
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rationale for highly efficient and outdoor-stable terpolymer solar cells†
Hua
Tang‡
 *a,
Zhihui
Liao
b,
Safakath
Karuthedath‡§
a,
Si
Chen
a,
Heng
Liu
c,
Jafar I.
Khan
a,
Maxime
Babics
a,
Wenchao
Yang
a,
Maryam
Alqurashi
a,
Yakun
He
adf,
Julien
Gorenflot
a,
Jiaming
Huang
e,
Gang
Li
e,
Stefaan
De Wolf
a,
Xinhui
Lu
c,
Christoph J.
Brabec
df,
Frédéric
Laquai
a and
Shirong
Lu
*b
*a,
Zhihui
Liao
b,
Safakath
Karuthedath‡§
a,
Si
Chen
a,
Heng
Liu
c,
Jafar I.
Khan
a,
Maxime
Babics
a,
Wenchao
Yang
a,
Maryam
Alqurashi
a,
Yakun
He
adf,
Julien
Gorenflot
a,
Jiaming
Huang
e,
Gang
Li
e,
Stefaan
De Wolf
a,
Xinhui
Lu
c,
Christoph J.
Brabec
df,
Frédéric
Laquai
a and
Shirong
Lu
*b
aKAUST Solar Center, Physical Sciences and Engineering Division (PSE), Materials Science and Engineering Program (MSE), King Abdullah University of Science and Technology (KAUST), Thuwal, 23955-6900, Kingdom of Saudi Arabia. E-mail: hua.tang.1@kaust.edu.sa
bDepartment of Material Science and Technology, Taizhou University, Taizhou 318000, P. R. China. E-mail: lushirong@tzc.edu.cn
cDepartment of Physics, The Chinese University of Hong Kong, New Territories, Hong Kong 999077, P. R. China
dFriedrich-Alexander-Universität Erlangen-Nürnberg, Materialien für Elektronik und Energietechnik (i-MEET), Martensstraße 7, 91058 Erlangen, Germany
eDepartment of Electronic and Information Engineering, The Hong Kong Polytechnic University, Hung Hum, Kowloon, Hong Kong SAR, P. R. China
fForschungszentrum Jülich GmbH, Helmholtz-Institut Erlangen Nürnberg für Erneuerbare Energien (HI ERN), Immerwahrstraße 2, 91058 Erlangen, Germany
First published on 14th March 2023
Abstract
Random terpolymerization is an effective approach to achieving highly efficient and outdoor-stable terpolymer photovoltaics. However, the working principle behind this remains unclear. Herein, we report spectroscopic, morphological, and computational results and conclude the previous work on terpolymer research to unveil their structure–property relations and elucidate key rules for high-efficiency and outdoor-stable terpolymer photovoltaics. More specifically, introducing a strong electron-deficient or electron-rich third moiety is suggested to enable broadened absorption with minimized non-radiative voltage losses. Besides, the third component should manipulate the D/A miscibility towards a thermodynamically more stable morphology. However, the content of the third component should be small to maintain molecular orientation and exciton diffusion length in conjunction with optimized phase-separation and crystallinity, in turn reducing bimolecular carrier recombination. Obeying these rules, terpolymer solar cells based on the parent donors D18 and PM6 with enhanced power conversion efficiency (PCE) and excellent outdoor stability are demonstrated. Our findings provide a rationale for explaining and achieving high-performance and outdoor-stable terpolymer photovoltaics, paving the path to commercialization.
Broader contextIn the last decade, the development of non-fullerene acceptors (NFAs), especially those of the ITIC family and the Y-series has triggered a rapid advance in the performance of organic photovoltaics (OPV), endowing OPV to arise as a promising contender for 3rd generation photovoltaic technologies. In the current stage, the commercialization of OPV is the first priority to be considered by the research community. Meanwhile, this technology highly relies on the polymer donor:NFA (D:A) bulk hetero-junction (BHJ) framework to guarantee efficient charge generation and collection as well as long-term (outdoor) stability to enable high-performance and outdoor-stable devices. Thus, advancing NFAs matched with (photo)stable polymeric donors is of utmost importance to improve competitiveness and facilitate the commercialization of OPV. Random terpolymerization is a proven effective approach to address the aforementioned issues. In this study, we identify a set of rules for high-performance and outdoor-stable terpolymer solar cells and propose a rationale to explain the working principle behind them, pushing them closer to commercial realization. |
Introduction
The development of non-fullerene acceptors (NFAs), especially those of the ITIC family1–3 and the Y-series,4,5 which combine tunable energy levels and near-infrared (NIR) absorption has triggered a rapid advance in the performance of organic photovoltaics (OPV), endowing OPV to arise as a promising contender for 3rd generation photovoltaic technologies.6–12 Meanwhile, polymer donors have continued to play a crucial role since, when paired with NFAs, the polymer donor:NFA (D:A) bulk hetero-junction (BHJ) requires a balance between phase-separation and domain crystallinity to ensure efficient charge generation and collection, resulting in high power conversion efficiencies (PCEs).13–21 However, commercialization of OPV requires not only high efficiency, but also long-term (outdoor) stability, specifically in typical PV operating conditions such as continuous illumination, heat, and day-to-night temperature changes.22,23 NFAs are prone to aggregate when excess energy such as light and heat is supplied.24,25 Importantly, the stability of polymer:NFA OPV is predominantly governed by the polymer donor:NFA BHJ framework.9,26,27 In this regard, advancing NFAs matched with (photo)stable polymer donors is of utmost importance to improve the competitiveness and facilitate the commercialization of OPV.Random terpolymerization is an emerging strategy to tailor polymer donors toward enhanced photovoltaic performance and long-term stability.28,29 Improvements originate from the incorporation of a second donor or acceptor moiety, which modifies the polymer structure from a D–A type copolymer to a D1–A–D2–A or D–A1–D–A2 type terpolymer.30–37 The second donor (or acceptor) moiety influences the optical absorption, energetics, and blend morphology.30 Yang and co-workers demonstrated that incorporating as little as 5% of a second donor moiety thieno[3,2-b]thiophene (TT) into the polymer PBDB-T increases the absorption coefficient and optimizes the thin film morphology. This enhanced both short-circuit current density (JSC) and fill factor (FF) in both PC71BM and m-ITIC-based terpolymer donor devices. However, the introduction of excessive TT has been detrimental to the photovoltaic performance of both PC71BM and m-ITIC-based terpolymer donor devices.
To unleash the full potential of terpolymer photovoltaics, improving the JSC and FF only is not sufficient, since the open-circuit voltage (VOC) needs to be maximized as well. Yang et al. reported that incorporating 5% of a second electron-deficient building block (iBDD-Si) into the donor polymer PM6 lowers the energy level of the highest occupied molecular orbital (HOMO), increases the absorption coefficient, and optimizes the molecular packing, thereby enhancing the device PCE from 15.15% to 17.01% due to simultaneously improved VOC, JSC, and FF.32
In addition to PCE enhancements, random terpolymerization can also improve the stability of solar cells. Kettle and co-workers designed and synthesized a terpolymer (P3HT:PTB7-Th) system, namely, P1.37 Devices based on P1:PCBM exhibited remarkably improved photostability compared to P3HT:PCBM and PTB7-Th:PCBM devices, indicating that the random terpolymerization strategy has the potential to deliver more stable OPV. However, clear design rules for terpolymer photovoltaics that lead to enhanced PCE and stability, as well as the precise mechanism leading to the efficiency enhancement are unclear at present.
Herein, we summarized the representative best-performing terpolymer solar cells in the past 5 years (Table S1, ESI†) and identified key rules for highly efficient and outdoor-stable terpolymer photovoltaics. Besides, we elucidate these key rules by demonstrating two series of terpolymer photovoltaic devices based on the parent donor polymers D18 and PM6. These rules are: (i) introducing a strong electron-deficient or electron-rich moiety that broadens absorption and down-shifts the HOMO level, enhancing the JSC and reducing energy losses; (ii) using small contents of the third component to maintaining the molecular orientation and exciton diffusion length while optimizing phase-separation and domain crystallinity to reduce the bimolecular carrier recombination, thereby enhancing the FF; (iii) manipulating the D/A miscibility by introducing a third comonomer to obtain a more thermodynamically stable thin film morphology, in turn leading to improved outdoor stability. To elucidate the key rules, we selected the D18 and PM6-based copolymers with 5% strong electron-deficient moiety of ester-substituted thiazole (E-TZ) and found that the third component can effectively blue-shifted the absorption, down-shifted the HOMO level, and fine-tuned the morphology, in turn leading to a reduced bimolecular carrier recombination and enhanced JSC, VOC, and FF. Furthermore, the introduction of 5% TZ (the abbreviation of E-TZ) improved the morphological stability, resulting in improved outdoor operating stability of D18-based terpolymer photovoltaics under the harsh local outdoor conditions at KAUST in Saudi Arabia (highest/lowest air temperature during daytime/nighttime: 48 °C/22 °C). Our work unveils the working principle of terpolymer photovoltaics and provides a rationale to explain and achieve highly efficient and outdoor-stable terpolymer photovoltaics.
