
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improving the efficiency and stability of perovskite solar cells using π-conjugated aromatic additives with differing hydrophobicities†
Ran
Wang
 *a,
Amal
Altujjar
a,
Nourdine
Zibouche
bc,
Xuelian
Wang
a,
Ben F.
Spencer
ad,
Zhenyu
Jia
a,
Andrew G.
Thomas
ad,
Muhamad Z.
Mokhtar
ae,
Rongsheng
Cai
a,
Sarah J.
Haigh
a,
Jennifer M.
Saunders
a,
M. Saiful
Islam
f and
Brian R.
Saunders
*a
*a,
Amal
Altujjar
a,
Nourdine
Zibouche
bc,
Xuelian
Wang
a,
Ben F.
Spencer
ad,
Zhenyu
Jia
a,
Andrew G.
Thomas
ad,
Muhamad Z.
Mokhtar
ae,
Rongsheng
Cai
a,
Sarah J.
Haigh
a,
Jennifer M.
Saunders
a,
M. Saiful
Islam
f and
Brian R.
Saunders
*a
aDepartment of Materials, University of Manchester, MECD(A), Manchester, M1 7HL, UK. E-mail: ran.wang-11@postgrad.manchester.ac.uk; Brian.Saunders@manchester.ac.uk
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK
cDepartment of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK. E-mail: n.zibouche@lancaster.ac.uk
dPhoton Science Institute, The Henry Royce Institute, University of Manchester, Manchester, M13 9PL, UK
eDepartment of Water and Environmental Engineering, Faculty of Civil Engineering, Universiti Teknologi Malaysia, 81310 Johor Bahru, Johor, Malaysia
fDepartment of Materials, University of Oxford, Oxford, OX1 3PH, UK
First published on 11th May 2023
Abstract
Perovskite solar cells (PSCs) continue to excite the research community due to their excellent power conversion efficiency (PCE) and relative ease of preparation. Additive engineering has played a decisive role in improving PSC performance and stability. In particular, π-conjugated aromatic additives (CAAs) offer key advantages such as high charge transport. However, the roles of hydrophobicity and structure in determining CAA performance as additives are still being established. Here, we investigate the effects of two coumarin additives on the PCE and stability of PSCs based on Cs0.05(FA0.85MA0.15)0.95Pb(I0.85Br0.15)3 perovskite. The CAAs are coumarin methacrylate (CMA) and coumarin hydroxyethyl (CHE) and were added to the precursor perovskite solutions prior to film deposition with CMA being more hydrophobic than CHE. These additives increase the best PCE of 19.15% for the control to 21.14% and 21.28% for the best devices containing CHE and CMA, respectively. The stability of the devices with the additives are far superior to that of the control (CAA-free) system. The time lengths required for the PCE to decrease to 80% of the initial value for CMA- and CHE-containing devices are 98 and 38 days, respectively, compared to only 14 days for the control. The moisture and thermal stabilities of the systems containing CMA are markedly improved compared to those containing CHE and the control. Our results show that the extents of binding to Pb2+ and passivation increase as the coumarin's hydrophobicity increases which decreases recombination. Our findings show that adding CAAs with increasing hydrophobic character to the precursor perovskite solution is useful for achieving high-performance and long-term stable PSCs.
Broader contextPerovskite solar cells (PSCs) have attracted great interest and excitement as potentially low-cost alternatives to silicon solar cells for power generation. PSCs satisfy the key pillars for new solar energy technology commercialization of efficiency and low cost. Unfortunately, PSC stability still remains well below that of commercial silicon solar cells. Including additives in the precursor solutions used to prepare PSCs has proved to be a very useful technique to improve both efficiency and stability of the devices. Such an approach is also amenable to future scale up. Of all the different types of additives used for PSCs, π-conjugated aromatic additives (CAAs) have intrinsic properties that especially favour their inclusion such as facile charge transport. Here, we investigate how substituents of CAA affect PSC properties, including stability and efficiency. We show that substituents of CAA that improve additive hydrophobicity are desirable for enhanced PSC performance and discuss the relations that underpin this finding. |
Introduction
Organic–inorganic metal hybrid perovskites are regarded as highly promising photovoltaic materials due to their excellent optoelectronic performance and ability to enable low-cost solution processable technologies.1–4 Their advantageous optoelectronic properties include high absorption lengths, low exciton binding energies and high carrier mobility.5–7 Perovskite solar cells (PSCs) have emerged as one of the most promising 3rd generation solar cell candidates, not only because of their excellent power conversion efficiency (PCE) of 25.7%8,9 but also their scalable preparation.10,11 Despite extensive research and development efforts, PSCs are still faced with relatively poor stability.12–15 Additive engineering has proven to be successful in improving both the PCE and stability of PSCs,16 which is due in large part to the passivation of defects17 and decreasing the migration of mobile ions.18,19 Indeed, π-conjugated aromatic additives (CAAs) offer a range of potential advantages for PSCs because their delocalized electrons enable charge to easily pass at perovskite interfaces and their planarity favors the alignment of neighboring molecules at interfaces.20 They can also passivate Lewis acids (such as Pb) by donating electrons21–23 and act as interfacial compatibilizers between the perovskite and the hole transport matrix (HTM).20Whilst many studies have used CAAs in PSCs,20 very few have included them in the precursor perovskite solution. Wei et al. included 3-phenyl-2-propen-1-amine iodide (PPEAI) as an antisolvent additive and reported that this small-molecule CAA provided conductive channels between neighboring perovskite grains.24 Wu et al. used 2-amidinopyrimidine in a similar manner to increase PSC efficiency.12 Several studies have used CAAs as interfacial modifiers between the perovskite and HTM.25,26 In other cases, CAAs have been used to modify the interface between the perovskite and electron transport matrix (ETM).22,27 In such cases the CAAs were very hydrophobic and not likely to be soluble in the polar aprotic solvents used for perovskite precursor solutions, such as DMF and DMSO. In contrast, the present study uses two coumarins as CAAs that are able to be included in the perovskite precursor solution. We use two structurally similar CAAs with differing hydrophobicities to elucidate the role of this parameter and the structural differences on PSC performance and stability. This approach has the advantages of providing a simpler, potentially more scalable, construction method that enables the additives to distribute throughout the perovskite layer as well as the bottom and top perovskite interfaces.
Coumarins belong to the benzopyrone family and contain a benzene ring fused with a pyrone ring.28 Here, coumarin methacrylate (CMA) and coumarin hydroxyethyl (CHE) additives are investigated for planar films and PSCs (Fig. 1a and b). We hypothesized that these CAAs would allow charge to pass more easily at interfaces.22,29 Moreover, by introducing the methacrylate group via CMA the hydrophobicity is increased. CMA contains carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) functional groups at two positions (Fig. 1b) and such functional groups are well known to bind to Pb and passivate perovskites.30 The other single bonded O atoms have a lone pair of electrons, which can bond with the under-coordinated Pb, in principle. In contrast, CHE contains a terminal hydroxyl (OH) group and only one CO group (Fig. 1b) and was expected to be less hydrophobic than CMA.
O) functional groups at two positions (Fig. 1b) and such functional groups are well known to bind to Pb and passivate perovskites.30 The other single bonded O atoms have a lone pair of electrons, which can bond with the under-coordinated Pb, in principle. In contrast, CHE contains a terminal hydroxyl (OH) group and only one CO group (Fig. 1b) and was expected to be less hydrophobic than CMA.
 | ||
| Fig. 1 (a) Depiction of the method used to prepare the perovskite films. CBZ is chlorobenzene. (b) Structures of CMA and CHE. (c) UV-visible spectra of water or CBZ solutions containing CMA or CHE. Each additive was placed in CBZ/water mixtures, shaken thoroughly and the solvents allowed to phase separate prior to analysis of each layer. The calculated CBZ/water partition coefficients (PCBZ/water) are shown. (d) Water contact angle results for perovskite films prepared using CMA or CHE. The concentrations of the latter (wt%) are shown. (e) Water contact angles from (d). | ||
There have only been two reports of coumarins used in PSCs to the best of our knowledge. Chen et al. used the coumarin dye 8GFF (Disperse Yellow 82) as an additive for PSCs and reported a best PCE of 19.16%.31 Liu et al. included Coumarin 343 dye in their devices and achieved a champion PCE of 20.9%.32 8GFF and Coumarin 343 have UV-visible maximum absorption values (λmax) at wavelengths of 43033 and 446 nm,34 respectively. In contrast, the coumarins selected for this investigation absorb in the UV region of the spectrum where photon-to-current conversion by the perovskite is negligible. This enables CHE and CMA to be used at much higher concentrations than Coumarin 34332 without significantly contributing to parasitic absorption.
