electrode. Graph (A) and (B) are raw potentiometric traces without temperature correction. In this case, the nitrate concentration of the calibrant was estimated at 161.8 μM by ion chromatography. The nitrate concentration calculated from graph (C) can be seen in Fig. 7. | ||
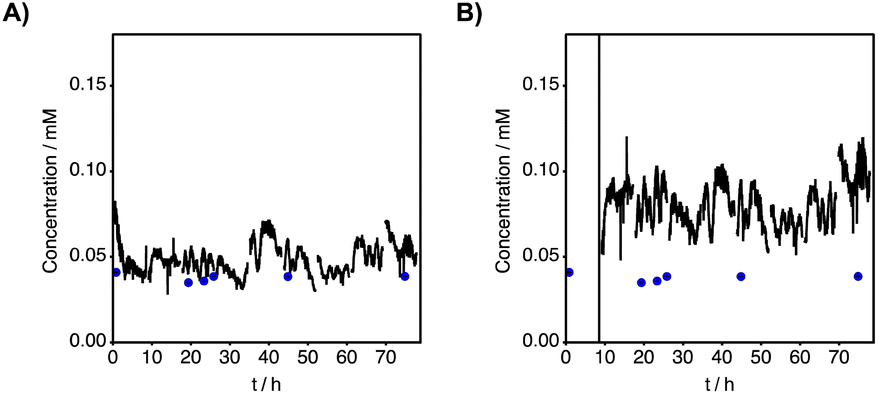 | ||
| Fig. 7 Converted potentiometric data using eqn (7) to get the concentration of an unknown sample. (A) Results of the nitrate-selective electrode versus the symmetrical reference. (B) Results of the nitrate-selective electrode versus the Ag/AgCl reference. On both graphs, blue dots represent the nitrate concentration obtained by ion chromatography from punctual sampling. Error bars represent the standard deviation (n = 3). | ||
To confirm the reliability of the system, the water from the Arve River was punctually sampled throughout the testing period and analysed by ion chromatography to allow cross-correlation. The potential readings obtained with the probe were converted to concentration values using eqn (7). It should be noted here that the parameter EMFcal was always updated after a calibration cycle to use the latest value. Throughout the cycles, the average value for EMFcal was more reproducible when using the symmetrical reference (EMFcal, symmetry = −22.0 ± 1.6 mV) compared to that obtained when using the Ag/AgCl reference (EMFcal, Ag/AgCl = −16.7 ± 2.2 mV). The same conclusion can be drawn when estimating the ΔEMF between EMFcal and the EMF reading before the calibration process. The average ΔEMF when measuring against the symmetrical reference was found to be equal to ΔEMFsymmetry = −27.5 ± 1.2 mV compared to ΔEMFAg/AgCl = −23.6 ± 2.0 mV, obtained using the traditional reference element. Both average ΔEMF values differ by 3.9 mV, which will have an impact on the observed nitrate concentration value.
Fig. 7 presents the converted raw data, extracted from the probe, into a molar nitrate concentration. The implementation of the in-line calibration step allowed one to have direct access to the concentration of nitrate ions instead of activity. The blue dots on the graph indicate the concentration of nitrate obtained by ion chromatography used as the reference method. From Fig. 7A and B it can be seen that the concentration levels obtained using the symmetrical reference correlate better with the measured levels by ion chromatography. The fluctuation of the signal is also minimised when using the symmetrical reference since interferences, such as temperature drift or convection, affect both plasticised membranes in a similar manner. A more stable signal correlates with the trend seen in the different ion chromatography (IC) measurements over time. The average nitrate concentration measured by ion chromatography is 38.2 ± 2.1 mM. Individual nitrate concentrations recorded by IC can be found in Table S6.† Sampling more often would have been an advantage for better cross-correlation between IC data and the probe, but it was unfortunately not possible due to the complicated access to the anchoring point of the probe. When performed, sampling was conducted manually, thus stressing the need for better measuring systems and sampling facilities. The average nitrate concentration observed using the probe for the same times at which these samples were taken was 44.0 ± 3.5 mM when measured against the symmetrical reference element. This value increases to 77.4 ± 6.4 mM when using Ag/AgCl as the reference, which is unacceptable. This overestimated concentration was measured with the Ag/AgCl electrode and is presented in Fig. 7B is due to a lower and biased measured ΔEMF than expected. A smaller ΔEMF suggests a smaller difference between the concentration of the sample and calibrant, thus the overestimation. Such high deviations can cause issues during a monitoring campaign, expressing the need for better reference elements.