Results and discussion
The molecular structures of D18, the acceptor moiety (TZ), the terpolymer donors (D18(5% TZ), D18(10% TZ), D18(50% TZ)), and Y6 are shown in Fig. 1a. The corresponding PM6 and PM6-based terpolymer donor structures are shown in Fig. S1 (ESI†). First, we studied the effect of incorporation of the second acceptor moiety (TZ) on the polymer properties in neat thin films.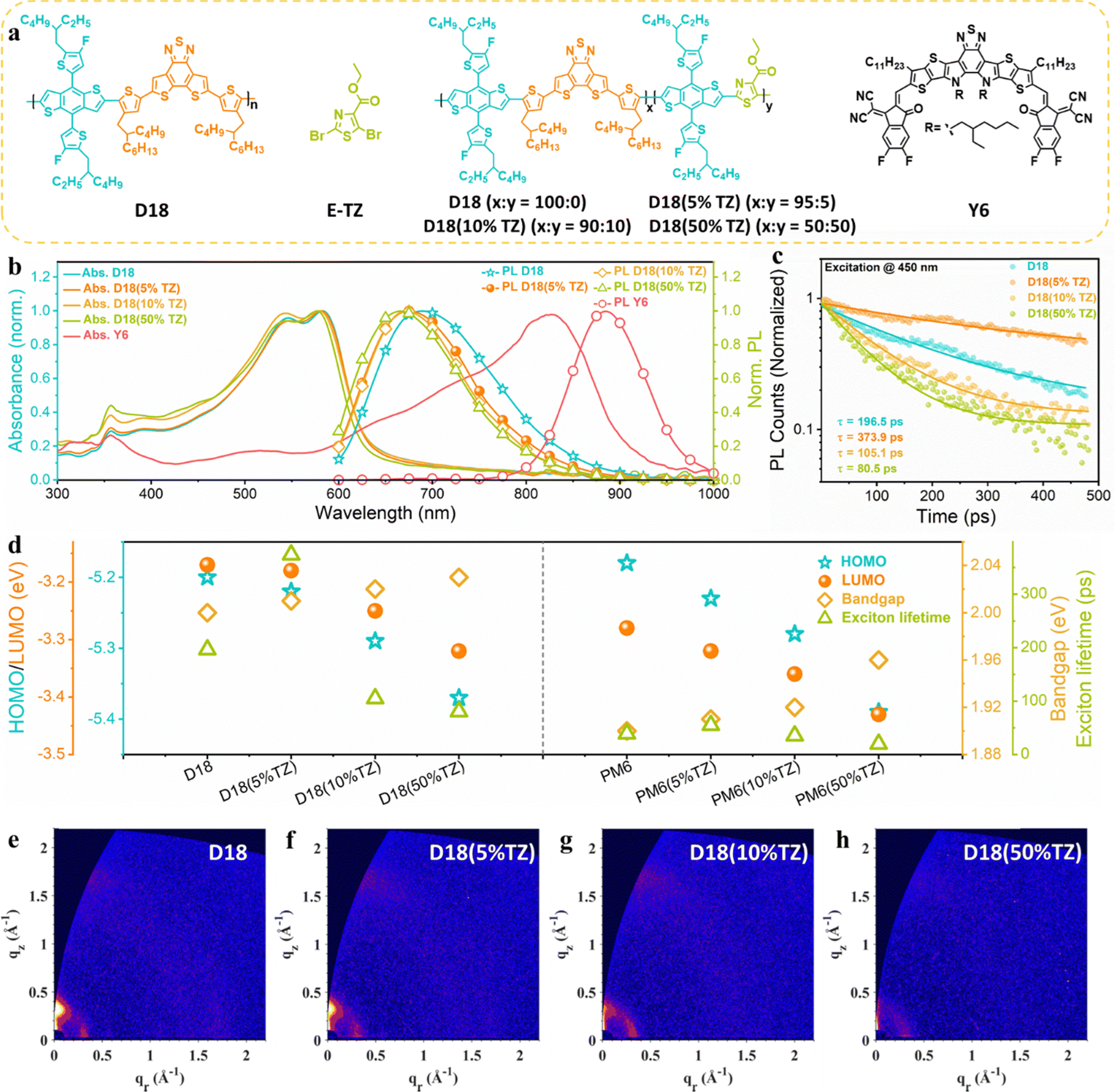 | ||
| Fig. 1 Evolution of terpolymer properties when incorporating different fractions of TZ. (a) Molecular structures of TZ, D18, D18(5% TZ), D18(10% TZ), D18(50% TZ), and Y6. (b) Normalized absorption and photoluminescence (PL) spectra, (c) TRPL of the neat D18-based terpolymers. (d) Comparison of D18 and PM6-based terpolymers in terms of HOMO, LUMO, bandgap, and PCE. (e–h) 2D GIWAXS patterns of neat terpolymer thin films. | ||
The normalized UV-Vis absorption and steady-state photoluminescence (PL) spectra are shown in Fig. 1b. The introduction of the second acceptor moiety (TZ) into the polymer backbone slightly blue-shifts both the UV-Vis absorption and PL spectra. More specifically, when increasing the content of TZ from 5% to 50% in D18, the optical bandgaps of the terpolymers increase from 2.01 eV (D18(5%TZ)) to 2.03 eV (D18(50%TZ)), as estimated at the intersection of the normalized UV-Vis absorption and PL spectra of the neat polymer and terpolymer films (Fig. S4, ESI†).
The HOMO levels of D18, the D18-based terpolymers, and NFA used in this work were determined by photo-electron spectroscopy in air (PESA). Unlike cyclic voltammetry (CV) obtain the energy level from oxidation and reduction potential of materials, often in solution, PESA can quantify the HOMO level on thin-film surface.38,39 The introduction of TZ gradually lowers the HOMO levels of the D18-based terpolymers due to the high electron-deficiency of TZ.35,36 The lowest unoccupied molecular orbital (LUMO) levels were approximated based on the measured HOMO levels and optical bandgaps (from the intersection of normalized absorption and PL) of the corresponding materials (Fig. S7a, ESI†). To confirm the generality of broadened absorption spectra and deepened HOMO levels caused by the introduction of the electron-deficient moiety, 5% to 50% TZ were also incorporated in PM6 as parent copolymer. As shown in Fig. S3, S5 and S7b (ESI†), PM6-based terpolymers exhibited a virtually similar evolution as the D18-based terpolymers.
Next, the PL lifetime of neat D18 and D18-based terpolymers was characterized by time-resolved photoluminescence (TRPL) spectroscopy. The data analysis details have been described in our previous works.40,41 As presented in Fig. 1c, the D18(5%TZ) neat polymer thin film had a PL lifetime of 373.9 ps, which is higher than that of the parent polymer D18 (196.5 ps) in neat films, implying that terpolymerization with a small fraction (∼5%) of a second acceptor can enhance the exciton lifetime, which could be attributed to the finely optimized crystal structure possesses fewer defects. However, when further increasing the TZ content to D18(10%TZ) and D18(50%TZ), the PL lifetime was reduced, implying that excessive TZ may result in severer defects. We note that the PL lifetime of neat PM6 and PM6-based terpolymers was increased with the incorporation of TZ (Fig. S9, ESI†), indicating that exciton lifetimes are generally increased by the strong electron-deficient TZ moiety. Fig. 1d summarizes the energy levels, optical bandgaps, and exciton lifetimes of terpolymers based on D18 and PM6, demonstrating the impact of introducing a second strong electron-deficient moiety into the polymer backbone.
To decipher the role of the TZ moiety on the exciton diffusion lengths, we fitted the TRPL kinetics in the ps–ns time range with a rate equation accounting for monomolecular exciton recombination into the ground state and bimolecular exciton–exciton annihilation (EEA).42 Typically, EEA leads to a fast intensity-dependent decay of the exciton population in the initial time range below 100 ps. Fig. S10 (ESI†) shows the fitting results, which confirm the presence of both bimolecular EEA and exciton recombination. The fitting parameters extracted from fitting the experimental data are presented in Table S5 (ESI†). While the absolute values of the exciton diffusion length cannot be determined without measuring fluence-dependent kinetics, we can still estimate the ratios between the exciton diffusion lengths of D18(5%TZ), D18(10%TZ), and D18(50%TZ) and compare to that of D18, and we find 1.1, 0.8 and 0.8, respectively. The results imply that the incorporation of 5% TZ has little impact on the exciton diffusion length. In fact, the enhanced PL lifetime can facilitate exciton dissociation.43,44
Next, the molecular orientation and crystallinity of the donor materials in thin films were probed by grazing-incidence wide-angle X-ray scattering (GIWAXS). As shown in Fig. 1e–h and Fig. S8, Table S2 (ESI†), the terpolymers exhibit virtually the same molecular orientation despite the introduction of the TZ moiety. However, their crystallinity was reduced by increasing the TZ content from 5% to 50% in the parent polymer D18. We conclude that the terpolymerization strategy retains the polymers’ molecular orientation while gradually reducing their crystallinity. Thus, based on the results from GIWAXS and TRPL, introducing a small fraction of a second acceptor moiety leads to optimized morphology and enhanced PL lifetimes without sacrificing the materials’ exciton diffusion length.
Then, we evaluated the impact of terpolymerization on the solar cell device performance. The conventional device architecture of glass substrate/indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene):polystyrene sulfonate (PEDOT:PSS, 4083)/BHJ active layer/PNDIT-F3N/Ag (Fig. S11, ESI†) was employed. The current density–voltage (J–V) characteristics of D18:Y6 and D18-based terpolymer:Y6 devices are presented in Fig. S12a (ESI†) and the detailed photovoltaic parameters are shown in Table 1. The D18:Y6 device exhibits a PCE of 17.2% with a VOC of 0.853V, a JSC of 27.17 mA cm−2, and a fill factor (FF) of 74.1%. Increasing the TZ content from 5% to 50% improved the VOC of the D18-based terpolymer:Y6 devices due to the lowering of the HOMO level in the terpolymers. Furthermore, the D18-based terpolymer:Y6 devices exhibited enhanced JSC and FF when introducing 5% TZ in D18. Incorporating larger TZ contents, however, significantly reduced JSC and FF, indicating a trade-off exists between improving VOC and reducing the device photocurrent (JSC and FF). The external quantum efficiency (EQE) spectra of the D18:Y6, D18(5%TZ):Y6, D18(10%TZ):Y6, and D18(50%TZ):Y6 devices are presented in Fig. S12b (ESI†). All four devices exhibit very similar EQE spectra. Integrating the device EQE resulted in JSC values of 26.2, 26.3, 25.5, and 22.5 mA cm−2 for the D18:Y6, D18(5%TZ):Y6, D18(10%TZ):Y6, and D18(50%TZ):Y6 devices, respectively, well in line with the experimentally obtained JSC values.