In this study we show that CMA is more hydrophobic than CHE and that this difference, together with relative strengths of bonding interactions, strongly affect the optoelectronic and device properties of the perovskite films and PSCs. Both coumarins increase the PCE of the PSCs. The champion CMA modified device achieves a high open circuit voltage of 1.15 V and a PCE of 21.28%. The champion CHE modified device had a PCE of 21.14%. In contrast, the best additive-free device had a PCE of 19.15%. Inclusion of the CMA or CHE into the PSC strongly increased device stability, with CMA providing the greatest stability. The CMA-based device has a very high shelf-life stability at room temperature with 98 days being required to reach 80% of the initial PCE (t80). In contrast the t80 values for PSCs containing CHE or no additive are 38 days and 14 days, respectively. The results of this study show that increasing the hydrophobicity of a precursor-soluble CAA such as a coumarin via substitution is beneficial for both PSC efficiency and stability.
Results and discussion
Measurements on hydrophobic properties
A solvent partition measurement was developed to demonstrate the difference in the hydrophobicities of CMA and CHE. Accordingly, CMA or CHE were added to a solvent mixture containing equal volumes of chlorobenzene (CBZ) and water. Each mixture was shaken and then the CBZ and water phases were allowed to separate. The concentration of each additive in the CBZ and water phase was measured using UV-visible spectroscopy for each phase (Fig. 1c) as well as spectra measured (Fig. S1, ESI†) for calibration curve construction (Fig. S2, ESI†). The ratio of the additive concentrations in CBZ to that in water is the CBZ/water partition coefficient (PCBZ/water). The PCBZ/water values for CMA and CHE are 46.6 and 5.58 (Fig. 1c), respectively. These data demonstrate that CMA is more hydrophobic than CHE. This trend is confirmed by the calculated logarithms of the octanol/water partition coefficients for CMA and CHE which are 3.08 and 1.54, respectively.35Fig. 1c also shows that CMA and CHE have λmax values of 320 nm, which implies that these CAAs will not contribute significantly to parasitic absorption in PSCs.Perovskite films were prepared by adding CMA or CHE to the precursor solution prior to spin coating as depicted in Fig. 1a. Fig. 1d and e show that the water contact angle on the perovskite surfaces increases from 55° to 72° when the CMA concentration increases from 0 to 1.0%. This trend indicates that CMA increases the perovskite surface hydrophobicity. In contrast, the contact angle decreases from 55° to 49° when the CHE concentration in the precursor solutions increases from 0 to 1.0%. In the latter case, the perovskite surface becomes more hydrophilic. Furthermore, CMA has a methacrylate group (Fig. 1b) which is polymerizable in principle. An 1H NMR spectroscopy investigation (Fig. S3, ESI†) showed that CMA did not undergo polymerization and behaved as a single molecule additive under the conditions used in this study (see discussion and text in the ESI†). The 13C NMR spectra for CMA and CHE are also shown in Fig. S4 (ESI†).
Device performance and stability
Planar n–i–p PSCs were constructed (Fig. 2a) to investigate the effect of introducing CMA or CHE in the precursor solution on performance. The J–V curves for the champion devices are shown in Fig. 2b. We investigated additive concentrations of 0.3, 0.5 and 1.0%. The champion CMA- and CHE-based devices were both prepared using 0.3% of additive and they had a PCE of 21.28% and 21.14%, respectively. In contrast, the champion control device PCE was 19.15% (The photovoltaic parameters measured are shown in Table S1, ESI†). The best PCE reported in this study is close to the average of the best PCE values reported for PSCs prepared using similar perovskite compositions as shown in Table S2 (ESI†). Our devices are not designed for maximizing the PCE, but to evaluate the effects of two structurally related CAAs on efficiency and stability. The stabilized power outputs and the short-circuit current density (Jsc) data (Fig. 2c) agree with those obtained from J–V measurements (J–V curves and stabilized power outputs are shown for all of the devices in Fig. S5a and b of the ESI†). EQE spectra were measured for the devices (Fig. S5c, ESI†) and the integrated Jsc values calculated from the respective EQE data agree with the Jsc values from the J–V curves to within 10%. We calculated the average EQE values from the spectra (Fig. S5d, ESI†) which show maximum values for the devices containing 0.3% CMA and CHE. Furthermore, the average EQE values are highest for the CMA-based devices. Box plots for the PCE data for all of the devices are shown in Fig. 2d (Box plots for Voc, Jsc and FF are shown in Fig. S6, ESI†). Overall, these data confirm that 0.3% is the optimum concentration for both CMA and CHE and that both coumarins improve the PCE.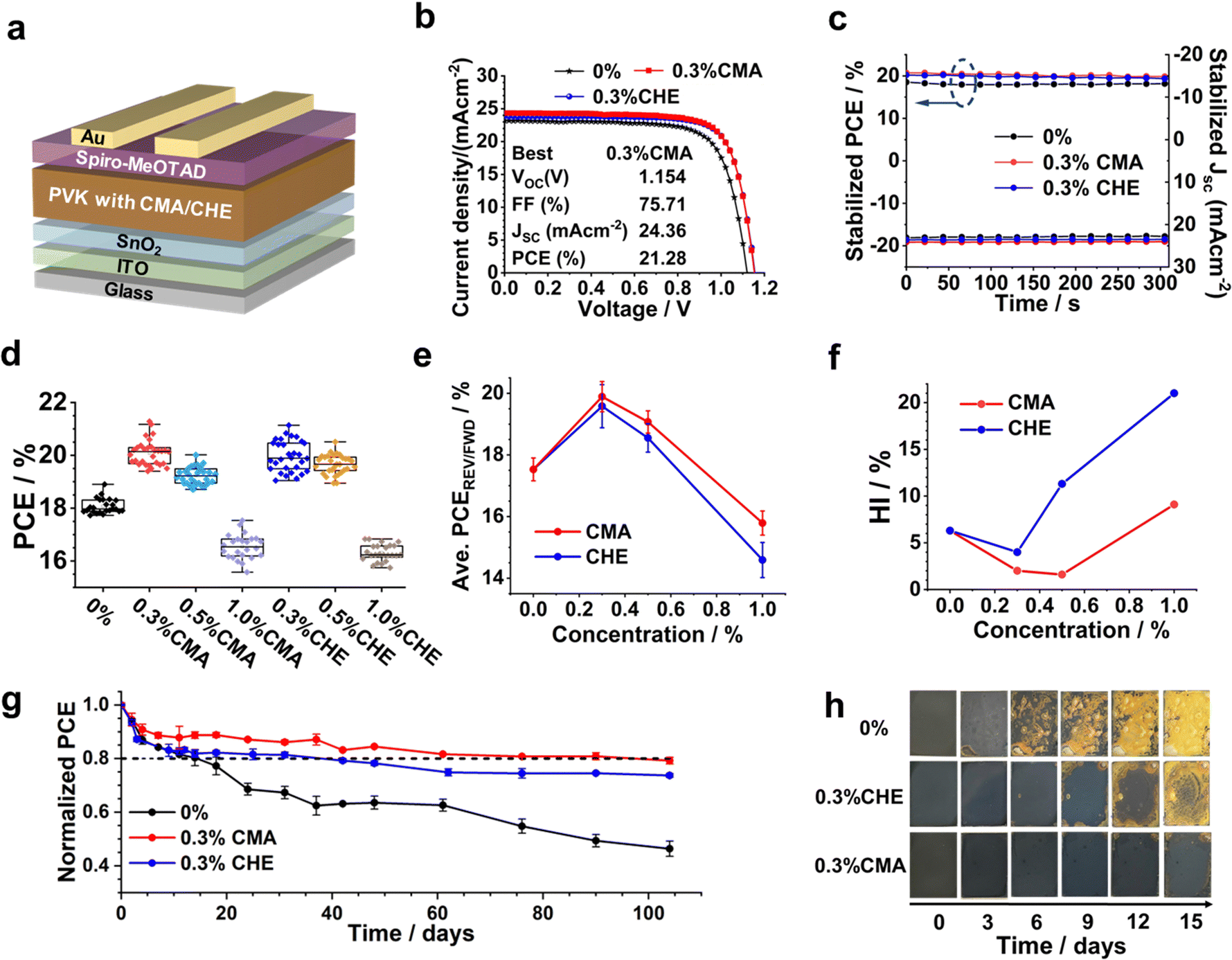 | ||
| Fig. 2 (a) Device architecture used. (b) J–V curves of champion devices for 0% and 0.3% CMA and 0.3% CHE devices. Parameters for the best device from the study are shown. (c) Stabilized power output and photocurrent for 0% and the 0.3% devices measured at the maximum power point. (d) Box plots for all of the PCE data obtained using the reverse sweeps. (e) Average PCEs using data from the reverse and forward sweeps. (f) Hysteresis index values for the devices. (See also Table S1, ESI.†) (g) Shelf-life stability of non-encapsulated devices stored in air at room temperature at 45% RH in the dark. (h) Photographs from a moisture stability challenge study for different perovskite films stored in the dark at a RH of ∼90% at 25 °C for 15 days. Film dimensions: 15 mm × 20 mm. | ||