The potentiometric response of the electrodes was tested before the immersion in the real sample and after the testing period, to confirm that they were still functional. It can be seen in Fig. S7† that after the experiment in the Arve River, the E0 value has indeed shifted and the lower detection limit shifted from −5.2 to −4.8 in nitrate logarithmic activity. This change is not significant for our experiments as the natural nitrate concentration found in the Arve River is in both cases still in the linear response range. It is assumed that this effect is due to biofouling at the surface of the electrode. For longer deployment periods, a new strategy to avoid biofouling will have to be implemented. Several options could be envisioned, such as replacing the stainless-steel mesh by copper mesh that is known to have antifouling properties42 or by in situ electrochemical generation of chlorine43 using the metallic mesh as the electrode. The latter strategy is mainly used in seawater, where the concentration of salt is high, but recently attempts with lower amounts of NaCl44 have been undertaken and could be applied to the case of freshwater. For improved results, both of these options could be combined with ion-selective membrane surface modification with silver nanoparticles45 for increased anti-biofouling properties. To prevent microbial growth and biofouling in the compartment where the reference electrodes are located, a piece of copper mesh could be implemented between the calibrant bag and tubing inlet. By avoiding biofouling of the electrodes and using solar panels to power the probe, maintenance should only be required to refill the calibrant bag and exchange the electrodes. In a previous study, it was shown that nitrate-selective electrodes were functional for at least 3 weeks.34 The main limiting factor in this case is the lifetime and lower detection limit of the ion-selective electrodes.
Conclusions
Finding new ways to easily estimate the quality of freshwater environments is crucial for the needs of our society. This new fully integrated potentiometric probe offers the prospect of long-term nitrate monitoring with minimal required maintenance. Owing to its compact and portable design, this instrument is not only limited to sampling stations, but could be potentially deployed anywhere using a boat or a buoy. The use of an ion-selective electrode of the same batch as the measuring ones and placing it in a reference compartment allowed one to test the concept of electrochemical symmetry under real conditions. It was demonstrated here that the symmetrical reference element offers superior performances as it frees the signal from unwanted noise and generates more accurate results. For further applications, one could consider not using a Ag/AgCl reference element but only a symmetrical reference. The implementation of an in-line calibration procedure enables the user to obtain a direct estimation of the nitrate levels in freshwater systems without having to analyse a sample beforehand. The intermittent measurement of a solution of a fixed concentration also offers an additional control of drifts occurring in ion-selective electrodes. Using different types of sensors, this instrument may be adapted to measure other types of analytes and be used for multi-analyte quantification. It is believed that this work based on a narrow timeframe (80 h) may serve as a basis for long-term deployment measurements.Author contributions
T. F.: conceptualisation, investigation, methodology visualisation and writing – original draft. T. C.: resources and methodology. S. J.: resources and software. E. Z.: conceptualisation and methodology. P. D.: conceptualisation and methodology. E. B.: conceptualisation, funding acquisition, methodology, supervision and writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This project was funded by the Eurostars Project E! 113301-AQISE.References
- Council Directive 91/676/EEC of 12 December 1991 concerning the protection of waters against pollution caused by nitrates from agricultural sources, https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=celex%3A31991L0676, accessed 26.07.2022 Search PubMed.
- CIPEL, https://www.cipel.org/catalogue/rapport-scientifique/, accessed 25.07.2022.
- R. Michalski, Ion Chromatography as a Reference Method for Determination of Inorganic Ions in Water and Wastewater, Crit. Rev. Anal. Chem., 2006, 36, 107–127 CrossRef CAS.
- E. Murray, P. Roche, M. Briet, B. Moore, A. Morrin, D. Diamond and B. Paull, Fully automated, low-cost ion chromatography system for in situ analysis of nitrite and nitrate in natural waters, Talanta, 2020, 216, 120955 CrossRef CAS PubMed.
- W. L. Daniel, M. S. Han, J.-S. Lee and C. A. Mirkin, Colorimetric Nitrite and Nitrate Detection with Gold Nanoparticle Probes and Kinetic End Points, J. Am. Chem. Soc., 2009, 131, 6362–6363 CrossRef CAS.
- G. Harrow, XXXVII.—A rapid method of estimating nitrates in potable waters, J. Chem. Soc., Trans., 1891, 59, 320–323 RSC.
- E. Murray, E. P. Nesterenko, M. McCaul, A. Morrin, D. Diamond and B. Moore, A colorimetric method for use within portable test kits for nitrate determination in various water matrices, Anal. Methods, 2017, 9, 680–687 RSC.
- J. H. Wetters and K. L. Uglum, Direct spectrophotometric simultaneous determination of nitrite and nitrate in the ultraviolet, Anal. Chem., 1970, 42, 335–340 CrossRef CAS.
- F. A. J. Armstrong, Determination of Nitrate in Water Ultraviolet Spectrophotometry, Anal. Chem., 1963, 35, 1292–1294 CrossRef CAS.
