
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA practical perspective on the potential of rechargeable Mg batteries†
J. Alberto
Blázquez
 *a,
Rudi R.
Maça
a,
Olatz
Leonet
a,
Eneko
Azaceta
a,
Ayan
Mukherjee
b,
Zhirong
Zhao-Karger
cd,
Zhenyou
Li
cd,
Aleksey
Kovalevsky
e,
Ana
Fernández-Barquín
a,
Aroa R.
Mainar
a,
Piotr
Jankowski
f,
Laurin
Rademacher
g,
Sunita
Dey
h,
Siân E.
Dutton
i,
Clare P.
Grey
h,
Janina
Drews
g,
Joachim
Häcker
g,
Timo
Danner
cg,
Arnulf
Latz
cgj,
Dane
Sotta
k,
M. Rosa
Palacin
l,
Jean-Frédéric
Martin
k,
Juan Maria García
Lastra
f,
Maximilian
Fichtner
cd,
Sumana
Kundu
mn,
Alexander
Kraytsberg
mn,
Yair
Ein-Eli
mn,
Malachi
Noked
*b and
Doron
Aurbach
*b
*a,
Rudi R.
Maça
a,
Olatz
Leonet
a,
Eneko
Azaceta
a,
Ayan
Mukherjee
b,
Zhirong
Zhao-Karger
cd,
Zhenyou
Li
cd,
Aleksey
Kovalevsky
e,
Ana
Fernández-Barquín
a,
Aroa R.
Mainar
a,
Piotr
Jankowski
f,
Laurin
Rademacher
g,
Sunita
Dey
h,
Siân E.
Dutton
i,
Clare P.
Grey
h,
Janina
Drews
g,
Joachim
Häcker
g,
Timo
Danner
cg,
Arnulf
Latz
cgj,
Dane
Sotta
k,
M. Rosa
Palacin
l,
Jean-Frédéric
Martin
k,
Juan Maria García
Lastra
f,
Maximilian
Fichtner
cd,
Sumana
Kundu
mn,
Alexander
Kraytsberg
mn,
Yair
Ein-Eli
mn,
Malachi
Noked
*b and
Doron
Aurbach
*b
aCIDETEC, Basque Research and Technology Alliance (BRTA), Parque Científico y Tecnológico de Gipuzkoa, Paseo Miramón, 196, 20014, Donostia-San Sebastián, Spain. E-mail: ablazquez@cidetec.es
bDepartment of Chemistry, Bar Ilan University, Ramat Gan, 5290002, Israel
cHelmholtz Institute Ulm (HIU) Electrochemical Energy Storage, D-89081, Ulm, Germany
dInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), D-76021, Karlsruhe, Germany
eIsrael Institute of Metals, Technion R&D foundation LTD., Technion City, Haifa, 3200003, Israel
fDepartment of Energy Conversion and Storage, Technical University of Denmark, Kgs. Lyngby, Denmark
gInstitute of Engineering Thermodynamics, German Aerospace Center (DLR), Pfaffenwaldring 38-40, D-70569, Stuttgart, Germany
hDepartment of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK
iCavendish Laboratory, University of Cambridge, JJ Thomson Avenue, Cambridge, CB3 0HE, UK
jInstitute of Electrochemistry, Ulm University, D-89081, Ulm, Germany
kCEA-LITEN, 17 Rue des Martyrs, 38054 Grenoble Cedex 9, France
lInstitut de Ciéncia de Materials de Barcelona, ICMAB-CSIC, Campus de la UAB, 08193 Bellaterra, Catalonia, Spain
mDepartment of Materials Science and Engineering, Technion-Israel Institute of Technology, Haifa, 3200003, Israel
nGrand Technion Energy Program (GTEP), Technion-Israel Institute of Technology, Haifa, 3200003, Israel
First published on 14th April 2023
Abstract
Emerging energy storage systems based on abundant and cost-effective materials are key to overcome the global energy and climate crisis of the 21st century. Rechargeable Magnesium Batteries (RMB), based on Earth-abundant magnesium, can provide a cheap and environmentally responsible alternative to the benchmark Li-ion technology, especially for large energy storage applications. Currently, RMB technology is the subject of intense research efforts at laboratory scale. However, these emerging approaches must be placed in a real-world perspective to ensure that they satisfy key technological requirements. In an attempt to bridge the gap between laboratory advancements and industrial development demands, herein, we report the first non-aqueous multilayer RMB pouch cell prototypes and propose a roadmap for a new advanced RMB chemistry. Through this work, we aim to show the great unrealized potential of RMBs.
Broader contextToday Li-ion batteries (LIBs) are considered the battery technology of reference for many current and promising applications such as transport electrification or renewable energy storage. Despite the good performances of LIBs, they are expected to face resource supply-chain challenges due to the relatively low natural abundancy of lithium (Li) and the geographically uneven distribution worldwide. Shifting towards fully non-Li rechargeable batteries may open an effective way to overcome such challenges. Rechargeable magnesium batteries (RMBs) constitute a paradigmatic example of such promising, alternative non-Li energy storage systems, following pioneering efforts and breakthroughs from world-wide research teams. The potential to use metallic Mg anodes in rechargeable batteries brings important advantages in terms of energy density, cost, safety, sustainability, and lower material supply risk due to the natural abundancy of Mg. Despite the important advances in the RMB literature, all the reported studies are still limited to the laboratory scale and coin-cell configuration, where many practical and industrial aspects of RMB are neglected. In this context, pouch cell configuration is a better platform to optimize components, and it represents a crucial step towards an application ready battery cell design. Herein, in this paper we present a critical perspective of the most promising materials and cell components for the development of high-TRL RMBs with competitive performances. The feasibility and great untapped potential of possible advanced RMB chemistry is highlighted. A roadmap for the development of mature RMBs that can reach an energy density of up to 160 W h kg−1 is outlined. |
1. Introduction
Over the last decade, the growing markets of zero-emission electromobility, large-scale stationary storage for renewable energy production, and portable consumer electronics have increased the demand for Li-ion battery cells exponentially, and they are forecasted to do so for at least another decade.1 This unsustainable growth is predicted to cause cell shortages due to severe Li-ion battery raw-material bottlenecks, especially lithium, nickel, and cobalt, owing to their limited amount, uneven geopolitical distribution (particularly Co), and the lack of political stability of some of the countries mining the resources.2,3 Therefore, even old chemistries with moderate to poor performance, such as lead–acid batteries, are being considered as an alternative for stationary applications, to relieve the pressure on the electromobility market.4In this context, the emergence of environmentally friendly battery technologies made of abundant, low-cost materials incurring low supply risk and exhibiting high energy density and performance is direly needed to achieve the set climate change goals.5–7 Rechargeable magnesium batteries (RMBs) are a very promising battery technology candidate on account of the high specific capacity (2205 mA h g−1), high volumetric capacity (3832 mA h cm−3), and low reduction potential (−2.37 V vs. SHE) of magnesium.8–10 Moreover, magnesium is the 8th most abundant element in the Earth's crust, is non-toxic and safe for handling in ambient air, has a low atomic weight, and is less inclined than other metals to dendrite formation during plating/stripping reactions,7,11–13 despite more recently the electrochemical growth of fractal Mg dendrites from Grignard reagents has been observed in symmetric cells.14 Additionally, in terms of the environmental friendliness of the entire supply chain, recycling reduces the demand for primary magnesium by up to 50%, unlike currently impractical lithium recycling (<1%).15,16 Although the feasibility of RMB was demonstrated at the laboratory scale by Aurbach's group in 2000,17 its low technology readiness level (TRL) (1–3)18 is still one of its major drawbacks, mainly because of challenges related to the lack of high-TRL electrolyte solutions and intercalation compounds. Since then, the scientific community has been focused on Li-ion battery research, with limited attention to divalent alternatives. Nowadays, the raised awareness of the limitations of lithium-ion batteries and growing market demand have led to an increase in the number of studies and intellectual property rights (IPR) petitions related to RMB components among alternative chemistries (Fig. 1). For the sake of comparison, the number of publications regarding lithium batteries has been included in the ESI† (Fig. SI1). Despite these developments, studies have still been limited to the laboratory scale and based on the coin-cells configuration, where many practical and industrial aspects of RMB are neglected. In this context, the pouch cells configuration is a better platform to optimize components, and it represents a crucial step towards an application-ready battery. Besides, large scale Mg-metal batteries have the potential to be integrated in a simpler battery packs (BP) reducing the cost and their production will demand a greener infrastructure. Additionally, this novel technology could be manufactured using the actual LIB production lines19,20 The conventional state-of-the-art RMB model, utilizes a thick (100 μm) pure magnesium metal anode, a Chevrel-phase (CP) Mo6S8 cathode on a nickel current collector, and an “all phenyl complex” (APC) electrolyte solution consisting of phenylmagnesium chloride (C6H5MgCl), aluminum chloride (AlCl3), and tetrahydrofuran (THF). In this system, the components are fairly compatible with each other and able to provide practical capacities of up to 70 mA h gCP−1 in the laboratory-scale coin cell configuration without side reactions.21 Although this system is functional, to the best of our knowledge, the technology has never been adapted to a larger size pouch cell configuration for cell optimization toward industrial use; this is due to the various disadvantages of each component, which are not suitable for mass production or are not economically viable.
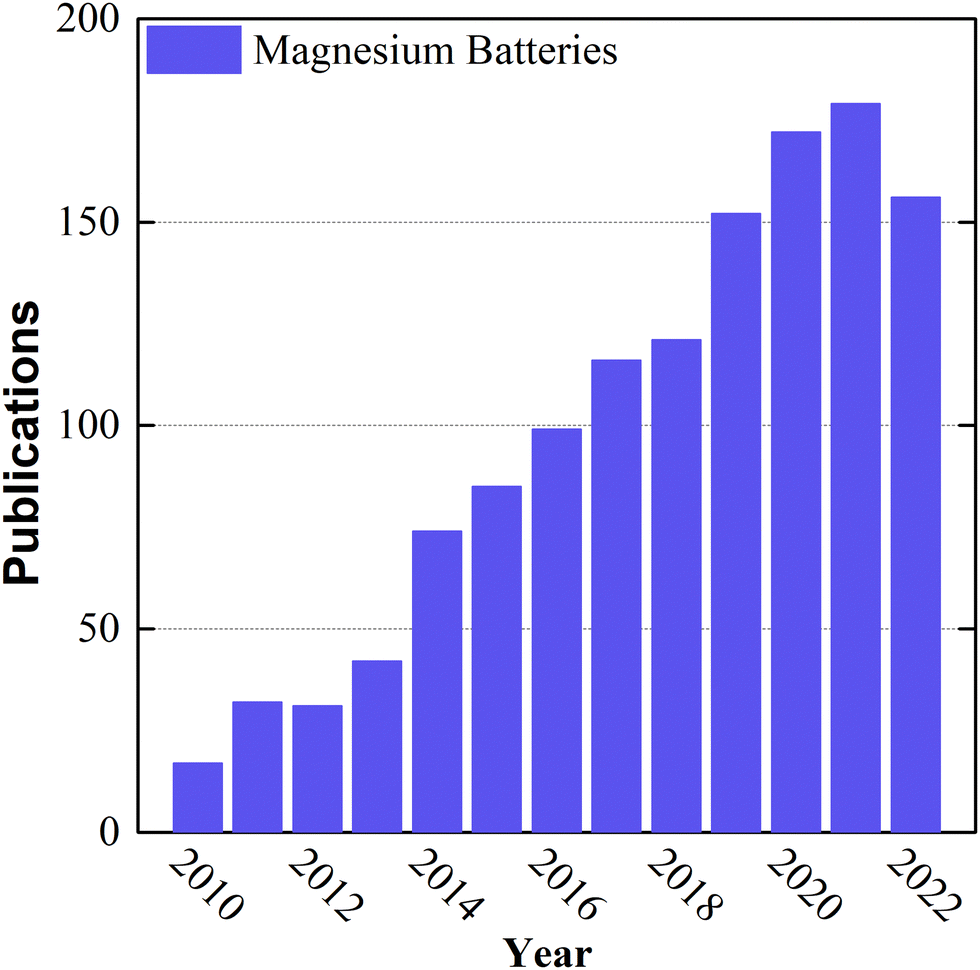 | ||
| Fig. 1 Number of publications per year, including the words “magnesium batteries” in their title, during the last two decades, based on data taken from the Web of Science. | ||
Herein, we present a critical perspective of the most promising materials and cell components for the development of high-TRL RMB which we have evaluated their potential in industrial-level battery cells. Xiu et al.22 have reported previously single layer Mg-based pouch cells, but with the same format as coin cells. However, in the perspective work described herein, for the first time, magnesium batteries were tested in real pouch cell prototypes comprising conventional pure Mg metal-foil anodes, APC electrolyte solution and CP cathodes in an effort to demonstrate an RMB at a high TRL. The differences between pouch cell format and lab-scale coin cell components were explained, and the impact on energy and mass was calculated. Finally, the impact of some of the most novel materials reported in the literature, such as a thin foil of magnesium alloy AZ31, a Mg[B(hfip)4]2/DME electrolyte solution, and the VS4 cathode active material was examined. The latter may enable RMB having a much higher energy density than the first generation, which is based on CP cathodes. The feasibility and great untapped potential of possible advanced RMB chemistry is highlighted. A roadmap for the development of mature RMB that can reach an energy density of up to 150 W h kg−1 is outlined.
