
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn universal oxygen electrode for reversible solid oxide electrochemical cells at reduced temperatures†
Jun Hyuk
Kim‡
a,
Dongyeon
Kim‡
b,
Sejong
Ahn‡
c,
Kyeong Joon
Kim§
 b,
SungHyun
Jeon
c,
Dae-Kwang
Lim
cd,
Jun Kyu
Kim¶
c,
Uisik
Kim
cd,
Ha-Ni
Im
b,
Bonjae
Koo
e,
Kang Taek
Lee
*bf and
WooChul
Jung
*cf
b,
SungHyun
Jeon
c,
Dae-Kwang
Lim
cd,
Jun Kyu
Kim¶
c,
Uisik
Kim
cd,
Ha-Ni
Im
b,
Bonjae
Koo
e,
Kang Taek
Lee
*bf and
WooChul
Jung
*cf
aDepartment of Chemical Engineering, Hongik University, Seoul, Republic of Korea
bDepartment of Mechanical Engineering, KAIST, Daejeon, Republic of Korea. E-mail: leekt@kaist.ac.kr
cDepartment of Materials Science and Engineering, KAIST, Daejeon, Republic of Korea. E-mail: wcjung@kaist.ac.kr
dKorea Electric Power Corp. Research Institute, Daejeon 34056, Republic of Korea
eSchool of Chemistry and Energy, Sungshin Women's University, Seoul, Republic of Korea
fKAIST Institute for the NanoCentury, Daejeon, Republic of Korea
First published on 12th July 2023
Abstract
Solid oxide electrochemical cells (SOCs) are promising energy storage and conversion devices that represent a facile and sustainable route for converting chemical fuels into electricity and vice versa on demand. In particular, the discovery of oxygen-electrode materials that exhibit excellent reactivity and durability is the key to related device technology. Here, we present Ta-doped BaCoO3−δ catalysts that exhibit record-breaking electrode performance in both fuel cell and electrolysis operations, which were demonstrated with two types of SOCs using oxygen ion- and proton-conducting electrolytes. The introduction of pentavalent Ta ions allows the parent oxide to maintain a cubic perovskite structure with high symmetry, which substantially improves its phase stability and electronic and ionic conductivity, and even catalytic activity for oxygen reduction and evolution. Hence an assortment of best-performing fuel cell and electrolysis cell results are recorded; for example, a proton-conducting SOC conveying a peak power density of 2.26 W cm−2 at 650 °C. The design principle of the versatile electrode presented in this study not only allows for the realization of high-performance SOCs but also contributes to a broader-real world impact: a multipurpose, standardized electrode of high demand will result in significant cost reductions for related electrochemical devices.
Broader contextAs the climate and environmental crisis deepens, concerns about the development of clean energy sources are rapidly increasing. Solid oxide electrochemical cells (SOCs) offer appealing opportunities, due to their high efficiency, low carbon emission, good tolerance against fuel impurities, excellent modularity, and capability for reversible operation. Although some technologies have reached commercialization, the broad market penetration of SOCs requires continuous material breakthrough to meet the productivity and economic demands. Particularly, finding an active and reliable oxygen-electrode material is at the heart to realize SOCs with practical performances, as the electrocatalytic reactions of oxygen (i.e., oxygen reduction and evolution) are notoriously sluggish. Here, we tackle this hurdle by developing a universal oxygen-electrode composed from Ta-stabilized barium cobalt perovskite oxides (BaCo1−xTaxO3−δ) that is applicable to both oxygen- and proton-conducting cells, as well as for electricity and hydrogen production. Such that, high-performance SOCs that greatly outrival previous records were enabled. Our findings indicate that our developed materials are highly promising candidates for a universal oxygen-electrode, which can substantially accelerate the large-scale application of SOC technologies. |
Introduction
At the dawn of a hydrogen economy, electrochemical cells consisting of ceramic compartments are expected to have a transformative impact on society due to their high energy-conversion efficiencies at low-to-intermediate temperatures (400–750 °C).1–4 While polymer electrolyte-based equivalents suffer from poor reversibility, as the active catalysts for oxygen reduction and evolution reactions are different (Pt and IrO2, respectively, which are both scarce, expensive, and highly volatile in cost),5,6 these ceramic cells, otherwise known as solid oxide electrochemical cells (SOCs), are fabricated with cheaper and scalable materials, and can often reversibly operate. Therefore, SOCs are set to play a decisive role in the field of energy storage and conversion technologies.7–10 Even compared to batteries, SOCs possess unique advantages which include specialized long-term and large-scale storage. Once converted, H2 can be stored either as a gas or a liquid for an indefinite period of time and has higher gravimetric and volumetric specific energy densities (∼0.7 kW h L−1 at ∼70 MPa in gaseous form) than those of lithium-ion batteries (∼0.3 kW h L−1 in the same conditions).7,11,12 Moreover, unlike batteries, SOC storage capacity is not limited by the weights of the active materials needed to assemble the units, alleviating scaling cost challenges.Currently, there are two types of SOCs: those with oxygen ion-conducting electrolytes, and those with proton-conducting analogs. Historically, SOCs based on oxygen ion conductors have been the mainstream and cornerstones of many industrial-grade products.13,14 However, the intrinsically high activation energy for oxygen ion conduction has shifted research attention further toward the use of proton conductors.15–18 Although proton-conducting SOCs are promising, the corresponding accessories for stack fabrication may not reach the level of commercialization.19 Currently, it is nearly impossible to predict which type of SOC will predominate the future market. In such a transition period, fuel cell developers in the industry may require a “universal” catalyst that can function exceptionally in both oxygen ion- and proton-conducting devices, so that they can adapt smoothly and balance the profit to expand options for large-scale SOC applications. However, finding such catalysts remains a tough challenge.
Herein, we highlight frontier opportunities for designing and enabling new universal oxygen electrocatalysts based on BaCoO3−δ, with an emphasis on tailoring the cubic perovskite structure with appropriate dopants. Replacing Co with Ta effectively promotes the formation of a nearly-ideal cubic perovskite structure, which results in rapid ionic and electronic conductivity and oxygen electrocatalytic kinetics. Exceptional power densities (1.03 and ∼1.14 W cm−2 at 550 °C with Gd0.2Ce0.8O2−δ (GDC)- and BaZr0.4Ce0.4Y0.1Yb0.1O3−δ (BZCYYb)-based cells, respectively), outstanding water electrolysis current densities (1.1 A cm−2 at 1.3 V and 550 °C with the BZCYYb-based cell) and remarkable stabilities (>700 h in fuel cell mode, >350 h in electrolysis mode, 100 h of reversible operations, and 10 rapid thermal cycles) were demonstrated, which are among the best values ever reported. Our findings indicate that Ta-stabilized barium cobalt perovskite oxides (BaCo1−xTaxO3−δ) are highly promising universal oxygen-electrode materials, regardless of the SOC type. The development of such single electrode materials compatible with multi-cell types and functions will enable production at scale, simplified manufacturing operations, and cost reductions, which contribute to the accelerated commercialization of SOC technologies.