| Active layer | E g (eV) | V OC (V) | E CT (eV) | ΔVOC,rad (V) | ΔVOC,nr (V) | J sc (mA cm−2) | J EQE b (mA cm−2) | FF (%) | PCEa (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Statistical data obtained from 10 devices. | |||||||||
| D18:Y6 | 1.447 | 0.853 | 1.439 | 0.330 | 0.256 | 27.17 | 26.2 | 74.1 | 17.2(17.0) |
| D18(5% TZ):Y6 | 1.450 | 0.860 | 1.442 | 0.350 | 0.232 | 27.27 | 26.3 | 77.9 | 18.3(18.0) |
| D18(10% TZ):Y6 | 1.455 | 0.871 | 1.450 | 0.355 | 0.224 | 26.07 | 25.5 | 73.4 | 16.7(16.5) |
| D18(50% TZ):Y6 | 1.465 | 0.905 | 1.465 | 0.369 | 0.192 | 23.38 | 22.5 | 60.5 | 12.8(12.4) |
The photovoltaic performance improvement of D18-based terpolymer photovoltaics is primarily due to increased VOC and FF. We first focus on the mechanism behind the VOC enhancement in D18-based terpolymer:Y6 devices. Electroluminescence (EL) and sensitive external quantum efficiency (EQEPV) measurements were carried out on D18:Y6, D18(5%TZ):Y6, D18(10%TZ):Y6, and D18(50%TZ):Y6 blends (Fig. 2a–e). As presented in Table 1 and Fig. S6 (ESI†), the blends’ optical bandgaps (Eg) were determined at the intersection of the normalized steady-state absorption and PL emission spectra of the D18:Y6, D18(5%TZ):Y6, D18(10%TZ):Y6, and D18(50%TZ):Y6 blends. The incorporation of TZ (5%, 10%, and 50%) into the backbone monotonically increased Eg from 1.447 eV (D18) to 1.450 eV, 1.455 eV, and 1.465 eV. The charge-transfer (CT) state energy (ECT) of D18:Y6 and D18-based terpolymer:Y6 devices was estimated by fitting the EL data to equation 5 shown in the ESI.† The introduction of 5%, 10%, and 50% TZ monotonically increases ECT from 1.439 eV to 1.442 eV, 1.450 eV, and 1.465 eV, respectively. The total photovoltaic energy loss limiting the Voc in devices consists of both radiative and non-radiative recombination losses (ΔVOC,rad and ΔVOC,nr), which can be estimated by equations 7–10, demonstrated in the ESI.† The introduction of TZ (5%, 10%, and 50%) increases ΔVOC,rad from 0.330 V to 0.350 V, 0.355 V, and 0.369 V, while reducing the ΔVOC,nr from 0.256 V to 0.232 V, 0.224 V, and 0.192 V, respectively. The enhanced VOC from terpolymers implies that the introduction of a second strong electron-deficient moiety (TZ) has a positive impact on the non-radiative recombination losses in turn increasing the VOC of terpolymer photovoltaics. However, to support the general versatility of the approach to incorporate another electron-deficient moiety (TZ) to enhance the VOC, TZ of different content was introduced to the donor polymer PM6. Clearly, the introduction of TZ improves the VOC from 0.839 V in PM6:Y6 to 0.871 V in PM6(5% TZ):Y6 to 0.900 V in PM6(50% TZ):Y6 (Table S4, ESI†), supporting the effectiveness of terpolymerization with TZ as a common approach to enhance the VOC of D18 and PM6-based terpolymer photovoltaics.
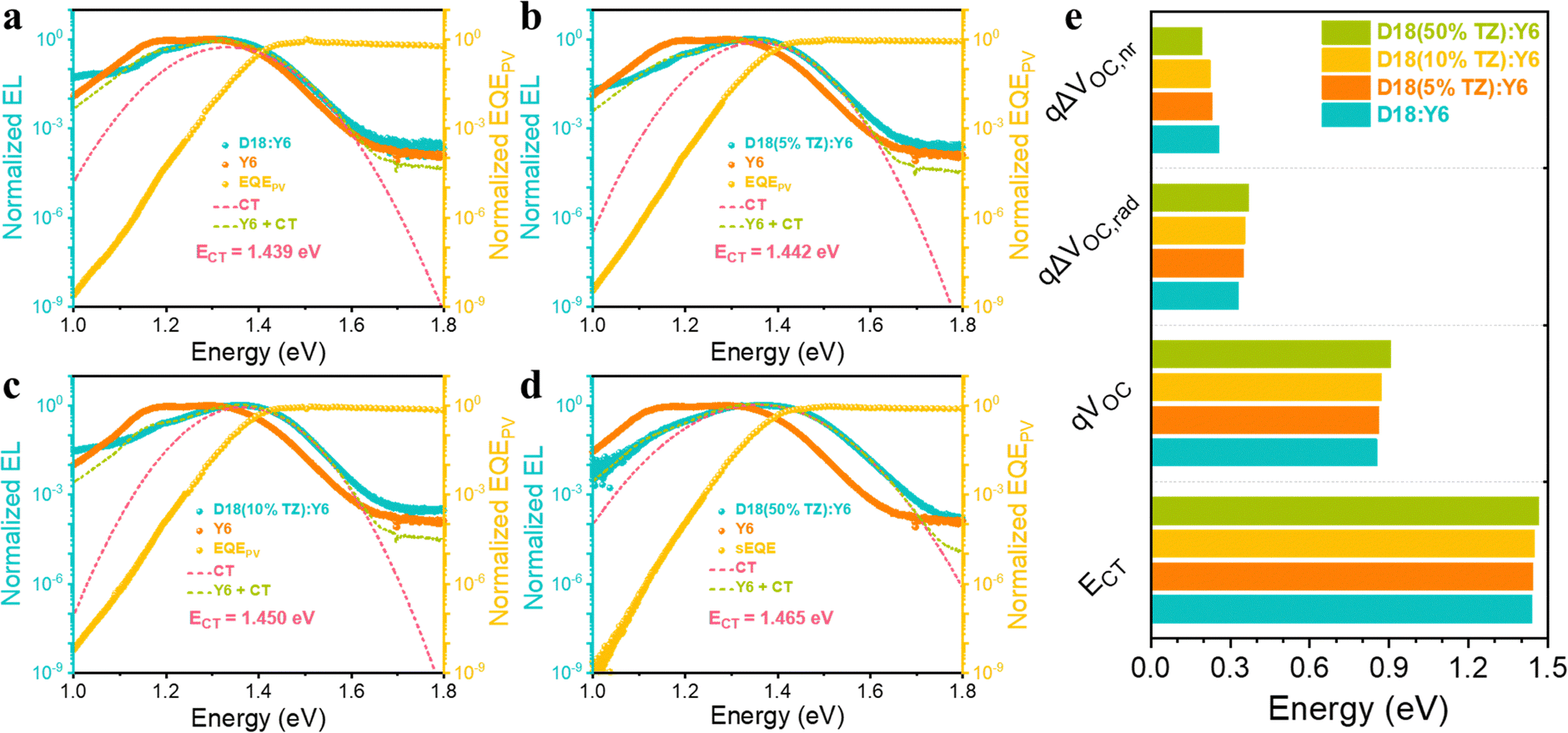 | ||
| Fig. 2 Random terpolymerization minimizes energy losses. Normalized EL and EQEPV spectra of: (a) D18:Y6, (b) D18(5%TZ):Y6, (c) D18(10%TZ):Y6, and (d) D18(50%TZ):Y6 devices. (e) Comparison of in ECT, qVOC, ΔVOC,rad, and ΔVOC,nr of D18:Y6, D18(5%TZ):Y6, D18(10%TZ):Y6, and D18(50%TZ):Y6 devices. | ||
Next, we investigated the FF enhancement and studied the exciton and charge carrier dynamics to better understand the photophysics of TZ-based terpolymers. The impact of TZ on the exciton dynamics in the D18-based terpolymer:Y6 BHJ active layer was studied by TRPL and PL quenching efficiency (PLQE) measurements. Here, the PLQE is obtained as (1 − τblend/τneat), where τblend and τneat represent the lifetimes of the blend and neat materials, respectively. The fitting details have been described in our previous works.40,41 We parameterized the PL decay using a bi-exponential decay function to extract the weighted-average lifetimes. The PL of neat Y6 and blend films of D18:Y6, D18(5%TZ):Y6, D18(10%TZ):Y6, and D18(50%TZ):Y6 were measured following pulsed laser excitation at 720 nm. The corresponding normalized TRPL data are shown as 3D plots in Fig. 3a–d with a clear PL emission peak from Y6 at ∼880 nm. For a closer analysis, the PL transients of the respective blends were tracked at the maxima of their respective emission peaks, as depicted in Fig. 3e. The incorporation of 5% TZ reduced the PL lifetime from 37.1 to 29.8 ps, while the PLQE remained similar (95.7% vs. 96.6%) implying efficient exciton quenching in either. However, increasing the TZ content led to longer PL lifetimes and concurrently reduced PLQE in D18(10%TZ):Y6 with 97.4 ps and 88.8% and D18(50%TZ):Y6 with 212.0 ps and 75.6%, indicating the incorporation of TZ in excess of 5% reduces the exciton quenching efficiency. These results are in line with the observed change of the JSC and EQE from devices, indicating that the additional acceptor moiety (TZ) influences exciton quenching in terpolymer photovoltaics.