2. Electrolytes
The electrolytes are arguably one of the most important components in a battery because of their continuous contact with all the cell components. An ideal electrolyte solution has high thermal, chemical, and electrochemical stability, allowing efficient reversible magnesium plating/stripping and ion diffusion. Importantly, for the Mg electrodes to exhibit a fully reversible behavior, no side reactions must occur. In contrast to the case of the Li- and Na-metal anodes, on which surface films formed by side reactions may behave like solid electrolyte interphases (the SEI model), any such surface film formed on Mg electrodes is prone to block the ion flow, leading to their deactivation. “All phenyl complex” APC electrolyte solutions, developed more than two decades ago,23 are nowadays among the most widely used solutions in the RMB field. They comprise the reaction products of Lewis base C6H5MgCl with Lewis acid AlCl3 in THF; due to the trans-metalation reaction between the Al and Mg cores, which exchange ligands, complex solutions containing MgCl(THF)5+ cations and AlClx(C6H5)4−x− anions are formed. These solutions allow the fully reversible deposition/dissolution of Mg and exhibit wide electrochemical windows (>3 V) (Fig. 2).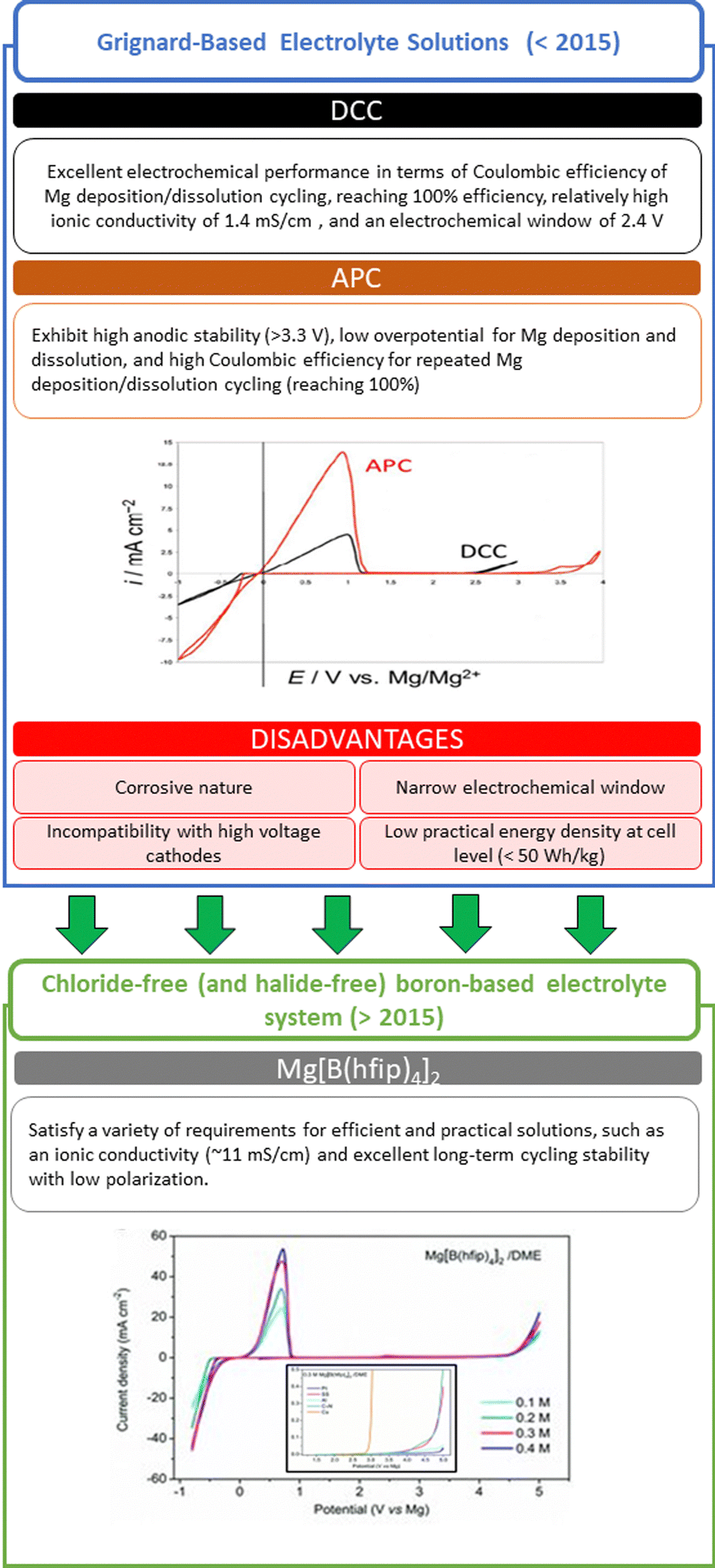 | ||
| Fig. 2 Summary and evolution of electrolyte solutions.3,35 | ||
The starting point in the search for suitable Mg electrolytes were Grignard reagent solutions comprising RMgX (X = Cl, Br) or MgR2 Lewis bases and ethereal solvents like THF. Building on these foundations, the first milestones on the way to advanced APC electrolyte solutions were the development of Mg(BR4)2 ether solutions containing organo–borate anions by Gregory et al.24 and the development of organo–chloro–aluminate anions Mg(AlClxR4−x)2 complexes by Aurbach et al.,7 (where R is an organic group, such as methyl, ethyl, and butyl). The next step was to use ethereal solutions containing products of reactions between Grignard Lewis bases and Lewis acids, such as AlCl3 or AlClxR3−x.25 These electrolyte solutions, containing organo–halo–aluminate Mg salts such as Mg(AlCl3R)2 and Mg(AlCl2RR′)2, demonstrate a room-temperature conductivity comparable to that of Li salts solutions at moderate salt concentrations (0.3–0.5 M) in THF or polyether solvents. Mixing Bu2Mg and EtAlCl2 in THF in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio afforded the best-performing solution, having an ionic conductivity of 1.4 mS cm−1 and anodic stability up to 2.2 V; it was later called the dichloro complex (DCC).17,25,26 However, even the best DCC electrolyte solution does not satisfy the requirements of high voltage, high-energy RMB applications for a wide electrochemical stability window (>3.5 V), chemical stability, and safety.27 During studies on advanced Grignard-based electrolyte solutions, it was understood that β-H elimination reaction, that is, the elimination of a hydrogen attached to a β-carbon (the second carbon of an alkyl ligand bound to the metal center), was the cause of the limited electrochemical window. In advanced Grignard-based electrolyte solutions such as APC, the organic ligand phenyl is directly bonded to a magnesium atom (e.g., PhMgCl) and no hydrogen is present on the α-carbon, thus not susceptible to β-H elimination. This enables APC solutions to exhibit high anodic stability (>3 V vs. Mg); they have become a benchmark RMB electrolyte in which most of the validation and feasibility studies are being carried out, including in the first part of this study. Despite the electrochemical stability window of the magnesium-metal-anode/APC-electrolyte pair, which extends to 3.3 V, the low overpotential for magnesium deposition, and their moderate 2 mS cm−1 ionic conductivity at room temperature, the ability of APC solutions to provide good performance with high-voltage transition-metal-oxide cathodes (such as VOx materials) is still limited.25 Attias et al.28 conducted a comprehensive study, comparing most of previously reported electrolyte solutions. In their study, the only electrolyte with CE higher than 99% for Mg deposition dissolution, was found to be DCC (di-chloro complex solutions comprising the reaction products of Bu2Mg and EtMgCl2 in THF).
:2 molar ratio afforded the best-performing solution, having an ionic conductivity of 1.4 mS cm−1 and anodic stability up to 2.2 V; it was later called the dichloro complex (DCC).17,25,26 However, even the best DCC electrolyte solution does not satisfy the requirements of high voltage, high-energy RMB applications for a wide electrochemical stability window (>3.5 V), chemical stability, and safety.27 During studies on advanced Grignard-based electrolyte solutions, it was understood that β-H elimination reaction, that is, the elimination of a hydrogen attached to a β-carbon (the second carbon of an alkyl ligand bound to the metal center), was the cause of the limited electrochemical window. In advanced Grignard-based electrolyte solutions such as APC, the organic ligand phenyl is directly bonded to a magnesium atom (e.g., PhMgCl) and no hydrogen is present on the α-carbon, thus not susceptible to β-H elimination. This enables APC solutions to exhibit high anodic stability (>3 V vs. Mg); they have become a benchmark RMB electrolyte in which most of the validation and feasibility studies are being carried out, including in the first part of this study. Despite the electrochemical stability window of the magnesium-metal-anode/APC-electrolyte pair, which extends to 3.3 V, the low overpotential for magnesium deposition, and their moderate 2 mS cm−1 ionic conductivity at room temperature, the ability of APC solutions to provide good performance with high-voltage transition-metal-oxide cathodes (such as VOx materials) is still limited.25 Attias et al.28 conducted a comprehensive study, comparing most of previously reported electrolyte solutions. In their study, the only electrolyte with CE higher than 99% for Mg deposition dissolution, was found to be DCC (di-chloro complex solutions comprising the reaction products of Bu2Mg and EtMgCl2 in THF).
Other electrolytes have been proposed as alternatives to APC. One of them is hexamethyldisilazide magnesium chloride (HMDS MgCl), a non-nucleophilic Hauser-base-derived electrolyte, which was reported by Liebenow et al.29 This work constituted a major milestone in electrolyte solutions development, demonstrating the oxidative stability of weak Al–R bonds in an all inorganic salt. Muldoon and coworkers30 extended the anodic stability of the systems to 3.3 V by adding AlCl3. Adding such a strong Lewis acid induces Lewis acid–base reactions similar to those discussed above and thus changes the composition and structure of the ions in solution, forming more stable solution species, which found particularly wide use in magnesium–sulfur systems.26,30,31 An electrolyte with a fully inorganic salt that contains chloride species was proposed by Doe et al., who mixed common ethereal solutions of AlCl3 and MgCl2 to prepare solutions of magnesium–aluminum–chloride complexes (MACC).32 Recently, Canepa et al. refined the structural components in this electrolyte solution and their effect on stability, coulombic efficiency, and aging/conditioning.33 They found that aluminum from the solution is deposited on the magnesium-metal anode in the early cycles, which reduces coulombic efficiency. However, in return, this behavior promotes the stabilization of charged species (MgCl+ and AlCl4−) dissolved in the electrolyte solutions through a pre-treatment called “conditioning” (repeated Mg deposition/dissolution cycling that cleans the solutions of contaminants) and leads to a smoother plating/stripping of Mg in consecutive cycles.33 Nevertheless, despite all the conditioning steps and improvements in coulombic and plating/stripping efficiencies, the anode remains unstable above 3.1 V.
Furthermore, fluorinated alkoxide-based electrolytes present solution conductivity of 3.5 mS cm−1 and anodic stability of 3.2 V vs. Mg2+.34
A common denominator among all the electrolyte solutions mentioned above is the existence of corrosive chloride species, which limits not only the electrolyte stability but also the choice of current collectors and cathode active materials.35,36 Hence, RMB electrolyte solutions have been intensively studied to find chloride-free magnesium salt solutions that exhibit high conductivity and promote fully reversible Mg plating/stripping processes, enabling the use of high-voltage/capacity transition-metal-oxide cathodes.37–40
A class of electrolytes based on magnesium borohydride Mg(BH4)2, the first chloride-free (and halide-free) boron-based electrolyte system, was expected to circumvent this incompatibility between the different components.41,42 Although this electrolyte was proven to promote reversible Mg plating/stripping, its very poor anodic stability (<1.8 V) is not in line with the energy density requirement of the RMB technology.41,43 However, this was a steppingstone for the development of non-nucleophilic family of electrolytes.30,43,44 A key breakthrough was made recently by Fichtner's group, with the successful synthesis of magnesium tetrakis(hexafluoroisopropyloxy)borate Mg[B(hfip)4]2, which brought new prospects for the cyclability of RMBs and the selection of current collectors and cathode active materials.38,45 Indeed, this salt may be used with dimethoxy ethane (DME)—an ether-based solvent that has high anodic stability (>4.5 V)—without corrosive Cl component, and electrolytes based on Mg[B(hfip)4]2 satisfy a variety of requirements for efficient and practical solutions, such as an ionic conductivity (∼11 mS cm−1) more than five folds that of conventional APC electrolyte solutions and excellent long-term cycling stability with low polarization.11,38,46–49 The uniqueness of this salt originates from its branched carbon structure, which allows the weak coordination of Mg cations, while the presence of proton on the α carbon and the fluorinated β carbon provide oxidative stability (Fig. 2).
As a result, this electrolyte opened up new possibilities to achieve high energy densities through its compatibility with lightweight aluminum current collectors and high-specific-capacity cathode materials, its resistance to side reactions with Mg-metal anodes, and its high anodic stability, suitable for use at high voltage. In addition, the demonstrated stability of this electrolyte toward polysulfide species formed during battery cycling opened the path for research on rechargeable Mg metal–sulfur batteries.38
In principle, solid Mg2+-conducting electrolytes can offer a number of advantages over liquid electrolytes. All-solid Mg-ion cells may offer higher level of safety as they do not contain volatile and flammable components and thus, are not prone to leakage and ignition; the ease of manufacturing of cells of all conceivable shapes is also apparent.
To be able to utilize a specific Mg2+-conducting material as solid electrolyte for Mg-ion cells, it should – first and foremost – hold Mg2+ ionic conductivity (σionic) matching the typical ionic conductivity of liquid Mg2+ electrolyte at ambient temperatures. Conductivities of current liquid Mg2+-electrolytes roughly corresponds with Li+-electrolyte conductivities, and are of the order of a few mS cm−1;50 the best reported Mg2+σionic is 11 mS cm−1.51
The other requirements are:
• Low electronic conductivity (regarding solid Li+-electrolyte, a typical (σionic/σelectronic) ratio ∼2 × 106).52
• Wide electrochemical stability window.
• Satisfactory mechanical properties.
• Being non-corrosive toward common battery components (cell housing, current collectors, etc.).
• Non-flammability and thermal stability.
• Mg2+ ion transference number close to one.
Currently, most materials, which can satisfy the above requirements, may be divided into three groups:53–55
• Metal–organic frameworks – based solid-state electrolytes (MOFs):56–58
MOFs present three-dimensional scaffold comprised of metal ions connected by organic ligands; the void space inside the scaffold may be filled with a Mg-salt solution, thus featuring a 3D system of interconnected ionic channels of a few Angstroms in diameter. These emerging Mg2+-electrolytes look promising alternatives to the liquid electrolytes, while the best reported conductivity of such electrolyte is 1.0 mS cm−1.57
The study of these electrolytes are at an early stage, though, and the information on Mg2+-transference numbers of MOF-based electrolytes are scarcely available; the best reported Mg2+ transference number of the electrolyte of this class is just 0.47.59 Also, the electrolytes contain some flammable liquid solvents, thus the cells with such electrolytes are expected to be flammable.