Results and discussion
Design and physical characterization of the materials
One of the prominent mixed-conducting oxides is perovskite, provided with the chemical formula of ABO3. The structure of perovskite affords great flexibility in the substitution of A- and B-sites, allowing for easier tuning of the desired properties; in this case, mixed conduction of oxygen ions and protons to be incorporated in oxygen- and proton-conducting SOCs, respectively. Here, placing Ba on the A-site has many advantages. First, the larger ionic radii of Ba (e.g., Ba: 161 pm > Sr: 144 pm > La: 136 pm > Pr: 99 pm) can lower the activation energy for oxygen ion migration in the crystal lattice, which is advantageous for securing sufficient oxygen ion conductivity at reduced temperatures.20,21 Secondly, the low electronegativity of Ba improves the overall oxide-ion basicity, facilitating the formation of protonic defects and improving proton conductivity.22 Moreover, the terrestrial abundance of Ba exceeds that of other A-site substituents (Ba: 425 ppm > Sr: 370 ppm > La: 30 ppm > Pr: 8.2 ppm), and the price of Ba is significantly lower than that of other A-site substituents (e.g., Ba: ∼0.25 USD kg−1 < La: ∼4.78 USD kg−1 < Sr: ∼6.53 USD kg−1 < Pr: 103 USD kg−1),23 which supports the scalability of the catalytic materials based on Ba.For B-site cations, redox-active transition metals, such as Co, Fe, Ni, and Mn, are typically used to achieve sufficient electronic conductivity and catalytic activity. Here, we chose Co as the major B-site cation because its strong metal 3d and O 2p hybridization character are claimed to improve the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) catalytic activities.24,25
Unfortunately, the oxide of pure BaCoO3−δ does not crystallize with cubic symmetry. Fig. 1(a) shows the representative diffraction patterns of the as-synthesized BCO obtained through a conventional solid-state reaction calcined at 1200 °C. Fig. 1(a) clearly shows that oxides purely composed of Ba and Co fail to structure cubic perovskites, without additional B-site dopants. Rather, the structure was found to be face-sharing hexagonal (BaCoO2.6) with a space group of P63/mmc (PDF #: 01-071-2453, a = b = 5.6710 Å, c = 28.5450 Å), in which structure-aligned vacancies impaired the transport properties of mobile charge carriers within the lattice.26
 | ||
| Fig. 1 X-ray diffraction (XRD) patterns of (a) as-synthesized barium cobaltites (BaCoO3−δ, BCO) without B-site dopants and (b) Ta substituted barium cobaltites (BaCo1−xTaxO3−δ, with X = 0.1, 0.2, 0.3, and 0.4). BCT10, BCT20, BCT30 and BCT40 denote the oxides having 10, 20, 30, and 40 mol% Ta in B-sites, respectively. It is noted that Si powder was deliberately added to serve as a reference peak (Si(111): 28.44°) for a more accurate analysis amongst the fabricated samples. (c) A high-resolution transmission electron microscopy (HRTEM) image of BCT20, (inset) fast Fourier transform (FFT) pattern. (d) Atomically resolved HAADF image of BCT20 and Elemental EDS maps for Ba, Co, Ta and overlaid. (e) Thermogravimetric profiles and (f) Raman spectra of BCT10, BCT20, BCT30 and BCT40. (g) The ratio of the thermally-expanded lattice parameters to the lattice parameters at 100 °C, Δa/a0, of BCO, BCT10, and BCT20 as a function of temperature. | ||
In order to improve charge-carrier mobility, a highly symmetric lattice (ideally cubic for a single perovskite) is preferred, as it enables facile rotation of corner-sharing oxygen octahedra, which supports isotropic transport and a greater overlap of atomic orbitals for small-polaron transport.27,28 Moreover, a symmetric lattice results in shorter O–O bond distances that are beneficial for proton hopping and mobility.24 In order to produce a symmetric cubic structure, an additional stabilizing element at the B-site was introduced to create vacancy disorder. Among several candidates (including Ta5+, Nb5+, and Mo6+), the stabilizing element of pentavalent Ta was chosen. A number of beneficial effects can be attributed to Ta doping. First, tantalum oxide is known to be a highly corrosion-resistant material that may effectively enhance the chemical stability of the resulting perovskite.29,30 Second, tantalum has a fixed valence state (+5), which is effective in mitigating the coefficient of thermal expansion. Finally, the relatively lower electronegativity of Ta compared to other stabilizing elements (Ta: 1.5, Nb: 1.6, Mo: 2.16) may create additional oxygen vacancies and protonic defects favorable for oxygen catalysis.31,32
Crystal structures of BaCo1−xTaxO3−δ (X = 0.1, 0.2, 0.3, and 0.4) powders were analyzed using powder X-ray diffraction (XRD), which revealed that Ta doping successfully promoted the formation of cubic BCO perovskites (Fig. 1(b)). BCT10, BCT20, BCT30, and BCT40 denote the oxides with 10, 20, 30, and 40 mol% Ta in the B-sites, respectively. Further Rietveld refinement results (Fig. S1 and Table S1, ESI†) showed that 10 mol% Ta doping is not enough to fully achieve the cubic perovskites, though no apparent hexagonal structure footprint could be assigned after 20 mol% or higher Ta doping was performed. The deduced lattice parameters for the isostructural BCT20, BCT30, and BCT40 oxides were a = 4.085, 4.076, and 4.071 Å, respectively. High-resolution transmission electron microscopy (HR-TEM) analysis and the corresponding SAED pattern of the prepared oxides (Fig. S2–S6, ESI†) were consistent with the XRD refinement outcome. Notably, streaking in SAED patterns of BCT10 implied that the twin or stacking faults were formed in the process of transitioning from an edge-sharing hexagonal to a corner-sharing cubic structure (Fig. S3, ESI†). The observation of such defects is clear evidence of our claim that Ta doping promotes the phase transition from hexagonal to cubic structures. For samples having Ta 20% or higher, such streaking is hardly observed, signifying pure cubic phase formation. A representative HR-TEM image of cubic BCT20 is shown in Fig. 1(c).
Fig. 1(d) shows the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and EDS mapping of BCT20, resolving the atomic structure for cubic perovskite with different metal cations occupying the A and B sublattices. Particularly, the HAADF image and EDS map prove that Ta is very randomly (homogenously) distributed. Ta is located exactly at the Co site and is consistently observed in both the HADDF image and EDS map. We note here that, in HADDF analysis focusing on the top surface, Ta EDS shows a relatively bright part where the Ta atom exists (as Ta exists only 20%), which is a typical result supporting the homogeneous distribution. Additional EELS and EDS analysis can be found in Fig. S7 (ESI†).
The actual stoichiometry of the resulting powders was analyzed using inductively-coupled plasma-optical emission spectroscopy (ICP-OES) measurements (Table S2, ESI†), and the results confirmed virtually identical compositions to the intended compositions.
As the size of the Ta5+ ion (64 pm) is similar to that of Co3+ (61 pm), the shrinkage in lattice parameters at higher Ta content is mainly due to the reduction of oxygen vacancies in the BCO lattice by the pentavalent Ta dopant. Thermogravimetric analysis (TGA) indicated that the weight change of the sample (i.e., oxygen loss) between 20 °C and 900 °C decreased precipitously as the Ta content increased (BCT10: 2.265%, BCT20: 1.926%, BCT30: 1.259%, and BCT40: 1.021%). Such results further support that the oxygen vacancies gradually decrease with an increase in Ta dopants (see Fig. 1(e)).
Fig. 1(f) shows the Raman spectra of BaCo1−xTaxO3−δ (for X = 0, 0.1, 0.2, 0.3, and 0.4). Note that the Raman spectra of undoped BCO are reminiscent of typical brownmillerite crystals such as SrCoO2.5 and Ba2In2O5.33,34 Specifically, the band appearing at approximately 608 cm−1 can be assigned to the structural feature arising from the Ag mode of alternating Co tetrahedrons and octahedrons, a sign of aligned, immobile oxygen vacancies.33 By looking at the Raman spectra of BCT10, it is apparent that this band feature lingers on – the replacement of 10 mol% Co by Ta is not sufficient to fully achieve a cubic perovskite character. Instead, the pure perovskite modes (at around 530 and 670 cm−1) were only available after more than 20 mol% of Co was superseded.35 However, excessive Ta doping (>30 mol% Ta) hampers the perovskite breathing mode (A1g, [transition metal (TM)-O6]) at approximately 670 cm−1, indicating the loss of mobile oxygen vacancies.36 These results show that the BCT20 catalyst performed best (as will be demonstrated later) due to the high mobile oxygen vacancy concentrations achieved without the formation of a deleterious hexagonal phase.
Because of the fixed valence properties of Ta5+ (X-ray photoelectron spectroscopy results focusing on the Ta 4f band are shown in Fig. S8, ESI†), its doping can possibly lower the thermal expansion coefficient (TEC) of the perovskites.37 A high-temperature XRD experiment was conducted to characterize the phase structure of BCO, BCT10, and BCT20 (Fig. S9–S11, ESI† respectively). Between room temperature and 700 °C, all the investigated oxides seemed to maintain their original structures (i.e., complementary DSC results displayed no observable thermal phase transition peak, Fig. S12, ESI†), while the XRD peaks shifted to lower angles as the temperature increased. Thus, the expansion of the unit cell parameters as a function of temperature was refined (and plotted in Fig. S13, ESI†). Accordingly, the ratio of the thermally-expanded lattice parameters to the lattice parameters at 100 °C (Δa/a0) is shown in Fig. 1(g). Significantly, this ratio clearly decreased when the Ta concentration increased. As for the Ta-doped samples, the lattice expansion appears to have different slopes at around 400 °C, presumably due to the spin-state reorientation of Co ions.38 Nonetheless, the TEC for each of the catalysts was calculated by fitting the slope from 373 to 973 K. As a result, the TEC for BCT20 was found to be as low as 10.4 × 10−6 K−1, signifying the positive role of Ta in mitigating a rise in the TEC. Indeed, the TEC of the undoped BCO was 22.7 × 10−6 K−1, nearly two times higher than that of BCT20.38 As the TEC values are reported with widely used oxygen-ion and proton-conducting electrolytes are in the range of 8–12 × 10−6 K−1,39 the insignificant TEC mismatch identified in BCT20 is propitious for accomplishing excellent thermomechanical compatibility with the SOC electrolytes, as will be presented in the later section.
