
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal–iodine batteries: achievements, challenges, and future
Leiqian
Zhang
ab,
Hele
Guo
c,
Wei
Zong
b,
Yunpeng
Huang
 b,
Jiajia
Huang
*d,
Guanjie
He
e,
Tianxi
Liu
b,
Johan
Hofkens
cf and
Feili
Lai
*ac
b,
Jiajia
Huang
*d,
Guanjie
He
e,
Tianxi
Liu
b,
Johan
Hofkens
cf and
Feili
Lai
*ac
aState Key Laboratory of Metal Matrix Composites, School of Materials Science and Engineering, Shanghai Jiao Tong University, Shanghai, 200240, P. R. China. E-mail: feililai@sjtu.edu.cn
bKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, International Joint Research Laboratory for Nano Energy Composites, Jiangnan University, Wuxi, 214122, P. R. China
cDepartment of Chemistry, KU Leuven, Celestijnenlaan 200F, Leuven 3001, Belgium. E-mail: feili.lai@kuleuven.be
dSchool of Chemical Engineering, Zhengzhou University, Zhengzhou 450001, P. R. China. E-mail: huangjiajia@zzu.edu.cn
eChristopher Ingold Laboratory, Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
fDepartment of Molecular Spectroscopy, Max Planck Institute for Polymer Research, Ackermannweg 10, Mainz 55128, Germany
First published on 8th September 2023
Abstract
Metal–iodine batteries (MIBs) are becoming increasingly popular due to their intrinsic advantages, such as a limited number of reaction intermediates, high electrochemical reversibility, eco-friendliness, safety, and manageable cost. This review details past attempts and breakthroughs in developing iodine cathode-based (rechargeable) metal battery technology, to arrive at a comprehensive discussion and analysis of the battery's working mechanisms and fundamental challenges. Especially, the realization of available rechargeable MIBs relies heavily on the joint action of the battery components. We therefore cover here the progress starting from electrodes, electrolytes, and separator/interlayer requirements to introduce various types of MIBs and finally a critical analysis of the status quo, allowing us to gain insight into the roadblocks that still exist in MIBs. Also, we collect and compare the electrochemical performance of MIBs by category with listing their actual active material loading species and cell fabrication parameters. Finally, we conclude with recommendations for future strategies to leverage current advances in battery engineering, characteristics, and computational designs, all of which enable MIBs to reach their full potential in the energy age ultimately.
 Leiqian Zhang | Leiqian Zhang, graduated from Zhengzhou University in 2022 with a master's degree. Currently, he is pursuing his PhD degree at Jiangnan University. His research interests mainly focus on electrode and electrolyte design for iodine-based batteries. |
 Jiajia Huang | Jiajia Huang received his PhD degree in polymeric chemistry and physics from Sun Yat-Sen University in 2014. Currently, he is an associate professor at the School of Chemical Engineering, Zhengzhou University. His research focuses on functional polymers and carbon-based materials and their applications in adsorption separation of metal ions and electrochemical energy storage. |
 Feili Lai | Feili Lai received his PhD degree from the Max Planck Institute for Colloids and Interfaces/Potsdam University in 2019. His current interests include the design and synthesis of low-dimensional solids for energy storage and conversion applications, smart polygels, and theoretical calculations. |
Broader contextThe use of reversible redox reactions of chalcogens (O, S, and Se) and halogens (Cl, Br, and I) for energy storage has a long history due to their high theoretical capacity and abundance in feedstocks. Particularly, iodine has low toxicity, high reactivity, and few intermediates, leading to generally better electrochemical properties (e.g., rate and cycling stabilities) than other elements. In addition, iodine also permits the creation of metal–iodine batteries (MIBs) by flexible pairing with different types of metal anodes, which readily caters to diverse forms of the electrochemical energy storage market. However, a number of issues, including iodine species dissolution, sluggish kinetics, anode corrosion/passivation, and dendrite growth, often lead to rapid capacity degradation, poor Coulombic efficiency, and even short-circuiting, greatly impeding the further practical application of MIBs. This review details past attempts and breakthroughs in developing iodine cathode-based (rechargeable) metal battery technology, while also presenting key innovations, deficiencies, and possible solutions for batteries. It is hoped that through this review article, further interest in MIBs will emerge, and efforts to make them useful for energy storage in the future will be triggered. |
1 Introduction
The heightened global attention toward the energy crisis and global warming has triggered urgent investigations into carbon-free energy sources, such as wind and solar energy.1,2 Especially, the recent surge in gas prices caused by Russia–Ukraine war appears to have accelerated the decarbonization process of the entire energy system from fossil-fuel-based power generation.3 It is anticipated that, by 2050, nearly 27% of global energy consumption will come from renewable sources.4 However, carbon-free energy sources usually have the characteristics of variability, uncertainty, and location-specificity. To make carbon-free energy compatible with our daily lives, efficient, flexible, and readily available electrical energy storage (EES) systems are essential.5,6 The development of lithium-ion battery (LIB) technology has revolutionized the way people live and brought energy into the era of portability. Despite these advancements, with the advent of electric vehicles and an increasing demand for large-scale energy storage solutions, the prices of raw materials for LIBs are skyrocketing. For instance, as of February 2023, the price of battery-grade lithium carbonate increased by over 14 times, from around $5816 ton−1 to approximately $88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 ton−1 in China (https://www.ccmn.cn). As a result, there is a significant research effort underway to develop new types of rechargeable battery systems as an adjunct or substitute to LIBs.
800 ton−1 in China (https://www.ccmn.cn). As a result, there is a significant research effort underway to develop new types of rechargeable battery systems as an adjunct or substitute to LIBs.
Typically, the conversion reactions of chalcogens (O, S, and Se) and halogens (Cl, Br, and I) have received significant attention because of their high theoretical storage capacity and abundance of raw materials (Table 1).7–9 Through flexible matching with different types of metal anodes (e.g., Li, Na, K, Mg, Al, Zn, and Fe), the as-prepared metal–element batteries can be well adapted for the speedy development of the electrochemical energy storage market.9 Among them, metal–oxygen and metal–sulfur batteries are regarded as the most promising battery systems due to their outstanding advantages in energy density and price. However, the inherent instability and complexity of their intermediate products, including but not limited to polysulfide and peroxide/superoxide, pose great challenges for their applications.10,11 On the other hand, the use of halogens for energy storage can be traced back to 1884.12,13 Charles Renard employed a 435 kg zinc-chlorine battery to power the 52 m-long airship La France for flying about 8 km. Despite this, the high toxicity, high corrosiveness, and volatility of chlorine make chlorine-based solutions fundamentally unsuitable for large-scale energy storage.14,15 Like chlorine, bromine also has been extensively studied for over a century.14,15 Although it has low reactivity compared to chlorine, its inherent liquid nature, severe corrosiveness, and high toxicity also make it unusable in portable configurations.16–18
| O | S | Se | Cl | Br | I | |
|---|---|---|---|---|---|---|
| Price ($ Kg−1) | Available from the air | 0.15 | 31.3 | 0.04 (NaCl) | 2.5 | 41 |
| Abundance (wt%, in the Earth's crust) | 47 | 0.047 | 5 × 10−6 | 0.017; 19 g L−1 (ocean) | 2.1 × 10−4; 65 mg L−1 (ocean) | 4 × 10−5; 55 μg L−1 (ocean) |
| Specific capacity (mA h g−1) | 3860 (Li–O2); 820 (Zn–O2) | 1672 (S2−/S0) | 675 (Se2−/Se0) | 755 (Cl−/Cl0) | 335.5 (Br−/Br0) | 211 (I−/I0); 422 (I−/I+) |
| Potential (V vs. SHE) | 2.96 (Li–O2); 1.65 (Zn–O2) | −0.57 (S2−/S8) | −0.67 (Se2−/Se0) | 1.36 (Cl−/Cl0) | 1.08 (Br−/Br0) | 0.62 (I−/I0); 1.07 (I0/I+) |
| Safety | High | High | High | Low | Moderate | High (essential element) |
| Cycle life | Low | Moderate | Moderate | Moderate | Moderate | High (up to 50000 cycles) |
| Rate capacity | Low | Low | Moderate | Low | Moderate | High (up to 100C) |
Significantly, metal–iodine batteries (MIBs) are gaining momentum in a wide variety of battery systems, including Li–I2, Na–I2, K–I2, Zn–I2, Mg–I2, Al–I2, and Fe–I2 batteries.19–25 This popularity can be attributed to the distinctive properties of iodine: (1) as an essential trace element in the human body, iodine is significantly safer, more stable, and more eco-friendly compared to chlorine and bromine.12 Intriguingly, in late 1972, Li–I2 primary cells were widely applied as a power source in cardiac pacemakers;26 (2) iodine has high abundance in oceans (50–60 μgiodine Locean−1) and relatively low cost (∼$41 kg−1), allowing its large-scale application;12,27 (3) iodine possesses a high theoretical volumetric capacity (1040 mA h cm−3), gravimetric capacity (211 mA h g−1), and high redox potential (0.62 V vs. standard hydrogen electrode (SHE)), indicating a possible high energy density when it is coupled with an anode; and (4) although iodine has low solubility in water (∼0.29 giodine kgwater−1), it can spontaneously bond with iodide ions to form highly soluble triiodide, thereby significantly increasing its solubility (up to 8–12 M) and reaction kinetics.28 Given these interesting properties, MIBs are poised to be a promising alternative to LIBs. It is high time for a systematic and up-to-date review of metal–iodine batteries.
In this review article, we first offer a historical perspective on MIBs to enable the reader to easily understand their development. Next, the electrochemistry principles, and the fundamental issues inherent in MIBs are discussed and analyzed in depth. Therefore, in Section 2, we further present a comprehensive overview of MIBs from the perspectives of iodine host, electrolyte, interlayer, separator, and anode, with a focus on how they achieve advanced electrochemical performance during battery operation. Also, advanced computational methods and in situ characterization techniques used in MIBs are summarized in Section 3. On this basis, Section 4 summarizes the key innovations and design guidelines of these strategies, and also suggests the corresponding deficiencies and possible solutions. Besides, the placement of various MIBs is discussed and analyzed in combination with the actual situation, while the future development and application of MIBs are also put forward. We hope that through this review article, more interest in metal–iodine batteries will be generated, and efforts to make them useful in energy storage in the future will be sparked.
1.1 Historical perspective (–2023): a myriad of concepts
Iodine (Greek: ioeides, violet) was accidentally discovered by Bernard Courtois in 1811 and named by Humphrey Davy in 1813.45 For a long time thereafter, iodine was mainly used to treat and prevent goiter. It was not until 1900 that Crotogino began to study the electrochemical properties of iodine.46 And in 1968, the first primary solid-state lithium–iodine (Li–I2) cell was conceived by workers at the Catalyst Research Corporation (Fig. 1).47 Then, in late 1972, it was introduced into commercial medical devices as a power source for cardiac pacemakers. Commonly, such a cell is composed of a lithium anode, lithium iodide, and a poly(2-vinylpyridine)–iodine (P2VP–I2) cathode. Among them, lithium iodide, as a solid-state electrolyte, is formed in situ by the direct contact between the lithium anode and P2VP–I2, while increasing in thickness with battery discharge. However, its poor lithium-ion conductivity (ca. 10−7 S cm−1 at room temperature), and increased thickness lead to poor rate capability of the cell (60 μA cm−2 at room temperature).47,48 Therefore, intensive efforts have been devoted to improving the discharge rate and the low-temperature performance of lithium–iodine cells by designing advanced active cathodes or increasing the anode surface area.48–51 Weinstein et al. first surveyed the electrochemical solid-state rechargeable Li–I2 cells with the polyvinylpyrrolidone (PVP)–I2 complex as the cathode in 2008.52 Especially, with the rise of hybrid electric vehicles and modern mobile life, a novel all solid-state rechargeable Li–I2 thin film battery was first proposed by Liu and co-workers in 2011.48 In this system, the Li–I2 battery is fabricated with a solid I2-LiI(3-hydroxypropionitrile)2 cathode and a Li anode, in which the rechargeability is made possible through the ionic conductivity of I− anions. Despite its limited current density (about 20 μA cm−2), this battery sparked considerable interest in rechargeable Li–I2 batteries. In the years that followed, Li–I2 battery technology continued to achieve breakthroughs. As of 2022, state-of-the-art Li–I2 batteries can endure more than 10000 cycles at 50C (1C = 211 mA g−1) with an iodine loading of 80 wt%.53 However, the development of other alkali metal batteries (Na–I2 and K–I2) has been slow in comparison with Li–I2 batteries. Until 2015, Ingersoll et al. showed a prototype of a high-temperature sodium–iodine battery based on NaSICON ceramics, and an inorganic NaI/AlCl3-based catholyte.54 Furthermore, Gong et al. also proposed a room-temperature Na–I2 battery with iodine quantum dots decorated reduced graphene oxide as the free-standing cathode in 2016.55 On the other hand, due to the higher activity and larger atomic radius of K metal, the K–I2 battery was not realized until 2018.56
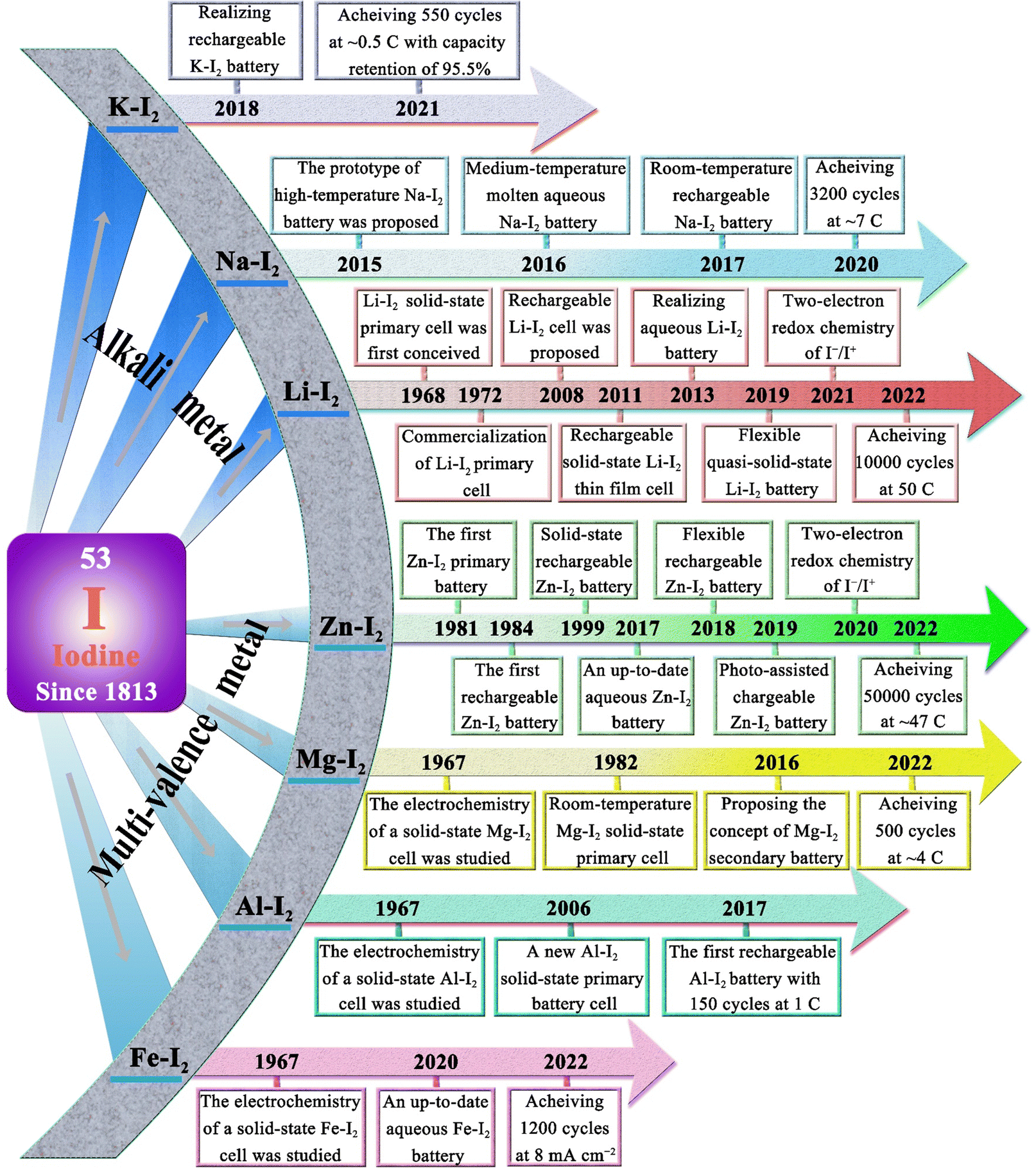 | ||
| Fig. 1 Development timeline of various MIBs.18,19,21,24,25,28,42,48,52–59,61,62,68–78 | ||
In terms of multivalent MIBs, the Zn–I2 battery enjoys the most extensive research due to the high operability and suitable redox potential of Zn metal. In 1981, the pioneers Yamamoto et al. built the first primary Zn–I2 cell by employing polymer–iodine adducts as the cathode and Zn metal as the anode.57 Then, they further succeeded in fabricating a Zn–I2 secondary battery in 1984 by introducing an additional ion-exchange membrane and carbon powder conductor into the battery structure.58 Despite this, the assembled battery undergoes only approximately 300 cycles due to the evaporation of aqueous electrolytes and partial degradation or corrosion of positive and negative electrodes. It is notable that the progress of Zn–I2 battery research slowed down after the emergence of LIB technology. In 2017, with the development of Zn–I2 flow batteries, Li et al. reported an up-to-date Zn–I2 battery with iodine/microporous carbon as the cathode and ZnSO4 as the electrolyte, marking the beginning Zn–I2 battery research.59 By January 2023, more than 100 articles on Zn–I2 batteries have been published (data from Web of Science). Furthermore, the rise of Zn–I2 batteries has brought attention to Fe–I2 batteries because of their high reversibility, abundant resources, low costs, and non-toxicity of iron anodes.19,60 In 2020, Bai and colleagues reported the first rechargeable Fe–I2 battery that showed an excellent cycle stability of 100% after 550 cycles at 2000 mA g−1.19 Besides, Wu et al. recently further improved the lifecycle of Fe–I2 batteries to 1200 cycles using a polyaniline cathode and a mixed electrolyte (5 M FeCl2 and 1 M ZnI2), which confirmed the promising potential of Fe–I2 batteries as a low-cost, eco-friendly, and safe large-scale EES system.61 Apart from Zn/Fe–I2 batteries, the Mg–I2 battery was also studied in 1967 by Gutmann and co-workers.62 As compared to traditional intercalation cathodes in Mg-ion batteries, the conversion-type iodine cathode could well bypass the sluggish cation dissociation and solid-state ion diffusion process caused by high charge densities of Mg2+.63,64 Nevertheless, the high susceptibility of Mg to contaminants (e.g., water, oxygen, carbon dioxide, protic residues, and polysulfide) results in a strong tendency to form a compact passivating surface film, which greatly blocks the electrochemical reaction on the Mg anode.65 Consequently, the development of Mg–I2 batteries lag far behind that of Zn–I2 batteries due to practical limitations. As the discovery of new electrolytes always plays a key role in the advancement of battery technology, Li et al. first presented the concept of a Mg–I2 secondary battery using ionic liquid as the electrolyte in 2016.21 However, the long-term cycling performance data for this system was not initially available. Fortunately, a rechargeable Mg–I2 battery was then implemented by Tian et al. in 2017, which exhibited a remarkable capacity retention of 94.6% after 120 cycles at 0.5C, demonstrating the feasibility of Mg–I2 secondary batteries.66 Similar to the Mg–I2 battery, the use of a conventional aluminum anode in an Al–I2 battery is problematic due to the formation of ionic and electrically insulating passivation layers.67 While research on Al–I2 batteries can be traced back to 1967,62 the rechargeable Al–I2 battery was developed by Tian et al. in 2017.25
1.2 Working principle
Commonly, MIBs are a type of conversion battery system, in which the storage of electric energy is based on the reversible redox reaction between a metal anode and an iodine cathode. Most MIBs operate in a neutral or mild-acidic electrolyte, as iodine undergoes spontaneous disproportionation in an alkaline medium.79 Therefore, an ideal redox event in the anodic region of MIBs can be represented using the following equation:| M ↔ Mn+ + ne− | (1) |
The iodine cathode, on the other hand, involves various reaction processes that can be classified into the following five categories. (1) Intermittent two-step conversion reaction (I− ↔ I3−, I3− ↔ I2). This process is common in MIBs that utilize organic electrolytes, such as lithium bis(trifluoromethane sulfonyl) imide (LiTFSI) in 1,3-dioxolane/tetra-ethylene glycol dimethyl ether or 1,3-dioxolane/1,2-dimethoxyethane.79,80 Some special gel electrolytes (e.g., poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide)) in aqueous Zn–I2 cells also lead to the formation of the above mechanism.81,82 As shown in Fig. 2a, its typical cyclic voltammetry curve contains two pairs of redox peaks, for which the electrochemical reaction is essentially the reversible redox reaction of I− ↔ I3− and I3− ↔ I2. Interestingly, Li et al. recently suggested that the peaks situated at high potential relate to the new chemistry of the I0 ↔ I+ pair instead of I3− ↔ I2, when using 0.1 M LiCl as an additive in the electrolyte of Li–I2 cells.18 This controversial view makes it necessary for future work to ascertain the transformation mechanism of iodine in organic electrolytes in detail. (2) One-step incomplete iodine redox process (I− ↔ I3−). This process is typically observed in MIBs with aqueous electrolytes (Fig. 2b). It is worth noting that when there is an excess production of I2 at high charging states, I3− can be converted into higher-order polyiodide ions (I5−, I7−, etc.). This is due to the continuous combination of I2 and polyiodide during cycling, which results in the formation of Ix− in an aqueous solution. Significantly, the distinction between type (1) and type (2) mechanisms may be due to the solubility disparity of iodine species in different electrolytes (aqueous electrolytes: I− (high), I3− (high), and I2 (low); tetraglyme electrolytes: I− (low), I3− (high), and I2 (high)), which would affect the formation and stabilization of iodine intermediates during redox processes.66 Despite this, in-depth studies are urgently needed to reveal the nature of the electrolyte affecting the reaction mechanism of MIBs. (3) One-step one-electron conversion process (I− ↔ I0). This mechanism is currently only found in aqueous Zn–I2 batteries (Fig. 2c). Possible explanations for this reaction mechanism can be attributed to the following reasons: (1) the rapid electron supply of iodine host materials (e.g., Nb2CTX flakes) enables a forceful one-step conversion between the I− ion and I2 element;83 (2) the appropriately sized pores (e.g., carbon cloth with a typical micropore size of around 6.3 Å) of iodine host material make it difficult for the distorted octahedral configuration of Zn(I3)2 (minimum diameter of 9.5 Å) to reside inside the material's channel;72,84 (3) specific electrolytes (e.g., high concentrations of ZnCl2 and eutectic electrolytes) can hinder the formation of I3− by restricting the amount of free H2O, leading to difficulties in the binding between I− ions and I2.40,85,86 It is remarkable that, due to the absence of polyiodide, this mechanism is more conducive to the cycling stability of MIBs. (4) Two-step two-electron conversion process (I− ↔ I0, I0 ↔ I+). The corresponding cyclic voltammetry curve in Fig. 2d displays two processes of I− ↔ I0 and I0 ↔ I+. In particular, the energy density of MIBs is significantly increased by the high redox potential (up to 1.07 V vs. SHE) of the I0 ↔ I+ couple.40,74 Nevertheless, this process requires to be implemented in a very harsh environment, since I+ is very unstable in aqueous electrolytes and could be further oxidized to form irreversible IO3−.40,87 Remarkably, the addition of nucleophilic species (e.g., halide, cyanide, and amines) into the electrolyte can stabilize I+ ions through the formation of charge-transfer complexes.40 Besides, electrolyte activity towards I+ ions is also an essential factor that impacts the conversion process's reversibility. Therefore, Zou et al. chose acetonitrile which is inert to I+, as a diluent to promote the ion transport and tune the activity of H2O in the electrolyte solution, thereby realizing 6000 cycles in a Zn–I2 battery based on two-electron conversion.40 Despite these strict conditions, this high voltage redox chemistry potentially represents a new avenue into MIBs. (5) Asymmetric iodine conversion process (oxidation: I− ↔ I3−, I3− ↔ I2; reduction: I2 ↔ I−). This process has only been observed in limited instances, specifically in the Zn–I2 cell using a high-conductivity iota-carrageenan gel electrolyte (Fig. 2e).88 The factors contributing to its occurrence are still unknown and necessitate further research.
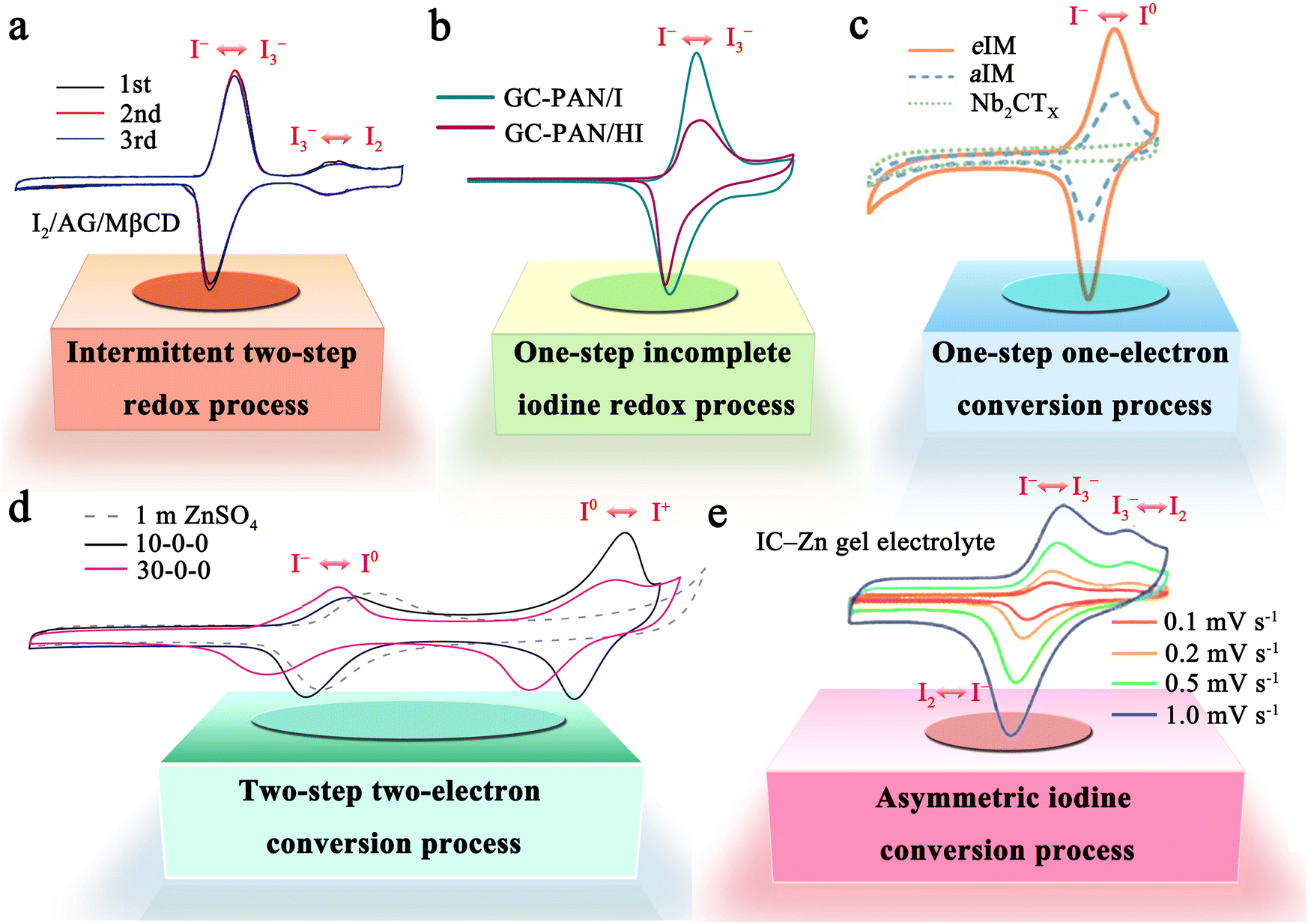 | ||
| Fig. 2 Five representative reaction mechanisms of iodine cathodes in MIBs. (a) An intermittent two-step redox process (I− ↔ I3−, I3− ↔ I2). Reprinted with permission from ref. 89. Copyright 2020 Elsevier. (b) One-step incomplete iodine redox process (I− ↔ I3−). Reprinted with permission from ref. 27. Copyright 2022 Wiley. (c) One-step one-electron conversion process (I− ↔ I0). Reprinted with permission from ref. 83. Copyright 2021 Wiley. (d) Two-step two-electron conversion process (I− ↔ I0, I0 ↔ I+). Reprinted with permission from ref. 40. Copyright 2021 Springer Nature. (e) Asymmetric iodine conversion process (oxidation: I− ↔ I3−, I3− ↔ I2; reduction: I2 ↔ I−). Reprinted with permission from ref. 88. Copyright 2023 Royal Society of Chemistry. | ||
1.3 Fundamental issues
Throughout the development history and internal reaction mechanisms of MIBs described above, several fundamental issues have greatly hindered the widespread success of rechargeable MIBs. The foremost problem is the shuttle effect of iodine species, i.e., iodine and polyiodide, formed during the charge/discharge cycling process. These species dissolve uncontrollably into the electrolyte and subsequently diffuse towards the anode side. The outcomes include severe capacity degradation, rapid self-discharge, and low Coulombic efficiency of the battery (Fig. 3a).90 To address this issue, it is noteworthy that the iodine host materials in MIBs should strongly interact with iodine species, leading to great enhancement in the thermal stability of iodine species in composites.53 Thus, the thermal instability of iodine species (iodine is volatile even at room temperature) may be considered as a less important issue in MIBs. Another critical issue for MIBs is the insulating property of iodine that inevitably results in poor redox kinetics, low energy/iodine utilization, and high polarization (Fig. 3b).83,91 In addition, the long-term cycle performance of the whole battery also depends heavily on the metal anode. Initially, the anode has minimal impact on battery performance, but as non-uniform plating/stripping of metal anodes (except Mg) causes dendrite growth, there is a risk of a short circuit, creating a severe safety hazard for the battery system (Fig. 3c). Besides, the formation of dendrites can result in “dead” metal, that is, dendrites fall off into the electrolyte, making it unsuitable for reuse, which commonly leads to rapid deterioration of battery performance. Meanwhile, the side reactions occurring between the metal anodes and electrolytes also frequently cause the corrosion and passivation of metal anodes; this not only consumes fresh metals and limited electrolytes, but also increases the impedance with severely restricted subsequent reaction of anodes, resulting in poor plating/stripping CE (Fig. 3d).92 Significantly, corrosion/passivation and dendrite growth interact and promote each other, accelerating the end of cell life.93 Extensive efforts that surround the above fundamental question have been carried out on various metal–iodine systems. Especially, the design of the iodine host, anode, separator, and electrolyte commonly affects kinetics (power), lifetime, and safety, while also tuning the limits of cell voltage and energy density. To account for these, we sort out and analyze MIBs from the above aspects based on available research results in the following sections systematically and comprehensively.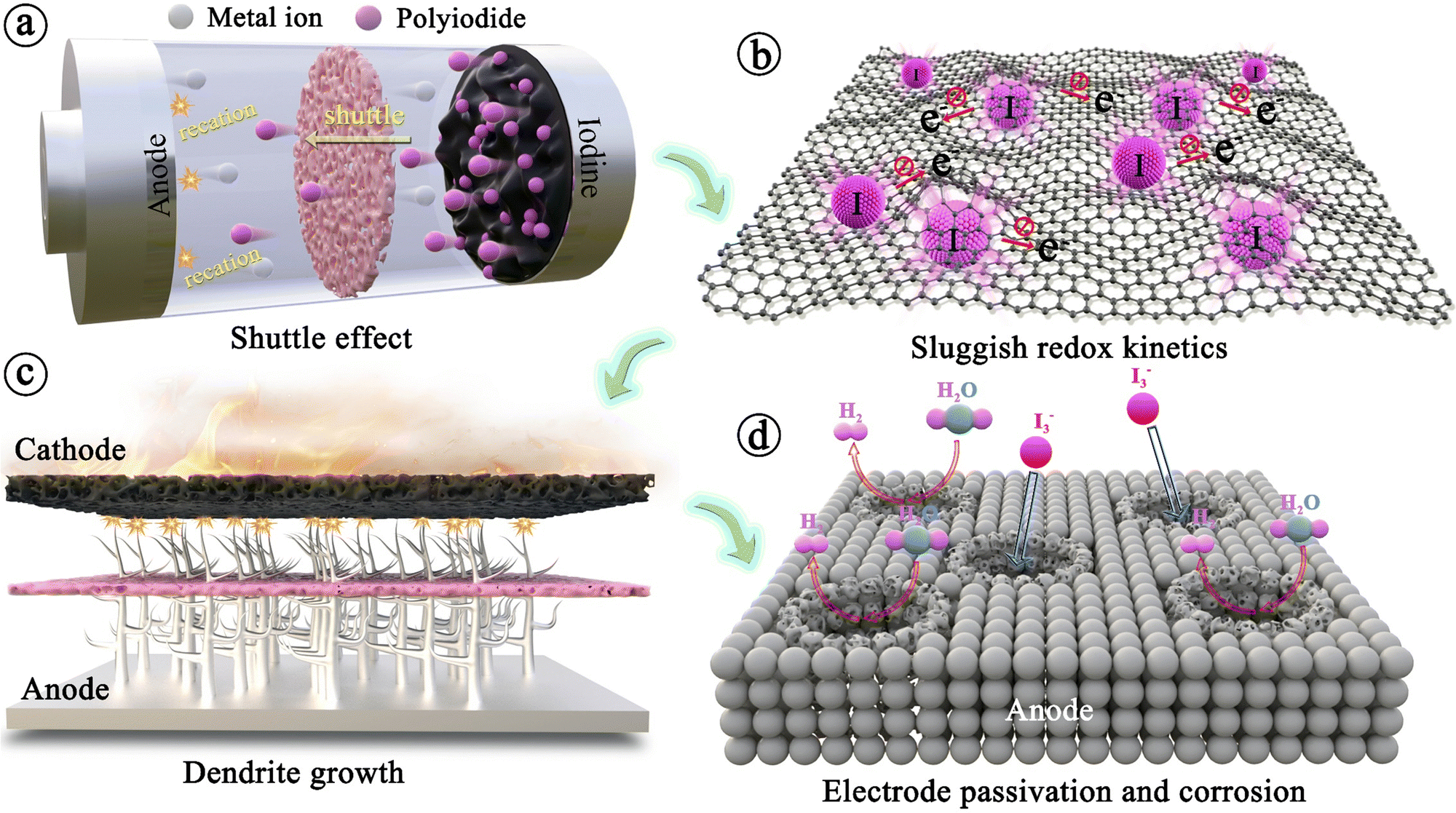 | ||
| Fig. 3 Four fundamental issues faced by metal–iodine batteries. (a) The shuttle effect of iodine species. (b) Intrinsic insulation of iodine. (c) Dendrite growth on anodes. (d) Passivation and corrosion of metal anodes. | ||
2 Metal–iodine batteries
2.1 Lithium–iodine battery
Tables 2–17 summarizes the electrochemical performance, fabrication parameters, and cell-testing conditions for metal–iodine batteries (MIBs) using different strategies, from 2011 to 2023. Among various MIBs, Li–I2 batteries are at the forefront of development. This is closely related to the keen interest in the research of lithium-based batteries (e.g., Li–O2 and Li–S batteries). Undoubtedly, it is also inseparable from the inherent benefits of Li–I2 batteries. One advantage of Li–I2 cells lies in their iodine/iodide redox chemistry that results in an open-circuit voltage of ∼3.5 V for Li/Li+. Additionally, the limited number of intermediates and rapid iodine conversion reaction enable the state-of-the-art Li–I2 battery to achieve over 10000 cycles with a high-power density of 29 kW kg−1. Nonetheless, the aforementioned fundamental problems in Fig. 3 still plague the development of Li–I2 batteries. Through a combination of tabularized numbers and the detailed discussion below, we hope to make it easier for readers to understand the development and progress of Li–I2 batteries. In particular, the specific capacities of batteries in this article are calculated based on the mass of iodine, unless otherwise stated.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|
| 1, year; 2, cathode; 3, electrolyte formula; 4, the mass loading of iodine at the cathode (wt%); 5, the area loading of iodine at the cathode (mg cm−2). Evaluation of Li–I2 cell performance: 6, current density (C, 1C = 211 mA g−1); 7, cycle number; 8, capacity retention (%); 9, reversible capacity (mA h g−1iodine). 10, reference. PVP, polyvinyl pyrrolidone; CNT, carbon nanotube; AC, activated carbon; LiTFSI, LiN(CF3SO2)2; EC, ethylene carbonate; DOL, 1,3-dioxolane; DME, dimethoxyethane; DMC, dimethyl carbonate; DMA, dimethylacetamide; EMC, ethyl methyl carbonate; TEGDME, tetraethylene glycol dimethyl ether. | |||||||||
| 2011 | I2/conductive carbon black | 1 M LiPF6 in EC/EMC/DMC (1:1:1, v/v/v) |
24 | ∼1.2 | ∼24 | 1000 | 71.4 | 125 | 94 |
| 2015 | I2/nanoporous carbon cloth | 1 M LiTFSI/1 wt% LiNO3 in DOL/DME (1:1, v/v) |
22 | 5.6 | 0.5 | 300 | ∼64.8 | 195 | 95 |
| 2017 | TiC0.7N0.3/liquid I2 | 1 M LiNO3/0.04 M I2/0.02 M LiI in DMA | — | 1 | 10 mA cm−2 | 7000 | — | 100 | 111 |
| 2017 | I2/N-doped carbon hollow folded hemisphere | 1 M LiTFSI/1 wt% LiNO3 in DOL/DME (1:1, v/v) |
32 | — | ∼5 | 300 | 84 | — | 102 |
| 2017 | I2/3D bio-foam carbon | 1 M LiTFSI/1 wt% LiNO3 in DOL/DME (1:1, v/v) |
30–40 | ∼1–1.5 | 10/4 | 500 | 94 | — | 96 |
| 2017 | I2/active graphene | 1 M LiTFSI/0.2 M LiNO3 in DOL/TEGDME (1:1, v/v) |
56 | 1–1.4 | 1 | 500 | 73.8 | 161 | 79 |
| 2017 | I2/N,P co-doped porous graphitic carbon | 1 M LiTFSI/1 wt% LiNO3 in DOL/DME (1:1, v/v) |
— | 2 | ∼2.4 | 2000 | 84.5 | ∼250 | 101 |
| 2018 | PVP–I/N-doped hollow carbon sphere–I2 | 1 M LiTFSI/1 wt% LiNO3 in DOL/DME (1:1, v/v) |
∼35 | — | 4 | 400 | 86 | — | 103 |
| 2018 | I2/β-cyclodextrin | 1 M LiTFSI/1 wt% LiNO3 in DOL/DME (1:1, v/v) |
15 | ∼0.9 | 0.1 | 300 | ∼93.7 | 164 | 80 |
| 2018 | LiI/meso–micro porous carbon polyhedrons | 4.5 M LiTFSI/0.1 M LiNO3 in DOL/DME (1:1, v/v) |
30 (LiI) | ∼1–1.2 | 2 | 800 | ∼83.2 | 200.1 | 107 |
| 2018 | Activated carbon cloth/PVP–I | 1 M LiTFSI/1 wt% LiNO3 in DOL/DME (1:1, v/v) |
∼10–11 | 1.6–1.8 | 8 | 2400 | ∼81 | 240 | 98 |
| 2018 | PVP–I | 1 M LiTFSI/1 wt% LiNO3 in DOL/DME (1:1, v/v) |
13 | 2 | 2 | 1100 | 79.1 | 190.6 | 109 |
| 2019 | LiI/N,P co-doped carbon cloth | 1 M LiTFSI/1 wt% LiNO3 in DOL/DME (1:1, v/v) |
— | ∼0.2 (LiI) | 10 | 2000 | 95.8 | 200.2 | 97 |
| 2020 | I2/active graphene/methyl-beta-cyclodextrin | 1 M LiTFSI/0.2 M LiNO3 in DOL/TEGDME (1:1, v/v) |
51 | ∼0.5–0.8 | 1 | 500 | 87.3 | ∼194.4 | 89 |
| 2020 | I2/active graphene/PVP | 1 M LiTFSI/0.2 M LiNO3 in DOL/TEGDME (1:1, v/v) |
49 | ∼0.5–0.8 | 1 | 500 | 90.1 | ∼200.1 | 89 |
| 2020 | I2/active graphene/amylose corn starch | 1 M LiTFSI/0.2 M LiNO3 in DOL/TEGDME (1:1, v/v) |
50 | ∼0.5–0.8 | 1 | 500 | 79.7 | ∼178.6 | 89 |
| 2021 | Graphene oxide/CNT/few-layer iodine nanosheets | 1 M LiTFSI/1 wt% LiNO3 in DOL/DME (1:1, v/v) |
41 | 1.0–1.5 | ∼9.5 | 1000 | — | 93.2 | 108 |
| 2021 | N-containing porous carbon/carbon fiber cloth/PVP–I | 0.5 M LiTFSI/0.5 M LiNO3 in DOL/DME (1:1, v/v) |
8 | — | 1 | 3000 | 79.7 | ∼119.6 | 264 |
| 2021 | LiI/active graphene | 1 M LiTFSI/0.2 M LiNO3 in DOL/TEGDME (1:1, v/v) |
40 (LiI) | ∼0.4–0.6 | ∼0.95 | 1000 | 93.7 | 232.0 | 265 |
| 2022 | Methylamine hydroiodide | 1 M LiTFSI in DOL/DME (1:1, v/v) |
80 | — | 5 | 10000 |
98.3 | 207.6 | 53 |
| 2022 | I2/MOF derived carbon/carbon cloth | 1 M LiTFSI/1 wt% LiNO3 in DOL/DME (1:1, v/v) |
∼44 | 1–2 | 10 | 3500 | 91 | ∼257.5 | 99 |
| 2022 | I2@AC/N719-dye/TiO2 | 1 M LiTFSI/3 wt% LiNO3 in DOL/TEGDME | ∼40 | 1.0–1.6 | ∼0.47 | 20 | — | — | 100 |
| 2022 | I2/hydrogen bonded organic framework@Ti3C2Tx | 1 M LiTFSI/1 wt% LiNO3 in DOL/DME (1:1, v/v) |
44.3 | ∼0.93 | 0.2 | 300 | ∼79.1 | ∼260 | 146 |
2.1.1.1 Carbon-based iodine host. Generally, it is not feasible to directly employ iodine as a cathode due to the high solubility of iodine in organic electrolytes and inherent insulating properties. Therefore, it is crucially important to have an effective host that can effectively store iodine and promote its utilization for high-performance MIBs. Table 2 presents critical data pertaining to hosts for Li–I2 batteries, where carbon-based materials have been used extensively as hosts for iodine and are considered highly advantageous. This can be attributed to their three prominent characteristics: (1) the abundant pores and high specific surface area of carbon-based hosts can provide strong adsorption, decreasing the solubility of active iodine species in organic electrolytes and thereby improving the stability of batteries; (2) the capacitive properties of carbon-based hosts can provide additional capacity and increase the energy density of rechargeable Li–I2 batteries; and (3) carbon-based materials normally have high conductivity and excellent electronic transfer ability, thus leading to improved utilization of iodine species despite their inherent insulating properties. Given these, Wang et al. first employed conductive carbon black borrowed from Li–S batteries as an iodine host, which exhibited a lifetime of up to 1000 cycles and a capacity retention of 71.4% at a current density of ∼24C (Fig. 4a).94 Subsequently, carbon cloth, 3D bio-foam carbon, and active graphene were also investigated as iodine hosts.79,95,96 Since free-standing carbon cloth is commercially available, has moderate iodine species binding, and can compound well with other active materials as a carrier, it is currently often used as a current collector in MIBs to provide dual iodine fixation.97–99 On the other hand, Li–I2 batteries exhibit a light-assisted charging behavior after combining the N719-dye@TiO2 photo-electrode with the iodine@activated carbon cathode (Fig. 4b).100 Despite this, the performance of these carbon-based hosts is less than satisfactory (≤1000 cycles), as their inertness leads to low interaction with iodine species.