• Polymer gel-based solid electrolytes (SGPE) and polymer – solid filler-based electrolytes:54,60,61
– The polymer-based gel electrolytes are comprised of polymer matrix (such polymers as PEO, PVA, PVDF, PPO – poly(propylene oxide), PVP – poly(vinyl pyrrolidone)), etc. and are usually plasticized by liquid electrolyte with a dissolved Mg2+-salt. This plasticizer/liquid forms an interconnected channels system within a polymer matrix, and Mg2+-ions are transporting along these channels.
– The other type of polymer-based gel electrolyte comprised of SGPE with dispersed nano-sized ceramic fillers, such as SiO2, TiO2, ZnO, Al2O3, MgO, MgAl2O4, etc.; the composite SGPE – ceramic electrolyte provide enhanced Mg2+-conductivity transference number as compared with pure SGPE.
While the best values of σionic for SGPE and filled SGPE are as high as 11 mS cm−1,62 the challenging point is that the transference numbers of these electrolytes are usually low, between 0.66 and 0.26.
Low transference number results in a concentration polarization in the electrolyte, which increases cell overpotential; the problem is aggravated under high current rate conditions. As the matter of fact, there is an inherent trade-off between electrolyte conductivity σion and the cation transference number t+. For instance, Li-ion model cells reaches SOC = 0.75 at 2C current rate and 4.2 V cutoff if σion = 4 mS cm−1 and t+ = 1, while in the case of t+ = 0.4 a SOC = 0.75 may be reached under the same current rate and cutoff voltage only if σion = 10 mS cm−1.63,64
The other problematic point of the SGPE and filled SGPE is their relatively low mechanical strength and thermal stability, as well as their limited fire safety.
• Ceramic-based solid electrolytes:16,54
Much of these materials are purely cationic conductors having transference number t+ close to one and are non-flammable. Here, we extensively elaborate on this type of materials and its sub-groups.
Hydride-based Mg2+-solid electrolytes (Mg(BH4)2, Mg3(BH4)4(NH2)2, etc.)53,65,66
Hydride-based compounds have hydrogen atoms in the polyanions (such as [BH4]−), which facilitates the polyanions rotation, thus assisting an effective Mg2+ ion transport through the paddle-wheel mechanism. This circumstance gives all the grounds to suggest that this type of materials can have high σion. However, so far, the studied materials have ambient temperature conductivity σion just around 10−5 S cm−1, which is not enough for all-solid Mg2+-ion battery cell applications.Oxide-based ceramics
The most part of the oxide-based materials exhibits ambient temperature conductivity markedly below σionic of liquid Mg2+-electrolyte at ambient temperatures, which is the most problematic aspect of these materials. Up to now, the highest reported σionic of this type of material is 0.7 mS cm−1 (for MgI2–Mg3(PO4)2 Mg2+-solid electrolyte).67Chalcogenide-based ceramics
In essence, chalcogenide-based Mg2+-solid ceramic electrolytes are in many respects similar to oxide-based Mg2+-conductive ceramics, just having different anionic scaffold, namely S, Se or Te instead of oxygen. Conductivity is the process of Mg2+-ion jumping from one locally-equilibrium position into a similar neighbouring position and the less energy is required for such a jump (viz. activation energy Em), the higher conductivity is. In turn, Em is inversely proportional to the volume per anion in the case of isostructural scaffolds, so Em is decreasing in the order O− → S− → Se−→ Te−;68 it is therefore reasonable to expect that chalcogenide ceramic-based Mg2+-electrolytes can have an outstandingly high conductivity.As a matter of fact, chalcogenide ceramics are very promising Mg2+-solid state electrolytes; the materials have transference number close to one, combining it with high conductivity and advantageous mechanical properties, the materials are safe and fire-resistant, and their manufacturing is relatively simple.53 Surprisingly, very few electrochemical studies have been reported on chalcogenide –based Mg2+-solid ceramic electrolytes; among these, two MgS–P2S5–MgI2 glass–ceramic systems with low ambient temperature σionic53 and MgSc2Se4 with a fair Mg2+-conductivity of ∼0.1 mS cm−169,70 were reported. The problematic aspect was a relatively high electronic conductivity; the (σionic/σelectronic) ratio for MgSc2Se4 was found to be ∼2.5 × 103. The feature was attributed to the inherent property of electronic energy spectra of MgSc2Se4 crystal structure (the material is a semiconductor with ∼2.15 eV bandgap), so was attempted to suppress the electronic conductivity of the ceramic by variation of selenium content (selenium deficient and selenium rich MgSc2Se4 ceramic), and by aliovalent substitution of scandium with titanium and cerium. The attempts were not successful, though, and it was found that Mg2+-conductivity changes in parallel with electronic conductivity in the case of aliovalent substitutions, namely the decrease of the electronic conductivity accompanies with the essential decrease of ionic conductivity.70
The high electronic conductivity clouds the prospects of a practical implementation of chalcogenide ceramic-based Mg2+-solid state electrolytes; this circumstance become a vigorous driving force for in-depth study of the nature of the electronic conductivity in ternary spinel chalcogenide MgSc2Se4 by Kundu et al.8 Mg–Sc–Se ceramics were prepared by high-temperature sintering of the elemental (Mg, Sc, and Se) powder mix, followed by thermal-treatment of the resultant ceramics. It was reported that the morphology of the ceramic strongly depends on the sintering and thermal-treatment modes and a clear correlation between the morphology and electronic conductivity of the ceramic was established.
Particularly, it was found that electron jumping between nano-sized conductive inclusions, which are distributed inside the electronically insulating matrix of MgSc2Se4, makes the major contribution into the electronic transport in the Mg–Sc–Se ceramic material (the Berthelot mechanism of conductivity). These electronically conductive areas comprise of a mix of metallic-type electronic conductors Sc and ScSe, and the presence and distribution of these inclusions depends on the Mg–Sc–Se ceramic preparation and thermal-treatment modes (see Fig. 3).
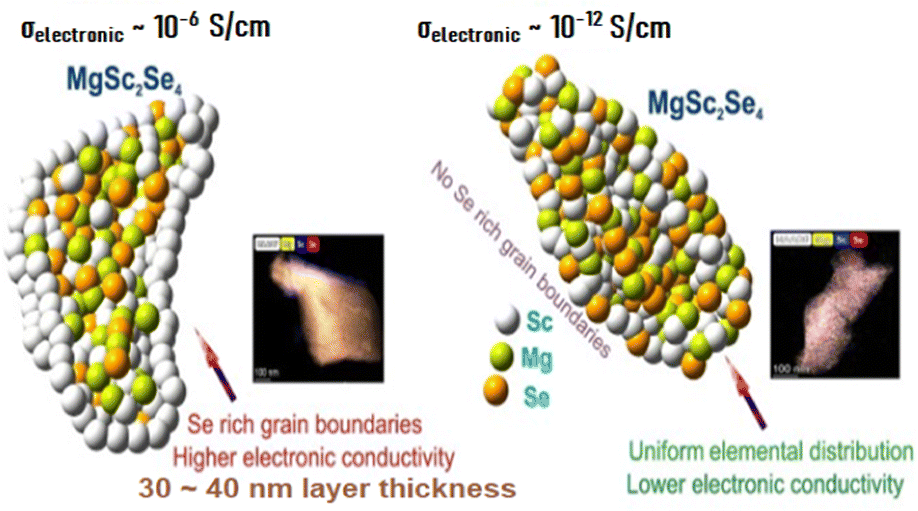 | ||
| Fig. 3 (left) Model of Se-rich grain boundaries and associated TEM analysis and a model of a uniform distribution of Mg, Se, and Sc and associated TEM analysis (right). | ||
The authors of the study were able to eliminate the formation of the above nano-conductive inclusions in the Mg–Sc–Se ceramic matrix by manipulating preparation and thermal-treatment modes of the material, reducing the electronic conductivity by six orders of magnitude; the resulting MgSc2Se4 ceramic demonstrated low electronic conductivity of ∼10−12 S cm−1 without compromising σionic of the solid electrolyte having (σionic/σelectronic) ratio ∼108.
Thus, this work reveals a synthetic method controlling the preparation conditions, eliminating the formation of nano-conductive inclusions, and restricting electronic conductivity of the chalcogenide ceramic – based Mg2+-solid electrolytes, without compromising their high σionic.
It should be noted, however, that despite notable progress the development of the chalcogenide-based Mg2+-solid electrolytes are at an early stage, and there is still much of work to be conduct on these materials before they can practically be used in rechargeable Mg-ion cells.
Table 1 provides a summary of the above discussion, describing the state-of-the-art ionic and electronic conductivities of the different solid Mg2+-conducting electrolytes (and related parameters), compared with state-of-the-art liquid Mg2+-conducting electrolyte.
| SoA electrolyte system | Transfer Nu. | RT ionic conductivity [mS cm−1] | Electronic conductivity [mS cm−1] | Ref. |
|---|---|---|---|---|
| Liquids | ≥0.5 | ≥11 | NA | 51 and 71 |
| Solids | ||||
| MOF | ≥0.47 | ≥1.0 | N/A | 57 and 59 |
| Polymers | ||||
| Polymer gel | ≥0.73 | ≥1.0 | N/A | 54 and 60 |
| Polymer + filler | ≥0.66 | ≥4.8 | N/A | 54 and 61 |
| Ceramics | ||||
| Hydrides | ≥0.95 | ≥1.3 × 10−2 | ≥3 × 10−9 | 65, 72 and 73 |
| Oxides | ≥1 | ≥0.7 | Unspecified low values | 16, 67 and 74 |
| Chalcogenides | N/A | ≥0.1 | ≥10−12 | 8 |
In summary, as mentioned above the Mg[B(hfip)4]2 electrolyte allows the use of lighter current collector and the development of high energy density cathode materials, which allow reversible two-electron redox with synergetic cationic–anionic contribution in RMB.
3. Anodes
Most RMB studies focus on high-energy cathodes and electrolytes, but anode materials (namely, thin Mg-foil anodes) are also an important factor in achieving high-energy densities.75–78 A thin foil of pure magnesium metal is naturally the most desired anode, as it may be used as a self-standing anode without the need for an additional current collector.21,79 The latter feature leads to a significantly higher energy density by avoiding the weight of an additional current collector, which is necessary with Prussian blue analogues, polyanionic compounds, organic compounds, spinel oxides, and insertion-type anode materials in general.80,81 On the other hand, adverse interactions between a reactive metal like Mg and the electrolyte solution components can rapidly compromise the reversibility of the electrochemical system by promoting the formation of passivating/blocking layers that are not penetrable by the double-charged Mg ions.11,16,80,82 This limitation related to Mg electrochemistry is at odds with the case of the Li electrochemistry, in which surface films on electrodes (in most cases) behave as solid electrolyte interphases (SEIs), and the singly charged Li ions can easily migrate through surface films comprising ionic Li compounds.83 To avoid the formation of charge-blocking passivation layers on magnesium, the electrolyte solution should be based on ether solvents (not reactive to Mg metal) and may contain chloride ions as a desirable component; chloride ions in ethereal solvents seem to interfere with the passivation of Mg anodes and facilitate the transport of Mg ions from heavily solvated states to the electrode surface (Mg deposition on the anode side and Mg intercalation on the cathode side).25,27,84 For this reason, conventional RMB technology—including the first part of this work—still utilizes the same combination of Mg-metal anodes and chloride-containing ethereal electrolyte solutions as the first publication more than 20 years ago.17,31,85Besides detrimental passivation phenomena, the mechanical properties of Mg metal also cause a limitation to its use.16,21 In order to ensure their high energy density, magnesium batteries require the use of very thin Mg-metal foil anodes. The preparation of a desirably thin Mg foil anode is very difficult because pure Mg metal is not ductile enough; the hexagonal close-packed (hcp) structure makes it challenging to process Mg metal mechanically below 200 °C.79 Although the industrial manufacturing of ultra-thin (<100 μm) Mg foils is possible, it is not economically scalable to the level required for battery production.21,86,87 Thus, the use of pure-Mg metal anodes in batteries should be avoided on the account of their poor mechanical properties. To circumvent this problem, some researchers investigated binary and ternary Mg alloys that have better mechanical properties than pure Mg yet exhibit reasonable electrochemical properties; alloys that have two or more metal species may provide efficient magnesium hosting while offering new properties that the pure Mg metal is unable to provide, including high electronic conductivity, buffering of volumetric changes, and even the advantage of compatibility with different types of electrolyte solutions (i.e., lower reactivity than Mg metal).79,88
Theoretically, 3A–5A group metals aluminum (Al), gallium (Ga), indium (In), tin (Sn), bismuth (Bi), lead (Pb) and metalloids silicon (Si), germanium (Ge), and antimony (Sb) can create binary Mg alloys (MgxMy) with high specific capacities at low alloying potentials.88 However, depending on the electrolyte used and the alloying/dealloying reaction of the candidate, electrochemical activity may be low, which is indeed the case for magnesium alloys with Ge, Si, and Sb. In the case of Mg alloys containing Sn, Pb, or In, some of the problems identified so far are the inevitable side reactions that lead to limited cycle life, the extreme volumetric expansion, and slow kinetics caused by the high Mg2+ diffusion barrier, along with possible difficulties in scalability82,89–93 Alternatively, binary Mg alloys Mg3Bi2, Mg2Si, and Mg2Ga5 may be directly used as anode materials without any host.82,88,94 Bismuth alloys of magnesium metal have attracted some attention owing to their availability, and their reported theoretical volumetric capacity (3783 mA h cm−3), being comparable to that of the Mg-metal anode (3833 mA h cm−3), and their high Mg-cation diffusivity, which make them highly advantageous.95 However, their theoretical specific gravimetric capacity (385 mA h g−1) is similar to that of the commercial graphite anode (372 mA h g−1) in state-of-the-art Li-ion batteries; therefore, the voltage and low capacity of these RMB systems will render the energy density of the cells too low to be attractive.88 Moreover, these alloys exhibit extreme volumetric expansion (100–300%), or low electrochemical drive of formation, making them unattractive as anodes in Mg cells. Various strategies (such as nanoscaling, nano-/micro-structuring, and film casting), and morphologies(crystalline and amorphous) were employed in the last decade to solve some of these issues, but the challenges remain.96 As for other alloys, aluminum is the most common addition as a cheap solution to improve the strength of magnesium, as well as to lower the corrosion rate and improve the anode utilization efficiency.81,97 Other metals, such as zinc (Zn) and gadolinium (Gd), were also reported to improve the workability of pure magnesium metal, although more research is needed for their use in rechargeable magnesium batteries to be considered.81,86,98
Multi-element alloys (>2 metals) have been investigated in the search for a promising anode candidate that offers a balance between the electrochemical and mechanical properties, many different binary alloys having failed to behave adequately as self-standing RMB anodes.88 It is widely thought that adding inactive elements as well as electrochemically active ones might be key to improving electrochemical reversibility and mechanical stability simultaneously.88 In particular, adjusting the alloy for lower volumetric changes can avoid the cracking and pulverization effects also seen in Li–metal alloys.42,99 Because magnesium is a widely used metal, a plethora of studies and reports have been published on alloying compounds and their mechanical properties and effects on corrosion.81 However, a recent computational study focused on cast alloys, which have good processibility and thus scalability, reported that only a few of the studied elements were beneficial for the electrochemical process of plating and stripping.100 Besides, Yang et al. collected several studies which reported the influence of microstructure and composition of Mg alloys on their mechanical properties, ductility, and strength. These kinds of properties might profoundly affect the performance of different alloy anode materials for RMB.81 Among a wide range of combinations, ternary Mg–Al–Zn alloys are one of the most studied types, owing to their known properties, production methods, refinement, and widespread use in the automotive and aerospace industries.101,102 The AZ31 alloy (96% Mg, 3% Al, 1% Zn) in the AZ series has attracted special interest because it has a much better ductility than pure magnesium metal. The presence of aluminum and zinc affects the bulk and surface structure, reduces surface reactivity and corrosion.21,102–104 The better ductility enables to prepare effectively very thin and elastic Mg foil anodes (very important for achieving high energy density). Additionally, AZ Mg alloy anodes were reported to have high utilization efficiency and excellent discharge capacity, increasing the energy density of RMB.79 These benefits are also due to the total low percentage of dopant (4%) allowing the electrochemical and mechanical properties to be modified while maximizing the amount of active magnesium in the alloy to provide high specific capacities, unlike other AZ series alloys. In other studies, AZ31 alloy itself was doped with small amounts of other elements; however, the change in corrosion potential was less than 0.04 V.97 Recent studies shown the electrochemical and surface chemistry behavior of AZ31 Mg alloy thin foil anodes during Mg dissolution and deposition process is comparable to that of pure Mg foil anodes.21 Therefore, AZ31 Mg alloy is considered one of the most promising self-standing magnesium-alloy anodes, demonstrating superior electrochemical stability and mechanical processibility for the large production of ultrathin layers, and it is expected to accelerate the maturation of the next-generation high energy density RMB technology.21 It is important to note that the electrochemical behavior of thin foil electrodes made of AZ31 Mg alloy, is expected to be very similar to that of pure Mg foils (thus ensuring an optimal energy density for the resulting Mg batteries). The combination of performance and processability makes AZ31 Mg alloy the anode material of choice for Mg battery cells (Fig. 4).