Electrochemical performance evaluation
To determine the oxygen electrocatalytic activity, symmetric cells with an electrode|electrolyte|electrode configuration were devised and measured using electrochemical impedance spectroscopy (EIS). For these half-cell tests, two different electrolytes were used: Sm0.2Ce0.8O2−δ (SDC) and BaZr0.4Ce0.4Y0.1Yb0.1O3−δ (BZCYYb). Each electrolyte renders different conduction characteristics, as the major charge carriers are different (O2− and H+, respectively). By using two different electrolytes, we can test whether the electrochemical performance of our developed materials is competent in both electrolytes. As the conducting media are different, the specific reactions occurring in each cell are distinctive. For example, the ORR and OER occurring on the SDC (O2− conductor) can be represented as non-proton-concerting reactions as follows (Fig. 2(a)):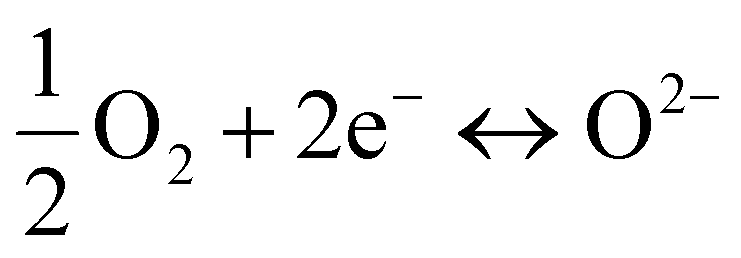 | (1) |
 | ||
| Fig. 2 (a) Schematics of a symmetric cell supported with Sm0.2Ce0.8O2−δ (SDC). (b) Electrochemical impedance spectroscopy (EIS) of BaCo0.8Ta0.2O3−δ (BCT20) oxygen-electrode at 450–650 °C measured with oxygen-ion-conducting SDC symmetric cells in dry air (pO2 = 0.21 atm). (c) Performance comparison of the BCT20 oxygen-electrode over several benchmark oxygen-electrodes measured in an oxygen-conductor-based symmetric cell configuration. Note: La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF),10 BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY),38 PrBa0.8Ca0.2Co2O5+δ (PBCC),10 Sr0.9Ce0.1Fe0.8Ni0.2O3−δ (SCFN),41 Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF),42 PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF),43 Ba0.95Ag0.05Co0.8Nb0.1Ta0.1O3−δ (e-BACNT),32 SrCo0.8Nb0.1Ta0.1O3−δ (SCNT),31 and Ba0.5Sr0.5Co0.6Fe0.2Zr0.1Y0.1O3−δ (BSCFZY).44 (d) Arrhenius plots of the Dchem and kchem for BCT20 at 450–650 °C. (e) Electrical conductivities of BCO, BCT10, BCT20, BCT30, and BCT40 in the temperature range of 300–800 °C. (f) Schematics of symmetric cell supported with BaZr0.4Ce0.4Y0.1Yb0.1O3−δ (BZCYYb). (g) EIS of the BCT20 oxygen-electrode at 450–650 °C measured with proton-conducting BaZr0.4Ce0.4Y0.1Yb0.1O3−δ (BZCYYb) symmetric cells in humidified air (pO2 = 0.21 atm, pH2O = 0.03 atm). (h) Performance comparison of BCT20 oxygen-electrode relative to several benchmark oxygen-electrodes measured in a protonic conductor-based symmetric cell configuration. Note: La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF),47 BaCo0.4Fe0.4Zr0.2O3−δ (BCFZ),47 BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY),16 BaGd0.8La0.2Co2O6−δ (BGLC, pO2 = 0.23 atm),48 Pr2NiO4+δ,49 Gd0.3Ca2.7Co3.82Cu0.18O9−δ (GCCCO),50 BaCo0.7(Ce0.8Y0.2)0.3O3−δ (BCCY, 2.5 vol% H2O/air),51 Ba0.95Ag0.05Co0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY-Ag). (i) Thermogravimetric profiles of BCT20. The sample was heated in dry air and cooled down in humidified air (pH2O = 0.03 atm). (j) Proton concentration of BCT20. | ||
Whereas the ORR and OER occurring on BZCYYb (H+ conductor) can be represented as proton-concerting reactions (Fig. 2(f)):
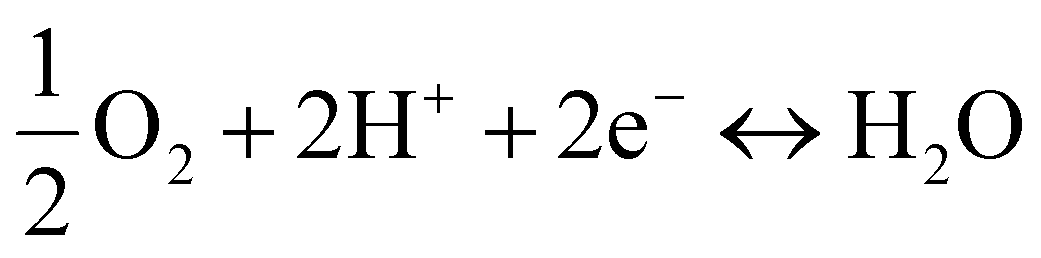 | (2) |
As more reaction participants are present in protonic conductors, they comprise more complicated elementary reactions,40 hence the reported electrode polarization resistances (Rp) in proton-conducting half-cells are generally greater than those in oxygen-conducting half-cells.
Nonetheless, we first measured the impedance spectra of the BaCo1−xTaxO3−δ electrodes, for X = 0, 0.1, 0.2, 0.3, and 0.4, on the oxygen ion-conducting SDC electrolyte in the temperature range 450–650 °C (the full sets of impedance arcs are shown in Fig. S14–S18 (ESI†), and the corresponding Arrhenius plots are shown in Fig. S19, ESI†). Representative Nyquist plots of BCT20 are shown in Fig. 2(b) (a cross-sectional SEM image of the SDC symmetric cell is shown in Fig. S20(a), ESI†). The impedance spectra showed a depressed arc, and the corresponding Rp value of the electrode is extracted from the difference between the high- and low-frequency intercepts to the real axis of the impedance loop. Among the selected compositions, BCT20 outperformed all the other compositions, as envisaged with previous Raman characterizations. Specifically, the area-specific resistance (ASR) of the BCT20 oxygen electrode in dry air at 650, 600, 550, and 500 °C were only ∼0.005, 0.013, 0.031, and 0.087 Ω cm2, respectively. Even at 450 °C, the polarization loss of the electrode was less than 1 Ω cm2 (0.29 Ω cm2).
Indeed, the exceptional activities found with BCT20 outranked all the previously reported state-of-the-art oxygen-electrodes, as shown in Fig. 2(c),10,31,32,38,41–44 including Nb and Ta co-doped SCO perovskites (SCNT), which exhibited ∼0.16 Ω cm2 at 500 °C,31 self-assembled nano-composites of Sr0.9Ce0.1Fe0.8Ni0.2O3−δ (SCFN),41 Ba0.5Sr0.5Co0.6Fe0.2Zr0.1Y0.1O3−δ (BSCFZY),44 and Ba0.95Ag0.05Co0.8Nb0.1Ta0.1O3−δ (e-BACNT) electrodes decorated with ex-solved Ag nanocatalysts.32 A stability test in synthetic air at 500 °C was also performed using the half-cell configuration, and excellent stability was demonstrated for 100 h (Fig. S21, ESI†).