Whilst the use of 0.3% CMA and CHE additives gives improved PCE, Voc, FF and Jsc values compared to the 0% system (Fig. 2d and Fig. S6, ESI†), the differences between the performance of the devices prepared using the two additives is difficult to distinguish at a glance when only the reverse sweep data are considered (Fig. 2d). Therefore, we show the average PCE values that include the forward and reverse data in Fig. 2e. Fig. S7 (ESI†) shows the values for Voc, FF and Jsc calculated using both forward and reverse sweep data. These data show that the average PCE, Voc and Jsc values are larger for the CMA devices compared to the CHE devices. This is due in part to the much higher hysteresis index (HI) values for the CHE devices as shown in Fig. 2f. The HI values are 4.0% for the 0.3% CHE device and 6.3% for the 0% control device. Whereas, the HI is only 2.0% for the 0.3% CMA device. Furthermore, the HI is a minimum of 1.6% for the 0.5% CMA device. Hence, a pronounced decrease of the mobile ion concentration occurs for PSCs containing low CMA concentrations.36,37
We investigated the shelf-life stability of the highest performing systems (0.3% CMA and 0.3% CHE) as well as the control (see Fig. 2g). The device stability is characterized in terms of the time required for the PCE to decrease to 80% of the initial PCE (i.e., t80). The values of t80 for the 0%, 0.3% CHE and 0.3% CMA devices are 14, 38 and 98 days, respectively, indicating that both coumarins increased device stability. Notably, CMA was by far the most effective at increasing device stability. The thermal stability for these devices at 85 °C was also investigated (see Fig. S8, ESI†). The t80 values for the 0%, 0.3% CHE and 0.3% CMA devices at 85 °C are ∼5, 15 and much greater than 30 days, respectively. These results demonstrate improved thermal stability using the coumarins and, especially, CMA. We also investigated the room temperature moisture stability of the perovskite films at 90% RH. Images for the films are shown in Fig. 2h and UV-visible spectra recorded for these films are shown in Fig. S9, ESI.† Both coumarins improved the moisture stability compared to the control and the CMA film had the best moisture stability. The stability of the devices was also studied using 100 mW cm−2 illumination in laboratory air without encapsulation. The data (Fig. S10, ESI†) show that the stability of the devices decreased in the order 0.3% CMA > 0.3% CHE > 0% (control). Indeed, the degradation rate for the 0.3% CMA device was less than half of that of the control (Fig. S10b, ESI†).
Top-view SEM images for the films are shown in Fig. S11a–g (ESI†). There is a slight increase of the average grain size from 395 nm (for the additive-free, 0% film) to 417 and 435 nm, for the 0.3% CHE and 0.3% CMA films, respectively. The grain size subsequently decreases for 0.5% and 1.0% concentrations of both CMA and CHE (Fig. S12, ESI†). The grain size distributions are shown in Fig. S13 of the ESI.† Generally, the widths of the size distributions are smaller for the CMA samples than those containing CHE which may indicate that CMA was more effective in producing uniform grain growth. The perovskite grain size increases with decreasing nucleation density.38 Therefore, the use of 0.3% CMA decreases nucleation density. The cross-sectional images of PSCs prepared with different concentrations of CMA/CHE are shown in Fig. S14a–g (ESI†). The perovskite film thickness increases slightly from 490 nm for the 0% control to 503 nm and 515 nm, respectively, for the 0.3% CHE- and CMA-based systems (Fig. S15, ESI†), before decreasing again for the 1.0% systems. The perovskite films prepared using 0.3% CMA or CHE have well-defined single grains in the vertical direction that extend from the HTM to the ETM. These large grains are more prevalent for the CMA films and benefit charge transport. The perovskite layers prepared using 1.0% of CMA or CHE have noticeably smaller grains in the vertical direction and, hence, more grain boundaries. This would be expected to increase defect density and decrease device PCE as shown in Fig. 2d.
UV-visible absorption spectra of perovskite films with different concentrations of CMA or CHE additives are shown in Fig. S16a (ESI†). The absorbance measured at 733 nm increases with additive concentration (Fig. S16b, ESI†) and is highest for the 0.3% CMA system before decreasing at higher CMA concentrations. A similar trend is apparent for the CHE systems. However, the absorbance values are higher for the CMA films compared to the CHE-based systems. These trends follow from the film thickness values (Fig. S15, ESI†) and grain size (Fig. S12, ESI†). Hence, 0.3% CMA most effectively increased light harvesting due to thicker films with larger grains.
To investigate potential passivation of the perovskite by the coumarins, steady state PL spectra were recorded for the films (see Fig. 3a and Fig. S17a, ESI†). The PL intensity reached a maximum value (Fig. S17b, ESI†) and the wavelength at the maximum PL intensity (λmax) was a minimum value (Fig. S17c, ESI†) for the 0.3% CMA and 0.3% CHE films. Both of these trends indicate passivation of defects39 occurred with CMA and CHE. As explained by Salado et al.39 a passivating additive can decrease the shallow trap density at the grain surface which causes a blue shift. Notably, the PL maximum was highest for the 0.3% CMA system. The lifetime of the charge carriers was probed using time-resolved PL (TRPL) spectroscopy. Data are shown in Fig. 3b and Fig. S17d (ESI†). The equation used to fit the TRPL data is40,41
 | (1) |
 | ||