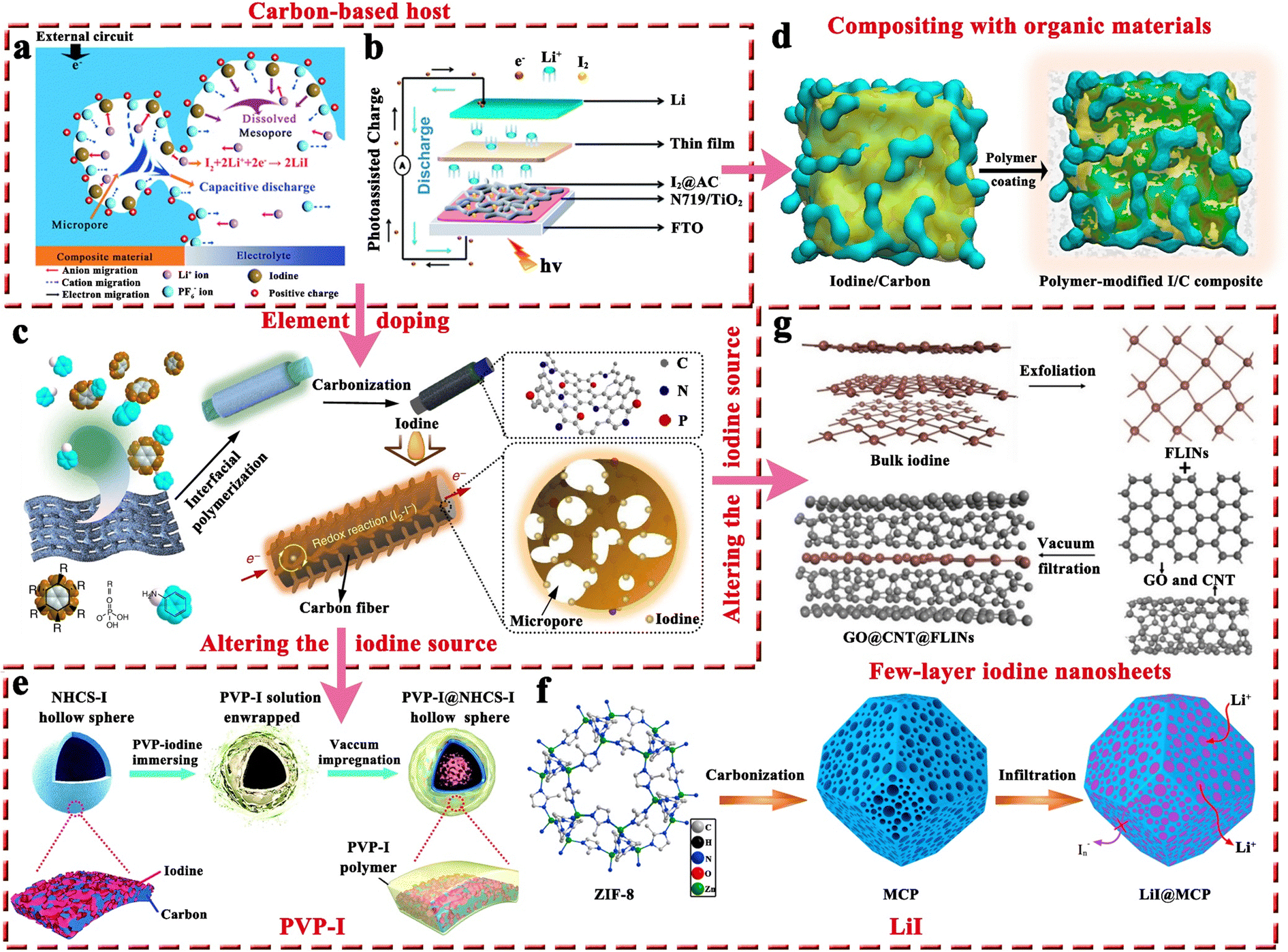 | ||
| Fig. 4 (a) Schematic illustration of the discharge mechanism of iodine in the iodine@conductive carbon black composite in Li–I2 batteries. Reprinted with permission from ref. 94. Copyright 2011 Royal Society of Chemistry. (b) Schematic diagram of the structure of a photo-assisted rechargeable Li–I2 battery. Reprinted with permission from ref. 100. Copyright 2022 Royal Society of Chemistry. (c) Schematic diagram of the preparation process of HPMC-NP@iodine composite. Reprinted with permission from ref. 101. Copyright 2017 Springer Nature. (d) Schematic description of the fabrication process for the polymer-modified I2@carbon composite. Reprinted with permission from ref. 89. Copyright 2020 Elsevier. (e) Schematic of the “top-down” synthesis process of N-doped carbon sphere@PVP–I composite. Reprinted with permission from ref. 103. Copyright 2018 Royal Society of Chemistry. (f) Schematic of the synthesis process of LiI@meso–micro porous carbon polyhedrons composite. Reprinted with permission from ref. 107. Copyright 2017 Elsevier. (g) Schematic illustration of the fabrication process of graphene oxide@carbon nanotubes@FLINs film. Reprinted with permission from ref. 108. Copyright 2021 Wiley. | ||
In this regard, several approaches have been developed to enhance the performance of carbon-based hosts. One basic approach is elemental doping, which involves incorporating polar heteroatoms like nitrogen and phosphorus atoms into carbon materials. Theoretically, these heteroatoms with abundant lone pair electrons would change the inertness of carbon materials, thereby effectively enhancing the interaction between the carbon support and iodine species with rapid charge transfer. As reported by Lu's group, Li–I2 batteries fabricated from nitrogen and phosphorus co-doped hierarchically porous graphitic carbon (HPMC-NP) hosts could achieve a high capacity retention of 84.5% after 2000 cycles at a current density of 500 mA g−1 (Fig. 4c).101 Furthermore, given the carbon cloth as the current collector, Lai's group prepared a free-standing oxygen-doped porous carbon-coated carbon cloth@iodine host by in situ carbonization of the metal organic framework (MOF) precursor. As a result, the as-prepared Li–I2 batteries could maintain an ultrahigh capacity retention of 91% after 3500 cycles at 10C, which well proves that heteroatomic doping is an effective method to improve the performance of Li–I2 batteries. However, hollow carbon spheres do not show a significant improvement in cell cycle stability performance (≤500 cycles) after heteroatom doping.102,103 A plausible explanation is that iodine or polyiodide tends to detach from the hollow carbon surface during the cycling process, leading to a rapid loss of active species.104 Therefore, it is recommended to refrain from utilizing carbon materials with hollow structures when hosting iodine. Nonetheless, if unavoidable, such materials' cavities should not exceed the size of the iodide, allowing for better confinement of the active substance within the hollow structure.105,106
In addition to the aforementioned solution, organic materials capable of chemisorption to capture iodine have also been reported to enhance the electrochemical performance of carbon-based hosts (Fig. 4d).89 Commonly, porous carbons are highly conductive, but their weak physical adsorption abilities can poorly restrict iodine species, particularly on cathodes with a high iodine loading amount.89 On the other hand, rich polar functional groups in organic materials can lead to strong chemisorption to trap dissolved iodine species, but generally, they have poor electrical conductivity. Given all these facts, organic material-coupled porous carbon is a promising iodine host for the development of advanced Li–I2 batteries. Zhang et al. employed three water-soluble nonionic polymers, including methyl-beta-cyclodextrin (MβCD), polyvinylpyrrolidone (PVP), and amylose corn starch (ACS), as the functional adsorption coatings of carbon-based hosts (active graphene).89 As a result, Li–I2 batteries using these polymer-modified iodine/carbon composites as cathodes showed enhanced electrochemical performance in terms of cycling stability and Coulombic efficiency (CE). However, the organic–inorganic composites prepared in the related follow-up studies did not show dramatic breakthroughs in battery electrochemical performance due to the lack of further systematic studies on the polymer properties. Encouragingly, with the introduction of quaternary ammonium groups into the polymer structure, the polymer-based Zn–I2 cell has achieved over 17000 cycles with 100% capacity retention due to the strong electrostatic interaction between the quaternary ammonium groups and the iodine species.27 To this end, it is desirable to combine rationally designed polymers and carbon materials for realizing high-capacity, high-rate, and high-stability Li–I2 batteries.
Altering the form in which iodine is present in carbon-based entities is another path to improvement. In this context, polyvinylpyrrolidone complexed iodine (PVP–I) (Fig. 4e) and inorganic LiI (Fig. 4f) are frequently used as iodine sources, while 2D few-layer iodine nanosheets (Fig. 4g) are rarely reported due to their additional preparation process, including sonication, and centrifugation103,107,108 In detail, the advantages of PVP–I are as follows: (1) the heteroatoms (oxygen and nitrogen) in the PVP chain enable the PVP to complex with iodine, thus reducing the solubility of iodine in electrolyte and improving its thermal stability;109 (2) PVP can bind strongly to carbon surfaces in aqueous solutions as it provides a strong thermodynamic driving force that eliminates the hydrophobic interface.104 Benefiting from these, the Li–I2 battery with nitrogen-containing porous carbon plate arrays grown on carbon fiber cloth as the host and PVP–I as the iodine source, can be cycled 3000 times at 1C with a capacity retention of 79.7%. But a thought-provoking issue is that the iodine content in commercial PVP–I is usually lower than 20 wt%, which greatly reduces the actual energy density of the whole battery and makes PVP–I unsuitable for real applications. On this basis, Zhang et al. directly used the iodine@activated graphene composites as the matrix, while employing PVP as the coating.89 Despite achieving a 49 wt% iodine content in the composite, the performance for the as-prepared Li–I2 battery dropped drastically (500 cycles with a capacity retention of 90.1% at 1C). This suggests that the ability of PVP to restrict iodine species is not as strong as expected. All in all, PVP–I, as an iodine source, may be somewhat limited in a practical sense due to its low iodine content and moderate interaction with iodine species.
On the other hand, LiI is also used as a replacement for iodine. One of the main reasons is that LiI is the discharge product that reduces cathode volume expansion (about 49% increase in volume from I2 to LiI) during cycling to forestall pulverization of the active components.107 Also, with LiI as an iodine source, the resulting Li–I2 battery is therefore compatible with Li-free metal electrodes, ensuring the battery's safety. To be noted, the theoretical capacity drops but is only about 5% when I2 (211 mA h g−1) is replaced with LiI (200 mA h g−1) as the iodine resource. Given all these facts, Wu et al. first demonstrated the performance of the Li–I2 cell with LiI confined in meso–micro porous carbon polyhedrons as the cathode (Fig. 4f),107 which displayed a cycling life of 800 times at 2C with a capacity retention of 83.2%. Particularly, Li et al. further combined LiI with nitrogen and phosphorus co-doped carbon cloth, enabling the as-assembled Li–I2 cell to be cycled more than 2000 times with an excellent capacity retention of 95.8%.97 Hence, LiI can be regarded as a promising alternative to thermally labile iodine.
As compared to PVP–I and LiI which have a role in mitigating iodine species shuttling, two-dimensional few-layer iodine nanosheets (FLINs) are primarily used to obtain high-rate iodine-based batteries (Fig. 4g). This is because FLINs are ultra-thin and have a large aspect ratio, making them highly uniform when dispersed in carbon-based materials. Since iodine is equivalent to active sites in carbon materials capable of desorbing/adsorbing zinc ions, FLINs will therefore not only provide sufficient active sites but also shorten the ion paths, thereby effectively enhancing the rate capability of batteries.108,110 Qian and her team reported the application of FLINs in Li–I2 batteries for the first time, which showed a long life of 1000 cycles with a capacity retention of 93.2% at 9.5C. Even at a high current density of ∼38C, the FLINs-based cell was capable of exhibiting a reversible capacity of ∼96 mA h g−1, which fully demonstrated the feasibility of FLINs in achieving fast cathode kinetics. In consideration of the excellent rate capability and feasible preparation procedure of FLINs, more extensive studies on FLINs in MIBs as the iodine source are desirable in the future.
2.1.1.2 Other iodine hosts (titanium carbonitrides, MXenes, and organic polymer/salts). Apart from carbon-based materials, high-performance Li–I2 batteries have also been constructed using other iodine hosts, including titanium carbonitrides, MXenes, and organic polymers/salts. Among them, titanium carbonitrides with ceramic properties are highly stable and corrosion-resistant, making them an interesting class of electrically conducting materials, which were then introduced by Anju et al. into Li–I2 batteries as an iodine host (Fig. 5a).111 By adjusting the C/N ratio, they found that TiC0.7N0.3 possessed a work function of 4.85 eV that is close to the potential of the redox couple (I3−/I−, 4.9 eV), thus considering that TiC0.7N0.3 has favorable adsorption characteristics for iodine species. Density functional theory (DFT) calculations also demonstrated the favorable interaction between TiC0.7N0.3 and the iodine/iodide couple, which may originate from the overlap of the d-band of Ti and the p-orbital of iodine. As a result, the as-prepared Li–I2 battery exhibited an outstanding performance over 7000 cycles with negligible capacity decay. However, follow-up research on this type of material is lacking. In addition to titanium carbide, a flexible hydrogen-bonded organic framework (HOF) linked to Ti3C2Tx MXene complexes (HOF@Ti3C2Tx) has been recently reported to serve as the iodine host. However, the performance of the I2/HOF@Ti3C2Tx cathode was less than satisfactory, showing a short lifetime of 300 cycles corresponding to a capacity retention of 79.1%. Despite this, from this single report, one cannot yet conclude that such materials are not suitable for MIBs, as Nb2CTx MXene has shown excellent performance (23
000 cycles with 80% capacity retention) in a Zn–I2 battery.83 The exploration of organic polymer materials is more extensive compared to titanium carbonitrides and MXenes (Fig. 5b).80 But these attempts have been restricted to a handful of well-known polymers, such as starch, PVP, and β-cyclodextrin, lacking a systematic study of polymers. In this connection, it is not surprising that Li–I2 batteries based on polymers exhibited suboptimal performance.80 The utilization of organic iodized salts as alternatives to organic polymer materials may be a potentially viable solution for several reasons: (1) the compact structures of organic iodized salts allow them to load more iodine-active substances and (2) organic iodized salts inherit the advantages of polymer materials, including lightweight, chemical abundance, sustainability, cost-effectiveness, tunable structure, and abundant active sites (Fig. 5c and d). Recently, Li et al. employed methylamine hydroiodide with an iodine content of 80 wt% for the first time as the cathode for Li–I2 batteries, achieving an ultra-high capacity retention of 98.3% over 10000 cycles at 5C (Fig. 5e); this can be attributed to the robust chemical interaction between iodides and the positively charged organic group (–NH3+).53 This result fully demonstrates the great potential of organic iodized salts in Li–I2 batteries, although the development of organic iodized salts for MIBs is still in its infancy. Some key issues still remain to be investigated, such as the structure–activity relationship between the functional group, chain length, and spatial structure of organic iodide salts and the electrochemical properties of cells, as well as the compatibility of organic iodide salts with electrolytes. The rapid advancement of research on organic iodized salts is expected to realize high-capacity, high-stability, and low-cost MIBs.
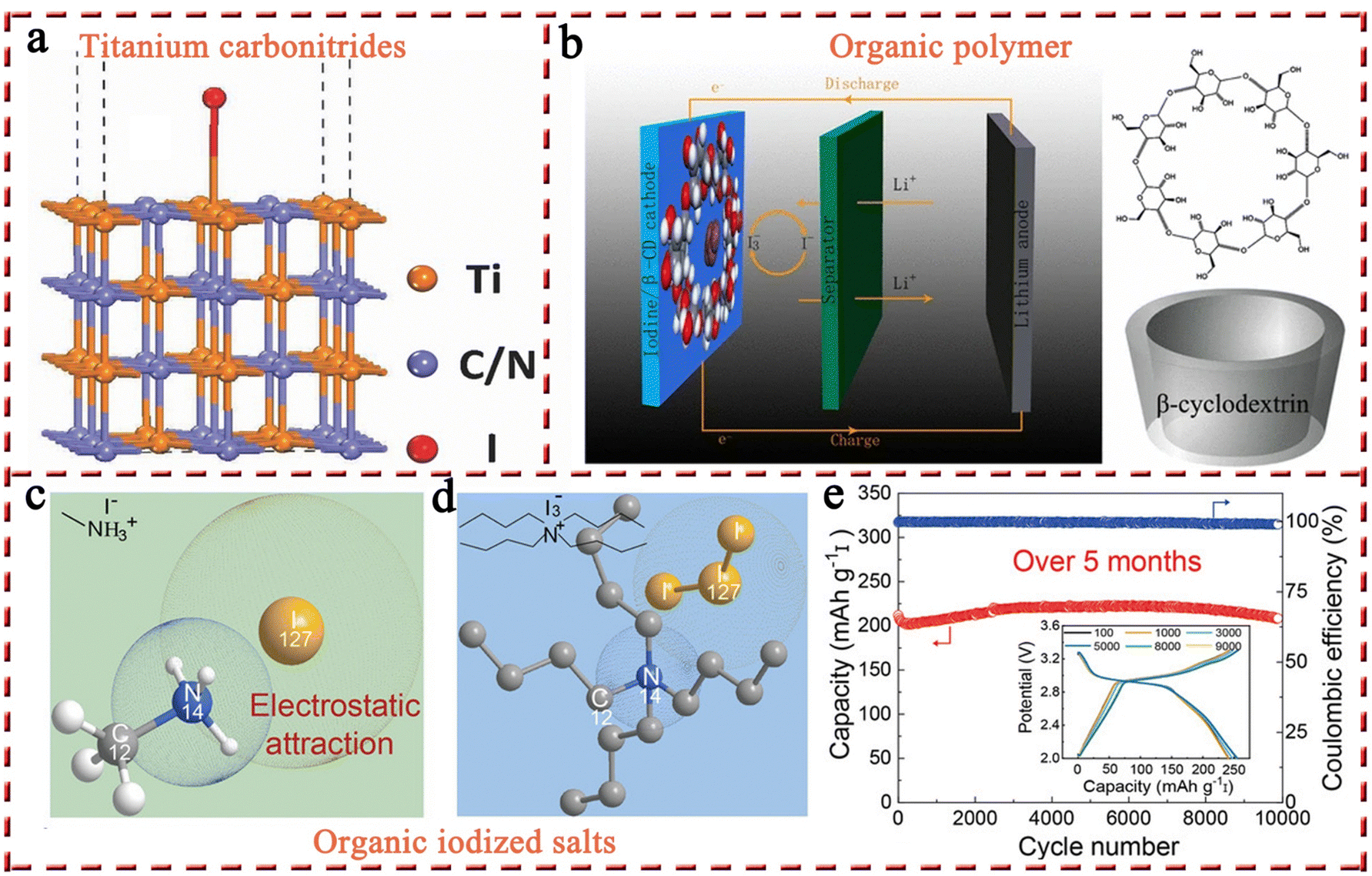 | ||
| Fig. 5 (a) The relaxed slab of (100) (titanium carbonitride) 2D surface unit cell with iodine extended periodically along x and y directions. Reprinted with permission from ref. 111. Copyright 2017 Wiley. (b) Schematic illustration of a Li–I2 battery with the iodine@β-cyclodextrin composite cathode. Reprinted with permission from ref. 80. Copyright 2018 Springer Nature. Schematic showing the electrostatic interactions between iodine species and (c) methylamine, and (d) tetrabutylammonium. (e) Long-term stability of Li–I2 cell with the methylamine hydroiodide cathode at 5C. Inset: Corresponding voltage profiles of the cell from the 100th to the 10000th cycle. Reprinted with permission from ref. 53. Copyright 2022 Wiley. | ||
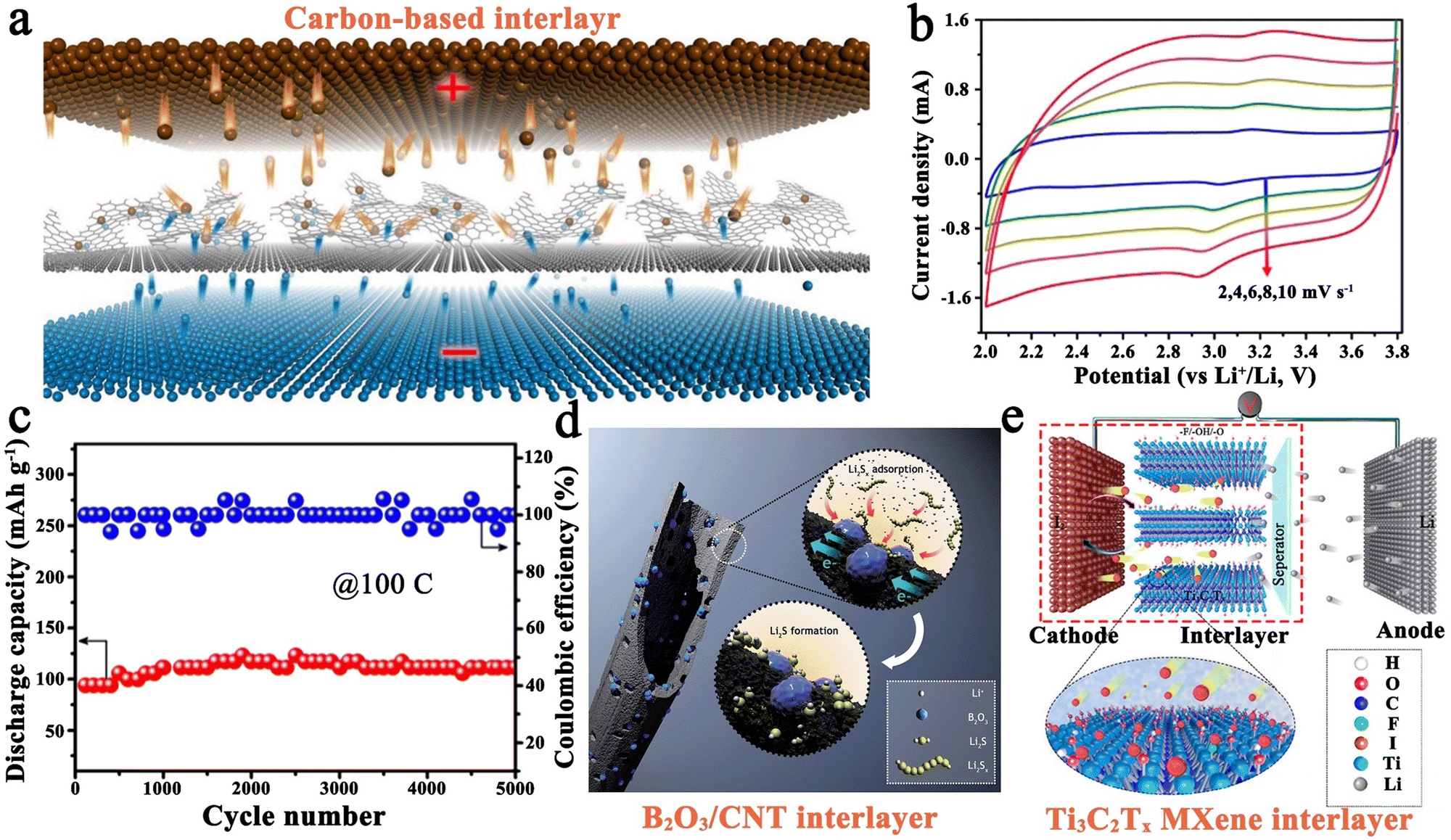 | ||
| Fig. 6 (a) Schematic construction of a pseudo-capacitive Li–I2 battery with a carbon-based interlayer. (b) Characteristic CV curves at various scan rates, and (c) long-term cycling performance (100C) of a Li–I2 battery with the carbon-based interlayer. Reprinted with permission from ref. 43. Copyright 2018 Elsevier. (d) Schematic diagram for the working mechanism of B2O3 nanocrystal-modified CNT. Reprinted with permission from ref. 115. Copyright 2016 Royal Society of Chemistry. (e) Schematic illustration for the reaction mechanism of a Li–I2 battery with the MXene interlayer. Reprinted with permission from ref. 23. Copyright 2020 American Chemical Society. | ||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|
| 1, year; 2, interlayer; 3, cathode; 4, electrolyte formula; 5, iodine loading (mg cm−2). Evaluation of Li–I2 cell performance: 6, current density (C, 1C = 211 mA h g−1); 7, cycle number; 8, capacity retention (%); 9, reversible capacity (mA h g−1iodine). 10, reference. CB, carbon black; PC, propylene carbonate. | |||||||||
| 2016 | B2O3-modified CNT | I2/superconducting CB | 1 M LiPF6 in PC/EC/DEC (1:4:5, v/v/v) |
∼1 | ∼100 | 5000 | — | 140.7 | 115 |
| 2018 | N-doped graphene | I2/superconducting CB | 1 M LiPF6/0.1 M LiTFSI in EC/DMC (1:1, v/v) |
∼1 | 100 | 5000 | — | 111.3 | 43 |
| 2018 | CNT | I2/super-P CB | 1 M LiPF6/0.1 M LiTFSI in EC/DMC (1:1, v/v) |
1 | 100 | 5000 | 100 | 100 | 114 |
| 2020 | Honeycomb-like carbon | I2/superconducting CB | 0.5 M LiTFSI/0.1 M LiNO3 in DOL/DME (1:1, v/v) |
∼1.1 | 50 | 4000 | — | 120.2 | 113 |
| 2020 | Ti3C2Tx MXene foam | I2@N-doped graphene | 1 M LiTFSI/0.2 M LiNO3 in DOL/DME (1:1, v/v) |
∼2.5 | 2 | 1000 | 78 | 80 | 23 |
Commonly, carbon matrices can assume the role of functional interlayers in Li–I2 cells. For example, Wu et al. employed carbon nanotubes (CNTs) as interlayers, which demonstrated a high capacity of 100 mA h g−1 and a CE value of ∼100% after 5000 cycles at an astonishing rate of 100C.114 Moreover, nitrogen-doped graphene nanosheets and honeycomb-like carbon materials from coffee extract were also reported as functional interlayers.43,113 Benefiting from the successful incorporation of electrochemical conversion reaction and pseudo-capacitive process into Li–I2 batteries, the as-fabricated batteries achieved a high power density output (≥50C) and an ultra-long cycling life (≥4000 cycles) (Fig. 6c). In addition to the carbon-based interlayers, the B2O3@CNT composite interlayer was also investigated (Fig. 6d).115 Coincidentally, the as-prepared Li–I2 battery showed a high reversible capacity of 140.7 mA h g−1 after 5000 cycles at 100C. Such excellent cycling stability of Li–I2 cells is mainly attributed to the strong interaction between the ultra-small B2O3 nanocrystals and iodine species. Despite this, the iodine content in this cell is relatively low (∼1 mg cm−2), leading to the significantly restricted energy density of the cell. Therefore, Sun et al. proposed to employ Ti3C2Tx MXene as an interlayer to implement a Li–I2 cell with high iodine loading because of the unique hexagonal lattice symmetry, surface chemistry, and metallic nature of MXene materials (Fig. 6e).23 As a result, the Ti3C2Tx-based battery can be cycled stably for over 1000 cycles at 2C with a cathode loading mass of 5.2 mg cm−2 (the actual iodine loading is about 2.5 mg cm−2). However, the performance of the cell under this condition is much worse than that of the battery with low iodine loading. More data and experiments are needed to establish a conclusive link between iodine loading and battery performance for this specific design.
All in all, constructing interlayers seems to enhance the performance of high-rate and stable Li–I2 batteries compared to the iodine hosts discussed above, indicating a positive effect of interlayers on battery performance. However, some problems and phenomena are worthy of consideration. (1) The introduction of interlayers actually increases the battery's inactive component, which may reduce the actual energy density and increase assembly cost. Thus, it is necessary to evaluate the pros and cons of the battery in terms of cost and performance after introducing the functional interlayers. (2) Compared with iodine host materials, there is still a lack of systematic research on interlayers. More breakthroughs for the improvement of interlayers are expected in the future. (3) The iodine host and the functional interlayer may be complementary in terms of both structure and function, that is, the iodine host can retain most of the polyiodide species well, while the functional interlayer can serve as an additional adsorption layer to prevent polyiodide diffusion, provide pseudo-capacitance, and catalyze the charge/discharge electrochemistry of the iodine redox couple. Interesting phenomena may arise by combining the iodine host and interlayer, which are worthy of further investigation.
2.1.3.1 Conventional organic liquid electrolytes. The electrolyte is critical to battery performance as it enables batteries to efficiently build a complete and functional circuit, much like blood in the human body.116–118Table 4 in the section modified electrolyte for Li–I2 batteries provides the relevant battery information and performance details. In general, the most commonly used electrolyte formulation in Li–I2 batteries is 1 M lithium bis-(trifluoromethane sulfonyl) imide (LiTFSI) with 1 wt% LiNO3 in 1,3-dioxolane (DOL)/1,2-dimethoxyethane (DME) (1
:1, v/v); this is similar to Li–S batteries due to the same problem (i.e., shuttle effect) that exists in both types of batteries.8 Particularly, the addition of LiNO3 into the ether-based electrolyte will help in stabilizing the anode surface and preventing electrolyte decomposition.95,119 In their study, Zhao et al. employed three different electrolytes, including 1 M LiPF6 in ethylene carbonate (EC)/ethyl methyl carbonate (EMC)/dimethyl carbonate (DMC) (1:1:1 by volume), 1 M LiTFSI in DOL/DME (1:1 by volume), and 1 M LiTFSI in DOL/DME (1:1 by volume) with 1 wt% LiNO3 addition, to investigate the relationship between electrochemical properties of Li–I2 batteries and electrolyte formulations (Fig. 7a).95 As a result, the cell using 1 M LiTFSI in DOL/DME with 1 wt% LiNO3 addition showed negligible self-discharge even after five days, thanks to the formation of passive films on the Li surface through the addition of LiNO3 in ether solvent. Besides, a few works used tetra-ethylene glycol dimethyl ether (TEGDME) as a solvent instead of DME, but the underlying reason was not known. A plausible explanation, although, is that TEGDME is capable of effectively solubilizing and dissociating lithium salt and polyiodide due to its abundant glycolate structures.120,121 Overall, there still remains a lack of in-depth understanding of electrolytes for Li–I2 batteries, which are inherited more from the Li–S battery system. Nonetheless, Li et al. recently discovered novel two-electron redox chemistry of iodine (i.e., I− ↔ I0 ↔ I+) by adding LiCl into the DOL/DME electrolyte system, where the Cl− ion can fully activate and stabilize the deactivated I+ ions by forming I+Cl− complexes (Fig. 7b).18 To this end, the Li–I2 battery can double the theoretical capacity to 422 mA h g−1, while obtaining a remarkable improvement in energy density of over 200%. With ammonium methyl iodide as the cathode, the as-prepared Li–I2 battery exhibited excellent cycle stability for over 1300 cycles with a low capacity decay ratio of 20% at 2.0 A g−1. This work endows Li–I2 batteries with new vitality as they exhibit attractive energy density (1324 W h kg−1 based on iodine), and more achievable multi-electron reactions compared to Li–S and Li–O2 batteries. However, in such a system, it is puzzling that the redox peaks ascribed to the I0 ↔ I+ process coincide with those of I3− ↔ I2 transition in conventional Li–I2 batteries, although the intensity of the former peak is much higher (Fig. 7c). One possible reason is that the I0 ↔ I+ process actually also occurs in conventional Li–I2 batteries without the use of LiCl-containing electrolyte. Of course, this urgently needs to be further clarified in future work.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|
| 1, year; 2, electrolyte formula; 3, cathode; 4, iodine loading (mg cm−2). Evaluation of Li–I2 cell performance: 5, current density (C, 1C = 211 mA h g−1); 6, cycle number; 7, capacity retention (%); 8, reversible capacity (mA h g−1iodine). 9, reference. KB, ketjenblack; PVDF, polyvinylidene fluoride; PEO, poly(ethylene oxide); PP13TFSI, N-methyl-N-propylpiperidinium bis(trifluoromethanesulfonyl) imide. | ||||||||
| 2011 | LiI(3-hydroxypropionitrile)2 (LiI(HPN)2) | I2/LiI(HPN)2 | — | 20 μA cm−2 | ∼5 | — | — | 48 |
| 2013 | Anodolyte, 1 M LiPF6 in EC/DMC (3:7, v/v); catholyte, 0.08 M I2/1 M KI/0.03 M LiI in aqueous solution |
Super P/PVDF | — | 2.5 mA cm−2 | 100 | 99.6 | ∼207 | 28 |
| 2014 | Anodolyte, P(EO)20LiTFSI/PP13TFSI; catholyte, 0.2 M I2/1 M KI in aqueous solution | Super P/PVDF | — | 1 mA cm−2 | 50 | 97 | ∼190 | 266 |
| 2014 | Anodolyte, 1 M LiPF6 in EC/DMC (3:7, v/v); catholyte, 0.1 M I2/1 M KI in aqueous solution |
Vertically aligned multi-walled CNT | — | 2.5 mA cm−2 | 200 | ∼89.8 | 185 | 125 |
| 2015 | LiI(HPN)2 | I2/LiI(HPN)2 film paper | — | 0.25 mA cm−2 | 15 | — | — | 131 |
| 2016 | Anodolyte, 1 M LiTFSI in EC/DMC (3:7, v/v); catholyte, 0.2 M I2/2 M KI in aqueous solution |
α-Fe2O3/fluorine-doped tin oxide coated glass | — | 0.3 mA cm−2 | 22 | ∼99 | 180 | 127 |
| 2019 | NaNO3 particles in pentaerythritol-tetraacrylate-based gel polymer electrolyte | Ti3C2Tx/carbon cloth/I2 | — | 0.5 | 1000 | 85 | 280.5 | 70 |
| 2021 | 1 M LiTFSI/0.2 M LiNO3/0.1 M LiCl in DOL/DME (1:1, v/v) |
Ammonium methyl iodide/carbon cloth | ∼1.9 | ∼9.5 | 1300 | 80 | ∼274 | 18 |
| 2021 | U-shaped garnet Li6.4La3Zr1.4Ta0.6O12 ceramic electrolyte | Molten CsI/LiI eutectic salt | ∼174 (LiI) | 3 | 2000 | ∼100 | ∼180.2 | 132 |
| 2022 | PEO/LiTFSI (18:1) on the Li1.5Al0.5-Ge1.5(PO4)3 |
I2@KB/PEO/LiTFSI/carbon cloth | 0.5 | 1 | 9000 | 84.1 | 112.9 | 128 |
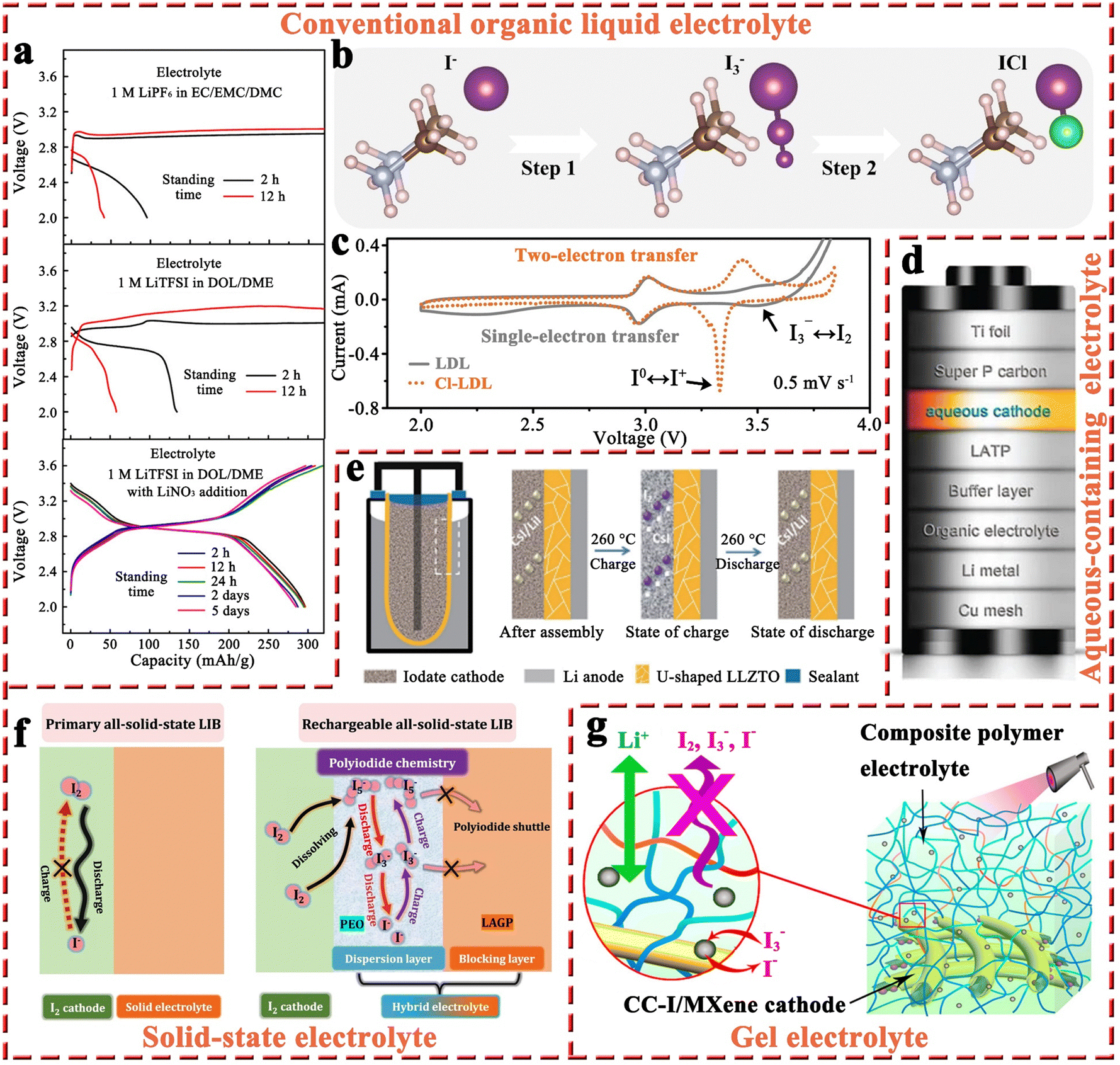 | ||
| Fig. 7 (a) Discharge and charge curves of Li–I2 batteries with the electrolytes of 1 M LiPF6 in EC/EMC/DMC, 1 M LiTFSI in DOL/DME, and 1 M LiTFSI in DOL/DME with 1 wt% LiNO3 addition after different standing times at 0.5C. Reprinted with permission from ref. 95. Copyright 2015 American Chemical Society. (b) The possible conversion process from I− to I+. (c) CV profiles of Li–I2 cells with or without two-electron redox chemistry at 0.5 mV s−1. Reprinted with permission from ref. 18. Copyright 2021 Wiley. (d) Schematic diagram for the composition of an aqueous Li–I2 battery. Reprinted with permission from ref. 28. Copyright 2013 Springer Nature. (e) Schematic diagram for the structure of Li–I2 batteries with LLZTO electrolyte and its working mechanism. Reprinted with permission from ref. 132. Copyright 2021 Springer Nature. (f) Schematic illustration for the reaction mechanism of all-solid-state Li–I2 batteries at the cathode/electrolyte interface with or without the “confined dissolution” strategy. Reprinted with permission from ref. 128. Copyright 2022 Springer Nature. (g) Schematic compositions for the MXene-wrapped carbon cloth–iodine cathode and composite polymer electrolyte. Reprinted with permission from ref. 70. Copyright 2019 American Chemical Society. | ||
2.1.3.2 Organic/aqueous mixed electrolytes. As compared to organic electrolytes, aqueous solutions generally have high ionic conductivity and can lead to fast redox reactions in electrochemical cells. Additionally, the high solubility of the I3−/I− (over 11 mol L−1 with a lithium salt) redox couple in aqueous solution also ensures that Li–I2 batteries can deliver superior energy density.122 Furthermore, the electron-transfer rate for the I3− reduction reaction in an aqueous solution is superior to that in organic mediums, such as acetonitrile, dimethyl sulfoxide, N,N-dimethylformamide, and propylene carbonate (PC).123–125 Therefore, Byon's group proposed the concept of a Li–I2 battery with an aqueous cathode. As shown in Fig. 7d, the aqueous Li–I2 battery consists of a Li composite anode (i.e., Cu mesh/Li metal/organic electrolyte/buffer layer), a ceramic separator (Li2O–Al2O3–TiO2–P2O5, LATP), an aqueous cathode, and a current collector (i.e., super P carbon/Ti foil).28 The as-prepared Li–I2 cell showed that the primary redox reactions occur in the potential region of 3.5–3.7 V with about 100 cycles. Furthermore, they employed vertically aligned carbon nanotubes (VACNT) instead of super P/polyvinylidene fluoride as the current collector.125 Because the binder-free VACNTs eliminated unnecessary mass and prevented parasitic corrosive reactions arising from polymer binders, the Li–I2 cell exhibited improved cycling performance over 200 cycles with negligible capacity decline. In addition, stemming from the rise of the “all-in-one” concept (i.e., the integrated power system with energy conversion and storage functions),126 they also demonstrated a self-charging aqueous Li–I2 cell using a hematite electrode, which enables a facile charging process with the aid of harnessed photoenergy.127 However, the resulting battery exhibited a very short lifespan of about 30 cycles.