 | ||
| Fig. 4 SEM images of the surface of pristine (a) Mg alloy AZ31 thin film, and (b) cross-section SEM image of ultrathin AZ31 foil.21 | ||
4. Cathodes
The conventional state-of-the-art RMB, which was developed two decades ago, uses a CP Mo6S8 cathode prepared through the high-temperature synthesis of CuMo3S4 followed by the electrochemical removal or chemical leaching of copper.17 The CP contains three-dimensional channels facilitating Mg2+ transport and a unique metallic electronic structure providing easy accommodation of electrons.105,106 The charge of the magnesium cations is balanced by sulfur atoms in the structure, with a screening cloud that enables weak electrostatic interactions and 2+ charge shielding, leading to a low migration barrier (570 meV) and fast diffusion (10−12 cm2 s−1) at room temperature in comparison with other materials.75,76 However, its low average working voltage of 1.1 V (an upper intercalation voltage range at 1.3–1.1 V into the CP “inner ring”, and an intercalation plateau at around 1.05 V into its “outer ring” accommodation sites) in combination with its low theoretical (129 mA h g−1) and practical capacity (approximately 70–80 mA h g−1vs. Mg/Mg2+, ∼112 mA h g−1vs. Li/Li+, measured in a coin cell configuration) makes it a non-competitive cathode.17,107–109 Consequently, in this study, a CP cathode is only utilized to establish an electrochemical performance reference of the current conventional RMB technology; for a cell to reach the desired high energy density, the CP cathode should be replaced with a high working-voltage/capacity alternative.Among all the cathode materials proposed, only a few groups of candidates demonstrate adequate properties (Fig. 5), and further study is needed to find a suitable cathode for practical RMB.110,111 So far, transition metal oxides and sulfides (TMOs and TMSs), layered transition metal chalcogenides (TMCs), layered and spinel oxides, Prussian blue analogues (PBAs), vanadates, silicates, polyanionic phosphates, and even organic compounds have been studied as potential secondary magnesium battery cathodes.11,12,15,16,24,25,43,112–118 TMOs (i.e., Co3O4, Mn2O3, Mn3O4, etc.) constitute one of the most studied groups in state-of-the-art Li-ion batteries on account of the high capacities they can provide due to the crystal lattices which contains tunnels and pathways for intercalation.119 In RMBs, according to early studies, TMOs can generate relatively high capacity and working voltages, although reversibility is hampered by the interaction of the double charge of magnesium with the crystal lattice.120,121 Moreover, while conventional APC solutions—containing chlorides—present compatibility problems with these materials, new high-stability boron-based electrolytes might spark a new research avenue for the use of TMOs in RMB.24,105
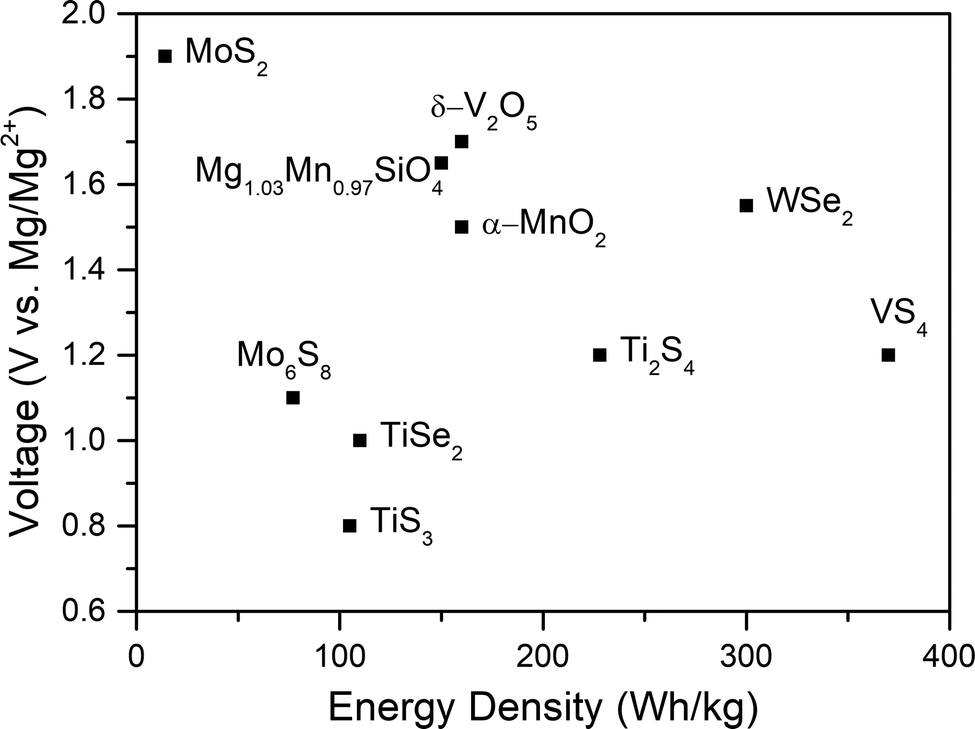 | ||
| Fig. 5 Voltage (vs. Mg/Mg2+) and energy density (calculated based on the cathode parameters only) of several cathode materials that may be relevant to RMBs.111 | ||
On the other hand, chalcogenides have better properties, thanks to S2− and Se2− having a larger ionic size than O2−, thus providing wider channels.122 Additionally, the weak coulombic interactions in the sulfur layers of their structure ease the ionic mobility of doubly charged magnesium ions and the Mg2+ desolvation in electrode/electrolyte interfaces.50,75 For cathodes such as TiS2, TiS3, or Ti2S4, the working voltages are in fact lower than in CP cathodes, although the theoretical capacities and energy densities they can provide are slightly higher.76 Another molybdenum chalcogenide alternative to Mo6S8 is MoS2. It has a higher working voltage (1.9 V) than most cathodes and has a theoretical capacity of 223 mA h g−1. However, its cyclability is inferior to that of CP.77
Promising studies have explored selenides, such as WSe2, which can provide high working voltages (1.5 V) and high specific discharge capacities (203 mA h g−1).106 However, this material loses ∼20% of its initial capacity after a mere 50 cycles. Therefore, despite their many advantages, the cyclability of chalcogenides needs to be improved for them to be used in practical RMBs.43,123
PBAs, with a formula of AxMA[MB(CN)6]·zH2O (A is an alkali metal, MA and MB are transition metals), are the subject of a vast body of literature, in particular studying the intercalation of different valency cations and of some small molecule solvents in their open framework with 3D diffusion channels.40,43,124 The reason for the successful insertion of a wide variety of cations into PBAs lies in the coordination of triple-bonded cyanide groups (CN−) with the metal ions and the opening of the structure caused by the increase in the separation between transition metal ions (Ni, Cu, Fe, etc.).43,125 Nickel hexacyanoferrate (NiHCF) has been shown to intercalate Mg2+ ions successfully, although at low capacities and in the presence of a cell with flooded electrolyte. Moreover, the structural crystal water in PBAs and the capacity contribution originating from the inserted electrolyte solvent molecules prevent a full understanding of the working mechanism of these cathodes.10,125
Among the many different cathode materials used for intercalation, polyanionic compounds are some of the most studied.40,78 These compounds are mostly sought after because of their stable 3D network structure, high voltage, and weak electrostatic interaction with the mobile cation, a set of properties that, in principle, make them good RMB cathode candidates.78,126 In particular, olivine-type silicates (MgMSiO4, M = Fe, Mn, Co) can intercalate magnesium at a high working voltage and capacity (Mg1.03Mn0.97SiO4: ∼1.6 V vs. Mg/Mg2+, ∼210 mA h g−1) and have a structural ability to ease volumetric expansion.106 NASICON-structured phosphates (NASICON = Na super ionic conductor), such as Mg0.5Ti2(PO4)3 and MgZr4(PO4)6, were also studied.122 The first composition, Mg0.5Ti2(PO4)3, was only studied for its magnesium intercalation, and though it has Mg in its structure, it is probable that it cannot be used in a full magnesium battery because of the initial oxidation state of titanium (Ti4+).122 As for MgZr4(PO4)6, it is only promising at high temperatures (∼800 °C), where its ionic conductivity is high (6.9 × 10−3 S cm−1) because of the formation of a secondary phase that decreases the grain boundary resistance.122
Vanadium and vanadates provide an important advantage due to their flexible oxidation states, keeping local electroneutrality and lowering the Mg2+ diffusion barriers.76,122 V2O5, VS2, and VS4 are among many compositions that have been studied for RMB.76,115,122 One of the motivations for the extensive study of V2O5 polymorphs is their theoretical energy density of 660 W h kg−1, which depends on the V5+/V3+ redox couple.122 Specifically, α-V2O5 was reported to reversibly intercalate magnesium into its structure with a specific capacity of 280 mA h g−1.127 A recent computational study was carried out to clarify the intercalation mechanism of Mg2+ in α-V2O5 under equilibrium conditions; it concluded that the intercalation through the formation of δ-MgxV2O5 (0 < x < 1) is hindered kinetically, while a metastable insertion path of Mg in α-V2O5, leads to an ε-Mg0.5V2O5 phase, that is more consistent with experimental data.128,129 Other studies have confirmed that the phase transformation rearranges the a-axis layer stacking, changing the cation coordination for intercalation sites.129 Moreover, the Mg2+ migration is calculated to be faster and energetically more favorable in the δ phase, so methods for the solid-state synthesis of δ-MgV2O5 and its electrochemical demagnesiation have been seen as a way to enhance the sluggish kinetics and low performance of V2O5.128
As one of the most promising cathode materials, VS4 has attracted a lot of attention for its one-dimensional chain-like crystal structure along the c-axis composed of V4+ and sulfur dimers (S2−2) bound by weak van der Waals forces.130,131 The weak forces of interaction within the material and the repeating chain distance (5.83 Å, (110) plane for monoclinic VS4), which is much larger than the diameter of Mg2+ cations (1.44 Å), allow easy Mg-ion transportation through the open channels and support good electrochemical performance.75,111,132,133 Additionally, the VS4 structure was previously reported to present a Peierl distortion (dimerizations) due to its one-dimensional chain structure; in such structures, a perfect crystal is electronically unstable and goes through a natural lattice periodicity reordering to allow the electrons to sit at lower energy.115 This lattice distortion also introduces new bandgaps with a smaller energy difference (∼1.0 eV in case of VS4) that provides high electronic conductivity.132,134 In terms of electrochemistry, VS4 has been shown to exhibit an initial capacity of 250 mA h g−1 at C/12, while a VS4/rGO composite has shown an extraordinarily high specific capacity of approximately 330 mA h g−1 at a 100 mA g−1 current density, with an initial specific capacity of 408 mA h g−1111,115 Besides, in another recent study, VS4 nanodendrites have demonstrated a specific capacity of 74 mA h g−1 at a high current density of 500 mA g−1 after 800 cycles.75 Overall, these results show promising rate capability and cyclability, and most importantly, a near 5-fold improvement in practical energy density in VS4 cells in comparison to CP cells. Indeed, the minimum acceptable specific capacity and working voltage established by Gregory et al. is 230 mA h g−1 at 1.5 V, corresponding to a specific capacity of ∼315 mA h g−1 at 1.1 V, which is very similar to the experimental results with VS4.24,105,111
As stated previously, VS4 (Fig. 6) presents competent electrochemical performance, such as its high specific capacity and is a model material for reversible two-electron redox reaction with synergetic cationic–anionic contribution. Therefore, in this study, VS4 was chosen as the most promising cathode material for competitive advanced RMB pouch cell prototypes to be used in commercial storage applications in the near future.
