
The chemistry of proton carriers in high-performance lithium-mediated ammonia electrosynthesis†
Hoang-Long
Du
 ,
Karolina
Matuszek
,
Rebecca Y.
Hodgetts
,
Khang
Ngoc Dinh
,
Pavel V.
Cherepanov
,
Jacinta M.
Bakker
,
Douglas R.
MacFarlane
* and
Alexandr N.
Simonov
*
,
Karolina
Matuszek
,
Rebecca Y.
Hodgetts
,
Khang
Ngoc Dinh
,
Pavel V.
Cherepanov
,
Jacinta M.
Bakker
,
Douglas R.
MacFarlane
* and
Alexandr N.
Simonov
*
School of Chemistry, Monash University, Clayton, Victoria 3800, Australia. E-mail: douglas.macfarlane@monash.edu; alexandr.simonov@monash.edu
First published on 12th January 2023
Abstract
Electrochemical lithium-mediated nitrogen reduction can enable synthesis of ammonia from renewables in a distributed fashion on various scales, but its integration into electrolyser devices presents an ongoing challenge, in particular due to the lack of understanding of the interrelation between the performance and the proton transport parameters. Herein, we use a top-performance N2 electroreduction system with lithium bis(trifluoromethylsulfonyl)imide electrolyte to correlate the reaction metrics to the properties of the proton carrier, focusing on alcohols, a phosphonium cation, tetrahydrofuran, a Brønsted acid, ammonium and water. We demonstrate that productive carriers require optimised electrolyte compositions, which define the interplay of key reaction steps. Through this understanding, we achieved ammonia electrosynthesis with the phosphonium cation and iso-propanol at performance metrics close to those provided by the ethanol benchmark. Critically, we demonstrate that the latter undergoes irreversible degradation through reaction with oxidised solvent, which is not the case for the more robust iso-propanol and phosphonium cation proton carriers.
Broader contextAmmonia is a key precursor of fertiliser manufacturing for feeding the rapidly expanding world population and also a promising fuel for powering massive machinery and ships. To satisfy these requirements in the long-run, a scalable process for the distributed synthesis of ammonia from renewables is required. This can be achieved by a renewable-powered electrolysis combining the water or hydrogen oxidation at the anode with the nitrogen reduction reaction (NRR) at the cathode. Separating the two electrodes is a specifically designed electrolyte that facilitates movement of ions in the cell, including delivery of hydrogen ions generated at the anode to the cathode to complete the synthesis of ammonia from nitrogen gas. Conventionally used carriers of the hydrogen ions for the NRR, like ethanol, have been broadly questioned to be able to deliver stable, non-sacrificial operation on a practical timescale, but the mechanism of its degradation remained elusive. The present work explains this mechanism through detailed spectroscopic studies, and presents a scrutiny into the factors determining the effectiveness of the hydrogen ion transport during the NRR with the current top-performance electrolyte system. Finally, we demonstrate that iso-propanol can sustain operation at a close to 100% faradaic efficiency without being significantly converted to side-products – a finding of immediate practical significance for the development of the sustainable ammonia electrosynthesis technologies. |
Introduction
The lithium-mediated nitrogen reduction reaction (Li-NRR; Scheme 1) is a strong candidate as a future technology for the synthesis of green ammonia.1,2 Although the energy efficiency of the Li-NRR is clearly limited by the role of Li0/+ and Li3N in this process, it remains the only known pathway towards sustainable conversion of N2 to NH3 under mild conditions at high rates. One key requirement for the realisation of the Li-NRR in an electrolyser device is the provision of effective delivery of protons generated at the anode to the cathode by a robust shuttle, like the phosphonium/ylide system introduced in our recent work.3 The latter development presented the first demonstration of sustainable proton cycling in a complete ammonia electrosynthesis process from N2 and H2, as opposed to the possibly sacrificial protonation of Li3N by the much studied ethanol (EtOH) proton carrier.3–13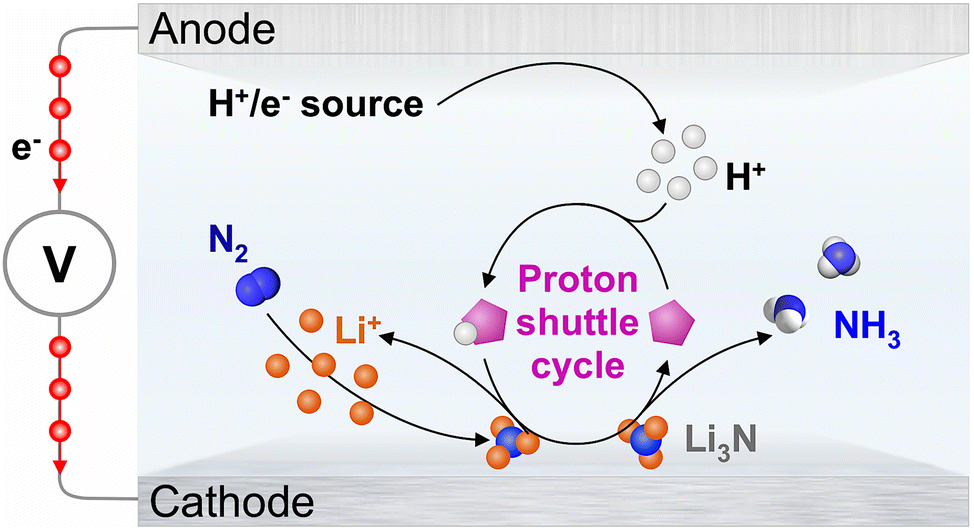 | ||
| Scheme 1 Lithium-mediated N2 reduction. In a fully-fledged sustainable process, the H+/e− source is H2, or most preferably, H2O. Under model conditions aiming to investigate the Li-NRR cathode process only, H+/e− are derived from the electrooxidation of solvent, most commonly tetrahydrofuran; however, oxidation of some proton carriers and/or electrolyte anions might also occur. | ||
A pronounced influence of the chemical nature of the proton carrier on the Li-NRR rate and current-to-ammonia (faradaic) efficiency (FE) were already noted in the early studies of the process.5,14 Recently, this was confirmed in the aforementioned report on the phosphonium/ylide system,3 and in an insightful study by Krishnamurthy et al.15 The latter work presented the first systematic analysis of the effects of the chemical nature of the proton carrier on the Li-NRR, introducing solvatochromic Kamlet–Taft parameters as descriptors of the capability of a compound to protonate the electrochemically generated Li3N to produce NH3. Through this analysis, n-butanol (n-BuOH) was found to provide better results than EtOH – the benchmark in the field that has been used to achieve the current best Li-NRR performance.12,16,17 However, the capability of EtOH to engage in genuine deprotonation–protonation cycling in the Li-NRR system has not been proven yet,15,16,18 in contrast to the phosphonium-ylide system.3 It has also been suggested that EtOH can be unproductively consumed through reactions with the solution components, although not under the actual Li-NRR conditions.18
The major limitation of the existing studies on proton carriers in the Li-NRR is the common use of an unstable lithium electrolyte like LiBF4,3,15 which cannot provide high faradaic efficiency close to 100% and practical yield rates on a reasonably extended timescale where steady-state conditions are clearly established.16 Thus, the outcomes of these previous studies were overshadowed by the detrimental influence of unproductive and poorly understood side-processes, which might have affected the Li-NRR to a more significant extent than the chemical nature of the proton source. To resolve this, an investigation into the effects of the proton carrier on the Li-NRR is required, using a highly productive, selective and robust system, such as the one based on a high-concentration bis(trifluoromethylsulfonyl)imide ([NTf2]−) electrolyte introduced in our recent work.16 This enabled robust operation at practical yield rates and faradaic efficiency approaching 100%, though with the use of EtOH as an apparent proton carrier. To support future translation of this process towards practical ammonia electrosynthesis, our present study delves into the chemistry of proton carriers for N2 electroreduction mediated by LiNTf2 using a wider family of compounds to establish relevant degradation mechanisms and key performance trends.