As a whole, aqueous Li–I2 batteries are still in the conceptual stage and confronted with a plenty of difficulties: (1) a complex battery structure is necessary to match the Li anode with the aqueous cathode. Whereas this not only increases battery costs and reduces battery energy density dramatically but also shortens battery life due to possible breakage of battery components during cycling and (2) given the introduction of aqueous electrolyte, the inherent safety problems associated with the metal anode are also more worrisome. Therefore, it is essential to assess the costs and benefits of aqueous Li–I2 batteries carefully in future research to ensure their practical feasibility.
2.1.3.3 Solid-state electrolytes. The utilization of non-flammable solid-state electrolytes can effectively improve the inherent safety of batteries, addressing concerns such as leakage, fire, and explosion commonly faced by liquid electrolytes.128–130 The primary all-solid-state Li–I2 battery was commercialized in 1972 to serve as the power source for cardiac pacemakers.26 Nevertheless, it can barely be recharged due to the poor Li-ion conductivity of LiI solid-state electrolyte and its continuous increase in thickness during discharge.47,48,131 Thanks to the vigorous development of solid-state electrolytes in battery systems (e.g., Li-ion batteries), Sun et al. introduced a U-shaped garnet Li6.4La3Zr1.4Ta0.6O12 (LLZTO) ceramic electrolyte into Li–I2 batteries.132 As shown in Fig. 7e, the battery comprises a molten lithium anode, LLZTO electrolyte, and a molten CsI/LiI eutectic salt (CLES) cathode with an operating temperature of 260 °C. During the charge/discharge process, the CLES cathode undergoes a highly reversible conversion between LiI and I2, while the Li anode is capable of exhibiting ultra-high stability due to its liquid form. Notably, the addition of CsI significantly reduces the melting point of LiI, allowing the cathode to form a liquid–solid interface with the LLZTO electrolyte, thereby maintaining good interfacial contact and low interfacial impedance. Given that the LLZTO electrolyte can also effectively separate the Li anode from the eutectic iodate cathode, the as-fabricated Li–I2 battery possessed a long-term lifetime of 2000 cycles with a negligible capacity decay at 3C, even with a LiI loading of 593 mg. However, this battery system requires an ultra-high operating temperature (260 °C) to maintain the melting characteristics of the anode and cathode, as well as the high ionic conductivity of LLZTO, which severely limits its application range. In response to this limitation, Cheng et al. proposed a “confined dissolution” strategy, that is, polyethylene oxide (PEO) was introduced into the iodine cathode as a dispersion layer to facilitate the dissolution of polyiodide, while adopting a single lithium-ion conductor Li1.5Al0.5Ge1.5(PO4)3 (LAGP) as a barrier layer to effectively avoid the shuttle effect of polyiodide (Fig. 7f).128 Benefiting from the utilization of the PEO dispersion layer with well-dissolved polyiodide, the all-solid-state Li–I2 battery can operate at 60 °C with a high specific capacity of 202 mA h g−1. Meanwhile, the PEO dispersion layer also had a low Young's modulus, which could lead to close contact between the LAGP and I2 cathode and facilitate fast Li-ion transport within the cell. On this basis, the battery exhibited an 84.1% capacity retention over 9000 cycles at 1C, which opens a new avenue for the development of rechargeable all-solid-state Li–I2 batteries.
Overall, the incompatibility between the solid iodine cathode and solid-state electrolyte has limited the development of all-solid-state Li–I2 batteries due to their sluggish reaction kinetics, low capacity, and poor rechargeability. The use of a molten iodine cathode and the “confined dissolution” strategy could greatly solve this fundamental problem. Furthermore, given the intrinsic safety and strong blocking ability for polyiodide, solid-state electrolytes may be the cornerstone for the rapid development of Li–I2 batteries in the future.
2.1.3.4 Gel electrolytes. Similar to solid-state electrolytes, the aim of using gel electrolytes is to replace liquid electrolytes, thus improving battery safety and restraining the diffusion of dissolved iodine species. In particular, gel electrolytes have higher ionic conductivity at ambient temperature and a more stable electrode/electrolyte interface compared to solid-state electrolytes.70 To this end, Tang et al. developed a flexible quasi-solid-state Li–I2 battery, which integrates a composite polymer electrolyte (CPE), composed of catalytic NaNO3 particles dispersed in a pentaerythritol-tetraacrylate-based (PETEA-based) gel, and a Ti3C2Tx MXene-wrapped carbon cloth–iodine (CC–I/MXene) cathode (Fig. 7g).70 As a result, the as-prepared Li–I2 quasi-solid-state battery exhibited superior long-term cycling stability with 85% capacity retention after 1000 cycles at 0.5C. This can be attributed to the abundant surface functional groups present in Ti3C2Tx MXene sheets, which effectively confine the iodine species within the cathode. Meanwhile, the CPE can greatly block the diffusion of iodine species and stabilize the Li anode without dendrite growth. On the other hand, the addition of NaNO3 particles into PETEA-based polymer matrix could enhance the conversion kinetics of iodine species. Because the
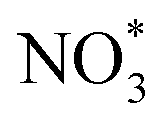 radical generated by the oxidation of NO3− can act as a reaction intermediate and is prone to be consumed instantly by I3− as described in the following equation:
radical generated by the oxidation of NO3− can act as a reaction intermediate and is prone to be consumed instantly by I3− as described in the following equation: 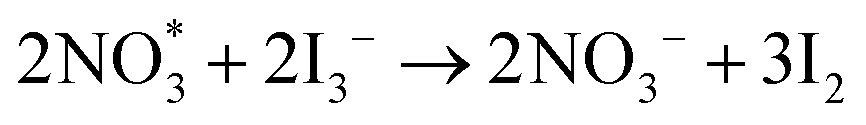 .133 In this regard, the LiNO3 additive in liquid electrolyte may play a dual role of inhibiting dendrite growth and promoting iodine/iodide conversion. Owing to the excellent mechanical properties (strength, flexibility, etc.) of gel electrolytes, the as-assembled quasi-solid-state battery also showed promising application prospects in flexible electronic devices. Nonetheless, high levels of liquid plasticizers, such as EC, DMC, DME, and TEGDME, are inevitably required to achieve a satisfactory ion conductivity value (>10−4 S cm−1 at 25 °C) in gel electrolytes, which usually results in a poor thermal stability and safety hazard during thermal runaway.134 At the same time, there is a lack of systematic research on gel electrolytes in Li–I2 batteries. Therefore, there is an urgent need to optimize the composition and structure of gel polymers in Li–I2 batteries for achieving better performance and extending the range of applications for this type of battery.
.133 In this regard, the LiNO3 additive in liquid electrolyte may play a dual role of inhibiting dendrite growth and promoting iodine/iodide conversion. Owing to the excellent mechanical properties (strength, flexibility, etc.) of gel electrolytes, the as-assembled quasi-solid-state battery also showed promising application prospects in flexible electronic devices. Nonetheless, high levels of liquid plasticizers, such as EC, DMC, DME, and TEGDME, are inevitably required to achieve a satisfactory ion conductivity value (>10−4 S cm−1 at 25 °C) in gel electrolytes, which usually results in a poor thermal stability and safety hazard during thermal runaway.134 At the same time, there is a lack of systematic research on gel electrolytes in Li–I2 batteries. Therefore, there is an urgent need to optimize the composition and structure of gel polymers in Li–I2 batteries for achieving better performance and extending the range of applications for this type of battery.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1, year; 2, anode; 3, electrolyte formula; 4, cathode. Evaluation of Li‖Li cell performance: 5, current density (mA cm−2) and areal capacity (mA h cm−2); 6, cycle time (h). Evaluation of Li–I2 cell performance: 7, current density (C, 1C = 211 mA h g−1); 8, cycle number; 9, capacity retention (%); 10, reversible capacity (mA h g−1iodineorLiI). 11, reference. |
||||||||||
| 2017 | Passivated Li by InI3/layered graphene paper | 1 M LiTFSI/1 wt% LiNO3/1 M LiI3 in DOL/DME (1:1, v/v) |
Carbon cloth | — | — | 1.5 | 100 | 89 | ∼80 | 138 |
| 2019 | Molten Li in N,P co-doped carbon cloth | 1 M LiTFSI/1 wt% LiNO3 DOL/DME (1:1, v/v) |
LiI/porous doped carbon | 3 (—) | 600 | 10 | 4000 | 99 | 197 | 136 |
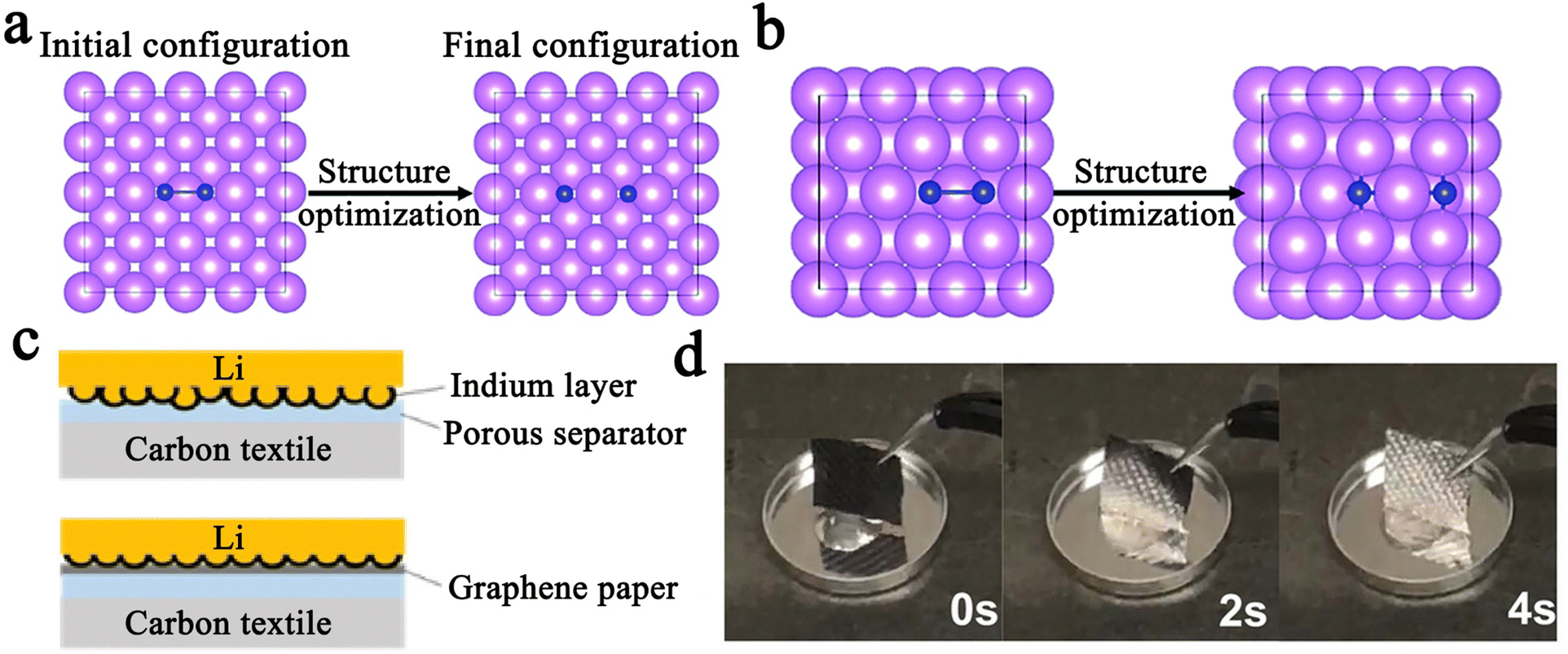 | ||
| Fig. 8 Initial and optimized atomistic configurations of I2 adsorption on the top sites of (a) (100) surface, and (b) (110) surface of Li. The violet spheres stand for Li atoms, while the blue spheres are I atoms. Reprinted with permission from ref. 137. Copyright 2018 Royal Society of Chemistry. (c) Graphs showing the combined effect of a pre-deposited indium layer with the graphene paper. Reprinted with permission from ref. 138. Copyright 2017 Elsevier. (d) Time-lapse images of molten lithium immersed into the N,P co-doped carbon cloth. Reprinted with permission from ref. 136. | ||
To sum up, the strategies proposed in Li–I2 batteries to protect the Li anode can be mainly classified into the following four categories: (1) adding LiNO3 to the electrolyte that can protect the Li anode; (2) using solid-state electrolytes based on the “molten electrode” or “confined dissolution” strategy, and gel electrolytes; (3) constructing a passivation layer (i.e., artificial SEI); and (4) preparing a 3D-stabilized Li metal anode. It is worth noting that these strategies can all be found in Li-based batteries. We therefore predict that lithium-based batteries will experience new breakthrough once the bespoke lithium anode problems are solved.
2.2 Sodium–iodine batteries
Li–I2 batteries possess the highest energy density among various MIBs due to their high voltage window. However, the scarcity, uneven distribution, and exorbitant price of lithium resources greatly limit the extensive utilization of Li–I2 batteries, especially in the presence of numerous other Li-based batteries. Thus, it is crucial to develop MIBs based on earth-abundant metal anodes. As a high-performance anode, Na has become the focus of researchers due to its highly abundant natural sources (2.74% in the Earth's crust), high theoretical specific capacity (1166 mA h g−1), and low potential (−2.71 V vs. SHE).140–142 Given this, a promising path to expand the application of MIBs is by replacing Li with Na as the anode. Tables 6 and 7 in the section on Na–I2 batteries provides the relevant battery information and performance. Research on Na–I2 batteries began with medium-temperature molten Na-based batteries,54,143 as similar high-temperature Na-S battery systems have been put into commercial production and tested for grid storage in Japan, France, and the United States.144 Ngersoll et al. demonstrated the prototype of a Na–I2 battery based on NaSICON ceramics and inorganic NaI/AlCl3-based catholyte.54 Zhu et al. further conducted a theoretical study on an aqueous Na–I2 system with a molten sodium anode under an operating temperature of 120 °C (Fig. 9a).143 Moreover, Holzapfel et al. realized a rechargeable molten-sodium system at approximately 100 °C using aqueous iodine/iodide solution as the catholyte and sodium-ion conductive NaSICON (Na3Zr2Si2.3P0.7O11.85) as the solid-state electrolyte.54 Despite this, room-temperature Na–I2 batteries are still eagerly pursued because of their less demanding and safer operation compared to the high-temperature cell systems.145| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|
| 1, year; 2, cathode; 3, electrolyte formula; 4, the mass loading of iodine at the cathode (wt%); 5, the area loading of iodine at the cathode (mg cm−2). Evaluation of Na–I2 cell performance: 6, current density (C, 1C = 211 mA g−1); 7, cycle number; 8, capacity retention (%); 9, reversible capacity (mA h g−1iodine). 10, reference. RGO, reduced graphene oxide; GO, graphene oxide; COFs, covalent organic frameworks; FEC, fluoroethylene carbonate. γ-Mo2N/mNC, N-doped hierarchical mesoporous carbon decorated with γ-Mo2N nanoparticles. | |||||||||
| 2016 | Iodine quantum dots decorated RGO | 1 M NaClO4 in EC/DEC (1:1, v/v) |
26 | ∼0.8 | ∼0.5 | 500 | ∼82 | 141 | 55 |
| 2017 | I2/3D bio-foam derives from C. Barometz spores | 1 M NaClO4 in PC | 30–40 | ∼1–1.5 | 6/2 | 500 | 91 | — | 96 |
| 2018 | PVP–I‖N-doped hollow carbon sphere–I2 | 1 M NaClO4 in PC | ∼35 | — | 4 | 400 | 73 | — | 103 |
| 2020 | CNT/GO/few-layer iodine nanosheets | 1 M NaClO4 in EC/DEC (1:1, v/v) |
∼44 | — | ∼24 | 5000 | — | 84.5 | 110 |
| 2020 | Fe2–O8–PcCu/I2 phthalocyanine copper COF | 1 M NaClO4 in EC/DEC (1:1, v/v) |
40 | — | ∼7 | 3200 | — | 150 | 24 |
| 2022 | I2/single primitive polypyrrole | 1 M NaClO4 in EC/DEC (1:1, v/v) |
48 | — | 1 | 400 | ∼52 (from 10th cycle) | ∼70 | 147 |
| 2022 | I2/hydrogen bonded organic framework@Ti3C2Tx | 1.0 M NaClO4/5.0% FEC in EC/DEC (1:1, v/v) |
44.3 | ∼0.93 | 0.2 | 200 | ∼100 | 207.6 | 146 |
| 2023 | I2@γ-Mo2N/mNC | 1.0 M NaClO4/5.0% FEC in EC/DEC (1:1, v/v) |
25 | 0.7–1.5 | 3.55 | 800 | — | 213.5 | 148 |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1, year; 2, anode; 3, electrolyte formula; 4, cathode. Evaluation of Na‖Na cell performance: 5, current density (mA cm−2) and areal capacity (mA h cm−2); 6, cycle time (h). Evaluation of Na–I2 cell performance: 7, current density (C, 1C = 211 mA h g−1); 8, cycle number; 9, capacity retention (%); 10, reversible capacity (mA h g−1iodineorLiI). 11, reference. SEI, solid electrolyte interface. |
||||||||||
| 2019 | NaI SEI | 1 M NaClO4 in EC/DEC with 5% FEC | I2/active carbon cloth with iodine of ∼1.5 mg cm−2 | 0.25 (0.75) | 500 | 2 | 2200 | 100 | 110 | 151 |
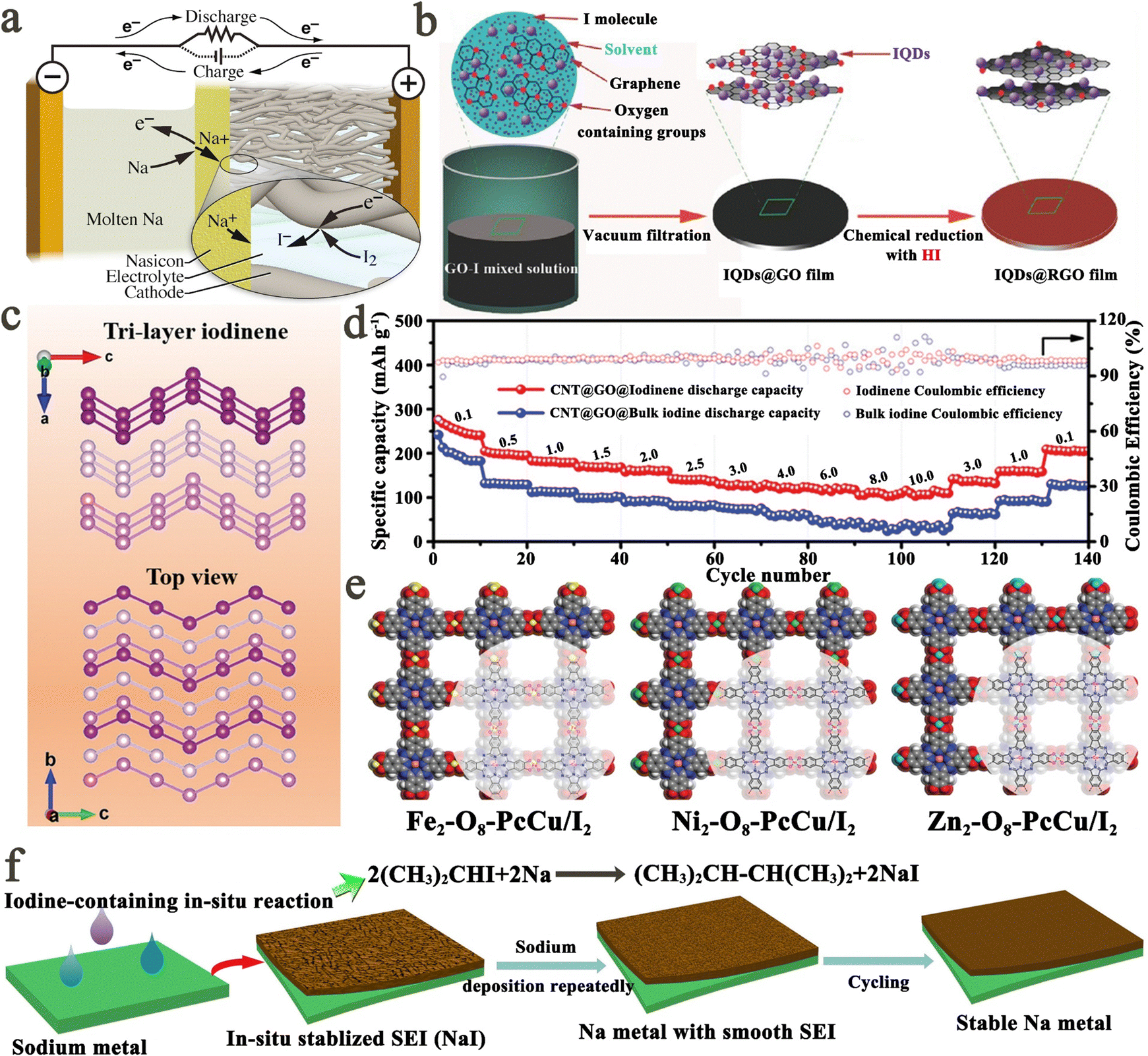 | ||
| Fig. 9 (a) Schematic diagram for the structure of a high-temperature Na–I2 secondary battery. Reprinted with permission from ref. 143. Copyright 2016 Elsevier. (b) Schematic diagram for the fabrication process of the IQDs@RGO cathode. Reprinted with permission from ref. 55. Copyright 2016 Wiley. (c) The structure of tri-layer iodine (the top and bottom parts present the front and top views, respectively). (d) Rate capabilities of Li–I batteries with bulk iodine and FLINs at various current densities from 0.1 to 10 A g−1. Reprinted with permission from ref. 110. Copyright 2020 Wiley. (e) Schematic modeling and chemical structures of Fe2–O8–PcCu, Ni2–O8–PcCu, and Zn2–O8–PcCu. Reprinted with permission from ref. 24. Copyright 2019 Wiley. (f) Schematic illustration for in situ construction of an ultra-thin NaI SEI layer on the Na metal anode. Reprinted with permission from ref. 151. Copyright 2018 Elsevier. | ||
Up to now, the commonly used electrolytes for realizing room temperature Na–I2 batteries are focused on 1 M NaClO4 in carbonate electrolytes (i.e., propylene carbonate, ethylene carbonate, and dimethyl carbonate). In this case, Gong et al. first demonstrated a room-temperature rechargeable Na–I2 battery by employing free-standing iodine quantum dots (IQDs)-decorated reduced graphene oxide (IQDs@RGO) as the cathode (Fig. 9b).55 Given that porous RGO acts as a conductive network, the loaded IQDs will favor intercalation and shorten the diffusion distance of Na+ ions. The RGO can also provide strong adsorption, restraining the free diffusion of iodine species. As a result, Na–I2 with the IQDs@RGO cathode exhibited 500 cycles at a current density of 100 mA g−1. However, the preparation of iodine quantum dots also results in a low iodine content (∼26 wt%) in the material. On the other hand, Qian et al. reported the successful preparation of FLINs via a sonochemical liquid-phase exfoliation approach, which can thus increase the iodine content in the material to about 44 wt% (Fig. 9c).110 Especially, with the loading of FLINs into the carbon nanotube-graphene oxide composite, the as-prepared Na–I2 battery showed an outstanding rate performance of 109.5 mA h g−1 at a high current density of 10 A g−1 (Fig. 9d). Therefore, the utilization of FLINs may be more suitable for implementing high-rate MIBs. Moreover, hollow carbon spheres and HOF@Ti3C2Tx were also investigated in Na–I2 batteries, but their performance was consistently poor as in Li–I2 batteries.96,103,146 On the other hand, attempts have been made to utilize ordered porous materials synthesized through self-assembly techniques as iodine hosts in Na–I2 batteries. For instance, Xiang et al. tried to employ ordered single primitive polypyrrole, poly(m-phenylenediamine), and polydopamine as iodine hosts but with poor performance (70 mA h g−1 after 400 cycles of the optimal polypyrrole/I2 cathode at 1C).147 This may be attributed to the excessively large pores (45 nm) of these materials, leading to inadequate confinement of iodine species. Therefore, when the pore size was reduced to 10–20 nm and γ-Mo2N was introduced as a catalyst for iodine conversion, the self-assembled porous materials demonstrated improved battery stability and capacity (213.5 mA h g−1 after 800 cycles at 3.55C).148 Notably, a novel fully conjugated phthalocyanine copper metal–organic framework (MOF) was introduced into Na–I2 batteries as the iodine host. The presence of atomically dispersed metal centers, abundant pore structures, and an extended π-conjugation structure makes it an ideal host for iodine. As a result, Wang et al. investigated three fully conjugated PcCu-MOFs (i.e., Fe2–O8–PcCu, Ni2–O8–PcCu, and Zn2–O8–PcCu) with 2, 3, 9, 10, 16, 17, 23, 24-octahydroxy phthalocyaninato copper as the building blocks and metal-bis(dihydroxy) complexes as the linkages (Fig. 9e).24 Benefiting from the strong polarization interaction between polyiodide and conjugated Fe-O4 centers, as well as more favorable polyiodide oxidation conversion on the Fe-O4 centers, the Na–I2 battery with a Fe2–O8–PcCu/I2 cathode exhibited superior long-term cycling stability up to 3200 cycles at 1.5 A g−1. This confirms the potential application of MOFs in MIBs.
From the perspective of sodium anodes, Na metal is unstable with carbonate electrolytes, which usually leads to the formation of a loose and fragile SEI. Meanwhile, the uneven plating/stripping behavior of Na+ during the cycling process would greatly induce the formation of Na dendrites that deteriorate the battery's performance.149,150 Hence, extensive efforts to stabilize metallic Na anodes are essential. As previously stated, the application of an “artificial” SEI layer of Li–In alloy on the Li surface will effectively minimize the side reaction and dendrite growth on Li anodes. Similarly, Tian et al. proposed a method of in situ construction of an ultrathin NaI SEI layer on the Na anode surface via the reaction between 2-iodopropane and Na metal (Fig. 9f).151 The resulting Na/Na symmetrical cells can be cycled for up to 500 h, while the as-assembled Na–I2 battery showed over 2200 cycles at 2C with an active carbon cloth@iodine cathode. However, the presence of LiI is generally believed to negatively affect the anode stability in Li–I2 batteries. This results from its loose structure that cannot prevent the anode surface from corrosion, also, it can easily react with soluble I2 to form LiI3.95,137 For this case, a possible explanation is that the NaI layer in the Na–I2 battery formed by the in situ reaction of 2-iodopropane with metallic Na is compact and even, which is conducive to achieving a uniform reaction between carbonate-based electrolyte and soluble iodine species, thereby improving the interfacial stability of Na anodes. In the comparative experiments performed by Tian et al., the morphology of the NaI layer prepared using diethyl carbonate (DEC) with 50 mM I2 is loose and inconsecutive, and as a result, the as-assembled battery showed poor performance. Besides, the NaI layer also exhibits a lower diffusion barrier (ionic conductivity: ∼3 × 10−2 S cm−1) compared to NaF (common inorganic component for dendrite inhibition in the Na-based SEI layer),151,152 enabling high-performance Na–I2 batteries. Therefore, achieving a uniform and dense NaI layer with high ionic conductivity could be a new approach toward a highly stable Na anode.
Compared to Li–I2 batteries, studies on Na–I2 batteries are few and far between. Benefiting from the commonality of iodine cathodes, however, strategies to improve iodine cathodes can be shared between both types of MIBs, as shown by the applications of FLINs in Li/Na–I2 batteries. On the other hand, room-temperature Na–I2 batteries typically use carbonate ester-based electrolytes derived from Na-ion batteries because of their ability to form more stable SEI films.149 However, Tian et al. discovered that treating Na with an I2-containing ether solvent can lead to the creation of a relatively dense NaI layer, which is beneficial for stabilizing the Na anode.151 In this regard, extensive investigation into new-type electrolyte formulations for Na–I2 batteries is required in the future. Besides, few studies have explored the issues related to the Na metal anode in Na–I2 batteries. It is anticipated that more work will be devoted to realizing advanced Na–I2 batteries in the future, as they exhibit advantages of high stability, low cost, and moderate energy density. Last but not least, the concept of high-temperature Na–I2 batteries is also worth exploring, considering the successful application of high-temperature Na-S batteries.