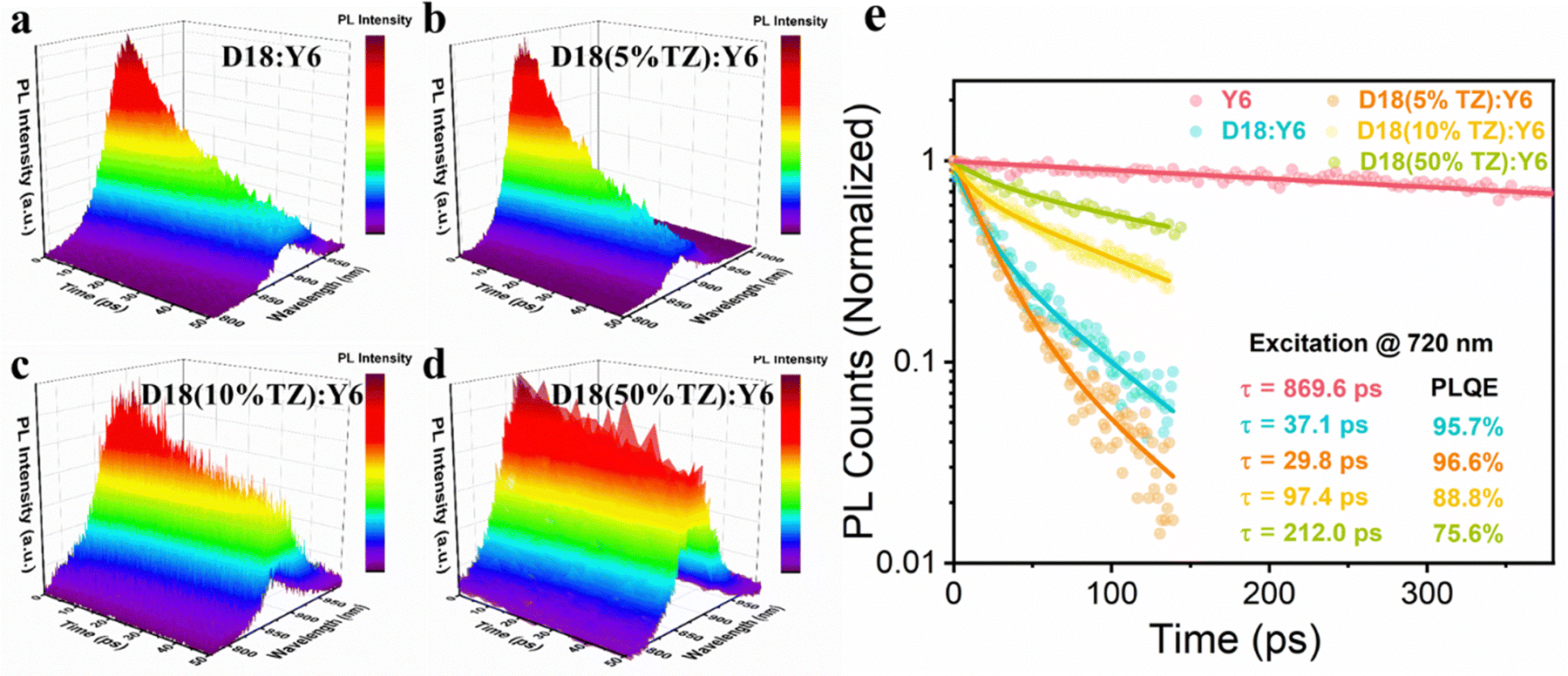 | ||
| Fig. 3 Time-resolved photoluminescence of terpolymer:Y6 thin films. TRPL spectra in 3D plots of (a) D18:Y6, (b) D18(5%TZ):Y6, (c) D18(10%TZ):Y6, and (d) D18(50%TZ):Y6 blend films. (e) TRPL kinetics of Y6 neat films and blend films of D18:Y6, D18(5%TZ):Y6, D18(10%TZ):Y6, and D18(50%TZ):Y6 tracked at the peak position of the PL and fits to the experimental data using a bi-exponential decay (solid line). | ||
To further reveal the photogenerated charge carrier dynamics, we performed transient absorption (TA) pump–probe measurements over a wide temporal (picoseconds to microseconds) and broad probe energy range (2.2–0.7 eV) using our home-built TA setup.45 Positive ΔT/T signals correspond to the material's ground state bleaching (GSB), while negative ΔT/T signals correspond to regions of photo-induced absorption (PA) from excited states.39,45,46Fig. 4a–c shows the ps–ns TA spectra of D18:Y6, D18(5%TZ):Y6, and D18(50%TZ):Y6 blend films after photoexcitation at 650 nm, where mainly Y6 molecules are excited (Fig. 1b). Soon after photoexcitation at 700 fs, a photo-induced absorption (PA) band has emerged in the 1.4–1.45 eV region (orange line in Fig. 4a–c) observed in all three blends, which we assigned to charge transfer states formed in Y6, in agreement with previous studies.47–50 Over time, the narrow band at 1.4 eV exhibited spectral broadening and a concomitant redshift. This spectral evolution indicates the formation of a new species, namely, charge carrier, observed by their charge-induced absorption.47 The TA spectra of neat donor and acceptor materials are given for comparison in the SI (Fig. S13 and S14, ESI†).
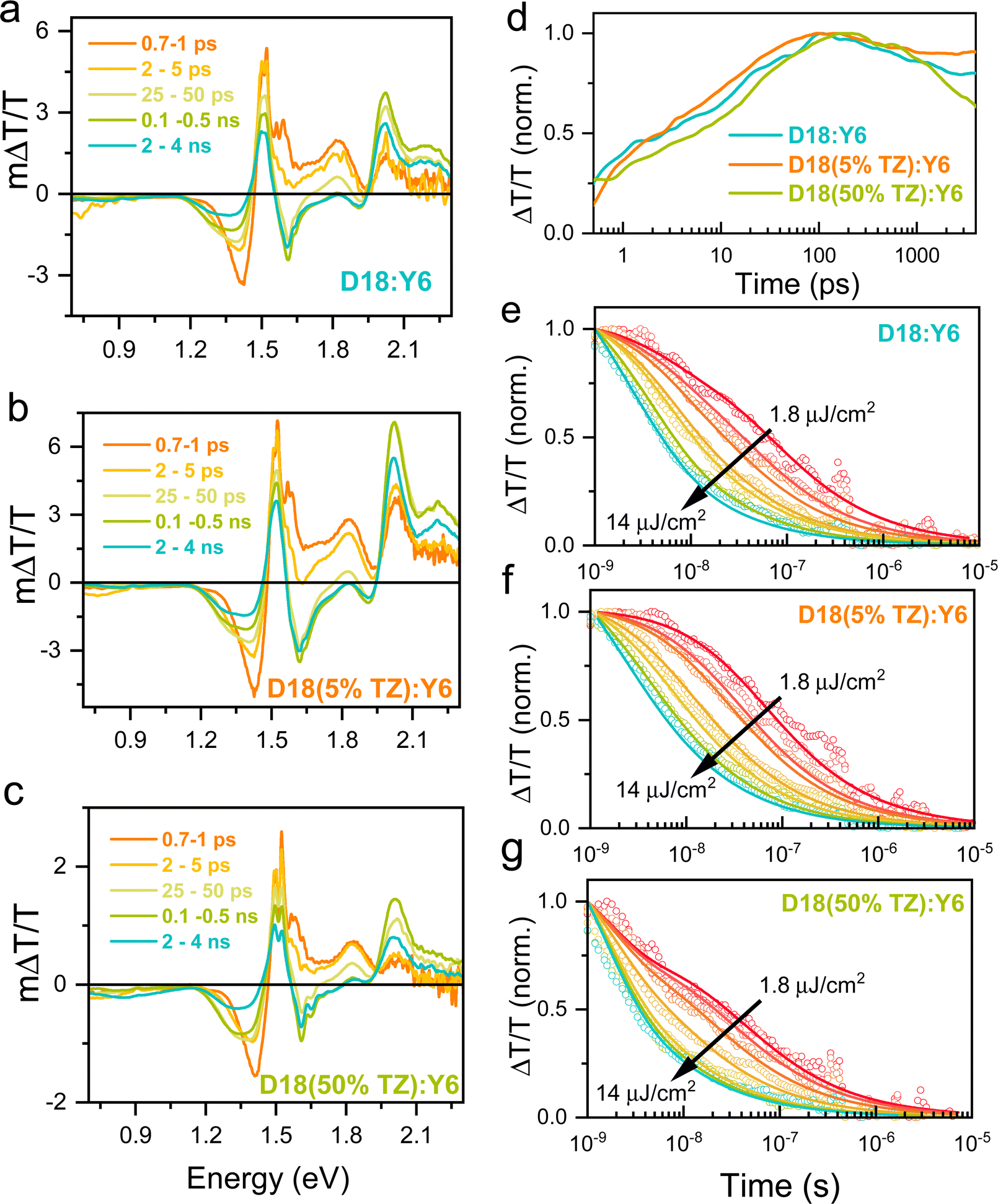 | ||
| Fig. 4 TA spectra and dynamics of terpolymer:Y6 films. ps–ns TA spectra of the (a) D18:Y6, (b) D18(5%TZ):Y6, and c D18(50%TZ):Y6 after photoexcitation at 650 nm (mainly excite Y6). (d) ps–ns TA kinetics of 1.23–1.3 eV spectral region (3.1 μJ cm−2) (a, b and c). (e–g) ns–μs TA kinetics of 1.23–1.3 eV (charge carriers) region for a wide range of fluences (symbols) and two-pool global fit (lines). | ||
The GSB band of the donor (1.9–2.1 eV) exhibited a slow rise up to 100 ps after an ultrafast charge generation. The exciton generated at the donor/acceptor interfaces can go for ultrafast charge generation, but the exciton generated far away from the D/A interface need diffuse through the pure phases. As a result, diffusion through pure phase delays the charge transfer/slows down the generation, but they do not decrease its efficiency because modern NFAs show long-range exciton diffusion length.51 In addition to larger diffusion length, Y6 exhibit a larger exciton lifetime, which is much larger than other NFAs51 larger than the maximum charge generation time reported here. Fig. 4d shows the integrated kinetics of the 1.22–1.3 eV (charge carrier-induced absorption) spectral region. The charge carrier generation rate for D18(5%TZ):Y6 blend films are faster than that of D18:Y6 and D18(50%TZ):Y6 at the same laser fluence, and in line with the exciton (fluorescence) lifetimes obtained by TRPL experiments (Fig. 3e). Fig. 4d compares the charge carrier dynamics of thin film blends. Here, a slightly faster charge generation but slower recombination was observed for D18(5%TZ):Y6 films (orange line), which is in line with the higher PCE of this blend.