Results and discussion
The bulk of the Li-NRR experiments were undertaken herein in a static N2 (15 bar) atmosphere in a single-compartment cell with a bare nickel wire electrode, while key performance tests used an improved configuration devoid of a gas-solution-electrode interface, which was recently demonstrated to decrease the FE16 (Fig. S2, ESI†). Protons were derived from electrooxidation of the tetrahydrofuran solvent at an auxiliary electrode. We have specifically avoided the use of H2 to generate H+ in order to test the oxidative stability of the proton carriers and exclude an interference of dihydrogen at the cathode. See ESI,† for extended experimental details.Except for the electrolyte optimisation studies, the LiNTf2 concentration was 2 M, which provides the highest Li-NRR performance reported so far with flat electrodes.16 Specifically, when using a bare Ni electrode and 0.1 M EtOH proton carrier, the reaction mediated by 2 M LiNTf2 produces ammonia at a rate of 230 ± 20 nmol s−1 cm−2 and FE of 86 ± 9% during 6 h experiments at −0.55 V vs. Li0/+app (apparent potential of the Li0/+ redox couple; hereinafter, all potentials are vs. this reference). With an isolated Ni electrode, the yield rate is 530 ± 20 nmol s−1 cm−2 and FE = 98 ± 2% during 24 h tests. These metrics were considered as a benchmark.
Chemical nature and concentration effects
Herein, we aimed to investigate a broad range of compounds strongly differing in their deprotonation–protonation abilities as potential proton carriers for the Li-NRR. Specifically, we examined C1–5 alcohols, the [NTf2]− and [eFAP]− (tris(pentafluoroethyl)trifluorophosphate) salts of [P6,6,6,14]+ (trihexyl(tetradecyl)phosphonium), a Brønsted acid HNTf2, water and NH4NTf2. The effects of the length of the alkyl chain on the effectiveness of alcohols has been noted before,5,15 but are yet to be investigated under high-productivity Li-NRR conditions, as are their electrochemically induced transformations during ammonia electrosynthesis. For the [P6,6,6,14]+ proton shuttle, the role of the anion of the phosphonium salt as well as the effects of the electrolyte solution composition on the performance have not been examined before. HNTf2 acid presents an extreme, benchmark, case of the highest proton activity, and might also, in theory, provide a buffer-like proton carrier system when combined with the LiNTf2 electrolyte. Water is the most desirable proton carrier in terms of the development of a complete ammonia electrosynthesis technology using N2 and H2O as reactants. Finally, NH4+ is of special interest as it might progressively accumulate in the electrolyte solution during the Li-NRR at concentrations exceeding that of the intentionally added proton carrier. For example, the NH3/NH4+ concentration produced after 6 h benchmark experiments with EtOH is as high as 0.15 ± 0.02 M, and NH4+ in particular, as a slightly acidic species, might contribute to the proton-based transformations within the system.First, we compared the performance of proton carriers at 0.1 M concentration, which provides the best results for the EtOH benchmark (Table S1, ESI†). In the initial voltammograms, EtOH produced the lowest Li0/Li3N oxidation charge (as indicated by the area of the peak between 0 and 1 V), except NH4NTf2, indicating that other carriers provide slower kinetics of protonation of the reduced lithium species (Fig. 1a). This might appear unexpected for HNTf2, indicating that the high-activity H+ in this case are consumed unproductively in other processes. When using H2O, the voltammetric oxidation peak shifted towards more positive potentials with cycling (Fig. S5a, ESI†), which suggests the formation of LiH and/or delithiation of the electrodeposited materials.