2.3 Potassium–iodine batteries
The redox potential of K/K+ (−2.93 V vs. SHE), compared to that of Na/Na+ (−2.70 V vs. SHE), is closer to that of Li/Li+ (−3.04 V vs. SHE) in nonaqueous electrolytes.153 Besides, potassium reserves in the Earth's crust show a high abundance of 2.09 wt%. These advantages suggest that potassium-based batteries might have high cost-efficiency and promising energy density, thus leading to the emergence of K–I2 batteries. Relevant battery information and performance for K–I2 batteries can be found in Tables 8 and 9. In 2018, Lu et al. first reported a rechargeable K–I2 battery by employing a free-standing I2/carbon cloth composite cathode and a metallic K anode (Fig. 10a).56 By combing in situ Raman and ex situ X-ray photoelectron spectroscopy studies, they confirmed a reversible conversion mechanism between I2, KI3, and KI species, which is similar to the one in Li–I2 batteries. And the as-prepared K–I2 battery showed over 500 charge/discharge cycles with a capacity retention of 71% at 100 mA g−1, demonstrating the feasibility of K–I2 batteries. Subsequently, a K–I2 battery with a mesoporous carbon (CMK-3)/iodine cathode was also investigated, which can be cycled for 300 times at 0.17 A g−1 (Fig. 10b).154 Notably, as the development of K–I2 cells is in its initial infant stage, the cathode materials used above are commonly derived from other MIBs. Especially, on comparison with Li/Na–I2 batteries, several issues associated with K–I2 batteries are exposed, including short lifespan and sluggish kinetics. This could be attributed to the large potassium ion radius, as well as the highly reactive potassium metal anode. Typically, a larger ionic radius means a difficult uptake/release process of K+ in the iodine cathode. This can also cause the iodine cathode to undergo a large volume change (about 106% increase in volume from I2 to KI) upon potassiation and depotassiation. Inevitably, this will drive severe pulverization of iodine hosts, especially carbon-based hosts that typically rely on physical effects to capture and reutilize iodine species. Furthermore, K-metal anodes with high reactivity are prone to dendrite formation and interactions with iodine species, leading to the formation of an unstable SEI layer.68,155,156 Given these facts, K–I2 batteries would perform poorly compared to Li/Na–I2 batteries. To address the above problems, Deng et al. employed KI as the iodine source to effectively alleviate the volume expansion of the iodine cathode.68 Meanwhile, to avoid the possible pulverization of iodine host material, a graphene-modified separator was further adopted to alleviate the shuttle effect, which also led to a dissolution–precipitation reaction mechanism at the iodine cathode (Fig. 10c). For the electrolyte, a 7 M potassium bis(fluorosulfonyl) imide (KFSI) dissolved in EC and DEC (volume ratio 1:1) electrolyte was chosen. As the polyiodide dissolution can be efficiently mitigated by reducing the number of free solvent molecules in the electrolyte, it could be a useful strategy to slow down the diffusion of polyiodide by increasing the viscosity of the electrolyte (Fig. 10d). As a result, the as-prepared K–I2 batteries can maintain a high-capacity retention of 95.5% after 550 cycles at 100 mA g−1, thus representing a state-of-the-art K–I2 battery (Fig. 10e). Nevertheless, these batteries have a shorter lifespan in comparison to Li/Na–I2 batteries. This may be attributed to easier dendrite growth in potassium anodes, thus making potassium anode protection critical for optimal performance.157,158 Combined with the characteristics of KI cathode and previous studies, strategies such as carbon-based, alloy, and transition metal compound anodes could be used to further improve the performance of K–I2 batteries.156 Overall, it is more challenging to achieve high-performance potassium–iodine batteries compared to lithium/sodium–iodine batteries, but perhaps that is what the science is about.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|
| 1, year; 2, cathode; 3, electrolyte formula; 4, the mass loading of iodine at the cathode (wt%); 5, the area loading of iodine at the cathode (mg cm−2). Evaluation of K–I2 cell performance: 6, current density; 7, cycle number; 8, capacity retention (%); 9, reversible capacity (mA h g−1iodine). 10, reference. | |||||||||
| 2018 | I2/carbon cloth | 0.5 M KPF6 in EC/DEC (1:1, v/v) |
44 | 4 | 0.1 A g−1 | 500 | 71 | 156 | 56 |
| 2019 | Mesoporous carbon (CMK-3) | 1 M KPF6 in PC/DEC (1:1, v/v) |
29 | — | 0.17 A g−1 | 300 | — | 126.3 | 154 |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|
| 1, year; 2, separator; 3, cathode; 4, electrolyte formula; 5, iodine loading (mg cm−2). Evaluation of K–I2 cell performance: 6, current density; 7, cycle number; 8, capacity retention (%); 9, reversible capacity (mA h g−1iodine). 10, reference. KFSI, KN(SO2F)2. | |||||||||
| 2021 | Graphene-modified separator | KI/KB | 7 M KFSI in EC/DEC electrolyte (1:1, v/v) |
∼1 (KI) | 0.1 A g−1 | 550 | 95.5 (form 5th cycle) | — | 68 |
 | ||
| Fig. 10 (a) Schematic diagram for the structure and discharge process of a rechargeable K–I2 secondary battery. Reprinted with permission from ref. 56. Copyright 2018 Elsevier. (b) The preparation process and working mechanism of CMK-3/iodine electrode in a K–I2 battery. Reprinted with permission from ref. 154. Copyright 2019 Elsevier. (c) Schematic diagram for the preparation process of a graphene-modified separator. (d) Digital images of color changes over time for 1 M and 7 M KFSI-EC/DEC electrolytes after adding KI3. (e) Long-term cycling performance of K–I2 cell at 100 mA g−1 with 7 M KFSI-EC/DEC electrolyte, graphene-modified separator, and KI@carbon cathode. Reprinted with permission from ref. 68. Copyright 2021 Elsevier. | ||
2.4 Zinc–iodine batteries
Aqueous zinc-ion batteries are one of the most promising sustainable energy storage technologies due to their safety, non-toxicity, and high theoretical capacity.159–162 Especially, the aqueous Zn–I2 battery has garnered significant attention as an ideal contender for large-scale energy storage applications. This can be ascribed to their many intrinsic merits: (1) both of zinc and iodine have high environmental tolerance. Both of them are stable in an atmospheric environment at room temperature, greatly facilitating battery assembly (e.g., without the need of a glovebox) and lowering the entry criteria for battery research. Besides, they show good compatibility with aqueous electrolytes (e.g., ZnSO4 aqueous solution), avoiding the usage of toxic, flammable, and expensive organic electrolytes and (2) Zn metal offers high theoretical volumetric and gravimetric capacities of 5855 mA h cm−3 and 820 mA h g−1, respectively, coupled with a low redox potential of −0.76 V (vs. SHE). When matched with an iodine cathode, a theoretical capacity of 211 mA h g−1 (even up to 422 mA h g−1) and a voltage window of ∼1.3 V can be obtained. Remarkably, this voltage range is close to that of water splitting (∼1.23 V), indicating that the side reaction of H2 evolution at the Zn anode can be effectively avoided to realize a highly stable Zn–I2 battery. Considering its low environmental testing and research necessities, coupled with its environmentally friendliness, safety, and cost-effectiveness, the research on Zn–I2 batteries is poised to experience rapid development. In particular, the high solubility of ZnI2 in water (up to 7 M) allows for the classification of Zn–I2 batteries into two configurations: static and flow (Fig. 11).163 The static configuration primarily loads iodine at the cathode and employs a small amount of electrolyte to achieve high energy density. This makes it well-suited for mobile electronic devices and flexible batteries. On the other hand, the flow configuration dissolves iodine in the electrolyte and stores energy through the reaction of active species in the flow electrolytes at both the cathode and anode. As a result, this configuration is more suitable for large-scale energy storage stations that provide low-cost service to the grid. In the subsequent section, we will delve into a detailed discussion on both types of Zn–I2 cells.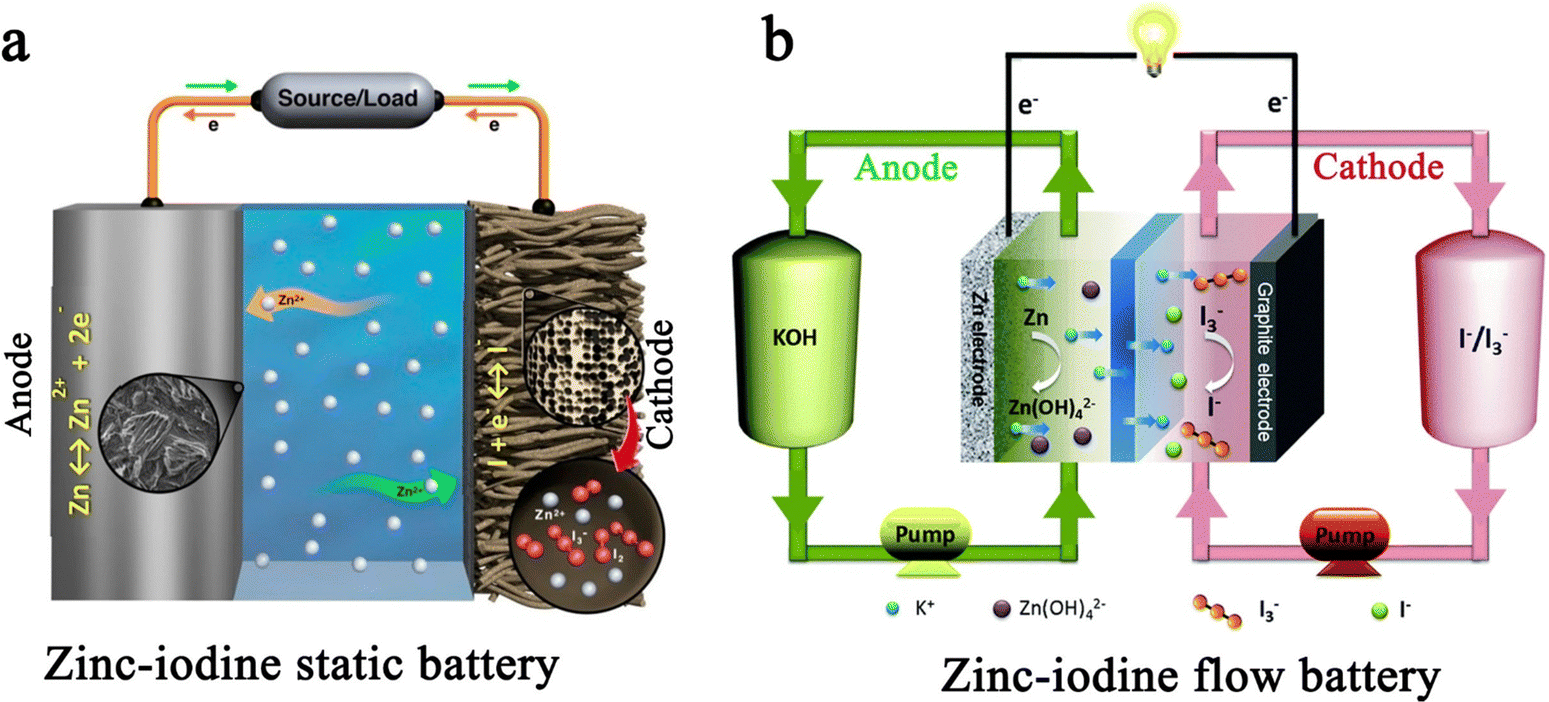 | ||
| Fig. 11 Schematic illustration of a typical (a) Zn–I2 static device. Reprinted with permission from ref. 59. Copyright 2017 American Chemical Society; and (b) Zn–I2 flow device. Reprinted with permission from ref. 164. Copyright 2018 Royal Society of Chemistry. | ||
2.4.1.1 Iodine hosts.
Carbon-based hosts. Carbon-based hosts, like those used in Li–I2 batteries, are commonly utilized in Zn–I2 batteries to prevent the shuttle effect caused by the dissolution and diffusion of iodine species. Table 10 in the iodine host section for Zn–I2 batteries presents relevant battery information and performance. In the early stage of Zn–I2 battery development, carbon cloth was extensively studied as an iodine host. Pan et al. found that a low E/I ratio (the ratio of the electrolyte volume to iodine mass) could improve the ability of active carbon fiber cloth to inhibit the dissolution of iodine species (Fig. 12a).59 This is because the lower E/I ratio favors a fast equilibrium between adsorption on the carbon surface and solvation in the electrolyte of iodine species, thereby reducing the total loss of iodine species. Li et al. also demonstrated that carbon cloth can act as a self-supporting, mechanically flexible cathode to construct cable-type Zn–I2 batteries.72 Particularly, carbon cloth was also revealed to avoid the generation of I3− intermediates (the main “culprit” behind the shuttle effect in Zn–I2 batteries), due to its small internal channels (typical micropore size of ∼6.3 Å), making it difficult for Zn(I3)2 (minimum diameter of 9.5 Å) to reside (Fig. 12b).72,165 As a result, carbon cloth was subsequently often adopted as a current collector of Zn–I2 batteries to improve the battery performance. In combination with the electrodeposition method (i.e., deposition of I− onto carbon cloth electrochemically), the Zn–I2 cell with the as-obtained carbon cloth electrode can stably cycle for 2000 cycles with 19% capacity decay at 1 A g−1.166 Intriguingly, light-assisted chargeable Zn–I2 cells can be realized by incorporating photo-electrodes (i.e., TiO2 and BiOI) onto carbon cathodes.73,167 In particular, a p-type BiOI photocathode is able to increase the discharge voltages (up to 1.49 V) of Zn–I2 batteries because the positive photovoltage of BiOI can be superimposed onto the I−/I3− redox pair during discharging.167 However, the stability and rate performance of this kind of battery are very poor and require further optimization and improvement. In addition to carbon cloth, other carbon materials, such as hollow carbon nanostraws,105 biomass (litchi-shell or gingko) derived hierarchically porous carbon,168,169 and ZIF-8 derived porous carbon,170 have also been reported. However, the non-polar nature of carbon weakens its adsorption to polyiodide polarities, causing these materials to have inferior properties.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|
| 1, year; 2, cathode; 3, electrolyte formula; 4, the mass loading of iodine at the cathode (wt%); 5, the area loading of iodine at the cathode (mg cm−2). Evaluation of Zn–I2 cell performance: 6, current density (C, 1C = 211 mA h g−1); 7, cycle number; 8, capacity retention (%); 9, reversible capacity (mA h g−1iodine). 10, reference. HAc/NaAc, acetic acid/sodium acetate. | |||||||||
| 2017 | I2/active carbon fiber cloth | 1 M ZnSO4 | 38 | — | 2 | 3000 | 90 | ∼128 | 59 |
| 2018 | I2/carbon cloth | 2 M ZnSO4 | — | 8.8 | ∼1 | 200 | 91 | 275 | 72 |
| 2018 | I2/nanoporous activated carbon cloth | 1 M ZnSO4 | — | 4.5 | 5 | 1500 | 90 | 160 | 165 |
| 2019 | S,N co-doped graphene foam | Anolyte, 0.05 M ZnSO4; catholyte, 0.1 M ZnI2/0.01 M I2 | — | — | ∼1 | 500 | 81 | — | 173 |
| 2019 | Super P carbon/Ti/TiO2 photo-electrode | 0.5 M ZnI2 | — | — | 0.1 mA cm−2 | 10 | — | — | 73 |
| 2020 | I2/N-doped porous carbon | 1 M ZnSO4 | 52 | 1.7 | 10 | 10000 |
80.9 | ∼144 | 171 |
| 2020 | I2/polyaniline | 2 M ZnSO4 | ∼33 | ∼0.7–1.2 | ∼7 | 700 | 79 | 160 | 186 |
| 2020 | I2/Co[Co1/4Fe3/4(CN)6] | 2 M ZnSO4 | 61.2 | — | ∼19 | 2000 | 80.2 | 165.6 | 190 |
| 2021 | Polypyrrole/carbon cloth | 0.5 M ZnI2 in 0.1 M HAc/NaAc buffer solution | — | — | ∼0.4 | 600 | — | — | 187 |
| 2021 | I2/porous carbon material ZC-mK2CO3 | 2 M Zn(CF3SO3)2/0.1 M ZnI2 | 50 | ∼1.0–1.4 | 50 | 35000 |
∼98.4 | 150 | 175 |
| 2021 | I2/N-doped carbon nanofiber | 2 M ZnSO4 | — | 0.8–1.0 | 5 | 10000 |
77 | — | 176 |
| 2021 | I2/Nb2CTX | 1 M ZnSO4 | 80–85 | — | ∼28 | 23000 |
80 | — | 83 |
| 2022 | I2/N-doped porous carbon nanocages | 2 M ZnSO4 | 50 | ∼1.0 | ∼24 | 3500 | — | — | 177 |
| 2022 | Polyiodide/the strong base resin-D296 | 2 M ZnSO4 | 40 | 1.5–2.0 | ∼15 | 17000 |
100 | 105.1 | 27 |
| 2022 | I2/graphitic/porous carbon microtubes | 1 M ZnSO4/0.1 M ZnI2 | 50 | 2.9–3.2 | ∼19 | 1000 | 86.8 | — | 174 |
| 2022 | Starch | Anolyte, 0.5 M ZnSO4/0.5 M Li2SO4; catholyte, 0.1 M I2/1 M LiI | — | — | ∼47 | 50000 |
90.5 | 75 | 42 |
| 2022 | Polyaniline/carbon felt | 0.75 M ZnCl2/1.5 M KI | — | — | ∼17 | 10000 |
75.7 | — | 188 |
| 2022 | I2/ZIF-8 derived porous carbon | 1 M ZnSO4 | 22 | 1.7 | ∼4 | 1000 | 72 | 99 | 267 |
| 2022 | I2/biomass derived hierarchically porous carbon | 1 M ZnSO4 | 41 | — | ∼1.4 | 800 | — | ∼70 | 169 |
| 2022 | I2/porous hollow carbon nanostraw | 0.5 M ZnSO4/0.5 M H2SO4 | ∼25 | — | ∼4.7 | 1500 | — | — | 105 |
| 2022 | I2/CMK-3 | 2 M ZnSO4 | 60 | 1.2–1.5 | ∼47 | 39000 |
∼80.6 | — | 268 |
| 2022 | I2/N-doped porous carbon@single Fe atom | 2 M ZnSO4 | 30 | 1 | 10 | 10000 |
— | 170 | 183 |
| 2022 | I2/carbon cloth | 2 M ZnSO4 | — | 1.45 | ∼4.7 | 2000 | 81 | ∼136 | 166 |
| 2022 | I2/ZIF-8-derived N-doped porous carbon | 1 M ZnSO4 | 58 | 6 | ∼47 | 10000 |
67 | ∼133 | 170 |
| 2022 | BiOI semiconductor photoelectrode | 10 mM ZnI2/1.6 mM I2 | — | — | — | — | — | — | 167 |
| 2023 | I2/N-doped litchi-shell derived porous carbon | 1 M ZnSO4 | 44 | 1.5 | ∼1.4 | 400 | — | — | 168 |
| 2023 | I2/N-doped hierarchical porous carbon | 1 M ZnSO4 | 61.6 | — | 5 | 10000 |
85.2 | 166.7 | 172 |
| 2023 | I2/carbon nanosheets derived from ZIF-8 | 2 M ZnSO4 | 40 | ∼2 | 20 | 6000 | 85 | — | 180 |
| 2023 | I2-loaded defect-rich graphene | 2 M ZnSO4/25 mM ZnI2 | 40 | 0.5–1 | ∼4.7 | 3500 | 88.1 | ∼230 | 185 |
| 2023 | W2N modified porous carbon polyhedron | 2 M ZnSO4 | — | 0.8–1 | 5 | 2000 | 85 | 170.3 | 178 |
| 2023 | PTCDA-derived porous carbon | 2 M ZnSO4 | 40 | — | ∼23.7 | 50000 |
55 | ∼90 | 269 |
| 2023 | I2/Fe single atom on mesoporous N-doped carbon | 2 M ZnSO4/0.04 M I3− | 76.72 | — | ∼23.7 | 50000 |
79.5 | 159.7 | 182 |
| 2023 | I2/LiVS2 | 2 M ZnSO4 | 60.7 | ∼1.5 | ∼23.7 | 30000 |
86 | — | 270 |
| 2023 | I2/Fe2N decorated porous N-doped carbon fibers | 2 M ZnSO4 | — | — | 2 | 1500 | 79 | 214 | 184 |
| 2023 | I2/Ni single atoms on hierarchical porous carbon | 2 M ZnSO4 | 41.7 | 11.6 | 10 | 10000 |
93.4 | 141 | 181 |
| 2023 | I2/porous oxidized salt-templated carbon | 2 M ZnSO4 | 26.6 | ∼0.3–0.5 | ∼4.7 | 10000 |
85.04 | — | 251 |
| 2023 | Pre-embedded Prussian blue with iodide ions | 1 M ZnSO4/5 mM KI | — | — | ∼19 | 1500 | 94 | 206 | 191 |
| 2023 | I2/N-doped nanocarbons with ∼2.5 nm pores | 1 M ZnSO4 | 60.8 | — | 5 | 10000 |
— | 178.8 | 179 |
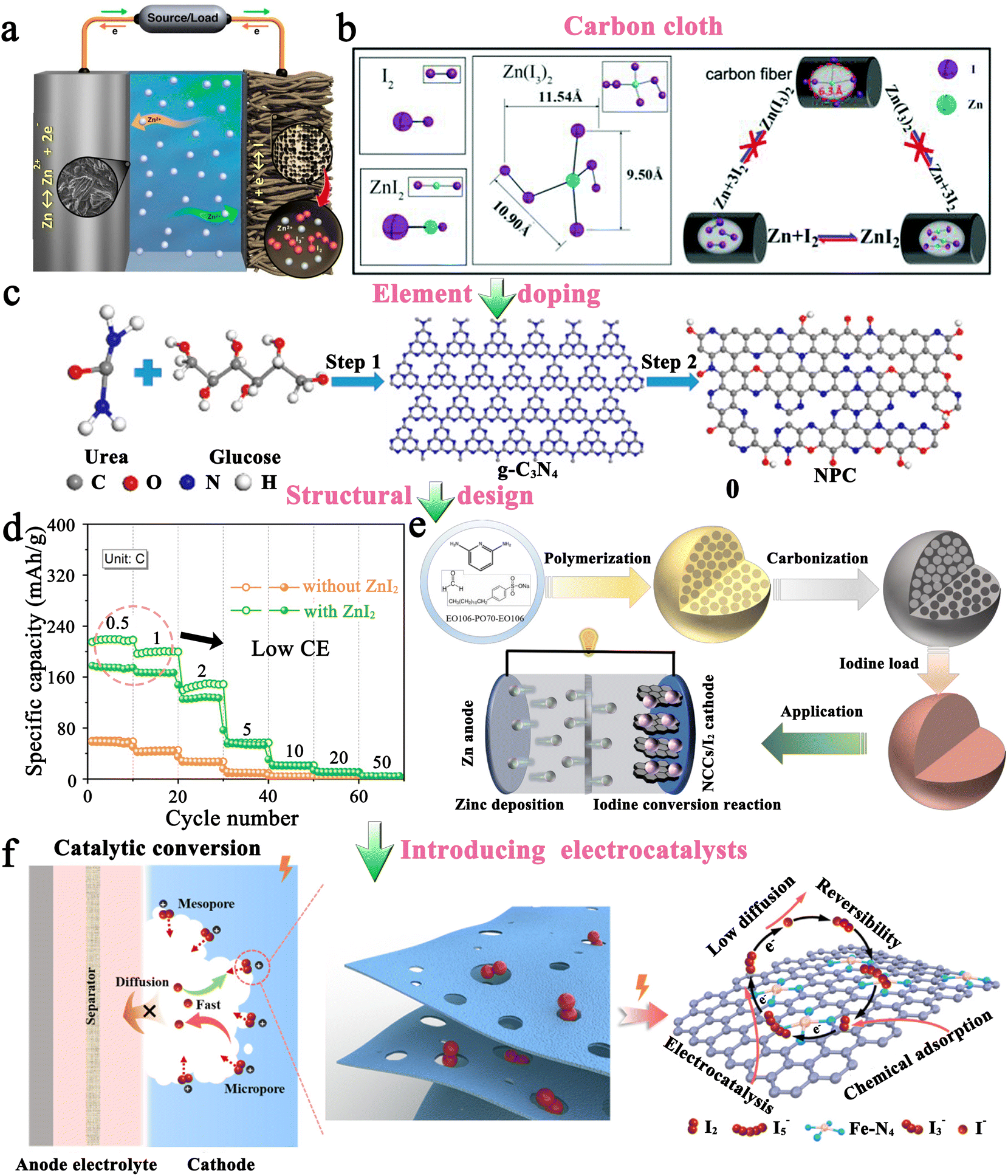 | ||
| Fig. 12 (a) Schematic diagram for the working process of Zn–I2 battery with active carbon fiber cloth@I2 cathode. Reprinted with permission from ref. 59. Copyright 2017 American Chemical Society. (b) The configurations of I2, ZnI2 and Zn(I3)2 molecules, and the proposed possible electrode reaction process in the micropores of carbon cloth. Reprinted with permission from ref. 72. Copyright 2018 Royal Society of Chemistry. (c) Schematic illustration for the synthesis process of a nitrogen-doped porous carbon host. Reprinted with permission from ref. 171. Copyright 2020 American Chemical Society. (d) Rate performance of Zn–I2 cells in 2 M Zn(CF3SO3)2 electrolyte with or without 0.1 M ZnI2 additive. Reprinted with permission from ref. 175. Copyright 2021 American Chemical Society. (e) Schematic synthesis for the nitrogen-doped porous carbon nanocages@I2 cathode in a Zn–I2 battery. Reprinted with permission from ref. 183. Copyright 2022 American Chemical Society. (f) Schematic diagram for the working mechanism of polyiodide adsorption/conversion on the porous carbon with the atomic bridging structure of metal–nitrogen–carbon. Reprinted with permission from ref. 177. Copyright 2022 American Chemical Society. | ||
Next, a heteroatom doping strategy has been proposed for Zn–I2 batteries, capable of fine-tuning the composition and physicochemical properties of carbon materials, resulting in increased surface polarity of the carbon crystal lattice. Yu et al. took the lead in achieving a breakthrough in carbon-based iodine hosts (Fig. 12c).171 With N-doped porous carbon as the iodine scaffold, they implemented a high-performance Zn–I2 battery that can be cycled 10000 times with a capacity retention of 81% at 10C. Especially, they discovered that graphitic-N exhibits the strongest adsorption of iodine, while pyridinic-N has the weakest adsorption of iodine. Furthermore, Liu et al. revealed that graphitic-N can promote the conversion of I− to I2; this is due to the charge redistribution effect resulting from the interaction of graphite N with iodine molecules, which increases the length of I–I intermolecular bonds, leading to the facile cleavage/formation of I–I bonds during cycling.170 Surprisingly, Gong et al. recently concluded that pyridinic-N is superior in adsorbing iodine species and catalyzing the electrochemical reactions.172 These conflicting findings call for additional studies with sufficient samples to understand the real role played by different-N species. In any case, it is essential for carbon materials to promote the electrochemical performance of Zn–I2 batteries by tuning the ratio of graphite-N to pyridine-N species. It is worth noting that the reported multi-component doping materials did not effectively enhance the electrochemical properties of batteries compared to those using a single N-doping. For example, Lu et al. reported S and N co-doped three-dimensional graphene foams that can be used as both cathodic and anodic matrixes.173 However, the as-assembled Zn–I2 batteries can only achieve 500 cycles with a capacity retention of 81% at 200 mA g−1. Coincidentally, Zn–I2 batteries with multiple heteroatoms (N, O, S, etc.) co-doped graphitic/porous carbon microtubes as the iodine host showed a capacity retention of 86.8% after 1000 cycles.174 One possible reason for this is that the electrolytes used in these systems contain ZnI2, which greatly promotes the polyiodide shuttling in the cathode, resulting in poor battery stability.95,175 As shown in Fig. 12d, the ZnI2 additive results in a low CE at low rates, indicating that the addition of ZnI2 actually leads to the formation of more water-soluble polyiodide via the process of I− + I2 → I3, promoting the shuttle effect of batteries. Nonetheless, the battery exhibits an enhanced capacity as the ZnI2 additive can realize a rapid transformation of iodine from the solid to liquid state and result in an improved iodine utilization.175 Therefore, the ZnI2 additive may be a double-edged sword, increasing the utilization of insulating iodine but also aggravating the shuttle effect of the battery. In terms of the structural design of heteroatom-doped carbon, N-doped porous carbon nanofibers, N-doped porous carbon nanocages, and tungsten nitride-modified porous carbon (ZIF-8) polyhedra have also been reported with excellent energy storage performance (Fig. 12e).176–178 Besides, as mentioned earlier, the control of pore size in carbon-based materials enables the I−/I2 process and prevents the polyiodide shuttle effect effectively. Thus, Hou et al. constructed an N-doped nanocarbon with a pore structure of approximately 2.5 nm meticulously.179 As anticipated, the resulting N-doped nanocarbon cathode, with an iodine content of 60.8 wt%, exhibited excellent long-term cycling stability, corresponding to a reversible specific capacity of 178.8 mA h g−1 after 10000 cycles at 5C. To further investigate the structure-electrochemical properties of heteroatom-doped carbon cathodes in Zn–I2 batteries, Sun et al. prepared four different configurations of porous carbon, namely, carbon nanosheets, carbon nanoshells, carbon skeletons, and carbon dodecahedra, by altering the additives with ZIF-8 as precursors.180 Despite having the highest heteroatom content, carbon dodecahedra/I2 did not exhibit the best electrochemical performance. In contrast, carbon nanosheets/I2 displayed superior performance due to its maximum total pore volume and suitable pore size (containing pores of 1.2–3 nm). This indicates that heteroatom content alone is insufficient in assessing material properties, while factors such as pore size, porosity, specific surface area, and morphology, are equally significant.
Based on the aforementioned approaches, the polyiodide shuttle effect in Zn–I2 batteries can be mitigated effectively. However, another pressing concern pertains to the sluggish cell kinetics caused by the high activation energy of iodine conversion and the insulating property of I2 (∼10−9 S cm−1).180 To address this, one effective solution lies in the incorporation of single atom catalysts (SACs) into carbon-based materials (Fig. 12f). This strategy exploits the activated and isolated catalytic metal atoms of SACs, which provide optimal utilization of catalytic sites for catalytic reactions. Consequently, a small amount of catalyst enables a potent catalytic effect, avoiding an increase in the inactive cathode material. Currently, investigations are focusing on Fe and Ni single atoms in Zn–I2 batteries, both exhibiting excellent catalytic properties.181–183 For instance, when Fe single atoms were introduced into N-doped porous carbon, the resulting composite could achieve an outstanding redox kinetics, corresponding to a specific capacity of 158 mA h g−1, even at 20C.183 This can be attributed to the decreased energy barrier for the iodine conversion process facilitated by the Fe single atoms. Similarly, Ma et al. realized an ultra-high specific capacity of 121 mA h g−1 at 50C by dispersing Ni single atoms within hierarchical N-doped porous carbon, affirming the beneficial role of SACs in facilitating the iodine redox process.181 On the other hand, transition metal oxides, sulfides, nitrides, and carbides are also regarded as excellent electrocatalysts. Among them, transition metal nitride (Fe2N) has been explored for use as an electrocatalyst in Zn–I2 batteries due to its favorable electronic conductivity and intrinsic catalytic activity. Ding et al. reported the fabrication of a Fe2N-decorated N-doped carbon fiber-based iodine host using electrospinning and carbonization techniques.184 The resulting Zn–I2 batteries delivered a specific capacity of 146 mA h g−1 at 10C, slightly inferior to that achieved with SACs. In addition to the above electrocatalysts, Niu et al. also proposed the concept of defect-induced catalysis to expedite the iodine conversion process.185 By introducing defects (specifically carbon seven-membered ring defect sites) in the N-doped graphene, the active sites were increased while effectively catalyzing the iodine conversion process. Consequently, Zn–I2 batteries exhibited impressive specific capacities of 278.4 mA h g−1 at 0.5 A g−1 and 131.9 mA h g−1 at 10 A g−1. Therefore, the incorporation of electrocatalytic functionality into carbon-based materials proves to be a viable approach for achieving fast cathodic kinetics.
Overall, future design of carbon-based hosts in Zn–I2 batteries may require the following key factors: (1) a large surface area to provide a large number of active sites to reserve and anchor iodine species; (2) suitable pore size to hinder the formation of Zn(I3)2; (3) heteroatom doping to enhance the interaction between carbon and iodine species, thereby resulting in increased stability and reversibility of battery; (4) modulation of the graphite-N/pyridine-N value to optimize the adsorption and catalytic capacity of the materials for iodine species; and (5) the introduction of electrocatalysts (e.g., metal single atoms, transition metal oxides/sulfides/nitrides/carbides), and defective engineering to further enhance the catalytic ability for efficient iodine conversion.
Organic polymer hosts. In addition to carbon-based hosts, organic compounds have recently been pursued in Zn–I2 batteries due to their abundant active sites, adjustable structure, sustainability, and cost-effectiveness. However, PVP, which has been commonly investigated in Li–I2 batteries, may not be suitable for Zn–I2 batteries because of its high solubility in water. Therefore, when designing organic iodine hosts for different MIBs, the compatibility of electrolytes and electrode materials must be considered to avoid possible problems, such as dissolution and side reactions. Conducting polymers, such as polyaniline (PANI) and polypyrrole (PPy), have been extensively studied in Zn–I2 batteries. In general, the PANI framework can confine polyiodide through the electrostatic interactions with non-protonated amine (–NH–) and protonated imine (–NH+
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ) functional groups (Fig. 13a).186 Besides, the doping/de-doping processes of polyiodide in the PANI framework could improve the conductivity of PANI, thereby promoting the redox kinetics. Likewise, PPy also exhibits a doping/de-doping mechanism of iodine species on its backbone.187 Significantly, PANI has higher capacity and stronger stability than PPy.188 This can be attributed to the micro-nano-porous structure of PANI (PPy with a dense structure), which can facilitate the contact of PANI with electrolytes and also provide more active adsorption sites to minimize the shuttling of polyiodide ions. However, the lifetime of PANI-based Zn–I2 batteries is relatively short (<1000 cycles). One boosting strategy is to compound PANI with carbon materials but this undoubtedly increases the content of inactive material and reduces the cell density (Fig. 13b).188 Another approach is to change the electrolyte formulation by using a high concentration of ZnCl2 (containing KI or ZnI2 as the iodine source) (Fig. 13c). In this case, I− ions exist in the form of [ZnIx(OH2)4−x]2−x (x = 2), which can prevent I2 from forming water-soluble polyiodide, thereby improving battery performance86,189 As a result, the as-prepared PANI-based Zn–I2 battery was able to maintain an ultrahigh capacity retention of 99.9% after 1000 cycles.189 Despite this, the adsorption capacity of PANI for polyiodide is not sufficient to realize Zn–I2 batteries with long-term service life. Zhang et al. took the lead in making a breakthrough in the polymer-based iodine host of Zn–I2 batteries.27 They proposed an inexpensive, facile, and universal quaternization engineering approach to regulate polyiodide. Benefiting from the construction of an “electric double layer” structure between quaternary ammonium cations and iodine anions, the polyiodide shuttling is significantly eliminated. Especially, quaternary ammonium cations enable the generation of a large energy barrier between I3− and I2, resulting in fast solution-based iodine chemistry (I−/I3−) for high-rate charge–discharge of batteries. The resulting cutting-edge Zn–I2 battery demonstrated ∼100% capacity retention over 17000 cycles at 3.2 A g−1, which greatly prolongs the life of polymer-based Zn–I2 batteries and effectively improves the rate performance of the battery (Fig. 13d). Besides, Qiao's group reported the utilization of starch as an iodine host.42 Owing to the unique double-helix structure of starch, various iodine species can be strongly confined within the helical chains through bonding effects (Fig. 13e and f). The resulting starch-based Zn–I2 battery delivered over 50000 cycles with a capacity retention of 90.5% at 10 A g−1. All these results well confirm the promising application of organic polymer hosts in Zn–I2 batteries, especially considering that long-lasting and high-rate Zn–I2 batteries have been achieved.
) functional groups (Fig. 13a).186 Besides, the doping/de-doping processes of polyiodide in the PANI framework could improve the conductivity of PANI, thereby promoting the redox kinetics. Likewise, PPy also exhibits a doping/de-doping mechanism of iodine species on its backbone.187 Significantly, PANI has higher capacity and stronger stability than PPy.188 This can be attributed to the micro-nano-porous structure of PANI (PPy with a dense structure), which can facilitate the contact of PANI with electrolytes and also provide more active adsorption sites to minimize the shuttling of polyiodide ions. However, the lifetime of PANI-based Zn–I2 batteries is relatively short (<1000 cycles). One boosting strategy is to compound PANI with carbon materials but this undoubtedly increases the content of inactive material and reduces the cell density (Fig. 13b).188 Another approach is to change the electrolyte formulation by using a high concentration of ZnCl2 (containing KI or ZnI2 as the iodine source) (Fig. 13c). In this case, I− ions exist in the form of [ZnIx(OH2)4−x]2−x (x = 2), which can prevent I2 from forming water-soluble polyiodide, thereby improving battery performance86,189 As a result, the as-prepared PANI-based Zn–I2 battery was able to maintain an ultrahigh capacity retention of 99.9% after 1000 cycles.189 Despite this, the adsorption capacity of PANI for polyiodide is not sufficient to realize Zn–I2 batteries with long-term service life. Zhang et al. took the lead in making a breakthrough in the polymer-based iodine host of Zn–I2 batteries.27 They proposed an inexpensive, facile, and universal quaternization engineering approach to regulate polyiodide. Benefiting from the construction of an “electric double layer” structure between quaternary ammonium cations and iodine anions, the polyiodide shuttling is significantly eliminated. Especially, quaternary ammonium cations enable the generation of a large energy barrier between I3− and I2, resulting in fast solution-based iodine chemistry (I−/I3−) for high-rate charge–discharge of batteries. The resulting cutting-edge Zn–I2 battery demonstrated ∼100% capacity retention over 17000 cycles at 3.2 A g−1, which greatly prolongs the life of polymer-based Zn–I2 batteries and effectively improves the rate performance of the battery (Fig. 13d). Besides, Qiao's group reported the utilization of starch as an iodine host.42 Owing to the unique double-helix structure of starch, various iodine species can be strongly confined within the helical chains through bonding effects (Fig. 13e and f). The resulting starch-based Zn–I2 battery delivered over 50000 cycles with a capacity retention of 90.5% at 10 A g−1. All these results well confirm the promising application of organic polymer hosts in Zn–I2 batteries, especially considering that long-lasting and high-rate Zn–I2 batteries have been achieved.
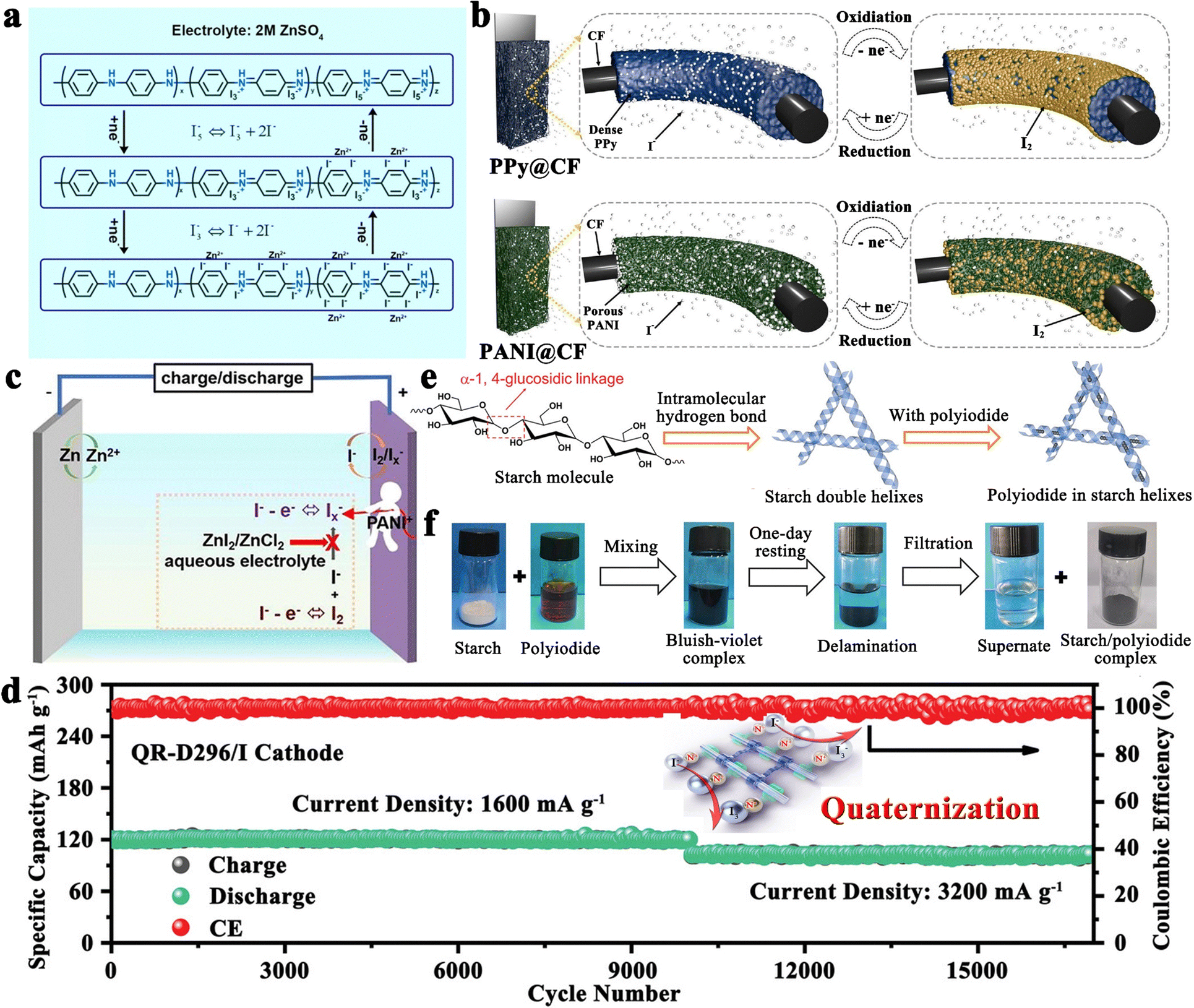 | ||
| Fig. 13 (a) Proposed redox mechanism of the PANI@I2 cathode in a Zn–I2 battery. Reprinted with permission from ref. 186. Copyright 2020 American Chemical Society. (b) Schematic diagram for the redox reaction of iodine species in PPy@carbon felt and PANI@carbon felt hosts. Reprinted with permission from ref. 188. Copyright 2022 Elsevier. (c) Schematic illustration for the working process of the PANI@I2 cathode using ZnI2/ZnCl2 electrolyte in a Zn–I2 cell. Reprinted with permission from ref. 189. 2021 Elsevier. (d) Long-term cycling performance of quaternized cathode at different C-rates. Reprinted with permission from ref. 27. Copyright 2022 Wiley. (e) Schematic diagrams for the chemical/double-helix structure of starch molecules and starch/polyiodide complex. (f) Photographs revealing the polyiodide capture processes by starch. Reprinted with permission from ref. 42. Copyright 2022 Wiley. | ||
Other types of iodine hosts. In addition to carbon-based hosts and organic polymer hosts, several other types of materials have been taken into consideration for their utilization as iodine hosts, including Prussian blue analogues (PBAs), Prussian blue (PB) and MXenes.83,190,191 Generally, PBAs possess adequate porosity, continuous and ordered micropore channels, and transition-metal species with electrochemical catalytic functions, which endow PBAs with the promising potential to act as iodine hosts. Accordingly, the rate capability of the Co[Co1/4Fe3/4(CN)6]/I2‖Zn cell can reach to 151.4 mA h g−1 even at 20 A g−1.190 Nevertheless, its cycle stability performance is not very prominent, with a capacity retention of 80.2% after 2000 cycles at 4 A g−1. This may be attributed to its uncontrolled phase transformation upon cycling process, resulting in structural failure.192,193 Similarly, the PB cathode with pre-embedded iodide ions had a short lifetime of 1500 cycles at 4 A g−1.191 On the other hand, MXenes could also be considered as iodine hosts on account of their exceptional electrical conductivity, abundant surface terminations (
O, –F, –OH, etc.), periodic layered microstructure, and arrayed interlayer gap. Li et al. used the 211-type Nb-based MXene (Nb2CTx) as a proof-of-concept for incorporation with I2.83 As a result, the obtained I2@Nb2CTx MXene electrode exhibited a lifespan of 23000 cycles at 6 A g−1, corresponding to a capacity retention of 80%. The emergence of these materials has greatly expanded the selection of iodine hosts. Importantly, the concept of catalyzing the iodine reduction reaction could be popularized more widely, as it commonly leads to batteries with extremely high-rate capability, even as high as 20 A g−1 (PBAs).