Such an exemplary performance from the BCT20 electrode can be associated with the intrinsic nature of the material, such as rapid oxygen surface exchange and bulk diffusivity. Therefore, electrical conductivity relaxation (ECR) was analyzed to determine the surface exchange coefficient (kchem) and the bulk diffusion coefficient (Dchem) for the electrode. The ECR, with a rectangular bar pellet of BCT20, was measured between pO2 = 0.21 and 1 atm. Significantly, the deduced kchem and Dchem values of BCT20 were ∼6.4 × 10−3 cm s−1 and ∼1.8 × 10−4 cm2 s−1 (χ2 = 0.99) at 600 °C (Fig. 2(d)). Hence, one can assume that the exceptional performance of the BCT20 electrode can be attributed to facile O2 surface exchange kinetics, coupled with rapid ion diffusion. Moreover, an additional O2-temperature-programmed desorption (TPD) test showed that O2 desorption of the BCT20 sample started to occur at a relatively low temperature of ∼290 °C, implying that Ta doping and cubic perovskite crystallization significantly improved the weakly bonded oxygen concentrations, which is beneficial for a fast surface oxygen exchange rate (Fig. S22, ESI†). The electronic conductivities of BaCo1−xTaxO3−δ (for X = 0, 0.1, 0.2, 0.3, and 0.4) were also measured, the results of which are presented in Fig. 2(e). The figure shows that the electrical conductivity decreased as the Co content decreased and that the conductivity of BCT20 was determined to be 1–12 S cm−1 between 350–800 °C.
The oxygen-electrode can be easily poisoned by impurity gases such as CO2 and H2O in the atmosphere, resulting in significant degradation of the electrochemical activity. To assess the impact of Ta doping on the chemical stability of the electrodes, EIS measurements were conducted in humidified air (3 vol% H2O) with varying concentrations of CO2 (1, 3, and 5 vol%) at 600 °C (Fig. S23(a), ESI†). As a result, the BCT20 sample was proven to be more tolerant toward CO2 gas than the BCO sample. Given that the CO2 concentration in the ambient atmosphere is only ∼0.4%, it can be concluded that BCT20 has adequate tolerance towards such acidic gases. Even in 5 vol% CO2 exposure, the ASR increase found in BCT20 samples was only ∼0.09 Ω cm2, which is a significantly smaller value than that found in BCO samples (∼0.6 Ω cm2).
Moreover, the CO2-poisoned electrochemical activity of the BCT20 electrode was regenerated to some extent after ceasing the CO2 feed. These findings demonstrate the effectiveness of the Ta doping strategy in enhancing the chemical stability of the oxygen-electrode toward air impurities.45
To further understand the mechanism by which the examined electrodes are able to tolerate CO2 poisoning, additional Raman spectroscopy analysis was performed by annealing the samples in a 2% CO2 atmosphere for 18 h. Raman spectroscopy is a powerful technique for depicting the chemical state of the electrode surface (Fig. S23(b), ESI†).3,10,46 Such that, a sharp peak at ∼1059 cm−1 was assigned to the BCO samples, indicating secondary –CO3 formation. However, the BCT20 samples showed no noticeable surface changes, signifying their superior chemical tolerance against CO2.
Next, we conducted another half-cell test of the BCT20 electrode over the BZCYYb protonic electrolytes (Fig. 2(g)). ASR values of 0.12, 0.23, 0.39, 0.65, and 1.2 Ω cm2 were recorded in wet air (pH2O = 0.03 atm) at 650, 600, 550, 500, and 450 °C, respectively. When compared with state-of-the-art oxygen-electrode materials designed for protonic devices, the performance of BCT20 compares favorably to other materials reported (Fig. 2(h)).16,47–51 For example, the ASR values of BCT20 are comparable to those of the self-assembled nano-composite of BCCY,44 or water-mediated Ag ex-solved Ba0.95Ag0.05Co0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY-Ag).52 The activation energy measured for the BCT20 sample was 0.64 eV, which is, to the best of our knowledge, one of the lowest electrode activation energy reported for a protonic symmetric cell. The results suggest that the BCT20 electrode will be favored at lower temperatures because of its low activation energy, and is therefore ideally suited for operations in proton-conducting SOCs.
The superior activity of BCT20 in protonic electrolytes implies bulk proton incorporation into the electrode, including the entire electrode surface acting as active reaction sites. The water uptake in perovskite oxides is largely determined by the basicity of the oxide ions, and the low electronegativity of Ba can enhance proton uptake in the perovskite structure. Therefore, the extent of water uptake by BCT20 was evaluated using TGA (Fig. 2(i)). To this end, the sample mass was first recorded in dry air conditions by heating the sample to 800 °C (see the red line in Fig. 2(i)), before another set of weight data was recorded as the sample was cooled in humidified air (pH2O = 0.03 atm, see the blue line in Fig. 2(i)). The clear mass deviation between the two atmospheres can be attributed to water uptake into the bulk BCT20 perovskite crystal by the hydration reaction, as follows:52
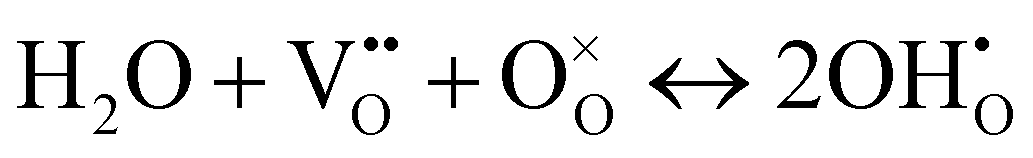 | (3) |
Thereby, meaningful amounts of proton concentration, ranging from 4.83 mol% at 300 °C to 1.57 mol% at 700 °C, were found (Fig. 2(j)), implying that BCT20 is a triple-conducting oxide. Significantly, the proton concentration found in BCT20 (2.36 mol% at 500 °C) is comparable to that in the state-of-the-art triple-conducting oxides (Table 1).53–55
| Triple-conducting oxides | Proton concentration (mol%) | Experimental conditions |
|---|---|---|
| Note: PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF), PrBa0.8Ca0.2Co2O5+δ (PBCC), BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY). | ||
| BCT20 (this work) | 2.36 | 500 °C, pO2 = 0.21 atm, pH2O = 0.03 atm |
| PBSCF54 | 2 | 500 °C, pO2 = 0.19atm, pH2O = 0.02 atm |
| PBCC53 | 0.39 | 500 °C, pO2 = 0.21 atm, pH2O = 0.03 atm |
| BCFZY55 | 0.21 | 500 °C, pO2 = 0.043 atm, pH2O = 0.095 atm |
For the impedance data, it is suggested that each frequency range represents different electrochemical steps in the Nyquist plot. Hence, to assess the Ta-doping effect, a distribution of relaxation time (DRT) analysis was conducted to deconvolute the impedance data into specific frequency ranges. The DRT plots of the BCT20 electrode based on the SDC (Fig. S24(a), ESI†) and BZCYYb (Fig. S24(b), ESI†) electrolytes showed three distinct peaks: high-frequency (HF), mid-frequency (MF), and low-frequency (LF). The literature suggests that the HF peak is attributed to charge transfer across the interface, the MF peak is related to oxygen surface exchange, and the LF peak is associated with mass transfer.56,57 Our DRT results are consistent with the previous literature, as the HF peak shows independence toward pO2 variation, whereas the MF peak is significantly affected by pO2 change. In this light, when the DRT functions of BCT20 and BCO were compared (Fig. S24(c) and (d), ESI†), it was found that Ta stabilization had a significant impact on improving the charge transfer process (HF) and surface exchange kinetics (MF).