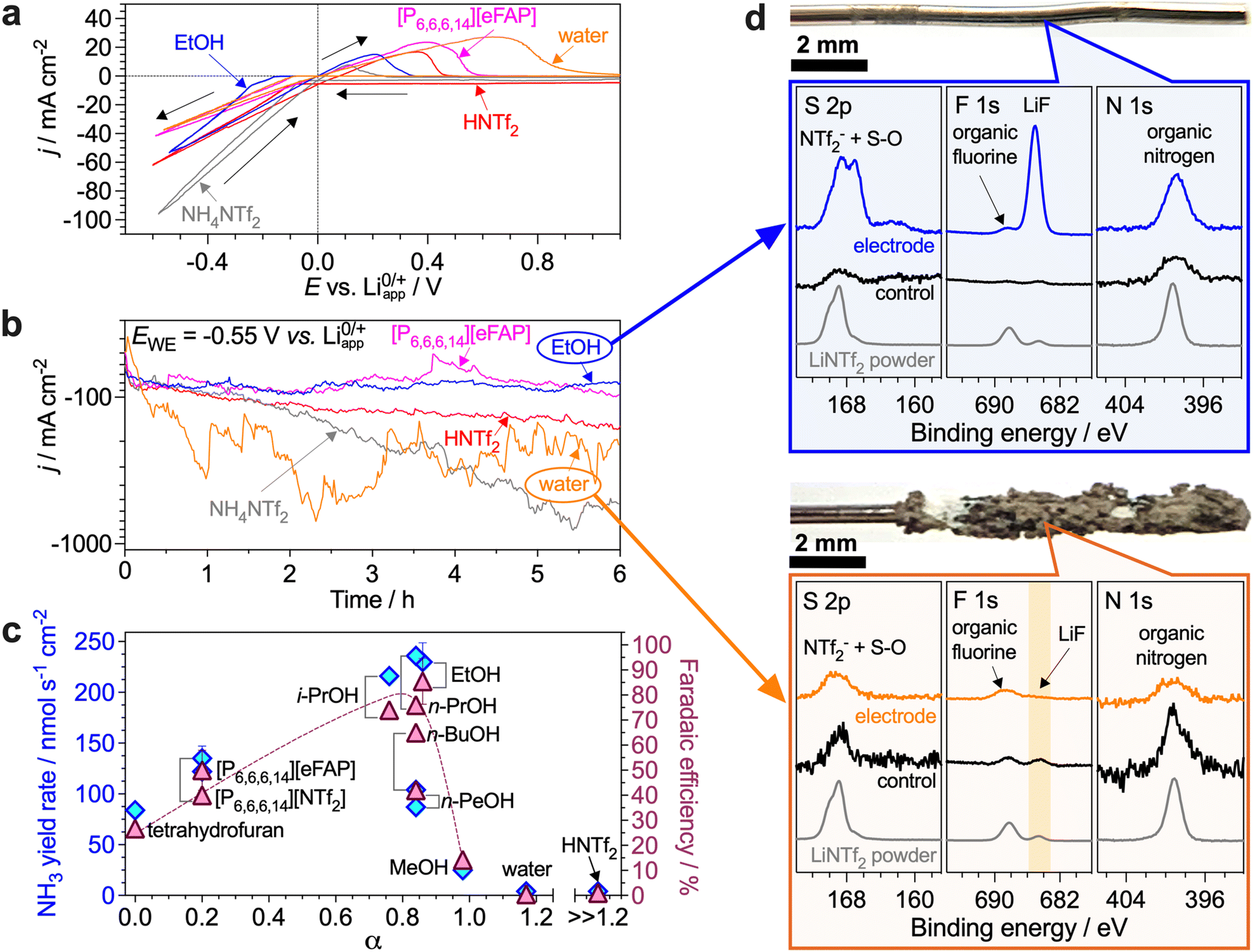 | ||
| Fig. 1 Li-NRR performance vs. chemical nature of the proton source. (a) Cyclic voltammetry (0.020 V s−1; 20th cycle; arrows show scan direction) and (b) chronoamperometry at −0.55 V vs. Li0/+app (note the logarithmic current density scale) with 0.1 M EtOH (blue), NH4NTf2 (grey), [P6,6,6,14][eFAP] (magenta), HNTf2 (red) and H2O (orange) as proton carrier. (c) NH3 yield rate (diamonds) and Li-NRR faradaic efficiency (triangles) plotted against Kamlet–Taft hydrogen bond donation parameter15,21 of the proton carrier (except NH4NTf2) added at 0.1 M concentration (except for the tetrahydrofuran data, which represent a solution with no additional proton carrier). (d) Electrode photographs along with S 2p, F 1s and N1 s XPS data of electrodes after the tests with 0.1 M EtOH (blue) and 0.1 M water (orange) shown in panel b (“control” data were collected for an electrode kept in contact with the corresponding electrolyte solution with no potential applied). Extended XPS data are shown in Fig. S19–S24 (ESI†). Experiments were undertaken in a single-compartment cell using a bare Ni wire electrode (0.15 cm2) and stirred tetrahydrofuran solutions containing 0.1 M of the proton carrier along with 2 M LiNTf2 and saturated with N2 at 15 bar (static atmosphere). | ||
Under potentiostatic conditions (hereinafter, at −0.55 V), the current density remained stable with the phosphonium and ethanol proton carriers, but not with HNTf2, NH4NTf2 and especially H2O where the reductive current substantially increased during the experiments before plateauing at high values (Fig. 1b). Measurements of the open-circuit potential of the working electrode after these tests demonstrated that only EtOH enables effective protonation of Li3N under the examined conditions, as evidenced by rapid (<1 min) relaxation to values significantly more positive than 0 V (Fig. S7, ESI†). XPS revealed that the electrodes derived from these experiments were coated with a visually-undetectable nanometre-scale layer of LiF and reduced [S–O] species19 (Fig. 1d and Fig. S19, ESI†). The electrode potential with the other proton carriers did not relax, even after 15 min (Fig. S7, ESI†), indicating that significant deposits of Li0/Li3N/LiH were formed and remained unprotonated.
Voltammograms recorded after potentiostatic Li-NRR with 0.1 M H2O revealed substantial suppression of the lithium-mediated reduction (Fig. S5a, ESI†). This is attributed to the formation of a substantial deposit on the electrode (Fig. 1d) containing lithium (hydr)oxide and hydride detected by XPS20 (Fig. S20, ESI†). Similar results were obtained for 0.1 M HNTf2 (Fig. S21, ESI†). Consistent with this degradation, ammonia was formed at low yield rates and minuscule FE in these experiments (Fig. 1c).
The very low Li-NRR performance provided by NH4NTf2 (Table S1, ESI†) indicates not only the inability of the NH4+/NH3 system to sustain the process, but also a pernicious effect of the presence of the protonated form of the reaction product at the start of the experiments. Most likely, NH4+ provides excessive proton activity which induces reductive degradation of the electrolyte solution components, similar to H2O and HNTf2. Formation of LiH and/or delithiation was revealed by significant broadening of the voltammetric oxidation peak towards positive potentials after potentiostatic tests with 0.1 M NH4NTf2 (Fig. S6a, ESI†), which was corroborated by XPS (Fig. S22, ESI†). These results also raise an important question on the ammonia vs. ammonium state of the Li-NRR product, which requires a separate, future study.
With both phosphonium salts examined, NH3 was produced at respectable yield rates, but the faradaic efficiencies were lower than with EtOH (Fig. 1c) due to the consumption of charge for the formation of visible, yet not very thick, coatings on the electrode (Fig. S23 and S24, ESI†). These undesired processes presumably arise from un-balanced transport of Li+, N2 and proton carrier, likely resulting from the increased viscosity and decreased ionic conductivity of the [P6,6,6,14]+-based solutions (Fig. S4, ESI†). Importantly, no P 2p signals were detected on the electrodes after experiments with [P6,6,6,14]+ confirming the robustness of the cation (Fig. S23, ESI†).