| Fig. 3 (a) Steady-state and (b) time-resolved PL spectra for selected perovskite films. (c) Variation of the decay time with CMA or CHE concentration. The variation of the (d) open-circuit voltage and (e) short-circuit current density with incident light intensity. (f) Data from SCLC measurements for electron only devices. (g) J1/2vs. V plots for the electron-only devices using the data from (f). (h) Cut-off region (hν = 21.22 eV) and valence band edge region UPS spectra for perovskite films prepared with 0%, 0.3% CMA and 0.3% CHE. (i) Energy level diagrams for selected systems (see text). Ecbm, EF and Evbm are the conduction band minimum, Fermi level and valence band maximum energies, respectively. | ||
The parameter τ1 is due to fast charge carrier quenching by Shockley–Read–Hall (SRH) recombination.43,44 The decay time τ2 is the slow decay process from defect-assisted charge recombination at the surface and/or grain boundaries.45 The average carrier lifetime (τavg) is calculated using:46
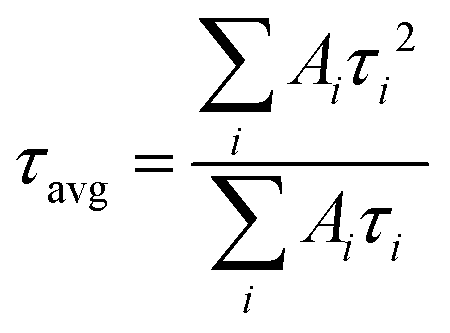 | (2) |
To further explore the charge recombination mechanism light intensity-dependent measurements for Voc and Jsc were performed for the 0%, 0.3% CMA and 0.3% CHE devices. The light intensity dependence of Voc (Fig. 3d) was analyzed using:47
 | (3) |
To quantify the trap density within the films space-charge-limited current (SCLC) analysis was performed using electron-only (ITO/SnO2/perovskite/PC61BM/Au) devices. The SCLC curves (Fig. 3f) consist of the ohmic contact region (with voltage exponent, n = 1), Child's law region (n = 2), and trap-filled limit region (n > 3).51Fig. 3f shows the dark current–voltage characteristics of representative devices and the trap-filled limit voltage (VTFL) values. The trap density (Nt) is calculated using52
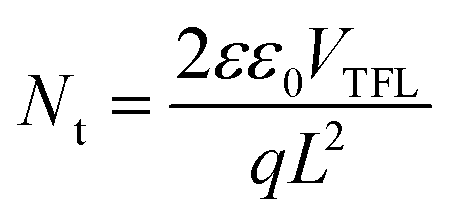 | (4) |
The electron mobility (μe) is calculated from the n = 2 region of the data shown in Fig. 3f using55
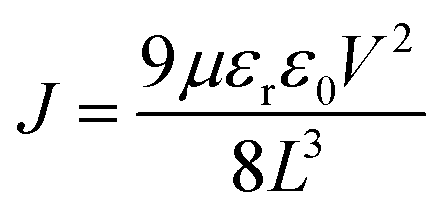 | (5) |
To further probe the effects of the coumarins on perovskite energy levels UPS data were measured for the 0%, 0.3% CHE and 0.3% CMA films (Fig. 3h). Tauc plots for the films (Fig. S19, ESI†) provided the band gaps that were used for the calculations of the energy levels. Fig. 3i shows the inclusion of 0.3% CMA leads to the upward shift of the Fermi level (EF) and the device becoming increasingly n-type. An increase of the valence band maximum (Evbm) for the 0.3% CMA system also occurs indicating better energy level alignment between the perovskite and Spiro-OMeTAD, which should improve carrier extraction. Well-matched energy levels may reduce the energy offset for hole transfer,57 which in turn, can increase Voc.58,59 and is consistent with the Voc values shown in Fig. S6 and S7 of the ESI.† However, it is noted that such an effect is not always observed.60 Furthermore, there is no correlation between the photocurrents for the three systems (Table S1, ESI†) and the valence band offsets between perovskite and Spiro. It follows that the energy level changes that are apparent in Fig. 3i had no significant effect on charge transfer between the perovskite/Spiro interface. This finding is consistent with the work of Belisle et al.60
Atomistic insights into coumarin–perovskite interactions
The results above consistently show that both coumarins passivate the perovskite, which implies strong interactions between each of the coumarins and the perovskite. To gain atomistic insights into such coumarin–perovskite interactions we employed DFT calculations (see the Experimental section for computational methods). Since the precise molecular structures of these CMA and CHE coumarins are not available, we first examined different conformations for each of these additives. The lowest energy structures for CMA and CHE were determined and are shown in Fig. 4a and b, respectively. The most energetically stable CMA conformation is planar while the CHE conformation is slightly bent. More details regarding CMA are provided in Fig. S20 and Table S4 (ESI†). We found that CMA has a higher calculated dipole moment (6.9 D) compared to CHE (6.3 D).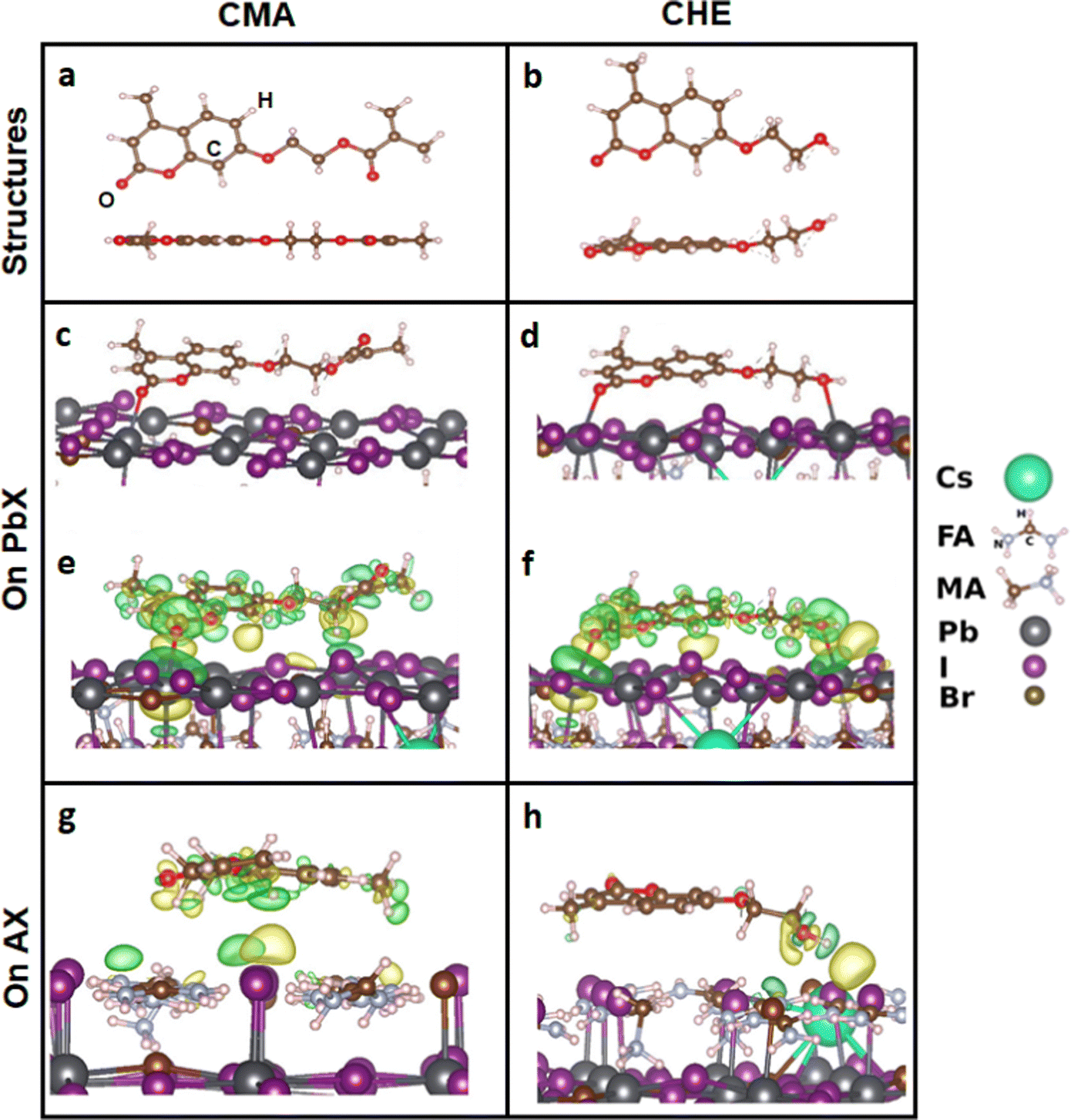 | ||
| Fig. 4 Lowest energy structures for (a) CMA and (b) CHE. The formation of Pb–O bonds (c and d) and charge transfer surfaces (e and f) for CMA and CHE on PbX (X = I or Br) perovskite surfaces. (g) and (h) show bonding to the AX surfaces. The yellow and green regions are electron-rich and electron-poor orbitals, respectively. | ||
Next, we modelled the interaction of the most favourable CMA and CHE structures on the perovskite surface. We considered five different configurations for the adsorbed CMA or CHE on two different terminations of the (001) perovskite surface: one with PbI/Br (Pb/X) termination and the second with FACs/I/Br (A/X) termination. (X represents I and/or Br; whereas, A corresponds to FA and/or Cs.) We focused on the (001) surface as it has been shown to be one of the most stable and studied perovskite halide surfaces.61 The interface models used in this study are shown in Fig. S21 and S22 of the ESI,† and the calculated relative energies of the configurations with respect to the most favourable configuration on each surface are shown in Table S5 (ESI†).
The results show that the arrangement of CMA on the Pb/X terminated surface with the lowest energy, and hence the most favorable, is Configuration 2. Overall analysis indicates that Configuration 2 is the most favored arrangement of these CMA and CHE coumarin additives on the perovskite films and is shown in Fig. 4c and d. This result can be rationalised by the large contact area that Configuration 2 confers when CMA or CHE are in this partly planar arrangement on the perovskite surface compared to the other configurations. This degree of contact allows greater favourable interactions between the atomic species of the coumarins and those of the perovskite surfaces. We mention that the shape of the CMA additive is no longer fully planar after its adsorption on the surface due to the local strain imposed by the bonding interactions between the atomic species of the coumarin and the perovskite.