- B. S. Gentle, P. S. Ellis, M. R. Grace and I. D. McKelvie, Flow analysis methods for the direct ultra-violet spectrophotometric measurement of nitrate and total nitrogen in freshwaters, Anal. Chim. Acta, 2011, 704, 116–122 CrossRef CAS PubMed.
- P. J. Milham, A. S. Awad, R. E. Paull and J. H. Bull, Analysis of plants, soils and waters for nitrate by using an ion-selective electrode, Analyst, 1970, 95, 751–757 RSC.
- B. Müller, K. Buis, R. Stierli and B. Wehrli, High spatial resolution measurements in lake sediments with PVC based liquid membrane ion-selective electrodes, Limnol. Oceanogr., 1998, 43, 1728–1733 CrossRef.
- J. Schwarz, H. Kaden and G. Pausch, Development of miniaturized potentiometric nitrate- and ammonium selective electrodes for applications in water monitoring, Fresenius. J. Anal. Chem., 2000, 367, 396–398 CrossRef CAS PubMed.
- F. Zuther and K. Cammann, A selective and long-term stable nitrate sensor, Sens. Actuators, B, 1994, 19, 356–358 CrossRef CAS.
- N. Le Bris, P. M. Sarradin, D. Birot and A. M. Alayse-Danet, A new chemical analyzer for in situ measurement of nitrate and total sulfide over hydrothermal vent biological communities, Mar. Chem., 2000, 72, 1–15 CrossRef CAS.
- H. W. Jannasch, K. S. Johnson and C. M. Sakamoto, Submersible, Osmotically Pumped Analyzer for Continuous Determination of Nitrate in situ, Anal. Chem., 1994, 66, 3352–3361 CrossRef CAS.
- N. Pankratova, G. A. Crespo, M. G. Afshar, M. C. Crespi, S. Jeanneret, T. Cherubini, M.-L. Tercier-Waeber, F. Pomati and E. Bakker, Potentiometric sensing array for monitoring aquatic systems, Environ. Sci.: Processes Impacts, 2015, 17, 906–914 RSC.
- L. Rieger, H. Siegrist, S. Winkler, E. Saracevic, R. Votava and J. Nadler, In situ measurement of ammonium and nitrate in the activated sludge process, Water Sci. Technol., 2002, 45, 93–100 CrossRef CAS PubMed.
- T. Le Goff, J. Braven, L. Ebdon and D. Scholefield, Automatic continuous river monitoring of nitrate using a novel ion-selective electrode, J. Environ. Monit., 2003, 5, 353–358 RSC.
- S. Capelo, F. Mira and A. M. de Bettencourt, In situ continuous monitoring of chloride, nitrate and ammonium in a temporary stream: Comparison with standard methods, Talanta, 2007, 71, 1166–1171 CrossRef CAS.
- J. V. Capella, A. Bonastre, R. Ors and M. Peris, A step forward in the in-line river monitoring of nitrate by means of a wireless sensor network, Sens. Actuators, B, 2014, 195, 396–403 CrossRef CAS.
- YSI, https://www.ysi.com/parameters/nitrate, accessed 26.07.2022.
- D. J. Yuan, A. H. C. Anthis, M. G. Afshar, N. Pankratova, M. Cuartero, G. A. Crespo and E. Bakker, All-Solid-State Potentiometric Sensors with a Multiwalled Carbon Nanotube Inner Transducing Layer for Anion Detection in Environmental Samples, Anal. Chem., 2015, 87, 8640–8645 CrossRef CAS PubMed.
- A. Mimendia, J. M. Gutiérrez, J. M. Alcañiz and M. del Valle, Discrimination of Soils and Assessment of Soil Fertility Using Information from an Ion Selective Electrodes Array and Artificial Neural Networks, Clean: Soil, Air, Water, 2014, 42, 1808–1815 CAS.
- J. Choosang, A. Numnuam, P. Thavarungkul, P. Kanatharana, T. Radu, S. Ullah and A. Radu, Simultaneous Detection of Ammonium and Nitrate in Environmental Samples Using on Ion-Selective Electrode and Comparison with Portable Colorimetric Assays, Sensors, 2018, 18, 3555 CrossRef PubMed.
- Y. Soda and E. Bakker, Ionic strength-independent potentiometric cation concentration sensing on paper using a tetrabutylammonium-based reference electrode, Sens. Actuators, B, 2021, 346, 130527 CrossRef CAS.
- W. Gao, X. Xie and E. Bakker, Direct Potentiometric Sensing of Anion Concentration (Not Activity), ACS Sens., 2020, 5, 313–318 CrossRef CAS PubMed.