Among the alcohol proton carriers tested, iso-propanol (i-PrOH) and n-propanol (n-PrOH) provided similar electrochemical behaviour to that found with ethanol, although current transients during potentiostatic tests with n-PrOH produced initially faster reduction followed by deceleration of the process (Fig. S9a–c, ESI†). Although relaxation of the open-circuit potential after the Li-NRR with C3-alcohols commenced relatively quickly (∼1–2 min), an intermediate state with an open-circuit potential of ca. 1–1.5 V was observed and attributed to small amounts of LiH formed (Fig. S9e, ESI†). Nevertheless, the overall similarities in the electrochemical behaviour of i-PrOH, n-PrOH and EtOH were mirrored in the similarly high Li-NRR metrics (Fig. 1c).
Methanol (MeOH), n-BuOH and n-pentanol (n-PeOH) provided lower reduction rates, along with suppressed oxidative stripping processes in voltammetry (Fig. S9b–d, ESI†). MeOH also produced an extensive amount of reduced Li-species on the electrode surface, which were not removed even after 15 min of relaxation (Fig. S9e, ESI†). A less significant deposit, which was fully protonated after ca. 11 min, was found for n-PeOH, while the evolution of the open-circuit potential after the Li-NRR with n-BuOH was similar to that for i-/n-PrOH though demonstrated the presence of higher amounts of LiH (Fig. S9e, ESI†). Again, the electrochemical observations were reflected in the productivity of the Li-NRR, which was low for MeOH and mediocre for n-BuOH and n-PeOH (Fig. 1c and Table S1, ESI†).
Without a dedicated proton source added, the reduction rate of lithium was still significant notwithstanding the excessive formation of reduced Li-based compounds on the electrode surface (see "0 M" data in Fig. S7a, S8a, and S8e ESI†). Since effective H+ transport by tetrahydrofuran molecules is unlikely, we have undertaken NMR analysis of the electrolyte solutions after 6 h Li-NRR, which revealed the emergence of a characteristic [CH3] but not carbonyl/carboxyl signals (Fig. S32, ESI†). This might be ascribed to the cathodic ring-opening of tetrahydrofuran producing an alcohol, most likely n-BuOH. Hence, the measurable rates of the Li-NRR observed in these experiments are likely sustained by the in situ generated alcohol proton carrier.
A recent study by Krishnamurthy et al. demonstrated a lack of correlation between the Li-NRR metrics and the pKa of a proton source,15 as also observed herein (Fig. S10, ESI†). As alternative descriptors, Krishnamurthy et al. suggested Kamlet–Taft parameters reflecting the hydrogen bond donating (α) and accepting (β) ability.15 Indeed, a correlation between the Li-NRR performance achieved with the compounds examined herein and their α parameters seems to exist, with an apparent maximum achieved at α ≈ 0.8–0.9 typical for alcohols (Fig. 1c), though the correlation between the FE and β is less obvious (Fig. S11, ESI†). However, neither these parameters nor pKa can separate and explain the wide-ranging performance of the alcohols.
Thus, and as expected, the chemical nature of a proton carrier strongly affects the Li-NRR, with some compounds undersupplying H+ (e.g. [P6,6,6,14]+) and others delivering detrimentally excessive amounts of protons (e.g. HNTf2) under the conditions examined. Another obvious approach to tune the proton activity near the electrode surface is to alter the concentration of the proton carrier (cPC), which effects are investigated in Section S3 (ESI†) with the key conclusions summarised below.
Changing cPC of EtOH, n-PrOH, i-PrOH, [P6,6,6,14]+ and HNTf2 produced qualitatively similar bell-shaped dependencies of the Li-NRR FE and yield rate (Fig. S18, ESI†). Obviously, this reflects the lack of protons to convert Li3N into NH3 and Li+ at low concentrations, and oversupply of H+ promoting its reduction rather than the Li-NRR at high concentrations, respectively. Optimal cPC in terms of both Li-NRR performance metrics was the same (0.1 M) for the examined alcohols, confirming similarities in their proton donating properties. For HNTf2, the highest FE (ca. 40%) and yield rate (70 ± 30 nmol s−1 cm−2) were achieved at different concentrations (Fig. S18, ESI†), which highlights the limitations of this system due to the excessive acidity of HNTf2.
The performance of the [P6,6,6,14]+ carrier could not be improved by changing the cPC with respect to the results obtained at 0.1 M (Fig. S18, ESI†). In our recent study undertaken in parallel with the present work, we have ascribed this to the insufficiently effective mass-transport of this bulky proton carrier in the more viscous electrolyte solution.16 Hence, improved results might emerge from decreasing the amount of LiNTf2 – an approach that was further examined herein (Fig. S15 and Table S2, ESI†). The highest NH3 yield rate of ca. 170 nmol s−1 cm−2 (ca. 66% FE) was achieved at 1.5 M LiNTf2, but the best faradaic efficiency of 75 ± 4% (at 140 ± 3 nmol s−1 cm−2) was produced with 1 M LiNTf2. Importantly, the faradaic efficiency provided by 0.1 M EtOH at 1 M LiNTf2 was only ca. 45%,16 indicating that phosphonium cation is the preferred proton carrier under these conditions, not ethanol. Even better results with the [P6,6,6,14]+ and LiNTf2 electrolyte are likely to be achievable through further optimisation of the concentrations of both components and/or the use of faster-diffusing phosphonium cations with shorter alkyl chains, which is the focus of our ongoing studies.
We emphasise that the apparent optimal proton donors and concentrations thereof identified above are only relevant to the conditions employed herein, including the chemical nature of the solvent and electrolyte, concentrations of other key species, viz. N2 and LiNTf2, temperature and mass-transport conditions. In other words, each Li-NRR system will require separate optimisation of the protonation conditions.