We have also computed and analyzed the charge density difference between the most stable configuration of the coumarins and the perovskite surfaces, as shown in Fig. 4. On the Pb/X terminated surface, the charge density distribution is mainly found between the oxygen atoms of the coumarins and Pb atoms of the perovskite that are close to each other (Fig. 4e and f), indicating bond formation between these species. Indeed, the Pb–O bond distances are found to be 2.47 Å for CMA and 2.51/2.86 Å for CHE. On the A/X terminated surface, the charge density difference shows that the coumarins essentially interact with the A-site organic cations including weak hydrogen bonding (Fig. 4g and h). The calculated binding energies for CMA/CHE on the Pb/X and A/X terminated surfaces are found to be −1.89/−1.84 eV and −1.23/−0.88 eV, respectively, indicating that the coumarins bind most strongly to the Pb/X inorganic cage. The data also show that charge transfer occurs most strongly from CMA to the perovskite, which correlates with the change in the Fermi level observed experimentally (Fig. 3i). Therefore, these findings show that the preferential planar arrangement of CMA and CHE additives can indeed passivate the perovskite surface via strong Pb/X surface-coumarin interactions including Pb–O bond formation, which minimize the defect density at the surface. In addition, both CMA and CHE will likely prevent the interaction of the perovskite with air and moisture, which will confer a better resistance to the ambient environment, enhancing the film stability.
X-Ray photoelectron spectroscopy (XPS) characterization was used to investigate the interactions between the coumarins and perovskite. Fig. 5a shows that the under-coordinated Pb0 signal that is evident for the 0% system is either absent or mostly absent for the 0.3% CHE and 0.3% CMA systems. (A small fraction could be buried under the tails in the peaks for CHE and CMA.) Furthermore, after CMA inclusion the peaks for Pb 4f7/2 and Pb 4f5/2 at 138.2 and 143.1 eV shift to lower binding energies by 0.36 eV. In the case of CHE, the shift also occurred, but was only by 0.11 eV. These results strongly imply: (1) binding of both CME and CHE to Pb and (2) a stronger binding of CMA with Pb compared to CHE. These results further support the DFT modelling results on coumarin–perovskite interactions in Fig. 4 and show that both coumarins bind to Pb. Interestingly, the peaks for I 3d3/2 and 3d5/2 (Fig. 5b) shifted to lower binding energy by ∼0.6 eV for CMA. However, no such shift occurred for CHE. This may indicate better coordination of the iodine ions by CMA compared to CHE. Taken together, these data show that CMA can better bind to the perovskite.
 | ||
| Fig. 5 XPS spectra for (a) Pb 4f and (b) I 3d core levels of the control perovskite and films containing CMA or CHE. (c) Digital photographs of various perovskite films after different times of annealing at 70 °C (shown). The concentrations of CMA and CHE were 1.0%. (d) Variation of the (001) peak full width at half maximum height with CMA and CHE concentration. FTIR spectra showing the (e) CO and (f) O–H stretching regions. | ||
An interesting question concerns the distribution of the CAAs within the perovskite films. We addressed this question in two ways. Firstly, we used high angle annular dark field (HAADF) scanning transmission electron microscopy (STEM) imaging with energy dispersive X-ray spectroscopy (EDS) to probe the elemental distribution within the perovskite films. The films were prepared under dilute precursor conditions using 1.0% CMA or in the absence of added CMA. Because CMA contains five oxygen atoms (Fig. 1b) we used oxygen elemental mapping to understand the CMA distribution. The imaging of the 1.0% CMA sample show oxygen is present (Fig. S23d, ESI†) and located on the perovskite grains (Fig. S23e, ESI†). In contrast, no oxygen could be detected for the control system (Fig. S24d, ESI†). These data support our view that CMA (and CHE) adsorbed onto the grain surfaces. Furthermore, the PL data measured for the buried interface (Fig. S18, ESI†) imply that CMA and CHE are also present at, or in the vicinity of, the buried perovskite interface. Together these data indicate that the CAA additives are distributed at the surfaces of the grains throughout the films.
Additional evidence for perovskite-coumarin binding
Crystallization rate tests were conducted to determine whether CMA or CHE could control perovskite crystal growth (see Fig. 5c). The delay in achieving the black colour shows that the crystallization time was increased from 6 s for the 0% control film to 20 s in the presence of 1.0% CHE or 1.0% CMA. Hence, both CMA and CHE decrease the perovskite crystallization rate, which implies that both coumarins bind to the growing perovskite. The X-ray scattering patterns for the as-prepared films (Fig. S25, ESI†) show an increase in PbI2 conversion when the coumarins are present. Moreover, the full-width at half-maximum height (FWHM) is a minimum for the 0.3% CHE and 0.3% CMA systems (Fig. 5d). This trend agrees with the grain size changes recorded from SEM (Fig. S12, ESI†). In addition, the (001) scattered intensity is highest for the 0.3% CMA-based film (Fig. S25b, ESI†). These data provide evidence for an interaction of CMA and CHE wherein the crystallization rate is decreased and the crystal size increased.We used FTIR spectroscopy to further probe the interaction between PbI2 and CHE as well as CMA. The spectra recorded over the whole FTIR range are shown in Fig. S26, ESI.†Fig. 5e shows that the stretching vibration of the CO group of CMA occurs at 1714 cm−1. When PbI2 was mixed with CMA, the peak moves to 1708 cm−1. This red-shift is due to CO binding to Pb2+,62,63 and supports the simulation data (Fig. 4c). The position of the CO band for CHE at 1682 cm−1 moves to 1702 cm−1 when mixed with PbI2 which is an indication of a changed environment. Furthermore, the O–H stretch for CHE at 3455 cm−1 shifts to 3438 cm−1 when mixed with PbI2 (Fig. 5f). This indicates a direct interaction between the CHE O–H group and Pb and supports the DFT findings of Fig. 4d.
Underlying mechanism explanation
The results presented have provided insights into the improved performance and stability of the PSCs containing 0.3% CMA or 0.3% CHE as well as the greater overall benefit provided by CMA. There is evidence from FTIR, XPS and DFT that the CO groups from CMA and CHE coordinate to Pb2+. The benefit from this effect is optimum at 0.3% CMA or CHE. However, at concentrations of 0.5 or 1.0%, it is likely that excessive CMA or CHE deposited at the perovskite grains impede charge transport. Moreover, the above data show that CMA more strongly binds to Pb2+ than CHE which is proposed to be due to the higher dipole moment calculated from DFT analysis. The more effective binding by CMA decreases the defect density and recombination, which increased Voc and the PCE.
We applied the Student's t-test to the Jsc data for the CMA and CHE based devices measured at the same concentrations. When data from the reverse and forward sweeps are compared for each system, the analysis shows that the Jsc values for CMA are significantly higher than those for CHE (see discussion in ESI† and Table S6). The Jsc difference contributed to the greater PCE for the CMA-based devices compared to those prepared using CHE in this work.
The contact angle data (Fig. 1e) showed that CMA provided a protective hydrophobic layer. The stability of the films and devices benefits from the enhanced defect passivation of the perovskite surface by CMA and the good moisture resistance afforded by the hydrophobic groups. The moisture stability of perovskites is dependent on access to defects because water can bind firmly to the defect surface through hydrogen bonds, thereby accelerating the degradation.64 Notably, the device containing 0.3% hydrophilic CHE had better moisture stability than the 0% film (Fig. 2h), which is likely to benefits from the bonding of the OH groups in CHE with the perovskite as evidenced from the DFT simulation results. Moreover, the moisture-resistance of the perovskite layer for this system is likely due to the protective barrier affect provided by bound CHE as noted above. Nevertheless, CMA provided the best stability due to stronger bonding to the perovskite and greater hydrophobicity.
An important question emanating from this study is why the PCEs for the CMA devices are only slightly higher than that for those prepared using CHE devices (Fig. 2e) whereas the stabilities for the former systems are far greater than those for the latter (Fig. 2g and h). To address this question we calculated the maximum number of CHE and CMA layers on the top and bottom of a nominal cubic perovskite grain that extended from the ETM to the HTM (see discussion and Fig. S27 in the ESI†). We estimate that there are a maximum of about 6 layers for both 0.3% CMA and 0.3% CHE systems. Accordingly, the relatively insulating nature of CMA and CHE (compared to the perovskite) will tend to impede charge transport to the ETM and HTM. This may be a reason why the Jsc values are not greatly different; although, Jsc is larger for the CMA as discussed above. Moreover, it is the nature of the hydrophobicity differences for these CMA and CHE multilayers that provide a far greater difference to water penetration. The latter will be more difficult for CMA multilayers and provides much stronger stability enhancement for the CMA-based systems.