Demonstration with oxygen ion-conducting SOCs
The exceptional activity of BCT20 was examined using Gd0.2Ce0.8O2−δ (GDC20) electrolyte-based button cells with oxygen-ion-conducting characteristics (Fig. 3(a) and (b)). SEM revealed the surface morphology of the BCT20 oxygen-electrode, with the material demonstrating good porosity capable of facile gas diffusion (Fig. 3(c)). The oxygen-electrode was tightly bonded to the GDC20 electrolyte with a thickness of ∼8.3 μm.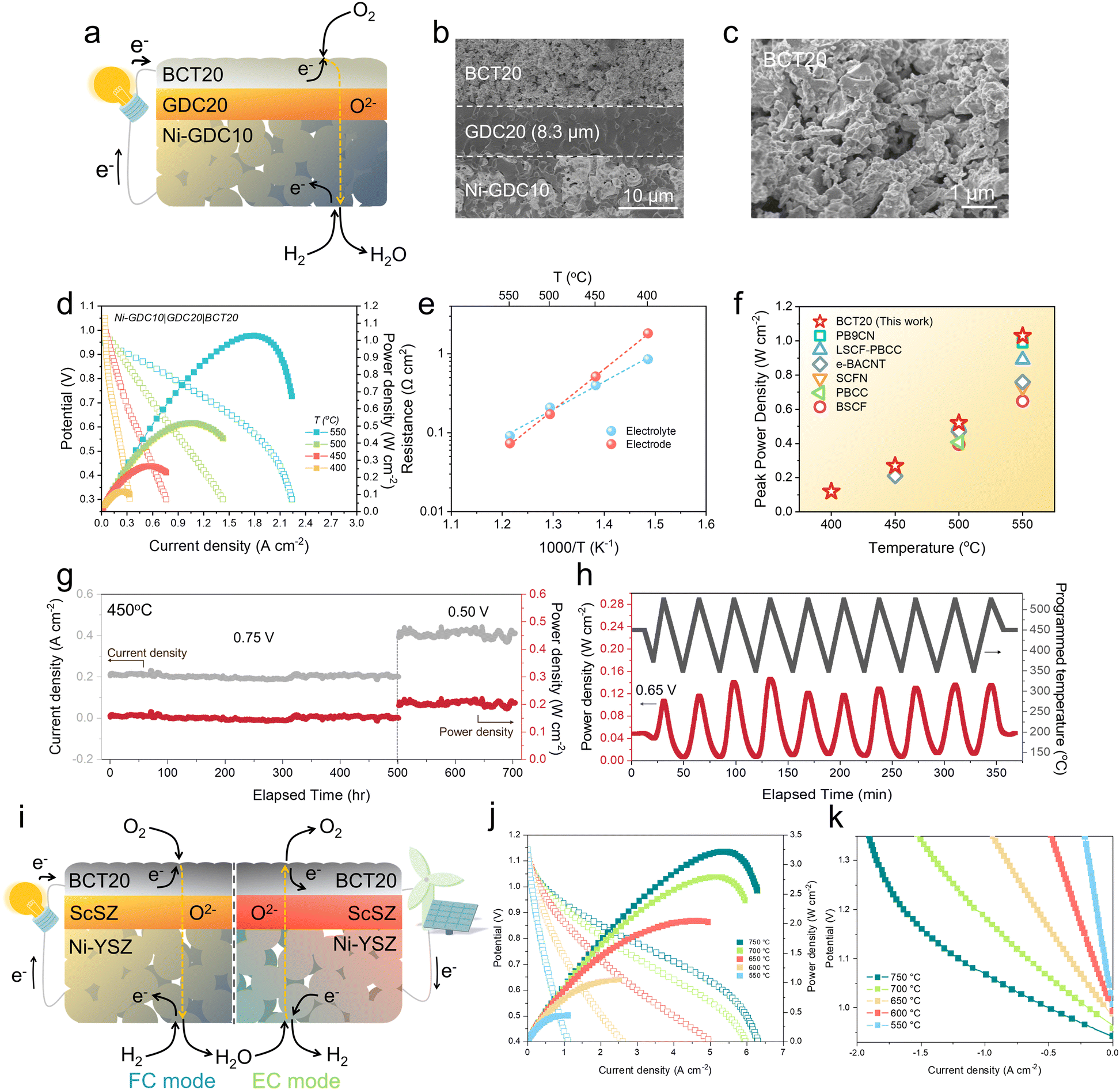 | ||
| Fig. 3 Performance demonstration with button cells with oxygen-ion-conducting electrolytes: (a) Schematics of the oxygen-ion-conducing SOC based on SDC electrolyte. (b) Cross-sectional SEM image of the single cell. (c) Scanning electron microscope (SEM) image depicting porous oxygen-electrode microstructures of BCT20. (d) Typical I–V–P curves of a single cell with a configuration of Ni-Gd0.1Ce0.9O2−δ (GDC10)|Gd0.2Ce0.8O2−δ (GDC20)|BaCo0.8Ta0.2O3−δ (BCT20) at temperature range of 400–550 °C. (e) Arrhenius plots of electrode and electrolyte resistances. (f) Comparison of peak power density of BCT20 over recently reported oxygen-ion-conducting SOCs. Note: PrBa0.9Co1.96Nb0.04O5+δ (PB9CN),58 PrBa0.8Ca0.2Co2O5+δ infiltrated La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF-PBCC),56 Ba0.95Ag0.05Co0.8Nb0.1Ta0.1O3−δ (e-BACNT),32 Sr0.9Ce0.1Fe0.8Ni0.2O3−δ (SCFN),41 PrBa0.8Ca0.2Co2O5+δ (PBCC),10 Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF).42 (g) Long-term stability data measured with Ni-Sm0.2Ce0.8O2−δ (SDC)|SDC|BCT20 at 450 °C. (h) Thermal cycling of SDC-based single cell between ∼350 and ∼525 °C. A constant voltage load of 0.65 V is applied during the cycling. (i) Schematics of the oxygen-ion-conducing SOC based on the ScSZ electrolyte. (j) Typical I–V–P curves of a single cell with a configuration of Ni-YSZ fuel electrode |Ni-ScSZ functional layer|ScSZ electrolyte|Sm0.075Nd0.075Ce0.85O2−δ (SNDC) interlayer|BCT20 oxygen electrode at temperature range of 550–750 °C. (k) Typical I–V curves of a BCT20 single cell with ScSZ elelctrolyte under humidified H2 (50 vol% H2O/H2) on the fuel electrode and dry air on the oxygen electrode at temperature range of 550–750 °C. | ||
Fig. 3(d) shows the typical polarization curves of the single cells measured in the temperature regime of 400–550 °C (EIS is shown in Fig. S25 (ESI†) and respective Arrhenius plots are presented in Fig. 3(e)). Incredibly, the single cell comprising the BCT20 oxygen-electrode reached a peak power density of as high as 1.03 W cm−2 at 550 °C, performing very highly among the oxygen-ion-conducting SOCs at such low temperatures (Fig. 3(f)).10,32,41,42,56,58 Indeed, a similarly-configured SOC with a BSCF oxygen-electrode could only muster a peak power density of 0.63 W cm−2 at 550 °C.42 Even at 400 °C, the highly-active BCT20 material enabled a notable peak power density of 0.12 W cm−2. Although the demonstrated power densities are impressive as of present, it is worth mentioning that there is room for further improvement, which can be accomplished either by delicate nanoscale oxygen-electrode microstructure control or by reducing the thickness of the electrolytes.59 Given the excellent performance outcomes above, employment of the BCT20 oxygen-electrode may significantly reduce the operating temperature to lower than 500 °C, which opens up opportunities for future low-temperature SOC technologies.
High oxygen-electrode activity may be obtained at the expense of its chemical stability. To evaluate it, the reliability of the BCT20 oxygen-electrode was examined in long-term stability tests using an SDC-based single cell, with a Ni-SDC|SDC|BCT20 configuration. The microstructure of the SDC-based cell (Fig. S26, ESI†) was almost the same as that of the GDC-based single cell. Thus, an excellent peak power density of 1.27 W cm−2 at 600 °C was delivered by the BCT20 oxygen-electrode (Fig. S27, ESI†). Fig. 3(g) illustrates results from a long-term stability test measured at 450 °C. For the first 500 h, the cell was operated under a constant voltage load of 0.75 V, before the applied voltage was switched to 0.5 V to operate for another 204 h. For more than 700 h of continuous operation, the current density and power generation of the single cell displayed stable behavior, indicating that the BCT20 is a highly durable and reliable oxygen-electrode material for practical operations.
Although it is not frequently reported in lab-scale reports, the thermal cyclability of the fuel cell is also of high significance to guarantee the service life of the power generation system.44,52,60 In particular, a SOC stack can experience thermal shock during the rapid start-up process, restarting after a hot standby, periodic maintenance, or shutdown emergencies. While the thermal stress during the aforementioned process can be attributed to various solid-state components, such as interconnects, seals, and manifolds,61 the electrode–electrolyte interface of the cell is also worthy of investigation. In an earlier part of this work, we derived the TEC (10.4 × 10−6 K−1) of BCT20 via high-temperature XRD analysis. The small TEC difference discovered between the developed oxygen-electrode and oxygen-conducting electrolyte (in this case, SDC, 12.3 × 10−6 K−1) seems attractive for consolidating a strong electrode–electrolyte interfacial contact. To verify this theory, the cell was subjected to 10 rapid ramping cycles between ∼350 °C and ∼525 °C, while a constant voltage of 0.65 V was applied to the cell (Fig. 3(h)). The programmed heating and cooling rates were ∼10 °C min−1, and the testing furnace was purposely opened to allow ambient air in, to satisfy the fast cooling rates. Notably, the cell completely recovered its performance upon completion of the thermal cycling (Fig. S28, ESI†), demonstrating the excellent thermomechanical compatibility between the BCT20 oxygen-electrode and SDC electrolyte.