Electrolyte solution transformations
Since the phosphonium shuttle has been investigated in detail previously3 and given the promising results provided by C3 alcohol carriers (Fig. 1c), the latter along with the benchmark EtOH were the focus of further investigations herein.Little is known about the fate of the alcohol proton shuttles during the Li-NRR and the extent to which they are consumed. Although EtOH electrooxidation is negligible in the LiNTf2 tetrahydrofuran solutions up to ca. 3.9 V,22 potentials of the auxiliary electrode during the high-performance Li-NRR tests here can exceed 5 V (Fig. S9, S12–S14, ESI†). This induces significant colouration of the solution (Fig. 2a), which is likely due to electrooxidation of tetrahydrofuran,3,12 although transformations involving EtOH cannot be excluded. Indeed, 1H NMR analysis of the electrolysed solutions revealed a new signal at 1.2 ppm, the identity of which we aimed to establish through the experiments described below.
 | ||
| Fig. 2 Transformations of the 2 M LiNTf2 + 0.1 M EtOH tetrahydrofuran solutions during the Li-NRR. (a) Photographs, UV-vis spectra (optical pathlength 10 μm), and (b) 1H NMR analysis of the 2 M LiNTf2 with and without 0.1 M EtOH electrolyte solutions in THF after 6 h potentiostatic experiments at −0.55 V vs. Li0/+app with a bare Ni wire electrode (0.15 cm2) under 15 bar Ar (green and wine) and N2 (blue and purple). Experiments were undertaken in a single- (green and blue) and two-compartment cell (provided by a P3 frit; wine and purple) configuration under stirring of the working electrolyte solution; WE and AE notations indicate working and auxiliary compartment electrolytes from the two-compartment cell. Data for the 2 M LiNTf2 tetrahydrofuran solution with no EtOH added in the auxiliary compartment under N2 are shown as orange curves (data for Ar were very similar and are not shown). Grey, green and pink shadings in panel b highlight NMR signals from tetrahydrofuran, EtOH and 2-ethoxytetrahydrofuran (labelled as “side product”), respectively. (c) Photographs of the auxiliary electrode compartment for the experiment with a P3 frit, and chronoamperometric data for experiments at −0.55 V vs. Li0/+app under 15 bar Ar (green and wine) or N2 (blue and purple) without (green and blue) and with (wine and purple) P3 frit. This frit does not provide perfect separation of the solutions, which produces colouration of the working electrolyte solutions in the two-compartment experiments. | ||
First, potentiostatic tests were undertaken with the working and auxiliary electrodes separated with a low-porosity frit and with no EtOH added to the auxiliary compartment (Fig. 2c and Fig. S16, ESI†), contrasting the single-compartment experiments above. The use of the two-compartment configuration suppressed the colouration of the working solution under both N2 and Ar, but produced intense-black solutions in the auxiliary compartment with enhanced absorption at >350 nm (Fig. 2a and c). Hence, major degradation pathways are associated with the oxidation processes.
The 1H NMR triplet at 1.2 ppm was not detected in the two-compartment experiments when EtOH was not added to the auxiliary electrode chamber (Fig. 2b). Formation of this side-product was also suppressed during experiments in the undivided cell under Ar (Fig. 2b, ESI†), indicating that degradation processes in the absence of N2 are different. UV-vis absorption also suggests that the quantity and diversity of the side-products were higher in Ar (Fig. 2a). This suggests facilitated decomposition of the electrolyte solution when excessive amounts of Li0/LiH are accumulated, additionally emphasising the importance of achieving 100% FE for stable operation of the Li-NRR.