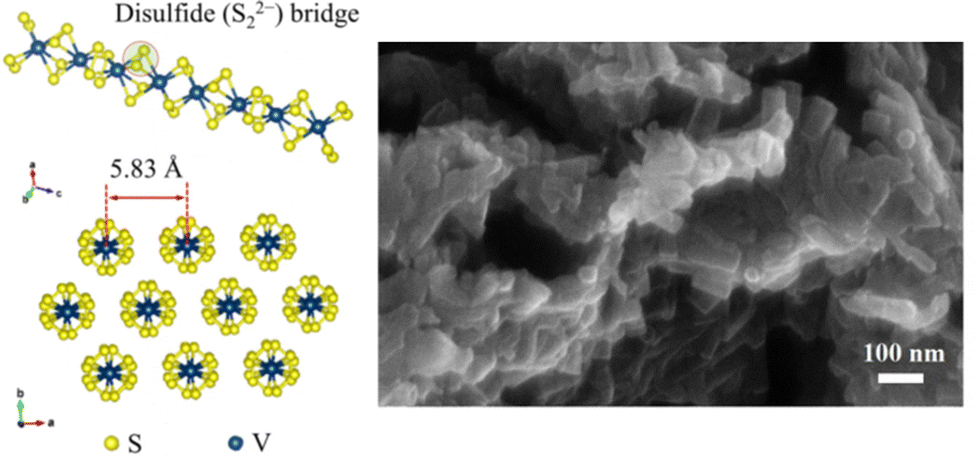 | ||
| Fig. 6 Molecular structure of material VS4/rGO and SEM image with a higher magnification.111 | ||
5. Experimental section
High-purity Mg metal foil (99.9%, 100 μm) was provided by Gelon LIB group and Chevrel Phase (CP) Mo6S8 by the American NEI Corp. (1 kg). Pouch cell cathodes were processed in a pilot plant line using CP/carbon black (C45)/PVDF (90:5:5) at an 11.8 mgCP cm−2 loading (1.6 mA h cm−2) on both sides of a nickel current collector (Gelon LIB group, 20 μm), while coin cell cathodes on nickel current collectors were produced in our laboratory by the doctor blade technique at a 3.5 mgCP cm−2 loading.
Pouch cell prototypes consisted of five pure-magnesium metal-foil anodes (44 × 61 mm) and four double-sided cathodes (43 × 60 mm) stacked with a double-layer polyolefin-based separator in between, unless indicated otherwise.
The 2025-type coin cells consisted of one layer of a magnesium metal foil anode, a single-side coated cathode, and a layer of Whatman glass fiber (GF/F) in between the electrodes. The assembly of coin cells was done in a high-purity (99.999%) Ar-filled glovebox (MBraun, Germany) under ideal conditions (O2 < 1 ppm, H2O < 1 ppm), whereas pouch cells were assembled in a dry room (dew point −50 °C) using standard pouch cell packaging foil (Targray) and nickel tabs (Gelon LIB group). The assembly technique of the scaled-up conventional RMB pouch cells was the same used for conventional lithium-ion batteries.
The pouch cells were sealed under vacuum conditions in the above-mentioned glovebox. APC electrolyte solutions were prepared following procedures described elsewhere.17,38 The amounts of electrolyte solution used were 100 μL in each coin cell and 3 mL in each pouch cell. Prior to each cycle life test,10 activation cycles were applied.
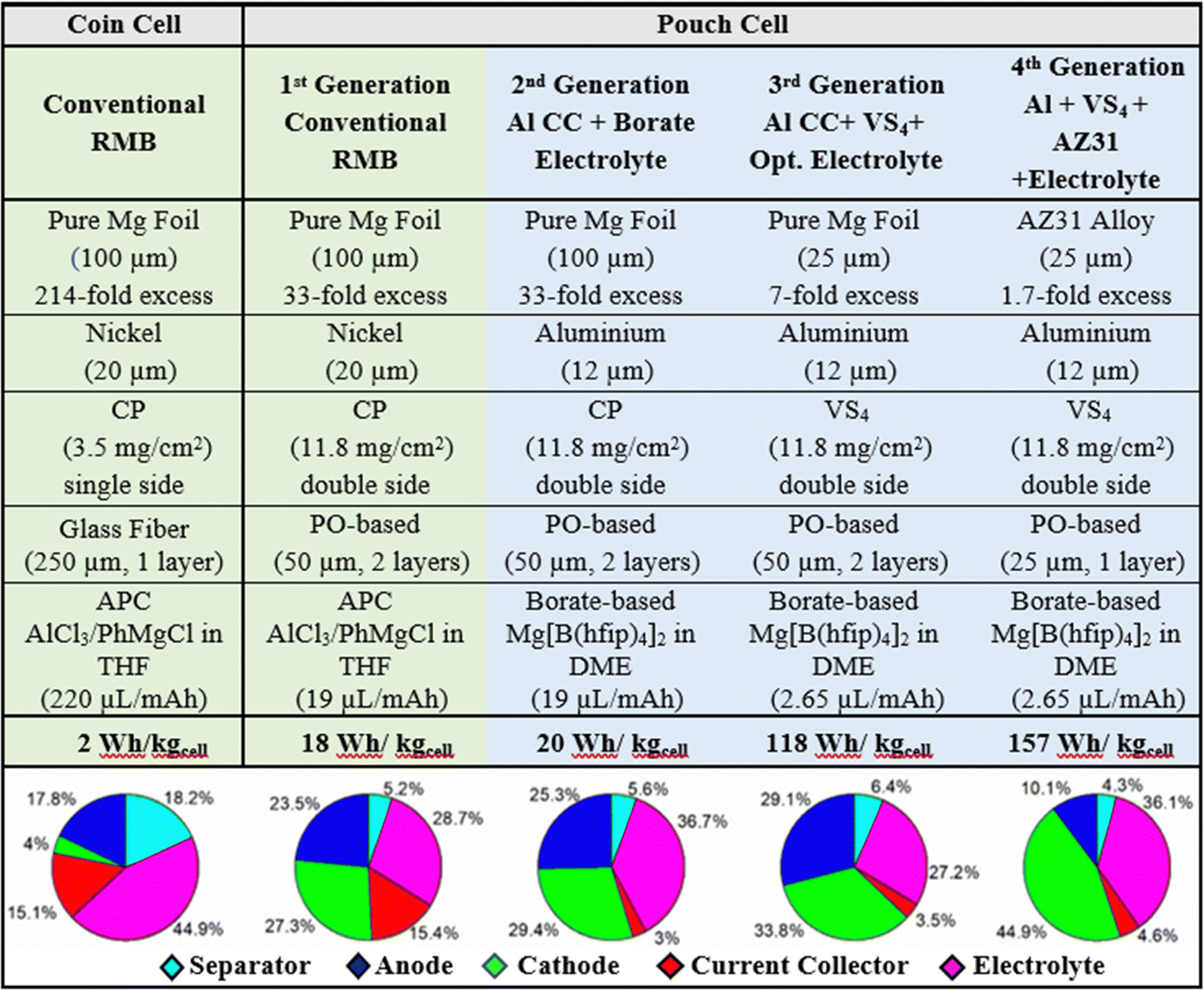
The calculation spreadsheets for the energy density and mass distribution values given in Table 2 of the article are in the ESI.†
|
6. Design and properties
Materials research experiments for energy storage applications are habitually performed in a coin cell setup, an economical small-scale configuration that necessitates only small amounts of active material and is adapted to the materials discovery, characterization, and performance-testing stage. However, the testing parameters and results thus obtained do not always accurately reflect the working parameters of practical cells, and the cells may show differences in the ratio of active materials in the electrode formulation and in active material loading, positive/negative electrode balancing, efficient components selection, electrolyte amount, and other aspects of an industrial battery design.In this section, the differences between coin cells and pouch cells are highlighted, and further impacts of the pouch cell modifications with novel materials on energy density, mass distribution, and battery design are estimated.
6.1. Conventional RMB based on a CP cathode/standard APC electrolyte solution/pure Mg metal anode
The cathode current collector accounts for a large portion of the final weight of the battery. It is well established in the literature that aluminum current collectors are a standard for high-voltage cathodes in Li-ion batteries because of their lightness (densityAl = 2.7 g cm−3) and the already established methods to produce thin foils (12 μm).142 The compatibility between the electrolyte solution and the current collector is also essential for the reversibility and good cycle life of rechargeable batteries.39
As mentioned earlier, aluminum cannot be utilized in conventional RMBs because of corrosive chloride species in APC electrolyte solutions; thus, nickel current collectors are used for electrochemical stability reasons, although they are thicker and intrinsically heavier (20 μm, densityNi = 8.9 g cm−3).26
When all these components, their properties, and amounts are factored in, they cumulatively generate very poorly optimized coin cells with flooded electrolyte solutions, poor anode/cathode balancing, and weight inefficiencies, leading to a poor energy density (2 W h kg−1); this is despite the fact that RMBs assembled in coin cells show comparable specific capacities to those reported in the literature (60 mA h gCP−1) and can be cycled more than 200 times with coulombic efficiencies above 96% (Fig. 7). The mass distributions of above-mentioned coin cells are given in Table 2 (see Experimental details in the ESI†). Accordingly, almost half of the battery mass (44.9%) comes from the flooded APC electrolyte used to wet the thick GF separator (18.2%). In parallel, the excessively thick magnesium-metal anode represents 17.8% of the total mass of the battery prototype in the coin cell configuration. The low loading of CP active material on the cathodes only contributes 4% of the total mass of the battery, and the thick nickel current collector corresponds to 15.1% of the battery mass. Even when the contributions of the binder and the carbon additive is included, the electrochemically inactive nickel current collector component makes up a disproportionate 79% percent of the total cathode weight.
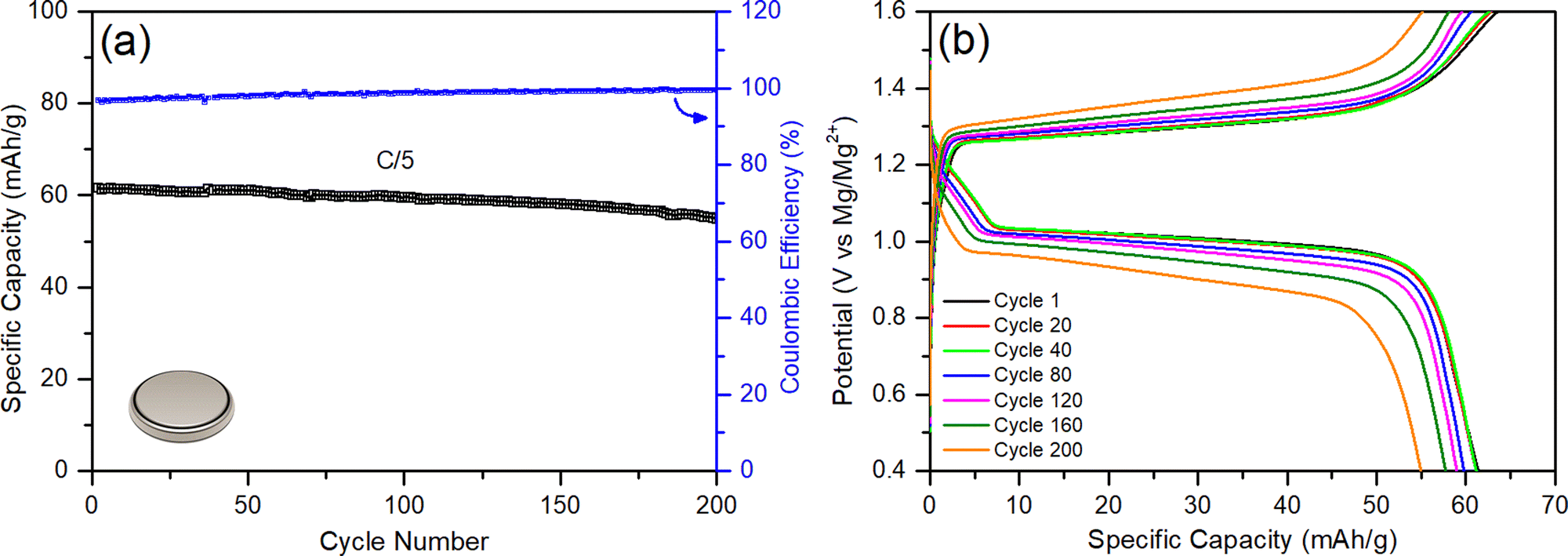 | ||
| Fig. 7 Conventional RMB-chemistry coin cell: (a) galvanostatic cycling performance at C/5, and (b) voltage profile. | ||
Clearly, all these issues must be addressed for practical RMB pouch cells design to be achieved for the first time.
The GF separator in the coin cell configuration was replaced with a double layer of a thin PO-based separator; this decreased the associated mass percentage from 18.2% to 5.2%, thanks to the great difference between the unit weights of the GF (17 mg cm−2) and PO separator (single layer, 3 mg cm−2). This change also allows the amount of APC electrolyte used in the cells to be decreased dramatically, from 44.9% to 28.7% of the total mass.
The format was changed from a single-sided coating with a low loading of material (3.5 mgCP cm−2) to a double-sided coating with a high loading of material (11.8 mgCP cm−2); this translated into a change in electrolyte–volume/capacity ratio from 214 μL mA−1 h−1 in the coin cell setup to 19 μL mA−1 h−1 in the pouch cell setup. Additionally, the increase in active material loading and the doubling of surface coverage by the active material had a direct impact on the current collector/total-cathode mass ratio, which improved from 79% to 36%. Another ratio pointing to an optimized configuration of the components is the anode/cathode capacity ratio, which must be balanced to get the best performance out of the used materials: this ratio was improved from a 214-fold excess of anode active material (Mg) to a 33-fold excess, even though the anode/cathode layer ratio was changed from 1/1 in coin cells to 5/4 in pouch cells. Overall, the combined mass of the electrolyte solution and the electrochemically inactive separator was decreased, and the relative amount of cathode active material was dramatically increased. As shown in Table 2, the optimization of these three components in relation to each other has the most drastic effect on the mass distribution.