To prove that the BCT20 electrode is compatible with other oxygen-conducting electrolytes rather than ceria, an oxygen-conducting electrolyte of scandium-stabilized zirconia (ScSZ) was additionally used in order to fabricate reversible oxygen-conducting SOCs (Fig. 3(i)). In the fuel cell (FC) mode, The I–V–P characteristics of the BCT20 single cell employing the ScSZ electrolyte were measured over a temperature range of 550–750 °C, as shown in Fig. 3(j). Throughout the entire temperature range, the BCT20 single cell exhibited an open-circuit voltage exceeding 1.1 V. Furthermore, the maximum power densities that the BCT20 cell achieved were 3.21, 2.78, 2.04, 1.05, and 0.45 W cm−2 at 750, 700, 650, 600, and 550 °C, respectively. The electrolysis cell (EC) mode performance of the BCT20 single cell using the ScSZ electrolyte was then measured between 550–750 °C, in humidified H2 (50 vol% H2O/H2) on the fuel-electrode side and dry air on the oxygen-electrode side (Fig. 3(k)). The current densities of the BCT20 single cell at 750, 700, 650, 600, and 550 °C were 1.85, 1.39, 0.83, 0.42, and 0.19 A cm−2, respectively (at 1.3 V). Based on the results obtained from the FC and EC modes, we have determined that the BCT20 oxygen electrode exhibits excellent performance and compatibility with the ScSZ electrolyte-based SOCs.
Demonstration with proton-conducting SOCs
Next, we implemented BCT20 on reversible proton-conducting SOCs (Fig. 4(a)). Fig. 4(b) shows a representative cross-sectional SEM image of the BCT20 single cell, configured as Ni-BZCYYb|BZCYYb|BCT20. A pulsed laser deposition (PLD)-BCT20 layer was inserted for better contact between the oxygen-electrode and electrolyte (Fig. 4(c)). Thereby, proton-conducting SOCs with remarkably high performances were enabled, achieving peak power densities of 2.26, 1.64, 1.14, 0.76, and 0.45 W cm−2 at 650, 600, 550, 500, and 450 °C, respectively. To the best of our knowledge, a peak power density of >2 W cm−2 has rarely been achieved for proton-conducting SOCs operating at 650 °C (Fig. 4(i), left).7,16,50–54,62 It is worth mentioning that even without the PLD processing, a notable peak power density of 1.83 W cm−2 at 650 °C was achieved with the BCT20 oxygen-electrode alone (Fig. S29, ESI†), with excellent long-term durability for 500 hours (Fig. S30, ESI†).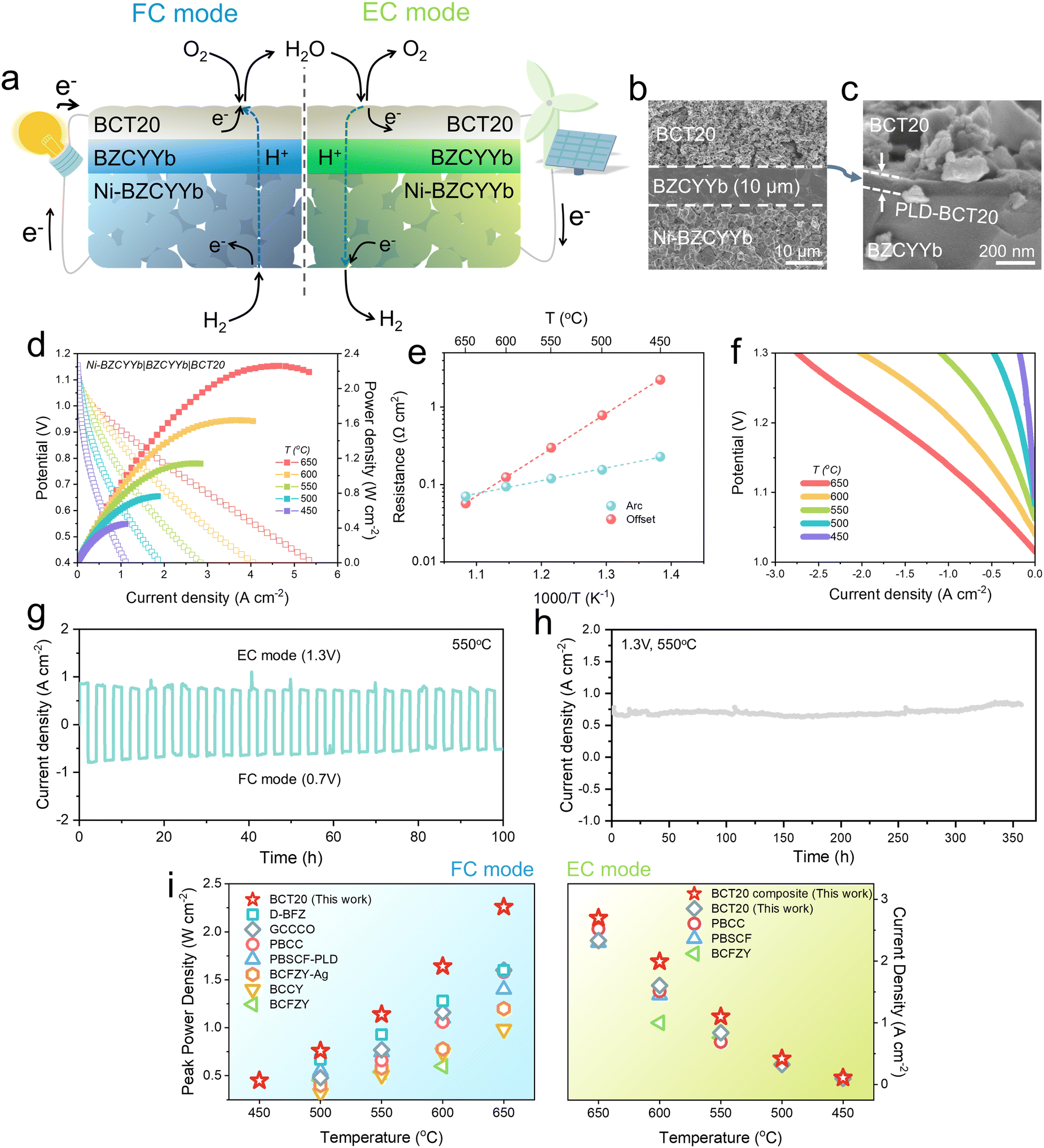 | ||
| Fig. 4 Performance demonstration with button cells with proton-conducting electrolytes: (a) Schematics of the reversible proton-conducing SOC. (b) Cross-sectional SEM image of the single cell. (c) Scanning electron microscope (SEM) image depicting pulsed laser deposited-BaCo0.8Ta0.2O3−δ (PLD-BCT20) layer between oxygen-electrode–electrolyte interface. (d) Typical I–V–P curves of a single cell with a configuration of Ni-BaZr0.4Ce0.4Y0.1Yb0.1O3−δ (BZCYYb)|BZCYYb|BCT20 at temperature range of 450–650 °C. (e) Arrhenius plots of electrode and electrolyte resistances. (f) Typical I–V curves of a single cell with humidified H2 (3 vol% H2O/H2) in the fuel-electrode and humidified air (3 vol% H2O/air) in the oxygen-electrode. (g) Reversible operation of the proton-conducing SOC. The operation was switched between electrolysis cell (EC) and fuel cell (FC) modes at 1.3 V and 0.7 V, respectively, at 550 °C. (h) Long-term stability of a proton-conducing SOC measured in electrolysis mode at 1.3 V at 550 °C. (i) Performance comparison of BCT20 over state-of-the-art proton-conducing SOCs (left: peak power density at FC mode, right: current density at 1.3 V at EC mode). Note: Ba0.875Fe0.875Zr0.125O3−δ (D-BFZ),62 Gd0.3Ca2.7Co3.82Cu0.18O9−δ (GCCCO),50 PrBa0.8Ca0.2Co2O5+δ (PBCC),53 PrBa0.5Sr0.5Co1.5Fe0.5O5+δ with PLD layer (PBSCF-PLD),54 PBSCF,8 Ba0.95Ag0.05Co0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY-Ag),52 BaCo0.7(Ce0.8Y0.2)0.3O3−δ (BCCY),51 BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY).7,16 | ||
The exceptional performance demonstrated here can therefore be attributed to the highly efficient BCT20 oxygen-electrode, as the arc resistance measured at OCV is significantly lower than that of other reported proton-conducting SOCs (Fig. 4(e) and Fig. S31, ESI†). For instance, the electrode resistance obtained in our cell was only ∼0.12 Ω cm2 at 600 °C, which is approximately two times lower than the values obtained with other proton-conducting SOCs, such as in PBCC-based (0.24 Ω cm2) or BCCY-based (0.25 Ω cm2) cells.51
The water-splitting ability of the BCT oxygen-electrode in the electrolysis mode was also investigated. Fig. S32 (ESI†) depicts the typical polarization curves of the single cell measured in the temperature range of 450–650 °C, in humidified H2 (3 vol% H2O/H2) in the fuel-electrode and humidified air (3 vol% H2O/air) in the oxygen-electrode. Notable current densities of 2.33, 1.60, and 0.84 A cm−2 were reached at 1.3 V at 650, 600, and 550 °C, respectively. The faradaic efficiencies of hydrogen production were then analyzed using gas chromatography, the results of which are shown in Fig. S33 (ESI†).