- R. Athavale, C. Dinkel, B. Wehrli, E. Bakker, G. A. Crespo and A. Brand, Robust Solid-Contact Ion Selective Electrodes for High-Resolution In Situ Measurements in Fresh Water Systems, Environ. Sci. Technol. Lett., 2017, 4, 286–291 CrossRef CAS.
- N. Pankratova, M. Cuartero, T. Cherubini, G. A. Crespo and E. Bakker, In-Line Acidification for Potentiometric Sensing of Nitrite in Natural Waters, Anal. Chem., 2017, 89, 571–575 CrossRef CAS PubMed.
- M. Cuartero, N. Pankratova, T. Cherubini, G. A. Crespo, F. Massa, F. Confalonieri and E. Bakker, In Situ Detection of Species Relevant to the Carbon Cycle in Seawater with Submersible Potentiometric Probes, Environ. Sci. Technol. Lett., 2017, 4, 410–415 CrossRef CAS.
- D.-H. Jung, H.-J. Kim, J. Y. Kim, S. H. Park and W. J. Cho, Water Nitrate Remote Monitoring System with Self-Diagnostic Function for Ion-Selective Electrodes, Sensors, 2021, 21, 2703 CrossRef CAS PubMed.
- M. Cuartero, G. Crespo, T. Cherubini, N. Pankratova, F. Confalonieri, F. Massa, M.-L. Tercier-Waeber, M. Abdou, J. Schäfer and E. Bakker, In Situ Detection of Macronutrients and Chloride in Seawater by Submersible Electrochemical Sensors, Anal. Chem., 2018, 90, 4702–4710 CrossRef CAS PubMed.
- T. Forrest, E. Zdrachek and E. Bakker, Thin Layer Membrane Systems as Rapid Development Tool for Potentiometric Solid Contact Ion-selective Electrodes, Electroanalysis, 2020, 32, 799–804 CrossRef CAS.
- E. Zdrachek, T. Forrest and E. Bakker, Solid-Contact Potentiometric Cell with Symmetry, Anal. Chem., 2022, 94, 612–617 CrossRef CAS PubMed.
- P. Damala, E. Zdrachek, T. Forrest and E. Bakker, Unconditioned Symmetric Solid-Contact Electrodes for Potentiometric Sensing, Anal. Chem., 2022, 94, 11549–11556 CrossRef CAS PubMed.
- E. Zdrachek, T. Forrest and E. Bakker, Symmetric cell for improving solid-contact pH electrodes, Anal. Chim. Acta, 2023, 1239, 340652 CrossRef CAS PubMed.
- M. Guzinski, J. M. Jarvis, P. D'Orazio, A. Izadyar, B. D. Pendley and E. Lindner, Solid-Contact pH Sensor without CO2 Interference with a Superhydrophobic PEDOT-C14 as Solid Contact: The Ultimate “Water Layer” Test, Anal. Chem., 2017, 89, 8468–8475 CrossRef CAS PubMed.
- P. C. Meier, Two-parameter debye-hückel approximation for the evaluation of mean activity coefficients of 109 electrolytes, Anal. Chim. Acta, 1982, 136, 363–368 CrossRef CAS.
- E. Bakker, Determination of Unbiased Selectivity Coefficients of Neutral Carrier-Based Cation-Selective Electrodes, Anal. Chem., 1997, 69, 1061–1069 CrossRef CAS.
- T. Forrest, L. Höfler and E. Bakker, Dialysis membranes as liquid junction materials: Simplified model based on the phase boundary potential, J. Electroanal. Chem., 2022, 904, 115886 CrossRef CAS.
- A. J. Bard, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, 2 edn, 2001 Search PubMed.
- I. B. Gomes, M. Simões and L. C. Simões, Copper Surfaces in Biofilm Control, Nanomaterials (Basel), 2020, vol. 10 Search PubMed.
- V. C. Pinto, P. J. Sousa, E. M. F. Vieira, L. M. Gonçalves and G. Minas, Antibiofouling strategy for optical sensors by chlorine generation using low-cost, transparent and highly efficient electrodes based on platinum nanoparticles coated oxide, Chem. Eng. J., 2021, 404, 126479 CrossRef CAS.
- X. Wang, M. Sun, Y. Zhao, C. Wang, W. Ma, M. S. Wong and M. Elimelech, In Situ Electrochemical Generation of Reactive Chlorine Species for Efficient Ultrafiltration Membrane Self-Cleaning, Environ. Sci. Technol., 2020, 54, 6997–7007 CrossRef CAS PubMed.
- L. Qi, T. Jiang, R. Liang and W. Qin, Polymeric membrane ion-selective electrodes with anti-biofouling properties by surface modification of silver nanoparticles, Sens. Actuators, B, 2021, 328, 129014 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2em00341d |
| This journal is © The Royal Society of Chemistry 2023 |