Further, NMR investigation was undertaken using both regular and deuterated reactants, which observations are described in Section S4 (ESI†). These experiments indicate that (i) formation of the side product occurs when the auxiliary electrode is accessible to both EtOH and tetrahydrofuran, (ii) transformation of ethanol preserves the [CH3CH2O] structural motif, (iii) ethanol is not oxidised to aldehyde, carboxylic acids or C1 products, (iv) transformation of the solvent results in the formation of a compound containing [O–CH–O] functionality (Fig. 3a, b and Fig. S26, ESI†). These conclusions narrow down the possible side products to pentanal diethyl acetal and 2-ethoxytetrahydrofuran (Fig. 3c). Acetal can be formed via reaction between ethanol and butyraldehyde, and the latter may be the product of the electrochemically induced ring opening of tetrahydrofuran; however, no 13C NMR evidence for the presence of carbonyl species could be found (Fig. S29, ESI†). Formation of 2-ethoxytetrahydrofuran is much more probable and the NMR data obtained here are in agreement with the AIST database23 (Fig. 3a and Fig. S26, ESI†). This product is produced via tetrahydrofuralynation,24–26viz. a reaction of EtOH with a carbocation formed upon oxidation of tetrahydrofuran.
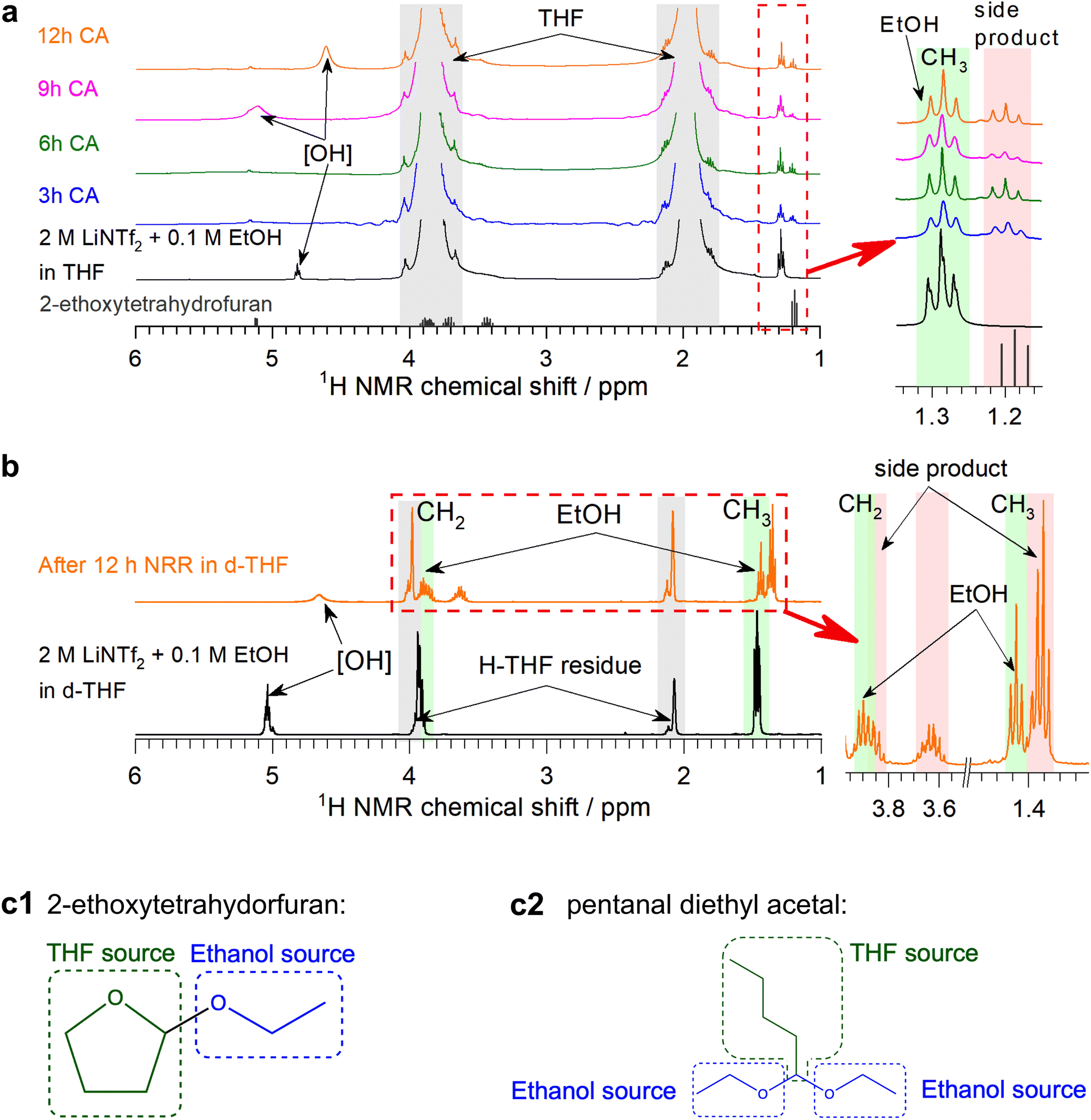 | ||
| Fig. 3 1H NMR analysis of fresh and tested 2 M LiNTf2 + 0.1 M EtOH electrolyte solutions. (a) Spectra of tetrahydrofuran-h8 (grey lines show data for 2-ethoxytetrahydrofuran from the AIST database23) and (b) tetrahydrouran-d8 solutions recorded before (black) and after 3 (blue), 6 (green), 9 (magenta) and 12 h (orange) of electroreduction at −0.55 V vs. Li0/+app using a bare Ni wire electrode (0.15 cm2) under continuous stirring and 15 bar N2 pressure in a single-compartment configuration; insets show enhanced plots of peaks contained in red boxes highlighting the signals associated with EtOH and side product. Except for the peaks of d6-benzene standard, no signals and changes in the spectra were observed beyond 7 ppm; hence, this region is omitted in the figure. (c) Most plausible side products 2-ethoxytetrahydrofuran and pentanal diethyl acetal with ethanol and tetrahydrofuran sources highlighted with blue and green, respectively. | ||
Formation of the side-product majorly occurred during the initial ca. 3 h and complete degradation of EtOH was not observed during longer experiments (Fig. 3a, b and Fig. S26, ESI†). This is consistent with the apparent ethanol turnover numbers with respect to the amount of NH3 formed being greater than 1 in the 12 h experiments herein and greater than 4 in our previous study.16 Hence, EtOH might be capable of operating as a genuine proton shuttle for the Li-NRR, but with sacrificial consumption occurring via processes induced by electrooxidation of the solvent.
Improved proton carrier stability
Significant degradation of EtOH during the Li-NRR likely renders this proton carrier unsuitable for practical ammonia synthesis, which urged us to investigate if this limitation also applies to the C3 alcohols shown in Fig. 1 to be productive. After 6 h potentiostatic Li-NRR, n-PrOH also suffered partial tetrahydrofuralynation (Fig. S30, ESI†), but the relative intensity of the corresponding 1H NMR triplet with respect to the [CH3]-group of n-PrOH was much lower (ca. 0.1) than that found for EtOH (ca. 0.5). When using i-PrOH, hard to resolve 1H NMR signals emerged after 6 h experiments, but their intensity was less than 0.05 of the half intensity corresponding to the major signal from the two CH3 groups of i-PrOH (Fig. 4a). Moreover, 13C NMR could not detect any side products formed from the C3 alcohols (Fig. S31, ESI†). Thus, n-PrOH and especially i-PrOH are significantly less prone to the partial degradation as compared to EtOH.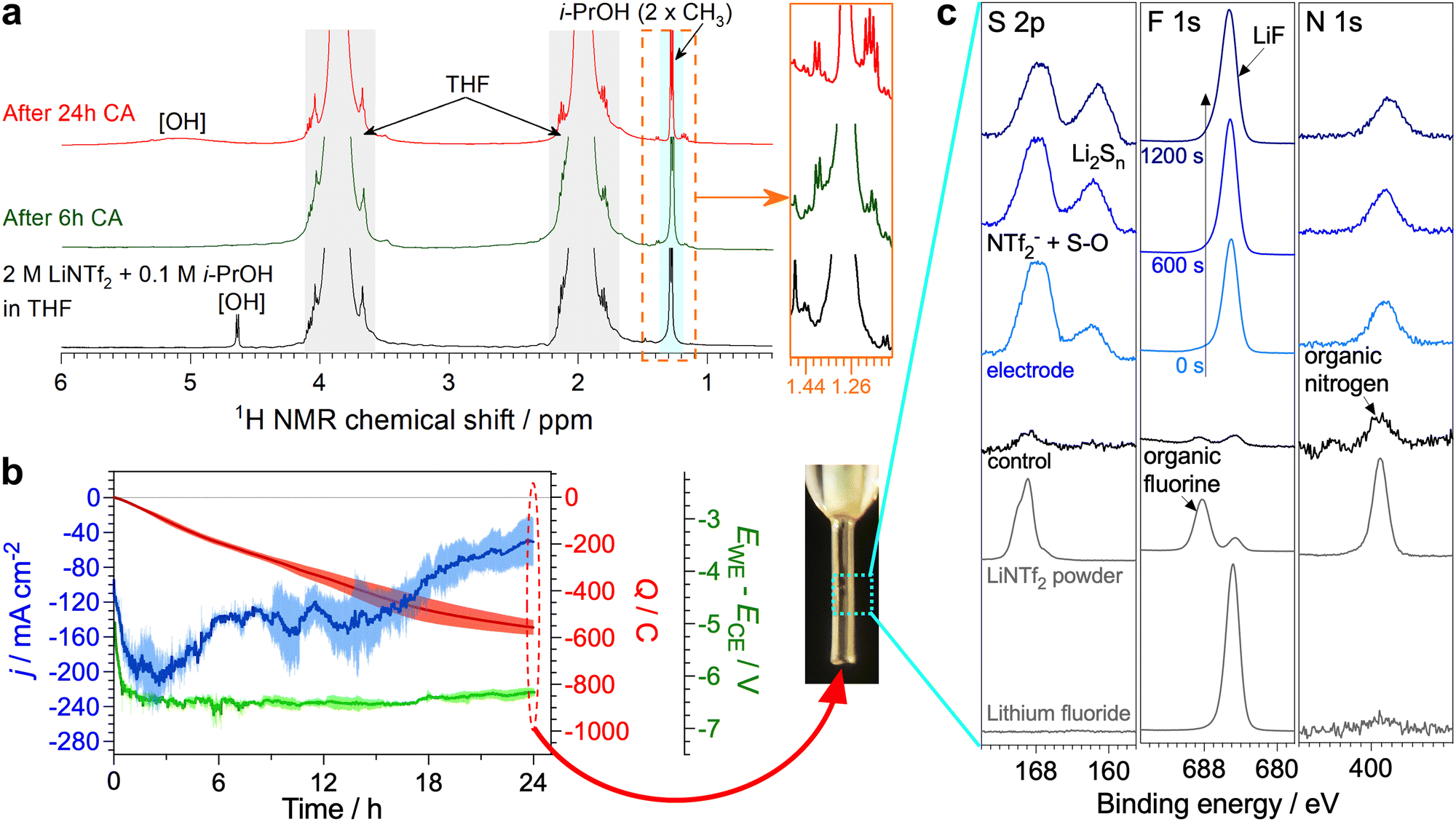 | ||
| Fig. 4 Long-term Li-NRR experiment with 2 M LiNTf2 and 0.1 M i-PrOH. (a) 1H NMR spectra before (black) and after the Li-NRR at −0.55 V vs. Li0/+app undertaken using a bare nickel wire electrode (0.15 cm2) for 6 h (green) and isolated nickel wire electrode (0.05 cm2) for 24 h (red); inset shows enhanced plots of side product. (b) Evolution of the current density (blue), cell potential (green) and charge passed (red) during chronoamperometric reduction of an isolated nickel wire (0.05 cm2) in stirred 2 M LiNTf2 + 0.1 M i-PrOH tetrahydrofuran solutions at −0.55 V vs. Li0/+app in a single-compartment cell; data are shown as mean (lines) and standard deviation (shading) derived from n = 3 independent repeats of the experiment; horizonal grey line shows j = 0 mA cm−2. (c) Electrode photograph along with S 2p, F 1s and N 1s XPS data for electrodes after the Li-NRR test shown in panel b; control data (black) were collected for an electrode kept in contact with the corresponding electrolyte solution with no potential applied; reference data are shown as grey curves; time (0, 600 and 1200 s) indicates the duration of argon ion etching of the electrode. | ||