Importantly, the water electrolysis rate can be further improved, by fabricating a BCT20-BaZr0.1Ce0.7Y0.1Yb0.1O3−δ composite electrode (weight ratio of 65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35), presumably due to advanced hydration capability of BaZr0.1Ce0.7Y0.1Yb0.1O3−δ. Thereby, with a composite electrode, superior current densities of ∼2.70, 1.99, and 1.1 A cm−2 were achieved at 1.3 V at 650, 600, and 550 °C, respectively (Fig. 4(f) and (i)). Again, the results from the water electrolysis mode are comparable or often outrival the previous records (Fig. 4(i), right).7,8,16,53
:35), presumably due to advanced hydration capability of BaZr0.1Ce0.7Y0.1Yb0.1O3−δ. Thereby, with a composite electrode, superior current densities of ∼2.70, 1.99, and 1.1 A cm−2 were achieved at 1.3 V at 650, 600, and 550 °C, respectively (Fig. 4(f) and (i)). Again, the results from the water electrolysis mode are comparable or often outrival the previous records (Fig. 4(i), right).7,8,16,53
The cell was then put through a reversible operation between the EC mode and FC mode, by sequentially alternating the voltage between 1.3 V and 0.7 V, respectively (Fig. 4(g)). Each mode lasted for 2 h, and a total of 25 cycles (FC and EC modes) were tested for 100 h at 550 °C. As such, the SOC showed fairly stable behavior, indicating that excellent reversibility was achieved with the BCT20 electrode. After the reversible operation, the cell was operated in the EC mode by setting the voltage at 1.3 V (Fig. 4(h)). Over 357 h of continuous water electrolysis at 550 °C, stable current densities were drawn from the cell, indicating that BCT20 is a highly durable and reliable oxygen-electrode material for realistic water splitting operations.
Conclusions
In summary, the synthesized highly active and durable oxygen-electrode materials, Ta-stabilized barium cobaltites (BaCo1−xTaxO3−δ) demonstrated exceptional SOC performance at low to intermediate temperatures. The doping strategy of pentavalent Ta was proved effective in promoting the phase stability of the cubic perovskites, and the 20 mol% Ta substituted sample (BCT20) achieved record-level electrochemical performances; the electrode was efficient in both ORR and OER and was not significantly influenced by the conducting-type of electrolyte. The BCT20 electrode even enabled stable SOC operations for both the FC and EC modes for hundreds of hours. The systematic investigation shown in this study indicates that BCT20 bodes well for its use as a next-generation SOC oxygen-electrode for large-scale energy storage and conversion applications.Author contributions
K. T. L. and W. J. conceived and supervised the project. J. H. K. designed the material compositions and performed the following experiments: electrochemical performance evaluation of oxygen-conducting SOC tests and TEM, Raman, and TGA analyses. D. K. performed proton-conducting SOC tests. S. A. synthesized the materials and conducted XRD analysis, half-cell measurements for oxygen conductors, and ECR. K. J. K. assisted with the proton-conducting SOC tests. S. J. performed the half-cell measurements of the proton conductors. D. L. assisted in electrical conductivity measurements and ECR. J. K. K. performed the pulsed laser deposition. U. K. assisted with the TGA experiments for water uptake. H. L. assisted in the electrolyte powder fabrication. B. K. conducted XPS analysis. J. H. K., K. T. L., and W. J. contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (Ministry of Science and ICT (MSIT)) (No. 2021M3H4A1A01002695 and No. 2021M3H4A3A02086497).References
- Y. Chen, B. deGlee, Y. Tang, Z. Wang, B. Zhao, Y. Wei, L. Zhang, S. Yoo, K. Pei, J. H. Kim, Y. Ding, P. Hu, F. F. Tao and M. Liu, Nat. Energy, 2018, 3, 1042–1050 CrossRef CAS
.
- E. D. Wachsman and K. T. Lee, Science, 2011, 334, 935–939 CrossRef CAS PubMed
- J. H. Kim, M. Liu, Y. Chen, R. Murphy, Y. Choi, Y. Liu and M. Liu, ACS Catal., 2021, 11, 13556–13566 CrossRef CAS
- D. Kim, I. Jeong, K. J. Kim, K. T. Bae, D. Kim, J. Koo, H. Yu and K. T. Lee, J. Korean Ceram. Soc., 2022, 1–22 Search PubMed
- Y. Wang, D. Y. Leung, J. Xuan and H. Wang, Renewable Sustainable Energy Rev., 2016, 65, 961–977 CrossRef CAS
- Z. Pu, G. Zhang, A. Hassanpour, D. Zheng, S. Wang, S. Liao, Z. Chen and S. Sun, Appl. Energy, 2021, 283, 116376 CrossRef CAS
- C. Duan, R. Kee, H. Zhu, N. Sullivan, L. Zhu, L. Bian, D. Jennings and R. O’Hayre, Nat. Energy, 2019, 4, 230 CrossRef CAS
- S. Choi, T. C. Davenport and S. M. Haile, Energy Environ. Sci., 2019, 12, 206–215 RSC
- D. Kim, J. W. Park, M. S. Chae, I. Jeong, J. H. Park, K. J. Kim, J. J. Lee, C. Jung, C.-W. Lee and S.-T. Hong, J. Mater. Chem. A, 2021, 9, 5507–5521 RSC
- J. H. Kim, S. Yoo, R. Murphy, Y. Chen, Y. Ding, K. Pei, B. Zhao, G. Kim, Y. Choi and M. Liu, Energy Environ. Sci., 2021, 14, 1506–1516 RSC
- A. Sartbaeva, V. Kuznetsov, S. Wells and P. Edwards, Energy Environ. Sci., 2008, 1, 79–85 RSC
- D. Teichmann, W. Arlt, P. Wasserscheid and R. Freymann, Energy Environ. Sci., 2011, 4, 2767–2773 RSC
- P. Wray, Am. Ceram. Soc. Bull., 2010, 89, 25 CAS
-
B. M. Goodman, N. M. Bosowski and T. W. Sanford, Doctoral dissertation, Worcester Polytechnic Institute, 2011
- L. Yang, S. Wang, K. Blinn, M. Liu, Z. Liu, Z. Cheng and M. Liu, Science, 2009, 326, 126–129 CrossRef CAS PubMed
- C. Duan, J. Tong, M. Shang, S. Nikodemski, M. Sanders, S. Ricote, A. Almansoori and R. O’Hayre, Science, 2015, 349, 1321 CrossRef CAS PubMed
- C. Duan, R. J. Kee, H. Zhu, C. Karakaya, Y. Chen, S. Ricote, A. Jarry, E. J. Crumlin, D. Hook, R. Braun, N. P. Sullivan and R. O’Hayre, Nature, 2018, 557, 217–222 CrossRef CAS PubMed
- H. An, H.-W. Lee, B.-K. Kim, J.-W. Son, K. J. Yoon, H. Kim, D. Shin, H.-I. Ji and J.-H. Lee, Nat. Energy, 2018, 3, 870–875 CrossRef CAS
- L. Q. Le, C. H. Hernandez, M. H. Rodriguez, L. Zhu, C. Duan, H. Ding, R. P. O'Hayre and N. P. Sullivan, J. Power Sorces, 2021, 482, 228868 CrossRef CAS
- Z. Shao and M. O. Tadé, Chem. Soc. Rev., 2016, 37, 1568 Search PubMed
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef
- R. Zohourian, R. Merkle, G. Raimondi and J. Maier, Adv. Funct. Mater., 2018, 28, 1801241 CrossRef
- https://en.institut-seltene-erden.de/current-prices-of-strategic-metals/ .