The first pouch cell RMB prototypes were prepared by implementing only these design changes and their electrochemical performance was characterized by galvanostatic cycling at C/10. The cells demonstrate an energy density of 18 W h kg−1 (a 9-fold improvement) and a 100% energy-density retention even after 200 cycles (Table 2 and Fig. 8(a)). The coulombic efficiencies of the cells were initially >97% and improved to 99% during cycling. The voltage profiles in Fig. 8(b) present excellent reversibility and show kinetics that are characteristic of a Mg2+ intercalation reaction into the “inner ring” and “outer ring” accommodation sites of the CP cathode structure.7,108 Although the charging curves show a small overpotential (0.1 V) for the plating reaction and end with a slope for Mg2+ deintercalation, the results obtained with these pouch cells are in very good agreement with the results presented above for the coin cells and are in line with the kinetics data described in the literature.143,144 Besides, no relevant changes in the temperature of the cells were detected by external thermocouples that were attached to them. This 9-fold improvement in energy density can be considered a great achievement, especially in the first pouch cell prototypes ever presented in the literature; nonetheless, these cells are far from meeting accepted industry standard, and the cells’ design need further optimization to reach a cheap, environmentally friendly and well-performing battery system that can penetrate the energy storage markets.
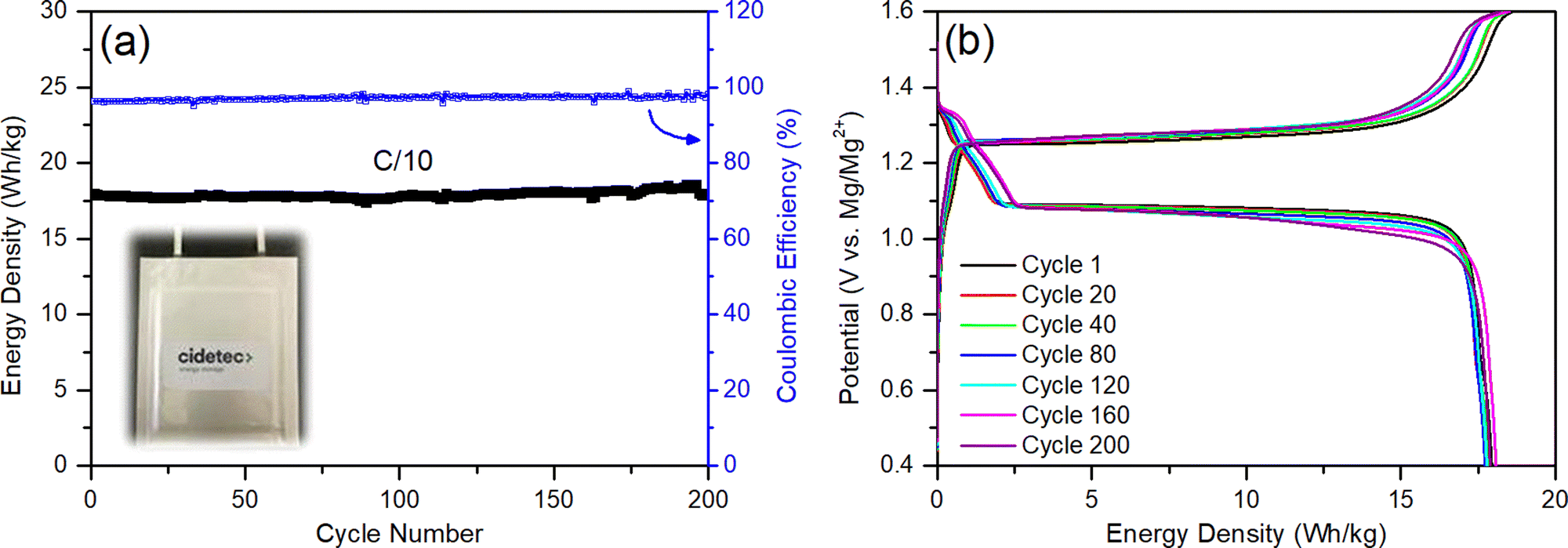 | ||
| Fig. 8 (a) Energy density and coulombic efficiency of the first generation conventional RMB pouch cell over 200 cycles. (b) Voltage profiles at C/10 for cycles 1, 20, 40, 80, 120, 160, and 200. | ||
6.2. Impact analysis of advanced materials on RMB performance and the total mass distribution in the cells
Pure-magnesium metal-foil anodes, standard APC electrolyte solutions, and CP cathodes on nickel-foil current collectors were utilized to establish the conventional RMB technology (which can be considered the most reasonable benchmark) as a reference to evaluate the electrochemical performance and mass distribution in the prototype cells. However, even after a considerable optimization of the cell design, it is clear that the battery components have their limitations, which restrict the cell performance from reaching a level suitable for practical applications. Therefore, a higher energy density in such cells requires the replacement of the conventional RMB technology components presented above and the adoption of advanced RMB chemistries.After collecting solid data related to first generation RMB prototypes in practical pouch cells configuration, loaded with most appropriate reference materials, demonstrating stable cycling performance we move to the second stage of this work and related paper. Based on solid data that we have, related to alternative components that can take the RMB technology further and upgrade it considerably in terms of energy density, we can estimate the performance of much more advanced models of RMB, in similar pouch cells (same configuration as described in Section 6.1). We show below how the possible use of new and novel components improves the energy density of RMB technology, thus making it more practically important.
A solution comprising the Mg[B(hfip)4]2 salt in DME, a thin AZ31 magnesium alloy foil anode, and a VS4 cathode were chosen for the final step in order to design more advanced RMB prototypes (in pouch cells similar to that described above). The components chosen for the next step are expected to have a less corrosive nature, higher stability, better self-standing properties, easier mechanical processibility, higher practical capacity, and improved cyclability.
To evaluate the impact of these changes, the cell components, mass distributions, and resulting performance were discussed in detail: the step-by-step replacement of each component was considered and the effect on the cell parameters and performance were estimated. In order to ensure that the methodology used in the simulation of the energy density of the different versions of magnesium batteries made, it has been decided to use this tool with a reference battery system based on LFP/graphite (Table S6, ESI†). The energy density values obtained for this base line system (135 W h kg−1) agrees with the values reported in the state of the art.145,146
In such a case, the estimations show a net 82% decrease in the mass of current collector, on account of the large difference between the densities of aluminum and nickel and the possibility of producing thin aluminum foils. Although the replacement of the electrolyte solution leads to an increase in mass because the new borate-based electrolyte solution (3 mL in the pouch cells) has a higher density (by 18%) than the APC benchmark solution; the mass of the average pouch cell sees an overall decrease of 7% from the combined changes of the solution and the cathode current collector. As a result of the estimated changes in mass, the calculated mass distribution of the main components of the cell is ca. 29.4%, 25.3%, 5.6%, 36.7%, and 3% for the cathode, anode, separator, electrolyte solution, and Al current collector, respectively, as presented in Table 2.
Compared to the first-generation RMB pouch cells, the current-collector/total-cathode mass ratio (36%) is 9.3% lower in the second-generation pouch cells, indicating an improvement in weight efficiency through a decrease in the mass of the inactive components.
Overall, the above-described changes implemented in the second-generation RMB pouch cells (using the same CP cathode material) should lead to an energy density of 20 W h kg−1, a 7% enhancement over that of the first-generation cells.
Other implications of increasing the cathode capacity are an improvement of the anode/cathode capacity balancing and solution–volume/capacity ratio. The previous 33-fold excess of pure-magnesium metal-anode capacity decreases to 7-fold when VS4/rGO is used, indicating a more efficient use of the cell weight. In addition, the amount of electrolyte solution (3 mL, 19 μL mA−1 h−1vs. CP capacity) was decreased to 1.935 mL, converging to the amount seen in industrial Li-ion batteries (2.65 μL mA−1 h−1vs. VS4 capacity). While the optimization of the amount of electrolyte solution was estimated to decrease the net cell weight by 13%, compounding it with an improvement in capacity by the change of cathode resulted in a calculated pouch cell energy density of 118 W h kg−1 (Table 2), in what can be termed as the third-generation cell in the framework of the systematic studies described herein.
The substitution of the anode component should reduce the Mg excess with respect to the cathode capacity to a more realistic 69%. Further reduction of excess magnesium may be achieved by decreasing the thickness of the alloy (only a 35% anode capacity excess should be seen when using 20 μm thick Mg anodes). To further diminish the mass percentage of the inactive components, the two layers of PO-based separator (50 μm) between each stack may be reduced to one (25 μm). In this sense, Table 2 contains the estimated energy density of our 4th generation RMB pouch cell prototype, whose calculations are explained in detail in the ESI.† Consequently, the calculations show the following estimated mass distribution for the final, 4th generation RMB prototypes (in pouch cells): ca. 44.9%, 10.1%, 4.3%, 36.1%, and 4.6% for the VS4 cathode, AZ31 alloy anode, PO-based separator, borate-based electrolyte solution, and aluminum foil current collector, respectively (Table 2).
The overall impact of the changes carried out between the first conventional RMB pouch cells and the estimated advanced chemistry in the 4th generation RMB pouch cells is emphasized in Table 3. The total weight is lowered by 40%, mainly thanks to the substitution of the current collector, the optimization of the electrolyte, and the reduction in anode thickness. In addition, the excess magnesium with respect to cathode capacity is reduced significantly (95%) by changing the cathode active material, the anode type and its thickness. The electrolyte–solution–volume/capacity ratio is also decreased by 86% by reducing the amount of electrolyte in the cells down to industrial battery levels and by using an active material that has a high specific capacity at the cathode side. The substitution of the heavy nickel current collector with an aluminum current collector reduced the current-collector/total-cathode mass ratio by 74%. Hence, by implementing all these changes, the practical energy density of the 4th generation RMB pouch cells (without taking into account the case) should reach values of approximately 157 W h kg−1, an 855% enhancement compared to the first-generation pouch cell of this work.
| Conventional RMB (1st gen) | Adv. RMB (4th gen) | Impact (%) | |
|---|---|---|---|
| Weight (g) | 10 | 6 | −40 |
| Excess Mg vs. cathode capacity | 30-fold | 1.7-fold | −95 |
| Electrolyte/capacity ratio (μL mA−1 h−1) | 18 | 3 | −86 |
| Current coll./total cathode mass | 35% | 9% | −74 |
| Energy density (W h kg−1) | 18 | 157 | +855 |
Importantly, these calculations are based on the conservative capacity values reported for VS4 cells.111 If coin cell capacity values can be realized in pouch cells, as was the case for assembled CP/APC/Mg pouch cell prototypes, the resulting energy density of next-generation RMB could thus reach impressively high values. Alternatively, if a cathode material with a higher working voltage than VS4 (ideally >2.0 V) can be used, advanced RMB would easily become more commercially relevant and even be able to compete with other post-lithium battery technologies, such as Na-ion batteries, in the field of stationary storage technologies. This is now made plausible (yet calls for experimental validation) by the use of high capacity/voltage TMO cathodes in the presence of the new Mg borate-based electrolyte solutions, which avoids the use of chloride moieties present in previously used solutions.
7. Conclusions
In summary, we have demonstrated a conventional-chemistry pouch-cell RMB prototypes for the first time, using a Mg-metal anode, a CP cathode, and an APC electrolyte solution. To overcome the electrochemical limitations of these materials, we proposed a step-by-step strategy to replace the components with a laboratory-validated aluminum current collector, a Mg[B(hfip)4]2/DME electrolyte solution, a VS4 cathode, and a very thin AZ31 magnesium alloy anode to reach a competitive, high-energy density, advanced RMB chemistry.The RMB prototypes based on the conventional chemistry was used as a reference to compare the total mass, mass distribution ratio and electrochemical performance when estimating the impact of systematic changes of components. The battery design factors such as excess magnesium with respect to the cathode capacity, the electrolyte–volume/capacity ratio, the current-collector/total-cathode mass ratio and the mass distribution and energy density of the modified pouch cells were carefully monitored and optimized.
The conservative estimation of the pouch cell energy densities revealed that by using the most advanced materials in the current literature related to RMB, it should be possible to reach a remarkable energy density approaching 160 W h kg−1. Moreover, given the properties of the new chloride-free Mg borate-based electrolyte solutions, this can be enhanced further by utilizing high-voltage/capacity cathodes based on TMOs that were found active with Mg ions (e.g., VOx and MoOx oxides). Therefore, through this work, we aim to emphasize the considerable but yet-to-be-realized potential of rechargeable magnesium batteries. We encourage further efforts to develop high-energy cathodes and thin Mg-alloy anodes and then to use more industrially relevant approaches to achieve cost-effective, sustainable, and environmentally friendly advanced chemistries for RMBs in the near future. A final important note is related to cycle life: achieving a prolonged cycle life is critically important for RMB that are supposed to be used for load leveling and large energy-storage applications. Although we did not address this critical aspect herein, thanks to the use of ethereal solutions, which are not reactive to Mg metal and cathodes which red-ox potentials do not challenge the anodic stability of the electrolyte solutions available today (<3 V vs. Mg), this goal may be achieved and well demonstrated in further studies of advanced RMB in the near future (extensive work is in progress by several groups throughout the world).
Author contributions
Rudi R. Maça: writing original draft, review and edit, conceptualization, investigation, methodology, calculations, formal analysis. J. Alberto Blázquez: conceptualization, methodology, supervision, review and edit, project administration, funding acquisition. Olatz Leonet and Ana Fernández-Barquín: investigation, supervision, review. Eneko Azaceta: investigation. Alexey Kovalevsky: AZ31 investigation, materials supply. Jean Frederic Martin and Dane Sotta: chevrel Phase cathode material supply. Zhirong Zhao-Karger and Zhenyou Li: boron-based electrolyte salt synthesis and supply. Yair Ein-Eli: solid-electrolyte part original draft, supervision. Doron Aurbach: conceptualization, supervision, writing, review and edit. Malachi Noked: conceptualization. Ayan Mukherjee, Aroa R. Mainar, Piotr Jankowski, Laurin Rademacher, Joachim Häcker, Sunita Dey, Siân E. Dutton, Sumana Kundu, Claire P. Grey, Rosa Palacin, Juan Maria García Lastra, Maximilian Fichtner contributed to the review and edit of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by European Union's Horizon 2020 research and innovation program under the FET Proactive call with grant agreement no 824066 via the “E-MAGIC” project.Notes and references
- S. Bobba, S. Carrara, J. Huisman, F. Mathieux, C. Pavel and I. European Commission. Directorate-General for Internal Market, Critical raw materials for strategic technologies and sectors in the EU: a foresight study.