2.4.1.2 Electrolyte. Extensive research has also been carried out on electrolytes for Zn–I2 batteries. Table 11 in the modified electrolyte section for Zn–I2 batteries presents the relevant battery information and performance. An important breakthrough lies in the realization of the two-electron iodine redox reaction (i.e., I− ↔ I0 ↔ I+). Typically, I+ is highly unstable in aqueous solutions and can be further oxidized to form electrochemically irreversible IO3−. However, electrophilic I+ will be activated and stabilized by forming charge-transfer complexes through strong interactions with nucleophile species, such as halides, cyanides, and amines.40,87,194–197 For the first time, Li et al. investigated the effects of different salt components (i.e., 2 M ZnSO4 electrolyte with/without one of KF, NaF, NH4F, KCl, NaCl, and NH4Cl salt) on the activation and stability of I+ ions in Zn–I2 batteries.74 As shown in Fig. 14a, with the introduction of F− (Cl−) ions, an obvious redox peak pair related to the redox of I0/I+ appeared at approximately 1.84/1.66 V (1.74/1.66 V), indicating the successful activation of I0/I+ conversion. Intriguingly, the high-potential I0/I+ reaction in chloride-containing salt electrolytes is more reversible compared to the electrolytes containing F− ions. On the other hand, cations also affect the stability of I0/I+ conversion. Among three different cations (K+, Na+, and NH4+), the K+ cation is recognized to raise the redox potential of the I−/I0 transition due to its lower conversion energy barrier compared to the Na+ and NH4+ ions.74 In view of this, a mixed electrolyte with 2 M ZnCl2 and 1 M KCl was chosen to realize the two-electron iodine redox reaction in Zn–I2 batteries. By using the Ti3C2I2 MXene as the cathode and Zn foil as the anode, the as-assembled Zn–I2 battery showed a cyclic lifespan of over 2800 cycles with 80% capacity retention, as well as an excellent rate of 126 mA h g−1 (207 mA h g−1) at 5 A g−1 (0.5 A g−1) (based on the mass of Ti3C2I2). However, the charge transfer complexes may be at risk of hydrolysis in aqueous media.196,198,199 Because ICl would be hydrated by water (donating its lone pair of electrons) to form H2OICl due to the unsaturated coordination sphere of iodine in ICl.198,200 Then, the deprotonation or the disproportionation of the H2OICl hydrate yields HIO, via. H2OICl → HIO + HCl (deprotonation), or H2OICl → H2OI+ + Cl− → HIO + HCl (disproportionation) (total reaction: ICl +H2O → HIO + HCl). Moreover, HIO intermediates could be further decomposed in acidic environments, via. 2HOI → HIO2 + HI, and HOI + HIO2 → HIO3 + HI (total reaction: 3HOI → HIO3 + 2HI).196,199 Especially, in the presence of light or metal catalysts, HIO intermediates would be decomposed via another pathway, i.e., 2HIO ↔ 2HI + O2, and 2HIO + 2IH ↔ 2I2 + 2H2O (total reaction: 4HIO ↔ 2I2 + 2H2O + O2).40,201 Therefore, Zou et al. recently achieved a more reversible I0/I+ conversion by considering the hydrolysis issue of ICl.40 They found that the I0/I+ process could be activated even in a diluted 1 M ZnCl2 solution, despite its low reversibility. Upon increasing the ZnCl2 concentration to 30 M, the I0/I+ conversion will be fully activated and exhibit high reversibility due to the reduction of free water and the strong coordination of Cl− to I+. Nevertheless, such a high concentration of ZnCl2 electrolyte resulted in poor battery stability, which may be attributed to the formation of non-electroactive Zn–I clusters ([ZnIx(OH2)4−x]2−x (x = 2)) during the cycling process and the low Cl− activity that cannot greatly suppress I+ hydrolysis (Fig. 14b). Notably, Hong et al. revealed that the Zn–I clusters formed by high concentrations of chloride salts can effectively inhibit the shuttling of polyiodide; this is due to the low concentration level of free iodide ions in such an electrolyte, which prevents iodine to combine with iodide ions to form water-soluble polyiodide (Fig. 14c).86 In this respect, it is still worth considering whether the formation of Zn–I clusters contributes to cell instability in two-electron iodine redox reactions. One more thing to note is that ZnCl2 is a strong Lewis acid that can form [ZnCl2(OH)]− anions and is therefore highly corrosive. Consequently, 30 M ZnCl2 electrolyte may cause severe corrosion to the battery structure, causing a decline in battery capacity. Given this, a hybrid electrolyte of 19 M ZnCl2 + 19 M LiCl + 8 M acetonitrile was formulated. LiCl, which possesses high solubility and miscibility with ZnCl2, can not only provide an extra Cl− source but also further regulate the coordination between Zn2+, H2O, and Cl− due to the unique solvation properties of lithium ions. Additionally, acetonitrile can act as a diluent to facilitate ion transport while also reducing water activity due to its inertness. In this case, the Zn–I2 battery revealed a reversible capacity of 420 mA h g−1 after 6000 cycles with a capacity fading of only 0.003% per cycle at 2 A g−1, indicating noteworthy competitiveness against vanadium-based/manganese-based oxides.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|
| 1, year; 2, electrolyte formula; 3, cathode; 4, iodine loading (mg cm−2). Evaluation of Zn–I2 cell performance: 5, current density (C, 1C = 211 mA h g−1); 6, cycle number; 7, capacity retention (%); 8, reversible capacity (mA h g−1iodine). 9, reference. EG, ethylene glycol; PPO, poly(propylene oxide); PEG, poly(ethylene glycol). | ||||||||
| 2020 | 2 M ZnCl2/1 M KCl | Ti3C2I2 MXene | — | ∼14 | 2800 | 80 | — | 74 |
| 2020 | Anolyte, 0.5 M ZnSO4/PEO53–PPO34–PEO53; catholyte, 0.1 M I2/1 M KI/PEO53–PPO34–PEO53 | Carbon cloth | — | 1 | 500 | 94.3 | 198 | 82 |
| 2020 | Anolyte, 0.5 M ZnSO4 in PEO53–PPO34–PEO53; catholyte, 0.2 M I2/porous MOF gel/carbon cloth | — | — | 1 | 1500 | 95.8 | 184.9 | 81 |
| 2021 | ZnCl2/LiCl/acetonitrile (19:19:8, m/m/m) |
I2/PAC carbon | 0.48–0.8 | ∼9.5 | 6000 | 82 | 420 | 40 |
| 2021 | Alginate-based polyanionic hydrogel electrolyte | I2/AC | — | ∼9.5 | 2000 | 66.8 | 75.3 | 207 |
| 2022 | 25% PEG/1 M Zn(CF3SO3)2/1 M KI | Double carbon cloth | — | 7 mA·cm−2 | 1200 | ∼58 | — | 203 |
| 2022 | 1 M Zn(CF3SO3)2/4 M N-methylacetamide/0.5 M KI in 20% volume fractions of H2O | AC | — | 4 mA·cm−2 | 5000 | 98.7 | 2.2 mA h cm−2 | 85 |
| 2022 | 9 M ZnCl2/1 M ZnI2 | Polyaniline | — | 6 mA cm−2 | 1000 | 99.9 | — | 189 |
| 2022 | 1 M ZnI2 in starch aqueous solution | Carbon cloth | — | 10 mA cm−2 | 2000 | 92 | 0.72 mA h cm−2 | 271 |
| 2022 | Cross-linked polyacrylamide/2 M ZnSO4/0.5 M 1-methyl-3-n-propylimidazolium iodide polyelectrolyte | Hydrophilic O-containing CNT | — | 10 mA cm−2 | 2600 | 89.2 | ∼0.35 mA h cm−2 | 206 |
| 2023 | 2 M ZnSO4/0.2 M ZnI2/20% EG | Ordered porous graphene | — | ∼24 | 15000 |
97.6 | 144.1 mA h cm−3 | 204 |
| 2023 | Iota-carrageenan gel electrolyte | N,P co-doped porous carbon | ∼1.5 | 5 | 5000 | 91.9 | — | 88 |
| 2023 | 5 M KI/20 M ZnCl2 | Hydrothermal RGO | 22.5 | 50 mA cm−2 | 2000 | ∼100 | 2 mA h cm−2 | 202 |
| 2023 | Vermiculite nanosheets suspension electrolyte | I2/activated carbon cloth | 2.5–2.8 | 20 | 40000 |
∼100 | 149.8 | 205 |
| 2023 | 4 M ZnSO4/0.4 M LiI | 3D porous carbon | — | 2 | 5000 | 95 | — | 272 |
| 2023 | 5.9 mole per kilogram ZnCl2 + 0.20 M KI | I2/activated porous corncob carbon | 0.8–1.8 | ∼2.4 | 600 | — | — | 273 |
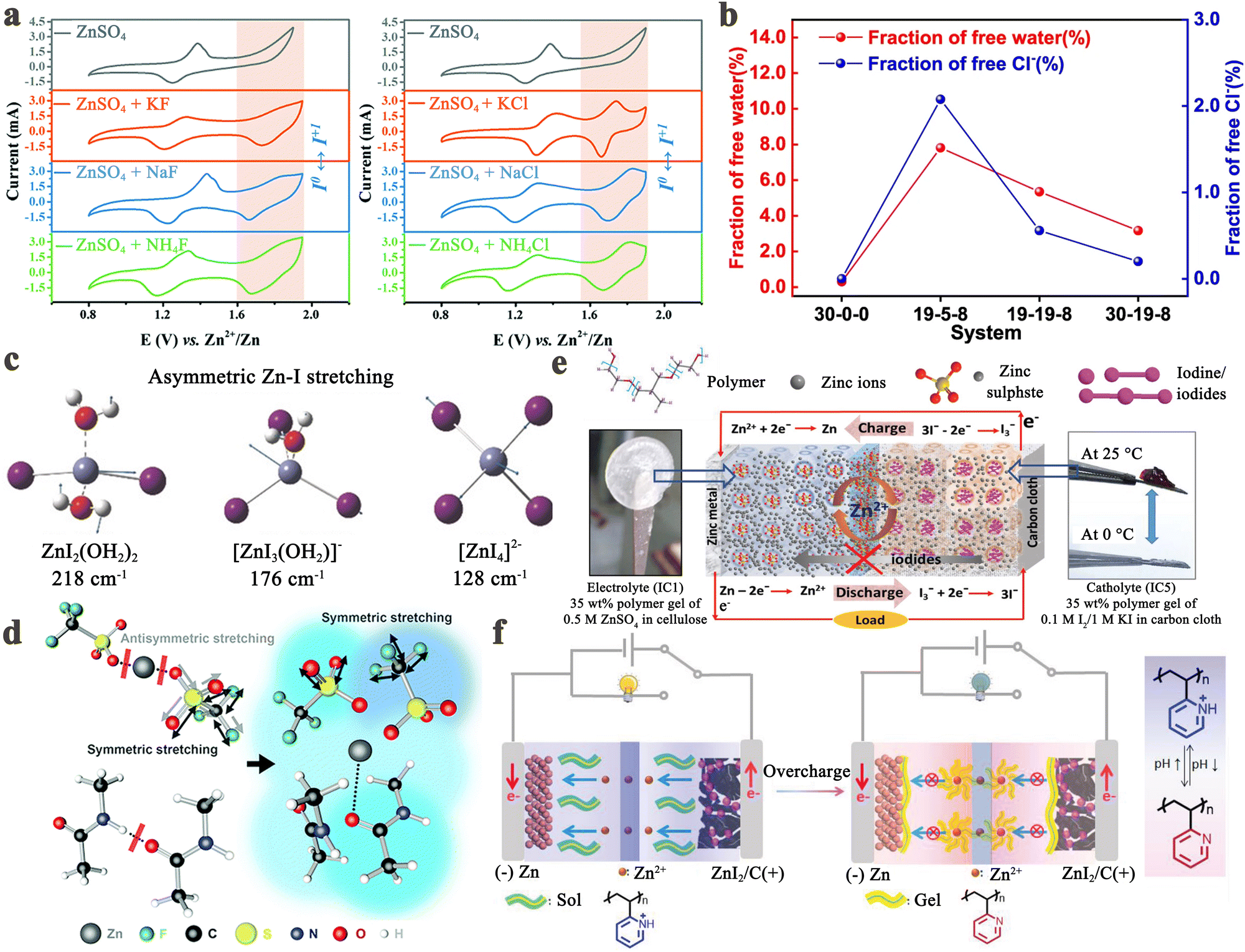 | ||
| Fig. 14 (a) CV curves of the Zn–I2 batteries in 2 M ZnSO4 electrolyte without or with various F−/Cl−ions additives (including KF, NaF, NH4F, KCl, NaCl, and NH4Cl salts). Reprinted with permission from ref. 74. Copyright 2020 Royal Society of Chemistry. (b) Summary of free water and Cl− contents in electrolytes with different formulations based on molecular dynamics simulations. The horizontal coordinate in the graph defines the specific molarity of ZnCl2, LiCl, and ACN in water. For example, 19-19-8 means 19 M ZnCl2 + 19 M LiCl + 8 M acetonitrile. Reprinted with permission from ref. 40. Copyright 2021 Springer Nature. (c) Depiction of major Raman modes of zinc iodide in aqueous solution from DFT calculations. H, white; O, red; Zn, light blue; I, purple. Reprinted with permission from ref. 86. Copyright 2019 Wiley. (d) The proposed formation mechanism of Zn(CF3SO3)2@N-methylacetamide eutectic solution. Reprinted with permission from ref. 85. Copyright 2022 Royal Society of Chemistry. (e) Schematic representation of the working mechanism of Zn–I2 battery with PEO53–PPO34–PEO53 gel electrolyte. Reprinted with permission from ref. 82. Copyright 2020 Wiley. (f) Schematic illustration of the self-protection function of a stimulus-responsive Zn–I2 battery. Reprinted with permission from ref. 208. Copyright 2020 Wiley. | ||
In addition to increasing the theoretical capacity of the battery, electrolytes have also been developed to serve multiple functions, including the reduction of iodine species diffusion, fabrication of flexible electronic devices, protection of the zinc anode, and prevention of battery overcharging. Of these functions, research into mitigating the shuttle effect of Zn–I2 batteries has received the most attention. In theory, the shuttle effect in Zn–I2 batteries arises primarily from the formation of water-soluble polyiodide (iodine is hardly soluble in water). Therefore, the aforementioned high concentration of chloride salts can effectively avoid the shuttle effect by inhibiting the formation of polyiodide. As reported by Ji et al., a Zn–I2 cell based on a hydrothermally RGO cathode and a highly concentrated ZnCl2 electrolyte (5 M KI/20 M ZnCl2) was able to achieve 2000 cycles without capacity decline even at an ultra-high iodine load of 25.33 mg cm−2.202 Nevertheless, the high cost brought by the highly concentrated electrolytes cannot be ignored. Given this, Yang et al. developed a novel electrolyte (i.e., 1 M Zn(CF3SO3)2 + 4 M N-methylacetamide + 0.5 M KI in 20% volume fractions of H2O) to circumvent the formation of I3− intermediates.85 In this case, Zn2+ in the eutectic solution has a unique double-shell solvated structure, that is, a compact inner layer composed of I− and CF3SO3−, and an outer layer including H2O and C3H7NO (Fig. 14d). This not only reduces the amount of free water effectively, but also screens the I− interaction with surrounding ions or molecules, thereby suppressing the formation of I3−. As a result, the as-assembled Zn–I2 battery performed 5000 cycles with a capacity retention of 98.7%. In such a system, however, a low concentration of active species (0.5 M KI) means a low energy density, suggesting that the protocol may need further refinement. On the other hand, the restriction of polyiodide diffusion, as a conventional method to mitigate the shuttle effect, is also achieved by electrolyte additives and gel electrolytes. Research on electrolyte additives is relatively scarce. Recently, polyethylene glycol (PEG400) and ethylene glycol (EG) additives have been reported to inhibit the formation and shuttling of I3− by complexing with I2.203,204 Additionally, Zhao et al. introduced vermiculite nanosheets (VS) into a 1 M ZnSO4 solution to form a VS suspended electrolyte.205 By leveraging the interaction between polyiodide and silica-oxygen bonds present in the VS, the dissolved polyiodide elegantly binds to the scattered VS particles in the bulk electrolyte, thus triumphantly alleviating the pernicious shuttle effect. For gel electrolytes, Soni's group found that the poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) (FF7, PEO53–PPO34–PEO53) gel electrolyte for Zn–I2 cells could lead to future inhibition of iodine species migration and prevent self-discharge (Fig. 14e).82 However, under such conditions, the battery had a short lifespan of only 500 cycles. Hence, they implanted a highly solubilized iodine-enriched metal–organic gel-enabled catholyte into PEO53–PPO34–PEO53, since MOFs can provide strong adsorption properties for polyiodide.81 Consequently, the cyclic life of Zn–I2 cells was significantly improved to 1500 cycles with a superior capacity retention of 95.8% at 1C. Notably, gel electrolytes with abundant electronegative groups, such as hydroxyl,206 carboxyl,207 and sulfate groups,88 are able to mitigate the shuttle effect through electrostatic repulsion. For example, Shang et al. achieved a long lifetime of 2000 cycles with a capacity retention of 66.8% at 2 A g−1 in a Zn–I2 battery by virtue of abundant –COOH groups in the alginate-based polyanionic hydrogel electrolyte.207 Benefiting from the flexible structure of gel electrolytes, Lin et al. recently employed the cross-linked polyacrylamide/2 M ZnSO4/0.5 M 1-methyl-3-n-propylimidazolium iodide (CPAM-Zn–I-0.5) polyelectrolyte to construct Zn–I2 microbatteries.206 As a result, the as-fabricated micro-battery showed unprecedented volumetric and areal energy densities of 1647.3 mW h cm−3 and 2339.1 μW h cm−2, demonstrating the great potential of Zn–I2 cells in the field of micro-batteries. Besides, a novel Zn–I2 battery with fast overcharge self-protection capability could be obtained based on poly(2-vinylpyridine) (P2VP) additives (Fig. 14f).208 As the pH changes in overcharged Zn–I2 batteries can induce a reversible and fast transition of the P2VP-based aqueous electrolyte from a conductive soluble state to a gel state with high internal resistance, which in turn results in a rapid shutdown of the battery under overcharge conditions and prevents continued battery damage.
All in all, in developing electrolytes for zinc–iodine batteries, researchers have focused on three main areas: (1) activating and stabilizing the I0/I+ process to improve the battery energy density with the key points on stabilizing I+ ions and inhibiting water activity; (2) inhibiting the formation of water-soluble polyiodide (i.e., achieving I−/I2 conversion), which requires electrolytes capable of reducing the activity of I− ions to prevent them from combining with I2; and (3) mitigating the diffusion of water-soluble polyiodide through gel electrolytes and electrolyte additives (PEG400, EG). In addition, due to their flexibility, lack of liquid components, safety, and high energy density, Zn–I2 batteries utilizing gel electrolytes offer potential for use in micro-batteries and flexible wearable devices.
2.4.1.3 Separators and interlayers.
Separators. As one of the important constituents of batteries, the separator plays a crucial role in the performance of batteries. Table 12 in the modified separator section for Zn–I2 batteries presents the relevant information and performance. Typically, the separators in Zn–I2 batteries can assume the dual role of inhibiting polyiodide diffusion and protecting the zinc anode via their ion channel function. For instance, Yang et al. selected a Zn-BTC (Zn3(BTC)2) MOF that has high stability under aqueous conditions and a suitable porous structure, as the functional separator to realize high-performance Zn–I2 batteries.92 Attributed to the –COOH groups on the walls of Zn-BTC channels, the separator can selectively block the migration of I3− to the Zn anode through electrostatic repulsion.207,209 Additionally, the Zn-BTC separator also suppressed H2O-induced Zn corrosion by expelling out free water molecules from Zn2+ cations, thereby resulting in enhanced stability of the Zn anode (Fig. 15a and b). Therefore, the Zn–I2 battery achieved a long cycling lifetime of 6000 cycles with a capacity retention of 84.6% at 1.92 A g−1. Furthermore, Li et al. recently reported a zeolite separator based on a zeolite molecular sieve (Na12(AlO2)12(SiO2)12·xH2O) to effectively confine the cross-over of soluble I3− (Fig. 15c).90 Since the size of the porous framework (4 Å) inside the zeolite separator is smaller than that of I3− ions (5.14 Å), the as-prepared battery can effectively block the uncontrollable diffusion of I3−. As a result, the zeolite-based Zn–I2 battery achieves over 30
000 cycles at 4.0 A g−1 with a capacity retention of 91.0%. The cost-effective and easy-to-operate preparation process of the multifunctional zeolite separator makes this work potentially valuable for the construction of advanced Zn–I2 batteries. In addition, Hou's group also experimented with a single-sided ketone black modified cotton fiber separator, but its performance was relatively poor in a Zn–I2 cell with 2000 cycles, corresponding to a reversible capacity of 112 mA h g−1.210 All in all, limited work has been carried out on the modification of Zn–I2 battery separators. Currently, the design of separators mainly lies in the regulation of pores and surface chemistry. This requires that the separators can facilitate the desolvation of zinc ions (reducing free water molecules) and avoid the passage of polyiodide, while maintaining the high flux of ions.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1, year; 2, anode; 3, electrolyte formula; 4, cathode. Evaluation of Zn‖Zn cell performance: 5, current density (mA cm−2) and areal capacity (mA h cm−2); 6, cycle time (h). Evaluation of Zn–I2 cell performance: 7, current density (C, 1C = 211 mA h g−1); 8, cycle number; 9, capacity retention (%); 10, reversible capacity (mA h g−1iodineorLiI). 11, reference. PEDOT, poly(3,4-ethylenedioxythiophene); PSS, polystyrene sulfonate. |
||||||||||
| 2020 | Zn3(BTC)2 MOF membrane | Anolyte, 0.5 M ZnSO4/0.5 M Li2SO4; catholyte 0.5 M ZnSO4/1 M LiI/0.1 M I2 | KB/PTFE | 0.2 (0.2) | 3000 | ∼9 | 8000 | 84.6 | 85.1 | 92 |
| 2022 | Zeolite membrane of Na12(AlO2)12(SiO2)12·xH2O | Anolyte, 0.5 M ZnSO4/0.5 M Li2SO4; catholyte, 0.5 M ZnSO4/1 M LiI/0.1 M I2 | KB/PTFE | 1 (1) | ∼260 | ∼19 | 30000 |
91 | 44.5 | 90 |
| 2022 | PEDOT:PSS interlayer | 2 M ZnSO4 | I2/active carbon | 10 (5) | 550 | ∼47 | 20000 |
∼72 | — | 211 |
| 2023 | KB modified cotton fiber separator | 1 M ZnSO4 | I2/MOF-5-derived mesoporous carbon | — | — | ∼4.7 | 2000 | — | 112 | 210 |
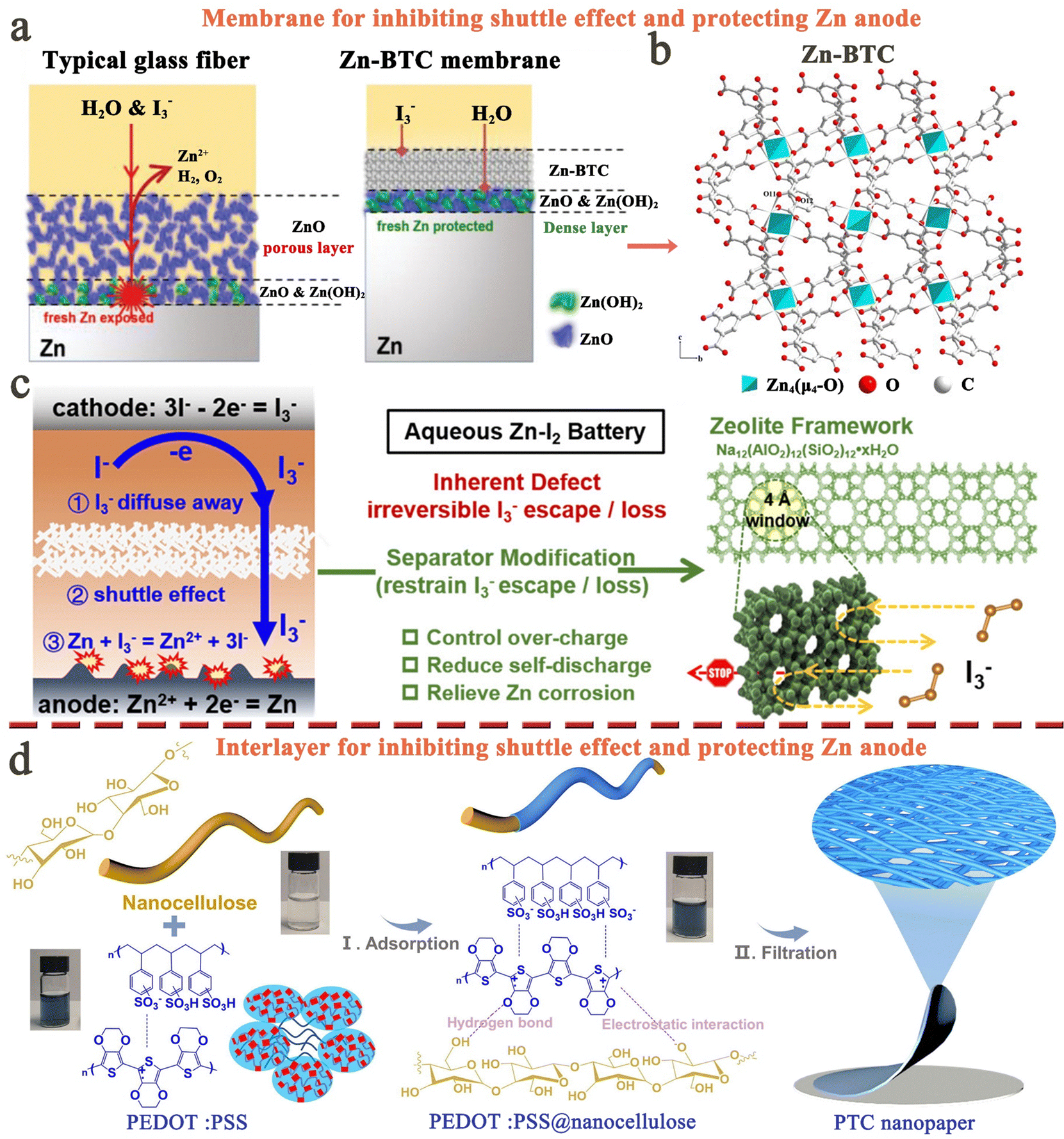 | ||
| Fig. 15 (a) Schematic illustration for the component evolution of the Zn anode with glass fiber separator and Zn-BTC membrane after being corroded by iodine species and water. Reprinted with permission from ref. 92. Copyright 2020 Wiley. (b) Schematic diagram for the structure of Zn-BTC. Reprinted with permission from ref. 209. Copyright 2008 Elsevier. (c) Schematic illustration for the critical problems in Zn–I2 batteries and the working mechanism of zeolite molecular sieve. Reprinted with permission from ref. 90. Copyright 2022 American Chemical Society. (d) Schematic diagram of the preparation process of PTC nanopaper. Reprinted with permission from ref. 211. Copyright 2022 Elsevier. | ||
Interlayers. In contrast to Li–I2 batteries, research on the interlayer in Zn–I2 cells is very scarce. Zhang et al. reported the first interlayer in Zn–I2 batteries, namely, the poly(3,4-ethylenedioxythiophene): polystyrene sulfonate (PEDOT: PSS)@nanocellulose (PTC) interlayer.211 The advantages of the PTC interlayer are as follows: (1) 3D nanofiber networks with uniformly distributed mesopores allow rapid Zn2+ transfer; (2) the abundant polar groups (–SO3H, –S, and –O) enable uniform Zn2+ flow, while suppressing polyiodide shuttling through the Coulomb repulsion and chemisorption effect; and (3) the inherent electrical conductivity facilitates homogeneous current distribution (Fig. 15d). Thus, the Zn–I2 battery presented an ultralong lifespan (>20
000 cycles) at 10 A g−1 with a capacity retention of ∼72%. It is worth noting that this interlayer does not provide as high of a pseudo-capacitance performance as that found in Li–I2 batteries. This may be due to the difference in the nature of interlayers (mainly using carbon-based interlayers in Li–I2 batteries) and cell systems. It is therefore desirable to perform more research on the different interlayers in Zn–I2 cells to elucidate the working mechanisms and design principles of interlayers.
2.4.1.4 Anodes. It is well known that zinc anodes encounter issues of zinc dendrite growth and hydrogen evolution reaction (HER) in zinc-ion batteries. However, the occurrence of polyiodide shuttling in Zn–I2 cell also tends to cause corrosion and passivation of the Zn anode, which further aggravates the instability of the Zn electrode.42,90,92 As revealed by Zhang et al., polyiodide can react with Zn to form water-soluble ZnI2, which leads to the formation of a honeycomb and bumpy structure on the Zn surface and eventually promotes rampant dendrite growth, causing rapid cell failure.212 Hence, appropriate strategies should be adopted to protect zinc anodes, such as those used in zinc-ion batteries (optimization of the intrinsic structure of Zn anodes, modification of the anode surface, and so on), for long-lasting Zn–I2 batteries.93Table 13 lists the information and performance of modified anode for Zn–I2 batteries. In optimizing the bulk structure of zinc anodes, Wang et al. first adopted the ZIF-8 MOF annealed at 500 °C (noted as ZIF-8-500) as the host material for zinc, enabling a Zn–I2 battery to achieve over 1600 cycles (Fig. 16a).213 However, this strategy focuses on regulating the transfer/reaction of Zn2+, resulting in inadequate prevention of corrosion of the Zn anode by polyiodide, limiting cell stability. Therefore, more efforts have been made by modifying the Zn anode surface to realize high-performance Zn–I2 cells, as the layer constructed on the Zn surface can serve a dual function of protecting the Zn anode from dendrite growth and HER, as well as inhibiting polyiodide corrosion. For instance, Su et al. reported a silicon-based anticorrosion film (Zn@Ts) prepared by the treatment of tetraethyl orthosilicate steam on a Zn disk, which can realize an ultra-long lifetime of 5000 h at 2 mA cm−2 (2 mA h cm−2) in a symmetric cell, and 20
000 cycles with a high capacity retention of 93% in a Zn–I2 full cell.214 This can be attributed to the fact that the Zn@Ts layer can promote a homogeneous Zn2+ flux, protect the Zn anode from aqueous electrolytes, and limit the damage induced by polyiodide to Zn (Fig. 16b and c). Similarly, a series of artificial SEIs were prepared by in situ reactions, including Zn@Sn (Zn@tin, prepared using SnCl4),215 Zn@disordered zinc silicate (Zn@ZSO, prepared using Na2SiO3),216 and phytic acid@Zn layer.217 Among them, the performance of the Zn@ZSO coating layer is optimal, which may be due to the disordered and compact nature of the ZSO interfacial layer that can eliminate grain boundaries inside the interphases, and thus realize excellent plating/stripping reversibility of Zn2+ (2500 h at 1 mA cm−2 (0.25 mA h cm−2)).216 In addition, Rei et al. proposed a novel approach for achieving uniform Zn deposition, that is, the utilization of Cu foil with in situ formed zincophilic Cu nanoclusters (prepared by treatment with trace amounts of I3− additives in the electrolyte) as an anode for current collection.218 In fact, however, these Cu nanoclusters cannot prevent the corrosion of zinc by polyiodide, thus leading to the short lifetime of the as-fabricated Zn–I2 cell (63.8% capacity retention after 200 cycles). Therefore, the protection strategies for Zn anodes should focus on improving the ability of Zn to resist polyiodide corrosion. On the other hand, some artificial coating layers, including zeolite-based cation-exchange,219 sulfonate-rich ion-exchange fibers,212 carboxymethyl chitosan cross-linked gel,220 RGO,221 zinc citrate-derived porous carbon,222 and biomass-based lignin layers,223 were also presented. Among them, the layers with ion exchange mechanisms generally had excellent properties (≥5000 cycles). This is because the negatively-charged groups/cavities in these materials can provide uniform and rapid flux of Zn2+, while preventing the penetration of polyiodide into the Zn anode through electrostatic repulsion (Fig. 16d). Thus, the resulting Zn–I2 battery by Lai's group can work over 6000 cycles with a high capacity retention of 90.2% at 3.2 A g−1.212 Apart from these methods, the aforementioned high-concentration salt electrolytes, gel electrolytes, and modified separators can not only effectively protect the zinc anode, but also avoid the free migration of polyiodide.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1, year; 2, anode; 3, electrolyte formula; 4, cathode. Evaluation of Zn‖Zn cell performance: 5, current density (mA cm−2) and areal capacity (mA h cm−2); 6, cycle time (h). Evaluation of Zn–I2 cell performance: 7, current density (C, 1C = 211 mA h g−1); 8, cycle number; 9, capacity retention (%); 10, reversible capacity (mA h g−1iodine). 11, reference. PTFE, poly tetra fluoroethylene; GC-PAN/I, N,N′-dimethyl-1,3-propanediamine-grafted, and triethylenetetramine-cross-linked acrylic fiber/iodide. | ||||||||||
| 2019 | Zn@ZIF-8-500 | Anolyte, 0.5 M Li2SO4/0.5 M ZnSO4; catholyte, 1 M LiI/0.1 M I2/0.5 M ZnSO4 | KB/PTFE | 1 (1) | 50 | ∼9.5 | 1600 | 97 | ∼117 | 213 |
| 2022 | Zinc citrate-derived porous carbon/Zn | 2 M ZnSO4 | I2/zinc citrate-derived porous carbon | 2 (1) | 400 | 12 | 3000 | 88.1 | ∼108 | 222 |
| 2022 | Biomass-based lignin layer | Anolyte, 0.5 M Li2SO4/0.5 M ZnSO4; catholyte, 1 M LiI/0.1 M I2/0.5 M ZnSO4 | KB/PTFE | 2 (2) | 650 | ∼28 | 35000 |
99.7 | 86.7 | 223 |
| 2022 | Sn coating layer | 2 M ZnSO4 | N,P co-doped porous carbon | 1 (1) | 900 | 5 | 1200 | 90.7 | ∼178 | 215 |
| 2022 | Zeolite-based cation-exchange protecting layer | 1 M ZnSO4 | AC | 2.5 (2.5) | 460 | ∼9.5 | 5600 | ∼92 | ∼141.8 | 219 |
| 2022 | Disordered zinc silicate artificial SEI | 2 M ZnSO4 with 0.05 KI | — | 1 (0.25) | 2500 | ∼9.5 | 10000 |
— | — | 216 |
| 2022 | Silicon-based films based on tetraethyl orthosilicate | Anolyte, 0.5 M Li2SO4/0.5 M ZnSO4; catholyte, 1 M LiI/0.1 M I2/0.5 M ZnSO4 | KB/PTFE | 2 (2) | 5000 | ∼28 | 20000 |
93 | 89 | 214 |
| 2022 | Phytic acid treated zinc | 2 M ZnSO4 | N,P co-doped carbon | 5 (1) | 500 | 5 | 5000 | 93.6 | 187.7 | 217 |
| 2022 | Zincophilic Cu nanocluster | 5 mM ZnI2/10 mM I2/2 M ZnSO4 | ZnI2/graphene/PVP heterostructure | 20 (1) | 800 (Zn‖modified Cu cell) | ∼4.7 | 200 | 63.8 | ∼77 | 218 |
| 2023 | Reduced graphene oxide | Anolyte, 1 M ZnSO4/0.5 M Na2SO4; catholyte, 2 M KI/0.5 M Na2SO4 | 3D functionalized graphene | 5 (1) | 80 | ∼24 | 2000 | 96.7 | — | 221 |
| 2023 | Sulfonate-rich ion-exchange fiber layer | 2 M ZnSO4 | GC-PAN/I | 1 (1) | 600 | ∼15 | 6000 | 90.2 | 84.6 | 212 |
| 2023 | Carboxymethyl chitosan crosslinked gel layer | 2 M ZnSO4/0.5 M KI | Adsorptive activated carbon on carbon fiber cloth | 1.25 (∼0.3) | 1300 | 5 mA cm−2 | 28000 |
100 | ∼1.04 mA h cm−2 | 220 |
 | ||
| Fig. 16 (a) Schematic illustration for the Zn plating process in the ZIF-8-500@Zn electrode. Reprinted with permission from ref. 213. Copyright 2019 Elsevier. (b) Schematic diagram of the Zn plating/striping behavior on the bare Zn and Zn@Ts electrode, and (c) protection of Zn foil using a silicon-based corrosion protection film. Reprinted with permission from ref. 214. Copyright 2022 Elsevier. (d) Schematic diagram of the brief synthesis process of a sulfonate-rich ion-exchange fiber layer and its role in achieving high performance Zn–I2 batteries. Reprinted with permission from ref. 212. Copyright 2023 Wiley. | ||
All in all, the protection of the zinc anode for Zn–I2 batteries should include the following aspects: (1) inhibiting the growth of zinc dendrites; (2) avoiding hydrogen evolution reaction; and (3) preventing polyiodide from migrating to the zinc anode. Modification of the anode surface has been proven to be effective in achieving these objectives, which generally involves two strategies, namely, in situ construction of artificial SEIs and coating layers by spin/scrape-coating. The advantage of the former lies in the ability to form uniform and strongly adherent layers, while the latter is easier to prepare on a large scale.224 Despite this, some design recommendations for modified layers can be described as follows: (1) rich in negative charges, which can regulate the transfer/reaction of Zn2+ and polyiodide through electrostatic interaction; (2) a uniform pore structure, which can result in a homogeneous and rapid Zn2+ flux; (3) zincophilic properties to reduce the overpotential of the Zn plating/stripping process; and (4) a disordered interphase to eliminate grain boundaries inside the interphases. Based on these guidelines, extensive efforts are desired in future to advance the Zn anode toward high stability.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|
| 1, year; 2, cathode; 3, catholyte (volume of electrolyte); 4, anolyte (volume of electrolyte); 5, membrane. Evaluation of Zn–I2 cell performance: 6, current density (mA cm−2); 7, cycle number; 8, energy efficiency (%); 9, reference. MIL-125-NH2, Ti8O8(OH)4[(O2C–C6H5–CO2)]6; UiO-66-CH3, (Zr6O4(OH)4[O2C–C6H2(CH3)2–CO2]6; ACN, acetonitrile; GF, graphite felt; CMC/PVA, carboxymethyl cellulose/polyvinyl alcohol. | ||||||||
| 2015 | GF | 3.5 M ZnI2 (—) | 3.5 M ZnI2 (—) | Nafion 115 | 10 | 40 | ∼80 | 16 |
| 2016 | MIL-125-NH2-modified GF | 2.5 M ZnI2 (—) | 2.5 M ZnI2 (—) | Nafion 115 | 30 | 45 | 64 | 239 |
| 2016 | UiO-66-CH3-modified GF | 2.5 M ZnI2 (—) | 2.5 M ZnI2 (—) | Nafion 115 | 30 | 50 | ∼64 | 239 |
| 2017 | GF | 5 M ZnI2 + 2.5 M ZnBr2 (1 mL) | 5 M ZnI2 + 2.5 M ZnBr2 (1 mL) | Nafion 117 | 10 | 20 | — | 227 |
| 2018 | GF | 6 M KI/3 M ZnBr2 (50 mL) | 6 M KI/3 M ZnBr2 (50 mL) | Porous polyolefin membrane | 80 | 1000 | 80 | 231 |
| 2018 | Graphite foil | 6 M KI/6 M I2 (10 mL) | 6 M KOH (—) | Nafion 117 | 10 | 70 | 80 | 164 |
| 2019 | MoS2-modified GF | 2 M ZnI2 (2.5 mL) | 2 M ZnI2 (2.5 mL) | Nafion 115 | 40 | 45 | ∼67.9 | 238 |
| 2019 | Carbon felt | 1.5 M NaI, 0.5 M ZnCl2, 1.0 vol% PEG200, 0.15 vol% HCl, 20 vol% PC (—) | 1.5 M NaI, 0.5 M ZnCl2, 1.0 vol% PEG200, 0.15 vol% HCl, 20 vol% PC (—) | Durapore membrane | 20 | 50 | ∼65 | 229 |
| 2019 | GF | 7.5 M KI/3.75 M ZnBr2 (7.2 mL) | 7.5 M KI/3.75 M ZnBr2 (—) | Polyolefin separator with 250 μm Nafion layer | 20 | 100 | ∼80 | 232 |
| 2020 | Heat-treated GF | 6.5 M NH4I/1.5 M NH4Cl (10 mL) | 3.25 M ZnCl2/1.5 M NH4Cl (—) | Nafion 117 | 10 | 2500 | 80 | 226 |
| 2020 | GF | 0.5 M KI/0.5 M I2 (15 mL) | 1 M ZnSO4 (15 mL) | CMC/PVA polyelectrolyte membrane | 10 | 300 | — | 234 |
| 2021 | GF | 1 M NH4Br/1 M ZnI2 (—) | 1 M NH4Br/1 M ZnI2 (—) | Nafion 115 | 40 | 100 | 85 | 225 |
| 2021 | Heat-treated GF | 5 M NH4I/2.5 M NH4Cl (—) | 2.5 M ZnCI2/2.5 M NH4Cl (—) | Daramic microporous polyethylene separators | 80 | 1100 | — | 237 |
| 2021 | GF | 6 M KI with 5.3 wt% PVP (—) | 0.5 M ZnCl2/3 M KCl (—) | Perfluorinated sulfonic acid membrane | 20 | 600 | 70 | 230 |
| 2022 | GF | 2 M KCl/0.5 M ZnBr2/1 M KI (10 mL) | 2 M KCl/0.5 M ZnBr2/1 M KI (10 mL) | Chitosan-starch composite membrane | 80 | 200 | 77.4 | 233 |
| 2022 | GF | 1 M ZnI2 + 7.5% ACN (—) | 1 M ZnI2 + 7.5% ACN (—) | Nafion 115 | 100 | 170 | ∼60 | 228 |
| 2023 | CNT with iron particles | 1 M KI/0.5 M ZnBr2/1 M KCl (—) | 1 M KI/0.5 M ZnBr2/1 M KCl (—) | Nafion 212 | 17 | 60 | ∼60 | 240 |
2.4.2.1 Electrolyte. Electrolyte engineering has become a mainstream research interest in developing high-performance ZIRFBs, as the iodine-active species in ZIRFBs are stored in electrolytes. By using a near-neutral 5.0 M ZnI2 electrolyte, Li et al. pioneered the preparation of high energy density ZIRFBs.16 However, only two-third of iodine is utilized in this system, because one-third of I− ions is coordinated to I2 to form I3−, resulting in this portion of iodine not being utilized for energy conversion. To address this issue, various electrolyte additives such as NH4Br,225 NH4Cl,226 and ZnBr2,227 were introduced to promote the formation of I2 and improve iodine utilization. This is because Br− and Cl− ions can stabilize free iodine by forming I2Br− and I2Cl− (2I− + Br− ↔ I2Br− − 2e−, E0 = 0.594 V vs. SHE; 2I− + Cl− ↔ I2Cl− − 2e−, E0 = 0.61 V vs. SHE) respectively, thereby releasing iodide ions for charge storage (Fig. 17a).226,227 Additionally, the introduction of NH4+ ions was also effective in mitigating the growth of zinc dendrites due to the electrostatic shielding effect formed by the complexation of NH4+ ions with Zn2+ ions (Zn2+ + xNH4+ + yCl− ↔ [Zn(NH3)xCly]2−y + xH+).226 As a result, the ZIRFB with a catholyte of 6.5 M NH4I/1.5 M NH4Cl can achieve a long cycle life of 2500 cycles at 10 mA cm−3, with an energy efficiency of ∼80% and an energy density of 137 W h L−1.226 On the other hand, the insolubility and electrical insulating properties of solid I2 in water can lead to the formation of iodine films during the cycling process, which not only blocks the pore structure of electrodes but also impedes the iodine redox process (Fig. 17b). As revealed by Zhao et al., the rate constant for the transition from I2 to I3− was only 10−6 mol (cm−2 s−1).228 To accelerate electrode kinetics, organic electrolyte additives such as propylene carbonate,229 acetonitrile,228 and polyvinylpyrrolidone230 were added to solubilize the iodine film. Indeed, the current density of ZIRFBs can reach up to more than 100 mA cm−2 when 1 M ZnI2 + 7.5 vol% acetonitrile is employed as the electrolyte.228 Despite this, the introduction of these organic solvents increased the risk of electrolyte cross-contamination, leading to low cell Coulombic efficiency (Fig. 17c). An alternative to these organic additives is KI, which is able to promote iodine solubility and simultaneously supply iodine ions.231,232 As shown in Fig. 17d, the ZIRFB can operate at a high current density of 180 mA cm−2 with an electrolyte of 2 M KI + 1 M ZnBr2.231 Furthermore, Zhang et al. attempted to increase the energy density of ZIRFBs by using an alkaline anolyte (containing 6 M KOH), which can lower the redox potential of zinc anodes to −1.26 V (Zn + 4OH− − 2e−→ Zn(OH)42−, E0 = −1.260 V), thereby increasing the overall voltage of the ZIRFB to 1.796 V (Zn + I3− + 4OH− → Zn(OH)42− + 3I−, E0 = 1.796 V) (Fig. 17e).164 Although the as-prepared ZIRFB exhibited a high-energy-density of 330.5 W h L−1, the battery demonstrated a short life of 70 cycles at 10 mA cm−2 due to the cross-contamination of Zn(OH)42−, I3−, and I−, as well as severe zinc dendrite growth and corrosion. Besides, the irreversible disproportionation reaction of iodine with OH− (6OH− + 3I2 → 5I− + IO3− + 3H2O) should also be considered. Therefore, the feasibility of using alkaline anolytes still requires critical assessment. Overall, the current development of electrolyte engineering is centered on the following areas, including the realization of the I− to I2 process, the promotion of iodine film dissolution, and the exploration of alkaline anolytes.