To monitor the charge carrier recombination dynamics and quantify the carrier recombination rates, we performed nanosecond–microsecond (ns–μs) TA spectroscopy on the same blends used for the ps–ns TA studies.39,45 The ns–μs TA spectra showed similar spectral features for all three thin film blends (see Fig. S14, ESI†), yet their kinetics differed. Here, the fluence-independent decay of the charge-induced absorption at early probe delay times indicates a contribution from geminate recombination of bound charge-transfer states, while the fluence-dependent decay indicates non-geminate recombination of free charges dominates at later times.52 To quantify the individual process yields and to determine the kinetic parameters we fitted the experimentally-determined dynamics to a previously reported two-pool model (solid lines in Fig. 4e–g). Details of the two-pool model and fit can be found in earlier works and in the ESI.†52,53 Here, we assumed the charge carrier cross-section to be 1 × 10−16 cm−2, that is, on the order of values typically determined for polymeric materials.54 The fit to the two-pool model revealed that spatially-separated charges are generated with a yield of 96% and 90% in D18(5%TZ):Y6 and D18:Y6 blend films (Table 2), respectively. Free-charge carriers can be extracted as photocurrent from a device if they escape non-geminate recombination. Interestingly, in D18(50%TZ):Y6 films, the yield of free charge carriers were found to be much lower (76%) (Table 2), in turn indicating that 24% of the initial charge carrier population is bound in charge-transfer states which subsequently recombine geminately. Clearly, it can be seen that carrier losses due to geminate charge recombination directly impact the obtained photovoltaic device performance.
| Films | f (%) | λ + 1 | γ (cm3)λ s−1 | β (cm3 s−1) |
|---|---|---|---|---|
| D18:Y6 | 90 ± 1 | 2.6 ± 0.02 | 1.17 × 10−21 | 5 × 10−12 |
| D18(5%TZ):Y6 | 96 ± 2 | 2.58 ± 0.01 | 1 × 10−21 | 2 × 10−12 |
| D18(50%TZ):Y6 | 76 ± 2 | 2.6 ± 0.01 | 1.65 × 10−21 | 7 × 10−12 |
Apart from a reduced fraction of geminate recombination, D18(5%TZ):Y6 exhibited also a further reduced non-geminate recombination coefficient (β), indicating that free charge carriers are longer-lived and thus can be extracted to a larger extent from the device. In contrast, D18:Y6 and D18(50%TZ):Y6 films exhibited higher β values, pointing to faster non-geminate recombination of free charge carriers and thus less charge extraction. Overall, our intensity-dependent TA results reveal not only the mechanism of carrier recombination but also the fraction of charge carrier losses. It appears that adding 5%TZ to the parent polymers reduced carrier losses in blends with Y6, compared to the control device D18:Y6 (Table 2). However, incorporating higher loadings of TZ into the polymer backbone reduced the charge generation efficiency and increases both geminate and non-geminate recombination of charge carriers, in turn resulting in reduced FF and JSC in devices.
In addition to the excited state dynamics, the molecular orientation, material crystallinity, and D/A phase-separation impact the device FF.55–63 GIWAXS on thin films was performed to determine the molecular orientation and crystallinity of D18:Y6 and D18-based terpolymer:Y6 thin films and their D/A phase-separation was assessed by grazing incidence small-angle X-ray scattering (GISAXS). As presented in Fig. 5a, b and i, the introduction of TZ caused no obvious change in the molecular orientation, implying the TZ-based terpolymerization does not affect the molecular packing and thus the charge transport pathways are also retained. The corresponding GIWAXS parameters are summarized in Table S3 (ESI†). The D18:Y6 BHJ film exhibits (100) lamellar peaks at qxy ≈ 0.31 Å−1 (coherence length (CCL) = 5.28 nm) and (010) π–π peaks at qz ≈ 1.80 Å−1 (CCL = 2.64 nm). In comparison, the D18(5%TZ):Y6, D18(10%TZ):Y6, and D18(50%TZ):Y6 systems present (100) lamellar peaks at qxy ≈ 0.31 Å−1 (CCL = 4.67, 4.25 and 3.95 nm, respectively) and (010) π–π peaks at qz ≈ 1.77 Å−1 (CCL = 2.55, 2.36 and 2.29 nm, respectively), showing a continuously reduced film crystallinity in both IP and OOP direction when increasing the TZ content. The reduced film crystallinity is consistent with the reduced charge carrier mobility found in D18-based terpolymer solar cells (Table S7, ESI†). However, the D18(5%TZ):Y6 device exhibits an enhanced FF, which can be ascribed to different and more optimal D/A phase-separation.35,58
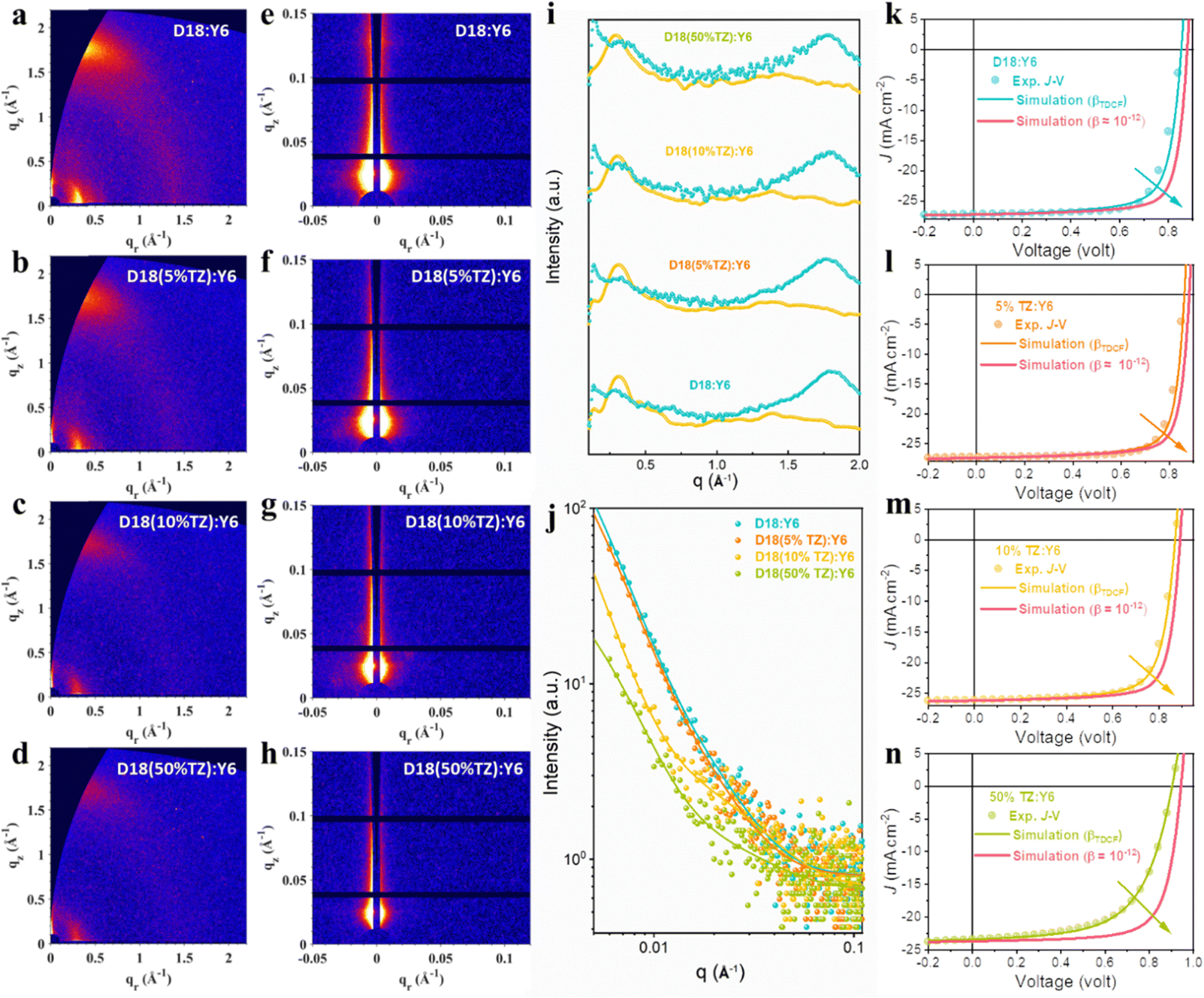 | ||
| Fig. 5 GIWAXS and GISAXS data of terpolymer:Y6 thin film blends and experimentally measured, as well as simulated device J–V characteristics. 2D GIWAXS patterns of (a) D18:Y6, (b) D18(5%TZ):Y6, (c) D18(10%TZ):Y6, and (d) D18(50%TZ):Y6 blend films. 2D GISAXS patterns of (e) D18:Y6, (f) 5%TZ:Y6, (g) 10%TZ:Y6, (h) 50%TZ:Y6 blend films. (i) The corresponding GIWAXS intensity profiles along the in-plane (IP) (yellow lines) and out-of-plane (OOP) (green lines). (j) The corresponding GISAXS intensity profiles and best-fitting curves along the in-plane. Experimentally measured (dots) and simulated (lines) current density–voltage (J–V) characteristics of (k) D18:Y6, (l) D18(5%TZ):Y6, (m) D18(10%TZ):Y6, and (n) D18(50%TZ):Y6 devices. | ||
GISAXS was carried out to evaluate the D/A phase-separation, and the 2D patterns are presented in Fig. 5e–h, and their corresponding IP intensity plots and fitting results based on the Debye–Anderson–Brumberger (DAB) model and fractal-like network are shown in Fig. 5j. XDAB refers to the intermixing D/A domain size and 2Rg represents the average pure domain size of the acceptor phase in this work for D18:Y6 and D18-based terpolymer:Y6 samples. The corresponding calculated results for XDAB and 2Rg are summarized in Table S4 (ESI†). The D18:Y6 and D18(5%TZ):Y6 films exhibited a nanoscale phase separation with an intermixed D/A domain size of ∼25 nm and a pure acceptor domain size of ∼20 nm (comparable to exciton diffusion length in NFAs), in conformity with the high FF. The D18(10%TZ):Y6 and D18(50%TZ):Y6 films exhibited a similar intermixed D/A domain size, but a smaller pure acceptor domain size of ∼12 nm, which may result in the reduced FF in devices.