To further demonstrate the capability of i-PrOH to sustain the high-performance Li-NRR, extended 24 h experiments with an improved electrode configuration with no gas-solution-electrode phase boundary present were undertaken. The reductive charge passed during these tests was 560 ± 40 C (Fig. 4b), which is slightly lower than 670 ± 20 C for similar experiments with EtOH16 and attributed to lower diffusion coefficient of the C3 alcohol. Most importantly, the Li-NRR faradaic efficiency provided by i-PrOH was as high as 96 ± 1%, viz. very close to 98 ± 2% found with EtOH; the corresponding ammonia yield rates were 430 ± 20 and 530 ± 20 nmol s−1 cm−2, respectively (Table S4, ESI†). The apparent turnover number of the i-PrOH carrier achieved in these experiments was ca. 2.
Finally, the NMR signals associated with possible side products after the 24 h tests with i-PrOH remained essentially unchanged after being formed in minuscule amounts at the initial stages of the test (Fig. 4a and Fig. S31b, ESI†). The surface of the electrode remained visually clean, and was functionalised with LiF, reduced [S–O] and polysulphide species that could be only detected by XPS (Fig. 4c). These species formed a very thin, nanometre-scale layer, as concluded from the detection of strong signals of the Ni metal electrode (Fig. S25, ESI†).
Conclusions
Protonation of the product of the Li+ and N2 co-reduction during the Li-NRR is a critical step towards the formation of ammonia, the optimisation of which can produce significant improvements in the performance. The present work shows that the optimal chemical nature and concentration of the proton carrier are unique for every set of the reaction conditions, and it might be challenging to establish universal selection rules. This is particularly important for the scale-up of the Li-NRR, since associated modifications to the cell geometry induce inevitable changes to the mass-transport of all the participating species, including the proton carrier, and thereby affect performance. Nevertheless, it can be suggested at this stage that C2–C4 alcohols and phosphonium cations present the most viable candidates as effective proton carriers for the Li-NRR under conditions currently examined in the field.Investigation of transformations of the EtOH proton carrier during the Li-NRR revealed that it is indeed consumed irreversibly, although not in the way previously hypothesised. This undesired consumption is not direct and not reductive, but is induced by sacrificial oxidation of tetrahydrofuran. Notwithstanding significant formation of 2-ethoxytetrahydrofuran during the Li-NRR, apparent turn-over numbers for ethanol higher than 1 are still achievable under conditions examined here. However, this might not be the case for practical flow Li-NRR cells under development. Theoretically, electrooxidation of tetrahydrofuran and therefore irreversible degradation of ethanol might be avoided if H2 is introduced as a proton/electron source. However, the hydrogen oxidation reaction presents its own challenges under the Li-NRR conditions where conventional platinum-based catalysts rapidly lose activity,22 which might eventuate in the potentials of the anode that are sufficiently positive to enable tetrahydrofuran oxidation. As an alternative solution, we demonstrate that iso-propanol does not undergo the tetrahydrofuralynation process to a significant extent and enables the Li-NRR with the faradaic efficiency approaching 100%.
This brings us to an important conclusion on the pernicious role of tetrahydrofuran in the degradation of the system components. While the highly robust phosphonium cation shuttle does not undergo tetrahydrofuralynation, there might be other mechanisms for degradation emerging through deeper oxidation and reductive decomposition of the solvent,24,27,28 even when a practical anode reaction like oxidation of H222 or H2O12 is in place. Thus, the search for an alternative, electrochemically stable solvent for the Li-NRR seems to present an important direction for future research. Importantly, the new solvent will likely require re-optimisation of the proton carriers in order to achieve high and stable performance.