 | ||
| Fig. 17 (a) Schematic representation of Br− ions as complexing agents for iodine stabilization. Reprinted with permission from ref. 227. Copyright 2017 Royal Society of Chemistry. (b) The working mechanisms of I− to I3− transition without (left) and with (right) a cosolvent in an aqueous solution. Reprinted with permission from ref. 228. Copyright 2022 Royal Society of Chemistry. (c) Efficiencies of ZIRFB at 20 mA cm−2 with the electrolyte of 1.5 M NaI, 0.5 M ZnCl2, 1.0 vol% PEG200 (polyethyleneglycoland), and 0.15 vol% HCl with 20 vol% propylene carbonate. Reprinted with permission from ref. 229. Copyright 2019 Elsevier. (d) Rate capability of ZIRFB with an electrolyte of 2 M KI + 1 M ZnBr2. Reprinted with permission from ref. 231. Copyright 2018 Wiley. (e) Open-circuit voltage of conventional and designed alkaline ZIRFBs. Reprinted with permission from ref. 164. Copyright 2018 Royal Society of Chemistry. (f) Polarization voltage curves for C50 (CMC:PVA = 50:50 wt%) and C75 (CMC:PVA = 75:25 wt%) cells show decent performance and cycling stability)at 10 mA cm−2 for the first cycle. Reprinted with permission from ref. 220. Copyright 2022 Elsevier. (g) Schematic diagram of ion convection caused by unbalanced hydraulic pressure in a ZIRFB. Reprinted with permission from ref. 237. Copyright 2021 Elsevier. (h) Cycling performance of ZIRFBs with the pristine graphite felt and the MOF-modified graphite felt electrodes. Reprinted with permission from ref. 239. Copyright 2016 American Chemical Society. (i) Charge–discharge curves of the pristine graphite felt, and the MoS2-modified graphite felt electrodes at 30 mA cm−2. Reprinted with permission from ref. 238. Copyright 2019 Royal Society of Chemistry. | ||
2.4.2.2 Separators. The cross-diffusion of soluble iodine species between the cathode and anode is recognized as a key challenge that hampers the advancement of ZIRFBs, as it can lead to severe self-discharge in batteries. Therefore, cation exchange membranes (e.g., Nafion membrane) have become an essential component of ZIRFBs to prevent cross-contamination of iodine species. Nonetheless, these ion exchange membranes are costly, typically accounting for 40% of the overall stack cost for flow batteries.233 This leads to the necessity of developing economical, highly selective, high-flux, and stable ion exchange membranes. In this regard, Tangthuam's group has attempted to develop a polyelectrolyte membrane consisting of carboxymethyl cellulose compounded with polyvinyl alcohol.234 Unfortunately, the membrane exhibited unsatisfactory performance, with a polarization voltage as high as 0.648 V (Fig. 17f). In view of this, Gao et al. successfully achieved a high-performance and low-cost ion-exchange membrane based on the “host–guest” chemistry between the membrane and iodine species.233 Specifically, they utilized a starch–chitosan complex as a host to confine iodine species (guest). This choice was motivated by the following reasons: (1) starch is one of the most abundant natural polymers with a cheap price (∼$0.3 kg−1); (2) starch possesses an internal hydrophobic cavity (∼5 Å in diameter) that can interact with iodine species via host–guest chemistry; and (3) chitosan can prevent starch from dissolving in water through hydrogen bonding, while its rich –OH groups facilitate interaction with iodine species and inhibit the shuttle effect. Consequently, the proof-of-concept membranes enabled ZIRFBs to be cycled for 200 cycles at 60 mA cm−3 without a significant capacity loss. However, it should be noted that the method based on the adsorption of iodine species by the membrane to prevent cross-contamination may result in the accumulation of iodine species within the membrane and eventual blockage of the membrane. Therefore, Donnan exclusion and size exclusion strategies are two main strategies for achieving high-performance ion exchange membranes.235,236 For example, in zinc–iodine static cells, long-cell cycling was often achieved by altering the charge properties of membranes and controlling their pore size.90,92 Nevertheless, research on membranes for ZIRFBs remains limited due to the excellent performance of commercially available Nafion membranes, although their high cost is a consideration. In addition, Mousavi et al. found that the increased viscosity and density of catholytes during battery cycling can result in elevated pressure within the cathode chamber of ZIRFBs (Fig. 17g).237 This will induce the transfer of iodine species from the cathode to the anode, leading to a significant decrease in cell capacity. An effective strategy to alleviate this problem is to maintain a balanced hydraulic pressure by adjusting the flow rate ratio of electrolytes (catholyte
:anolyte = 1:7), thereby avoiding convection of iodine species.
2.4.2.3 Cathodes. Generally, ZIRFB cathodes are mainly composed of graphite felts due to their high porosity (>90%), low cost, and excellent conductivity. However, the ZIRFBs exhibit limited energy efficiency at high current densities due to the sluggish redox kinetics of the I3−/I− process on the graphite felt surface. Therefore, some materials with electrocatalytic functions (e.g., MOFs and MoS2) have been employed to modify graphite felts.238,239 Among them, MOFs have been considered promising candidates for advanced adsorbents and catalysts because of their extraordinary surface area, tunable pore geometries, and diverse chemical compositions. Therefore, Li et al. explored the use of two MOF materials, including MIL-125-NH2 (Ti8O8(OH)4[(O2C–C6H5–CO2)]6 and UiO-66-CH3 (Zr6O4(OH)4[O2C–C6H2(CH3)2–CO2]6, loaded on graphite felts as electrocatalysts to accelerate the I−/I3− redox reaction.239 As a result, both MIL-125-NH2 and UiO-66-CH3 modified graphite felts exhibited improved energy efficiency compared to pristine graphite felts, with energy efficiency values increasing by approximately 6.4% and 2.7%, respectively, at 30 mA cm−2. This enhancement can be attributed to two factors: (1) the Lewis acid sites in the MOFs can act as catalytic sites to accelerate electron transfer and (2) the weaker Lewis acid sites in MIL-125-NH2 compared to UiO-66-CH3 facilitate the dissociation of electrons from the Ti atoms in MIL-125-NH2, thus better facilitating the iodine conversion reaction. Nevertheless, the poor chemical stability of MOFs is the major obstacle limiting its development, as UiO-66-CH3-modified graphite felts (good stability but poor catalytic performance) only proved to be stable for about 50 cycles (Fig. 17h). To overcome this limitation, Liu et al. sought to accelerate the iodine redox reaction using the defect engineering strategy. MoS2, which possesses a unique layered structure and semiconducting properties, was chosen as the electrocatalyst and in situ grafted onto the graphite felt surface.238 The resulting ZIRFB with a MoS2-modified graphite felt electrode showed higher energy efficiency (10.2%) compared to pristine graphite felts at 30 mA cm−2 (Fig. 17i). The excellent catalytic performance of MoS2 nanoplates can be attributed to three key factors: (1) the abundant defects (e.g., S vacancies) on the MoS2 nanoplates result in excellent catalytic activity; (2) the introduction of active S edges and S ligands in the MoS2 nanoplates provides more active sites than in pristine MoS2; and (3) the MoS2 nanoplates have vertically aligned distributions on the graphite felt surface which facilitates electrolyte diffusion. In addition to the modified graphite felt, Williams et al. also utilized carbon nanotubes with redox-active iron particles as the electrode, which aligns with the single-atom catalytic strategy employed in static batteries.240 However, this does not mean that the cathode strategies for static and liquid zinc–iodine batteries are highly generalizable. In static cells, the cathode needs to retain iodine species within the electrode as much as possible to avoid shuttle effects, whereas, in liquid-flow cells, the cathode requires high porosity for efficient iodine species mobility instead of high iodine retention ability. Nonetheless, the acceleration of the iodine conversion process by introducing electrocatalysts remains highly similar in both types of cells. Thus, the power density of a ZIRFB with an iron-functionalized carbon electrode can be increased by 66% compared to a ZIRFB with inert carbon. In general, the key to the cathode design of ZIRFBs lies in catalyzing the iodine conversion process for reducing battery polarization and promoting efficient utilization of iodine.
2.5 Iron–iodine batteries
The Fe–I2 battery section of Table 15 provides the relevant battery information and performance. Fe metal is the most abundant transition metal in the Earth's crust, and the most mass-produced metal commodity.241,242 Its two-electron transfer (Fe2+/Fe, −0.447 V vs. SHE) can provide specific and volumetric capacities of 958 mA h g−1 and 7530 mA h cm−3, respectively.61 All these advantages lead to the emergence of Fe–I2 batteries. In 2020, Bai et al. constructed a Fe–I2 battery using a I2@nitrogen-doped hierarchically porous carbon (N-HPC) composite as the cathode, modified Fe powder as the anode, and 1 M FeSO4 in aqueous solution as the electrolyte.19 The resulting battery exhibited 550 cycles with ∼100% capacity retention at 2 A g−1, demonstrating the feasibility of Fe–I2 batteries. However, the sudden failure of the battery at high-capacity retention and short cycle life suggests serious instability issues like dendrite growth and side reactions with aqueous electrolytes in the Fe anode. Wu et al. further improved the reversibility and stability of the Fe plating/stripping process by doping Zn into the Fe anode.61 In other words, the positively charged [ZnI]+ complexes introduced by adding ZnI2 to FeCl2 electrolytes, can adsorb on the negatively charged surface of Fe metal, thereby realizing the doping of Zn into the Fe lattice during deposition (Fig. 18a). With Zn doping, Fe2+ will deposit as homogeneous small particles, since its (110) plane is preferably grown parallel to the substrate. Accordingly, the lifespan of the Fe anode is greatly extended to over 2500 h at 1 mA cm−2 (1 mA h cm−2). When combined with the frequently used PANI cathode, the battery showed over 1200 cycles with a capacity retention of 99.8% at 8 mA cm−2, which is promising for large-scale energy storage applications (Fig. 18b). However, a relatively low CE of the cell is also caused by the introduction of ZnI2 additives. In addition, due to the relatively low potential of iron itself, the discharge plateau of the Fe–I2 battery is below 0.9 V (Fig. 18c). Despite this, owing to the unique advantages of Fe metal in terms of cost and abundance, Fe–I2 batteries have opportunities for large-scale energy storage applications. In general, research on Fe–I2 batteries should place more emphasis on expanding the basic understanding of Fe plating/stripping chemistry in aqueous solution, as it occupies the “short slab” position in battery performance, particularly with the rapid advancements in iodine cathodes.| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|
| 1, year; 2, anode; 3, electrolyte formula; 4, cathode; 5, iodine loading (mg cm−2). Evaluation of Fe–I2 cell performance: 6, current density; 7, cycle number; 8, capacity retention (%); 9, reversible capacity (mA h g−1iodine). 10, reference. | |||||||||
| 2020 | Modified Fe powder with ascorbic acid | 1 M FeSO4 | I2/N-doped hierarchically porous carbon | ∼2 | 2 A g−1 | 550 | 100 | — | 19 |
| 2022 | Fe foil | 5 M FeCl2/1 M ZnCl2 | Polyaniline | — | 8 mA cm−2 | 1200 | 99.8 | — | 61 |
 | ||
| Fig. 18 (a) Schematic illustration for the surface species of Fe electrodes in 5 M FeCl2, 5 M FeCl2 + 1 M ZnCl2, and 5 M FeCl2 + 1 M ZnI2 electrolytes. Electrochemical performance of the Fe–I2 battery using the 5 M FeCl2 + 1 M ZnI2 aqueous electrolyte and the PANI@iodine cathode: (b) long-term cycling performance at 8 mA cm−2, and (c) galvanostatic charge/discharge curves at different current densities. Reprinted with permission from ref. 61. Copyright 2021 Elsevier. Electrochemical performance of the Al–I2 battery with an active carbon cloth@PVP cathode: (d) long-term cycling performance at 1C, and (e) typical galvanostatic charge/discharge curves at different current densities. Reprinted with permission from ref. 78. Copyright 2018 Royal Society of Chemistry. (f) The typical adsorption sites of iodine on the Mg(0001) surface. Reprinted with permission from ref. 249. Copyright 2018 American Chemical Society. (g) Cycling stability of the Mg–I2 battery with the active carbon cloth@I2 cathode at 0.5C. (h) The typical discharge/charge curves of the Mg–I2 battery with a microporous carbon@iodine cathode at 0.25C. Reprinted with permission from ref. 66. Copyright 2017 Springer Nature. | ||
2.6 Aluminum–iodine batteries
The section on Al–I2 batteries in Table 16 presents pertinent performance data. Al has been exploited as an electrode material for batteries since the 1850s. This can be ascribed to its abundant availability, low cost, high theoretical capacity of 2980 A h kg−1, and deep standard electrode potential of 1.66 V (vs. SHE).243 Despite its advantages, in aqueous systems, the formation of a passive oxide film, corrosion, and the hydrogen evolution reaction in an aqueous environment limit the electrochemical reversibility of Al, seriously retarding its practical wide application.63,244 The development of novel electrolytes has historically been crucial for battery technology advancement.77 Recent research has shown that Al anodes can show high reversibility in Al2Cl7−-containing ionic liquids with apparently little dependence on solution composition.63,244 In this regard, Tian et al. successfully created an initial rechargeable Al–I2 battery using a PVP–I2 complex as the cathode and a room-temperature ionic liquid-based electrolyte (mixing AlCl3 with 1-ethyl-3-methylimidazolium chloride at a molar ratio of 1.3:1). The resulting Al–I2 battery provided a high capacity of >200 mA h g−1 at 0.2C and over 150 cycles at 1C. Nevertheless, due to PVP's inadequate iodine species adsorption and low conductivity, battery capacity and lifespan were jeopardized. Therefore, Zhang et al. further integrated active carbon cloth and PVP–I in the cathode, resulting in a long-term life of Al–I2 batteries with a capacity of 102.7 mA h g−1 after 1050 cycles at 1C (Fig. 18d).78 These findings prove that Al–I2 batteries hold promise for future development. Unfortunately, although the conversion-type iodine cathode is able to bypass the difficult dissociation and solid-state diffusion of Al3+ cations in intercalation-based electrode materials, the battery still suffers from sluggish electrochemical kinetics (<1C) and large battery polarization (>0.2 V) (Fig. 18e).63 This may be due to the slow transport of Al3+ cations within the bulk electrode as well as in the electrolyte.245 Meanwhile, the intense electrostatic interaction between the charge-rich Al3+ cation and iodine lattices could be another potential issue in limiting the rapid charging/discharging process.246 All in all, in-depth research on viable electrolytes is urgently needed to achieve high reversibility of aluminum anodes while maintaining high migration rates of Al3+ cations.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|
| 1, year; 2, anode; 3, electrolyte formula; 4, cathode; 5, iodine loading (mg cm−2). Evaluation of Al–I2 cell performance: 6, current density (C, 1C = 211 mA g−1); 7, cycle number; 8, capacity retention (%); 9, reversible capacity (mA h g−1iodine). 10, reference. | |||||||||
| 2017 | Al foil | AlCl3/1-ethyl-3-methylimidazolium chloride (1.3:1, m/m) |
PVP–I2 | 1.2 | 1 | 150 | 100 | — | 25 |
| 2018 | Al foil | AlCl3/1-ethyl-3-methylimidazolium chloride (1.3:1, m/m) |
Active carbon cloth/PVP–I2 | — | 1 | 1050 | — | 102.7 | 78 |
2.7 Magnesium–iodine batteries
The Mg–I2 battery section of Table 17 provides the relevant battery information and performance. Mg metal possesses a deep redox potential (−2.37 V vs. SHE), high abundance (1.5 wt% in Earth's crust), and high theoretical volumetric energy density (3833 mA h cm−3).66,247 Particularly, Mg does not deposit into detrimental morphologies in most electrolyte solutions, making Mg-based batteries safer than other batteries.63,64 Similar to Al-based batteries, however, the fatal problem for Mg-based batteries is the Mg anode that has a strong tendency to form insulating and passivating surface layers in most environments, which would block internal electrochemical reactions kinetically.64,65,248 Based on simulations and calculations, Liu et al. revealed that the Mg(0001) surface can provide strong chemical bonding interaction for I2 molecules, leading to the dissociation and reduction of I2 to I− anions under a low iodine coverage condition (Θ = 12.5%) (Fig. 18f).249 However, in contrast to Li, when the iodine coverage is increased to 100%, the Mg surface exhibits good resistance to surface iodization without iodide exfoliation.137 This indicates that the Mg anode could have good resistance to iodization-induced corrosion and self-discharge. Typically, the development of new-type electrolytes can effectively result a highly stable and reversible Mg anode. For example, Bertasi et al. proposed a new iodoaluminate ionic liquid-based electrolyte prepared by reacting 1-ethyl-3-methylimidazolium iodide with AlI3 in a 1:1 molar ratio.21 They suggested that a 2.9 V Mg–I2 secondary battery could be realized by coupling the ionic liquid-based electrolyte with an Mg anode, although the performance of the relevant battery was not presented. Fortunately, Tian et al. realized a rechargeable Mg–I2 battery by using activated carbon cloth@iodine as the cathode, and magnesium bis(trimethylsilyl)amide with aluminum chloride and magnesium chloride in tetraglyme as the electrolyte.66 The resulting Mg–I2 battery exhibited a high capacity retention of 94.6% after 120 cycles at a rate of 0.5C (Fig. 18g). In addition, Zhang et al. further realized over 500 cycles at 800 mA h g−1 with a decay rate of 0.18% per cycle in a Mg–I2 cell based on a PVP–I cathode, as the shuttle effect was effectively mitigated by the chemical interaction between iodine and PVP.76 The successful application of common iodine cathodes, such as PVP/I and carbon cloth@iodine, proves the feasibility of Mg–I2 batteries. Among the multivalent MIBs, the Mg–I2 battery holds a special position with the highest discharge plateau at ∼2.4 V, indicating its high energy density (Fig. 18h). Considering the advantages of affordability and abundant resources of Mg metal, Mg–I2 could have a broad application value in large-scale energy storage applications. Nonetheless, Mg–I2 batteries are still in the early stages of development due to the issues of magnesium anodes. Normally, the passivation layer formed on the Mg surface during electrodeposition would block the associated electrochemical reactions and Mg2+ diffusion channels at room temperature. In addition, the strong electrostatic interaction generated by the high charge density of Mg2+ cation also leads to sluggish kinetics inevitably. However, with the development of novel electrolytes that can resolve the stability and reversibility issues of Mg anodes, Mg–I2 batteries have high potential due to their advantages of cost-effectiveness, high energy density, and the absence of intercalation/deintercalation processes.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|
| 1, year; 2, anode; 3, electrolyte formula; 4, cathode; 5, iodine loading (mg cm−2). Evaluation of Mg–I2 cell performance: 6, current density (C, 1C = 211 mA g−1); 7, cycle number; 8, capacity retention (%); 9, reversible capacity (mA h g−1iodine). 10, reference. (HMDS)2Mg, magnesium bis(trimethylsilyl)amide. | |||||||||
| 2017 | Mg foil | (HMDS)2Mg/AlCl3/MgCl2 in TEGDME | Active carbon cloth/I2 | 1 | 0.5 | 120 | 94.6 | ∼180 | 66 |
| 2022 | Mg foil | (HMDS)2Mg/AlCl3/MgCl2 in TEGDME | PVP–I2 | ∼0.5 | 0.8 | 500 | 90 | — | 76 |
3 Advanced calculation and characterization in metal–iodine batteries
The primary focus of this review is on the material design of metal–iodine batteries (MIBs). However, it is also essential to provide a concise overview of advanced characterization and computational methods commonly employed in MIBs. These methods play a crucial role in bridging the gap between experimental findings and results, promoting the understanding and development of MIBs.3.1 Advanced calculation methods
Throughout history, computational chemistry has emerged as an indispensable tool for interpreting experimental results, predicting intrinsic properties, and investigating new chemical phenomena.250 In the realm of MIBs, various calculation methods have been developed, including density functional theory (DFT) calculations, molecular dynamics (MD) simulation, and finite element analysis. Here, we present a summary of their typical applications in the context of MIBs.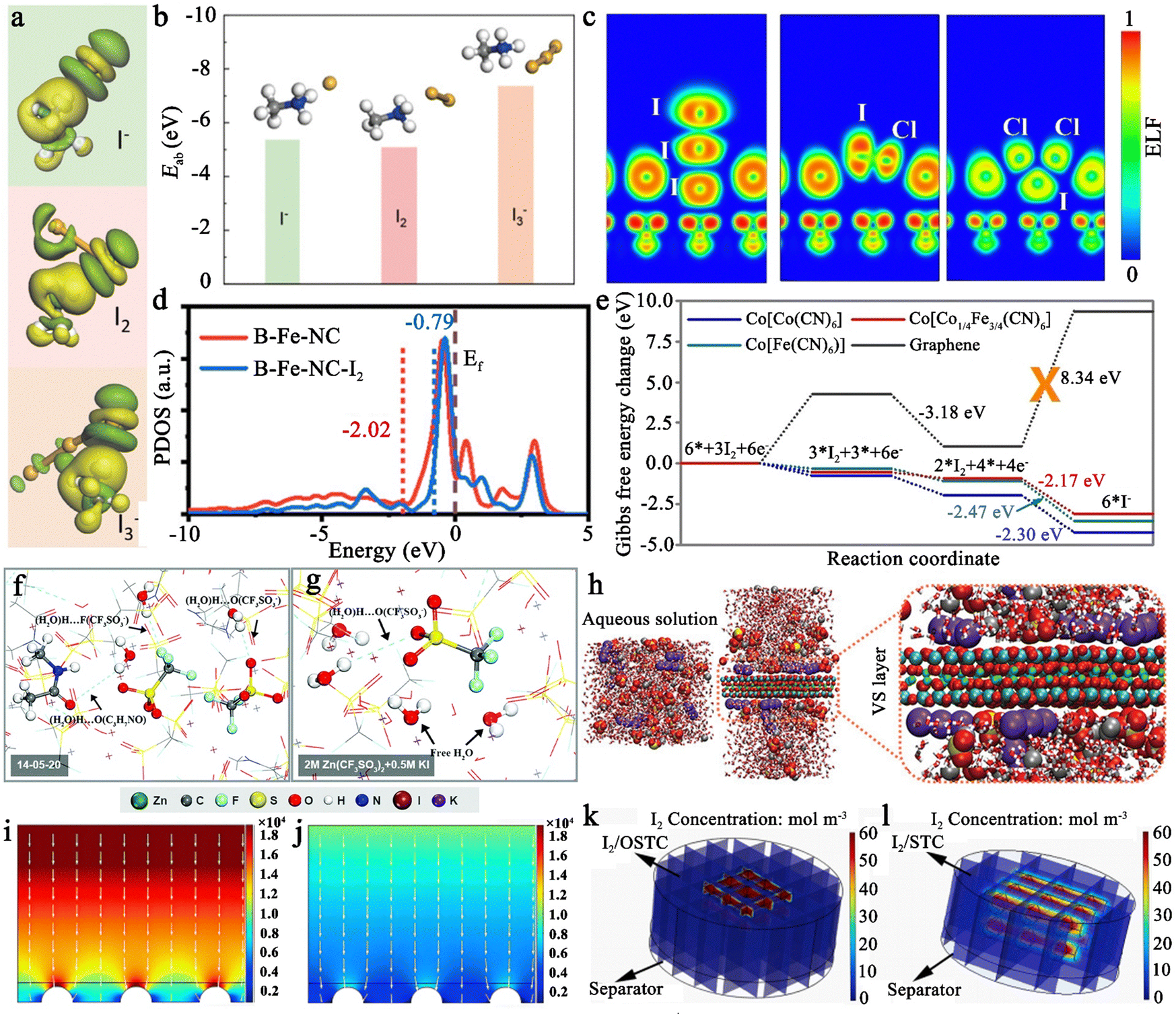 | ||
| Fig. 19 (a) Optimized charge-density-difference patterns of I−, I2, and I3− on CH6N+. (b) The calculated adsorption energy of iodine species (I−, I2, and I3−) on CH6N+. Reprinted with permission from ref. 53. Copyright 2022 Wiley. (c) Electron localization function of iodine redox products. Reprinted with permission from ref. 18. Copyright 2021 Wiley. (d) Partial density of states analysis for the d-band of Fe in B–Fe–NC and B–Fe–NC–I2. Reprinted with permission from ref. 183. Copyright 2022 American Chemical Society. (e) Gibbs free-energy diagrams of iodine reduction reactions on Co[Co(CN)6], Co[Fe(CN)6], Co[Co1/4Fe3/4(CN)6], and bilayer graphene. Reprinted with permission from ref. 190. Copyright 2020 Wiley. Local hydrogen bond distributions in (f) 14-05-20 eutectic solution, and (g) 2 M Zn(CF3SO3)2 + 0.5 M KI. Reprinted with permission from ref. 85. Copyright 2022 Royal Society of Chemistry. (h) MD simulations of iodine in 1 M ZnSO4 and vermiculite nanosheets electrolytes. I, Zn, S, O, H, Si, and Mg are represented by purple, gray, yellow, red, white, cyan, and green spheres, respectively. Reprinted with permission from ref. 205. Copyright 2023 Wiley. COMSOL Multiphysics simulations of the electric field distribution of (i) bare Zn and (j) Zn@ZSO anodes. Reprinted with permission from ref. 216. Copyright 2022 Elsevier. COMSOL Multiphysics simulations of I2 concentration distribution in (k) I2/OSTC, and (l) I2/STC models during the discharge process. Reprinted with permission from ref. 251. Copyright 2023 Elsevier. | ||
:4 with 0.5 M KI and 20% H2O).85 As shown in Fig. 19f and g, the F atoms of CF3SO3− and the carbonyl oxygen atoms of N-ACE in the eutectic solution offer additional sites capable of forming hydrogen bonds compared to the aqueous solution, indicating a low degree of free water in eutectic solution. Furthermore, by combining the radial distribution functions with the characterization results, they effectively demonstrated the double-shell structure of the eutectic solution, i.e., a tight inner layer consisting of I− and CF3SO3− and an outer layer including H2O and C3H7NO−. (2) Visualization of the affinity of materials for iodine species. The relatively macroscopic perspective provides insight into the adsorption of material towards iodide species. As illustrated in Fig. 19h, polyiodide is primarily concentrated near the vermiculite nanosheet layer, while showing a uniform distribution in a 1 M ZnSO4 aqueous solution.205 This effectively demonstrated that the vermiculite nanosheet layer can impede the shuttle effect of polyiodide in the electrolyte.