Next, carrier drift-diffusion simulations were used to reproduce the experimentally-measured J–V characteristics. We used optical constants, namely the refractive index n and extinction coefficient k, determined by spectroscopic ellipsometry measurements (Fig. S18, ESI†), hole and electron mobilities measured by SCLC experiments (Fig. S17, ESI†), the bimolecular charge carrier recombination coefficient (βTDCF) obtained from time-delayed collection field (TDCF) measurements (Fig. S16, ESI†), and the device stack architecture as input parameters (Fig. S11, ESI†). We note that the bimolecular recombination coefficient obtained from TA on thin film blends differs from the one determined by TDCF. This mismatch is largely due to the n-dependence of β as we explained in our previous work64 and ESI.† In fact, TDCF appears to give a better estimate of the carrier recombination coefficient in devices as found in our previous works and thus we used the recombination coefficient determined by TDCF for our device simulations.65 More details about the simulation can be found in the ESI,† and our previous works.40,65 As shown in Fig. S16 (ESI†), the βTDCF of D18:Y6, D18(5%TZ):Y6, D18(10%TZ):Y6, and D18(50%TZ):Y6 devices are 7 × 10−11, 5 × 10−11, 9 × 10−11 and 1 × 10−10 cm3 s−1, respectively. We note that the trend for βTDCF is in line with the change of βTA (Fig. 4e–g), confirming reduced carrier recombination when 5% TZ is incorporated into the polymer backbone, while recombination increases when introducing a higher content of TZ. Details of the simulation using the commercial software tool Setfos 5.2 (Fluxim AG) and the precise simulation procedure can be found in our previous works.40,45,65,66 The comparison of the simulated and experimentally-measured J–V characteristics of the terpolymer photovoltaics are presented in Fig. 5k–n, and the relevant photovoltaic parameters are listed in Table S8 (ESI†). The good match of the simulated and measured J–V data of D18:Y6, D18(5%TZ):Y6, D18(10%TZ):Y6, and D18(50%TZ):Y6 devices indicates that the input parameters (hole/electron mobilities, n, k, and βTDCF) determined by spectroscopic experiments are meaningful and capture the processes and their dynamics in operating solar cell devices. As the βTA of D18-based terpolymer photovoltaics from TA experiments was as low as ∼10−12 cm3 s−1, we used β ≈ 10−12 cm3 s−1 to observe its impact on the performance of D18-based terpolymer solar cells. Clearly, reducing β by one more order would enhance the PCE of D18:Y6, D18(5%TZ):Y6, D18(10%TZ):Y6, and D18(50%TZ):Y6 devices further to 18.6%, 19.0%, 17.9%, and 15.8%, respectively, showing that reducing the bimolecular recombination coefficient and improving carrier extraction is important to increase the PCE not only in D18-based terpolymer solar cells.
Long-term (outdoor) operational stability is of utmost importance to commercialize PV technology, and an important challenge for OPV. The devices need to sustain prolonged operation under solar irradiance and exhibit high tolerance to heat and temperature changes during day-night cycles.23 Thus, a thermodynamically stable morphology is required to obtain outdoor-stable OSCs. Previous works have demonstrated that D/A miscibility close to the percolation threshold of small molecules (fullerenes and NFAs) is key to obtaining a (meta)stable morphology.67,68 Here, the terpolymerization strategy is employed to fine-tune the D/A miscibility to obtain outdoor-stable OSCs with a thermodynamically stable morphology. The experimental details have been shown in ESI.† The outdoor stability tests for all four systems were performed in the hot and sunny climate at KAUST, Thuwal, Kingdom of Saudi Arabia, a location with high irradiance and temperatures. In fact, the irradiance was as high as 1039.5 W m−2 at noon (Fig. 6a) as measured by a calibrated pyranometer and the highest/lowest air temperatures during daytime/nighttime were 48 °C/22 °C (Fig. 6b). As shown in Fig. 6c, the figures-of-merit of the encapsulated D18:Y6, D18(5%TZ):Y6, D18(10%TZ):Y6 and D18(50%TZ):Y6 devices were tracked every 10 min for 32 days in total, presenting an uptrend of the PGD from sunrise to noon, followed a downtrend from noon to sunset. For a better comparison, the device performance obtained at noon was normalized (accounting also for the irradiance by dividing the irradiance measured at noon) to monitor the outdoor degradation (Fig. 6d). The devices based on D18:Y6 (χdonor–acceptor (D/A miscibility) of 59.9 × 10−2 K) and D18(5%TZ):Y6 (χdonor–acceptor of 50.5 × 10−2 K) (Table S10 and Fig. S21a, b, ESI†) exhibited a similar degradation in the first 4 weeks, however, the D18:Y6 devices demonstrated significantly lower device performance after 4 weeks. It appears the miscibility of 50.5 × 10−2 K in D18-based terpolymer:Y6 systems deliver a thermodynamically more stable thin film. Furthermore, the D18(10%TZ):Y6 with a χdonor–acceptor of 12.5 × 10−2 K and D18(50%TZ):Y6 with a χdonor–acceptor of 1.3 × 10−2 K (Table S10 and Fig. S21c, d, ESI†) exhibited much severer degradation in the first week, implying that the miscibility in D18-based terpolymer devices is different from that in D18(5%TZ):Y6 systems, resulting in less thermodynamically stable films. It is worth noting that the D18(5%TZ):Y6 system, which exhibited the thermodynamically most stable morphology, retained 92.6% initial performance after 32 days of harsh outdoor stressing in Saudi Arabia, which is among the best outdoor stabilities reported so far for 3rd generation photovoltaics.22,23,69–71
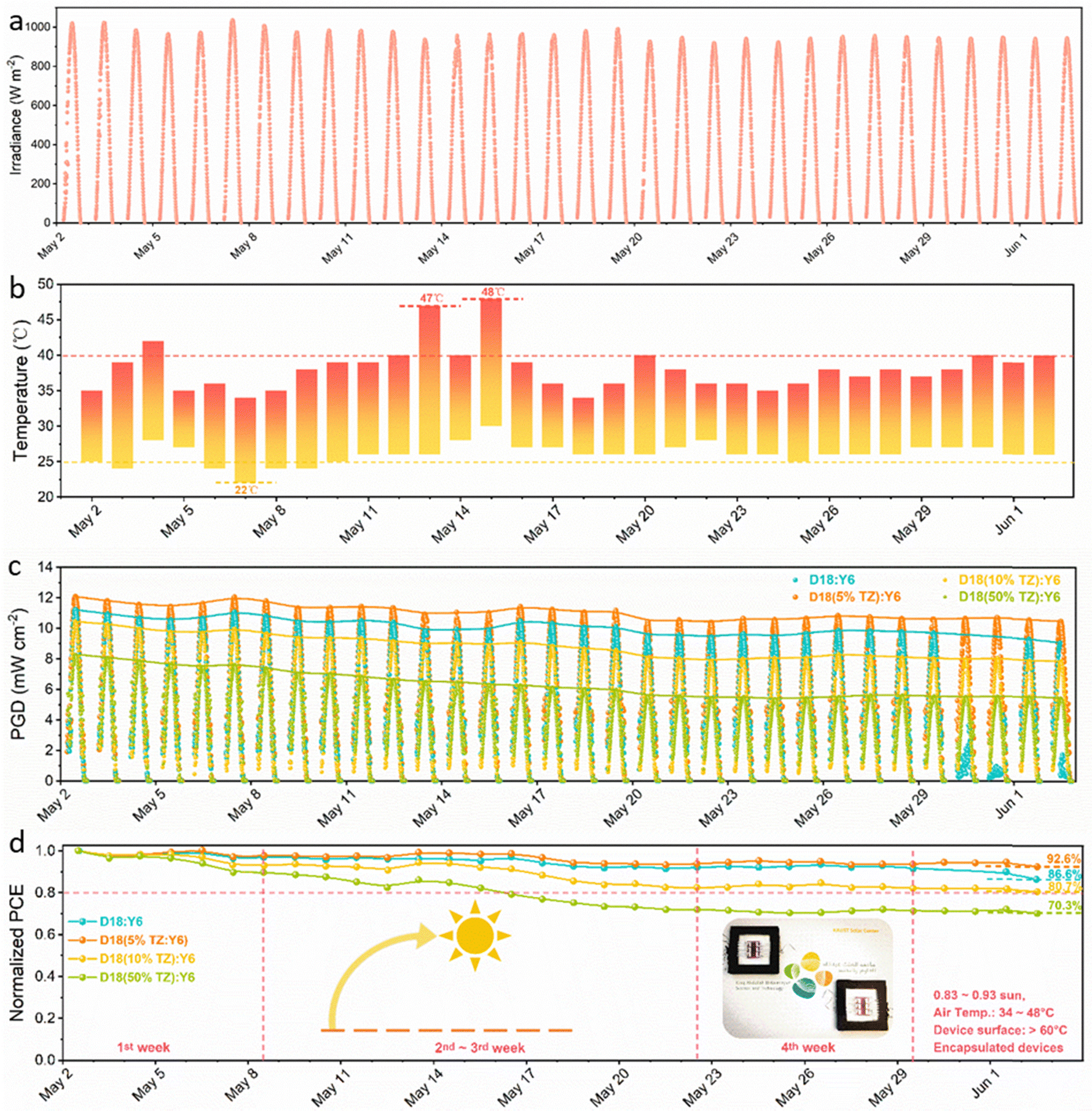 | ||
| Fig. 6 Outdoor stability of terpolymer photovoltaic device. (a) Solar irradiance, and (b) air temperature status during the test period. (c) Power generation density (PGD) of the encapsulated D18:Y6, D18(5%TZ):Y6, D18(10%TZ):Y6 and D18(50%TZ):Y6 devices obtained during the test period, and (d) the corresponding normalized encapsulated devices at noon. Note: the outdoor stability analysis was carried out on the KAUST campus, Thuwal, Kingdom of Saudi Arabia from May 2nd to Jun. 2nd. J–V curves were measured every 10 min during the daytime. | ||
To confirm that the D18(5%TZ):Y6 system is the most thermodynamically stable blend, hyperspectral PL imaging was performed on fresh and aged devices of all four systems tested outdoors. The normalized average PL spectra of the entire mapped area are depicted in Fig. S24a and b (ESI†). The D18 and D18-based terpolymers are very stable due to their intrinsic morphological stability, whereas, NFAs are prone to aggregate under light-soaking or heat exposure. Hence, by tracking the PL intensity of the D18/D18-based terpolymer and the Y6 acceptor domains, we can disentangle which D18/D18-based terpolymer:Y6 system is the most thermodynamically stable. As shown in Fig. S24a (ESI†), all four systems in fresh devices exhibited a stronger PL of the Y6 phase, indicating optimum blend morphology.65 After 32 days of outdoor testing, the D18:Y6, D18(5%TZ):Y6, D18(10%TZ):Y6, and D18(50%TZ):Y6 devices all exhibited significantly reduced PL intensity of the Y6 domains due to the aggregation of Y6,72,73 while the D18(5%TZ):Y6 system still showed comparably intense PL from the Y6 domains (Fig. S24b, ESI†), indicative of a more thermodynamically stable morphology and in line with the outdoor stability data showing 92.6% of the initial device performance was retained after 32 days.