- M. Papac, V. Stevanović, A. Zakutayev and R. O’Hayre, Nat. Mater., 2020, 1–13 Search PubMed
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Science, 2017, 358, 751 CrossRef CAS PubMed
- A. Jacobson and J. Hutchison, J. Solid State Chem., 1980, 35, 334–340 CrossRef CAS
- X. Liu, H. Zhao, J. Yang, Y. Li, T. Chen, X. Lu, W. Ding and F. Li, J. Membr. Sci., 2011, 383, 235–240 CrossRef CAS
- S. Tao and J. T. Irvine, Chem. Rec., 2004, 4, 83–95 CrossRef CAS PubMed
- J. Li, Z. Fu, B. Wei, C. Su, X. Yue and Z. Lü, J. Mater. Chem. A, 2020, 8, 18778–18791 RSC
- D. F. Taylor, Ind. Eng. Chem., 1950, 42, 639 CrossRef CAS
- M. Li, M. Zhao, F. Li, W. Zhou, V. K. Peterson, X. Xu, Z. Shao, I. Gentle and Z. Zhu, Nat. Commun., 2017, 8, 13990 CrossRef CAS PubMed
- J. H. Kim, J. K. Kim, H. G. Seo, D. K. Lim, S. J. Jeong, J. Seo, J. Kim and W. Jung, Adv. Funct. Mater., 2020, 2001326 CrossRef CAS
- A. Glamazda, K.-Y. Choi, P. Lemmens, W. S. Choi, H. Jeen, T. L. Meyer and H. N. Lee, J. Appl. Phys., 2015, 118, 085313 CrossRef
- J. Bielecki, S. F. Parker, D. Ekanayake, S. M. Rahman, L. Börjesson and M. Karlsson, J. Mater. Chem. A, 2014, 2, 16915–16924 RSC
- X. Li, K. Blinn, Y. Fang, M. Liu, M. A. Mahmoud, S. Cheng, L. A. Bottomley, M. El-Sayed and M. Liu, Phys. Chem. Chem. Phys., 2012, 14, 5919–5923 RSC
- E. Sediva, T. Defferriere, N. H. Perry, H. L. Tuller and J. L. Rupp, Adv. Mater., 2019, 31, 1902493 CrossRef PubMed
- M. Li, W. Zhou and Z. Zhu, ChemElectroChem, 2015, 2, 1331–1338 CrossRef CAS
- C. Duan, D. Hook, Y. Chen, J. Tong and R. O'Hayre, Energy Environ. Sci., 2017, 10, 176–182 RSC
- E. Y. Pikalova, V. Maragou, A. Demina, A. Demin and P. Tsiakaras, J. Power Sorces, 2008, 181, 199–206 CrossRef CAS
- D. Poetzsch, R. Merkle and J. Maier, J. Electrochem. Soc., 2015, 162, F939–F950 CrossRef CAS
- Y. Song, Y. Chen, M. Xu, W. Wang, Y. Zhang, G. Yang, R. Ran, W. Zhou and Z. Shao, Adv. Mater., 2020, 32, 1906979 CrossRef CAS PubMed
- Z. Shao and S. M. Haile, Nature, 2004, 431, 170–173 CrossRef CAS PubMed
- S. Choi, S. Yoo, J. Kim, S. Park, A. Jun, S. Sengodan, J. Kim, J. Shin, H. Y. Jeong, Y. Choi, G. Kim and M. Liu, Sci. Rep., 2013, 3, 2426 CrossRef PubMed
- J. H. Kim, K. Jang, D.-K. Lim, S. Ahn, D. Oh, H. Kim, J. Seo, P.-P. Choi and W. Jung, J. Mater. Chem. A, 2022, 10(5), 2496–2508 RSC
- Z. Luo, Y. Zhou, X. Hu, N. Kane, W. Zhang, T. Li, Y. Ding, Y. Liu and M. Liu, ACS Energy Lett., 2022, 7, 2970–2978 CrossRef CAS
- J. H. Kim, Z.-Y. Chern, S. Yoo, B. M. deGlee, J.-H. Wang and M. Liu, ACS Appl. Mater. Interfaces, 2019, 12, 2370–2379 CrossRef PubMed
- M. Shang, J. Tong and R. O'Hayre, RSC Adv., 2013, 3, 15769–15775 RSC
- R. Strandbakke, V. A. Cherepanov, A. Y. Zuev, D. S. Tsvetkov, C. Argirusis, G. Sourkouni, S. Prünte and T. Norby, Solid State Ionics, 2015, 278, 120–132 CrossRef CAS
- A. Grimaud, F. Mauvy, J. M. Bassat, S. Fourcade, M. Marrony and J. C. Grenier, J. Mater. Chem., 2012, 22, 16017–16025 RSC
- M. Saqib, I.-G. Choi, H. Bae, K. Park, J.-S. Shin, Y.-D. Kim, J.-I. Lee, M. Jo, Y.-C. Kim, K.-S. Lee, S.-J. Song, E. Wachsman and J.-Y. Park, Energy Environ. Sci., 2021, 14, 2472–2484 RSC
- Y. Song, Y. Chen, W. Wang, C. Zhou, Y. Zhong, G. Yang, W. Zhou, M. Liu and Z. Shao, Joule, 2019, 3(11), 2842–2853 CrossRef CAS
- J. H. Kim, J. Hong, D.-K. Lim, S. Ahn, J. Kim, J. K. Kim, D. Oh, S. Jeon, S.-J. Song and W. Jung, Energy Environ. Sci., 2022, 15(3), 1097–1105 RSC
- Y. Zhou, E. Liu, Y. Chen, Y. Liu, L. Zhang, W. Zhang, Z. Luo, N. Kane, B. Zhao, L. Soule, Y. Niu, Y. Ding, H. Ding, D. Ding and M. Liu, ACS Energy Lett., 2021, 6, 1511–1520 CrossRef CAS
- S. Choi, C. J. Kucharczyk, Y. Liang, X. Zhang, I. Takeuchi, H.-I. Ji and S. M. Haile, Nat. Energy, 2018, 1 Search PubMed
- R. Zohourian, R. Merkle and J. Maier, Solid State Ionics, 2017, 299, 64–69 CrossRef CAS
- Y. Chen, Y. Choi, S. Yoo, Y. Ding, R. Yan, K. Pei, C. Qu, L. Zhang, I. Chang and B. Zhao, Joule, 2018, 2(5), 938–949 CrossRef CAS
- Y. Song, J. Liu, Y. Wang, D. Guan, A. Seong, M. Liang, M. J. Robson, X. Xiong, Z. Zhang, G. Kim, Z. Shao and F. Ciucci, Adv. Energy Mater., 2021, 11, 2101899 CrossRef CAS
- W. Zhang, Y. Zhou, E. Liu, Y. Ding, Z. Luo, T. Li, N. Kane, B. Zhao, Y. Niu, Y. Liu and M. Liu, Appl. Catal., B, 2021, 299, 120631 CrossRef CAS
- Y. Zhu, W. Zhou, R. Ran, Y. Chen, Z. Shao and M. Liu, Nano Lett., 2016, 16, 512–518 CrossRef CAS PubMed
- Y. Zhang, B. Chen, D. Guan, M. Xu, R. Ran, M. Ni, W. Zhou, R. O’Hayre and Z. Shao, Nature, 2021, 591, 246–251 CrossRef CAS PubMed
- J. Pan, J. Yang, D. Yan, J. Pu, B. Chi and J. Li, Int. J. Hydrogen Energy, 2020, 45, 17927–17934 CrossRef CAS
- Z. Wang, Y. Wang, J. Wang, Y. Song, M. J. Robson, A. Seong, M. Yang, Z. Zhang, A. Belotti, J. Liu, G. Kim, J. Lim, Z. Shao and F. Ciucci, Nat. Catal., 2022, 5, 777–787 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee04108a |
| ‡ These authors contributed equally. |
| § Current address: Next Generation Battery R&D Center, SK on, 325, Expo-ro, Yuseong-gu, Daejeon, 34124, Republic of Korea. |
| ¶ Current address: Samsung Air Science Research Center, Samsung Advanced Institute of Technology (SAIT), Samsung Electronics Co., Ltd., Suwon 16678, Republic of Korea. |
| This journal is © The Royal Society of Chemistry 2023 |