- J. Betz, G. Bieker, P. Meister, T. Placke, M. Winter and R. Schmuch, Adv. Energy Mater., 2019, 9, 1803170 CrossRef.
- H. D. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265–2279 RSC.
- D. A. J. Rand and P. T. Moseley, Electrochemical Energy Storage for Renewable Sources and Grid Balancing, Elsevier Inc., 2015, pp. 201–222 Search PubMed.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- European Commission, European Climate Law, The European Parliament and the Council of the European Union, Brussels, 2021.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- S. Kundu, N. Solomatin, Y. Kauffmann, A. Kraytsberg and Y. Ein-Eli, Appl. Mater. Today, 2021, 23, 100998 CrossRef.
- R. Mohtadi, O. Tutusaus, T. S. Arthur, Z. Zhao-Karger and M. Fichtner, Joule, 2021, 5, 581–617 CrossRef CAS.
- A. Ponrouch and M. Rosa Palacín, Philos. Trans. R. Soc., A, 2019, 377, 20180297 CrossRef CAS PubMed.
- V. Bhaghavathi Parambath, Z. Zhao-Karger, T. Diemant, M. Jäckle, Z. Li, T. Scherer, A. Gross, R. J. Behm and M. Fichtner, J. Mater. Chem. A, 2020, 8, 22998–23010 RSC.
- S. Chakrabarty, J. A. Blázquez, T. Sharabani, A. Maddegalla, O. Leonet, I. Urdampilleta, D. Sharon, M. Noked and A. Mukherjee, J. Electrochem. Soc., 2021, 168, 080526 CrossRef CAS.
- M. Matsui, J. Power Sources, 2011, 196, 7048–7055 CrossRef CAS.
- R. Davidson, A. Verma, D. Santos, F. Hao, C. Fincher, S. Xiang, J. van Buskirk, K. Xie, M. Pharr, P. P. Mukherjee and S. Banerjee, ACS Energy Lett., 2019, 4, 375–376 CrossRef CAS.
- N. Bell, R. Waugh, D. Parker and K. Baker, Magnesium Recycling in the EU, 2017, pp. 1–79 Search PubMed.
- M. Fichtner, in Royal Society of Chemistry, ed. M. Fichtner, Royal Society of Chemistry, Ulm, 2019, pp. 1–16 Search PubMed.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- S. Ferrari, M. Falco, A. B. Muñoz-García, M. Bonomo, S. Brutti, M. Pavone and C. Gerbaldi, Adv. Energy Mater., 2021, 11, 2100785 CrossRef CAS.
- M. A. Hannan, S. B. Wali, P. J. Ker, M. S. A. Rahman, M. Mansor, V. K. Ramachandaramurthy, K. M. Muttaqi, T. M. I. Mahlia and Z. Y. Dong, J. Energy Storage, 2021, 42, 103023 CrossRef.
- Z. Zhang, S. Shui and Z. Editors, Green Energy and Technology Rechargeable Batteries Materials, Technologies and New Trends, 2015 Search PubMed.
- A. Maddegalla, A. Mukherjee, J. A. Blázquez, E. Azaceta, O. Leonet, A. R. Mainar, A. Kovalevsky, D. Sharon, J. F. Martin, D. Sotta, Y. Ein-Eli, D. Aurbach and M. Noked, ChemSusChem, 2021, 14, 4690–4696 CrossRef CAS PubMed.
- Y. Xiu, Z. Li, V. Bhaghavathi Parambath, Z. Ding, L. Wang, A. Reupert, M. Fichtner and Z. Zhao-Karger, Batteries Supercaps, 2021, 4, 1850–1857 CrossRef CAS.
- R. Attias, M. Salama, B. Hirsch, Y. Goffer and D. Aurbach, Joule, 2019, 3, 27–52 CrossRef CAS.
- T. D. Gregory, R. J. Hoffman and R. C. Winterton, J. Electrochem. Soc., 1990, 137, 775–780 CrossRef CAS.
- I. Shterenberg, M. Salama, Y. Gofer, E. Levi and D. Aurbach, MRS Bull., 2014, 39, 453–460 CrossRef CAS.
- C. B. Bucur, T. Gregory, A. G. Oliver and J. Muldoon, J. Phys. Chem. Lett., 2015, 6, 3578–3591 CrossRef CAS PubMed.
- I. Shterenberg, M. Salama, H. D. Yoo, Y. Gofer, J.-B. Park, Y.-K. Sun and D. Aurbach, J. Electrochem. Soc., 2015, 162, A7118–A7128 CrossRef CAS.
- R. Attias, B. Dlugatch, O. Blumen, K. Shwartsman, M. Salama, N. Shpigel and D. Sharon, ACS Appl. Mater. Interfaces, 2022, 14, 30952–30961 CrossRef CAS PubMed.
- C. Liebenow, Z. Yang and P. Lobitz, Electrochem. Commun., 2000, 2, 641–645 CrossRef CAS.
- H. S. Kim, T. S. Arthur, G. D. Allred, J. Zajicek, J. G. Newman, A. E. Rodnyansky, A. G. Oliver, W. C. Boggess and J. Muldoon, Nat. Commun., 2011, 2, 427 CrossRef PubMed.
- P. Wang and M. R. Buchmeiser, Adv. Funct. Mater., 2019, 29, 1905248 CrossRef CAS.
- R. E. Doe, R. Han, J. Hwang, A. J. Gmitter, I. Shterenberg, H. D. Yoo, N. Pour and D. Aurbach, Chem. Commun., 2014, 50, 243–245 RSC.
- P. Canepa, S. Jayaraman, L. Cheng, N. N. Rajput, W. D. Richards, G. S. Gautam, L. A. Curtiss, K. A. Persson and G. Ceder, Energy Environ. Sci., 2015, 8, 3718–3730 RSC.
- A. J. Crowe, K. K. Stringham and B. M. Bartlett, ACS Appl. Mater. Interfaces, 2016, 8, 23060–23065 CrossRef CAS PubMed.
- Z. Zhao-Karger and M. Fichtner, Front. Chem., 2019, 6, 1–12 RSC.
- J. Muldoon, C. B. Bucur, A. G. Oliver, J. Zajicek, G. D. Allred and W. C. Boggess, Energy Environ. Sci., 2013, 6, 482–487 RSC.
- J. Song, E. Sahadeo, M. Noked and S. B. Lee, J. Phys. Chem. Lett., 2016, 7, 1736–1749 CrossRef CAS PubMed.
- Z. Zhao-Karger, R. Liu, W. Dai, Z. Li, T. Diemant, B. P. Vinayan, C. Bonatto Minella, X. Yu, A. Manthiram, R. J. Behm, M. Ruben and M. Fichtner, ACS Energy Lett., 2018, 3, 2005–2013 CrossRef CAS.
- Z. Zhang, Z. Cui, L. Qiao, J. Guan, H. Xu, X. Wang, P. Hu, H. Du, S. Li, X. Zhou, S. Dong, Z. Liu, G. Cui and L. Chen, Adv. Energy Mater., 2017, 7, 1602055 CrossRef.
- F. Liu, T. Wang, X. Liu and L. Z. Fan, Adv. Energy Mater., 2021, 11, 2000787 CrossRef CAS.
- R. Mohtadi, M. Matsui, T. S. Arthur and S. J. Hwang, Angew. Chem., Int. Ed., 2012, 51, 9780–9783 CrossRef CAS PubMed.
- O. Tutusaus, R. Mohtadi, N. Singh, T. S. Arthur and F. Mizuno, ACS Energy Lett., 2017, 2, 224–229 CrossRef CAS.
- P. Canepa, G. Sai Gautam, D. C. Hannah, R. Malik, M. Liu, K. G. Gallagher, K. A. Persson and G. Ceder, Chem. Rev., 2017, 117, 4287–4341 CrossRef CAS PubMed.
- T. J. Carter, R. Mohtadi, T. S. Arthur, F. Mizuno, R. Zhang, S. Shirai and J. W. Kampf, Angew. Chem., Int. Ed., 2014, 53, 3173–3177 CrossRef CAS PubMed.
- Z. Zhao-Karger, M. E. Gil Bardaji, O. Fuhr and M. Fichtner, J. Mater. Chem. A, 2017, 5, 10815–10820 RSC.
- S. B. Son, T. Gao, S. P. Harvey, K. X. Steirer, A. Stokes, A. Norman, C. Wang, A. Cresce, K. Xu and C. Ban, Nat. Chem., 2018, 10, 532–539 CrossRef CAS PubMed.
- P. Jankowski, J. M. G. Lastra and T. Vegge, Batteries Supercaps, 2020, 3, 1350–1359 CrossRef CAS.
- J. Drews, T. Danner, P. Jankowski, T. Vegge, J. M. García Lastra, R. Liu, Z. Zhao-Karger, M. Fichtner and A. Latz, ChemSusChem, 2020, 13, 3599–3604 CrossRef CAS PubMed.
- J. Drews, P. Jankowski, J. Häcker, Z. Li, T. Danner, J. M. G. Lastra, T. Vegge, N. Wagner, K. A. Friedrich, Z. Zhao-Karger, M. Fichtner and A. Latz, ChemSusChem, 2021, 14, 4820–4835 CrossRef CAS PubMed.
- M. Rashad, M. Asif, Y. Wang, Z. He and I. Ahmed, Energy Storage Mater., 2020, 25, 342–375 CrossRef.
- Z. Zhao-Karger, R. Liu, W. Dai, Z. Li, T. Diemant, B. P. Vinayan, C. Bonatto Minella, X. Yu, A. Manthiram, R. J. Behm, M. Ruben and M. Fichtner, ACS Energy Lett., 2018, 3, 2005–2013 CrossRef CAS.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed.
- M. Guo, C. Yuan, T. Zhang and X. Yu, Small, 2022, 18, 2106981 CrossRef CAS PubMed.
- P. W. Jaschin, Y. Gao, Y. Li and S. Bo, J. Mater. Chem. A, 2020, 8, 2875–2897 RSC.
- M. Wang, Z. Wang, S. Zhao, J. Wang and S. Wang, Chin. J. Chem. Eng., 2017, 25, 1581–1597 CrossRef.
- R. Zhao, Y. Wu, Z. Liang, L. Gao, W. Xia, Y. Zhao and R. Zou, Energy Environ. Sci., 2020, 13, 2386–2403 RSC.
- S. Ma, L. Shen, Q. Liu, W. Shi, C. Zhang, F. Liu, J. A. Baucom, D. Zhang, H. Yue, H. bin Wu and Y. Lu, ACS Appl. Mater. Interfaces, 2020, 12, 43824–43832 CrossRef CAS PubMed.
- W. Xue, C. D. Sewell, Q. Zhou and Z. Lin, Angew. Chem., Int. Ed., 2022, 61, e2022065 Search PubMed.
- Y. Yoshida, K. Kato and M. Sadakiyo, J. Phys. Chem. C, 2021, 125, 21124–21130 CrossRef CAS.
- B. Park and J. L. Schaefer, J. Electrochem. Soc., 2020, 167, 070545 CrossRef.
- J. Sharma and S. Hashmi, Polym. Compos., 2019, 40, 1295–1306 CrossRef CAS.
- M. F. Bósquez-Cáceres, S. Hidalgo-Bonilla, V. Morera Córdova, R. M. Michell and J. P. Tafur, Polymers, 2021, 13, 24 CrossRef PubMed.
- K. M. Diederichsen, E. J. McShane and B. D. McCloskey, ACS Energy Lett., 2017, 2, 2563–2575 CrossRef CAS.
- P. Zhou, X. Zhang, Y. Xiang and K. Liu, Nano Res., 2022 DOI:10.1007/s12274-022-4833-1.
- Y. Pang, Y. Liu, J. Yang, S. Zheng and C. Wang, Mater. Today Nano, 2022, 18, 100194 CrossRef CAS.
- J. Cuan, Y. Zhou, T. Zhou, S. Ling, K. Rui, Z. Guo, H. Liu and X. Yu, Adv. Mater., 2019, 31, 1803533 CrossRef PubMed.
- A. H. Ahmad and F. S. A. Ghani, AIP Conf. Proc., 2009, 1136, 31–35 CrossRef CAS.
- Y. Gao, T. P. Mishra, S.-H. Bo, G. Sai Gautam and P. Canepa, Annu. Rev. Mater. Res., 2022, 52, 129–158 CrossRef.
- P. Canepa, S.-H. Bo, G. Sai Gautam, B. Key, W. D. Richards, T. Shi, Y. Tian, Y. Wang, J. Li and G. Ceder, Nat. Commun., 2017, 8, 1759 CrossRef PubMed.
- L.-P. Wang, Z. Zhao-Karger, F. Klein, J. Chable, T. Braun, A. R. Schür, C.-R. Wang, Y.-G. Guo and M. Fichtner, ChemSusChem, 2019, 12, 2286–2293 CrossRef CAS PubMed.
- D. Hubble, D. E. Brown, Y. Zhao, C. Fang, J. Lau, B. D. McCloskey and G. Liu, Energy Environ. Sci., 2022, 15, 550–578 RSC.
- K. Kisu, S. Kim, M. Inukai, H. Oguchi, S. Takagi and S. Orimo, ACS Appl. Energy Mater., 2020, 3, 3174–3179 CrossRef CAS.
- Y. Pang, Z. Nie, F. Xu, L. Sun, J. Yang, D. Sun, F. Fang and S. Zheng, Energy Environ. Mater., 2022, e12527 Search PubMed.
- K. Kajihara, H. Nagano, T. Tsujita, H. Munakata and K. Kanamura, J. Electrochem. Soc., 2017, 164, A2183 CrossRef CAS.