Conclusions
This study has compared two new CAAs, i.e., CMA and CHE, that are free of parasitic absorption, which enabled them to be added to the precursor solution (simplifying processing) at relatively high concentration compared to related studies32 and revealed the effects of CAA hydrophobicity on PSC performance and stability. Three key results emerge. First, the CO functional group in CMA and CHE coordinates with Pb and passivates defects on the perovskite surface and grain boundaries throughout the perovskite films. This inhibits non-radiative recombination as determined by the PL measurements. Second, the alignment of binding energy levels also improves the extraction of charge-carriers and contributes to the increase of the Voc. These effects contributed to the PCE of 21.28% for the champion CMA-modified devices and 21.14% for the best CHE-modified devices, whereas the additive-free system had a maximum PCE of 19.15%. Third, we also successfully fabricated devices containing hydrophobic CMA with high shelf-life, moisture and thermal stabilities. This system achieved a t80 value of 98 days when stored in ambient air. The increased hydrophobicity of CMA provided a decisive stability advantage.
Overall, our results show that hydrophobic CAAs, such as CMA are good choices for improving PSC device performance. An unexpected result is that CHE also improved device stability even though it increased the perovskite hydrophilicity; such protection was attributed to effective binding of CHE to the perovskite. The study demonstrates that the substituents of CAAs can be used to enhance the performance and stability of PSCs, with the greatest enhancement from substituents that increase the overall hydrophobicity. We believe that selecting CAAs that were soluble in the perovskite precursors enabled these benefits to be conferred throughout the perovskite films.
Experimental
Materials
4-Methylumbelliferone (98%), K2CO3 (99%), CHCl3 (99%), triethylamine (TEA), methacryloyl chloride (97%), 2-bromoethanol (95%), dimethylformamide (DMF, 99.8%), dimethyl sulfoxide (DMSO, 99.9%), CBZ (99.8%), PbBr2 (99.999%), CsI (99.999%), Spiro-OMeTAD (99%), bis(trifluoromethane)sulfonimide lithium salt (LiTFSI, 99.95%), Co(III) TFSI salt (FK 209, 98%), acetonitrile (ACN, 99.9%) and 4-tert-butylpyridine (t-BP, 96%) were all purchased from Sigma-Aldrich. ITO glass substrates, formamidinium iodide (FAI, 99.5%), and methylammonium bromide (MABr, 99.5%) were purchased from Ossila Limited. PbI2 (99.99%) was purchased from Tokyo Chemical Industry UK Ltd. All materials were used as received. SnO2 (15% in H2O) was a colloidal dispersion supplied by Alfa Aesar. All water used was de-ionized and of ultra-high purity.Synthesis of CHE
The synthesis of CHE (7-(2-hydroxyethoxy)-4-methylcoumarin) followed our earlier study.65 Firstly, 4-methylumbelliferone (4.50 g, 22.7 mmol) and K2CO3 (7.00 g 45.4 mmol) was dissolved in dry DMF (45 mL) at 90 °C under an N2 atmosphere for 30 min. Then, 2-bromoethanol (2.52 mL, 34.0 mmol) was slowly added dropwise. The reaction mixture was then refluxed overnight under N2. After cooling to room temperature, the mixture was poured into water at 5 °C (700 mL) and then stored at this temperature for 6 h. The white product was then collected and dried in vacuum at room temperature for 48 h. CHE (4.25 g, 85.0% yield) was stored with a desiccator (over silica gel) in the dark until required. δH (400 MHz, DMSO): 7.72–7.65 (1 H, m, Hd), 7.01–6.95 (2 H, m, Hc,e), 6.21 (1 H, q, J 1.3, Ha), 4.94 (1 H, s), 4.10 (2 H, t, J 4.9, Hf), 3.75 (2 H, t, J 4.9, Hg), 2.40 (3 H, d, J 1.3, Hb).Synthesis of CMA
For the synthesis of CMA (7-(2-methacryloyloxyethoxy)-4-methylcoumarin), CHE (4.00 g, 20.4 mmol) and TEA (4.00 g, 44.2 mmol) were dissolved in CHCl3 (64.0 mL). After stirring for 20 min under N2, methacryloyl chloride (4.00 g, 43.0 mmol) was slowly added dropwise to the stirred solution at 0 °C. The solution was then allowed to warm to room temperature and stirred for a further 18 h. The solution was diluted with CH2Cl2 (200 mL) and washed twice with of brine (200 mL). The solution was then dried over MgSO4 (40 g) overnight. The solution was concentrated and the product recrystallized from ethanol to obtain a white solid. CMA (2.82 g, 48% yield) was stored in a desiccator over silica gel in the dark until required. δH (400 MHz, DMSO): 7.70 (1 H, d, J 8.8, Hd), 7.05 (1 H, d, J 2.5, Hc), 7.01 (1 H, dd, J 8.8, 2.5, He), 6.23 (1 H, d, J 1.3, Ha), 6.04 (1 H, m, J 2, 1.0, Hh), 5.71 (1 H, p, J 1.6, Hh), 4.46 (2 H, m, Hf), 4.38 (2 H, m, Hg), 2.40 (3 H, d, J 1.3, Hb), 1.88 (3 H, t, J 1.0, Hi).Fabrications of perovskite solar cells
The PSCs used here are structured as follows: glass/ITO/SnO2/perovskite/Spiro-OMeTAD/Au. The pervoskite used was Cs0.05(FA0.85MA0.15)0.95Pb(I0.85Br0.15)3. Glass coated with ITO was successively washed in detergent solution, and deionized water, then dried with N2 flow and treated them with UV ozone for 15 min. SnO2 precursor solution (2.67 wt%) was obtained by diluting 15% of the as-supplied colloidal solution. The diluted solution was spin-coated onto the cleaned glass/ITO substrates at 3000 rpm for 30 s and then annealed at 150 °C for 30 min to form a compact SnO2 ETM (∼30 nm thickness). The Glass/ITO/SnO2 substrates were then treated with UV ozone again for 15 min and transferred to a glove box.All operations were carried out in the glove box. The precursor solution (1.4 M) was prepared by dissolving appropriate amounts of CsI (0.013 g, 0.050 mmol), FAI (0.139 g, 0.81 mmol.), MABr (0.016 g, 0.143 mmol.), PbBr2 (0.056 g, 0.153 mmol.) and PbI2 (0.406 g, 0.880 mmol., which was 4 mol% in excess) in DMF/DMSO (v/v 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Different concentrations of CMA or CHE were added directly to the precursor and mixed with stirring at 55 °C for 2 h. To prepare the 0.3% solutions, CHE or CMA (4.0 mg) were mixed with the other solid precursors before adding DMF (568 μL)/DMSO (142 μL). The precursor solution was prepared and subsequently spin-coated in two steps: 1000 rpm for 10 s followed by 6000 rpm for 20 s. Then, 10 s before the end, CBZ (200 μL) was drop-coated as an antisolvent. The substrates were annealed at 110 °C for 50 min to produce perovskite films. The Spiro-OMeTAD solution was prepared by dissolving Spiro-OMeTAD (85.7 mg) and additives in CBZ (1000 μL). Li-TFSI solution (22 μL of 520 mg mL−1 solution in ACN), Co(III) TFSI salt (18 μL of 375 mg mL−1 solution in ACN), and t-BP (32 μL) were added to the Spiro solution. The HTM (∼250 nm thickness) was prepared by spin-coating using 3000 rpm for 30 s. Finally, a gold layer (80 nm) was thermally evaporated on top of the HTL as an electrode.
:1). Different concentrations of CMA or CHE were added directly to the precursor and mixed with stirring at 55 °C for 2 h. To prepare the 0.3% solutions, CHE or CMA (4.0 mg) were mixed with the other solid precursors before adding DMF (568 μL)/DMSO (142 μL). The precursor solution was prepared and subsequently spin-coated in two steps: 1000 rpm for 10 s followed by 6000 rpm for 20 s. Then, 10 s before the end, CBZ (200 μL) was drop-coated as an antisolvent. The substrates were annealed at 110 °C for 50 min to produce perovskite films. The Spiro-OMeTAD solution was prepared by dissolving Spiro-OMeTAD (85.7 mg) and additives in CBZ (1000 μL). Li-TFSI solution (22 μL of 520 mg mL−1 solution in ACN), Co(III) TFSI salt (18 μL of 375 mg mL−1 solution in ACN), and t-BP (32 μL) were added to the Spiro solution. The HTM (∼250 nm thickness) was prepared by spin-coating using 3000 rpm for 30 s. Finally, a gold layer (80 nm) was thermally evaporated on top of the HTL as an electrode.