Conclusion
This study summarized a set of rules for high-performance and outdoor-stable terpolymer solar cells and proposed a rationale to explain the working principle behind them. We combined spectroscopic, morphological, and computational approaches to elucidate the key rules for highly efficient and outdoor-stable terpolymer solar cells based on parent polymer donor D18 and PM6. We found that the device's JSC is enhanced and energy loss is reduced in terpolymer devices when incorporating another strong electron-deficient moiety that broadens the material's optical absorption and lowers the HOMO level of the terpolymer. Enhanced device FFs were found when the fraction of the third component of the terpolymer was 5% only, which retained the original D/A framework with delicately optimized phase-separation that reduced the bimolecular charge carrier recombination and further improved the device FF. Highly outdoor-stable terpolymer devices were obtained due to the incorporation of a third component which fine-tuned the D/A miscibility and led to a thermodynamically more stable morphology. Our findings systematically unveiled the photovoltaic performance and outdoor stability enhancement by monitoring the exciton/charge carrier dynamics and the blends’ thin film morphology, providing a rationale for highly efficient and outdoor-stable terpolymer solar cells, pushing them closer to commercial realization.Author contributions
Conceptualization, H. T.; methodology and critical data curation, H. T.; material syntheses, Z. L.; TA spectroscopy, S. K.; E. L., S. C., and M. A.; TDCF, H. T., and S. C.; simulation, H. T., W. Y., and S. C.; TRPL spectroscopy, J. K., outdoor stability test, M. B.; GIWAXS/GISAXS test, H. L.; other data curation, J. H.; writing – original draft, H. T.; writing – review & editing, H. T., S. K., Y. H., G. J., F. L., and S. L.; supervision, G. J., G. L., S. W., X. L., C. B., F. L., and S. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication is based upon work supported by the King Abdullah University of Science and Technology (KAUST) Office of Research Administration (ORA) under Award No: OSR-CARF/CCF-3079. S. L. thanks the research grant from the National Youth Thousand Program Project (R52A199Z11), Chongqing Funds for Distinguished Young Scientists (cstc2020jcyj-jqX0018), Chongqing talent plan (CQYC201903008) and General Program of National Natural Science Foundation of China (62074149). D. H. thanks the research grant from Chongqing Funds for Distinguished Young Scientists (cstc2021jcyj-msxm0139). G. L. thanks the Hong Kong RGC Senior Research Fellowship Scheme (SRFS2223-5S01). H. T. acknowledges Prof. Derya Baran for her fruitful discussion and valuable suggestion for revising the draft.References
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- Y. Lin, Q. He, F. Zhao, L. Huo, J. Mai, X. Lu, C. J. Su, T. Li, J. Wang, J. Zhu, Y. Sun, C. Wang and X. Zhan, J. Am. Chem. Soc., 2016, 138, 2973–2976 CrossRef CAS PubMed.
- Y. Lin and X. Zhan, Mater. Horiz., 2014, 1, 470 RSC.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- Y. Yang, ACS Nano, 2021, 15, 18679–18682 CrossRef CAS PubMed.
- H. Tang, C. Yan, J. Huang, Z. Kan, Z. Xiao, K. Sun, G. Li and S. Lu, Matter, 2020, 3, 1403–1432 CrossRef.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- P. Cheng, G. Li, X. Zhan and Y. Yang, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS.
- H. Tang, J. Lv, K. Liu, Z. Ren, H. T. Chandran, J. Huang, Y. Zhang, H. Xia, J. I. Khan, D. Hu, C. Yan, J. Oh, S. Chen, S. Chu, P. W. K. Fong, H. Chen, Z. Xiao, C. Yang, Z. Kan, F. Laquai, S. Lu and G. Li, Mater. Today, 2022, 55, 46–55 CrossRef CAS.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS PubMed.
- J. Hou, O. Inganas, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed.
- H. Tang, H. Chen, C. Yan, J. Huang, P. W. K. Fong, J. Lv, D. Hu, R. Singh, M. Kumar, Z. Xiao, Z. Kan, S. Lu and G. Li, Adv. Energy Mater., 2020, 10, 2001076 CrossRef CAS.
- G. Li, W.-H. Chang and Y. Yang, Nat. Rev. Mater., 2017, 2, 17043 CrossRef CAS.
- Y. Yang, W. Chen, L. Dou, W.-H. Chang, H.-S. Duan, B. Bob, G. Li and Y. Yang, Nat. Photonics, 2015, 9, 190–198 CrossRef CAS.
- D. Qian, Z. Zheng, H. Yao, W. Tress, T. R. Hopper, S. Chen, S. Li, J. Liu, S. Chen, J. Zhang, X. K. Liu, B. Gao, L. Ouyang, Y. Jin, G. Pozina, I. A. Buyanova, W. M. Chen, O. Inganas, V. Coropceanu, J. L. Bredas, H. Yan, J. Hou, F. Zhang, A. A. Bakulin and F. Gao, Nat. Mater., 2018, 17, 703–709 CrossRef CAS PubMed.
- Z. Zhou, S. Xu, J. Song, Y. Jin, Q. Yue, Y. Qian, F. Liu, F. Zhang and X. Zhu, Nat. Energy, 2018, 3, 952–959 CrossRef CAS.
- P. Cong, Z. Wang, Y. Geng, Y. Meng, C. Meng, L. Chen, A. Tang and E. Zhou, Nano Energy, 2023, 105, 108017 CrossRef CAS.
- Y. Zhang, Y. Ji, Y. Zhang, W. Zhang, H. Bai, M. Du, H. Wu, Q. Guo and E. Zhou, Adv. Funct. Mater., 2022, 32, 2205115 CrossRef CAS.
- Q. Guo, Q. Guo, Y. Geng, A. Tang, M. Zhang, M. Du, X. Sun and E. Zhou, Mater. Chem. Front., 2021, 5, 3257–3280 RSC.
- X. Zhu, S. Liu, Q. Yue, W. Liu, S. Sun and S. Xu, CCS Chem., 2021, 3, 1070–1080 CrossRef CAS.
- W. Liu, S. Xu, H. Lai, W. Liu, F. He and X. Zhu, CCS Chem., 2022, 1–15, DOI:10.31635/ccschem.022.202201963.
- J. Liu, E. Aydin, J. Yin, M. De Bastiani, F. H. Isikgor, A. U. Rehman, E. Yengel, E. Ugur, G. T. Harrison, M. Wang, Y. Gao, J. I. Khan, M. Babics, T. G. Allen, A. S. Subbiah, K. Zhu, X. Zheng, W. Yan, F. Xu, M. F. Salvador, O. M. Bakr, T. D. Anthopoulos, M. Lanza, O. F. Mohammed, F. Laquai and S. De Wolf, Joule, 2021, 5, 3169–3186 CrossRef CAS.
- M. Babics, M. De Bastiani, A. H. Balawi, E. Ugur, E. Aydin, A. S. Subbiah, J. Liu, L. Xu, R. Azmi, T. G. Allen, A. U. Rehman, T. Altmann, M. F. Salvador and S. De Wolf, ACS Energy Lett., 2022, 7, 1604–1610 CrossRef CAS.
- L. Duan and A. Uddin, Adv. Sci., 2020, 7, 1903259 CrossRef CAS PubMed.
- P. Cheng and X. Zhan, Chem. Soc. Rev., 2016, 45, 2544–2582 RSC.
- B. Yang, S. Zhang, S. Li, H. Yao, W. Li and J. Hou, Adv. Mater., 2019, 31, 1804657 CrossRef PubMed.
- G. Li, C. W. Chu, V. Shrotriya, J. Huang and Y. Yang, Appl. Phys. Lett., 2006, 88, 253503 CrossRef.
- J. Wu, G. Li, J. Fang, X. Guo, L. Zhu, B. Guo, Y. Wang, G. Zhang, L. Arunagiri, F. Liu, H. Yan, M. Zhang and Y. Li, Nat. Commun., 2020, 11, 4612 CrossRef CAS PubMed.
- X. Guo, Q. Fan, J. Wu, G. Li, Z. Peng, W. Su, J. Lin, L. Hou, Y. Qin, H. Ade, L. Ye, M. Zhang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 2322–2329 CrossRef CAS PubMed.
- M. Jeong, S. Chen, S. M. Lee, Z. Wang, Y. Yang, Z.-G. Zhang, C. Zhang, M. Xiao, Y. Li and C. Yang, Adv. Energy Mater., 2018, 8, 1702166 CrossRef.
- H. Sun, T. Liu, J. Yu, T.-K. Lau, G. Zhang, Y. Zhang, M. Su, Y. Tang, R. Ma, B. Liu, J. Liang, K. Feng, X. Lu, X. Guo, F. Gao and H. Yan, Energy Environ. Sci., 2019, 12, 3328–3337 RSC.
- M. Jeong, J. Oh, Y. Cho, B. Lee, S. Jeong, S. M. Lee, S. H. Kang and C. Yang, Adv. Funct. Mater., 2021, 31, 2102371 CrossRef CAS.
- X. Xu, K. Feng, Z. Bi, W. Ma, G. Zhang and Q. Peng, Adv. Mater., 2019, 31, 1901872 CrossRef PubMed.
- T. Wang, R. Sun, M. Shi, F. Pan, Z. Hu, F. Huang, Y. Li and J. Min, Adv. Energy Mater., 2020, 10, 2000590 CrossRef CAS.
- Z. Liao, D. Hu, H. Tang, P. Huang, R. Singh, S. Chung, K. Cho, M. Kumar, L. Hou, Q. Chen, W. Yu, H. Chen, K. Yang, Z. Kan, F. Liu, Z. Xiao, G. Li and S. Lu, J. Mater. Chem. A, 2022, 10, 7878–7887 RSC.
- Z. Liao, K. Yang, L. Hou, J. Li, J. Lv, R. Singh, M. Kumar, Q. Chen, X. Dong, T. Xu, C. Hu, T. Duan, Z. Kan, S. Lu and Z. Xiao, Macromolecules, 2020, 53, 9034–9042 CrossRef CAS.
- P. Tyagi, S.-C. Hua, D. R. Amorim, R. M. Faria, J. Kettle and M. Horie, Org. Electron., 2018, 55, 146–156 CrossRef CAS.
- J. Bertrandie, J. Han, C. S. P. De Castro, E. Yengel, J. Gorenflot, T. Anthopoulos, F. Laquai, A. Sharma and D. Baran, Adv. Mater., 2022, 34, 2202575 CrossRef CAS PubMed.