Author contributions
HLD conceived of and undertook electrochemical and UV-vis experiments, and co-wrote the manuscript. KM and RYH collected and analysed NMR data. KND and PVC collected and analysed XPS data. JMB synthesised ionic liquids. DRM and ANS conceived of the experiments, directed the project and co-wrote the manuscript.Conflicts of interest
HLD, DRM and ANS are inventors on an Australian provisional patent application that covers aspects of the work reported here, and which has been licensed to Jupiter Ionics Pty Ltd. DRM and ANS have minority equity ownership, as well as management and consulting roles, in Jupiter Ionics Pty Ltd.Acknowledgements
The authors acknowledge funding of this work by the Australian Research Council (Discovery Project DP200101878, Future Fellowship to ANS FT200100317) and the Australian Renewable Energy Agency (“Renewable Hydrogen for Export” project 2018RND/009 DM015). Monash X-ray platform and Monash Analytical platform are acknowledged for providing access to the physical characterisation and spectroscopic facilities. The authors are also grateful to Dr B. Guan and Dr C. Priest from the University of South Australia for the provision of a low-pathlength quartz cuvette.References
- D. R. MacFarlane, J. Choi, B. H. R. Suryanto, R. Jalili, M. Chatti, L. M. Azofra and A. N. Simonov, Adv. Mater., 2020, 32, 1904804 CrossRef CAS PubMed
.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. Ferrero Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS
- B. H. Suryanto, K. Matuszek, J. Choi, R. Y. Hodgetts, H.-L. Du, J. M. Bakker, C. S. Kang, P. V. Cherepanov, A. N. Simonov and D. R. MacFarlane, Science, 2021, 372, 1187–1191 CrossRef CAS PubMed
- S. Z. Andersen, M. J. Statt, V. J. Bukas, S. G. Shapel, J. B. Pedersen, K. Krempl, M. Saccoccio, D. Chakraborty, J. Kibsgaard, P. C. K. Vesborg, J. Nørskov and I. Chorkendorff, Energy Environ. Sci., 2020, 13, 4291–4300 RSC
- A. Tsuneto, A. Kudo and T. Sakata, J. Electroanal. Chem., 1994, 367, 183–188 CrossRef CAS
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed
- L.-F. Gao, Y. Cao, C. Wang, X.-W. Yu, W.-B. Li, Y. Zhou, B. Wang, Y.-F. Yao, C.-P. Wu, W.-J. Luo and Z.-G. Zou, Angew. Chem., Int. Ed., 2021, 60, 5257–5261 CrossRef CAS PubMed
- K. Kim, Y. Chen, J.-I. Han, H. C. Yoon and W. Li, Green Chem., 2019, 21, 3839–3845 RSC
- K. Li, S. G. Shapel, D. Hochfilzer, J. B. Pedersen, K. Krempl, S. Z. Andersen, R. Sažinas, M. Saccoccio, S. Li, D. Chakraborty, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, ACS Energy Lett., 2022, 7, 36–41 CrossRef CAS
- K. Li, S. Z. Andersen, M. J. Statt, M. Saccoccio, V. J. Bukas, K. Krempl, R. Sažinas, J. B. Pedersen, V. Shadravan, Y. Zhou, D. Chakraborty, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Science, 2021, 374, 1593–1597 CrossRef CAS PubMed
- N. Lazouski, Z. J. Schiffer, K. Williams and K. Manthiram, Joule, 2019, 3, 1127–1139 CrossRef CAS
- N. Lazouski, M. Chung, K. Williams, M. L. Gala and K. Manthiram, Nat. Catal., 2020, 3, 463–469 CrossRef CAS
- P. V. Cherepanov, M. Krebsz, R. Y. Hodgetts, A. N. Simonov and D. R. MacFarlane, J. Phys. Chem. C, 2021, 125, 11402–11410 CrossRef CAS
- A. Tsuneto, A. Kudo and T. Sakata, Chem. Lett., 1993, 851–854 CrossRef CAS
- D. Krishnamurthy, N. Lazouski, M. L. Gala, K. Manthiram and V. Viswanathan, ACS Cent. Sci., 2021, 7, 2073–2082 CrossRef CAS PubMed
- H.-L. Du, M. Chatti, R. Y. Hodgetts, P. V. Cherepanov, C. K. Nguyen, K. Matuszek, D. R. MacFarlane and A. N. Simonov, Nature, 2022, 609, 722–727 CrossRef CAS PubMed
- S. Li, Y. Zhou, K. Li, M. Saccoccio, R. Sažinas, S. Z. Andersen, J. B. Pedersen, X. Fu, V. Shadravan, D. Chakraborty, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Joule, 2022, 6, 2083–2101 CrossRef CAS PubMed
- R. Sažinas, S. Z. Andersen, K. Li, M. Saccoccio, K. Krempl, J. B. Pedersen, J. Kibsgaard, P. C. K. Vesborg, D. Chakraborty and I. Chorkendorff, RSC Adv., 2021, 11, 31487–31498 RSC
- C. Xu, B. Sun, T. Gustafsson, K. Edström, D. Brandell and M. Hahlin, J. Mater. Chem. A, 2014, 2, 7256–7264 RSC
-
D. W. Jeppson, J. L. Ballif, W. W. Yuan and B. E. Chou, Lithium literature review: lithium's properties and interactions, USA, 1978 Search PubMed
- J. M. Padró and M. Reta, J. Mol. Liq., 2016, 213, 107–114 CrossRef
- R. Y. Hodgetts, H.-L. Du, T. D. Nguyen, D. MacFarlane and A. N. Simonov, ACS Catal., 2022, 12, 5231–5246 CrossRef CAS
- AIST: Integrated Spectral Database System of Organic Compounds, https://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi.
- L. Troisi, C. Granito, L. Ronzini, F. Rosato and V. Videtta, Tetrahedron Lett., 2010, 51, 5980–5983 CrossRef CAS
- B. Das, M. Krishnaiah, V. S. Reddy and K. Laxminarayana, Helv. Chim. Acta, 2007, 90, 2163–2166 CrossRef CAS
- R. Baati, A. Valleix, C. Mioskowski, D. K. Barma and J. R. Flack, Org. Lett., 2000, 2, 485–487 CrossRef CAS PubMed
- A. N. Dey and E. J. Rudd, J. Electrochem. Soc., 1974, 121, 1294 CrossRef CAS
- D. Aurbach, M. L. Daroux, P. W. Faguy and E. Yeager, J. Electrochem. Soc., 1988, 135, 1863–1871 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee03901j |
| This journal is © The Royal Society of Chemistry 2023 |