Characterization
1H and 13C NMR spectra were measured using a B400 Bruker Avance III HD400-Cyan spectrometer. XPS was conducted on an ESCA2SR spectrometer (Scienta Omicron GmbH) using monochromatic Al Kα radiation (1486.6 eV, 25 mA emission at 300 W, 1 mm spot size) at a base vacuum pressure of ∼1 × 10−9 mbar. UPS was characterized by a vacuum UV source producing HeI (21.2 eV, 60 W, FOCUS GmbH) and an Argus electron energy analyzer (Scienta Omicron GmbH) with a bias voltage of 20.2 V and a pass energy of 5.0 eV. SEM images were obtained with Tescan Mira 3 SC. The morphology and chemical composition of the films were characterized by scanning transmission electron microscopy (HR-STEM) and energy dispersive X-ray spectroscopy (EDS) using a Thermo Fisher Titan STEM (G2 80–200) equipped with a Cs probe corrector (CEOS) operating at 200 kV. The TEM samples were prepared by depositing dilute perovskite precursor solutions (0.28 M) in the presence or absence of 1.0 wt% CMA spin coated onto a copper grid coated with an amorphous carbon films on 300 mesh Cu grids. XRD data were acquired with an XRD5 – PANaytical X'Pert Pro X-ray diffractometer. FTIR was recorded using a Nicolet 5700 spectrometer. FTIR samples were prepared by mixing PbI2, CMA or CHE with DMF/DMSO mixture and stirring at 55 °C for 2 h, and then evaporating the solvent at 110 °C to obtain a dry powder. UV-visible spectra were measured using an Agilent Cary 60 UV-Vis spectrophotometer. Steady-state PL was measured by an Edinburgh Instrument FLS980. The excitation wavelength was 470 nm. TRPL data were also measured using the FLS980 and an excitation wavelength of 405 nm. All UV-visible, PL and TRPL spectroscopy measurements were recorded using glass/ITO/SnO2 substrates. For all sample measurements, the light was incident on the film side. Contact angle measurements were performed with a drop of water (60 μL) on the film surface using a Kruss DSA100. J–V curves were generated by a Keithley 2420 source meter simulating AM 1.5G sunlight (100 mW cm−2) in an Abet solar simulator. The instrument was calibrated and corrected for spectral irradiance mismatch using a certified Oriel Si reference cell. The device area is 0.079 cm2. Unless otherwise stated, PCE and other data discussed in this article are from reverse scans. For all stability tests, two devices were used per system. A Keithley 2420 source meter was also used for measuring the SCLC characteristics of electron only devices. A Newport QuantX-300 instrument was used for external quantum efficiency (EQE) measurements. The Long-term light soaking test was performed under full AM 1.5 sun-equivalent white LED arrays. All devices were in ambient air. PCE was measured in the same way as the J–V curves.DFT simulation
We carried out density functional theory (DFT) calculations as implemented in the Quantum ESPRESSO package.66,67 For the reaction energies, the Kohn–Sham wave-functions and energies are calculated with the functional GGA-PBE68,69 for electron exchange and correlation, using a plane-wave basis, with energy and charge density cutoffs of 30 and 300 Ry, respectively. The Grimme dispersion correction DFT-D3,70,71 was used to account for the dispersion corrections. Ultrasoft pseudopotentials are used to describe the core–valence interactions.72 The structural relaxation is performed until the force on each atom is smaller than 0.01 eV Å−1. For the geometry optimization, a Gamma-point and a 4 × 4 × 1 k-point sampling for the Brillouin zone integration are used for the interface and bulk systems, respectively, following the Monkhorst–Pack scheme.73 The bulk system contains 418 atoms to model the stoichiometry (Cs0.05(FA0.85MA0.15)0.95Pb(I0.85Br0.15)3) used in experiment (Fig. S21, ESI†). The perovskite surfaces are obtained by cutting along the 001 direction, with PbI/Br (Pb/X) and FAI/Br (A/X) terminations. To fit in the different configurations of the coumarin structures in the simulation box, the vacuum region was set to at least 16 Å. Such DFT based methods have been applied to related studies on perovskite halide materials.74–77Author contributions
R. Wang was responsible for film and device construction and measurement as well data analysis and contributed to the writing. A. Altujjar helped construct and measure the solar cells and devices for EQE and SCLC as well as conduct of the PL studies. N. Zibouche conducted the DFT studies and helped edit the manuscript. M. S. Islam assisted with DFT interpretation and writing of the manuscript. X. Wang and J. M. Saunders assisted with the NMR experiments and monomer synthesis as well as data interpretation. B. F. Spencer and A. G. Thomas obtained and helped interpret the XPS and UPS data. Z. Jia helped measure and interpret the XRD data. M. Z. Mokhtar assisted with solar cell construction and measurement. R. Cai helped with STEM/HAADF and S. J. Haigh helped with data interpretation. B. R. Saunders conceived of the study, assisted with data interpretation and the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the EPSRC for funding (EP/R020590/1). M. S. Islam and Nourdine Zibouche gratefully acknowledge an EPSRC Grant (EP/R020485/1) and ARCHER2 rescources via membership of the UK's HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/R029431). Electron microscopy access was supported by the Henry Royce Institute for Advanced Materials, funded through EPSRC grants EP/R00661X/1, EP/S019367/1, EP/P025021/1 and EP/P025498/1.References
- J. J. Yoo, G. Seo, M. R. Chua, T. G. Park and Y. Lu,
et al.
, Nature, 2021, 590, 587–593 CrossRef CAS PubMed
.
- J. Tian, Q. Xue, Q. Yao, N. Li and C. J. Brabec,
et al.
, Adv. Energy Mater., 2020, 10, 2000183 CrossRef CAS
- J. Jeong, M. Kim, J. Seo, H. Lu and P. Ahlawat,
et al.
, Nature, 2021, 592, 381–385 CrossRef CAS PubMed
- J. Y. Kim, J.-W. Lee, H. S. Jung, H. Shin and N.-G. Park, Chem. Rev., 2020, 120, 7867–7918 CrossRef CAS PubMed
- J. Huang, Y. Yuan, Y. Shao and Y. Yan, Nat. Rev. Mater., 2017, 2, 1–19 Search PubMed
- Y. Lei, Y. Chen, Y. Gu, C. Wang and Z. Huang,
et al.
, Adv. Mater., 2018, 30, 1705992 CrossRef PubMed
- M. Shao, T. Bie, L. Yang, Y. Gao and X. Jin,
et al.
, Adv. Mater., 2022, 34, 2107211 CrossRef CAS PubMed
- National Renewable Energy Laboratory, “Best research-cell efficiency chart” (2022), http://www.nrel.gov/pv/cell-efficiency.html.
- Z. Li, B. Li, X. Wu, S. A. Sheppard and S. Zhang,
et al.
, Science, 2022, 376, 416–420 CrossRef CAS PubMed
- T. Bu, J. Li, H. Li, C. Tian and J. Su,
et al.
, Science, 2021, 372, 1327–1332 CrossRef CAS PubMed
- Q. Jiang, J. Tong, Y. Xian, R. A. Kerner and S. P. Dunfield,
et al.
, Nature, 2022, 611, 278–283 CrossRef CAS PubMed
- Y. Wu, Q. Wang, Y. Chen, W. Qiu and Q. Peng, Energy Environ. Sci., 2022, 15, 4700–4709 RSC
- D. Liu, D. Luo, A. N. Iqbal, K. W. P. Orr and T. A. S. Doherty,
et al.
, Nat. Mater., 2021, 20, 1337–1346 CrossRef CAS PubMed
- S. Chen, X. Dai, S. Xu, H. Jiao and L. Zhao,
et al.
, Science, 2021, 373, 902–907 CrossRef CAS PubMed
- Y. Wang, T. Wu, J. Barbaud, W. Kong and D. Cui,
et al.
, Science, 2019, 365, 687–691 CrossRef CAS PubMed
- S. Bai, P. Da, C. Li, Z. Wang and Z. Yuan,
et al.
, Nature, 2019, 571, 245–250 CrossRef CAS PubMed
- Y. Deng, S. Xu, S. Chen, X. Xiao and J. Zhao,
et al.
, Nat. Energy, 2021, 6, 633–641 CrossRef CAS
- P. Calado, A. M. Telford, D. Bryant, X. Li and J. Nelson,
et al.
, Nat. Commun., 2016, 7, 1–10 Search PubMed
- D. W. Ferdani, S. R. Pering, D. Ghosh, P. Kubiak and A. B. Walker,
et al.
, Energy Environ. Sci., 2019, 12, 2264–2272 RSC
- Y. Lao, S. Yang, W. Yu, H. Guo and Y. Zou,
et al.