- S. Karuthedath, J. Gorenflot, Y. Firdaus, N. Chaturvedi, C. S. P. De Castro, G. T. Harrison, J. I. Khan, A. Markina, A. H. Balawi, T. A. D. Pena, W. Liu, R. Z. Liang, A. Sharma, S. H. K. Paleti, W. Zhang, Y. Lin, E. Alarousu, D. H. Anjum, P. M. Beaujuge, S. De Wolf, I. McCulloch, T. D. Anthopoulos, D. Baran, D. Andrienko and F. Laquai, Nat. Mater., 2021, 20, 378–384 CrossRef CAS PubMed.
- J. I. Khan, M. A. Alamoudi, N. Chaturvedi, R. S. Ashraf, M. N. Nabi, A. Markina, W. Liu, T. A. Dela Peña, W. Zhang, O. Alévêque, G. T. Harrison, W. Alsufyani, E. Levillain, S. De Wolf, D. Andrienko, I. McCulloch and F. Laquai, Adv. Energy Mater., 2021, 11, 2100839 CrossRef CAS.
- J. I. Khan, R. S. Ashraf, M. A. Alamoudi, M. N. Nabi, H. N. Mohammed, A. Wadsworth, Y. Firdaus, W. Zhang, T. D. Anthopoulos, I. McCulloch and F. Laquai, Sol. RRL, 2019, 3, 1900023 CrossRef.
- S. Chandrabose, K. Chen, A. J. Barker, J. J. Sutton, S. K. K. Prasad, J. Zhu, J. Zhou, K. C. Gordon, Z. Xie, X. Zhan and J. M. Hodgkiss, J. Am. Chem. Soc., 2019, 141, 6922–6929 CrossRef CAS PubMed.
- A. Classen, C. L. Chochos, L. Lüer, V. G. Gregoriou, J. Wortmann, A. Osvet, K. Forberich, I. McCulloch, T. Heumüller and C. J. Brabec, Nat. Energy, 2020, 5, 711–719 CrossRef CAS.
- D. B. Riley, P. Meredith, A. Armin and O. J. Sandberg, J. Phys. Chem. Lett., 2022, 13, 4402–4409 CrossRef CAS PubMed.
- S. Karuthedath, Y. Firdaus, R. Z. Liang, J. Gorenflot, P. M. Beaujuge, T. D. Anthopoulos and F. Laquai, Adv. Energy Mater., 2019, 9, 1901443 CrossRef.
- H. Tang, C. Yan, S. Karuthedath, H. Yin, Y. Gao, J. Gao, L. Zhang, J. Huang, S. K. So, Z. Kan, F. Laquai, G. Li and S. Lu, Sol. RRL, 2020, 4, 1900528 CrossRef CAS.
- M. B. Price, P. A. Hume, A. Ilina, I. Wagner, R. R. Tamming, K. E. Thorn, W. Jiao, A. Goldingay, P. J. Conaghan, G. Lakhwani, N. Davis, Y. Wang, P. Xue, H. Lu, K. Chen, X. Zhan and J. M. Hodgkiss, Nat. Commun., 2022, 13, 2827 CrossRef CAS PubMed.
- Y. Wang, M. B. Price, R. S. Bobba, H. Lu, J. Xue, Y. Wang, M. Li, A. Ilina, P. A. Hume, B. Jia, T. Li, Y. Zhang, N. Davis, Z. Tang, W. Ma, Q. Qiao, J. M. Hodgkiss and X. Zhan, Adv. Mater., 2022, 34, 2206717 CrossRef CAS PubMed.
- G. Zhou, M. Zhang, J. Xu, Y. Yang, T. Hao, L. Zhu, L. Zhou, H. Zhu, Y. Zou, G. Wei, Y. Zhang and F. Liu, Energy Environ. Sci., 2022, 15, 3483–3493 RSC.
- R. Wang, C. Zhang, Q. Li, Z. Zhang, X. Wang and M. Xiao, J. Am. Chem. Soc., 2020, 142, 12751–12759 CrossRef CAS PubMed.
- Y. Firdaus, V. M. Le Corre, S. Karuthedath, W. Liu, A. Markina, W. Huang, S. Chattopadhyay, M. M. Nahid, M. I. Nugraha, Y. Lin, A. Seitkhan, A. Basu, W. Zhang, I. McCulloch, H. Ade, J. Labram, F. Laquai, D. Andrienko, L. J. A. Koster and T. D. Anthopoulos, Nat. Commun., 2020, 11, 5220 CrossRef CAS PubMed.
- S. Karuthedath, J. Gorenflot, A. Melianas, Z. Kan, M. Kemerink and F. Laquai, J. Phys. Chem. Lett., 2020, 11, 2838–2845 CrossRef CAS PubMed.
- I. A. Howard, R. Mauer, M. Meister and F. Laquai, J. Am. Chem. Soc., 2010, 132, 14866–14876 CrossRef CAS PubMed.
- S. Karuthedath, Y. Firdaus, A. D. Scaccabarozzi, M. I. Nugraha, S. Alam, T. D. Anthopoulos and F. Laquai, Small Struct., 2022, 3, 2100199 CrossRef CAS.
- D. Hu, Q. Yang, H. Chen, F. Wobben, V. M. Le Corre, R. Singh, T. Liu, R. Ma, H. Tang, L. J. A. Koster, T. Duan, H. Yan, Z. Kan, Z. Xiao and S. Lu, Energy Environ. Sci., 2020, 13, 2134–2141 RSC.
- C. Yan, T. Liu, Y. Chen, R. Ma, H. Tang, G. Li, T. Li, Y. Xiao, T. Yang, X. Lu, X. Zhan, H. Yan, G. Li and B. Tang, Sol. RRL, 2019, 3, 201900377 Search PubMed.
- X. Dong, K. Yang, H. Tang, D. Hu, S. Chen, J. Zhang, Z. Kan, T. Duan, C. Hu, X. Dai, Z. Xiao, K. Sun and S. Lu, Sol. RRL, 2019, 4, 1900326 CrossRef.
- J. Lv, H. Tang, J. Huang, C. Yan, K. Liu, Q. Yang, D. Hu, R. Singh, J. Lee, S. Lu, G. Li and Z. Kan, Energy Environ. Sci., 2021, 14, 3044–3052 RSC.
- C. Yan, H. Tang, R. Ma, M. Zhang, T. Liu, J. Lv, J. Huang, Y. Yang, T. Xu, Z. Kan, H. Yan, F. Liu, S. Lu and G. Li, Adv. Sci., 2020, 7, 2000149 CrossRef CAS PubMed.
- H. Tang, T. Xu, C. Yan, J. Gao, H. Yin, J. Lv, R. Singh, M. Kumar, T. Duan, Z. Kan, S. Lu and G. Li, Adv. Sci., 2019, 6, 1901613 CrossRef CAS PubMed.
- D. Hu, Q. Yang, Y. Zheng, H. Tang, S. Chung, R. Singh, J. Lv, J. Fu, Z. Kan, B. Qin, Q. Chen, Z. Liao, H. Chen, Z. Xiao, K. Sun and S. Lu, Adv. Sci., 2021, 8, 2004262 CrossRef CAS PubMed.
- J. Huang, Z. Ren, Y. Zhang, K. Liu, H. Zhang, H. Tang, C. Yan, Z. Zheng and G. Li, Adv. Funct. Mater., 2021, 31, 2010172 CrossRef CAS.
- C. Yan, J. Qin, Y. Wang, G. Li and P. Cheng, Adv. Energy Mater., 2022, 12, 2201087 CrossRef CAS.
- S. Karuthedath, A. Melianas, Z. Kan, V. Pranculis, M. Wohlfahrt, J. I. Khan, J. Gorenflot, Y. Xia, O. Inganäs, V. Gulbinas, M. Kemerink and F. Laquai, J. Mater. Chem. A, 2018, 6, 7428–7438 RSC.
- D. Hu, H. Tang, S. Karuthedath, Q. Chen, S. Chen, J. I. Khan, H. Liu, Q. Yang, J. Gorenflot, C. E. Petoukhoff, T. Duan, X. Lu, F. Laquai and S. Lu, Adv. Funct. Mater., 2023, 33, 2211873 CrossRef CAS.
- A. H. Balawi, Z. Kan, J. Gorenflot, P. Guarracino, N. Chaturvedi, A. Privitera, S. Liu, Y. Gao, L. Franco, P. Beaujuge and F. Laquai, Sol. RRL, 2020, 4, 2000181 CrossRef CAS.
- L. Ye, B. A. Collins, X. Jiao, J. Zhao, H. Yan and H. Ade, Adv. Energy Mater., 2018, 8, 1703058 CrossRef.
- K. Zhou, J. Xin and W. Ma, ACS Energy Lett., 2019, 4, 447–455 CrossRef CAS.
- M. De Bastiani, E. Van Kerschaver, Q. Jeangros, A. Ur Rehman, E. Aydin, F. H. Isikgor, A. J. Mirabelli, M. Babics, J. Liu, S. Zhumagali, E. Ugur, G. T. Harrison, T. G. Allen, B. Chen, Y. Hou, S. Shikin, E. H. Sargent, C. Ballif, M. Salvador and S. De Wolf, ACS Energy Lett., 2021, 6, 2944–2951 CrossRef CAS.
- S. A. Gevorgyan, M. V. Madsen, B. Roth, M. Corazza, M. Hösel, R. R. Søndergaard, M. Jørgensen and F. C. Krebs, Adv. Energy Mater., 2016, 6, 1501208 CrossRef.
- Y. Zhang, I. D. W. Samuel, T. Wang and D. G. Lidzey, Adv. Sci., 2018, 5, 1800434 CrossRef PubMed.
- Y. Zhang, K. Liu, J. Huang, X. Xia, J. Cao, G. Zhao, P. W. K. Fong, Y. Zhu, F. Yan, Y. Yang, X. Lu and G. Li, Nat. Commun., 2021, 12, 4815 CrossRef CAS PubMed.
- X. Du, T. Heumueller, W. Gruber, O. Almora, A. Classen, J. Qu, F. He, T. Unruh, N. Li and C. J. Brabec, Adv. Mater., 2020, 32, 1908305 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee00350g |
| ‡ HT and SK contributed equally to this work. |
| § Present address: Institute of Materials Research, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen, 518055 China. |
| This journal is © The Royal Society of Chemistry 2023 |