3.2 Advanced in situ characterization techniques
In situ characterization techniques have advanced the monitoring of changes in physicochemical properties of electrode materials, electrolytes, and separators in real-time during the charging and discharging processes. This holds significant implications in elucidating the working mechanism of batteries, while providing crucial theoretical foundations and experimental strategies for developing innovative materials of MIBs. Various in situ characterization techniques have been developed in the field of MIBs, including in situ Raman spectroscopy, ultraviolet-visible (UV-vis) spectroscopy, X-ray diffraction (XRD), and optical microscopy (OM). In the following, we present an overview of the typical applications of these techniques in MIBs: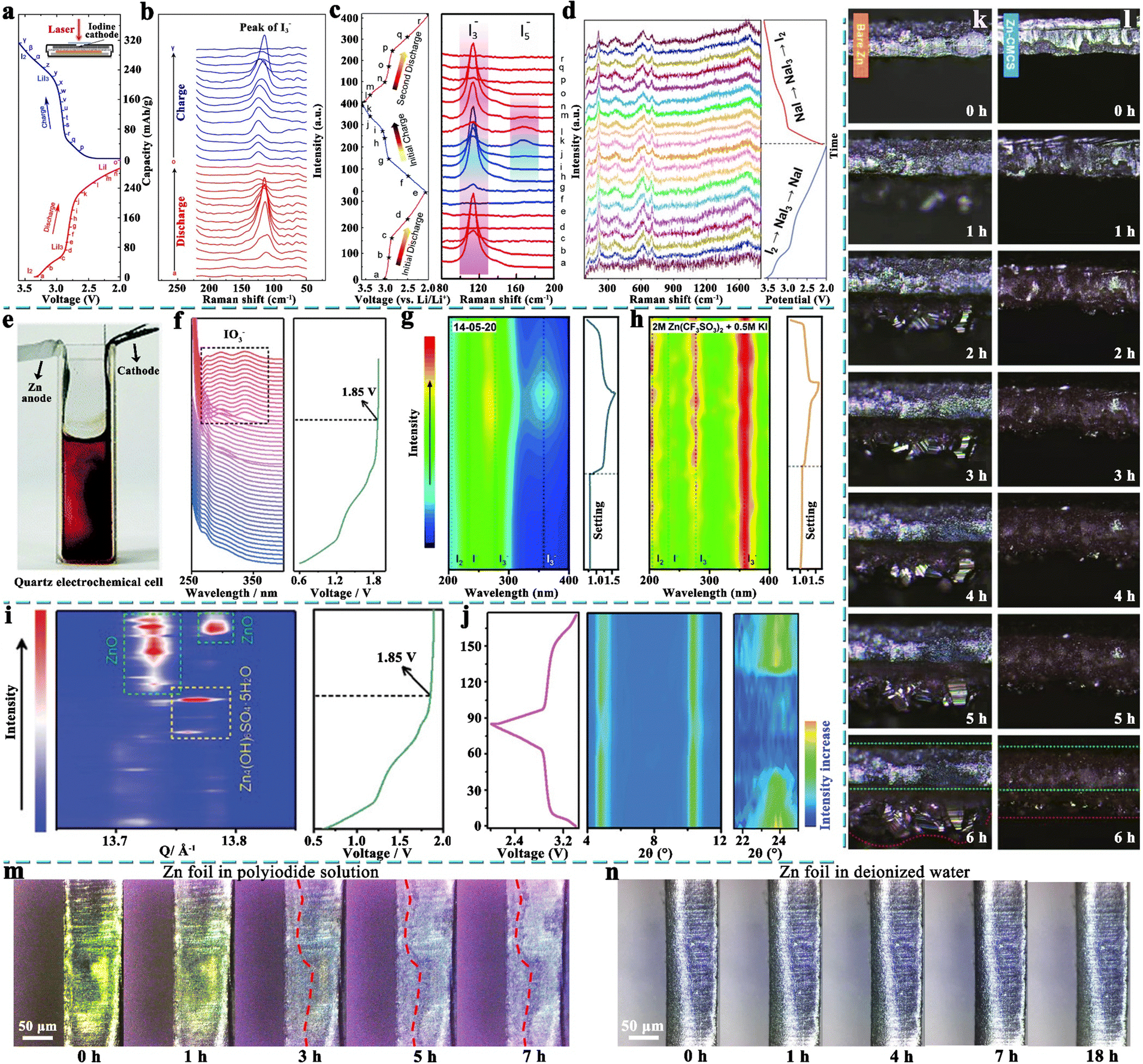 | ||
| Fig. 20 Analysis of Li–I2 batteries using in situ Raman with (a) corresponding charge and discharge curves (inset: schematic diagram of an in situ Raman cell), and (b) Raman spectra of a Li–I2 battery at different voltages. Reprinted with permission from ref. 95. Copyright 2015 American Chemical Society. (c) Discharge and charge curves of an in situ Raman cell with ACC/PVP–I2 as the cathode, and Raman spectra corresponding to the marked points in the curves. Reprinted with permission from ref. 98. Copyright 2018 Royal Society of Chemistry. (d) In situ Raman analysis of the I2/HOF@Ti3C2Tx cathode and corresponding charge/discharge curve. Reprinted with permission from ref. 146. Copyright 2022 Wiley. (e) Schematic of an in situ UV cell. Reprinted with permission from ref. 85. Copyright 2022 Royal Society of Chemistry. (f) The operando UV-vis spectroscopy of electrolyte in a Zn–I2 cell with a I2/N-doped carbon cathode during the charging process. Reprinted with permission from ref. 208. Copyright 2020 Wiley. In situ UV-vis absorption spectra of Zn–I2 batteries using (g) 14-05-20 eutectic electrolyte, and (h) 2 M Zn(CF3SO3)2 + 0.5 M KI electrolyte. Reprinted with permission from ref. 85. Copyright 2022 Royal Society of Chemistry. (i) The operando synchrotron XRD analysis of anode composition changes during charging of Zn–I2 cells. Reprinted with permission from ref. 208. Copyright 2020 Wiley. (j) The operando XRD characterization of Fe2–O8–PcCu/I2 electrode during the charge/discharge process. Reprinted with permission from ref. 24. Copyright 2019 Wiley. In situ OM observation of Zn2+ deposition behavior on (k) Zn-CMCS, and (l) bare Zn. Reprinted with permission from ref. 220. Copyright 2023 Wiley. Corrosion processes of Zn foil in (m) polyiodide solution, and (n) deionized water observed by in situ OM. Reprinted with permission from ref. 212. Copyright 2023 Wiley. | ||
4 Summary and outlook
4.1 A full retrospective for metal–iodine batteries
So far, we have thoroughly reviewed the current development status of various metal–iodine batteries (MIBs) comprehensively. Some problems are commonly faced by MIBs, including the shuttling of iodine species and the irreversibility of metal anodes. Focusing on these questions, a great deal of work has been carried out based on iodine hosts, electrolytes, separators/interlayers, and anodes. Therefore, we further summarize the key innovations and design guidelines of these strategies in implementing advanced MIBs (Fig. 21), which will be discussed in detail below.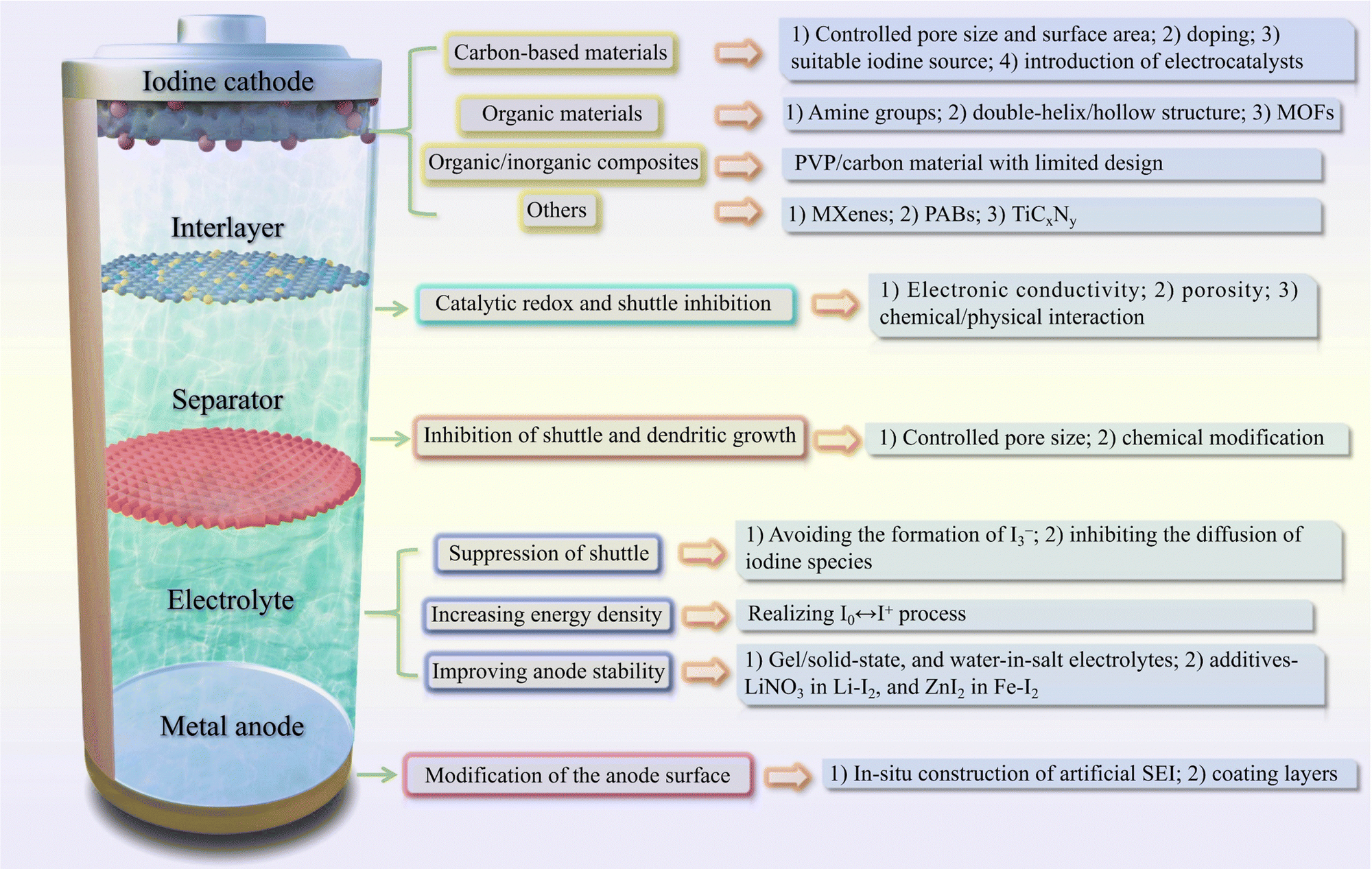 | ||
| Fig. 21 Summary of strategies for implementing advanced metal–iodine batteries. | ||
(1) Suppression of polyiodide shuttling to improve battery stability. This can be achieved in two ways, i.e., avoiding the formation of polyiodide, and inhibiting the diffusion of iodine species in electrolytes. In theory, the first method is only applicable for aqueous batteries because iodine hardly dissolves in water but has high solubility in organic solvents, which will lead to shuttle effects. Typically, electrolyte systems, including PEG400-containing electrolytes, N-methylacetamide-containing eutectic electrolytes, and high-concentration chloride-containing salt electrolytes, are capable of fulfilling the former function, where the key to the success of these electrolytes is to prevent the bonding of iodine and iodide ions.85,86,203 For instance, with the formation of the [ZnIx(OH2)4−x]2−x superhalide ions or double-shell solvated structure (the inner layer consists of I− and CF3SO3−, and the outer layer includes H2O and C3H7NO), the electrostatic interaction between iodide ions and surrounding ions/molecules will be greatly reduced, thereby resulting in the low concentration of polyiodide.85,86 Especially, with a high concentration of ZnCl2 electrolyte (5 M KI/20 M ZnCl2), Ji et al. achieved an advanced Zn–I2 battery with 2000 cycles without capacity decline, even under an ultra-high iodine load of 25.33 mg cm−2.202 Besides, the complexation between PEG400 and iodine can also inhibit the reaction of iodine with I− to form polyiodide.203 On the other hand, both solid-state and gel electrolytes exhibit the function of restricting the shuttle effect of iodine species due to the restricted solubility of iodine species in these electrolytes. In terms of solid-state electrolytes, it is a key factor to achieve high battery performance by keeping iodine species in the liquid state, since solid-phase transformation would lead to sluggish reaction kinetics and poor battery rechargeability. For example, by introducing a polyethylene oxide layer to dissolve a large amount of polyiodide, more than 6000 cycles were achieved in Li1.5Al0.5Ge1.5(PO4)3 solid-state electrolyte at a relatively low temperature of 60 °C.128 For gel electrolytes, there are two approaches to achieve efficient shuttling inhibition, including integration with other materials capable of polyiodide confinement (e.g., MXenes, MOFs, and carbon cloth) and structural modification. Tang et al. integrated Ti3C2Tx MXene-wrapped carbon cloth–iodine with a composite gel polymer electrolyte to obtain a Li–I2 battery with 85% capacity retention after 1000 cycles.70 By immobilizing negatively charged polyanionic chains on the hydrogel skeleton, Shang et al. effectively blocked I3− from shuttling and resulted in a capacity retention of 66.8% after 2000 cycles in a Zn–I2 battery.207
(2) Improvement of battery energy density. Commonly, chloride-containing salt electrolytes can achieve this purpose by I0/I+ conversion (theoretical capacity up to 422 mA h g−1). One of the key factors in such a system is the formation of I+Cl− complexes to achieve the stabilization of I+. Also, inert organic solvents (e.g., acetonitrile) can be added to aqueous electrolytes to reduce the activity of water for alleviating irreversible reactions of I+ ions in aqueous solutions.40 Notably, the difference in iodine cathode composition seems to determine the complexity and reversibility of I0/I+ conversion. For example, when the Ti3C2I2 MXene cathode was used, the concentration of ZnCl2 rapidly decreased from 19 M (PAC carbon@I2 cathode) to 2 M, although the high-concentration ZnCl2 was more favorable for I+ stabilization.40,74 However, there is still a lack of systematic studies on how to exploit the synergy between the cathode and electrolyte to achieve highly stable I+ ions. On the other hand, the energy density of Li–I2 batteries can be improved by using aqueous electrolytes due to the enhanced charge–discharge voltage platform (∼3.5 V).125 The high cost, high complexity, short lifespan, and low safety of this system, however, are significantly hindering its further development. In the case of the Zn–I2 flow battery, the electrolyte containing Br− and Cl− ions can release I3− into I2 by forming I2Br− and I2Cl−, thereby increasing iodine utilization and boosting battery capacity.226,227 It is worth noting that Br− ions are preferred over Cl− ions due to the higher corrosiveness of the latter. Additionally, the adoption of an alkaline anolyte, containing 6 M KOH, allows for the reduction of the redox potential of the zinc anode to −1.26 V, leading to an increase in the total voltage of the Zn–I2 flow cell to 1.796 V.164 However, the feasibility of using an alkaline anolyte still needs to be evaluated critically due to the shortened battery life cause by cross-contamination (Zn(OH)42−, I3−, and I−), severe zinc dendrite growth, and irreversible disproportionation reaction (6OH− + 3I2 → 5I− + IO3− + 3H2O).
(3) Improvement of anode stability. Gel/solid-state electrolytes and water-in-salt electrolytes are generally accompanied by positive functions to improve the stability of metal anodes. In addition, some electrolyte additives, such as LiNO3, are also considered to be effective in improving anode stability since they can form stable passive films on the anode surface. Particularly, given the introduction of ZnI2 in aqueous FeCl2 electrolyte, the formation of [ZnI]+ species allows the doping of Zn into the Fe lattice during the reduction process, resulting in uniform deposition of Fe anodes.61 Besides, NH4+ ions are also considered to be effective in slowing down the growth of zinc dendrites due to the electrostatic shielding effect caused by the complexation of NH4+ ions with Zn2+ ions.226 For Mg/Al–I2 batteries, the properties of electrolytes are particularly important as they greatly determine the degree of reversibility of the metal anodes. At present, Mg–I2 batteries use magnesium bis(trimethylsilyl)amide/AlCl3/MgCl2 in a tetraethylene glycol dimethyl ether electrolyte, while Al–I2 batteries employ a AlCl3/1-ethyl-3-methylimidazolium chloride electrolyte.
4.2 Existing bottlenecks and solutions
Despite most MIBs being in their infancy, Li/Zn–I2 batteries have been developed for a long time. As the fundamental issues of batteries (e.g., stability and rate performance) are continuously improved, some bottlenecks are gradually exposed that will be discussed below.Numerous efforts also have been undertaken to develop high-performance iodine hosts, including carbon-based/organic materials, MXenes, and PABs. Regardless of this, the content (unit: wt%) and loading (unit: mg cm−2) of iodine on the cathode jointly determine the areal capacity and energy density of battery devices. For instance, in lithium-ion batteries, an areal capacity of over 4 mA h cm−2 is required to power an electric vehicle.8,254–256 However, most Li–I2 batteries operate at around 3.0 V, which is lower than the 3.5 V of Li-ion batteries and results in an urgent requirement for Li–I2 batteries to have higher areal capacities (>5 mA h cm−2) in order to compete with state-of-the-art lithium-ion batteries. In this regard, the loading of iodine on the cathode must be as high as ∼24 mg cm−2 based on the theoretical capacity of iodine (211 mA h g−1). When assuming a total iodine content of 50 wt% in the cathode, an unachievable value of iodine loading of 48 mg cm−2 would be observed. Therefore, there is an urgent need to investigate and develop highly loaded and high-content iodine cathodes. However, despite great strides in lifetime made by Li/Zn–I2 batteries, their iodine content and loading are typically below 50 wt% and 3 mg cm−2 (Fig. 22a and b), respectively, which are far below the practical requirements. Encouragingly, on the way to achieving high iodine-containing cathodes, organic iodized salts and Nb-based MXene (Nb2CTx) materials have been able to achieve an iodine content of more than 80 wt%.53,83 Combining a hydrothermal reduced graphene oxide cathode with a high concentration of ZnCl2 electrolyte (5 M KI/20 M ZnCl2), Li et al. also realized 2000 cycles without capacity decline even under an ultra-high iodine load of 25.33 mg cm−2.202 These works could contribute significantly to the commercialization of Li/Zn–I2 batteries. Besides, Li/Zn–I2 batteries, as an emerging technology, may draw valuable insights and research approaches from the relatively mature research field of metal-sulfur batteries due to their high similarity.8 On another note, photo-assisted chargeable MIBs have been reported under the concept of “all-in-one”.73,100 However, these self-powered devices are generally at the proof-of-concept stage with poor performance. Although most studies demonstrated the synchronous conversion and storage of energy, they lacked insights into the ion and electron migration and storage involved in the process, as well as the investigations into battery failures. Relying on advanced characterization techniques, calculations, and simulations, in-depth and detailed working/failure mechanisms for MIBs batteries-contained practical “all-in-one” devices are expected to be explored in the future (Fig. 22c).
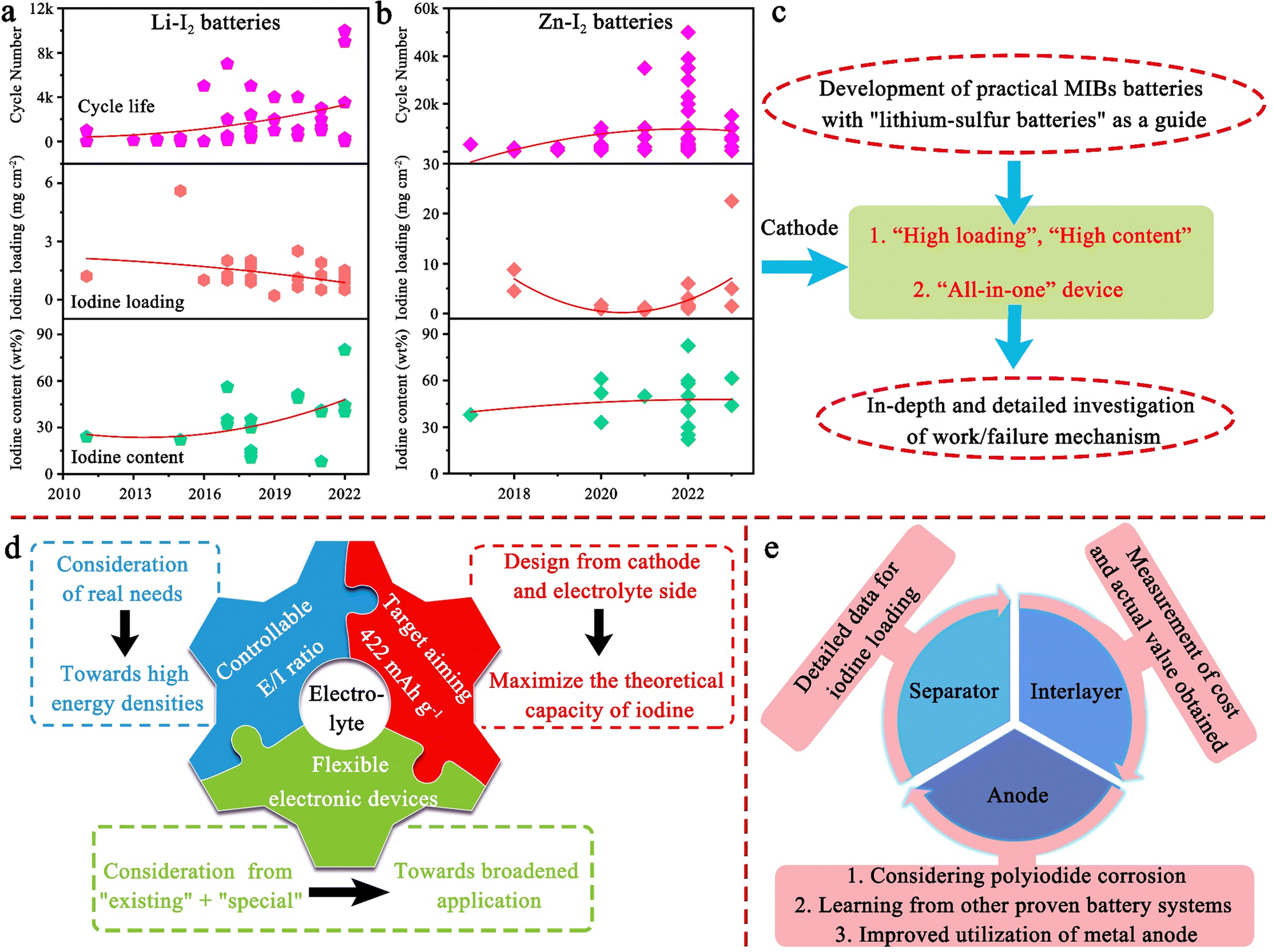 | ||
| Fig. 22 Plot of cycle life, iodine load, and iodine content versus time in (a) Li–I2 and (b) Zn–I2 batteries. Recommended guidelines for the development of (c) cathode, (d) electrolyte, and (e) separator/interlayer/anode in MIBs. | ||
:anolyte = 1:7), thereby preventing the convection of iodine species. However, this is clearly not suitable for static cells. Moreover, static cells also need to consider the tap density of cathodes and cannot hold a large amount of catholyte. Therefore, practical metal–iodine static batteries may not be compatible with an iodine-containing catholyte. It is more desirable to incorporate a significant amount of iodine into the cathode uniformly, to achieve high energy densities.
On the other hand, some electrolytes, such as chlorine-containing electrolytes, have been reported to achieve I0/I+ conversion, which substantially increases the theoretical capacity of iodine-based batteries to 422 mA h g−1.18,40 Such an ideal capacity is highly desirable in MIBs, as it can effectively reduce the iodine loading required in Li–I2 batteries to ∼10 mg cm−2. However, maintaining high reversibility of I+ ions while suppressing the shuttle of polyiodide under high iodine loading remains problematic. Although the use of high concentration of chloride salts can stabilize I+ ions, this approach not only greatly increases battery costs, but also demands high corrosion resistance of the cell structure. One feasible method may be to modify the iodine host so that the cathode is also conducive to the stabilization of I+ ions, thereby reducing the need for electrolytes.74 In Mg/Al–I2 batteries, the high charge density of Mg2+/Al3+ makes it challenging for chloride ions to dissociate in electrolytes, resulting in the low activity of chloride ions.63 This could make I0/I+ conversion difficult to implement in these batteries even with a high concentration of chlorine salts. Therefore, an objective and comprehensive understanding of I0/I+ conversion is required in future work on MIBs. Solid-state/gel electrolytes generally provide higher security than liquid electrolytes. Besides, they can inhibit the shuttle of iodine species effectively and be used to develop a variety of flexible electronic devices. However, they often suffer from short cycle life and poor rate performance.134,258 A good solution is a combination of “existing” + “special”, i.e., using other proven battery system solutions, such as Li/Zn-ion batteries, with due consideration to improving iodine utilization and suppressing polyiodide shuttling in MIBs. A vivid example is the “confined dissolution” strategy proposed by Chen and co-workers, who used the traditional NASICON-type ceramic electrolyte in combination with a polyethylene glycol layer (dissolving iodine species) to achieve a high-performance all-solid-state Li–I2 battery with over 9000 cycles.128
000 cycles in Zn–I2 batteries) and rate capability (up to 100C). Therefore, they should further move towards practicality while considering the relationship between cost and performance.
The stability of metal anodes usually plays a vital role in battery performance during the long cycling process. Maintaining anode stability has become a common problem in metal-based batteries, and several effective solutions have emerged, including anode internal structure optimization,259 anode surface coating modification,260 multifunctional separator designs,92 and electrolyte modification,261 which can also be applied to MIBs. It is important to note that the adopted strategy should be able to protect anodes from iodine species and avoid problems such as polyiodide corrosion and battery self-discharge. For example, Ren et al. used a Li–In alloy layer to promote the stability of the Li anode while also imparting a graphene paper layer on the anode to inhibit polyiodide shuttling.138 Besides, improving the utilization of used metal anodes is necessary to speed up the practical application of MIBs. In this regard, the authors should provide key information in detail, including the amount of metal anode, electrolyte usage, and iodine loading, which would be beneficial for the objective evaluation of the actual battery performance.
4.3 Future prospects and recommendations
Reviewing the state-of-the-art rechargeable MIBs, it can be found that MIBs are on the rise as novel energy storage systems, especially Li/Zn–I2 batteries. In order to achieve the continued rapid development of MIBs, it is necessary to accurately understand the placement of different MIBs. Given the commonality of iodine cathodes, MIBs differ more significantly in terms of the anode. Therefore, Fig. 23a provides an intuitive insight into the advantages and disadvantages of MIBs by counting the differences in anodes (price, abundance, specific capacity, volumetric capacity, and redox potential) as well as the overall performance of MIBs (stability and rate). The data are typically processed by percentage conversion based on the obtained maximum value of a certain condition. For example, given that the prices of lithium and iron are ∼19.2 $ kg−1 (the highest value in terms of cost) and ∼2.4 $ kg−1, respectively, the obtained equivalent values for lithium and iron are thus 100 and 12.5, respectively. Unsurprisingly, Li–I2 batteries show a huge advantage in energy density due to the high redox potential and specific capacity of Li metal anodes. Currently, the state-of-the-art Li–I2 battery can possess an energy density of ∼1324 W h kg−1iodine (based on I−/I+ conversion) and a discharge voltage plateau of 3.42 V.18 As a class of conversion-type batteries, Li–I2 batteries have faster rates than Li-ion batteries, with a capability of up to 100C.43 Meanwhile, owing to the highly reversible iodine conversion reaction and few intermediates, Li–I2 batteries generally have long lifetimes of thousands of cycles (even up to 10000 cycles) and high capacity retention.53 These merits make Li–I2 batteries fully complementary to LIBs for alleviating problems associated with the shortage of battery resources such as Co and Ni. However, the low abundance and high price of Li metal make Li-based batteries theoretically unsuitable for the future large-scale energy storage market. Alternatively, Na–I2 batteries may be a promising system to compensate for the deficiencies of lithium-based batteries. Na–I2 batteries can possess a discharge plateau of ∼2.7 V and long lifetimes (up to 5000 cycles) with outstanding rate capability.110,151 As the commercialization of sodium-ion batteries continues and results in the gradual maturation of the sodium-based battery industry, Na–I2 batteries are demonstrating promising applications in a wide range of batteries. On the other hand, the advantages of K–I2 batteries lie in their high abundance and deep redox potential. However, their short lifespan and low safety, caused by own defects (K metal is too reactive), could seriously limit their wide application like Li/Na–I2 batteries, making research on them likely to remain in a long-term fundamental state.68,154 In terms of multivalent MIBs, Zn–I2 batteries are currently the most promising battery system. Typically, Zn–I2 batteries have demonstrated a durable life of over 10000 cycles and an outstanding rate performance of over 10C.27,42,83,175 With the realization of I0/I+ conversion, the Zn–I2 cell is able to deliver an energy density of ∼750 W h kg−1 (based on iodine) with two discharge plateaus of 1.83 V (I0/I+) and 1.29 V (I−/I0), demonstrating its sufficient advantages over manganese/vanadium-based Zn-ion batteries.40 By combining the advantages of safety, environmental friendliness, low cost, and high operability, Zn–I2 batteries show significant promise as one of the lead-acid battery replacements, and are applied in large-scale energy storage systems. Moreover, Zn–I2 flow batteries, with a remarkable theoretical capacity of 268 A h L−1, hold significant potential for practical applications.232 On the other hand, the high abundance and ultra-low cost of Fe/Mg/Al–I2 batteries have also aroused a passion for research. However, the intrinsic nature of these metals also severely limits the performance of the as-fabricated metal–iodine batteries. Although, Fe–I2 batteries can show over 1200 cycles and a rate of ∼7.6C, their low discharge plateau (<0.9 V) and the instability of the Fe anode in aqueous electrolytes may hinder their further investigation. For Mg/Al–I2 batteries, the anodes commonly tend to form insulating and passivation layers during the cycling process, resulting in short battery life and slow plating/stripping processes. In addition, the high charge density of Mg2+/Al3+ further inevitably leads to sluggish battery kinetics. As a result, the cycle life of Mg/Al–I2 batteries is inferior with a rate performance generally lower than 1C.25,76 On account of this, for these types of iodine batteries, extensive research should continue to focuson addressing the inherent problems of metal anodes.
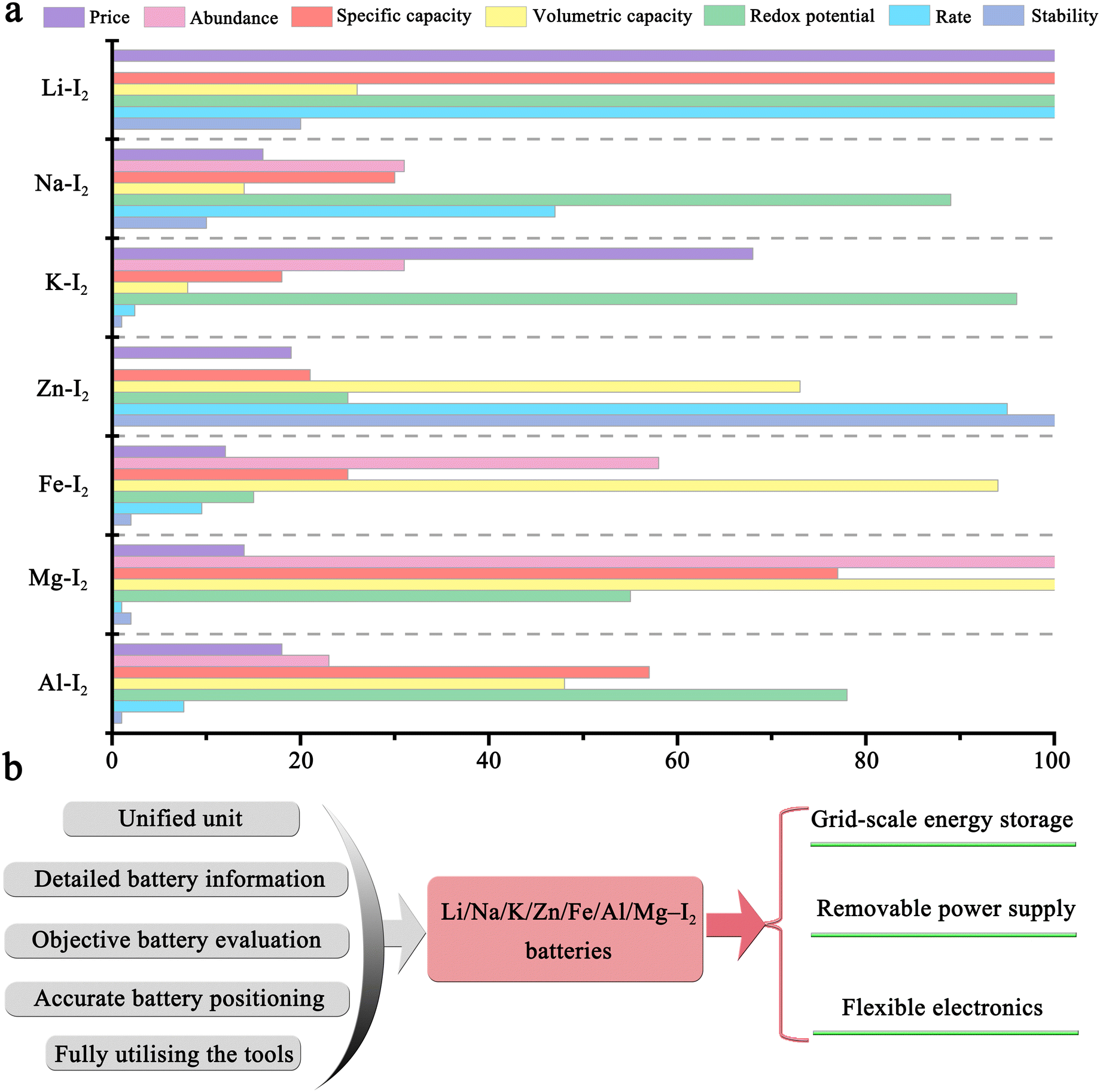 | ||
| Fig. 23 (a) Schematic diagram for visual comparison of different MIBs in terms of price, abundance, specific capacity, volumetric capacity, redox potential, rare, and stability. (b) Proposed suggestions and prospects for the continuous high-quality and rapid development of MIBs in the future. | ||
In order to maintain the rapid development of metal–iodine batteries, Fig. 23b presents some recommendations. (1) Uniform unit should be used (1C = 211 mA g−1 as the rate unit). Detailed cell information, including the amount of metal anodes, electrolyte usage, E/I ratio, iodine loading, negative/positive ratio, needs to be provided. These are helpful for readers to evaluate the actual working conditions of the battery and avoid misplaced guidance. (2) The overall volumetric energy density of iodine cathodes should be provided. According to the iodine content and the associated percentage of inactive material, the gravimetric energy density of iodine cathodes is easily obtained. However, despite the importance of volumetric energy densities of batteries in practical applications (e.g., it is impractical to install batteries with low volumetric energy density for space-constrained electric vehicles), studies (except for flow batteries) seldom refer to the volumetric energy density of iodine cathodes. Thus, it is necessary to provide relevant volumetric energy density of iodine cathodes, for driving enthusiasm in pursuing practically usable metal–iodine batteries. (3) Cell lifespan should be evaluated at low current rates below 2C. Because the high C-rate implies a fast charging and discharging process that constrains the diffusion of polyiodide dissolved in the electrolyte (electric field effect), which in turn will cover up the inhibitory effect of materials on the shuttle effect.262,263 (4) The performance of materials should be evaluated from both the “local” and “system” perspective. For example, when attempting to build a separator to inhibit polyiodide shuttling and achieve highly stable MIBs, tests should first be performed using a catholyte containing iodide, as well as a cathode that does not adsorb polyiodide; this can be a good reflection of the electrochemical performance of the material from the “local” perspective. Then, from the “system” aspect, the application of common (carbon cloth and active carbon) or state-of-the-art cathode materials to test the cells gives a good indication of the progressiveness of this work. Benefiting from this, readers can effectively understand the advancement of the work through literature review. (5) Accurate battery placement is essential to ensure that the batteries can meet market demand during the process of development, instead of accumulating useless data. For example, when defining the Zn–I2 battery as a safe, green alternative to the “lead-acid battery” system, we should also design Zn–I2 batteries according to “lead-acid battery” specifications, rather than positioning them to compete with the existing lithium-ion batteries. (6) Taking advantage of late movers. As different battery systems have both differences and commonalities, the development of MIBs should not be restricted to itself. For instance, the design of anodes and cathodes of Li–I2 batteries can draw upon some advanced concepts and methods from lithium-ion batteries and Li–S batteries, to reduce unnecessary “detours”. (7) Development of advanced characterization technologies, such as in situ Raman, X-ray diffraction, and ultraviolet-visible spectroscopy. These methods would help to discover the real working mechanism, reaction process, and existing problems of batteries, so as to design materials with advanced properties. (8) The utilization of systematic theoretical calculations, such as density functional theory calculation, molecular dynamics simulation, and COMSOL Multiphysics simulation, which allows us to establish theoretical foundations and provide guidance for experiments. (9) Combination with artificial intelligence (e.g., machine learning). This allows us to access knowledge quickly, enable interdisciplinarity, and bring more innovation to our work. All these would drive the rapid development of MIBs with continuous improvement of competitiveness. Desirably, by matching different anodes, iodine-based batteries can adapt to various complex and changeable application environments, that is, high-safety and green zinc–iodine batteries can be applied to smart home energy storage and flexible electronic devices; cheap and resourceful iron/aluminum/magnesium–iodine batteries can be used for grid-scale energy storage; and high-energy-density lithium–iodine batteries can power electric-driven vehicles. All in all, we believe that metal–iodine batteries will occupy a prominent place in a variety of battery systems in the future.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors sincerely acknowledge the financial support from the National Natural Science Foundation of China (no. 52161135302, no. 52233006, and no. 52211530489). F. L. acknowledges the financial support from the Research Foundation-Flanders (FWO, grant no. 1298323N). J. H. acknowledges the financial support from the Research Foundation-Flanders (FWO, grant no. G0F2322N, G983.19N, G0A5817N, and ZW15_09-G0H6316N, VS06523N), from the Flemish government through long-term structural funding Methusalem (CASAS2, Meth/15/04), and from the MPI as an MPI fellow.Notes and references
- E. C. Evarts, Nature, 2015, 526, S93–S95 CrossRef CAS PubMed.
- D. G. Kwabi, Y. Ji and M. J. Aziz, Chem. Rev., 2020, 120, 6467–6489 CrossRef CAS PubMed.
- B. Steffen and A. Patt, Energy Res. Soc. Sci., 2022, 91, 102758 CrossRef.
- U.S. Energy Information Administration, International Energy Outlook 2021, https://www.eia.gov/outlooks/ieo/climate.php.
- A. Azizivahed, A. Arefi, S. Ghavidel, M. Shafie-khah, L. Li, J. Zhang and J. P. S. Catalao, IEEE Trans. Sustain. Energy, 2020, 11, 662–673 Search PubMed.
- F. Cebulla, J. Haas, J. Eichman, W. Nowak and P. Mancarella, J. Cleaner Prod., 2018, 181, 449–459 CrossRef.
- H.-F. Wang and Q. Xu, Matter, 2019, 1, 565–595 CrossRef CAS.
- S. H. Chung and A. Manthiram, Adv. Mater., 2019, 31, e1901125 CrossRef PubMed.
- P. Li, C. Li, X. Guo, X. Li and C. Zhi, Bull. Chem. Soc. Jpn., 2021, 94, 2036–2042 CrossRef CAS.
- K. Chen, D. Y. Yang, G. Huang and X. B. Zhang, Acc. Chem. Res., 2021, 54, 632–641 CrossRef CAS PubMed.
- Q. Pang, X. Liang, C. Y. Kwok and L. F. Nazar, Nat. Energy, 2016, 1, 16132 CrossRef CAS.
- J. Ma, M. Liu, Y. He and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 12636–12647 CrossRef CAS PubMed.
- V. Viswanathan, A. H. Epstein, Y.-M. Chiang, E. Takeuchi, M. Bradley, J. Langford and M. Winter, Nature, 2022, 601, 519–525 CrossRef CAS PubMed.
- G. L. Soloveichik, Chem. Rev., 2015, 115, 11533–11558 CrossRef CAS PubMed.
- H. Lim, A. Lackner and R. Knechtli, J. Electrochem. Soc., 1977, 124, 1154 CrossRef CAS.
- B. Li, Z. Nie, M. Vijayakumar, G. Li, J. Liu, V. Sprenkle and W. Wang, Nat. Commun., 2015, 6, 6303 CrossRef CAS PubMed.
- P. Bai and M. Z. Bazant, Electrochim. Acta, 2016, 202, 216–223 CrossRef CAS.
- X. Li, Y. Wang, Z. Chen, P. Li, G. Liang, Z. Huang, Q. Yang, A. Chen, H. Cui, B. Dong, H. He and C. Zhi, Angew. Chem., Int. Ed., 2022, 61, e202113576 CrossRef CAS PubMed.
- C. Bai, H. Jin, Z. Gong, X. Liu and Z. Yuan, Energy Storage Mater., 2020, 28, 247–254 CrossRef.
- D. Lin and Y. Li, Adv. Mater., 2022, 34, e2108856 CrossRef PubMed.
- F. Bertasi, F. Sepehr, G. Pagot, S. J. Paddison and V. Di Noto, Adv. Funct. Mater., 2016, 26, 4860–4865 CrossRef CAS.
- K. Jayaramulu, D. P. Dubal, B. Nagar, V. Ranc, O. Tomanec, M. Petr, K. K. R. Datta, R. Zboril, P. Gómez-Romero and R. A. Fischer, Adv. Mater., 2018, 30, 1705789 CrossRef PubMed.
- C. Sun, X. Shi, Y. Zhang, J. Liang, J. Qu and C. Lai, ACS Nano, 2020, 14, 1176–1184 CrossRef CAS PubMed.
- F. Wang, Z. Liu, C. Yang, H. Zhong, G. Nam, P. Zhang, R. Dong, Y. Wu, J. Cho, J. Zhang and X. Feng, Adv. Mater., 2020, 32, 1905361 CrossRef CAS PubMed.
- H. Tian, S. Zhang, Z. Meng, W. He and W.-Q. Han, ACS Energy Lett., 2017, 2, 1170–1176 CrossRef CAS.
- C. Liang and C. Holmes, J. Power Sources, 1980, 5, 3–13 CrossRef CAS.
- L. Zhang, M. Zhang, H. Guo, Z. Tian, L. Ge, G. He, J. Huang, J. Wang, T. Liu, I. P. Parkin and F. Lai, Adv. Sci., 2022, 9, 2105598 CrossRef CAS PubMed.
- Y. Zhao, L. Wang and H. R. Byon, Nat. Commun., 2013, 4, 1896 CrossRef PubMed.
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1–21 CrossRef CAS.
- D. E. Harlov and L. Aranovich, The Role of Halogens in Terrestrial and Extraterrestrial Geochemical Processes: Surface, Crust, and Mantle, Springer International Publishing, Cham, 2018, pp. 1–19 DOI:10.1007/978-3-319-61667-4_1.
- S. Hou, L. Chen, X. Fan, X. Fan, X. Ji, B. Wang, C. Cui, J. Chen, C. Yang, W. Wang, C. Li and C. Wang, Nat. Commun., 2022, 13, 1281 CrossRef CAS PubMed.
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- X. Gu, T. Tang, X. Liu and Y. Hou, J. Mater. Chem. A, 2019, 7, 11566–11583 RSC.
- Y. Chen, J. Xu, P. He, Y. Qiao, S. Guo, H. Yang and H. Zhou, Sci. Bull., 2022, 67, 2449–2486 CrossRef CAS PubMed.
- G. Li, T. An, J. Chen, G. Sheng, J. Fu, F. Chen, S. Zhang and H. Zhao, J. Hazard. Mater., 2006, 138, 392–400 CrossRef CAS PubMed.
- Y. Jung, E. Hong, M. Kwon and J.-W. Kang, Chem. Eng. J., 2017, 312, 30–38 CrossRef CAS.
- X. Li, N. Li, Z. Huang, Z. Chen, Y. Zhao, G. Liang, Q. Yang, M. Li, Q. Huang, B. Dong, J. Fan and C. Zhi, ACS Nano, 2021, 15, 1718–1726 CrossRef CAS PubMed.
- A. A. Yaroshevsky, Geochem. Int., 2006, 44, 48–55 CrossRef.
- L. Zhang, M. Zhang, H. Guo, Z. Tian, L. Ge, G. He, J. Huang, J. Wang, T. Liu and I. P. Parkin, Adv. Sci., 2022, 9, 2105598 CrossRef CAS PubMed.
- Y. Zou, T. Liu, Q. Du, Y. Li, H. Yi, X. Zhou, Z. Li, L. Gao, L. Zhang and X. Liang, Nat. Commun., 2021, 12, 170 CrossRef CAS PubMed.
- S. H. Chung and A. Manthiram, Adv. Mater., 2019, 31, 1901125 CrossRef PubMed.
- S. J. Zhang, J. Hao, H. Li, P. F. Zhang, Z. W. Yin, Y. Y. Li, B. Zhang, Z. Lin and S. Z. Qiao, Adv. Mater., 2022, 34, 2201716 CrossRef CAS PubMed.
- Z. Su, Z. Wei, C. Lai, H. Deng, Z. Liu and J. Ma, Energy Storage Mater., 2018, 14, 129–135 CrossRef.
- N. Chen, W. Wang, Y. Ma, M. Chuai, X. Zheng, M. Wang, Y. Xu, Y. Yuan, J. Sun, K. Li, Y. Meng, C. Shen and W. Chen, Small Methods, 2023, 2201553, DOI:10.1002/smtd.202201553.
- L. Rosenfeld, J. Chem. Educ., 2000, 77, 984 CrossRef CAS.
- A. McKeown, Trans. Faraday Soc., 1922, 17, 517–521 RSC.
- A. A. Schneider, D. E. Harney and M. J. Harney, J. Power Sources, 1980, 5, 15–23 CrossRef CAS.
- F.-C. Liu, W.-M. Liu, M.-H. Zhan, Z.-W. Fu and H. Li, Energy Environ. Sci., 2011, 4, 1261–1264 RSC.
- J. Wiaux and R. Bannehr, J. Power Sources, 1987, 19, 37–43 CrossRef CAS.
- B. Scrosati and B. Owens, Solid State Ionics, 1987, 23, 275–278 CrossRef CAS.
- M.-L. Lin, Y.-X. Jin, Y.-Z. Zhang and Y.-F. Hou, J. Power Sources, 1985, 14, 173–177 CrossRef CAS.
- L. Weinstein, W. Yourey, J. Gural and G. Amatucci, J. Electrochem. Soc., 2008, 155, A590 CrossRef CAS.
- P. Li, X. Li, Y. Guo, C. Li, Y. Hou, H. Cui, R. Zhang, Z. Huang, Y. Zhao, Q. Li, B. Dong and C. Zhi, Adv. Energy Mater., 2022, 12, 2103648 CrossRef CAS.