- Y. Wang, Z. Liu, C. Wang, X. Yi, R. Chen, L. Ma, Y. Hu, G. Zhu, T. Chen, Z. Tie, J. Ma, J. Liu and Z. Jin, Adv. Mater., 2018, 30, 1802563 CrossRef PubMed.
- M. Mao, T. Gao, S. Hou and C. Wang, Chem. Soc. Rev., 2018, 47, 8804–8841 RSC.
- Z. Ma, D. R. MacFarlane and M. Kar, Batteries Supercaps, 2019, 2, 115–127 CrossRef.
- Q. Guo, W. Zeng, S. L. Liu, Y. Q. Li, J. Y. Xu, J. X. Wang and Y. Wang, Rare Met., 2021, 40, 290–308 CrossRef CAS.
- K. Jayasayee, R. Berthelot, K. C. Lethesh and E. M. Sheridan, Magnesium Batteries: Research and Applications, The Royal Society of Chemistry, 2020, pp. 114–141 Search PubMed.
- R. Attias, M. Salama, R. Pant, Y. Gofer and D. Aurbach, J. Phys. Chem. C, 2019, 123, 16577–16587 CrossRef CAS.
- Y. Yang, X. Xiong, J. Chen, X. Peng, D. Chen and F. Pan, J. Magnesium Alloys, 2021, 9, 705–747 CrossRef CAS.
- O. I. Malyi, T. L. Tan and S. Manzhos, J. Power Sources, 2013, 233, 341–345 CrossRef CAS.
- T. Gao, S. Hou, K. Huynh, F. Wang, N. Eidson, X. Fan, F. Han, C. Luo, M. Mao, X. Li and C. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 14767–14776 CrossRef CAS PubMed.
- H. Dong, O. Tutusaus, Y. Liang, Y. Zhang, Z. Lebens-Higgins, W. Yang, R. Mohtadi and Y. Yao, Nat. Energy, 2020, 5, 1043–1050 CrossRef CAS.
- R. Attias, M. S. Chae, B. Dlugatch, M. Oliel, Y. Goffer and D. Aurbach, ACS Catal., 2020, 10, 7773–7784 CrossRef CAS.
- D. Schloffer, S. Bozorgi, P. Sherstnev, C. Lenardt and B. Gollas, J. Power Sources, 2017, 367, 138–144 CrossRef CAS.
- Z. Wu and W. A. Curtin, Nature, 2015, 526, 62–67 CrossRef CAS PubMed.
- J. Niu, Z. Zhang and D. Aurbach, Adv. Energy Mater., 2020, 10, 2000697 CrossRef CAS.
- N. Singh, T. S. Arthur, C. Ling, M. Matsui and F. Mizuno, Chem. Commun., 2013, 49, 149–151 RSC.
- K. Periyapperuma, T. T. Tran, M. I. Purcell and M. N. Obrovac, Electrochim. Acta, 2015, 165, 162–165 CrossRef CAS.
- F. Murgia, E. T. Weldekidan, L. Stievano, L. Monconduit and R. Berthelot, Electrochem. Commun., 2015, 60, 56–59 CrossRef CAS.
- L. T. Ho, Appl. Phys. Lett., 1979, 35, 409–410 CrossRef CAS.
- T. S. Arthur, N. Singh and M. Matsui, Electrochem. Commun., 2012, 16, 103–106 CrossRef CAS.
- Z. Meng, D. Foix, N. Brun, R. Dedryvère, L. Stievano, M. Morcrette and R. Berthelot, ACS Energy Lett., 2019, 4, 2040–2044 CrossRef CAS.
- S. C. Jung and Y. K. Han, J. Phys. Chem. C, 2018, 122, 17643–17649 CrossRef CAS.
- R. Mohtadi, O. Tutusaus, T. S. Arthur, Z. Zhao-Karger and M. Fichtner, Joule, 2021, 5, 581–617 CrossRef CAS.
- K. Gusieva, C. H. J. Davies, J. R. Scully and N. Birbilis, Int. Mater. Rev., 2015, 60, 169–194 CrossRef CAS.
- J. Li, Y. Huang, F. Wang, X. Meng, L. Wan and Z. Dong, Mater. Sci. Eng., A, 2020, 773, 138877 CrossRef CAS.
- W. Lu, L. Sun, Y. Zhao, T. Wu, L. Cong, J. Liu, Y. Liu and H. Xie, Energy Storage Mater., 2021, 34, 241–249 CrossRef.
- S. Vincent, J. H. Chang and J. M. Garcia Lastra, Batteries Supercaps, 2021, 4, 522–528 CrossRef CAS.
- J. Ma, G. Wang, Y. Li, C. Qin and F. Ren, J. Mater. Eng. Perform., 2019, 28, 2873–2880 CrossRef CAS.
- O. Chusid, Y. Gofer, H. Gizbar, Y. Vestfrid, E. Levi, D. Aurbach and I. Riech, Adv. Mater., 2003, 15, 627–630 CrossRef CAS.
- A. Pardo, M. C. Merino, A. E. Coy, F. Viejo, R. Arrabal and S. Feliú, Electrochim. Acta, 2008, 53, 7890–7902 CrossRef CAS.
- G. L. Song, JOM, 2012, 64, 671–679 CrossRef CAS.
- A. el Kharbachi, O. Zavorotynska, M. Latroche, F. Cuevas, V. Yartys and M. Fichtner, J. Alloys Compd., 2020, 817, 153261 CrossRef CAS.
- Z. Guo, S. Zhao, T. Li, D. Su, S. Guo and G. Wang, Adv. Energy Mater., 2020, 10, 1903591 CrossRef CAS.
- K. Tang, A. Du, S. Dong, Z. Cui, X. Liu, C. Lu, J. Zhao, X. Zhou and G. Cui, Adv. Mater., 2020, 32, 1904987 CrossRef CAS PubMed.
- E. Levi, A. Mitelman, D. Aurbach and M. Brunelli, Chem. Mater., 2007, 19, 5131–5142 CrossRef CAS.
- K. Lu, S. Gao, R. J. Dick, Z. Sattar and Y. Cheng, J. Mater. Chem. A, 2019, 7, 6038–6044 RSC.
- T. Koketsu, J. Ma, B. J. Morgan, M. Body, C. Legein, W. Dachraoui, M. Giannini, A. Demortière, M. Salanne, F. Dardoize, H. Groult, O. J. Borkiewicz, K. W. Chapman, P. Strasser and D. Dambournet, Nat. Mater., 2017, 16, 1142–1148 CrossRef CAS PubMed.
- Z. Li, B. P. Vinayan, P. Jankowski, C. Njel, A. Roy, T. Vegge, J. Maibach, J. M. G. Lastra, M. Fichtner and Z. Zhao-Karger, Angew. Chem., Int. Ed., 2020, 59, 11483–11490 CrossRef CAS PubMed.
- X. Chen, X. Wang, Z. Wang, W. Yu and Y. Qian, Mater. Chem. Phys., 2004, 87, 327–331 CrossRef CAS.
- C. Peng, H. Lyu, L. Wu, T. Xiong, F. Xiong, Z. Liu, Q. An and L. Mai, ACS Appl. Mater. Interfaces, 2018, 10, 36988–36995 CrossRef CAS PubMed.
- R. Verrelli, A. P. Black, C. Frontera, J. Oro-Sole, M. E. Arroyo-De Dompablo, A. Fuertes and M. Rosa Palacín, ACS Omega, 2019, 4, 8943–8952 CrossRef CAS PubMed.
- S. Dey, J. Lee, S. Britto, J. M. Stratford, G. Cibin, E. N. Keyzer, S. J. Cassidy, M. Elgaml, M. T. Dunstan and C. P. Grey, J. Am. Chem. Soc., 2020, 142, 19588–19601 CrossRef CAS PubMed.
- M. Vincent, V. S. Avvaru, M. C. Rodríguez, M. Haranczyk and V. Etacheri, J. Power Sources, 2021, 506(1), 230118 CrossRef CAS.
- I. Gomez, O. Leonet, J. Alberto Blazquez, H. J. Grande and D. Mecerreyes, ACS Macro Lett., 2018, 7, 419–424 CrossRef CAS PubMed.
- B. Pan, J. Huang, Z. Feng, L. Zeng, M. He, L. Zhang, J. T. Vaughey, M. J. Bedzyk, P. Fenter, Z. Zhang, A. K. Burrell and C. Liao, Adv. Energy Mater., 2016, 6, 1600140 CrossRef.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- Q. Guo, W. Zeng, S. L. Liu, Y. Q. Li, J. Y. Xu, J. X. Wang and Y. Wang, Rare Met., 2021, 40, 290–308 CrossRef CAS.
- T. D. Gregory, R. J. Hoffman and R. C. Winterton, J. Electrochem. Soc., 1990, 137, 775–780 CrossRef CAS.
- A. Medina, C. Pérez-Vicente and R. Alcántara, Materials, 2021, 14, 7488 CrossRef CAS PubMed.
- Y. Liang, R. Feng, S. Yang, H. Ma, J. Liang and J. Chen, Adv. Mater., 2011, 23, 640–643 CrossRef CAS PubMed.
- R. Demir-Cakan, M. R. Palacin and L. Croguennec, J. Mater. Chem. A, 2019, 7, 20519–20539 RSC.
- R. Y. Wang, C. D. Wessells, R. A. Huggins and Y. Cui, Nano Lett., 2013, 13, 5748–5752 CrossRef CAS PubMed.
- M. Liu, A. Jain, Z. Rong, X. Qu, P. Canepa, R. Malik, G. Ceder and K. A. Persson, Energy Environ. Sci., 2016, 9, 3201–3209 RSC.
- H. D. Yoo, J. R. Jokisaari, Y. S. Yu, B. J. Kwon, L. Hu, S. Kim, S. D. Han, M. Lopez, S. H. Lapidus, G. M. Nolis, B. J. Ingram, I. Bolotin, S. Ahmed, R. F. Klie, J. T. Vaughey, T. T. Fister and J. Cabana, ACS Energy Lett., 2019, 4, 1528–1534 CrossRef CAS.
- G. Sai Gautam, P. Canepa, A. Abdellahi, A. Urban, R. Malik and G. Ceder, Chem. Mater., 2015, 27, 3733–3742 CrossRef CAS.
- R. Verrelli, A. P. Black, C. Pattanathummasid, D. S. Tchitchekova, A. Ponrouch, J. Oró-Solé, C. Frontera, F. Bardé, P. Rozier and M. R. Palacín, J. Power Sources, 2018, 407, 162–172 CrossRef CAS.
- S. Dey, J. Lee, S. Britto, J. M. Stratford, E. N. Keyzer, M. T. Dunstan, G. Cibin, S. J. Cassidy, M. Elgaml and C. P. Grey, J. Am. Chem. Soc., 2020, 142, 19588–19601 CrossRef CAS PubMed.
- Z. Li, S. Ding, J. Yin, M. Zhang, C. Sun and A. Meng, J. Power Sources, 2020, 451, 227815 CrossRef CAS.
- C. S. Rout, B. H. Kim, X. Xu, J. Yang, H. Y. Jeong, D. Odkhuu, N. Park, J. Cho and H. S. Shin, J. Am. Chem. Soc., 2013, 135, 8720–8725 CrossRef CAS PubMed.
- P. Gressier, I. Myung-hwan Whangbo, C. Alain Meerschaut and J. Rouxell, Inorg. Chem., 1984, 23, 1221–1228 CrossRef CAS.
- I. P. Batra, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 9162–9165 CrossRef CAS PubMed.
- H. Dong, Y. Liang, O. Tutusaus, R. Mohtadi, Y. Zhang, F. Hao and Y. Yao, Joule, 2019, 3, 782–793 CrossRef CAS.
- M. Kirchhöfer, J. von Zamory, E. Paillard and S. Passerini, Int. J. Mol. Sci., 2014, 15, 14868–14890 CrossRef PubMed.
- F. J. Günter, C. Burgstaller, F. Konwitschny and G. Reinhart, J. Electrochem. Soc., 2019, 166, A1709–A1714 CrossRef.
- H. Li, Joule, 2019, 3, 911–914 CrossRef CAS.
- S. Dörfler, H. Althues, P. Härtel, T. Abendroth, B. Schumm and S. Kaskel, Joule, 2020, 4, 539–554 CrossRef.
- S. Chen, C. Niu, H. Lee, Q. Li, L. Yu, W. Xu, J. G. Zhang, E. J. Dufek, M. S. Whittingham, S. Meng, J. Xiao and J. Liu, Joule, 2019, 3, 1094–1105 CrossRef CAS.
- D. Karabelli and K. P. Birke, Appl. Sci., 2021, 11, 7592 CrossRef CAS.
- P. Saha, M. K. Datta, O. I. Velikokhatnyi, A. Manivannan, D. Alman and P. N. Kumta, Prog. Mater. Sci., 2014, 66, 1–86 CrossRef CAS.
- K. Itaoka, I. T. Kim, K. Yamabuki, N. Yoshimoto and H. Tsutsumi, J. Power Sources, 2015, 297, 323–328 CrossRef CAS.
- Z. Li, T. Diemant, Z. Meng, Y. Xiu, A. Reupert, L. Wang, M. Fichtner and Z. Zhao-Karger, ACS Appl. Mater. Interfaces, 2021, 13, 33123–33132 CrossRef CAS PubMed.
- J. Hao, F. Yang, S. Zhang, H. He, G. Xia, Y. Liu, C. Didier, T. Liu, W. K. Pang, V. K. Peterson, J. Lu and Z. Guo, Des. Res., 2020, 117, 2815–2823 CAS.
- M. S. E. Houache, C. H. Yim, Z. Karkar and Y. Abu-Lebdeh, Batteries, 2022, 8(7), 70 CrossRef CAS.
- Z. Zhao-Karger, R. Liu, W. Dai, Z. Li, T. Diemant, B. P. Vinayan, C. Bonatto Minella, X. Yu, A. Manthiram, R. J. Behm, M. Ruben and M. Fichtner, ACS Energy Lett., 2018, 3, 2005–2013 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee04121a |
| This journal is © The Royal Society of Chemistry 2023 |