, Adv. Sci., 2022, 9, 2105307 CrossRef CAS PubMed
- W. Zhang, X. Li, X. Feng, X. Zhao and J. Fang, Chem. Eng. J., 2021, 412, 128680 CrossRef CAS
- Y. Lin, L. Shen, J. Dai, Y. Deng and Y. Wu,
et al.
, Adv. Mater., 2017, 29, 1604545 CrossRef PubMed
- H. Li, J. Shi, J. Deng, Z. Chen and Y. Li,
et al.
, Adv. Mater., 2020, 32, 1907396 CrossRef CAS PubMed
- F. Wei, B. Jiao, H. Dong, J. Xu and T. Lei,
et al.
, J. Mater. Chem. A, 2019, 7, 16533–16540 RSC
- B. Li, C. Zheng, H. Liu, J. Zhu and H. Zhang,
et al.
, ACS Appl. Mater. Interfaces, 2016, 8, 27438–27443 CrossRef CAS PubMed
- P.-L. Qin, G. Yang, Z.-W. Ren, S. H. Cheung and S. K. So,
et al.
, Adv. Mater., 2018, 30, 1706126 CrossRef PubMed
- F. Wu, W. Gao, H. Yu, L. Zhu and L. Li,
et al.
, J. Mater. Chem. A, 2018, 6, 4443–4448 RSC
- C. P. Kabb, C. S. O’Bryan, C. C. Deng, T. E. Angelini and B. S. Sumerlin, ACS Appl. Mater. Interfaces, 2018, 10, 16793–16801 CrossRef CAS PubMed
- L. Meng, C. Sun, R. Wang, W. Huang and Z. Zhao,
et al.
, J. Am. Chem. Soc., 2018, 140, 17255–17262 CrossRef CAS PubMed
- L. Chao, Y. Xia, X. Duan, Y. Wang and C. Ran,
et al.
, Joule, 2022, 6, 2203–2217 CrossRef CAS
- P. Chen, X. Yin, W.-H. Chen, L. Song and P. Du,
et al.
, Sustainable Energy Fuels, 2021, 5, 2266–2272 RSC
- S. Liu, R. Chen, X. Tian, Z. Yang and J. Zhou,
et al.
, Nano Energy, 2022, 106935 CrossRef CAS
- A. A. Atta-Eyison, J. Mater. Sci. Chem. Eng., 2020, 8, 11–19 CAS
- G. A. Reynolds and K. H. Drexhage, Optics Commun., 1975, 13, 222–225 CrossRef CAS
- SciFinder, American Chemical Society. Chemical Abstracts Service.
- C. Eames, J. M. Frost, P. R. F. Barnes, B. C. O’Regan and A. Walsh,
et al.
, Nat. Commun., 2015, 6, 7497 CrossRef CAS PubMed
- W. Zhu, S. Wang, X. Zhang, A. Wang and C. Wu,
et al.
, Small, 2022, 18, 2105783 CrossRef CAS PubMed
- S. S. Mali, J. V. Patil, D. W. Park, Y. H. Jung and C. K. Hong, Cell Rep. Phys. Sci., 2022, 100906 CrossRef CAS
- M. Salado, A. D. Jodlowski, C. Roldan-Carmona, G. de Miguel and S. Kazim,
et al.
, Nano Energy, 2018, 50, 220–228 CrossRef CAS
- Z. Xiong, X. Chen, B. Zhang, G. O. Odunmbaku and Z. Ou,
et al.
, Adv. Mater., 2022, 34, 2106118 CrossRef CAS PubMed
- R. Ma, J. Zheng, Y. Tian, C. Li and B. Lyu,
et al.
, Adv. Funct. Mater., 2022, 32, 2105290 CrossRef CAS
- Z. Wu, M. Jiang, Z. Liu, A. Jamshaid and L. K. Ono,
et al.
, Adv. Energy Mater., 2020, 10, 1903696 CrossRef CAS
- A. Kiligaridis, P. A. Frantsuzov, A. Yangui, S. Seth and J. Li,
et al.
, Nat. Commun., 2021, 12, 1–13 CrossRef PubMed
- V. Sarritzu, N. Sestu, D. Marongiu, X. Chang and S. Masi,
et al.
, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed
- T. Wu, R. Zhao, J. Qiu, S. Wang and X. Zhang,
et al.
, Adv. Funct. Mater., 2022, 32, 2204450 CrossRef CAS
- F. Li, X. Deng, F. Qi, Z. Li and D. Liu,
et al.
, J. Amer. Chem. Soc., 2020, 142, 20134–20142 CrossRef CAS PubMed
- D. Glowienka and Y. Galagan, Adv. Mater., 2022, 34, 2105920 CrossRef CAS PubMed
- T. Liu, J. Guo, D. Lu, Z. Xu and Q. Fu,
et al.
, ACS Nano, 2021, 15, 7811–7820 CrossRef CAS PubMed
- T. Singh, M. Ikegami and T. Miyasaka, ACS Appl. Energy Mater., 2018, 1, 6741–6747 CrossRef CAS
- D. Yang, R. Yang, K. Wang, C. Wu and X. Zhu,
et al.
, Nat. Commun., 2018, 9, 1–11 CrossRef PubMed
- Y. Lv, R. Yuan, B. Cai, B. Bahrami and A. H. Chowdhury,
et al.
, Angew. Chem., 2020, 132, 12067–12074 CrossRef
- Y. Huang, L. Li, Z. Liu, H. Jiao and Y. He,
et al.
, J. Mater. Chem. A, 2017, 5, 8537–8544 RSC
- Q. Han, S. H. Bae, P. Sun, Y. T. Hsieh and Y. Yang,
et al.
, Adv. Mater., 2016, 28, 2253–2258 CrossRef CAS PubMed
- H. Min, M. Kim, S.-U. Lee, H. Kim and G. Kim,
et al.
, Science, 2019, 366, 749–753 CrossRef CAS PubMed
- P. Li, Y. Zhang, C. Liang, G. Xing and X. Liu,
et al.
, Adv. Mater., 2018, 30, 1805323 CrossRef PubMed
- J. M. Ball and A. Petrozza, Nat. Energy, 2016, 1, 1–13 Search PubMed
- X. Zheng, H. Chen, Q. Li, Y. Yang and Z. Wei,
et al.
, Nano Lett., 2017, 17, 2496–2505 CrossRef CAS PubMed
- Z. Guo, A. K. Jena, G. M. Kim and T. Miyasaka, Energy Environ. Sci., 2022, 15, 3171–3222 RSC
- S. N. Habisreutinger, N. K. Noel, H. J. Snaith and R. J. Nicholas, Adv. Energy Mater., 2017, 7, 1601079 CrossRef
- R. A. Belisle, P. Jain, R. Prasanna, T. Leijtens and M. D. McGehee, ACS Energy Lett., 2016, 1, 556–560 CrossRef CAS
- J. Xue, R. Wang and Y. Yang, Nat. Rev. Mater., 2020, 5, 809–827 CrossRef CAS
- C. Ma and N.-G. Park, ACS Energy Lett., 2020, 5, 3268–3275 CrossRef CAS
- T. Zhu, L. Shen, S. Xun, J. S. Sarmiento and Y. Yang,
et al.
, Adv. Mater., 2022, 34, 2109348 CrossRef CAS PubMed
- M. M. Byranvand and M. Saliba, Sol. RRL, 2021, 5, 2100295 CrossRef CAS
- D. Lu, M. Zhu, S. Wu, W. Wang and Q. Lian,
et al.
, Polym. Chem., 2019, 10, 2516–2526 RSC
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau and M. Buongiorno Nardelli,
et al.
, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra and R. Car,
et al.
, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov and G. E. Scuseria,
et al.
, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef PubMed
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef
- B. Charles, J. Dillon, O. J. Weber, M. S. Islam and M. T. Weller, J. Mater. Chem. A, 2017, 5, 22495–22499 RSC
- K. Jayanthi, I. Spanopoulos, N. Zibouche, A. A. Voskanyan and E. S. Vasileiadou,
et al.
, J. Am. Chem. Soc., 2022, 144, 8223–8230 CrossRef CAS PubMed
- L. Lanzetta, T. Webb, N. Zibouche, X. Liang and D. Ding,
et al.
, Nat. Commun., 2021, 12, 2853 CrossRef CAS PubMed
- O. J. Weber, D. Ghosh, S. Gaines, P. F. Henry and A. B. Walker,
et al.
, Chem. Mater., 2018, 30, 3768–3778 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee00247k |
| This journal is © The Royal Society of Chemistry 2023 |