- M. Holzapfel, D. Wilde, C. Hupbauer, K. Ahlbrecht and T. Berger, Electrochim. Acta, 2017, 237, 12–21 CrossRef CAS.
- D. Gong, B. Wang, J. Zhu, R. Podila, A. M. Rao, X. Yu, Z. Xu and B. Lu, Adv. Energy Mater., 2017, 7, 1601885 CrossRef.
- K. Lu, H. Zhang, F. Ye, W. Luo, H. Ma and Y. Huang, Energy Storage Mater., 2019, 16, 1–5 CrossRef.
- T. Yamamoto, J. Chem. Soc., Chem. Commun., 1981, 187–188 RSC.
- T. Yamamoto, M. Hishinuma and A. Yamamoto, Inorg. Chim. Acta, 1984, 86, L47–L49 CrossRef CAS.
- H. Pan, B. Li, D. Mei, Z. Nie, Y. Shao, G. Li, X. S. Li, K. S. Han, K. T. Mueller, V. Sprenkle and J. Liu, ACS Energy Lett., 2017, 2, 2674–2680 CrossRef CAS.
- A. K. Manohar, S. Malkhandi, B. Yang, C. Yang, G. K. Surya Prakash and S. R. Narayanan, J. Electrochem. Soc., 2012, 159, A1209–A1214 CrossRef CAS.
- W. Wu, X. Yang, K. Wang, C. Li, X. Zhang, H.-Y. Shi, X.-X. Liu and X. Sun, Chem. Eng. J., 2022, 432, 134389 CrossRef CAS.
- F. Gutmann, A. M. Hermann and A. Rembaum, J. Electrochem. Soc., 1967, 114, 323 CrossRef CAS.
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656 CrossRef CAS.
- Z. Guo, S. Zhao, T. Li, D. Su, S. Guo and G. Wang, Adv. Energy Mater., 2020, 10, 1903591 CrossRef CAS.
- R. Attias, M. Salama, B. Hirsch, Y. Goffer and D. Aurbach, Joule, 2019, 3, 27–52 CrossRef CAS.
- H. Tian, T. Gao, X. Li, X. Wang, C. Luo, X. Fan, C. Yang, L. Suo, Z. Ma, W. Han and C. Wang, Nat. Commun., 2017, 8, 14083 CrossRef CAS PubMed.
- M. Li, J. Lu, X. Ji, Y. Li, Y. Shao, Z. Chen, C. Zhong and K. Amine, Nat. Rev. Mater., 2020, 5, 276–294 CrossRef CAS.
- L. Deng, M. Feng, R. Wang, Y. Yang, X. Niu, J. Zhang, L. Tan, J. Zhang, Y. Chen, L. Zeng, Y. Zhu and L. Guo, Energy Storage Mater., 2021, 41, 798–804 CrossRef.
- T. Yamamoto, J. Chem. Soc., Chem. Commun., 1981, 187–188 RSC.
- X. Tang, D. Zhou, P. Li, X. Guo, C. Wang, F. Kang, B. Li and G. Wang, ACS Cent. Sci., 2019, 5, 365–373 CrossRef CAS PubMed.
- O. N. Srivastava and R. A. Singh, Bull. Electrochem., 1999, 15, 372–375 CAS.
- Y. Li, L. Liu, H. Li, F. Cheng and J. Chen, Chem. Commun., 2018, 54, 6792–6795 RSC.
- Y. Man, Q. Hao, F. Chen, X. Chen, Y. Wang, T. Liu, F. Liu and N. Li, ChemElectroChem, 2019, 6, 5872–5875 CrossRef CAS.
- X. Li, M. Li, Z. Huang, G. Liang, Z. Chen, Q. Yang, Q. Huang and C. Zhi, Energy Environ. Sci., 2021, 14, 407–413 RSC.
- C. Johnson, R. J. Latham and R. G. Linford, Solid State Ionic, 1982, 7, 331–334 CrossRef CAS.
- Y. Zhang, D. Tao, F. Xu and T. Li, Chem. Eng. J., 2022, 427, 131592 CrossRef CAS.
- B. Xue, Z. Fu, H. Li, X. Liu, S. Cheng, J. Yao, D. Li, L. Chen and Q. Meng, J. Am. Chem. Soc., 2006, 128, 8720–8721 CrossRef CAS PubMed.
- S. Zhang, X. Tan, Z. Meng, H. Tian, F. Xu and W.-Q. Han, J. Mater. Chem. A, 2018, 6, 9984–9996 RSC.
- Q. Zhang, Z. Wu, F. Liu, S. Liu, J. Liu, Y. Wang and T. Yan, J. Mater. Chem. A, 2017, 5, 15235–15242 RSC.
- F.-s Cai, Y.-q Duan and Z.-h Yuan, J. Mater. Sci.: Mater. Electron., 2018, 29, 11540–11545 CrossRef CAS.
- H. K. Machhi, K. K. Sonigara, S. N. Bariya, H. P. Soni and S. S. Soni, ACS Appl. Mater. Interfaces, 2021, 13, 21426–21435 CrossRef CAS PubMed.
- K. K. Sonigara, J. Zhao, H. K. Machhi, G. Cui and S. S. Soni, Adv. Energy Mater., 2020, 10, 2001997 CrossRef CAS.
- X. Li, N. Li, Z. Huang, Z. Chen, G. Liang, Q. Yang, M. Li, Y. Zhao, L. Ma, B. Dong, Q. Huang, J. Fan and C. Zhi, Adv. Mater., 2021, 33, e2006897 CrossRef PubMed.
- C. Prehal, H. Fitzek, G. Kothleitner, V. Presser, B. Gollas, S. A. Freunberger and Q. Abbas, Nat. Commun., 2020, 11, 4838 CrossRef CAS PubMed.
- Y. Yang, S. Liang, B. Lu and J. Zhou, Energy Environ. Sci., 2022, 15, 1192–1200 RSC.
- J. J. Hong, L. Zhu, C. Chen, L. Tang, H. Jiang, B. Jin, T. C. Gallagher, Q. Guo, C. Fang and X. Ji, Angew. Chem., Int. Ed., 2019, 58, 15910–15915 CrossRef CAS PubMed.
- P. Beran and S. Bruckenstein, Anal. Chem., 1968, 40, 1044–1051 CrossRef CAS.
- Y. Tian, S. Chen, S. Ding, Q. Chen and J. Zhang, Chem. Sci., 2023, 14, 331–337 RSC.
- Q. Zhang, Y.-H. Zeng, S.-H. Ye and S. Liu, J. Power Sources, 2020, 463, 228212 CrossRef CAS.
- Z. Li, X. Wu, X. Yu, S. Zhou, Y. Qiao, H. Zhou and S. G. Sun, Nano Lett., 2022, 22, 2538–2546 CrossRef CAS PubMed.
- M. Xing, Z. Z. Zhao, Y. J. Zhang, J. W. Zhao, G. L. Cui and J. H. Dai, Mater. Today Energy, 2020, 18, 100534 CrossRef CAS.
- H. Yang, Y. Qiao, Z. Chang, H. Deng, P. He and H. Zhou, Adv. Mater., 2020, 32, e2004240 CrossRef PubMed.
- W. Du, E. H. Ang, Y. Yang, Y. Zhang, M. Ye and C. C. Li, Energy Environ. Sci., 2020, 13, 3330–3360 RSC.
- Y. L. Wang, Q. L. Sun, Q. Q. Zhao, J. S. Cao and S. H. Ye, Energy Environ. Sci., 2011, 4, 3947–3950 RSC.
- Q. Zhao, Y. Lu, Z. Zhu, Z. Tao and J. Chen, Nano Lett., 2015, 15, 5982–5987 CrossRef CAS PubMed.
- H. Wang, G. Zhang, L. Ke, B. Liu, S. Zhang and C. Deng, Nanoscale, 2017, 9, 9365–9375 RSC.
- K. Li, S. Chen, S. Chen, X. Liu, W. Pan and J. Zhang, Nano Res., 2018, 12, 549–555 CrossRef.
- Z. Meng, X. Tan, S. Zhang, H. Ying, X. Yan, H. Tian, G. Wang and W. Q. Han, Chem. Commun., 2018, 54, 12337–12340 RSC.
- Y. Lai, L. Wang and W. Chen, ChemElectroChem, 2022, 9, e202200022 CrossRef CAS.
- J. Li, H. Liu, K. Sun, R. Wang, C. Qian, F. Yu, L. Zhang and W. Bao, J. Mater. Chem. A, 2022, 10, 7326–7332 RSC.
- K. Lu, Z. Hu, J. Ma, H. Ma, L. Dai and J. Zhang, Nat. Commun., 2017, 8, 527 CrossRef PubMed.
- K. Li, B. Lin, Q. Li, H. Wang, S. Zhang and C. Deng, ACS Appl. Mater. Interfaces, 2017, 9, 20508–20518 CrossRef CAS PubMed.
- G. Zhang, H. Wang, S. Zhang and C. Deng, J. Mater. Chem. A, 2018, 6, 9019–9031 RSC.
- G. Zheng, Q. Zhang, J. J. Cha, Y. Yang, W. Li, Z. W. Seh and Y. Cui, Nano Lett., 2013, 13, 1265–1270 CrossRef CAS PubMed.
- L. Chai, X. Wang, Y. Hu, X. Li, S. Huang, J. Pan, J. Qian and X. Sun, Adv. Sci., 2022, 9, e2105063 CrossRef PubMed.
- Y. Xu, Y. Wen, Y. Zhu, K. Gaskell, K. A. Cychosz, B. Eichhorn, K. Xu and C. Wang, Adv. Funct. Mater., 2015, 25, 4312–4320 CrossRef CAS.
- Z. Wu, J. Xu, Q. Zhang, H. Wang, S. Ye, Y. Wang and C. Lai, Energy Storage Mater., 2018, 10, 62–68 CrossRef.
- C. Kuang, W. Zeng, M. Qian and X. Liu, ChemPlusChem, 2021, 86, 865–869 CrossRef CAS PubMed.
- Z. Meng, H. Tian, S. Zhang, X. Yan, H. Ying, W. He, C. Liang, W. Zhang, X. Hou and W. Q. Han, ACS Appl. Mater. Interfaces, 2018, 10, 17933–17941 CrossRef CAS PubMed.
- M. Qian, Z. Xu, Z. Wang, B. Wei, H. Wang, S. Hu, L. M. Liu and L. Guo, Adv. Mater., 2020, 32, e2004835 CrossRef PubMed.
- V. G. Anju, M. P. Austeria and S. Sampath, Adv. Mater. Interfaces, 2017, 4, 1700151 CrossRef.
- G. Li, F. Lu, X. Dou, X. Wang, D. Luo, H. Sun, A. Yu and Z. Chen, J. Am. Chem. Soc., 2020, 142, 3583–3592 CrossRef CAS PubMed.
- Z. Su, Z. Wei, C. Lai, H. Deng, Z. Liu and J. Ma, Energy Storage Mater., 2018, 14, 129–135 CrossRef.
- Z. Z. Wu, S. Y. Wang, R. Y. Wang, J. Liu and S. H. Ye, J. Electrochem. Soc., 2018, 165, A1156–A1159 CrossRef CAS.
- Z. Su, C.-J. Tong, D.-Q. He, C. Lai, L.-M. Liu, C. Wang and K. Xi, J. Mater. Chem. A, 2016, 4, 8541–8547 RSC.
- C. Gao, X. Li, G. Wei, S. Wang, X. Zhao and F. Kong, Compos. Commun., 2022, 33, 101226 CrossRef.
- J. Chen, J. Ma, B. Liu, Z. Li, X. Zhang, S. Sun, K. Lu, J. Yin, S. Chen, X. Zu, Z. Zhang, X. Qiu, Y. Qin and W. Zhang, Compos. Commun., 2023, 38, 101524 CrossRef.
- J. Yu, S. Liu, G. Duan, H. Fang and H. Hou, Compos. Commun., 2020, 19, 239–245 CrossRef.
- W. Li, H. Yao, K. Yan, G. Zheng, Z. Liang, Y. M. Chiang and Y. Cui, Nat. Commun., 2015, 6, 7436 CrossRef CAS PubMed.
- C. Barchasz, J.-C. Leprêtre, S. Patoux and F. Alloin, Electrochim. Acta, 2013, 89, 737–743 CrossRef CAS.
- F. Liu, S.-B. Cao, X.-G. Ou, Q.-Q. Hu and C.-M. Li, Adv. New Renewable Energy, 2020, 8, 143–150 Search PubMed.
- J. A. Dean, University of Tennessee, McGrawHill, Inc, Knoxville, 1999 Search PubMed.
- J. Coetzee and C. Gardner, Anal. Chem., 1982, 54, 2625–2626 CrossRef CAS.
- N. Papageorgiou, W. Maier and M. Grätzel, J. Electrochem. Soc., 1997, 144, 876 CrossRef CAS.
- Y. Zhao, M. Hong, N. Bonnet Mercier, G. Yu, H. C. Choi and H. R. Byon, Nano Lett., 2014, 14, 1085–1092 CrossRef CAS PubMed.
- C. Shao, Y. Zhao and L. Qu, SusMat, 2022, 2, 142–160 CrossRef CAS.
- G. Nikiforidis, K. Tajima and H. R. Byon, ACS Energy Lett., 2016, 1, 806–813 CrossRef CAS.
- Z. Cheng, H. Pan, F. Li, C. Duan, H. Liu, H. Zhong, C. Sheng, G. Hou, P. He and H. Zhou, Nat. Commun., 2022, 13, 125 CrossRef CAS PubMed.
- N. S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y. K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS PubMed.
- X. Yang, K. R. Adair, X. Gao and X. Sun, Energy Environ. Sci., 2021, 14, 643–671 RSC.
- F.-C. Liu, Z. Shadike, F. Ding, L. Sang and Z.-W. Fu, J. Power Sources, 2015, 274, 280–285 CrossRef CAS.
- B. Sun, P. Wang, J. Xu, Q. Jin, Z. Zhang, H. Wu and Y. Jin, Nano Res., 2021, 15, 4076–4082 CrossRef.
- R. Tomat and A. Rigo, J. Appl. Electrochem., 1986, 16, 8–14 CrossRef CAS.
- D. Zhou, D. Shanmukaraj, A. Tkacheva, M. Armand and G. Wang, Chem, 2019, 5, 2326–2352 CAS.
- X. Zhang, Y. Yang and Z. Zhou, Chem. Soc. Rev., 2020, 49, 3040–3071 RSC.
- K. Li, Z. Hu, J. Ma, S. Chen, D. Mu and J. Zhang, Adv. Mater., 2019, 31, e1902399 CrossRef PubMed.
- Z. Liu, W. Hu, F. Gao and H. Deng, J. Mater. Chem. A, 2018, 6, 7807–7814 RSC.
- Y. X. Ren, T. S. Zhao, H. R. Jiang, M. C. Wu and M. Liu, J. Power Sources, 2017, 347, 136–144 CrossRef CAS.
- M. J. Giammona, J. Kim, Y. Kim, P. Medina, K. Nguyen, H. Bui, G. O. Jones, A. T. Tek, L. Sundberg, A. Fong and Y. H. La, Adv. Mater. Interfaces, 2023, 10, 2300058 CrossRef CAS.
- P. K. Nayak, L. Yang, W. Brehm and P. Adelhelm, Angew. Chem., Int. Ed., 2018, 57, 102–120 CrossRef CAS PubMed.
- Y. Liu, J. Li, Q. Shen, J. Zhang, P. He, X. Qu and Y. Liu, eScience, 2022, 2, 10–31 CrossRef.
- C. Li, M. Qiu, R. Li, X. Li, M. Wang, J. He, G. Lin, L. Xiao, Q. Qian, Q. Chen, J. Wu, X. Li, Y.-W. Mai and Y. Chen, Adv. Fiber Mater., 2021, 4, 43–65 CrossRef.
- H. Zhu and R. J. Kee, Electrochim. Acta, 2016, 219, 70–81 CrossRef CAS.
- X. Lu, M. E. Bowden, V. L. Sprenkle and J. Liu, Adv. Mater., 2015, 27, 5915–5922 CrossRef CAS PubMed.
- H. Yingying, W. Xiangwei and W. Zhaoyin, Energy Storage Sci. Technol., 2021, 10, 781 Search PubMed.
- C. Guo, B. Han, W. Sun, Y. Cao, Y. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202213276 CrossRef CAS PubMed.
- L. Xiang, S. Yuan, F. Wang, Z. Xu, X. Li, F. Tian, L. Wu, W. Yu and Y. Mai, J. Am. Chem. Soc., 2022, 144, 15497–15508 CrossRef CAS PubMed.
- T. Zhang, F. Wei, Y. Wu, W. Li, L. Huang, J. Fu, C. Jing, J. Cheng and S. Liu, Adv. Sci., 2023, 10, 2301918 CrossRef CAS PubMed.
- Z. Lin, Q. Xia, W. Wang, W. Li and S. Chou, InfoMat, 2019, 1, 376–389 CrossRef CAS.
- H. Che, S. Chen, Y. Xie, H. Wang, K. Amine, X.-Z. Liao and Z.-F. Ma, Energy Environ. Sci., 2017, 10, 1075–1101 RSC.
- H. Tian, H. Shao, Y. Chen, X. Fang, P. Xiong, B. Sun, P. H. L. Notten and G. Wang, Nano Energy, 2019, 57, 692–702 CrossRef CAS.
- M. Xu, Y. Li, M. Ihsan-Ul-Haq, N. Mubarak, Z. Liu, J. Wu, Z. Luo and J. K. Kim, Energy Storage Mater., 2022, 44, 477–486 CrossRef.
- L. Zhang, L. Ge, G. He, Z. Tian, J. Huang, J. Wang, D. J. L. Brett, J. Hofkens, F. Lai and T. Liu, J. Phys. Chem. C, 2021, 125, 18604–18613 CrossRef CAS.
- M. Qian, M. Tang, J. Yang, W. Wei, M. Chen, J. Chen, J. Xu, Q. Liu and H. Wang, J. Colloid Interface Sci., 2019, 551, 177–183 CrossRef CAS PubMed.
- J. Ding, H. Zhang, W. Fan, C. Zhong, W. Hu and D. Mitlin, Adv. Mater., 2020, 32, e1908007 CrossRef PubMed.
- L. Ma, Y. Lv, J. Wu, C. Xia, Q. Kang, Y. Zhang, H. Liang and Z. Jin, Nano Res., 2021, 14, 4442–4470 CrossRef CAS.
- J. Xiong, M. Ye, Z. Wang, J. Chen, Y. Zhang, Y. Tang and C. C. Li, Chem. Eng. J., 2022, 442, 135927 CrossRef CAS.
- C. Wei, Y. Tao, H. Fei, Y. An, Y. Tian, J. Feng and Y. Qian, Energy Storage Mater., 2020, 30, 206–227 CrossRef.
- Z. Yang, B. Wang, Y. Chen, W. Zhou, H. Li, R. Zhao, X. Li, T. Zhang, F. Bu, Z. Zhao, W. Li, D. Chao and D. Zhao, Natl. Sci. Rev., 2023, 10, nwac268 CrossRef PubMed.
- H. Yang, T. Zhang, D. Chen, Y. Tan, W. Zhou, L. Li, W. Li, G. Li, W. Han, H. J. Fan and D. Chao, Adv. Mater., 2023, 35, 2300053 CrossRef CAS PubMed.
- R. Zhao, X. Dong, P. Liang, H. Li, T. Zhang, W. Zhou, B. Wang, Z. Yang, X. Wang, L. Wang, Z. Sun, F. Bu, Z. Zhao, W. Li, D. Zhao and D. Chao, Adv. Mater., 2023, 35, 2209288 CrossRef CAS PubMed.
- W. Zhou, M. Song, P. Liang, X. Li, X. Liu, H. Li, T. Zhang, B. Wang, R. Zhao, Z. Zhao, W. Li, D. Zhao and D. Chao, J. Am. Chem. Soc., 2023, 145, 10880–10889 CrossRef CAS PubMed.
- Z. Pei, Z. Zhu, D. Sun, J. Cai, A. Mosallanezhad, M. Chen and G. Wang, Mater. Res. Bull., 2021, 141, 111347 CrossRef CAS.
- J. Zhang, G. Jiang, P. Xu, A. Ghorbani Kashkooli, M. Mousavi, A. Yu and Z. Chen, Energy Environ. Sci., 2018, 11, 2010–2015 RSC.
- C. Bai, F. Cai, L. Wang, S. Guo, X. Liu and Z. Yuan, Nano Res., 2018, 11, 3548–3554 CrossRef CAS.
- L. Hang, W. Li, H. Wen, T. Zhang and G. Jiang, Chem. Eng. J., 2022, 443, 136230 CrossRef CAS.
- F. Chen, X. Chen, Q. Hao, X. Sun and N. Li, Nanoscale, 2022, 14, 15269–15274 RSC.
- Y. Ji, J. Xu, Z. Wang, M. Ren, Y. Wu, W. Liu, J. Yao, C. Zhang and H. Zhao, J. Electroanal. Chem., 2023, 931, 117188 CrossRef CAS.
- J. Xu, W. Ma, L. Ge, M. Ren, X. Cai, W. Liu, J. Yao, C. Zhang and H. Zhao, J. Alloys Compd., 2022, 912, 165151 CrossRef CAS.
- T. Liu, H. Wang, C. Lei, Y. Mao, H. Wang, X. He and X. Liang, Energy Storage Mater., 2022, 53, 544–551 CrossRef.
- D. Yu, A. Kumar, T. A. Nguyen, M. T. Nazir and G. Yasin, ACS Sustainable Chem. Eng., 2020, 8, 13769–13776 CrossRef CAS.
- Z. Gong, C. Song, C. Bai, X. Zhao, Z. Luo, G. Qi, X. Liu, C. Wang, Y. Duan and Z. Yuan, Sci. China Mater., 2022, 66, 556–566 CrossRef.
- K. Lu, H. Zhang, B. Song, W. Pan, H. Ma and J. Zhang, Electrochim. Acta, 2019, 296, 755–761 CrossRef CAS.
- S. Chai, J. Yao, Y. Wang, J. Zhu and J. Jiang, Chem. Eng. J., 2022, 439, 135676 CrossRef CAS.
- C. Chen, Z. Li, Y. Xu, Y. An, L. Wu, Y. Sun, H. Liao, K. Zheng and X. Zhang, ACS Sustainable Chem. Eng., 2021, 9, 13268–13276 CrossRef CAS.
- Y. He, M. Liu, S. Chen and J. Zhang, Sci. China: Chem., 2021, 65, 391–398 CrossRef.
- W. Liu, P. Liu, Y. Lyu, J. Wen, R. Hao, J. Zheng, K. Liu, Y. J. Li and S. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 8955–8962 CrossRef CAS PubMed.
- S. Chen, Y. He, S. Ding and J. Zhang, J. Phys. Chem. C, 2023, 127, 7609–7617 CrossRef CAS.
- Y. Hou, C. Zhu, Q. Wang, X. Zhao, K. Luo, Z. Gong and Z. Yuan, Chin. Chem. Lett., 2023, 108697, DOI:10.1016/j.cclet.2023.108697.
- J. Sun, H. Ma and D. Wang, J. Alloys Compd., 2023, 947, 169696 CrossRef CAS.
- L. Ma, G. Zhu, Z. Wang, A. Zhu, K. Wu, B. Peng, J. Xu, D. Wang and Z. Jin, Nano Lett., 2023, 23, 5272–5280 CrossRef CAS PubMed.
- X. Yang, H. Fan, F. Hu, S. Chen, K. Yan and L. Ma, Nano-Micro Lett., 2023, 15, 126 CrossRef CAS PubMed.
- M. Liu, Q. Chen, X. Cao, D. Tan, J. Ma and J. Zhang, J. Am. Chem. Soc., 2022, 144, 21683–21691 CrossRef CAS PubMed.
- S. Ding, Q. Chen, S. Chen, Y. Tian and J. Zhang, Chin. Chem. Lett., 2023, 34, 108232 CrossRef CAS.
- S. Niu, B. Zhao and D. Liu, ACS Appl. Mater. Interfaces, 2023, 15, 25558–25566 CrossRef CAS PubMed.
- X. Zeng, X. Meng, W. Jiang, J. Liu, M. Ling, L. Yan and C. Liang, ACS Sustainable Chem. Eng., 2020, 8, 14280–14285 CrossRef CAS.
- D. Lin, D. Rao, S. Chiovoloni, S. Wang, J. Q. Lu and Y. Li, Nano Lett., 2021, 21, 4129–4135 CrossRef CAS PubMed.
- X. Miao, Q. Chen, Y. Liu, X. Zhang, Y. Chen, J. Lin, S. Chen and Y. Zhang, Electrochim. Acta, 2022, 415, 140206 CrossRef CAS.
- W. Wu, C. Li, Z. Wang, H.-Y. Shi, Y. Song, X.-X. Liu and X. Sun, Chem. Eng. J., 2022, 428, 131283 CrossRef CAS.
- L. Ma, Y. Ying, S. Chen, Z. Huang, X. Li, H. Huang and C. Zhi, Angew. Chem., Int. Ed., 2021, 60, 3791–3798 CrossRef CAS PubMed.
- W. Gao, S. Cheng, Y. Zhang, E. Xie and J. Fu, Adv. Funct. Mater., 2023, 33, 2211979 CrossRef CAS.
- G. Kasiri, R. Trócoli, A. B. Hashemi and F. La Mantia, Electrochim. Acta, 2016, 222, 74–83 CrossRef CAS.
- B. Tang, L. Shan, S. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC.
- F. C. Anson and J. J. Lingane, J. Am. Chem. Soc., 1957, 79, 1015–1020 CrossRef CAS.
- Y. A. Fialkov, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1954, 3, 847–855 CrossRef.
- F. A. Philbrick, J. Am. Chem. Soc., 1934, 56, 1257–1259 CrossRef CAS.
- R. Whitaker, J. Ambrose and C. Hickam, J. Inorg. Nucl. Chem., 1961, 17, 254–256 CrossRef CAS.
- Y. L. Wang, J. C. Nagy and D. W. Margerum, J. Am. Chem. Soc., 1989, 111, 7838–7844 CrossRef CAS.
- Y. Bichsel and U. Von Gunten, Environ. Sci. Technol., 1999, 33, 4040–4045 CrossRef CAS.
- E. T. Urbansky, B. T. Cooper and D. W. Margerum, Inorg. Chem., 1997, 36, 1338–1344 CrossRef CAS PubMed.
- A. J. Downs and C. J. Adams, The Chemistry of Chlorine, Bromine, Iodine and Astatine: Pergamon Texts in Inorganic Chemistry, Elsevier, 1973, vol. 7 Search PubMed.
- Y. Ji, J. Xie, Z. Shen, Y. Liu, Z. Wen, L. Luo and G. Hong, Adv. Funct. Mater., 2023, 33, 2210043 CrossRef CAS.
- X. Qu, Y. Tang, X. He, J. Zhou, Z. Tang, W. Feng and J. Liu, J. Electrochem., 2022, 211026, DOI:10.13208/j.electrochem.211026.
- J. Zhang, Q. Dou, C. Yang, L. Zang and X. Yan, J. Mater. Chem. A, 2023, 11, 3632–3639 RSC.
- G. Chen, Y. Kang, H. Yang, M. Zhang, J. Yang, Z. Lv, Q. Wu, P. Lin, Y. Yang and J. Zhao, Adv. Funct. Mater., 2023, 33, 2300656 CrossRef CAS.
- X. Jin, L. Song, C. Dai, Y. Xiao, Y. Han, X. Li, Y. Wang, J. Zhang, Y. Zhao, Z. Zhang, N. Chen, L. Jiang and L. Qu, Adv. Mater., 2022, 34, 2109450 CrossRef CAS PubMed.
- W. Shang, J. Zhu, Y. Liu, L. Kang, S. Liu, B. Huang, J. Song, X. Li, F. Jiang, W. Du, Y. Gao and H. Luo, ACS Appl. Mater. Interfaces, 2021, 13, 24756–24764 CrossRef CAS PubMed.
- F. Wang, J. Tseng, Z. Liu, P. Zhang, G. Wang, G. Chen, W. Wu, M. Yu, Y. Wu and X. Feng, Adv. Mater., 2020, 32, 2000287 CrossRef CAS PubMed.
- L. Xu, E.-Y. Choi and Y.-U. Kwon, Inorg. Chem. Commun., 2008, 11, 1190–1193 CrossRef CAS.
- Y. Hou, F. Kong, Z. Wang, M. Ren, C. Qiao, W. Liu, J. Yao, C. Zhang and H. Zhao, J. Colloid Interface Sci., 2022, 629, 279–287 CrossRef PubMed.
- Y. Zhang, T. Zhao, S. Yang, Y. Zhang, Y. Ma and Z. Wang, J. Energy Chem., 2022, 75, 310–320 CrossRef CAS.
- L. Zhang, J. Huang, H. Guo, L. Ge, Z. Tian, M. Zhang, J. Wang, G. He, T. Liu, J. Hofkens, D. J. L. Brett and F. Lai, Adv. Energy Mater., 2023, 13, 2203790 CrossRef CAS.
- Z. Wang, J. Huang, Z. Guo, X. Dong, Y. Liu, Y. Wang and Y. Xia, Joule, 2019, 3, 1289–1300 CrossRef CAS.
- T.-T. Su, J.-B. Le, K. Wang, K.-N. Liu, C.-Y. Shao, W.-F. Ren and R.-C. Sun, J. Power Sources, 2022, 550, 232136 CrossRef CAS.
- Y. Tian, S. Chen, Y. He, Q. Chen, L. Zhang and J. Zhang, Nano Res. Energy, 2022, 1, e9120025 CrossRef.
- H. Peng, Y. Fang, J. Wang, P. Ruan, Y. Tang, B. Lu, X. Cao, S. Liang and J. Zhou, Matter, 2022, 5, 4363–4378 CrossRef CAS.
- S. Chen, Q. Chen, J. Ma, J. Wang, K. S. Hui and J. Zhang, Small, 2022, 18, e2200168 CrossRef PubMed.
- Y. Zhang, L. Wang, Q. Li, B. Hu, J. Kang, Y. Meng, Z. Zhao and H. Lu, Nano-Micro Lett., 2022, 14, 208 CrossRef CAS PubMed.
- W. Shang, Q. Li, F. Jiang, B. Huang, J. Song, S. Yun, X. Liu, H. Kimura, J. Liu and L. Kang, Nano-Micro Lett., 2022, 14, 82 CrossRef CAS PubMed.
- R. Yi, X. Shi, Y. Tang, Y. Yang, P. Zhou, B. Lu and J. Zhou, Small Struct., 2023, 4, 2300020 CrossRef CAS.
- H. X. Dang, A. J. Sellathurai and D. P. J. Barz, Energy Storage Mater., 2023, 55, 680–690 CrossRef.
- L. Yan, T. Liu, X. Zeng, L. Sun, X. Meng, M. Ling, M. Fan and T. Ma, Carbon, 2022, 187, 145–152 CrossRef CAS.
- K. Wang, J.-B. Le, S.-J. Zhang, W.-F. Ren, J.-M. Yuan, T.-T. Su, B.-Y. Chi, C.-Y. Shao and R.-C. Sun, J. Mater. Chem. A, 2022, 10, 4845–4857 RSC.
- J. Yang, R. Zhao, Y. Wang, Z. Hu, Y. Wang, A. Zhang, C. Wu and Y. Bai, Adv. Funct. Mater., 2023, 33, 2213510 CrossRef CAS.
- Q. P. Jian, M. C. Wu, H. R. Jiang, Y. K. Lin and T. S. Zhao, J. Power Sources, 2021, 484, 229238 CrossRef CAS.
- M. Mousavi, G. Jiang, J. Zhang, A. G. Kashkooli, H. Dou, C. J. Silva, Z. P. Cano, Y. Niu, A. Yu and Z. Chen, Energy Storage Mater., 2020, 32, 465–476 CrossRef.
- G.-M. Weng, Z. Li, G. Cong, Y. Zhou and Y.-C. Lu, Energy Environ. Sci., 2017, 10, 735–741 RSC.
- Y. Zhao, Y. Li, J. Mao, Z. Yi, N. Mubarak, Y. Zheng, J.-K. Kim and Q. Chen, J. Mater. Chem. A, 2022, 10, 14090–14097 RSC.
- S. Ito, M. Sugimasa, Y. Toshimitsu, A. Orita, M. Kitagawa and M. Sakai, Electrochim. Acta, 2019, 319, 164–174 CrossRef CAS.
- J. Yang, Y. Song, Q. Liu and A. Tang, J. Mater. Chem. A, 2021, 9, 16093–16098 RSC.
- C. Xie, H. Zhang, W. Xu, W. Wang and X. Li, Angew. Chem., Int. Ed., 2018, 57, 11171–11176 CrossRef CAS PubMed.
- C. Xie, Y. Liu, W. Lu, H. Zhang and X. Li, Energy Environ. Sci., 2019, 12, 1834–1839 RSC.
- L. Gao, Y. Ding, G. He and G. Yu, Small, 2022, 18, 2107055 CrossRef CAS PubMed.
- P. Tangthuam, J. Pimoei, A. A. Mohamad, F. Mahlendorf, A. Somwangthanaroj and S. Kheawhom, Heliyon, 2020, 6, e05391 CrossRef CAS PubMed.
- Z. Yuan, X. Liu, W. Xu, Y. Duan, H. Zhang and X. Li, Nat. Commun., 2018, 9, 3731 CrossRef PubMed.
- C. Zhang, L. Zhang, Y. Ding, S. Peng, X. Guo, Y. Zhao, G. He and G. Yu, Energy Storage Mater., 2018, 15, 324–350 CrossRef.
- M. Mousavi, H. Dou, H. Fathiannasab, C. J. Silva, A. Yu and Z. Chen, Chem. Eng. J., 2021, 412, 128499 CrossRef CAS.
- J. Liu, T. Ma, M. Zhou, S. Liu, J. Xiao, Z. Tao and J. Chen, Inorg. Chem. Front., 2019, 6, 731–735 RSC.
- B. Li, J. Liu, Z. Nie, W. Wang, D. Reed, J. Liu, P. McGrail and V. Sprenkle, Nano Lett., 2016, 16, 4335–4340 CrossRef CAS PubMed.
- A. A. Williams, R. K. Emmett and M. E. Roberts, Phys. Chem. Chem. Phys., 2023, 25, 16222–16226 RSC.
- X. Wu, A. Markir, Y. Xu, C. Zhang, D. P. Leonard, W. Shin and X. Ji, Adv. Funct. Mater., 2019, 29, 1900911 CrossRef.
- Y. Fang, Z. Chen, L. Xiao, X. Ai, Y. Cao and H. Yang, Small, 2018, 14, 1703116 CrossRef PubMed.
- L. Yu, F.-C. Liu and Z.-W. Fu, Electrochim. Acta, 2009, 54, 2818–2822 CrossRef CAS.
- Y. Zhang, S. Liu, Y. Ji, J. Ma and H. Yu, Adv. Mater., 2018, 30, 1706310 CrossRef PubMed.
- Z.-W. Fu, L. Yu, S.-C. Cheng, J. Yao and H. Li, Electrochem. Commun., 2007, 9, 1–5 CrossRef CAS.
- K. Zhang and Z. Jin, Energy Storage Mater., 2022, 45, 332–369 CrossRef.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- H. D. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265–2279 RSC.
- Z. Liu, X. Pu, W. Hu, F. Gao and H. Deng, J. Phys. Chem. C, 2018, 122, 28518–28527 CrossRef CAS.
- X. Chen, T. Hou, K. A. Persson and Q. Zhang, Mater. Today, 2019, 22, 142–158 CrossRef CAS.
- M. Chen, W. Zhu, H. Guo, Z. Tian, L. Zhang, J. Wang, T. Liu, F. Lai and J. Huang, Energy Storage Mater., 2023, 59, 102760 CrossRef.
- Y. Wu and N. Liu, Chem, 2018, 4, 438–465 CAS.
- R. E. Ciez and J. F. Whitacre, Nat. Sustainability, 2019, 2, 148–156 CrossRef.
- J. Zheng, D. Lv, M. Gu, C. Wang, J.-G. Zhang, J. Liu and J. Xiao, J. Electrochem. Soc., 2013, 160, A2288 CrossRef CAS.
- D. Lv, J. Zheng, Q. Li, X. Xie, S. Ferrara, Z. Nie, L. B. Mehdi, N. D. Browning, J. G. Zhang and G. L. Graff, Adv. Energy Mater., 2015, 5, 1402290 CrossRef.
- L. Luo and A. Manthiram, ACS Energy Lett., 2017, 2, 2205–2211 CrossRef CAS.
- M. Zhao, B. Q. Li, H. J. Peng, H. Yuan, J. Y. Wei and J. Q. Huang, Angew. Chem., Int. Ed., 2020, 59, 12636–12652 CrossRef CAS PubMed.
- C. Wang, K. Fu, S. P. Kammampata, D. W. McOwen, A. J. Samson, L. Zhang, G. T. Hitz, A. M. Nolan, E. D. Wachsman, Y. Mo, V. Thangadurai and L. Hu, Chem. Rev., 2020, 120, 4257–4300 CrossRef CAS PubMed.
- L. Fan, S. Li, L. Liu, W. Zhang, L. Gao, Y. Fu, F. Chen, J. Li, H. L. Zhuang and Y. Lu, Adv. Energy Mater., 2018, 8, 1802350 CrossRef.
- Z. Zhao, J. Zhao, Z. Hu, J. Li, J. Li, Y. Zhang, C. Wang and G. Cui, Energy Environ. Sci., 2019, 12, 1938–1949 RSC.
- J. Fu, X. Ji, J. Chen, L. Chen, X. Fan, D. Mu and C. Wang, Angew. Chem., 2020, 132, 22378–22385 CrossRef.
- G. Zampardi and F. La Mantia, Nat. Commun., 2022, 13, 687 CrossRef CAS PubMed.
- L. Yan, S. Zhang, Q. Kang, X. Meng, Z. Li, T. Liu, T. Ma and Z. Lin, Energy Storage Mater., 2023, 54, 339–365 CrossRef.
- L. Qiao, C. Wang and X. S. Zhao, ACS Appl. Energy Mater., 2021, 4, 7012–7019 CrossRef CAS.
- Q. Zhang, F.-H. Li, X.-X. Zhang, H.-B. Wang, F.-C. Li, Z.-Y. Guo and Z. Zhang, J. Electrochem. Soc., 2021, 168, 040522 CrossRef CAS.
- Y. Zhao, N. B. Mercier and H. R. Byon, ChemPlusChem, 2015, 80, 344–348 CrossRef CAS.
- J. Xu, J. Wang, L. Ge, J. Sun, W. Ma, M. Ren, X. Cai, W. Liu and J. Yao, J. Colloid Interface Sci., 2022, 610, 98–105 CrossRef CAS PubMed.
- Q. Guo, H. Wang, X. Sun, Y. N. Yang, N. Chen and L. Qu, ACS Mater. Lett., 2022, 4, 1872–1881 CrossRef CAS.
- Y. Wu, Y. Qian, C. Huang, Y. Zhang, Y. Yang, A. Hu, Q. Tang and X. Chen, Electrochim. Acta, 2023, 460, 142593 CrossRef CAS.
- Y. Du, R. Kang, H. Jin, W. Zhou, W. Zhang, H. Wang, J. Qin, J. Wan, G. Chen and J. Zhang, Adv. Funct. Mater., 2023, 2304811, DOI:10.1002/adfm.202304811.
- D. Zhao, Q. Zhu, Q. Zhou, W. Zhang, Y. Yu, S. Chen and Z. Ren, Energy Environ. Mater., 2022, e12522, DOI:10.1002/eem2.12522.
- Q. Chen, S. Chen and J. Zhang, J. Power Sources, 2023, 556, 232529 CrossRef CAS.
- W. Han and X. Li, J. Power Sources, 2023, 580, 233296 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |